

Metaplastic breast carcinoma (MBC) and uterine carcinosarcoma (UCS) are rare aggressive cancers, characterized by an admixture of adenocarcinoma and areas displaying mesenchymal/sarcomatoid differentiation. In this study, we investigate whether MBCs and UCSs harbor similar patterns of genetic alterations and mutational signatures using whole‐exome sequencing data. In addition, we examine whether the different histologic components of MBCs and UCSs are clonally related.

Keywords: breast cancer, carcinosarcoma, homologous recombination DNA repair, metaplastic, uterine cancer, whole‐exome sequencing
Abstract
Metaplastic breast carcinoma (MBC) and uterine carcinosarcoma (UCS) are rare aggressive cancers, characterized by an admixture of adenocarcinoma and areas displaying mesenchymal/sarcomatoid differentiation. We sought to define whether MBCs and UCSs harbor similar patterns of genetic alterations, and whether the different histologic components of MBCs and UCSs are clonally related. Whole‐exome sequencing (WES) data from MBCs (n = 35) and UCSs (n = 57, The Cancer Genome Atlas) were reanalyzed to define somatic genetic alterations, altered signaling pathways, mutational signatures, and genomic features of homologous recombination DNA repair deficiency (HRD). In addition, the carcinomatous and sarcomatous components of an additional cohort of MBCs (n = 11) and UCSs (n = 6) were microdissected separately and subjected to WES, and their clonal relatedness was assessed. MBCs and UCSs harbored recurrent genetic alterations affecting TP53, PIK3CA, and PTEN, similar patterns of gene copy number alterations, and an enrichment in alterations affecting the epithelial‐to‐mesenchymal transition (EMT)‐related Wnt and Notch signaling pathways. Differences were observed, however, including a significantly higher prevalence of FAT3 and FAT1 somatic mutations in MBCs compared to UCSs, and conversely, UCSs significantly more frequently harbored somatic mutations affecting FBXW7 and PPP2R1A as well as HER2 amplification than MBCs. Genomic features of HRD and biallelic alterations affecting bona fide HRD‐related genes were found to be more prevalent in MBCs than in UCSs. The distinct histologic components of MBCs and UCSs were clonally related in all cases, with the sarcoma component likely stemming from a minor subclone of the carcinoma component in the samples with interpretable chronology of clonal evolution. Despite the similar histologic features and pathways affected by genetic alterations, UCSs differ from MBCs on the basis of FBXW7 and PPP2R1A mutations, HER2 amplification, and lack of HRD, supporting the notion that these entities are more than mere phenocopies of the same tumor type in different anatomical sites.
Abbreviations
- CCF
cancer cell fraction
- CI
clonality index
- CNA
copy number alteration
- EMT
epithelial‐to‐mesenchymal transition
- HRD
homologous recombination DNA repair deficiency
- LST
large‐scale state transition
- MBC
metaplastic breast carcinoma
- NtAI
numerical telomeric allelic imbalance
- SNV
single nucleotide variant
- TCGA
The Cancer Genome Atlas
- TNBC
triple‐negative breast cancer
- UCS
uterine carcinosarcoma
- WES
whole‐exome sequencing
1. Introduction
Metaplastic breast carcinoma (MBC) is a rare histologic form of breast cancer, usually of triple‐negative phenotype, accounting for 0.2–5% of breast cancers [1]. These tumors are characterized by differentiation of malignant epithelium into squamous and/or mesenchymal elements, such as spindle, chondroid, osseous, or rhabdoid cells [1]. We and others have previously shown that the histologic heterogeneity of MBCs is paralleled by heterogeneity at the genomic and transcriptomic levels [2, 3, 4, 5, 6], and provided evidence that the histologically distinct components of each MBC are almost uniformly clonally related [7, 8, 9, 10, 11]. Given their clonal nature, it has been postulated that in MBCs with mesenchymal elements, epithelial‐to‐mesenchymal transition (EMT) may play a role in the development of the metaplastic component [12, 13, 14]. Consistent with this notion, these tumors are often transcriptomically classified as claudin‐low or mesenchymal‐like subtypes [4, 5], and display overexpression of cellular migration‐ and extracellular matrix formation‐related genes [4, 5, 15]. At the genetic level, MBCs are characterized by recurrent mutations affecting TP53 and genes related to the PI3K/AKT/mTOR, MAPK, Wnt, and Notch signaling pathways [2, 4, 8, 16, 17].
Uterine carcinosarcomas (UCSs), previously called malignant mixed Müllerian tumors (MMMTs), are rare aggressive tumors composed of high‐grade malignant carcinomatous and sarcomatous/mesenchymal elements, accounting for < 5% of uterine cancers and 15% of uterine cancer‐associated deaths in the United States [18, 19, 20]. The mesenchymal component of UCSs may consist of histologic elements native to the uterus (homologous) or of heterologous components, such as rhabdomyosarcoma or chondrosarcoma [20]. A number of studies have been conducted to identify pathways altered in UCSs and potential therapeutic targets. Akin to MBCs, UCSs have been found to harbor recurrent mutations affecting TP53 and the PI3K/AKT/mTOR signaling pathway [19] as well as mutations in chromatin remodeling and core histone genes [21, 22, 23].
Given their histologic similarities, we posited that MBCs and UCSs would constitute counterparts of the same tumor type in different anatomical sites, that these tumors would be underpinned by similar genetic alterations, and that the distinct histologic components of individual MBCs and UCSs would be clonally related. Hence, in this study, we have reanalyzed data previously published by our team [2] and The Cancer Genome Atlas (TCGA) [19] to compare the repertoire of genetic alterations and pathways altered in MBCs and UCSs. We have also sequenced independently microdissected carcinomatous and sarcomatous components of 11 MBCs and 6 UCSs to infer bioinformatically the chronology of the development of the histologically distinct components within MBCs and UCSs.
2. Materials and methods
2.1. Cases
This study was approved by the Institutional Review Boards (IRBs) of the authors' institutions, and patient consents were obtained as required by the protocols approved by the IRBs. This study is in compliance with the Declaration of Helsinki. Formalin‐fixed paraffin‐embedded (FFPE) tissue blocks of 11 MBCs (including 10 cases reported in Ng et al. [2]) and 6 fresh‐frozen (FF) UCSs were retrieved (Table S1). All 11 microdissected MBCs displayed a triple‐negative [i.e., estrogen receptor (ER), progesterone receptor (PR), and HER2‐negative] phenotype (Table S1). Samples were deidentified prior to analysis. All cases were reviewed by pathologists with expertise and experience in breast pathology (FCG, AV‐S, and JSR‐F) and gynecologic pathology (RM and RAS). The histologically distinct components of these MBCs and UCSs (i.e., epithelial and mesenchymal) were independently microdissected and subjected to whole‐exome sequencing (WES; Fig. 1).
Fig. 1.
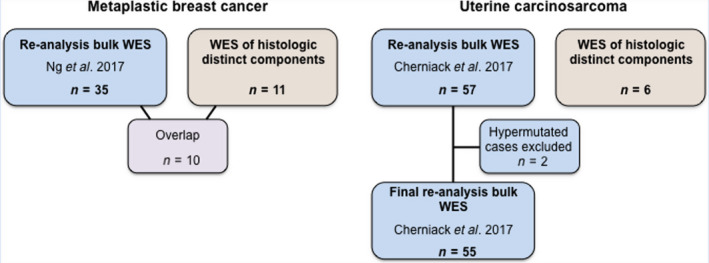
Schematic representation of the metaplastic breast carcinomas and uterine carcinosarcomas included in this study. WES data of metaplastic breast cancers (MBCs; n = 35) from Ng et al. [2] and uterine carcinosarcomas (UCSs; n = 55, n = 2 hypermutated cases were excluded) from Cherniack et al./The Cancer Genome Atlas [19] were reanalyzed. In addition, the epithelial and mesenchymal components of 11 MBCs, of which 10 overlapped with those from Ng et al. [2], and of 6 UCSs were separately microdissected and subjected to WES.
