

Summary
Conventional chromatin immunoprecipitation (ChIP) includes many steps that need to be optimized. Here, we have described a protocol of fractionation-assisted native ChIP (fanChIP) which combines subfractionation and native ChIP to purify protein/chromatin complexes applicable for analyses of both protein-protein and protein-DNA interactions within a short period of time. fanChIP is advantageous as subcellular fractionation removes chromatin-unbound materials before immunoprecipitation, and the chromatin fragmentation by micrococcal nuclease (MNase) in a mild condition enables one-step purification of intact protein/chromatin complexes.
For complete details on the use and execution of this protocol, please refer to Miyamoto et al. (2020).
Subject areas: Genomics, Molecular Biology, Chromatin immunoprecipitation (ChIP)
Graphical abstract
Highlights
-
•
A quick and easy protocol to perform chromatin immunoprecipitation
-
•
A method to capture protein/chromatin complexes in one-step purification
-
•
A crosslinking-free and sonication-free ChIP method
-
•
The purified complexes can be analyzed by deep sequencing and mass spectrometry
Conventional chromatin immunoprecipitation (ChIP) includes many steps that need to be optimized. Here, we have described a protocol of fractionation-assisted native ChIP (fanChIP) which combines subfractionation and native ChIP to purify protein/chromatin complexes applicable for analyses of both protein-protein and protein-DNA interactions within a short period of time. fanChIP is advantageous as subcellular fractionation removes chromatin-unbound materials before immunoprecipitation, and the chromatin fragmentation by micrococcal nuclease (MNase) in a mild condition enables one-step purification of intact protein/chromatin complexes.
Before you begin
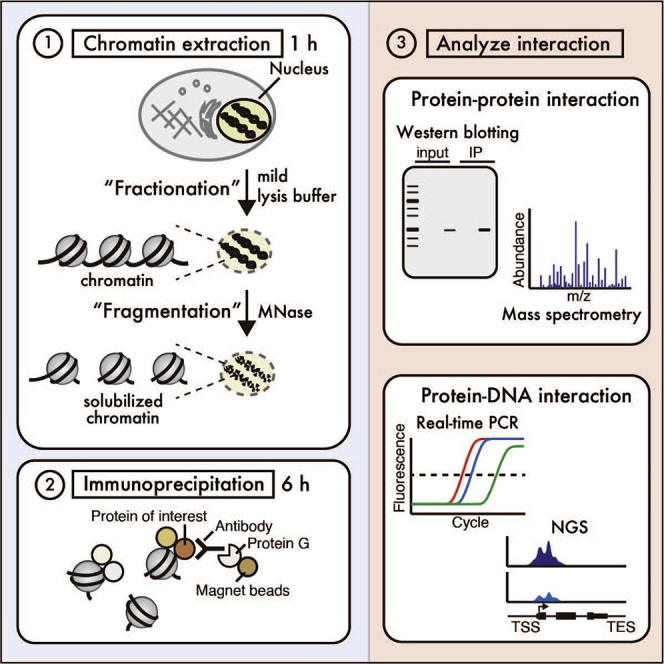
Mutations of epigenetic factors lead to misregulation of histone modification, DNA methylation, and chromatin remodeling, which have been implicated in the initiation and progression of many types of cancers (Dawson and Kouzarides, 2012). To understand the oncogenic mechanisms of causative epigenetic factors and identify therapeutic targets, it is important to understand their physiological functions and alterations in cancer cells. This is often achieved by identifying the protein-protein interactions on chromatin and its genomic localization in normal and cancer cells. We have developed a comprehensive method to capture protein-protein/DNA interactions on chromatin termed fanChIP (Okuda et al., 2014), which combines subcellular fractionation and the native ChIP method previously established by O'Neill and Turner (O'Neill and Turner, 2003). During our investigation of the molecular mechanism of acute leukemia caused by rearrangements of the Mixed Lineage Leukemia gene (MLL, also known as KMT2A), the fanChIP method has enabled characterization of leukemogenic transcriptional machinery nucleated by the MLL fusion protein and its genomic localization, thus accelerating to uncover the mechanism of MLL-associated leukemia and develop therapeutic strategies (Okuda et al., 2017, Okuda et al., 2014, Okuda et al., 2015, Miyamoto et al., 2020). This paper describes the detailed procedure for fanChIP, in which samples can be prepared for various detection systems such as western blotting (WB), Mass spectrometry, real-time quantitative PCR (qPCR) and next generation sequencing (NGS) with fewer processes compared to conventional ChIP (Figure 1).
Figure 1.

Scheme of fan-ChIP
In the fanChIP method, cultured cells are first lysed with mild lysis buffer to remove chromatin-unbound soluble fractions. Following this, the chromatin-containing fraction is treated with MNase to digest the chromatin to appropriate sizes compatible for IP. The fragmented chromatin is immunoprecipitated with an antibody against the protein of interest (POI). The immunoprecipitated materials can be immediately used for the analyses of protein-protein and protein-DNA interactions.
Compared to conventional ChIP, fanChIP offers several advantages, which are largely due to omission of the crosslinking step. (1) The immunoprecipitated sample is available for subsequent analyses within a day. (2) Masking of epitopes by crosslinking, which is often problematic, can be avoided. (3) The obtained chromatin can be applied for the detection of protein-protein interactions as well as protein-DNA interactions. The chromatin/protein complexes remain relatively intact in a mild extraction condition close to the physiological condition and can be purified by one-step affinity purification as described in our previous publications (Miyamoto et al., 2020, Okuda et al., 2015). (4) Importantly, combining subcellular fractionation and MNase digestion removes non-chromatin-bound proteins, which potentially eliminates false-positive interactions.
