

Abstract
Status epilepticus (SE) is a condition in which seizures are not self‐terminating and thereby pose a serious threat to the patient's life. The molecular mechanisms underlying SE are likely heterogeneous and not well understood. Here, we reveal a role for the RNA‐binding protein Fragile X‐Related Protein 2 (FXR2P) in SE. Fxr2 KO mice display reduced sensitivity specifically to kainic acid‐induced SE. Immunoprecipitation of FXR2P coupled to next‐generation sequencing of associated mRNAs shows that FXR2P targets are enriched in genes that encode glutamatergic post‐synaptic components. Of note, the FXR2P target transcriptome has a significant overlap with epilepsy and SE risk genes. In addition, Fxr2 KO mice fail to show sustained ERK1/2 phosphorylation induced by KA and present reduced burst activity in the hippocampus. Taken together, our findings show that the absence of FXR2P decreases the expression of glutamatergic proteins, and this decrease might prevent self‐sustained seizures.
Keywords: ERK signaling, FXR2P, glutamatergic synapses, kainic acid, status epilepticus
Subject Categories: Molecular Biology of Disease, Neuroscience, RNA Biology
Loss of RNA binding protein FXR2P reduces components of glutamatergic synapses and prevents the transition of kainate‐induced seizures to the self‐sustained status epilepticus.

Introduction
With a disease prevalence of about 1%, epilepsy is one of the most common neurological disorders (Pugliatti et al, 2007; Fiest et al, 2017). Despite the availability of many anti‐epileptic drugs, epilepsy is inadequately controlled in about a third of the cases (Pitkänen et al, 2016; Devinsky et al, 2018). The hallmark of the disease is the occurrence of sudden, transient, recurrent seizure episodes that result from neuronal hyperexcitability. Seizures are generally self‐limiting events, lasting a few minutes. If seizures last longer, they are called status epilepticus (SE). The International League Against Epilepsy (ILAE) (Trinka et al, 2015) defines SE as seizures that last more than 5–15 min depending on the types (tonic–clonic SE, focal SE, and absence SE). If SE lasts more than 60 min, it has long‐term consequences including neuronal injury, neuronal death, alteration of neuronal networks, and functional deficits (Trinka et al, 2015). SE is a major clinical emergency that poses a serious threat to the patient's life (on average 20% mortality rate) (Trinka & Kälviäinen, 2017).
During the last decade, next‐generation sequencing techniques have led to the identification of many monogenic epilepsies and epileptic encephalopathies (Thomas & Berkovic, 2014; Zhou et al, 2018). The genes responsible for these epilepsies encode for channel receptor, cell migration, cell metabolism, or RNA‐binding proteins (RBPs) (Toth, 2001). In recent years, the role of RBPs in disorders of neurodevelopment, neurodegeneration, and epilepsy has gained more interest and importance (Pernice et al, 2016; Conlon & Manley, 2017; Chen et al, 2019). RBPs are essential regulators of protein expression, and their loss or mutation can cause aberrant translation of different mRNAs (Achsel & Bagni, 2016; Hentze et al, 2018; Bagni & Zukin, 2019). In the case of epilepsy, genes controlling neuronal excitability that are under the control of such RBPs can manifestly affect seizure susceptibility. Fine‐tuned regulation of such epilepsy‐associated genes and pathways is therefore critical.
A prime example of RBP loss leading to epileptic seizures is the Fragile X Mental Retardation Protein (FMRP). Individuals with Fragile X syndrome (FXS) show increased seizure susceptibility and, likewise, mouse models for FXS display increased sensitivity to audiogenic induced seizures (Berry‐Kravis, 2002; Curia et al, 2013; Sethna et al, 2017). Indeed, the prevalence of seizures in individuals with FXS is around 14%, significantly higher than in the general population (Berry‐Kravis, 2002). This difference is thought to be due to elevated levels of ERK1/2 (extracellular kinase 1/2) phosphorylation (Curia et al, 2013; Sethna et al, 2017) and altered translation of specific mRNAs (Darnell et al, 2011; Fernández et al, 2013; Bagni & Zukin, 2019).
Another member of the Fragile X‐related family is the RBP Fragile X‐Related Protein 2 (FXR2P) (Zhang et al, 1995; Tamanini et al, 1999). FXR2 is encoded at the human locus 17p13.1, a hotspot for chromosomal rearrangements. The primary neurological phenotypes described in patients with de novo microdeletions and microduplications of this locus are intellectual disability and seizures (Komoike et al, 2010; Schluth‐Bolard et al, 2010; Belligni et al, 2012; Zeesman et al, 2012; Giordano et al, 2014; Kuroda et al, 2014). Interestingly, males with de novo 17p13.1 microduplications that included the FXR2 region can develop seizures (Belligni et al, 2012; Mooneyham et al, 2014; Maini et al, 2016). In contrast, no cases with a de novo 17p13.1 microdeletion that includes the FXR2 region in men have been reported with seizures. Given the large inter‐individual size variability for these microdeletions and microduplications, further studies are needed to clearly associate FXR2P with seizures.
Here, we investigated the contribution of FXR2P to seizure susceptibility. We used the Fxr2 KO model that displays several phenotypic traits similar to those observed in the Fmr1 KO model, like altered synaptic plasticity, hyperactivity, and impaired learning (Bontekoe et al, 2002; Spencer et al, 2006; Zhang et al, 2009). At the molecular level, a few studies in mice indicate that the absence of FXR2P negatively affects the expression of hippocampal glutamatergic proteins, such as PSD‐95 and GluA1 (Cavallaro et al, 2008; Fernandez et al, 2015; Guo et al, 2015). However, susceptibility to seizures has never been previously investigated in the Fxr2 KO mice. We found that the lack of FXR2P has a profound effect on SE. Fxr2 KO mice displayed less protracted SE upon treatment with kainic acid (KA), but not with pilocarpine administration. The seizure severity correlated with ERK1/2 phosphorylation in the hippocampus, suggesting a pathway‐specific protection toward prolonged seizures in Fxr2 KO mice. Furthermore, with a high‐throughput approach we identified the mRNAs specifically associated with FXR2P and found that the products of a large subset of these FXR2P‐bound mRNAs are located at glutamatergic synapses and mostly involved in glutamate receptor signaling. In addition, we found that the genes coding for these FXR2P targets share a significant overlap with epilepsy and SE risk genes. Notably, several glutamatergic receptors/proteins display lower expression levels in Fxr2 KO hippocampi, which may explain the reduced susceptibility to seizures as well as the downstream ERK1/2 phosphorylation at the late phase (Nateri et al, 2007; Chen et al, 2016b). Taken together, these data link FXR2P to the proper functioning of the glutamatergic signaling pathway and highlight a possible involvement of FXR2P in SE.
Results
Male patients with 17p13.1 microdeletion do not have seizures
To investigate whether FXR2P contributes to the 17p13.1 microdeletion and microduplication phenotype, we assessed the online database DECIPHER that contains information on copy number variation (CNV) cases with their respectively observed clinical symptoms (decipher.sanger.ac.uk) (Firth et al, 2009). A total of 58 male cases with 17p13.1 loci deleted or duplicated (< 10 Mb) were found. The percentage of individuals with seizures for the categories microdeletion and microduplication is summarized in Figure 1A. In microdeletions, cases with seizures are less frequent when the FXR2 gene is deleted compared with when the gene is present (0% versus 10.5%). In microduplications, cases with seizures are more frequent when the gene FXR2 is part of the duplication (30% versus 16.7%). From this exploratory analysis, one could speculate that FXR2 deletion might have a profound effect on seizure manifestation, even if this effect was observed only in male individuals, and not in females (Figure EV1). In addition, we used the Open Targets Platform that provides an association index of a certain gene with different diseases (value 0 equals to no association, value 1 equals to substantial evidence). When we searched for any possible association between the FXR2 gene and diseases of the central nervous system (CNS), we observed that early infantile epileptic encephalopathy shares a considerable association with the FXR2 gene (Figure 1B). Altogether these findings suggest a possible role of FXR2P in seizures in humans.