In addition, we retrieved the whole‐exome raw sequence data (BAM files) from 35 MBCs included in our previous study by Ng et al. [2] as well as from 57 UCSs reported by Cherniack et al. [19] (TCGA) from the NCI GDC portal (https://portal.gdc.cancer.gov/). Clinicopathologic characteristics of the MBCs and UCSs were retrieved from our previous study [2], Cherniack et al. [19], and from patient medical records (Table S1).
2.2. DNA extraction
For the microdissection of the distinct epithelial and mesenchymal components of a given MBC or UCS, we performed high‐molecular‐weight cytokeratin immunohistochemistry of the first and last sections as a guide. The distinct epithelial and mesenchymal components of MBCs (n = 11) were microdissected from 8‐µm‐thick representative FFPE sections with a needle under a stereomicroscope, as previously described [5, 24, 25]. For UCSs (n = 6), the distinct epithelial and mesenchymal components were microdissected from 8‐µm‐thick representative FF sections either with a needle under a stereomicroscope [5, 24, 25] or using laser microdissection, as previously described by our group [26], on a Leica LMD 6500 System (Leica Microsystems Inc., Buffalo Grove, IL, USA). All microdissections were performed by pathologists (FCG, ADP, NF, CM, and JSR‐F). Genomic DNA was extracted from tumor and matched normal tissues using the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD, USA), according to manufacturer's instructions, and quantified using the Qubit Fluorometer (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
2.3. WES and targeted amplicon resequencing
DNA samples from the histologically distinct components of each of the 11 MBCs and six UCSs and their respective normal samples were subjected to WES at MSKCC's Integrated Genomics Operation (IGO) following validated protocols [27, 28]. Sequencing data of the separately microdissected components, as well as of the 35 bulk MBCs [2] and 57 UCSs (TCGA) [19], were analyzed as previously described [27, 28] (Data S1). Mutation hotspots were determined according to Chang et al. [29]. A somatic mutation was defined as pathogenic if it affected a mutational hotspot or was deleterious/loss‐of‐function (in the case of tumor suppressor genes). For the 17 multicomponent cases, in addition to the identification of somatic mutations in individual samples, any mutation detected in one of the histological component of a given case was subsequently queried in the other matched component using samtools mpileup (v1.2) [30]. Allele‐specific copy number alterations (CNAs), tumor purity, and ploidy were obtained from the WES data using facets [31]. The cancer cell fractions (CCFs) of putative somatic mutations identified were computed using absolute (v1.0.6) [32], as previously described [27, 28]. The fraction of the genome altered was computed from the CNAs obtained from facets (Data S1).
Selected putative somatic mutations identified in MBCs (n = 11) and UCSs (n = 5) by WES were subjected to orthogonal validation using a custom‐designed AmpliSeq panel, as previously described [33]; 98% (444/451) of the nonsynonymous mutations subjected to orthogonal resequencing were validated in the MBCs and 97% (60/62) of the private nonsynonymous mutations were validated in the UCSs (Table S2). Somatic mutations that were not validated were excluded from the downstream analyses.
2.4. Microsatellite instability
The presence of microsatellite instability (MSI) was defined in the paired tumor‐normal WES data using MSIsensor [34], as previously described [35], and samples with MSIsensor scores ≥ 3.5 were considered MSI high [34].
2.5. Homologous recombination DNA repair defects and mutational signatures
Homologous recombination DNA repair deficiency (HRD) was assessed by defining large‐scale state transition (LST) scores, numerical telomeric allelic imbalance (NtAI) scores, mutational signature 3, microhomology‐mediated deletions, and the length of small deletions. LSTs and NtAIs were computed from the results of facets using the WES data according to Popova et al. [36] and Birkbak et al. [37], with a cutoff of ≥ 15 for LST high, as previously described [38]. Mutational signatures were inferred from both synonymous and nonsynonymous somatic mutations in MBCs and UCSs with at least 20 single nucleotide variants (SNVs) using DeconstructSigs [39] with default parameters, based on the set of mutational signatures represented in version 2 as part of COSMIC release v89 (https://cancer.sanger.ac.uk/cosmic/signatures_v2), as previously described [35]. All but two MBCs (META55 and META61) had ≥ 20 SNVs for mutational signature analysis, and the dominant mutational signature of a given case is reported. Given that tumors with deficient HR have been shown to have an enrichment for small deletions ≥ 5bp and microhomology‐mediated deletions [40, 41], the length of small deletions and the presence of deletions with microhomology were assessed in the samples analyzed, as described [35, 40, 41]. Finally, raw methylation data (Illumina Infinium MethylationEPIC BeadChips) from all 57 UCSs from TCGA [19] were retrieved from the TCGA NCI GDC portal (https://portal.gdc.cancer.gov/) and analyzed as previously described [42], and the methylation status of the promoter regions of RAD51C and BRCA1 in the UCSs was assessed.
2.6. Clonal relatedness
To infer the clonal relatedness between the histologically distinct components of each MBC (n = 11) and UCS (n = 6), we defined the ‘clonality index’ (CI) as the probability of two lesions sharing mutations not expected to have co‐occurred by chance based on a previously validated method [43] (Data S1).
2.7. Clonal decomposition
To define the clonal architecture and composition of the histologically distinct and independently microdissected components of the MBCs (n = 11) and UCSs (n = 6) included in this study, the somatic mutations identified were analyzed using pyclone [44]. Somatic mutations were excluded from the clonal decomposition analysis if they affected loci with (a) low total depth (< 20×) in the matched normal, (b) low total depth (< 50×) in any tumor component of a given case, (c) where the tumor variant allele fractions (VAFs) of both components of a given case were lower than five times the normal VAF, and (d) where the total tumor depth exceeds 1500× in any component of a given case. This usually corresponds to regions of the human genome with low‐sequence complexity (e.g., telomeres, centromeres, pseudogenes), which may lead to misaligned sequence reads and false‐positive mutations. Estimates of tumor purity and absolute copy numbers were obtained from the VAF of somatic mutations and Log2 ratios derived from WES data using absolute [32]. These were used as input for pyclone [44] with the beta‐binomial model, run through 20000 MCMC iterations with a burn‐in of 10000 iterations, total copy number prior, and a beta‐binomial precision value of 500, as previously described [43]. The resulting CCFs were used to categorize mutations as truncal or branch. Truncal mutations were defined as those displaying a modal clonal frequency/CCF in the clonally related mesenchymal and carcinoma components of a given case, whereas branch mutations were defined as all nontruncal mutations.
2.8. Pathway analyses
A DAVID pathway analysis was conducted based on genes affected by nonsynonymous somatic mutations, amplifications, or homozygous deletions [45]. Pathways found to be significantly enriched (P < 0.01) in MBCs or UCSs and previously curated and reported in Sanchez‐Vega et al. [46] were selected. The list of genes and interactions constituting the canonical versions of these pathways was retrieved from pathwaymapper [47].
2.9. Comparative and statistical analyses
For comparisons of MBCs and UCSs, hypermutated cases defined as those with ≥ 1000 somatic mutations were excluded [28]. Two of the 57 UCSs from TCGA but none of the MBCs were hypermutated. Comparisons of continuous and categorical variables were performed using the Mann–Whitney U and Fisher's exact tests, respectively, and adjusted for multiple testing using the false discovery rate (FDR), whenever appropriate. An FDR < 0.05 was considered statistically significant. All tests were two‐sided. Unless otherwise stated, all statistical analyses were performed using r/bioconductor (https://www.bioconductor.org/).