CRITICAL: Successful IP largely depends on the quality of the antibody. We recommend testing multiple IP-grade or ChIP-grade antibodies for individual target proteins. Checking the specificities and sensitivities of antibodies in advance by fanChIP-WB with appropriate positive controls (e.g,. HEK293T cells overexpressing the POI) is recommended to select better antibodies. By using knock-out cells for fanChIP-qPCR, one can confirm that the ChIP signal is due to the POI. We have used MLL-knockout HEK293T cells to ensure the specificity of anti-MLL antibody (Miyamoto et al., 2020). If the knock-out cells are unavailable, we recommend to perform ChIP-seq analyses with multiple antibodies for the same protein complex, which should show similar ChIP profiles.
Preparation of working solutions
Timing: 2–3 h
Prepare working solutions according to “working solution” in materials and equipment. We usually prepare the working solutions well ahead of the “Cell Harvesting” step, and store them at −20°C until use. They can also be prepared just before use and stored at 4°C.
CSK buffer, Complete
MNase buffer, Complete
Lysis buffer, Complete
fanChIP wash buffer
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| MNase | Sigma-Aldrich | Cat#N3755-200U |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#11873580001 |
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat#10003D |
| NaOH | FUJIFILM Wako | Cat#198-13765 |
| Tris base | Nacalai Tesque | Cat#35434-21 |
| PIPES | Nacalai Tesque | Cat#P6757 |
| EDTA·2Na·2H2O | Nacalai Tesque | Cat#15130-95 |
| EGTA | Nacalai Tesque | Cat#08907-42 |
| NaCl | Nacalai Tesque | Cat#31320-05 |
| MgCl2·6H2O | FUJIFILM Wako | Cat#132-00175 |
| CaCl2·2H2O | FUJIFILM Wako | Cat#031-00435 |
| NaHCO3 | FUJIFILM Wako | Cat#195-14515 |
| Na2HPO4·12H2O | FUJIFILM Wako | Cat#196-02835 |
| NaH2PO4·2H2O | Nacalai Tesque | Cat#31717-25 |
| KH2PO4 | FUJIFILM Wako | Cat#164-22635 |
| Sodium butyrate | FUJIFILM Wako | Cat#193-01522 |
| NP-40 (IGEPAL CA-630) | Sigma-Aldrich | Cat#I8896 |
| Triton X-100 | Sigma-Aldrich | Cat#93426 |
| SDS | Nacalai Tesque | Cat#08933-05 |
| DTT | FUJIFILM Wako | Cat#040-29223 |
| Sodium pyrophosphate | FUJIFILM Wako | Cat#195-03025 |
| NaF | FUJIFILM Wako | Cat#192-01972 |
| Sucrose | Nacalai Tesque | Cat#30406-25 |
| Phenol/chloroform/isoamyl alcohol (25:24:1) | NIPPON Genetics | Cat#311-90151 |
| Glycogen solution (20 mg/mL) | Nacalai Tesque | Cat#17110-11 |
| Sodium acetate (NaOAc, 3 M, pH 5.2) | NIPPON Genetics | Cat#316-90081 |
| Ethanol (99.5%) (referred to as 100% EtOH in this paper) | FUJIFILM Wako | Cat#057-00456 |
| MIDORIGreen Advance | NIPPON Genetics | Cat#NE-MG04 |
| Glycerol | FUJIFILM Wako | Cat#072-04945 |
| Lipofectamine 2000 transfection reagent | Thermo Fisher | Cat# 11668027 |
| Other | ||
| DynaMagTM-2 magnet | Thermo Fisher Scientific | Cat#12321D |
| Ice rack for 1.5 mL tubes | AXEL | Cat#2-4620-01 |
| UVP incubator (for 37°C incubation) | Analytik Jena | Cat#849-30005-5 |
| MACSmixTM Tube Rotator (for MNase digestion) | Miltenyi Biotec | Cat#130-090-753 |
| Rotator RT-50 (for IP) | TAITEC | Cat#0000165-000 |
| Cell lifter | Corning | Cat#3008 |
| NanoDrop One | Thermo Fisher Scientific | Cat#ND-ONE-W |
| Mupid-2plus | Takara Bio | Cat#M-2P |
| FAS-V (for detection of DNA electrophoresis) | NIPPON Genetics | Cat#FAS5 |
| Safe-Lock Tubes, 1.5 mL | Eppendorf | Cat#0030120086 |
| Safe-Lock Tubes, 2 mL | Eppendorf | Cat#0030120094 |
| 20 μL low binding Barrier Pipette Tips | Sorenson | Cat#2937T |
| 200 μL low binding Barrier Pipette Tips | Sorenson | Cat#2938T |
| 1000 μL low binding Barrier Pipette Tips | Sorenson | Cat#2955T |
| Freezer (−20°C) | Asahi Life Science | Cat#ALS-665HC |
| Deep freezer (−80°C) | Nihon Freezer | Cat#CLN-72UW |
| Centrifuge | TOMY | Cat#KITMAN-T24 |
| Vortex mixer | M&S Instruments Inc. | Cat#SI-0286 |
| Autoclave | TOMY | Cat#LSX-300 |
| Axygen® 200 μL Pipet Tips, Wide-Bore | Corning | Cat#T-205-WB-C-R-S |
| Axygen® 1000 μL Pipet Tips, Wide-Bore | Corning | Cat#T-1005-WB-C-R-S |
| Experimental models: cell lines | ||
| Adherent cells: e.g., HEK 293T | (Yokoyama et al., 2005) | Authenticated by JRCB cell bank in 2019 |
| Suspension cells: e.g., HB1119 | (Tkachuk et al., 1992) | N/A |
| Recombinant DNA (mammalian expression vectors) | ||
| Retroviral expression vectors: e.g., pMSCV-neo vectors | Clontech | Cat#634401 |
| Transient expression vectors: e.g., pCMV5 vectors | (Yokoyama et al., 2005) | N/A |
Note: Materials and devices required for downstream analyses (WB, qPCR, etc.) are not described here. Please check their availability before initiating the fanChIP procedure.