Figure 1. Frequency of seizures in males with 17p13.1 deletion/duplication.
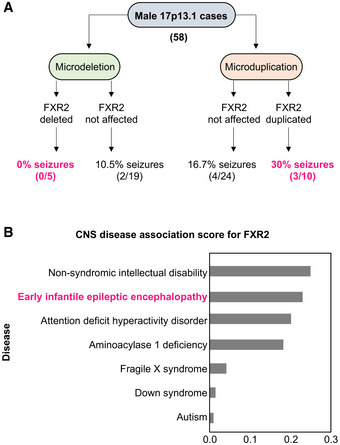
- 58 Males were described in DECIPHER of which 24 carried a microdeletion, and 34 carried a microduplication.
- Central nervous system (CNS) disease associations derived from the Open Target Platform. Association score ranges from 0 to 1, with 0 indicating no association, and 1 indicating full association. FXR2 has highest association with intellectual disability and epilepsy.
Figure EV1. Frequency of seizures in females with 17p13.1 deletion/duplication.
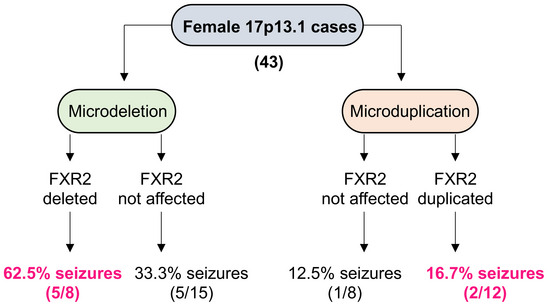
The analysis consists of 43 females described in the DECIPHER database and the CNV database from center of Medical Genetics in Antwerp (Belgium). 23 Patients carried a microdeletion of the 17p13.1 locus whereas 20 patients carried a microduplication.
Fxr2 KO mice show different susceptibility to seizure‐inducing convulsants
To investigate the contribution of FXR2P in regulating seizure susceptibility in vivo, we used two models of chemically induced acute seizures based on the administration of a neuro‐excitatory glutamatergic agonist (KA) and a muscarinic cholinergic agonist (pilocarpine) (Curia et al, 2008; Lévesque & Avoli, 2013; Lévesque et al, 2016; Jie et al, 2018). Upon administration of these compounds, both WT and Fxr2 KO animals display motor seizures (Figure 2A) (Racine, 1972; Janumpalli et al, 1998) that are thought to originate in the hippocampus (Nadler et al, 1981). In the case of KA administration, male Fxr2 KO mice showed a remarkable recovery within an hour, whereas WT mice continued seizure activity throughout the 2‐h observation period (Figure 2B). In fact, the epileptic profile (i.e., seizure class distribution) in Fxr2 KO mice is very different from WT mice, as Fxr2 KO mice had a Racine score of 0 during 80% of the experiment (Figure EV2A). None of the Fxr2 KO mice displayed motor SE at the end of the observation period, in contrast with 67% of the WT mice. This drastic difference in seizure susceptibility between WT and Fxr2 KO mice was also reflected in a lower cumulative score of seizure events in Fxr2 KO compared with WT mice (Figure 2C). Interestingly, female Fxr2 KO mice did not display resistance to KA‐induced seizures, coherent with the DECIPHER database (Figure EV2B and C). Therefore, we only considered males in all subsequent analyses.
Figure 2. Kainic acid‐ and pilocarpine‐induced epileptic seizures in WT and Fxr2 KO mice.
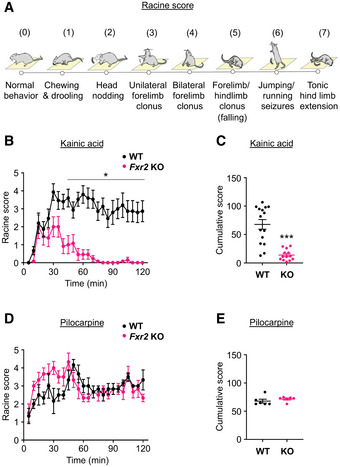
- The different stages of seizures based on increased severity.
- Average seizure score of KA‐treated WT and Fxr2 KO mice over time from t = 0 (injection time) to 2 h. *P < 0.05 compared with WT mice (two‐way Repeated Measures ANOVA; interaction effect of time and genotype; F 1,666 = 10.05, P = 0.0016; post hoc tests for indicated time points, P < 0.05). KA‐treated WT mice (n = 16), KA‐treated Fxr2 KO mice (n = 12). Data are presented as mean ± Standard Error of the Mean (SEM).
- Responsiveness to KA of WT (black, n = 16) and Fxr2 KO mice (magenta, n = 12). ***P < 0.001 compared with Fxr2 KO mice (two‐tailed Student's t‐test; t 26 = 5.72, P < 0.001). Data are presented as mean ± SEM.
- Average seizure score of pilocarpine‐treated WT and Fxr2 KO mice over time from t = 0 (injection time) to 2 h (two‐way Repeated Measures ANOVA, P = n.s.). Pilocarpine‐treated WT mice (n = 6), pilocarpine‐treated Fxr2 KO mice (n = 6). Data are presented as mean ± SEM.
- Responsiveness to pilocarpine (PC) of WT (n = 6) and Fxr2 KO mice (n = 6). (two‐tailed Student's t‐test, P = n.s.). Data are presented as mean ± SEM.
Figure EV2. Seizure distribution based on convulsant and gender.
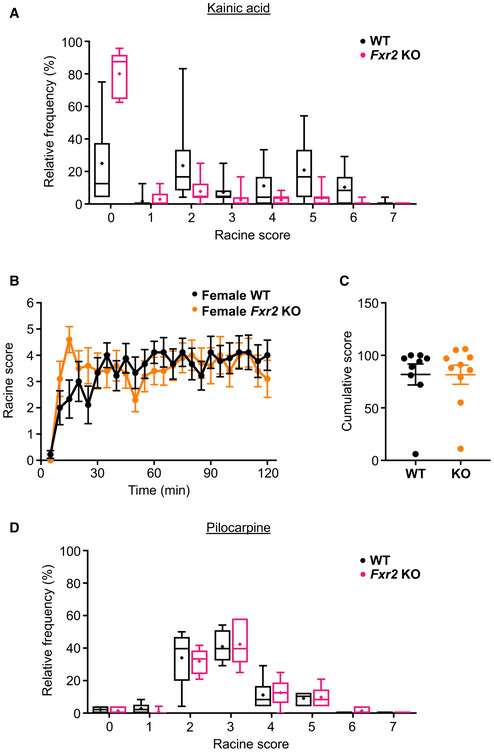
- Relative frequency of the different Racine scores in WT (black, n = 16) and Fxr2 KO (magenta, n = 12) upon KA administration (Chi‐square test; = 175.67, P < 0.001). Data are presented boxplots defined by median, quantiles 0.25 and 0.75. The length of the whiskers indicates 1.5 times the interquantile range (IQR).
- Average seizure score of female KA‐treated WT (n = 9) and Fxr2 KO mice (n = 10) over time from t = 0 (injection time) to 2 h. No genotype‐dependent differences observed (two‐way repeated measures ANOVA). Data are presented as mean ± SEM.
- Responsiveness to KA of female WT (black, n = 9) and female Fxr2 KO mice (orange, n = 10). KA‐treated WT mice (n = 9), KA‐treated Fxr2 KO mice (n = 10; two‐tailed Student's t‐test). Data are presented as mean ± SEM.