3. Results
3.1. Repertoire of somatic genetic alterations in MBCs and UCSs
Reanalysis of WES data from 35 MBCs reported in our previous study by Ng et al. [2] and of 55 nonhypermutated UCSs retrieved from TCGA [19] (Fig. 1) revealed that MBCs had a higher median number of somatic mutations and nonsynonymous somatic mutations than UCSs (MBCs: median of 2.9 (range 0.5–10) and 1.6 (range 0.25–5.4) of total and nonsynonymous somatic mutations per Mb, respectively; UCSs: median of 1.3 (range 0.7–7.9) and 0.8 (range 0.4–4.7) of total and nonsynonymous somatic mutations per Mb, respectively; P < 0.05; Mann–Whitney U‐test; Fig. 2A). Despite the higher mutational burden in MBCs, the repertoire of somatic mutations in MBCs and UCSs shared many similarities (Fig. 2B), including alterations affecting PIK3CA (29%, 10/35 MBCs vs 33%, 18/55 UCSs, P = 0.816, Fisher's exact test) and PTEN (14%, 5/35 MBCs vs 16%, 9/55 UCSs, P = 1, Fisher's exact test). Important differences were observed, however; MBCs more frequently displayed somatic mutations in FAT3 (26% vs 4%, P = 0.0028, Fisher's exact test), ABCA13 (14% vs 2%, P = 0.031, Fisher's exact test), FAT1, CHERP, and RYR1 (each, 11% vs 0%, P = 0.02; Fisher's exact test) than UCSs. Conversely, UCSs significantly more frequently harbored somatic mutations affecting FBXW7 (38% vs 0%, P < 0.01; Fisher's exact test) and PPP2R1A (27% vs 0%, P < 0.01; Fisher's exact test) than MBCs (Fig. 2B). In addition, although TP53 mutations were common in both tumor types, they were significantly more frequently found in UCSs than in MBCs (93% vs 69%; P = 0.004, Fisher's exact test).
Fig. 2.
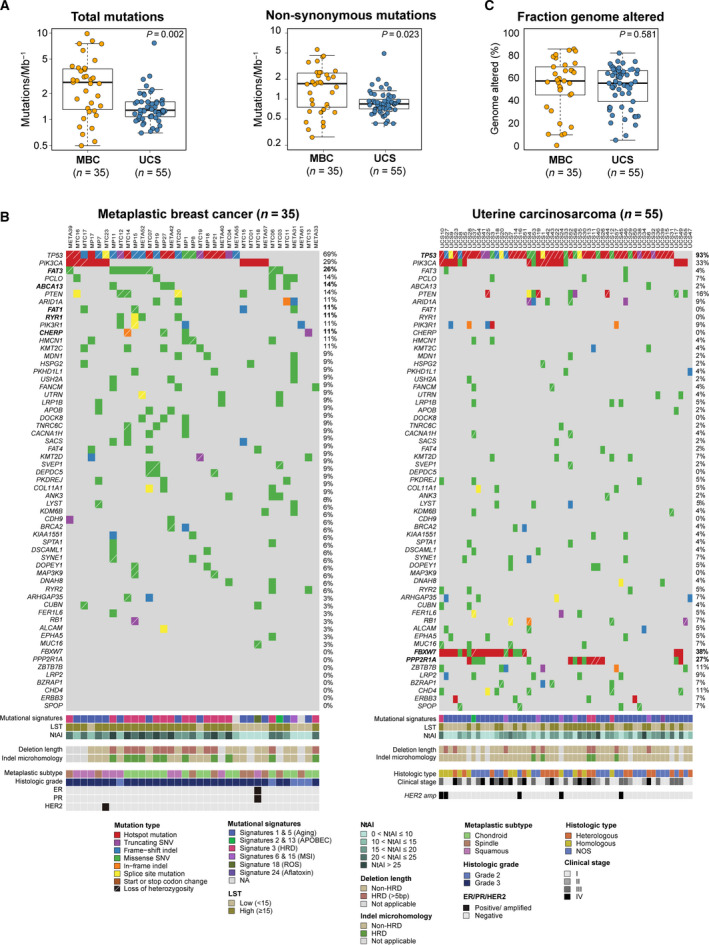
Repertoire of somatic mutations in metaplastic breast carcinomas and uterine carcinosarcomas. (A) Total number of somatic mutations and nonsynonymous somatic mutations per Mb in metaplastic breast cancers (MBCs) reanalyzed from Ng et al. [2] and uterine carcinosarcomas (UCSs) reanalyzed from The Cancer Genome Atlas (TCGA) [19]. Mann–Whitney U‐test employed. (B) Nonsynonymous somatic mutations identified in WES data from MBCs reanalyzed from Ng et al. [2], left, and UCSs reanalyzed from TCGA [19], right. Cases are shown in columns and genes in rows. Mutation types, mutational signatures, LSTs, NtAIs, small deletion length, small insertion and deletion (indel) microhomology, and clinicopathologic factors are color‐coded according to the legend. . (C) Fraction of the genome altered in MBCs reanalyzed from Ng et al. [2] and UCSs reanalyzed from TCGA [19]. Mann–Whitney U‐test employed.
MBCs and UCSs displayed high levels of copy number alterations (CNAs), with similar fractions of the genome altered (MBC, median 58%, range 0–81%; UCS, median 55%, range 5–82%, P = 0.581; Mann–Whitney U‐test; Fig. 2C, Fig. S1a). Recurrent CNAs included gains of 1q (43%, 15/35 MBCs; 28%, 16/55 UCSs), 3q (23%, 8/35 MBCs; 18%, 10/55 UCSs), and 8q (46%, 16/35 MBCs; 47%, 27/55 UCSs), and losses of 3p (20%, 7/35 MBCs; 19%, 11/55 UCSs) and 8p (34%, 12/35 MBCs; 37%, 21/55 UCSs), which did not differ between the MBCs and UCSs (all P > 0.05). In addition, we observed recurrent 8q12.1 and 8q24.1‐22 amplifications in both MBCs and UCSs, encompassing the CHCHD7 (9%, 3/35 MBCs; 9%, 5/55 UCSs), PLAG1 (9%, 3/35 MBCs; 9%, 5/55 UCSs), MYC (26%, 9/35 MBCs; 11%, 6/55 UCSs), and NDRG1 (23%, 8/35 MBCs; 9%, 5/55 UCSs) oncogenes (Fig. S1a). In contrast, however, while MBCs are generally of triple‐negative phenotype and only 1/35 (3%) of the MBCs studied here were HER2‐positive, 5/55 (9%) UCSs were found to display a HER2 amplification (P = 0.40, Fishers' exact test; Fig. 2B).
3.2. MBCs and UCSs harbor recurrent somatic genetic alterations affecting the p53, PI3K, Wnt, and Notch pathways
Given the similarities in the repertoire of somatic genetic alterations detected in MBCs and UCSs, we sought to compare the signaling pathways targeted by somatic genetic alterations in these tumors. A pathway analysis based on the somatic mutations and CNAs revealed an enrichment of genetic alterations targeting the canonical p53, PI3K/AKT/mTOR, Wnt, and Notch pathways, as defined by Sanchez‐Vega et al. [46], in both MBCs and UCSs (Fig. 3, Table S3); however, the target genes in these pathways varied according to the cancer type. The most frequently affected genes of the p53 signaling pathway were TP53 and MDM2/4 in both MBCs and UCSs (Fig. 3A); however, CDKN2A alterations were solely found in MBCs (14% MBCs vs 0% UCSs, P = 0.007, Fisher's exact test). Although PIK3CA (29% MBCs and 33% UCSs), PTEN (17% MBCs and 16% UCSs), and PIK3R1 (11% MBCs and 9% UCSs; all P > 0.05, Fisher's exact test) were PI3K signaling pathway components frequently affected by somatic mutations or CNAs in both MBCs and UCSs, other genes of the PI3K pathway such as PPP2R1A (27% UCSs vs 0% MBCs, P < 0.001; Fisher's exact test) and AKT2 (7% UCSs vs 0% MBCs, P = 0.154; Fisher's exact test) were affected exclusively in UCSs, whereas genetic alterations affecting AKT3 (9% MBCs vs 0% UCSs, P = 0.055; Fisher's exact test) and INPP4B (3% MBCs vs 0% UCSs, P = 0.389; Fisher's exact test) were uniquely found in MBCs (Fig. 3B, Fig. S1b).
Fig. 3.