Materials and equipment
Stock solutions
The stock solutions are required for preparing working solutions. Unless otherwise stated, they can be stored at 23°C–25°C.
| 10 M NaOH | 20g NaOH, fill up to 50 mL with double distilled H2O (ddH2O) |
| 2 M Tris-HCl, pH 7.5 | 242.2 g Tris base, ∼120 mL conc. HCl to pH 7.5, fill up to 1 L with ddH2O |
| 0.5 M Tris-HCl, pH 6.8 | 30.3 g Tris base, ∼40 mL 6 N HCl to pH 6.8, fill up to 500 mL with ddH2O) |
| 0.5 M PIPES, pH 6.8 | 15.1 g PIPES, 7–8 mL 10 N NaOH to pH 6.8, fill up to 100 mL with ddH2O |
| 0.5 M EDTA, pH 8.0 | 93.06 g EDTA·2Na·2H2O, 10 g NaOH to pH 8.0, fill up to 500 mL with ddH2O |
| 50 mM EGTA, pH 7.6 | 1.90 g EGTA, adjust pH with 10 M NaOH, fill up to 100 mL with ddH2O |
| 5 M NaCl | 292.2 g NaCl, fill up to 1 L with ddH2O |
| 1 M MgCl2 | 20.3 g MgCl2·6H2O, fill up to 100 mL with ddH2O |
| 1 M CaCl2 | 14.7 g CaCl2·2H2O, fill up to 100 mL with ddH2O |
| 1 M NaHCO3 | 8.40 g NaHCO3, fill up to 100 mL with ddH2O |
| 0.2 M Na2HPO4 | 71.63 g Na2HPO4·12H2O, fill up to 1000 mL with ddH2O |
| 0.2 M NaH2PO4 | 15.6 g NaH2PO4·2H2O, fill up to 500 mL with ddH2O |
| 0.2 M sodium phosphate buffer | 700 mL 0.2 M Na2HPO4, to pH 7.0 with 350–450 mL 0.2 M NaH2PO4 |
| 1 M sodium butyrate | 5.5 g sodium butyrate, fill up to 50 mL with ddH2O |
| 10% NP-40 | 10 mL NP-40 (IGEPAL CA630), 90 mL ddH2O, warm to 40°C∼50°C to dissolve NP-40 |
| 10% Triton X-100 | 10 mL Triton X-100, 90 mL ddH2O, warm to 40°C∼50°C to dissolve Triton X-100 |
| 10% SDS | 10 g SDS, fill up to 100 mL with ddH2O |
| 1 M DTT | 1.542 g DTT, fill up to 10 mL with ddH2O, store in aliquots at −20°C for up to 1 year |
| 1% bromophenol blue (BPB) | 1 g BPB, fill up to 100 mL with ddH2O |
| 1% xylene cyanol FF | 1 g xylene cyanol FF, fill up to 100 mL with ddH2O |
| 1U/μL MNase | reconstitute lyophilized MNase with an MNase storage buffer, store at −20°C |
Note: Gradually add NaOH pellets to 20 mL (approx.) of ddH2O in a 100 mL glass beaker with continuous stirring. (Add NaOH pellets by small degrees as excess heat can be generated upon NaOH dissolution.)
Note: Add EDTA·2Na·2H2O powder in 400 mL (approx.) of ddH2O in a 500 mL glass beaker. Gradually add 10 M NaOH with stirring to dissolve the EDTA powder and adjust the pH.
Note: Wear gloves while handling NaOH as it is harmful to the skin. Handle SDS and DTT in a draft chamber as they are toxic on inhalation.
2 × CSK buffer
| Reagent | Final concentration | Stock concentration | Amount in 250 mL |
|---|---|---|---|
| PIPES pH 6.8 | 20 mM | 0.5 M | 10 mL |
| NaCl | 200 mM | 5 M | 10 mL |
| MgCl2 | 6 mM | 1 M | 1.5 mL |
| EGTA pH 7.6 | 2 mM | 50 mM | 10 mL |
| ddH2O | Up to 250 mL |
Store at −20°C for up to 1 year.
2 × MNase buffer
| Reagent | Final concentration | Stock concentration | Amount in 1 L |
|---|---|---|---|
| Tris-HCl pH 7.5 | 100 mM | 2 M | 50 mL |
| MgCl2 | 8 mM | 1 M | 8 mL |
| CaCl2 | 2 mM | 1 M | 2 mL |
| ddH2O | Up to 1 L |
Store at −20°C for up to 1 year.
Lysis buffer, -DTT
| Reagent | Final concentration | Stock concentration | Amount in 1 L |
|---|---|---|---|
| Sodium phosphate pH 7.0 | 20 mM | 0.2 M | 100 mL |
| Sodium pyrophosphate10·H2O | 30 mM | 13.38 g | |
| NaCl | 250 mM | 5 M | 50 mL |
| NaF | 10 mM | 0.42 g | |
| EDTA pH 8.0 | 5 mM | 0.5 M | 10 mL |
| Glycerol | 10% | 100 mL | |
| NP-40 | 0.1% | 10% | 10 mL |
| ddH2O | Up to 1 L |
Store at −20°C for up to 1 year.