- Relative frequency of the different Racine scores in WT (n = 6) and Fxr2 KO mice (n = 6) upon pilocarpine administration (Chi‐square test). Data are presented boxplots defined by median, quantiles 0.25 and 0.75. The length of the whiskers indicates 1.5 times the interquantile range (IQR).
To address the question whether such a resistance to prolonged seizures in male Fxr2 KO mice could be observed in another model of epilepsy, WT and Fxr2 KO mice were treated with pilocarpine, which activates M1 muscarinic receptors. Under these conditions, all mice reached SE, irrespective of the genotype. Specifically, time‐lapse of seizures, seizure class distribution, and cumulative score showed no differences between WT and Fxr2 KO mice (Figures 2D and E, and EV2D). These findings suggest that the observed protective nature of the Fxr2 KO mice toward seizures is possibly receptor and/or signaling pathway specific.
Transcriptome‐wide identification of FXR2P‐bound mRNAs
We used an unbiased approach to identify the FXR2P brain regulon that could be involved in the observed epileptic phenotype. Specifically, we performed RNA immunoprecipitation (RIP) followed by second‐generation RNA sequencing (RIP‐seq) from the forebrain of adult mice. FXR2P was efficiently immunoprecipitated using an FXR2P antibody (1G2) (Darnell et al, 2009) that recognizes the C terminus of the protein (a.a. 414–658), which diverges between members of the FMRP protein family (Ferrari et al, 2007). PSD‐95 mRNA, a well‐established FXR2P target (Fernandez et al, 2015), was used as a control to verify the specificity of the RIP assay (Figure 3A). FXR2P RNA‐IP from three independent WT and Fxr2 KO animals were each subjected to amplification, library preparation, and sequencing (see Material and Methods). Upon read count normalization and threshold cutoff, we identified and considered 14,809 transcripts for further analysis. Fxr2 KO samples have significantly less normalized reads for PSD‐95 compared with WT (Figure 3B). Subsequently after ≥ 2 log2 fold change between WT and Fxr2 KO samples and appropriate FDR cutoff, we identified 488 genes that were differentially bound by FXR2P (Figure 3C, Dataset EV1). We then investigated in which ontologies these genes were most enriched. With respect to the cellular component, we identified that these genes were most enriched in ontologies for post‐synaptic density, neurons, and the glutamatergic synapse (Figure 3D, Dataset EV2). The latter, even though slightly lower in enrichment compared with the others, is in agreement with the large genotype effect we observed on seizures after administration of a glutamatergic agonist. Then, upon further investigating biological processes specifically involving glutamate, we found that the genes were most enriched for ontologies for the glutamatergic signaling pathway (Figure 3D, Dataset EV3). Taken together, our enrichment analysis highlights that the 488 FXR2 candidate genes are strongly associated with neuronal excitation.
Figure 3. Identification of FXR2P target mRNAs.
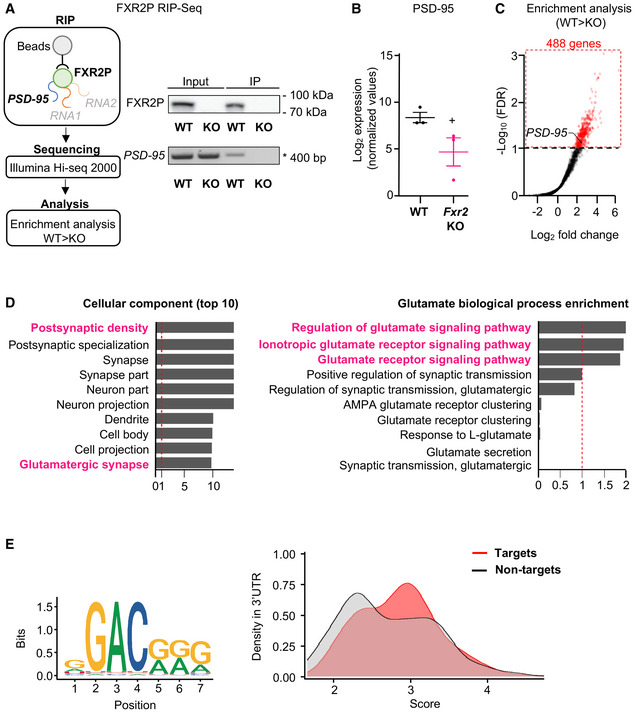
- Schematic representation of the experimental strategy for the RIP‐seq pipeline. Representative Western blot showing the FXR2P pull‐down. FXR2P was precipitated from WT and Fxr2 KO mice brain extracts using the 1G2 antibody (Fernandez et al, 2015) and analyzed by Western blotting. Input, 10%. PSD‐95 mRNA was used as a control to verify the specificity of the RIP‐seq.
- PSD‐95 mRNA had lower read counts in Fxr2 KO samples (n = 3) compared with WT samples (n = 3; +adjusted P < 0.1, two‐tailed Student's t‐test). Data are presented as mean ± SEM.
- Enrichment analysis of the 14,809 transcripts shows 488 genes (red) that pass the FDR threshold (adjusted P‐value cut‐off at 0.1).
- Left, gene ontology of the 488 FXR2P candidate mRNAs across the top 10 cellular components. Right, biological processes specific for glutamate are highlighted. −Log10 q‐values were normalized according to the maximum q‐value in the list of gene ontologies. The red dashed line indicates the threshold for significant enrichment in these gene ontologies.
- Left, FXR2P‐binding motif sequence as described by Ray et al (2013). Right, distribution of match scores between FXR2P target and non‐target mRNAs. FXR2P target mRNAs show on average higher match scores than FXR2P non‐target mRNAs (Kolmogorov–Smirnov test, P < 0.001).
To identify possible FXR2P binding sites, we analyzed the occurrence of the putative FXR2P‐binding sequences (Ray et al, 2013) in the FXR2P transcriptome (Figure 3C, 3′UTR of the 488 targets) using the RBPmap tool (http://rbpmap.technion.ac.il; Paz et al, 2014). In accordance with its highly degenerate nature (Ray et al, 2013; Figure 3E, left panel), the FXR2P‐binding motif is frequently found in the 3′UTRs of FXR2P target and non‐target mRNAs. Of note, higher‐scoring FXR2P‐binding sequences are overrepresented in target 3′UTRs with respect to an equally sized set of non‐target 3′UTRs (Figure 3E, right panel). A higher score corresponds to a stronger binding of FXR2P to the respective sequence, suggesting that the putative consensus sequence might contribute to the recognition of FXR2P targets. As for other RBPs, it can be hypothesized that the sequence domain of FXR2 is surrounded by additional cis‐acting elements that are recognized by other, cooperating RBPs (Achsel & Bagni, 2016). Additional studies are required to conclusively identify the bona‐fide FXR2 recognition domain/s.
FXR2P mRNA targets are involved in epilepsy
Given a potential role of FXR2P in increasing susceptibility to seizures and SE, we next compared the overlap of FXR2P target genes (488 targets, Dataset EV1) with risk genes for epilepsy and genes associated with SE. The list of epilepsy risk genes was compiled from several epilepsy gene databases, resulting in 1,089 targets (see also Material and Methods; Ran et al, 2015; Guo et al, 2017; Wang et al, 2017). We found a significant overlap of 75 genes shared between epilepsy genes and FXR2P candidate genes (15.4%, Figure 4A, Dataset EV4). Next, we used the String database to determine which of these 75 genes are closely interacting with each other and thereby define a tight network of proteins (http://string‐db.org). Protein interaction analysis was based on curated databases or literature that provide high confidence of inter‐protein actions such as activation, inhibition, binding, and reaction. Interestingly, 48% of the shared genes can be considered as part of a tight network of proteins (36 targets; Figure 4B). Furthermore, 12 out of 75 epilepsy genes were also associated with SE (Dataset EV4) (Bhatnagar & Shorvon, 2015). To validate these findings, we performed independent immunoprecipitation experiments followed by qPCR to detect a selection of eight out of the 36 epilepsy network mRNAs identified in this study, as well as eight FXR2P non‐targets. This analysis revealed a significant correlation between the enrichment values obtained in the RIP‐seq experiment and the RT–qPCR (Figure 4C).