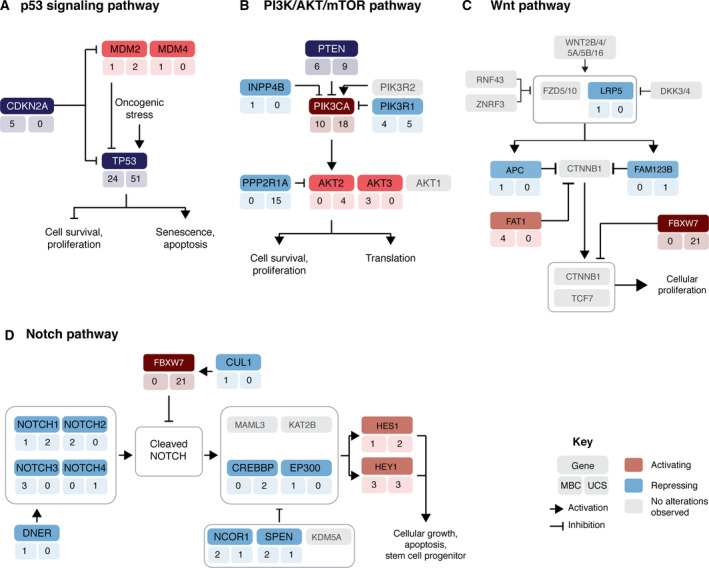
Metaplastic breast carcinomas and uterine carcinosarcomas harbor genetic alterations affecting similar signaling pathways. Frequency of activating (red) or loss‐of‐function (blue) somatic genetic alterations affecting genes in the canonical (A) p53, (B) PI3K/AKT/mTOR, (C) Wnt, and (D) Notch signaling pathways. The number of metaplastic breast cancers (MBCs, left) and uterine carcinosarcomas (UCSs, right) harboring a given somatic mutations or gene copy number alterations is depicted under the gene name. Pathways found to be significantly enriched (P < 0.01) in MCBs or UCSs and previously reported in Sanchez‐Vega et al. [46] are shown.
Several lines of evidence suggest that epithelial‐to‐mesenchymal transition (EMT)‐related processes might underpin MBCs and UCSs [2, 4, 12, 13, 14, 19, 48, 49]. Our analyses revealed that 43% (15/35) of MBCs and 53% (29/55) of UCSs harbored somatic genetic alterations affecting at least one gene of the canonical Wnt signaling pathway, of which 73% (11/15) of MBCs and 79% (23/29) of UCSs had at least one pathogenic mutation, amplification, or homozygous deletion (Fig. 3C). The Wnt pathway genes most frequently affected by somatic mutations or CNAs among MBCs and UCSs were ARID1A (11% MBCs vs 9% UCSs, P = 0.731, Fisher's exact test) and MYC (26% MBCs vs 11% UCSs, P = 0.08; Fisher's exact test). Importantly, however, genetic alterations affecting FBXW7 were found exclusively in UCSs (38% UCSs vs 0% MBCs, P < 0.01; Fisher's exact test), whereas FAT1 (11% MBCs vs 0% UCSs, P = 0.02; Fisher's exact test) and APC (3% MBCs vs 0% UCSs, P = 0.389; Fisher's exact test) were altered in MBCs but not in UCSs (Fig. 3C, Fig. S1b). Likewise, 43% (15/35) of MBCs and 56% (31/55) of UCSs harbored somatic genetic alterations affecting at least one gene of the canonical Notch signaling pathway, of which 73% (11/15) of MBCs and 81% (25/31) of UCSs were affected by at least one pathogenic mutation, amplification, or homozygous deletion (Fig. 3D). The genes of the Notch signaling pathway most frequently affected by genetic alterations in MBCs and UCSs were HEY1 (9% MBCs vs 5% UCSs), NOTCH1 (3% MBCs vs 4% UCSs), and HES1 (3% MBCs vs 4% UCSs; all P > 0.05, Fisher's exact test). Mutations affecting NOTCH2 (6%), NOTCH3 (9%), DNER (3%), EP300 (3%), and CUL1 (3%) were found in MBCs, whereas NOTCH4 (2%) alterations were only detected in UCSs (Fig. 3D, Fig. S1b).
3.3. MBCs more frequently display genomic features consistent with HRD than UCSs
MBCs have been reported to display frequent homologous recombination DNA repair (HRD) defects [2]. Hence, we sought to investigate whether the UCSs studied here would display similar genomic features suggestive of HRD or other biological processes that would result in genetic instability. Our analyses revealed the presence of a dominant mutational signature 3 associated with HRD in 45% (15/33) of MBCs. In contrast, only 7% of UCSs (4/55) displayed a dominant signature 3 (P < 0.001; Fisher's exact test). Instead, the majority (80%; 44/55) of UCSs displayed a dominant signature 1 or signature 5 [35], which have been ascribed to aging [50], compared to 42% (14/33) of MBCs (P < 0.001; Fisher's exact test; Figs 2A and 4A). Consistent with these findings, the median LST scores (24 vs 13, P < 0.002, Mann–Whitney U‐test), NtAI scores (21 vs 16, P = 0.029, Mann–Whitney U‐test), and deletion length of ≥ 5 bp (P = 0.008, Mann–Whitney U‐test) in MBCs were statistically significantly higher than those in UCSs (Fig. 4B–D). All MBCs (15/33) with a dominant mutational signature 3 displayed other genomic features suggestive of HRD, such as high LST scores (> 15), NtAI scores > 16, average small deletion length ≥ 5 bp, and deletions with microhomology in 100% (15/15), 80% (12/15), and 73% (11/15) of cases, respectively (Fig. 2B). The four UCSs displaying a dominant mutation signature 3 also had high LST scores, with two of them being associated with long deletions as well as deletions with microhomology (Fig. 2B).
Fig. 4.
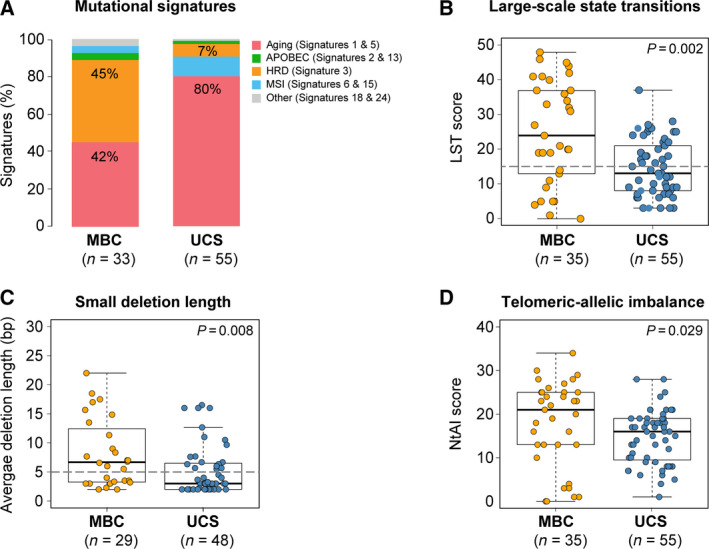
Genomic features of homologous recombination repair deficiency in metaplastic breast carcinomas and uterine carcinosarcomas. (A) Mutational signatures in metaplastic breast cancers (MBCs) from Ng et al. [2] and nonhypermutated uterine carcinosarcomas (UCSs) from TCGA [19] identified using DeconstructSigs [39]. Mutational signatures are color‐coded according to the legend and were only performed for samples ≥ 20 SNVs. (B) LST scores in MBCs from Ng et al. [2] and nonhypermutated UCSs from TCGA [19]. The gray line depicts the cutoff for LST high (≥ 15) [36]. (C) Small deletion length in MBCs from Ng et al. [2] and nonhypermutated UCSs from TCGA [19] according to Alexandrov et al. [40], which in HRD‐defective tumors has been found to be ≥ 5 nucleotides (gray line). (D) NtAI score in MBCs from Ng et al. [2] and nonhypermutated UCSs from TCGA [19] according to Morganella et al. [41]. Mann–Whitney U‐test was performed for comparisons in (B), (C), and (D). MBC, metaplastic breast cancer.
We next sought to identify the underlying genetic basis for HRD in the 45% of MBCs and 7% of UCSs displaying genomic features suggestive of HRD. Our analyses revealed that of the 15 MBCs with genomic features suggestive of HRD, 9 demonstrated biallelic inactivation of HRD‐related genes [38, 51]. Eight MBCs harbored germline mutations associated either with loss‐of‐heterozygosity or a second somatic mutation (BRCA1, n = 6; BRCA2, n = 1; and RBBP8, n = 1), and one MBC displayed a BRCA2 homozygous deletion (Table S4). None of the MBCs with a dominant aging‐related mutational signature were found to harbor biallelic genetic alterations in HRD‐related genes. Of the four UCSs displaying genomic features of HRD, UCS11 and UCS12 were found to harbor homozygous deletions in USP11 and FANCA, respectively (Table S4). In addition, analysis of the promoters of BRCA1 and RAD51C, whose methylation has been shown to be associated with HRD in breast and ovarian cancer [51], revealed that UCS10 and UCS12 displayed RAD51C promoter hypermethylation.