MNase storage buffer
| Reagent | Final concentration | Stock concentration | Amount in 10 mL |
|---|---|---|---|
| Tris-HCl pH 7.5 | 50 mM | 2 M | 250 μL |
| DTT | 1 mM | 1 M | 10 μL |
| NaCl | 50 mM | 5 M | 100 μL |
| Glycerol | 50% | 5 mL | |
| ddH2O | Up to 10 mL |
Store at −20°C for up to 1 year.
Working solution
PBS, pH 7.4
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 137 mM | 8 g |
| KCl | 2.7 mM | 0.2 g |
| KH2PO4 | 1.4 mM | 0.2 g |
| Na2HPO4·12H2O | 8 mM | 2.9 g |
| ddH2O | Up to 1 L |
Autoclave and store at 23°C–25°C for up to 6 months
CSK buffer, complete
| Reagent | Final concentration | Stock concentration | Amount in 50 mL |
|---|---|---|---|
| 2 × CSK buffer | 1 × | 25 mL | |
| Sucrose | 0.3 M | 5.15 g | |
| Triton X-100 | 0.5% | 10% | 2.5 mL |
| DTT | 0.5 mM | 1 M | 25 μL |
| Sodium butyrate | 5 mM | 1 M | 250 μL |
| ddH2O | Up to 50 mL |
Add 1 tablet of cOmplete protease inhibitor cocktail EDTA, and store it at −20°C for up to 1 year.
MNase buffer, complete
| Reagent | Final concentration | Stock concentration | Amount in 50 mL |
|---|---|---|---|
| 2 × MNase buffer | 1 × | 25 mL | |
| Sucrose | 0.3 M | 5.15 g | |
| DTT | 0.5 mM | 1 M | 25 μL |
| Sodium butyrate | 5 mM | 1 M | 250 μL |
| ddH2O | Up to 50 mL |
Add 2 tablets of cOmplete protease inhibitor cocktail EDTA, and store it at −20°C for up to 1 year.
Lysis buffer, complete
| Reagent | Final concentration | Stock concentration | Amount in 50 mL |
|---|---|---|---|
| Lysis buffer | 50 mL | ||
| DTT | 0.5 mM | 1 M | 25 μL |
Add 1 tablet of cOmplete protease inhibitor cocktail EDTA, and store it at −20°C for up to 1 year.
fanChIP wash buffer, complete
| Reagent | Final concentration | Stock concentration | Amount in 50 mL |
|---|---|---|---|
| MNase buffer, complete | X 0.5 | 25 mL | |
| Lysis buffer, complete | X 0.5 | 25 mL |
Add 1 tablet of cOmplete protease inhibitor cocktail EDTA, and store it at −20°C for up to 1 year.
Elution buffer
| Reagent | Final concentration | Stock concentration | Amount in 100 mL |
|---|---|---|---|
| NaHCO3 | 50 mM | 1 M | 5 mL |
| SDS | 1% | 10% | 10 mL |
| ddH2O | Up to 100 mL |
Store at 23°C–25°C for up to 6 months
2 × sample buffer
| Reagent | Final concentration | Stock concentration | Amount in 100 mL |
|---|---|---|---|
| Tris-HCl pH 6.8 | 125 mM | 0.5 M | 25 mL |
| SDS | 4% | 10% | 40 mL |
| 2-mercaptoethanol (2-ME) | 10 mL | ||
| Bromophenol blue | 0.02% | 1% | 2 mL |
| ddH2O | 23 mL |
Store at 23°C–25°C for up to 6 months.
6 × loading buffer
| Reagent | Final concentration | Stock concentration | Amount in 10 mL |
|---|---|---|---|
| Glycerol | 30% (v/v) | 3 mL | |
| Xylene cyanol FF | 0.25% | 1% | 2.5 mL |
| Bromophenol blue | 0.25% | 1% | 2.5 mL |
| ddH2O | 2 mL |
Store at 23°C–25°C for up to 6 months
Note: Wear gloves while handling 2-ME in a draft chamber as it is toxic.
Step-by-step method details
The procedure is illustrated in Figure 1.
Cell culture
The standard number of cells required for adherent cells (e.g., HEK 293T) is 1 × 107 and for suspension cells (e.g., leukemia cell lines) to prepare 2 mL of chromatin is 2 × 107. For HEK 293T cells, this is usually equivalent to a confluent culture in a 10 cm dish. Below is an example to perform fanChIP-WB and fanChIP-qPCR using HEK 293T cells transiently expressing POI.
-
1.
Two days prior to “Cell Harvesting”, seed HEK 293T cells on a 10 cm dish to achieve 80% confluency on the next day. This is usually achieved by seeding at a density of approximately 25%
-
2.
One day before “Cell Harvesting”, transfect expression vector carrying the gene of interest according to manufacturer's protocol(e.g., Lipofectamine 2000 transfection reagent: https://www.thermofisher.com/jp/en/home/references/protocols/cell-culture/transfection-protocol/lipofectamine-2000.html). Change media after 6–8 h of transfection.
Preparation of buffers/equipment
-
3.
Thaw the required buffers in icy water and keep them on ice until use. Make sure that the PBS, CSK buffer, MNase buffer, and lysis buffer are ready at the beginning of “Cell Harvesting”.
-
4.
Place the rotator (for MNase digestion) in a 37°C incubator to prewarm it
-
5.
Set the centrifugation temperature to 4°C.
Cell harvesting
In this step, cells are harvested for chromatin preparation. The methods are described for 1 × 107 adherent cells (cultured in a 10 cm dish) and 2 × 107 suspension cells to prepare 2 mL of chromatin. The volumes of buffers should be scaled up or down according to the amount of the starting material.
Note: Before starting the procedure, ensure that all required solutions are prepared and stored at an appropriate temperature. Use safe-lock tubes throughout the procedure to avoid leakage during incubation and rotation.