Figure 4. Association of FXR2P mRNA targets with epilepsy.
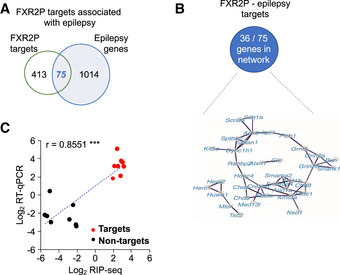
- Overlap between the 488 FXR2P targets and epilepsy risk genes (75 of 1,089; Fisher's exact test, P < 0.001).
- String database was used to classify FXR2P and epilepsy risk genes that are part of one network.
- Validation of RIP‐seq by RT–qPCR. Shown are the correlation of the enrichment observed in log2 RIP‐seq (X‐axis) and log2 RT–qPCR (Y‐axis) for eight epilepsy‐relevant FXR2P targets (red) and eight FXR2P non‐targets (black). Each point represents an individual mRNA, quantified using both methods (r = 0.8551, ***P < 0.001). mRNAs are normalized for the average of all the 16 mRNAs.
Among the 75 mRNAs shared between FXR2P targets and epilepsy‐risk genes, we next focused on selected mRNAs encoding proteins involved in glutamatergic signaling. We focused on the genes Grin2b and Grm5 encoding the glutamatergic receptors GluN2B and mGluR5, respectively, because aberrantly high levels of the glutamatergic receptors GluN2B and mGluR5 have been observed in patients with epilepsy and in different animal models for epilepsy (Nateri et al, 2007; Das et al, 2012; Kelly et al, 2018; Egbenya et al, 2018; Lam et al, 2019). Moreover, it was shown previously that GluN2B or mGluR5 inhibition can modulate the KA‐induced seizure activity (Chen et al, 2016a; Medina‐Ceja & García‐Barba, 2017). We therefore investigated the protein expression of GluN2B, mGluR5, GluK5 (kainate receptor 5, encoded by Grik5), and PSD‐95 (encoded by Dlg4, previously shown to be reduced in Fxr2 KO; Fernandez et al, 2015) in hippocampi of WT and Fxr2 KO mice. No difference was observed in GluK5 protein expression between WT and Fxr2 KO hippocampi (Figure 5A). In contrast, both GluN2B and mGluR5 protein expression were reduced by 20 and 40% in Fxr2 KO hippocampi, respectively, compared with WT (Figure 5A). Both GluN2B and mGluR5 are important players during the late phase of seizures. As such, these findings are consistent with the in vivo results obtained in KA‐treated WT and Fxr2 KO mice, where both groups showed early phase seizures, but only differed in late phase seizures (Figure 2B). In addition, PSD‐95, a protein affecting the GluN2B signaling (D'Mello et al, 2011; Lin et al, 2016), was significantly reduced in Fxr2 KO hippocampi (Figure 5A), likely buffering KA‐induced signaling. Interestingly, these three targets were not different at the level of mRNA, suggesting a difference in mRNA translation between WT and Fxr2 KO (Figure 5B).
Figure 5. Validation of glutamatergic FXR2P mRNA targets.
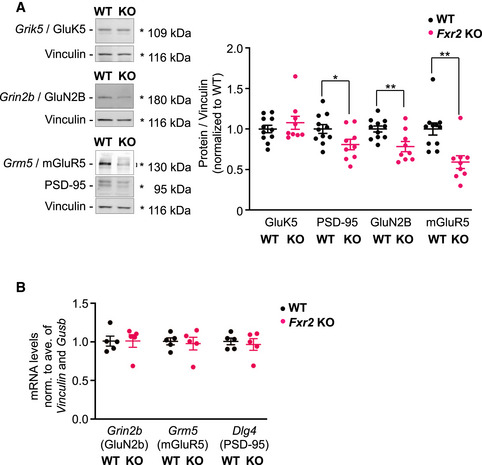
- Representative Western blot and quantification of GluK5, PSD‐95, GluN2B, and mGluR5 proteins. Endogenous GluK5, PSD‐95, GluN2B, and mGluR5 in Fxr2 KO mice (n = 9) compared with WT mice (n = 11). *P < 0.05, **P < 0.01, significant difference between WT and Fxr2 KO (two‐tailed Student's t‐test: GluK5: t 18 = 0.87, P = 0.394; PSD‐95: t 18 = 2.29, P < 0.05; GluN2B: t 18 = 3.09, P < 0.01; mGluR5: t 18 = 3.74, P < 0.01). Quantified bands are highlighted with an asterisk or squared bracket. Data are presented as mean ± SEM.
- Bar plot showing the mRNA levels of Grin2b, Grm5 and Dlg4, normalized for the average of Vinculin and Gusb mRNA levels in WT (n = 5) and Fxr2 KO (n = 5) hippocampi (two‐tailed Student's t‐test, P = n.s.). Data are presented as mean ± SEM.
To further investigate the involvement of the glutamatergic system, we investigated epileptiform bursting in Fxr2 KO disinhibited hippocampal slices. Bursting was induced by the removal of external magnesium, as previously described (Gong et al, 2014). As GABA receptors are known to impact epileptiform bursts (Gafurov & Bausch, 2013), we used picrotoxin to rule out any influence of GABA receptors on the recordings and focused only on the glutamatergic output (Newland & Cull‐Candy, 1992; Olsen, 2006). Under these conditions, Fxr2 KO hippocampal slices showed lower activity in terms of burst frequency and burst duration compared with WT hippocampal slices (Figure EV3, EV4). These results suggest that FXR2P deficiency alters, at the basal level, the strength of epileptiform bursts by affecting the excitatory network activity.
Figure EV3. Multi‐electrode array recording in WT and Fxr2 KO hippocampi.
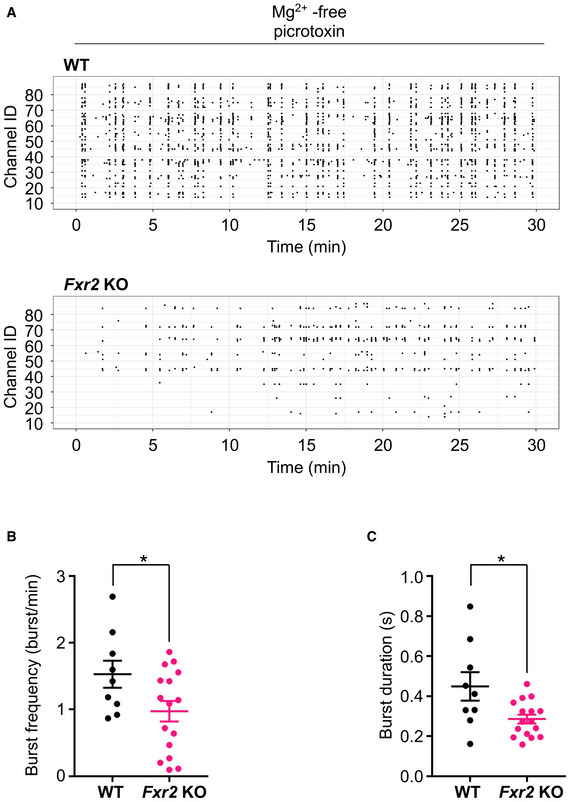
-
ARepresentative rasterplot showing burst activity from a WT (top) and Fxr2 KO (bottom) hippocampal slice recorded in a multi‐electrode array (MEA) preparation.