3.4. The epithelial and mesenchymal components of MBCs and UCSs are clonally related
There are multiple lines of evidence to support the contention that the different histologic components of MBCs and UCSs are clonally related [3, 7, 8, 9, 10, 19, 52], but there is also evidence to suggest that in a small subset of MBCs, the histologically distinct components may be genetically independent and/or collision tumors (e.g., case 5 from Geyer et al. [9]).
To define whether the histologically distinct components of MBCs and UCSs would be clonally related, we applied a previously validated approach to define clonal relatedness between tumor samples [43] (Data S1) based on the somatic mutations present in the histologically distinct microdissected components from 11 MBCs and 6 UCSs. Of these 11 MBCs, 10 were subjected to bulk WES previously described in Ng et al. [2] and reanalyzed in this study (Fig. 1; Fig. S2). This analysis revealed that the epithelial and mesenchymal components of all MBCs and UCSs studied here were clonally related, formally corroborating the notion that in the vast majority of MBCs and UCSs, the histologically distinct components originate from the same clone (Fig. 5A; Table S5).
Fig. 5.
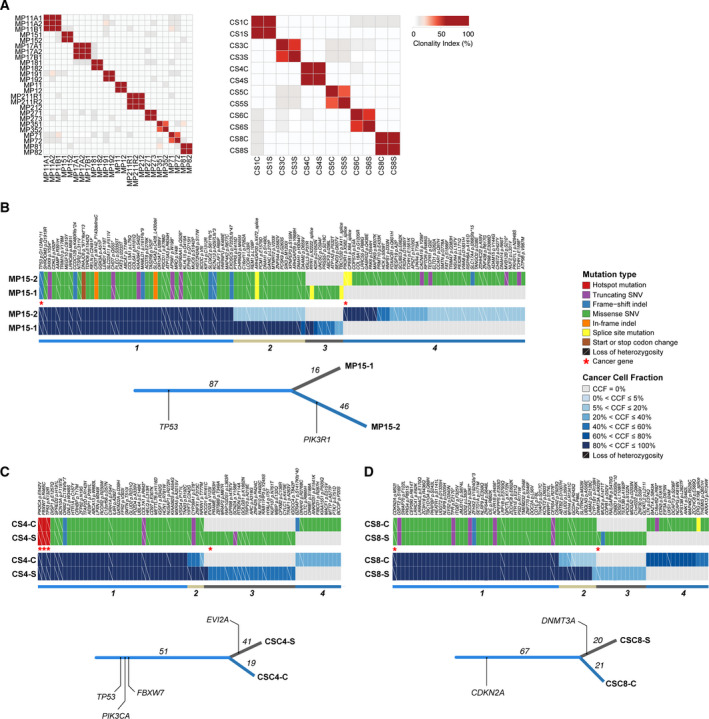
Clonal relatedness and decomposition of the epithelial and mesenchymal components of metaplastic breast carcinomas and uterine carcinosarcomas. (A) Clonality index of the epithelial and mesenchymal components of metaplastic breast cancers (MBCs, left) and of the epithelial and mesenchymal components of uterine carcinosarcomas (UCSs, right) subjected to WES based on somatic mutations. The histologic components are clonally related in all cases. (B) Cancer cell fractions (CCFs) of the somatic mutations identified in the epithelial and mesenchymal histologic components by WES in the metaplastic breast carcinoma MP15, (C) in the UCS CS4, and (D) UCS CS8. Mutations are grouped by their CCF as inferred by pyclone [44]. Cluster memberships are depicted below the heatmaps, and the corresponding phylogenetic trees are displayed. The length of the trunk and branches represent the number of shared and private somatic mutations identified in the different histologic components.
Given that in all MBCs and UCSs analyzed here, the histologically distinct components were clonally related and that, as a group, MBCs and UCSs were found to harbor genetic alterations affecting genes related to EMT, we posited that the mesenchymal component would stem from the epithelial component. Clonal decomposition using pyclone [44] revealed that in MBC15, a minor subclone of the ductal component became dominant in the mesenchymal (chondroid) component (Fig. 5B), consistent with the notion that in this case, the chondroid component originated from a minor subclone of the epithelial (i.e., ductal) component. Likewise, clonal decomposition of the six UCSs revealed evidence of clonal selection in CS4 and CS8 (Fig. 5C,D), in which the sarcoma component appeared to have stemmed from a minor subclone of the carcinoma. In the remaining MBCs and UCSs, the chronology of the development of the different components could not be inferred based on the sequencing results, given that no clonal enrichment in the carcinomatous or sarcomatous component was observed on the basis of mutations affecting protein‐coding genes and/or CNAs (Fig. S3a,b). No differences in the mutational signatures were observed between the two distinct histologic components in any given case (Table S5).
Among the truncal mutations across all 11 MBCs and 6 UCSs analyzed, TP53 somatic mutations were found to be clonal and truncal in all but 3 UCSs. In addition, UCS6 harbored a TP53 homozygous deletion (data not shown). These findings are supportive of the role of TP53 mutations as early drivers in the development of these cancers. No gene was found to be recurrently exclusively mutated in either the epithelial or mesenchymal components of the MBCs and UCSs analyzed (Fig. 5, Fig. S3a,b), suggesting that alterations other than somatic mutations or gene CNAs (e.g., epigenetic changes, somatic genetic alterations affecting regulatory elements) may account for the histologic diversity characteristic of these cancers.
4. Discussion
Here, we demonstrate that MBCs and UCSs harbor recurrent genetic alterations affecting TP53, PIK3CA, and PTEN, consistent with prior studies [2, 5, 14, 17, 19], and that these tumors display overall similar patterns of gene CNAs. Despite differences in the repertoire of somatic mutations observed between MBCs and UCSs, our analyses revealed an enrichment of genetic alterations affecting genes of the Wnt and Notch signaling pathways, which play pivotal roles in EMT [53, 54]. In fact, several of the genetic alterations that were distinct between MBCs and UCSs affected the same pathway (e.g., such as FAT1 and FBXW7, which were restricted to MBCs and UCSs, respectively, but signal through the Wnt pathway). In addition, we have also provided evidence that the histologically distinct components of MBC and UCS analyzed here were clonally related and that the mesenchymal components likely stemmed from the epithelial component in cases where the chronology of the development of the components could be inferred. Given that these tumors display recurrent alterations affecting Wnt, Notch, and other EMT‐related pathways, one could posit that EMT may play a role in the development of the histologic diversity that characterizes MBCs and UCSs.
Despite the molecular similarities, in particular the high frequency of TP53 mutations and high levels of chromosomal instability found between MBCs and UCSs, important differences were observed. In the datasets analyzed, MBC patients (median age 53, range 34–82) were significantly younger at diagnosis than UCS patients (median age 68, range 51–90; P < 0.0001, Mann–Whitney U‐test, Fig. S2c), which is consistent with the reported ages of diagnosis of MBCs and UCSs [11, 55]. Although MBCs were diagnosed at younger ages, we observed that 42% of cases had a dominant aging‐related mutational signature, akin to common‐type triple‐negative breast cancers [56, 57], and genomic features of HRD were present in 45% of the MBCs analyzed; conversely, only 7% of the UCSs were found to have HRD features, and 80% of the UCSs harbored dominant mutational signatures related to aging (i.e., mutational signatures 1 and 5). We further demonstrate that, in agreement with previous observations by our group [38] and others [51], biallelic alterations affecting canonical homologous recombination DNA repair‐related genes were the likely cause of HRD in the majority of MBCs and UCSs analyzed here. Furthermore, we identified RAD51C promoter hypermethylation in UCSs displaying HRD features (Table S4). Intriguingly, despite the evidence of HRD in MBCs, and unlike other forms of triple‐negative breast cancers, they appear to be resistant to conventional genotoxic chemotherapy [58]. As opposed to common forms of triple‐negative disease, where the rates of pathologic complete response (pCR) following neoadjuvant chemotherapy are > 40% [59], the reported pCR rates for MBCs range from 0% to 17% [11, 58, 60, 61]. Our findings may provide a molecular basis for this clinical conundrum, given that despite the high prevalence of HRD in MBCs, these tumors were found to display alterations in EMT‐related pathways, which may result in an intrinsic resistance to conventional genotoxic therapies [62]. Further studies are warranted to define the type of DNA repair defects playing a role in UCSs, given that based on WES analysis, the vast majority of UCSs displayed a dominant aging mutational signature, followed by HRD (i.e., signature 3 in 7% cases) and microsatellite instability (i.e., two cases excluded from the comparisons due to their hypermutated phenotype).