-
6.Adherent cells (e.g., HEK 293T cells)
-
a.Remove media and add 1 mL of PBS.
-
b.Detach the cells from the dish using a cell lifter (if it is difficult to detach the cells using a cell lifter, trypsinize, wash with PBS, and follow the procedure described for suspension cells).
-
c.Dissociate clumped cells by pipetting with P1000 pipette.
-
d.Transfer the suspension to a 2 mL centrifuge tube.
-
e.Spin down (400 × g, 5 min, 4°C) and remove the supernatant.
-
f.Resuspend the cells with 1 mL of ice-cold PBS.
-
g.Transfer the tube to a cold ice rack.
-
a.
-
7.Suspension cells (e.g., HB1119 cells)
-
a.Count the number of cells.
-
b.Transfer 2 × 107 cells to a 50 mL centrifuge tube.
-
c.Centrifuge the tube (400 × g, 5 min, 4°C) and remove the supernatant.
-
d.Resuspend the cells in 10 mL PBS.
-
e.Centrifuge the tube (400 × g, 5 min, 4°C) and remove the supernatant.
-
f.Resuspend the cells in 1 mL of ice-cold PBS, and transfer them to a 2 mL centrifuge tube.
-
a.
Extraction of chromatin fraction
In this step, chromatin samples are prepared for subsequent IP. From this step to the “elution” step, perform all procedures on ice or at 4°C to avoid protein degradation. Centrifuge tubes should be kept on a cold ice rack for short intervals between each procedure (See the procedures illustrated in Figure 2).
-
8.
Centrifuge the tubes containing the cell suspension (400 × g, 5 min, 4°C) and remove the supernatant.
-
9.
Add 1 mL of cold CSK buffer, and dissociate clamped cells completely by pipetting. This step solubilizes the cell membrane, permeabilizes the nuclear membrane, and liberates chromatin-unbound proteins.
-
10.
Incubate the lysates for 5 min on ice.
-
11.
Centrifuge the tubes (400 × g, 5 min, 4°C) and remove the supernatant.
-
12.
Add 1 mL of MNase buffer and pipet 5–10 times to resuspend the nuclei.
-
13.
Add the predetermined amount of MNase to the tubes [It is necessary to identify the optimal conditions for digestion by testing various amounts of MNase and the incubation time in advance (the steps 22–25). We recommend starting with 1 unit of MNase for 5 min of incubation].
-
14.
Set the tubes on a rotator in a 37°C incubator.
-
15.
Incubate at 37°C with rotation for the planned time period.
Alternatives: If the rotator is unavailable, a 37°C incubator or thermal block can be used. In this case, the tubes should be mixed every 1–2 min by gently inversing the tubes.
-
16.
Remove the tubes from the rotator, and immediately place them on a cold ice rack.
-
17.
Next, add 40 μL of EDTA (0.5 M, pH 8.0) to the tubes, and mix by inverting to completely stop MNase digestion.
-
18.
Add 1 mL of lysis buffer, and mix by pipetting five times.
-
19.
Incubate on ice for 5 min to solubilize proteins.
-
20.
Centrifuge tubes at 17,700 × g (or at maximum speed of the centrifuge) to pellet the insoluble materials (5 min, 4°C).
-
21.
Transfer the supernatant to a new 2 mL centrifuge tube as the “Chromatin fraction”, and store it at −80°C or proceed to check chromatin fragmentation (the steps 22–25).
Pause point: If you stored the samples at −80°C, you can pause at this point. However, frequent freeze-thaw cycles should be avoided as they may disrupt the protein-protein/DNA interactions of interest. Consider aliquoting the sample in volumes of 400 μL (for fanChIP-qPCR/seq) or 1 mL (for fanChIP-WB).
Figure 2.

Schema for “extraction of chromatin fraction”
Numbers correspond to the steps mentioned in the step-by-step method details.
Figure 3.

Chromatin fragmentation pattern using MNase
“chromatin fractions” were obtained from HEK 293T cells with different MNase treatment conditions. Purified DNA from the chromatin was electrophoresed in a 2% agarose gel at 100 V for 40 min. DNA was visualized with Midori Green Advance DNA staining solution. “n” indicates mono-nucleosome.
.
Checking chromatin fragmentation
In this step, the degree of chromatin digestion is checked.
Note: While handling DNA, use filter tips to avoid cross contamination.
-
22.
Transfer 50 μL of “Chromatin fraction” to a new 1.5 mL microcentrifuge tube.
-
23.
Add 50 μL of phenol/chloroform/isoamyl alcohol (25:24:1, PCI) to the tube and vortex vigorously.
-
24.
Centrifuge tubes at 17,700 × g (or at maximum speed of centrifuge machine, 10 min, 25°C)
After centrifugation, three layers consisting of top clear, middle white, and bottom yellow layers can be observed. Transfer 10 μL of the top layer to 2 μL of 6 × loading dye and mix.
-
25.
Electrophorese DNA in a 2% agarose gel.
Immunoprecipitation (primary antibody)
In this step, a specific antibody against your POI is added to the chromatin sample to form chromatin/antibody complexes. If the chromatin samples are stored at −80°C, they should be thawed on a cold ice rack in advance. This will take 10–30 min depending on the volume of stored samples. Set a rotator for antibody incubation in a refrigerator.
-
26.
Prepare 1.5 mL centrifuge tubes as the “input” samples. (For qPCR/NGS samples, they will be used in the step 50 for DNA extraction). Typically, we mix 20 μL of the chromatin sample and 180 μL of elution buffer in a tube as an input sample for fanChIP-qPCR/seq while we mix 60 μL of the chromatin sample and 60 μL of 2 × sample buffer for fanChIP-WB (10% input). Store the samples at −20°C.