-
B, CBurst frequency and burst duration. *P < 0.05, significant difference compared with WT slices. (n = 9 slices taken from 6 WT mice, 16 slices taken from 8 Fxr2 KO mice; two‐tailed Student's t‐test: burst frequency: t 23 = 2.17, P < 0.05; burst duration: t 23 = 2.71, P < 0.05). Data are presented as mean ± SEM.
Figure EV4. FXR2P and glutamatergic protein expression in KA‐treated WT and Fxr2 KO mice at different time intervals.
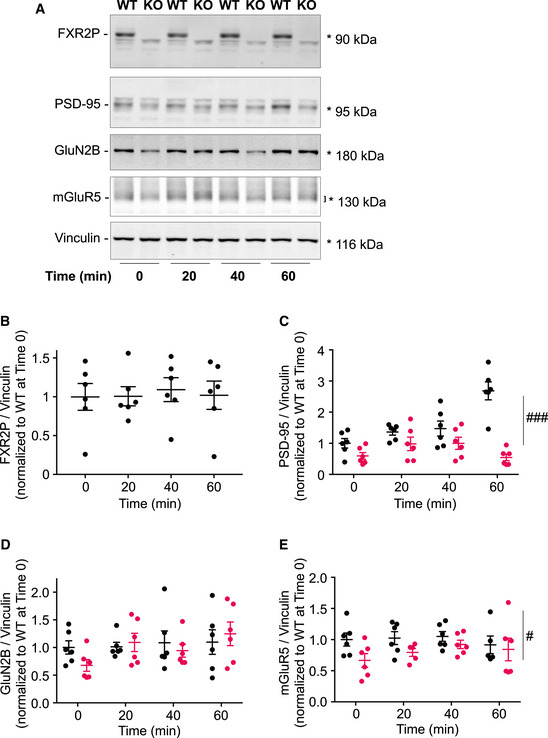
- Representative Western blot to detect the expression of FXR2P, PSD‐95, GluN2B, and mGluR5 proteins over different time intervals (0, 20, 40, and 60 min) in WT and Fxr2 KO hippocampi. Quantified bands are highlighted with an asterisk or squared bracket.
- Bar plot showing FXR2P expression over Vinculin in KA‐treated WT sacrificed at different time intervals (one‐way ANOVA). Data are presented as mean ± SEM.
- Bar plot showing PSD‐95 expression in control and KA‐treated WT and Fxr2 KO mice. ### P < 0.001, overall significant difference between WT and Fxr2 KO samples (two‐way ANOVA: Genotype effect, F 1,40 = 40.90, P < 0.001; Interaction of genotype and time, F 3,40 = 10.49, P < 0.001). Data are presented as mean ± SEM.
- Barplot showing GluN2B expression in control and KA‐treated WT and Fxr2 KO mice (two‐way ANOVA). Data are presented as mean ± SEM.
- Barplot showing mGluR5 expression in control and KA‐treated WT and Fxr2 KO mice. # P < 0.05, overall significant difference between WT and Fxr2 KO samples (two‐way ANOVA: Genotype effect, F 1,38 = 5.90, P = 0.02). Data are presented as mean ± SEM. Group sizes were n = 6 for each genotype and interval throughout the figure.
It has been shown that during SE, an imbalance of excitation and inhibition is produced through the internalization of GABA‐A receptors, but also through the externalization of glutamatergic receptors (i.e., AMPA and NMDA) to the synapse (Chen et al, 2007). In this perspective, overall reduced GluN2B and mGluR5 protein expression in Fxr2 KO mice may explain the reduced susceptibility to prolonged seizures upon KA administration, because this would result in less GluN2B and mGluR5 available to be externalized. We next investigated the involvement of the downstream pathway of GluN2B and mGluR5, and specifically ERK signaling because ERK1/2 has a predominant role in the initiation of convulsive activity (Nateri et al, 2007; Korotkov et al, 2017). Coherent with the in vivo data, we observed a differential ERK1/2 response upon KA treatment in WT compared with Fxr2 KO mice (Figure 6A). As expected, a significant increase in ERK1/2 phosphorylation levels was detected in WT mice upon KA administration (P < 0.001). On the contrary, KA‐treated Fxr2 KO mice did not show any difference compared with vehicle‐treated KO mice (Figure 6A). This lack of ERK1/2 phosphorylation was specific to KA treatment, as both pilocarpine‐treated WT and Fxr2 KO mice showed an increase in phosphorylated ERK1/2 compared with control animals (Figure 6B).
Figure 6. ERK‐mediated signaling in male WT and Fxr2 KO.
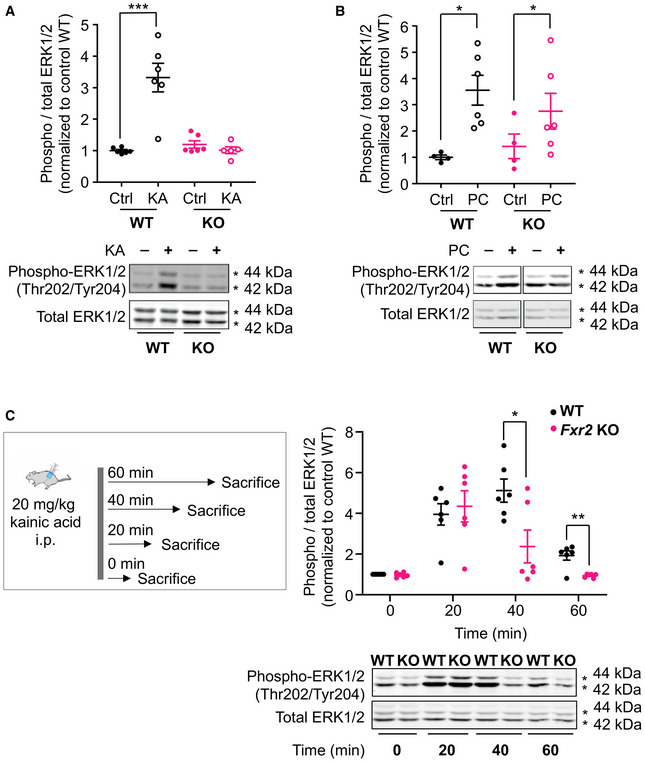
- Bar plot showing phospho‐ERK1/2 over total ERK1/2 levels in vehicle‐ and KA‐treated WT (closed and open black circles, vehicle‐treated WT n = 6; KA‐treated WT n = 6) and Fxr2 KO mice (closed and open magenta circles, vehicle‐treated Fxr2 KO n = 5; KA‐treated Fxr2 KO n = 6). ***P < 0.001 comparison between vehicle‐ and KA‐treated WT (two‐way ANOVA: interaction effect between genotype and treatment: F 1,19 = 24.66, P < 0.001). Data are presented as mean ± SEM.
- Phospho‐ERK1/2 levels in vehicle‐ and pilocarpine‐treated WT (n = 4–6) and Fxr2 KO mice (n = 4–6). Both genotypes responded with long‐lasting seizures upon pilocarpine administration *P < 0.05 (two‐way ANOVA: main effect for treatment: F 1,16 = 10.98, P < 0.01). Data are presented as mean ± SEM.
- Phospho‐ERK1/2 levels in control and KA‐treated WT (both n = 6) and Fxr2 KO mice (both n = 6) sacrificed at different time intervals (0, 20, 40, and 60 min). *adjusted P < 0.05, **adjusted P < 0.01 (two‐way ANOVA: main effect for genotype: F 1,40 = 5.99, P < 0.05; interaction effect genotype and time; F 3,40 = 4.11, P = 0.012; P < 0.05 and P < 0.01 with Bonferroni correction for 40 and 60 min, respectively). Data are presented as mean ± SEM.