While genomic features of HRD were rare in UCSs, we did identify a subset harboring HER2 amplification. The addition of trastuzumab to chemotherapy is now recommended for the treatment of HER2‐positive advanced or recurrent uterine serous carcinomas [63]. Given the clinically aggressive behavior of UCSs and limited treatment options [64], exploring targeting HER2 in this subset of HER2‐amplified UCSs may be warranted [65]. Likewise, therapeutic strategies based on synthetic lethality to target tumors with FBXW7 mutations have emerged [66, 67]; given the relatively high frequency of FBXW7 mutations in UCSs (30%), further studies testing this potential treatment strategy might be entertained.
Consistent with previous work by Joneja et al. [68], we found TP53 (69% this study, 56% Joneja et al.) and PIK3CA (29% this study, 23% Joneja et al.) to be the most commonly mutated genes in MBCs. Previous work by Hayes et al. [69] reported on the presence of identical frameshift WISP3 somatic mutations in five out of 27 MBCs; however, none of the MBCs studied here had mutations affecting WISP3 even after inspection and manual curation of the sequencing results. Furthermore, Krings and Chen [70] demonstrated that 25% of MBCs harbored TERT promoter mutations. TERT promoter mutations could not be investigated in this series as they are not included in the genomic footprint of the targeted WES panel utilized in this study. Further studies are required to confirm the frequency of TERT promoter mutations in this rare type of breast cancer.
Our clonal decomposition analysis revealed that the epithelial and mesenchymal components of MBCs and UCSs are clonally related and display marked genetic heterogeneity. We observed that the mesenchymal component of at least a subset of MBCs and UCSs stemmed from a subclone of the epithelial component, following a clonal selection evolutionary pattern. Nonetheless, in the majority of MCBs and UCSs analyzed, the epithelial and mesenchymal components appear to have diverged somewhat early in the evolution of the tumors. It is possible that the different histologic components of these tumors evolved from a common histologic precursor and acquired either genetic alterations affecting genes other than protein‐coding genes or epigenetic alterations that resulted in the acquisition of mesenchymal features.
Our study has important limitations. WES was the basis for the genomic characterization of the MBCs and UCSs and their microdissected histologically distinct components analyzed here. Although orthogonal high‐depth validation of the mutations employed for clonal decomposition was performed, WES data do not allow for the characterization of mutations affecting noncoding regulatory elements and structural variants. In addition, given the greater accuracy of whole‐genome sequencing (WGS) for the detection of HRD and its causes, the potential for the identification of defects in other DNA repair mechanisms, and the greater data density for clonal decomposition analyses, further WGS studies of larger series of these tumors are warranted. Finally, we cannot rule out FFPE‐based sequencing artifacts in the subset of FFPE MBCs analyzed; however, no biallelic genetic alterations in HRD‐related genes were identified in MBCs with a dominant aging‐related mutational signature, and no enrichment in aging‐related mutational signatures in FFPE vs fresh‐frozen MBCs was found.
5. Conclusions
Here, we demonstrate that MBCs and UCSs harbor recurrent somatic genetic alterations affecting TP53 and genes related to the PI3K, Wnt, and Notch pathways. The histologically distinct components present in MBCs and UCSs were found to be clonally related, and, at least in a subset of cases, the mesenchymal component likely originated from the epithelial component. Despite some differences in terms of specific genetic alterations between MBCs and UCSs, the pathways targeted by these alterations are remarkably similar in these tumors. Genomic features of HRD were found to be significantly more prevalent in MBCs than in UCSs, whereas known therapeutic targets, such as HER2 gene amplification and FBXW7 mutations, were found to be significantly more frequent in UCSs than MBCs. Hence, despite the histologic similarities and similar pathways being affected by somatic genetic alterations, MBCs and UCSs are more than mere phenocopies of the same tumors in different anatomical sites.
Conflict of interest
JS Reis‐Filho is a consultant of Paige.AI, REPARE Therapeutics and Goldman Sachs, a member of the Board of Directors of Grupo Oncoclinicas, a member of the scientific advisory board of Volition RX, Paige.AI, and REPARE Therapeutics, and an ad hoc member of the advisory boards of Roche Tissue Diagnostics, Novartis, Roche, Genentech, and InVicro, all outside the submitted work. NR Abu‐Rustum reports institutional grants from Stryker/Novadaq, Olympus, and GRAIL, outside the submitted work. The remaining authors have no conflicts of interest to declare.
Author contributions
BW and JSR‐F conceived the study. FCG, FP, RM, EB, HYW, RAS, AV‐S, and JSR‐F performed pathology review. FCG, FP, SP, ADP, NF, CM, and JSR‐F performed microdissection and DNA extraction. LF, ADCP, and DNB performed bioinformatics analysis. LM, LF, ADCP, DNB, FCG, NRA‐R, RM, EB, HYW, LN, RAS, AV‐S, JSR‐F, and BW interpreted data. LM, LF, ADCP, DNB, FP, JSR‐F, and BW wrote the first draft of the manuscript, which was read and approved by all authors.
Supporting information
Data S1. Supplementary Methods.
Fig. S1. Copy number alterations and somatic mutations affecting selected signaling pathways in metaplastic breast cancers and uterine carcinosarcomas.
Fig. S2. Representative micrographs of the histologic components of the metaplastic breast cancers subjected to bulk whole‐exome sequencing as well as to whole‐exome sequencing of the distinct microdissected components.
Fig. S3. Clonal decomposition of the epithelial and mesenchymal components of the metaplastic breast carcinomas and uterine carcinosarcomas.
Table S1. Clinico‐pathologic information of the 35 metaplastic breast cancers reanalyzed from Ng et al (Clin Cancer Res 2017), 57 uterine carcinosarcomas from Cherniack et al (TCGA, Cancer Cell 2017), 11 metaplastic breast cancers with separately analyzed histologic components (this study), and 6 uterine carcinosarcomas with separately analyzed histologic components (this study).
Table S2. Non‐synonymous somatic mutations identified in the epithelial and mesenchymal components of 11 metaplastic breast cancers and 6 uterine carcinosarcomas subjected to whole‐exome sequencing.
Table S3. DAVID pathway analysis in 35 metaplastic breast cancers from Ng et al. (Clin Cancer Res 2017) and 55 non‐hypermutated uterine carcinosarcomas from Cherniack et al (TCGA, 2017).
Table S4. Genetic alterations affecting homologous recombination genes in metaplastic breast carcinomas and uterine carcinosarcomas.
Table S5. Number of shared and unique mutations, mutational signatures and clonal relatedness index of the histologically distinct components of 11 metaplastic breast cancers and 6 uterine carcinosarcomas subjected to whole‐exome sequencing.
Acknowledgements
This study was funded by the Breast Cancer Research Foundation. SP is funded by the Swiss National Foundation (Ambizione PZ00P3_168165) and BW in part by Cycle for Survival and Stand Up To Cancer grants. The research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Lea A. Moukarzel, Lorenzo Ferrando, Arnaud Da Cruz Paula, and David N. Brown contributed equally to this study
Contributor Information
Jorge S. Reis‐Filho, Email: reisfilj@mskcc.org.
Britta Weigelt, Email: weigeltb@mskcc.org.