-
27.
Transfer 400 μL of chromatin sample to a new 1.5 mL centrifuge tube for “IP”. For the analysis of histone modifications, add 32 μL of 5 M NaCl to the tube (final NaCl concentration is approximately 550 mM). This high salt concentration induces dissociation of chromatin reader proteins and exposes the epitope.
-
28.
Add antibody to the “IP” tube. One microgram of antibody is the standard amount used for 400 μL of chromatin (final 2.5 μg/mL), but it should be empirically determined for your POI and antibody.
-
29.
Set the “IP” tube on a rotator placed in a refrigerator.
-
30.
Rotate for 5 h.
Note: Consider preparing a negative control. The use of normal IgG is one common method. If your POI is an exogenously expressed protein, the chromatin of untransfected cells can be a good negative control.
Note: You may use a bead-conjugated primary antibody (e.g., Anti-FLAG M2 Magnetic Beads) for IP-WB. In that case, skip the next step and proceed to “Elution”.
Immunoprecipitation (Dynabeads Protein G)
In this step, the Protein G beads are equilibrated and then added to the chromatin-antibody mix to purify the DNA/protein complex of your POI.
-
31.
Add 10 μL × (N + 1) [N is the number of samples] of Dynabeads Protein G suspension to a 1.5 mL centrifuge tube containing 1000 μL of fanChIP wash buffer (the “beads” tube).
-
32.
Place the “beads” tube on the DynaMag-2 Magnet tube holder, and wait until the beads are completely attached to the side of the magnet and the solution is cleared.
-
33.
Discard the cleared solution, and add 400 μL of fanChIP wash buffer to the “beads” tube.
-
34.
Resuspend the beads in the buffer by removing the upper plastic holder together with the tubes from the bottom magnet stand and inverting it repeatedly.
-
35.
Set the plastic holder on the magnet stand, and wait until the solution is clear.
-
36.
Repeat the steps 33–35 twice to equilibrate the beads.
-
37.
Discard the clear solution, and add 50 μL × (N + 1) of fanChIP wash buffer to the “beads” tube.
-
38.
Remove the “beads” tube from the plastic holder, and repeatedly invert the tube to suspend the beads.
-
39.
Place the “IP” tubes onto a cold ice rack from the rotator.
-
40.
Add 50 μL of the bead suspension to each “IP” tube.
-
41.
Set the tubes on a rotator, and rotate for another hour at 4°C.
Elution
In this step, the DNA/protein complexes of your POI attached to the beads are washed, and then eluted for further analyses.
-
42.
Place the “IP” tubes on the DynaMag-2 Magnet tube holder, and wash with 400 μL of fanChIP wash buffer five times as described in the steps 33–35.
-
43.
Discard the cleared solution and briefly centrifuge the “IP” tubes (400 × g, 30 s, 4°C) to spin down the residual wash buffer.
-
44.
Place the “IP” tubes on the DynaMag-2 Magnet tube holder.
-
45.
Carefully remove the residual wash buffer from the bottom of the tube.
-
46.
Add 40 μL (for WB) or 200 μL (for qPCR or NGS) of elution buffer, and break the pellet by pipetting five times.
-
47.
Place the “IP” tubes on the DynaMag-2 Magnet tube holder.
-
48.
For qPCR/NGS samples, carefully pipette the eluted sample into a new 1.5 mL centrifuge tube.
-
49.
For WB samples, transfer the eluted sample to a new 1.5 mL centrifuge tube containing 40 μL of 2 × sample buffer.
DNA extraction for ChIP-qPCR/seq
In this step, the samples for DNA analyses (e.g., qPCR, NGS) are prepared. DNA is purified from the eluted and input samples, and dissolved in TE buffer after EtOH precipitation.
-
50.
Add 200 μL of PCI (25:24:1) to the tube and vortex vigorously. Make sure to also perform the same procedure to the input samples from the step 26.
-
51.
Centrifuge tubes at 17,700 × g (or at maximum speed of the centrifuge machine, 10 min, 25°C).
-
52.
Dispense 20 μL of 3 M NaOAc and 2 μL glycogen solution (20 mg/mL) in a new 1.5 mL centrifuge tube during centrifugation.
-
53.
After centrifugation, carefully transfer the top layer (approximately 190 μL) to the tube with NaOAc/glycogen, and mix by vortexing.
-
54.
Add 500 μL of 100% EtOH and vortex vigorously. You can pause here and store the samples at −20°C.
-
55.
Centrifuge the tubes (17,700 × g, 10 min, 4°C).
-
56.
Carefully remove the supernatant so as not to disturb the white pellet.
-
57.
Add 500 μL of 70% EtOH and vortex vigorously.
-
58.
Centrifuge the tubes (17,700 × g, 5 min, 4°C).
-
59.
Carefully remove the supernatant so as not to disturb the white pellet.
-
60.
Briefly centrifuge tubes to spin down residual EtOH.
-
61.
Carefully remove EtOH with a P20 pipette.
-
62.
Add 400 μL (for qPCR) or 130 μL (for NGS) of TE buffer to the IP samples to solubilize DNA.
-
63.
Add 1000 μL of TE buffer to the input samples to prepare a 2% solution of input sample.
Expected outcomes
This protocol describes the procedures to obtain the DNA/protein complex of your POI formed on chromatin. The obtained samples can be applied for analyses of co-purified proteins and DNA using WB, mass spectrometry, qPCR, and NGS. Examples are shown in Figures 4 and 5(Miyamoto et al., 2020). For more details, refer to our previous publications (Okuda et al., 2017, Okuda et al., 2015, Okuda et al., 2014).
Figure 4.