Next, we investigated the kinetics of ERK1/2 phosphorylation after KA treatment (Figure 6C). We analyzed WT and Fxr2 KO at different time intervals (i.e., 20, 40, and 60 min after KA injection) and found that overall WT hippocampi displayed more robust and prolonged phosphorylation of ERK1/2 than Fxr2 KO hippocampi, in which phosphorylated ERK1/2 levels were back at baseline levels within 60 min (Figure 6C). Thus, ERK1/2 phosphorylation levels parallel the course of motor seizures.
Finally, we investigated whether the expression of FXR2P and of a few targets involved in the glutamatergic signaling change over time (Figure EV4). At first, we noticed that FXR2P expression in WT hippocampi did not change over time and was not affected by KA stimulation (Figure EV4A and B). The three glutamatergic FXR2P targets, namely PSD‐95, GluN2B, and mGluR5 showed different expression patterns. Specifically, PSD‐95 expression slightly increased over time in WT and remained reduced and unchanged in Fxr2 KO hippocampi (Figure EV4C). Second, GluN2B expression did not change over time in WT animals; however, its expression was initially low in Fxr2 KO but showed a slight increase in hippocampi upon 60 min KA stimulation (Figure EV4D). Finally, mGluR5 expression did not change over time upon stimulation in both genetic conditions, but was overall consistently lower in Fxr2 KO hippocampi (Figure EV4E). Altogether in Fxr2 KO hippocampi, the expression of GluN2B is restored upon 60 min stimulation, while PSD‐95 and mGluR5 levels remained reduced compared with WT hippocampi.
Taken together, these results point to a critical role of FXR2P governing glutamatergic‐mediated signaling that is responsible for ERK1/2 phosphorylation and late phase seizure propagation (Figure 7).
Figure 7. Model for FXR2P in SE.
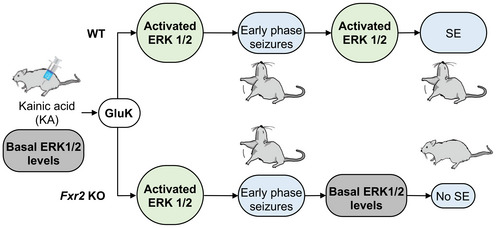
A global scheme that summarizes the role of FXR2P in SE, regulating key molecules of the glutamatergic synapses. KA activates the kainate receptors resulting in ERK1/2 phosphorylation and seizures in both WT and Fxr2 KO mice. While in WT mice, ERK1/2 phosphorylation and protracted seizures maintain, the absence of FXR2P affects glutamatergic signaling, ERK1/2 activity, and ultimately the maintenance of seizures.
Discussion
Here, we show that the absence of the RBP FXR2P protects from prolonged SE. Fxr2 KO mice displayed a specific mechanistic protection toward KA‐induced protracted seizures. Moreover, available data from the DECIPHER database showed no reports of seizures in male patients with 17p13.1 microdeletions with deleted FXR2 locus. The identification of a significant proportion of FXR2P‐interacting mRNAs annotated as cellular components of the glutamatergic synapse further supports the potential role of FXR2P in SE. Excessive activity or dysfunction of the ionotropic glutamate receptors kainate, AMPA and NMDA has been linked with epileptiform activity (Chapman, 2000; Vincent & Mulle, 2009; Di Bonaventura et al, 2017). In fact, we found that, in Fxr2 KO hippocampi, levels of mGluR5 and GluN2B glutamatergic receptors and of the PSD‐95 scaffolding protein involved in glutamatergic signaling were decreased under baseline conditions, and ERK1/2 phosphorylation remained low after KA administration. Thus, a reduced KA‐dependent glutamatergic signaling followed by lack of ERK1/2 phosphorylation in KA‐treated Fxr2 KO mice may lead to reduced excitation and failure to maintain SE (Figure 7).
The ILAE defines SE as “a condition resulting either from the failure of the mechanisms responsible for seizure termination or from the initiation of mechanisms, which lead to abnormally prolonged seizures (after time point t1). It is a condition, which can have long‐term consequences (after time point t2), including neuronal death, neuronal injury and alteration of neuronal networks” (Trinka et al, 2015). With this definition, t1 indicates when a seizure prolongs into SE, and t2 when SE may cause long term consequences.
Both WT and Fxr2 KO mice developed motor seizures of Racine stage 3 or above, suggesting similar propensity to develop seizures. However, while motor seizures persisted and developed into motor SE in the WT, SE does not occur in the Fxr2 KO mice.
Kainic acid acts through the kainate receptors (Falcón‐Moya et al, 2018), and we found that the only kainate receptor among the FXR2P targets, GluK5, was unchanged in WT and Fxr2 KO hippocampi, further substantiating that seizure initiation is not affected.
The maintenance of seizure activity is sustained by several glutamatergic receptors, i.e., AMPA, GluN2B, and mGluR5 (He et al, 2004; Chen et al, 2007; Chen et al, 2016b; Sethna et al, 2017). An increased relocation of NR2B‐containing NMDA receptors to the cell membrane was observed in hippocampal slices taken from rats that underwent 1 h of SE. These contribute to the continued/increased excitability of these neurons (Naylor et al, 2013; Wasterlain et al, 2013), because increased expression of GluN2B in humans and mice is linked with increased seizure susceptibility and epilepsy (Nateri et al, 2007; Frasca et al, 2011; Zhand et al, 2018), and GluN2B blockade is sufficient to reduce in vivo late phase seizures and induce in vitro depotentiation during epileptic activity (Hellier et al, 2009; Chen et al, 2016a). With respect to mGluR5, inhibition of this receptor was shown to reduce seizures in animal models, whereas mGluR5 activation exacerbates the seizure phenotype (Medina‐Ceja & García‐Barba, 2017; Kazim et al, 2017; Kelly et al, 2018). In Fxr2 KO hippocampi, we observed reduced protein levels of both GluN2B and mGluR5 receptors. Thus, the reduced severity of late phase SE (i.e., > 1 h) observed in Fxr2 KO mice and the reduced neuronal excitation in Fxr2 KO hippocampal slices may be attributed to a reduced expression of GluN2B and mGluR5. Moreover, membrane surface stability of NMDA receptors is warranted by a functional interaction with PSD‐95 (Won et al, 2016), and this can further reduce the efficacy of these receptors in promoting protracted seizures. Interestingly, initially reduced GluN2B expression could be restored through stimulation, possibly through FXR2P‐independent mechanisms such as p‐ERK1/2 (Nateri et al, 2007). However, PSD‐95 and mGluR5 expression remained low in Fxr2 KO hippocampus, even upon KA stimulation. PSD‐95, and other scaffolding proteins target of FXR2P such as Shank1, allow the communication between NMDA and mGluR5 and the downstream signaling molecules. Lower expression of these scaffolding proteins could also contribute to the reduced signaling leading to seizures.
AMPA receptors are also required to maintain SE (Chen et al, 2007). Two other reports observed reduced GluA1 subunit (encoded by Gria1) protein levels in the hippocampus in Fxr2 KO mice (Cavallaro et al, 2008; Guo et al, 2015). In our analysis, GluA1 mRNA was not significantly enriched upon FXR2P mRNP complex isolation, possibly because the whole forebrain was used, whereas the above studies were specifically performed in the hippocampus. Because the changes in GluN2B, mGluR5, and PSD‐95 were only detected at the protein level and not at the mRNA level, it can be hypothesized that FXR2P is responsible for the translation of this group of mRNAs encoding proteins affecting the glutamatergic synapse. This significantly reduced glutamatergic response may prevent long‐term maintenance of seizure activity.