References
- 1. Weigelt B, Eberle C, Cowell CF, Ng CK & Reis‐Filho JS (2014) Metaplastic breast carcinoma: more than a special type. Nat Rev Cancer 14, 147–148. [DOI] [PubMed] [Google Scholar]
- 2. Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, Lim RS, Natrajan R, Riaz N, Mariani O et al. (2017) The landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin Cancer Res 23, 3859–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cooper CL, Karim RZ, Selinger C, Carmalt H, Lee CS & O'Toole SA (2013) Molecular alterations in metaplastic breast carcinoma. J Clin Pathol 66, 522–528. [DOI] [PubMed] [Google Scholar]
- 4. Piscuoglio S, Ng CKY, Geyer FC, Burke KA, Cowell CF, Martelotto LG, Natrajan R, Popova T, Maher CA, Lim RS et al. (2017) Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. NPJ Breast Cancer 3, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weigelt B, Ng CK, Shen R, Popova T, Schizas M, Natrajan R, Mariani O, Stern MH, Norton L, Vincent‐Salomon A et al. (2015) Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected]. Mod Pathol 28, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gonzalez‐Martinez S, Perez‐Mies B, Carretero‐Barrio I, Palacios‐Berraquero ML, Perez‐Garcia J, Cortes J & Palacios J (2020) Molecular features of metaplastic breast carcinoma: an infrequent subtype of triple negative breast carcinoma. Cancers (Basel) 12, 1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lien HC, Lin CW, Mao TL, Kuo SH, Hsiao CH & Huang CS (2004) p53 overexpression and mutation in metaplastic carcinoma of the breast: genetic evidence for a monoclonal origin of both the carcinomatous and the heterogeneous sarcomatous components. J Pathol 204, 131–139. [DOI] [PubMed] [Google Scholar]
- 8. Avigdor BE, Beierl K, Gocke CD, Zabransky DJ, Cravero K, Kyker‐Snowman K, Button B, Chu D, Croessmann S, Cochran RL et al. (2017) Whole‐exome sequencing of metaplastic breast carcinoma indicates monoclonality with associated ductal carcinoma component. Clin Cancer Res 23, 4875–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, Savage K, Mackay A, Ashworth A & Reis‐Filho JS (2010) Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol 220, 562–573. [DOI] [PubMed] [Google Scholar]
- 10. Thompson L, Chang B & Barsky SH (1996) Monoclonal origins of malignant mixed tumors (carcinosarcomas). Evidence for a divergent histogenesis. Am J Surg Pathol 20, 277–285. [DOI] [PubMed] [Google Scholar]
- 11. Cimino‐Mathews A, Verma S, Figueroa‐Magalhaes MC, Jeter SC, Zhang Z, Argani P, Stearns V & Connolly RM (2016) A clinicopathologic analysis of 45 patients with metaplastic breast carcinoma. Am J Clin Pathol 145, 365–372. [DOI] [PubMed] [Google Scholar]
- 12. Lien HC, Hsiao YH, Lin YS, Yao YT, Juan HF, Kuo WH, Hung MC, Chang KJ & Hsieh FJ (2007) Molecular signatures of metaplastic carcinoma of the breast by large‐scale transcriptional profiling: identification of genes potentially related to epithelial‐mesenchymal transition. Oncogene 26, 7859–7871. [DOI] [PubMed] [Google Scholar]
- 13. Sarrio D, Rodriguez‐Pinilla SM, Hardisson D, Cano A, Moreno‐Bueno G & Palacios J (2008) Epithelial‐mesenchymal transition in breast cancer relates to the basal‐like phenotype. Cancer Res 68, 989–997. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y, Toy KA & Kleer CG (2012) Metaplastic breast carcinomas are enriched in markers of tumor‐initiating cells and epithelial to mesenchymal transition. Mod Pathol 25, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weigelt B, Kreike B & Reis‐Filho JS (2009) Metaplastic breast carcinomas are basal‐like breast cancers: a genomic profiling analysis. Breast Cancer Res Treat 117, 273–280. [DOI] [PubMed] [Google Scholar]
- 16. Hennessy BT, Gonzalez‐Angulo AM, Stemke‐Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C et al. (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial‐to‐mesenchymal transition and stem cell characteristics. Cancer Res 69, 4116–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCart Reed AE, Kalaw E, Nones K, Bettington M, Lim M, Bennett J, Johnstone K, Kutasovic JR, Saunus JM, Kazakoff S et al. (2019) Phenotypic and molecular dissection of metaplastic breast cancer and the prognostic implications. J Pathol 247, 214–227. [DOI] [PubMed] [Google Scholar]
- 18. McCluggage WG (2002) Malignant biphasic uterine tumours: carcinosarcomas or metaplastic carcinomas? J Clin Pathol 55, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR et al. (2017) Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 31, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wells M, Oliva E, Palacios J & Prat J (2014) Mixed epithelial and mesenchymal tumours. In WHO Classification of Tumours of Female Reproductive Organs, 4th edn. (Kurman RJ, Carcangiu M‐L, Herrington CS & Young RH, eds), pp. 148–151. International Agency for Research on Cancer, Lyon. [Google Scholar]
- 21. Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M et al. (2014) Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 5, 5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao S, Bellone S, Lopez S, Thakral D, Schwab C, English DP, Black J, Cocco E, Choi J, Zammataro L et al. (2016) Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial‐mesenchymal transition. Proc Natl Acad Sci USA 113, 12238–12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leskela S, Perez‐Mies B, Rosa‐Rosa JM, Cristobal E, Biscuola M, Palacios‐Berraquero ML, Ong S, Matias‐Guiu Guia X & Palacios J (2019) Molecular basis of tumor heterogeneity in endometrial carcinosarcoma. Cancers (Basel) 11, 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ng CK, Martelotto LG, Gauthier A, Wen HC, Piscuoglio S, Lim RS, Cowell CF, Wilkerson PM, Wai P, Rodrigues DN et al. (2015) Intra‐tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol 16, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geyer FC, Lambros MB, Natrajan R, Mehta R, Mackay A, Savage K, Parry S, Ashworth A, Badve S & Reis‐Filho JS (2010) Genomic and immunohistochemical analysis of adenosquamous carcinoma of the breast. Mod Pathol 23, 951–960. [DOI] [PubMed] [Google Scholar]
- 26. Piscuoglio S, Burke KA, Ng CK, Papanastasiou AD, Geyer FC, Macedo GS, Martelotto LG, de Bruijn I, De Filippo MR, Schultheis AM et al. (2016) Uterine adenosarcomas are mesenchymal neoplasms. J Pathol 238, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geyer FC, Li A, Papanastasiou AD, Smith A, Selenica P, Burke KA, Edelweiss M, Wen HC, Piscuoglio S, Schultheis AM et al. (2018) Recurrent hotspot mutations in HRAS Q61 and PI3K‐AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun 9, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pareja F, Brandes AH, Basili T, Selenica P, Geyer FC, Fan D, Da Cruz PA, Kumar R, Brown DN, Gularte‐Merida R et al. (2018) Loss‐of‐function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat Commun 9, 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, Chakravarty D, Phillips S, Kandoth C, Penson A et al. (2018) Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 8, 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R & Genome Project Data Processing S (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen R & Seshan VE (2016) FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi W, Ng CKY, Lim RS, Jiang T, Kumar S, Li X, Wali VB, Piscuoglio S, Gerstein MB, Chagpar AB et al. (2018) Reliability of whole‐exome sequencing for assessing intratumor genetic heterogeneity. Cell Rep 25, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC & Ding L (2014) MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics 30, 1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ashley CW, Da Cruz PA, Kumar R, Mandelker D, Pei X, Riaz N, Reis‐Filho JS & Weigelt B (2019) Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol Oncol 152, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Popova T, Manie E, Rieunier G, Caux‐Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal‐Zafrani B, Bollet M, Longy M et al. (2012) Ploidy and large‐scale genomic instability consistently identify basal‐like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72, 5454–5462. [DOI] [PubMed] [Google Scholar]