Application of fanChIP method for characterization of protein complexes on chromatin
FLAG-tagged Mixed Lineage Leukemia (MLL) (the residues 869-1252) was expressed in HEK 293T cells, and affinity-purified using the fanChIP method. Proteins co-purified with the MLL fragment were detected by silver staining (A). RNA Polymerase II (RNAP2) complex components were identified using mass spectrometry (B). The association was confirmed by western blotting (C). Data are obtained from Miyamoto et al. (2020).
Figure 5.

Application of fanChIP method for characterization of genomic localization of proteins
Chromatin localizations of the indicated proteins, histone markers, and RNAP2 in HB1119 leukemia cells were analyzed using fanChIP followed by deep-sequencing. Data are obtained from Miyamoto et al. (2020).
To identify the binding factors of POI, the co-purified proteins are analyzed using mass spectrometry. Subsequently, IP-WB analysis should be performed to evaluate whether the identified proteins are the true binding factors of POI. One example of characterizing protein complexes associated with MLL is shown in Figure 4 (Miyamoto et al., 2020).
To characterize the landscape of genomic localization of POI, the obtained DNA/protein complexes can be analyzed by NGS (fanChIP-seq). Data acquired with fanChIP-seq are shown in Figure 5 (Miyamoto et al., 2020). To obtain NGS-applicable samples, we typically shear the DNA before library preparation.
Limitations
Omission of the crosslinking step provides several advantages in fanChIP, but it might also lead to the loss of proteins which weakly associate with chromatin. Although we don’t have any specific evidence for this, proteins with a short residence time on chromatin are potentially subjected to this problem. MNase preferentially digests AT-rich naked DNAs, which can potentially generates a bias in particular open chromatin regions (Klein and Hainer, 2020). As previously pointed out by Teves and Henikoff (Teves and Henikoff, 2012), MNase-resistant genomic fractions can be retained in an insoluble pellet, which can also potentially generate some bias.
Lack of fixation might also facilitate the unwanted recruitment of DNA-repairing proteins onto MNase-digested sites, leading to artificial co-precipitation. If you are interested in the protein interaction involving DNA-repair proteins, you should take this into consideration and conduct another appropriate method to verify the results obtained by fanChIP.
Some epitopes are difficult to detect using specific antibodies in the native condition, presumably owing to the embedded nature of the epitope. For example, tri-methylated histone H3 lysine 27 and di-methylated histone H3 lysine 79 are not properly detected by the specific antibodies that give good results in crosslinked ChIP with a similar fractionation procedure termed as faxChIP (Okuda et al., 2017).
Although we see this as an advantage of fanChIP rather than a limitation, a significant difference exists in the localization patterns of RNA Polymerase II (RNAP2) between fanChIP and crosslinked ChIP (i.e., faxChIP). The ChIP seq profile of RNAP2, whose heptapeptide repeats in the C-terminal domain (CTD) are phosphorylated on Ser5 (RNAP2 Ser5-P), differs depending on the method used. The localization of RNAP2 Ser5-P accumulates at the transcription start sites (TSSs) in faxChIP as compared to that in fanChIP (Figure 6). We hypothesize that this is due to pausing of RNAP2 induced by chemical crosslinking. RNAP2, whose heptapeptide repeats in the CTD are not phosphorylated (RNAP2 non-P), remains enriched at TSSs. Thus, the fanChIP method can capture the difference between RNAP2 non-P and Ser5-P more clearly.
Figure 6.

Comparison between conventional crosslinked ChIP (faxChIP) and native ChIP (fanChIP)
The distribution pattern of RNAP2 is different between the two methods. RNAP2, whose heptapeptide repeats in the C-terminal domain (CTD) are phosphorylated on Ser5 (RNAP2 Ser5-P), tends to accumulate at transcription start sites (TSSs) in faxChIP (black), whereas it is more dispersed in fanChIP (blue). RNAP2, whose heptapeptide repeats in the CTD are not phosphorylated (RNAP2 non-P), is enriched at TSSs in fanChIP.
Troubleshooting
Problem 1
Chromatin is not optimally fragmented (step 25).
Potential solution
As shown in lane 4 of Figure 3, optimally fragmented chromatin gives ladder-like DNA bands within the range of 150–600 bp after electrophoresis. If your chromatin preparations give electrophoresis patterns such as lane #2, 3, or 7, the chromatin is insufficiently digested, and lane #10 indicates overly digested. You can increase or decrease the amount of MNase or the incubation time to obtain optimal chromatin samples.
In our experience, MNase reconstituted with an appropriate storage buffer is stable for at least three months if stored at −20°C. However, consider re-preparing MNase, if it does not work properly.
Problem 2
IP of POI (“bait” protein) cannot be detected by WB (step 49).
Potential solution
The use of a good antibody is critical for successful IP. We recommend the use of IP- or ChIP-grade antibodies, and check if your antibodies work well at least for whole cell lysates. If your POI is exogenously expressed using transient transfection or other relevant methods, check if the transduction process has been performed optimally. Other specific solutions are as follows:
If you cannot detect POI in the co-precipitates despite good detection in its “input” sample, the IP efficacy may be low. Increasing the amount of starting material/antibody, or extending the incubation time may increase the yield of IP. We routinely used 1 μg antibody for 400 μL of the chromatin sample prepared from HEK 293T cells. The incubation period for IP is a total of 6 h. Consider changing one (some) of these parameters.
Another possibility is the low abundance of your POI in the “Chromatin fraction”. If chromatin is insufficiently digested and remains insoluble, the majority of POI goes to the “pellet fraction”. In this case, your POI should be undetectable even in the “input” sample. Check whether chromatin is optimally fragmented and also check the abundance of POI in the “pellet fraction”. The latter can be done by solubilizing the pellet in elution buffer and analyzing it by WB. You can also check the presence of POI in the “CSK fraction” as it is possible that POI binds poorly to chromatin. In that case, it is difficult to further analyze using this method.