Protracted seizures are further maintained through phosphorylation of ERK1/2 downstream of glutamate receptors. ERK‐mediated signaling is an important pathway that regulates various intracellular functions and is implicated in epilepsy (Nateri et al, 2007; Curia et al, 2013; Chen et al, 2016b). Indeed, direct inhibition of ERK1/2 activity was shown to efficiently block seizure manifestation and phospho‐ERK1/2‐dependent transcriptomic changes (Glazova et al, 2015; Dorofeeva et al, 2017; Blüthgen et al, 2017). Phosphorylated ERK1/2 levels increased similarly in WT and Fxr2 KO hippocampi during the initial phase of SE. After 40 min however, ERK1/2 phosphorylation decreased in Fxr2 KO whereas it remained high in WT. This is consistent with the reduced expression of GluN2B and mGluR5 and, ultimately, with the reduced severity of SE. ERK1/2 is activated by upstream receptors, but in turn can stimulate NMDA receptor activity, initiating a self‐sustaining loop of neuronal hyperactivity (Nateri et al, 2007). This self‐sustaining loop may be compromised in Fxr2 KO mice.
While Grin2b and Grm5 are validated FXR2P mRNA targets and shared with epilepsy risk genes, it is possible that the absence of FXR2P has a more pleiotropic effect. Indeed, PSD‐95, albeit not in the list of epilepsy risk genes, is significantly reduced in Fxr2 KO hippocampi and has an impact on glutamatergic signaling, because it reduces the formation of functional GluN2B‐PSD95 complexes and decreases the physical connection between mGluR5 and GluN2B (Tu et al, 1999; Li et al, 2003; Bertaso et al, 2010; D'Mello et al, 2011; Won et al, 2016). Further, we noted that an additional 50 mRNAs that are FXR2 targets have a role in glutamatergic signaling (Figure 3D, Dataset EV2). Therefore, we conclude that dysregulation of multiple components of the glutamatergic synapse impacts the strength of glutamatergic signaling and contributes to the reduced seizure response.
Pilocarpine‐treated WT and Fxr2 KO mice were not different in SE and phosphorylated ERK1/2 levels, suggesting a protection toward a specific pathway involved in epilepsy. The FXR2P transcriptome did not show gene ontologies enriched for cholinergic synapses or cholinergic processes. Both KA and pilocarpine are used to model temporal lobe epilepsy, but they differ significantly in their mode of action (see review Lévesque et al, 2016). First, KA activates both kainate and AMPA receptors and affects the glutamatergic system, while pilocarpine activates muscarinic receptors and affects the cholinergic system. Second, KA and pilocarpine induce a different electrographic rhythm prior to the first seizure characterized as fast (gamma, 30–80 Hz) and slow (theta, 6–10 Hz) wave oscillations, respectively (Turski et al, 1989; Medvedev et al, 2000). Remarkably, gamma and theta oscillations activate GluN2 receptors through different mechanisms. The gamma oscillations induced by KA achieve it by strong post‐synaptic currents, whereas theta oscillations induced by pilocarpine trigger synchronized neuronal activity in optimized time windows and thus need less strong post‐synaptic responses (Larson & Munkácsy, 2015). This may explain why Fxr2 KO mice, which have weaker post‐synaptic responses, can progress to SE upon pilocarpine administration, but not upon KA.
Within the last decade, several RBPs have been identified and linked with epilepsy and seizures. The most‐studied RBP, FMRP, is linked to FXS, the most common known cause of inherited intellectual disability, where seizure susceptibility is hypothesized to be increased due to aberrant translation of a subset of mRNAs leading to ERK1/2 hyperphosphorylation (Berry‐Kravis et al, 2010; Sethna et al, 2014). Similarly, the absence of other RBPs (such as PUM2, RBFOX1, CELF4, BRUNOL4, among others) increase seizure susceptibility and spontaneous seizure occurrence, and were shown to regulate neuronal excitability in the limbic system (Yang et al, 2007; Wagnon et al, 2011; Follwaczny et al, 2017; Vuong et al, 2018). Whereas the loss of these RBPs decrease protein expression of inhibitory components and/or increase excitatory components leading to hyperexcitability, loss of FXR2P seems to produce primarily a negative regulation on the excitatory components, i.e., loss of FXR2P reduces expression of proteins that are involved in glutamate signaling in the hippocampus, leading to decreased excitation (Cavallaro et al, 2008). Interestingly, the absence of only one other RBP, i.e., HuR, has been reported to cause a similar, but less strong, protective effect on KA‐induced seizures (Skliris et al, 2015).
While epilepsy and SE are more frequently observed in men (Hesdorffer et al, 1998; Coeytaux et al, 2000), mutations in the X‐linked PCDH19 gene lead to early‐onset seizures specifically in women, leaving the male carrier largely unaffected (Dibbens et al, 2008). In this case, it is possibly due to tissue mosaicism of the X‐chromosome in women (Dibbens et al, 2008). FXR2, instead, is not located on the X chromosome but some of its targets, such as Hcfc1 and Huwe1 (Figure 4B and Dataset EV4), are in both mouse and human. Further studies are required to address this gender‐specific effect.
In conclusion, we provide evidence that Fxr2 KO mice do not undergo long‐lasting motor seizures. Even though we did not analyze long‐term consequences, we expect that the Fxr2 KO mice do not develop epilepsy, i.e., increased propensity to spontaneously seize long after the insult, which is normally observed after kainate‐induced SE (Lähteinen et al, 2002). This phenotypic trait may depend on reduced excitability due to reduced hippocampal glutamatergic signaling. In fact, Fxr2 KO hippocampi display reduced expression of glutamatergic components, including GluN2B and mGluR5. Also, coherently with the in vivo data, we detected a decreased global excitatory network activity in the Fxr2 KO hippocampi ex vivo, using the microelectrode array (MEA) that offers extracellular recording information on the network level with high spatial and temporal resolution. Our results provide a molecular and cellular framework for a mechanistic interpretation of the reduced susceptibility to prolonged SE and may also contribute to explain the epileptic phenotype reported in patients with the 17p13.1 deletion.
Materials and Methods
Animals
WT and Fxr2 <tm1Cgr> (129 * C57BL/6J * FVB genetic background) males were used in this study (Bontekoe et al, 2002). To acquire both WT and Fxr2 homozygous KO mice from the same litter, we paired Fxr2 heterozygous males with Fxr2 heterozygous females as a breeding scheme. Experimental mice were group‐housed in polyethylene cages (16 cm width × 28 cm length × 11 cm height; 448 cm2 living area) in groups of 3–5 mice and kept in a humidity and temperature‐controlled environment. The following primers were used to genotype the Fxr2 KO mouse colony: (i) 5′‐ACA‐GTT‐TCC‐TGC‐TTT‐ACA‐GTC‐CC‐3′, (ii) 5′‐AGG‐GGC‐TTC‐TGT‐GGT‐TGA‐CTG‐G‐3′ and (iii) 5′‐CTT‐TAC‐GGT‐ATC‐GCC‐GCT‐CCC‐GAT‐3′. Primers (ii) and (iii) amplified an Fxr2 WT allele of 773 base pairs. Primers (i) and (ii) amplified an Fxr2 KO allele of 220 base pairs, which binds to a Neo cassette that replaced exon 7 of the Fxr2 gene. Animal care was conducted conform to the institutional guidelines in compliance with international laws and policies (European Community Council Directive 86/609, OJa L 358, 1, December 12, 1987; National Institutes of Health Guide for the Care and Use of Laboratory Animals, US National Research Council, 1996) and approved by the Institutional Ethical Board at the University of Leuven (Belgium) and University of Lausanne (Switzerland, VD3151). The genetic background of the strain used throughout the study was also subjected to a genetic screening performed by Charles River (Charles River Genetic Testing Services, Wilmington, MA, USA). The background strain is > 90% FVB congenic.