- 37. Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman‐Colin C, Li Y, Greene‐Colozzi A, Iglehart JD et al. (2012) Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA‐damaging agents. Cancer Discov 2, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, Norton L, Weigelt B, Powell SN & Reis‐Filho JS (2017) Pan‐cancer analysis of bi‐allelic alterations in homologous recombination DNA repair genes. Nat Commun 8, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenthal R, McGranahan N, Herrero J, Taylor BS & Swanton C (2016) DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Boot A, Covington KR, Gordenin DA, Bergstrom E, Lopez‐Bigas N et al. (2018) The repertoire of mutational signatures in human cancer. bioRxiv. [Google Scholar]
- 41. Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M et al. (2016) The topography of mutational processes in breast cancer genomes. Nat Commun 7, 11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiang S, Weigelt B, Wen HC, Pareja F, Raghavendra A, Martelotto LG, Burke KA, Basili T, Li A, Geyer FC et al. (2016) IDH2 mutations define a unique subtype of breast cancer with altered nuclear polarity. Cancer Res 76, 7118–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, Ng CKY, Carniello JVS, Towers R, Giri DD et al. (2019) Lobular carcinomas in situ display intralesion genetic heterogeneity and clonal evolution in the progression to invasive lobular carcinoma. Clin Cancer Res 25, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard‐Cote A & Shah SP (2014) PyClone: statistical inference of clonal population structure in cancer. Nat Methods 11, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. da Huang W, Sherman BT & Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 46. Sanchez‐Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S et al. (2018) Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337. e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bahceci I, Dogrusoz U, La KC, Babur O, Gao J & Schultz N (2017) PathwayMapper: a collaborative visual web editor for cancer pathways and genomic data. Bioinformatics 33, 2238–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romero‐Perez L, Castilla MA, Lopez‐Garcia MA, Diaz‐Martin J, Biscuola M, Ramiro‐Fuentes S, Oliva E, Matias‐Guiu X, Prat J, Cano A et al. (2013) Molecular events in endometrial carcinosarcomas and the role of high mobility group AT‐hook 2 in endometrial carcinogenesis. Hum Pathol 44, 244–254. [DOI] [PubMed] [Google Scholar]
- 49. Castilla MA, Moreno‐Bueno G, Romero‐Perez L, Van De Vijver K, Biscuola M, Lopez‐Garcia MA, Prat J, Matias‐Guiu X, Cano A, Oliva E et al. (2011) Micro‐RNA signature of the epithelial‐mesenchymal transition in endometrial carcinosarcoma. J Pathol 223, 72–80. [DOI] [PubMed] [Google Scholar]
- 50. Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen‐Dale AL et al. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW et al. (2017) A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wada H, Enomoto T, Fujita M, Yoshino K, Nakashima R, Kurachi H, Haba T, Wakasa K, Shroyer KR, Tsujimoto M et al. (1997) Molecular evidence that most but not all carcinosarcomas of the uterus are combination tumors. Cancer Res 57, 5379–5385. [PubMed] [Google Scholar]
- 53. Zhan T, Rindtorff N & Boutros M (2017) Wnt signaling in cancer. Oncogene 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pastushenko I & Blanpain C (2018) EMT transition states during tumor progression and metastasis. Trends Cell Biol 29, 212–226. [DOI] [PubMed] [Google Scholar]
- 55. Arend R, Doneza JA & Wright JD (2011) Uterine carcinosarcoma. Curr Opin Oncol 23, 531–536. [DOI] [PubMed] [Google Scholar]
- 56. Nik‐Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC et al. (2016) Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, Xiao Y, Yu KD, Liu YR, Yu Y et al. (2019) Genomic and transcriptomic landscape of triple‐negative breast cancers: subtypes and treatment strategies. Cancer Cell 35, 428–440. e425. [DOI] [PubMed] [Google Scholar]
- 58. Hennessy BT, Giordano S, Broglio K, Duan Z, Trent J, Buchholz TA, Babiera G, Hortobagyi GN & Valero V (2006) Biphasic metaplastic sarcomatoid carcinoma of the breast. Ann Oncol 17, 605–613. [DOI] [PubMed] [Google Scholar]
- 59. Foulkes WD, Smith IE & Reis‐Filho JS (2010) Triple‐negative breast cancer. N Engl J Med 363, 1938–1948. [DOI] [PubMed] [Google Scholar]
- 60. Han M, Salamat A, Zhu L, Zhang H, Clark BZ, Dabbs DJ, Carter GJ, Brufsky AM, Jankowitz RC, Puhalla SL et al. (2019) Metaplastic breast carcinoma: a clinical‐pathologic study of 97 cases with subset analysis of response to neoadjuvant chemotherapy. Mod Pathol 32, 807–816. [DOI] [PubMed] [Google Scholar]
- 61. Nagao T, Kinoshita T, Hojo T, Tsuda H, Tamura K & Fujiwara Y (2012) The differences in the histological types of breast cancer and the response to neoadjuvant chemotherapy: the relationship between the outcome and the clinicopathological characteristics. Breast 21, 289–295. [DOI] [PubMed] [Google Scholar]
- 62. May CD, Sphyris N, Evans KW, Werden SJ, Guo W & Mani SA (2011) Epithelial‐mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 13, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, Chambers SK, Secord AA, Havrilesky L, O'Malley DM et al. (2018) Randomized phase II trial of carboplatin‐paclitaxel versus carboplatin‐paclitaxel‐trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J Clin Oncol 36, 2044–2051. [DOI] [PubMed] [Google Scholar]
- 64. Denschlag D & Ulrich UA (2018) Uterine carcinosarcomas – diagnosis and management. Oncol Res Treat 41, 675–679. [DOI] [PubMed] [Google Scholar]
- 65. Rottmann D, Snir OL, Wu X, Wong S, Hui P, Santin AD & Buza N (2020) HER2 testing of gynecologic carcinosarcomas: tumor stratification for potential targeted therapy. Mod Pathol 33, 118–127. [DOI] [PubMed] [Google Scholar]
- 66. Hinze L, Pfirrmann M, Karim S, Degar J, McGuckin C, Vinjamur D, Sacher J, Stevenson KE, Neuberg DS, Orellana E et al. (2019) Synthetic lethality of Wnt pathway activation and asparaginase in drug‐resistant acute leukemias. Cancer Cell 35, 664–676. e667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dolly SO, Gurden MD, Drosopoulos K, Clarke P, de Bono J, Kaye S, Workman P & Linardopoulos S (2017) RNAi screen reveals synthetic lethality between cyclin G‐associated kinase and FBXW7 by inducing aberrant mitoses. Br J Cancer 117, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Joneja U, Vranic S, Swensen J, Feldman R, Chen W, Kimbrough J, Xiao N, Reddy S, Palazzo J & Gatalica Z (2017) Comprehensive profiling of metaplastic breast carcinomas reveals frequent overexpression of programmed death‐ligand 1. J Clin Pathol 70, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hayes MJ, Thomas D, Emmons A, Giordano TJ & Kleer CG (2008) Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res 14, 4038–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krings G & Chen YY (2018) Genomic profiling of metaplastic breast carcinomas reveals genetic heterogeneity and relationship to ductal carcinoma. Mod Pathol 31, 1661–1674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary Methods.
Fig. S1. Copy number alterations and somatic mutations affecting selected signaling pathways in metaplastic breast cancers and uterine carcinosarcomas.
Fig. S2. Representative micrographs of the histologic components of the metaplastic breast cancers subjected to bulk whole‐exome sequencing as well as to whole‐exome sequencing of the distinct microdissected components.
Fig. S3. Clonal decomposition of the epithelial and mesenchymal components of the metaplastic breast carcinomas and uterine carcinosarcomas.
Table S1. Clinico‐pathologic information of the 35 metaplastic breast cancers reanalyzed from Ng et al (Clin Cancer Res 2017), 57 uterine carcinosarcomas from Cherniack et al (TCGA, Cancer Cell 2017), 11 metaplastic breast cancers with separately analyzed histologic components (this study), and 6 uterine carcinosarcomas with separately analyzed histologic components (this study).
Table S2. Non‐synonymous somatic mutations identified in the epithelial and mesenchymal components of 11 metaplastic breast cancers and 6 uterine carcinosarcomas subjected to whole‐exome sequencing.
Table S3. DAVID pathway analysis in 35 metaplastic breast cancers from Ng et al. (Clin Cancer Res 2017) and 55 non‐hypermutated uterine carcinosarcomas from Cherniack et al (TCGA, 2017).
Table S4. Genetic alterations affecting homologous recombination genes in metaplastic breast carcinomas and uterine carcinosarcomas.
Table S5. Number of shared and unique mutations, mutational signatures and clonal relatedness index of the histologically distinct components of 11 metaplastic breast cancers and 6 uterine carcinosarcomas subjected to whole‐exome sequencing.