Problem 3
Co-immunoprecipitation of “prey” protein cannot be detected by WB (step 49).
Potential solution
An efficient IP of bait protein is a prerequisite for capturing co-immunoprecipitation of prey protein. See also problem 2.
Another approach that may improve the efficacy of co-immunoprecipitation is the use of a low-salt buffer. To do so, skip the steps 18 and 19 of “Extraction of Chromatin Fraction”. After centrifugation (the step 20), the resultant supernatant mainly comprises MNase buffer and fragmented chromatin. Simply use this for subsequent steps. Use MNase buffer containing 20 mM EDTA (nucfrIP wash buffer) instead of fanChIP wash buffer for washing. This alternative method is called nucleosome-fraction IP (nucfrIP), and various associated factors have been successfully identified using this method (Okuda et al., 2017, Miyamoto et al., 2020). However, nucfrIP increases the possibility of false positive interactions due to the low salt condition. Additionally, some proteins are not solubilized, and they go to the pellet fraction.
Problem 4
Co-immunoprecipitated DNA cannot be amplified using PCR (step 63).
Potential solution
The factors to be considered are listed below.
-
1.
Similar to fanChIP-WB, the quality of the antibody is critical for successful fanChIP-qPCR.
-
2.
In qPCR, the primer set should be highly specific and sensitive. Check if the “input” sample is amplified well.
-
3.
If POI is a low abundance protein, it may not be able to pull down DNA above the detection limit of qPCR. Scale up the initial sample volume.
-
4.
If chromatin is over-digested, the majority of chromatin is too short to be amplified using PCR. See problem 1.
Antibodies for RNAP2 and active histone marks (e.g., H3K4me3 and H3K27ac) and primers for TSSs of housekeeping genes (e.g., ACTB and GAPDH) can be useful to evaluate the efficacy of fanChIP-qPCR. For example, RNAP2 must occupy the TSS of ACTB and GAPDH in any cell type, which can be confirmed using fanChIP-qPCR. Consider obtaining these materials in parallel with the antibodies and the primers of your interest.
Problem 5
High background or low signal/noise ratio in qPCR (step 63).
Potential solution
If unexpected signals are observed on the locus where the POI should not localize, it could be due to non-specific binding of the antibody. If the antibody detects
multiple and strong non-specific bands by WB using whole cell lysate or whole “chromatin fraction”, the antibody may not be suitable for ChIP. Other approaches are as follows:
-
1.
Use a low volume of antibody. Too much antibody potentially increases non-specific binding with only a moderate increase in the on-target signal.
-
2.
Check whether the chromatin is optimally fragmented. Poor fragmentation can also give a high background noise because longer chromatin may cause off-target binding of the antibody.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Akihiko Yokoyama (ayokoyam@ncc-tmc.jp).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
ChIP-seq data have been deposited under the accession numbers (DRA011231, DRA004872) in the DDBJ (DNA Data Bank of Japan) Sequencing Read Archive.
Acknowledgements
We thank Akinori Kanai for handling the NGS data. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI grants (16H05337 and 19H03694 to A.Y. and 19K16791 to R.M.) and in part by research funds from the Yamagata Prefectural Government, City of Tsuruoka, Dainippon Sumitomo Pharma Co. Ltd., and friends of Leukemia Research Fund.
Author contributions
Conceptualization, A.Y.; investigation, R.M. and A.Y.; writing – original draft, R.M.; writing – review and editing, A.Y.
Declaration of interests
A.Y. received a research grant from Dainippon Sumitomo Pharma Co. Ltd.
References
- Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Klein D.C., Hainer S.J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020;28:69–85. doi: 10.1007/s10577-019-09619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto R., Okuda H., Kanai A., Takahashi S., Kawamura T., Matsui H., Kitamura T., Kitabayashi I., Inaba T., Yokoyama A. Activation of CpG-rich promoters mediated by MLL drives MOZ-rearranged leukemia. Cell Rep. 2020;32:108200. doi: 10.1016/j.celrep.2020.108200. [DOI] [PubMed] [Google Scholar]
- O'Neill L.P., Turner B.M. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- Okuda H., Kanai A., Ito S., Matsui H., Yokoyama A. AF4 uses the SL1 components of RNAP1 machinery to initiate MLL fusion- and AEP-dependent transcription. Nat. Commun. 2015;6:8869. doi: 10.1038/ncomms9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H., Kawaguchi M., Kanai A., Matsui H., Kawamura T., Inaba T., Kitabayashi I., Yokoyama A. MLL fusion proteins link transcriptional coactivators to previously active CpG-rich promoters. Nucleic Acids Res. 2014;42:4241–4256. doi: 10.1093/nar/gkt1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H., Stanojevic B., Kanai A., Kawamura T., Takahashi S., Matsui H., Takaori-Kondo A., Yokoyama A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Invest. 2017;127:1918–1931. doi: 10.1172/JCI91406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves S.S., Henikoff S. Salt fractionation of nucleosomes for genome-wide profiling. Methods Mol. Biol. 2012;833:421–432. doi: 10.1007/978-1-61779-477-3_25. [DOI] [PubMed] [Google Scholar]
- Tkachuk D.C., Kohler S., Cleary M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- Yokoyama A., Somervaille T.C., Smith K.S., Rozenblatt-Rosen O., Meyerson M., Cleary M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ChIP-seq data have been deposited under the accession numbers (DRA011231, DRA004872) in the DDBJ (DNA Data Bank of Japan) Sequencing Read Archive.