In vivo seizure induction with kainic acid and pilocarpine
Over a 2‐h time course, seizure severity was scored based on a modified Racine method (Racine, 1972; Janumpalli et al, 1998): (0) normal behavior, (1) chewing and drooling, (2) head nodding, (3) unilateral forelimb clonus, (4) bilateral forelimb clonus, (5) forelimb and/or hindlimb clonus with falling, (6) running or jumping seizure, and (7) tonic hindlimb extension (Figure 2A). Seizures scored equal to or above three are convulsive and SE is defined as at least 5 min of uninterrupted convulsive seizures (Covolan & Mello, 2000; Sharma et al, 2018). The cumulative score was calculated as the sum of the maximal scores recorded with 5‐min intervals (total of 24 time points) following KA or pilocarpine injection (Bregola et al, 2002). Fxr2 KO (n = 12) and WT (n = 16) male mice of 8–12 weeks were injected intraperitoneally with 20 mg/kg KA (dissolved in PBS equivalent to 1% body weight; Tocris Bioscience, category number 0222/1) (Janumpalli et al, 1998; McCord et al, 2008; Schauwecker, 2010). Fxr2 KO and WT male mice (n = 6) were used as controls and were injected intraperitoneally with PBS vehicle. Pilocarpine, a muscarinic cholinergic agonist, was purchased at Sigma‐Aldrich (category number P6503). Fxr2 KO (n = 8) and WT (n = 9) male mice of 8–12 weeks were first injected intraperitoneally with 1 mg/kg of methylscopolamine (dissolved in PBS equivalent to 1% body weight; Sigma‐Aldrich, category number 28502) to minimize the peripheral effects of cholinergic stimulation. Afterward, mice were injected intraperitoneally with 100 mg/kg pilocarpine twice with 20 min between injections (Clasadonte et al, 2016). Fxr2 KO and WT male mice (n = 4) were used as controls and were injected intraperitoneally with PBS vehicle. To avoid mortality of the animals during the experiment as much as possible, we have included a humane endpoint—in agreement with the approved license for animal experimentation. Specifically, when a mouse experiences 15 consecutive minutes of Racine score 3 or above, the experiment will be prematurely terminated, the mouse euthanized and excluded from any further analyses (Fxr2 KO n = 2, WT n = 3). After the seizure experiment, a subset of six convulsant‐treated mice per genotype and all control mice were sacrificed by cervical dislocation, brains were removed and prepared for Western blot analyses.
To explore the changes of molecular markers over time during KA treatment, we additionally sacrificed WT and Fxr2 KO mice at different time intervals after KA injection, i.e., 20, 40, and 60 min. On each time interval, six mice of each genotype were used, and six untreated WT and Fxr2 KO mice served as baseline controls. Mice were sacrificed by cervical dislocation, brains removed and prepared for Western blot analyses.
Brain extract preparation
WT and Fxr2 KO mouse forebrains and hippocampi were lysated in 50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 0.3% NP‐40, 1 mM DTT in the presence of one tablet of protease inhibitors/10 ml of the solution, 40 U/ml RNAse‐out, for 10 min on ice and centrifuged at 12,000 g for 10 min at 4°C. The soluble fraction was used for Western blotting or immunoprecipitation (IP). For Western blotting, 20–25 µg of protein were loaded into a 10% sodium dodecyl sulfate‐polyacrylamide gel and subsequently were transferred onto a Polyvinylidene difluoride (PVDF) membrane (110 V at 4°C for 1 h 20 min). Membranes were blocked with 5% milk or bovine serum albumin (BSA) NCP‐Tween20 solution for 1 h and were subsequently incubated in NCP‐Tween20 solution with primary antibodies overnight. Table EV1 provides the antibodies and concentrations used in this study. Membranes were washed three times for 10 min with NCP‐Tween20 prior to 1 h incubation with a secondary antibody (anti‐mouse IgG and anti‐rabbit IgG coupled to Dylight 680 or Dylight 800, dilution 1:5,000). After a final series of three 10‐min washes, the membranes were scanned using an Odyssey infrared imager (Li‐COR Biosciences) and proteins were quantified using ImageQuant. PageRuler Prestained Protein Ladder (from Thermo Scientific, no. 26616) was used to evaluate the molecular weight of the proteins.
RNA immunoprecipitation and second‐generation sequencing
FXR2P immunoprecipitation (IP) followed by RNA isolation and sequencing was performed from mice forebrain cytoplasmic extracts (see brain extract preparation). Soluble lysates were incubated with protein‐G Dynabeads coated with antibodies that recognized FXR2P (1G2, Hybridoma Bank) for 1 h at 4°C. The beads were then washed with 50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.05% NP‐40, the complexes were incubated with 20 units of RNase‐free DNase I (15 min at 37°C) and further incubated with 0.1% SDS/0.5 mg/ml Proteinase K (15 min at 55°C) to remove DNA and proteins, respectively. The RNA from the immunoprecipitated elution as well as a portion of initial lysate (input) was extracted with phenol‐chloroform and ethanol precipitated. The RNAs were then converted to full‐length cDNAs and amplified using the SMARTer Ultra Low RNA Kit (Clontech).
The RNA libraries were prepared for sequencing using TrueSeq RNA Sample Prep (Illumina, CA, USA). The resulting cDNA was then subjected to DNA shearing and library preparation using Nextera XT DNA library prep kit (Illumina). The library was sequenced on an Illumina HiSeq2000 as single‐end reads of 50 bp. All preparations were performed by the Genomic Core facility of KU Leuven (Belgium). The sequenced reads were aligned to the latest reference assembly for mouse genome (GRCm38) using the STAR algorithm (Dobin et al, 2013). The number of reads per transcript was calculated with the open‐source HTSeq Python library (Anders et al, 2015).
All genes with a total expression higher than 30 reads per millions (RPM) in all samples were included in the analysis (14,809 genes). Read counts were normalized by library size using DESeq2, an R/Bioconductor package (Love et al, 2014). Differentially expressed genes between WT and Fxr2 KO samples were identified based on their weight in the differential pairwise expression analysis with the alternative hypothesis “greater than” and a false discovery rate (FDR)‐corrected P value of lower than 0.1 using the Benjamini–Hochberg adjustment. This adjusted P‐value of 0.1 is conventionally used in the field for multiple testing correction and is also recommended by the authors of the DESeq2 package. The identified candidate FXR2P mRNA targets were interrogated for statistically significant gene ontologies using GSEA (http://software.broadinstitute.org/gsea/index.jsp). As background gene list for the Gene Ontology term analysis, we used a total of 14,809 genes (i.e., genes that have a minimum of 30 reads in both WT and Fxr2 KO). For gene ontology enrichment, the top 10 “cellular components” and all the “biological processes” that contain the term glutamate were highlighted.
Real‐time qPCR
Total RNA was extracted from 50 μg brain lysate using 1 ml TRIzol reagent (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Before proceeding with the RT–qPCR, RNA concentration was determined with the NanoDrop2000 UV‐Vis Spectrophotometer (Thermo, USA). Total RNA was diluted to prepare aliquots of 700 ng/10 μl and used in RT–qPCR for 1 h 30 min at 37°C, using random primers, M‐MLV enzyme (Invitrogen), buffer 5× M‐MLV reaction buffer, RNase OUT and dNTPs, according to manufacturer's instructions (Invitrogen). cDNA was diluted 1:50 and 5 μl cDNA was used for each 15 μl qPCR. RT–qPCRs were performed on a Bio‐Rad CXF instrument with the SYBR‐green PCR mix (Roche, Switzerland). For each gene, the primer pairs were designed using the NCBI primer Blast algorithm (http://www.ncbi.nlm.nih.gov/tools/primer‐blast; Table EV2). Primer pairs were synthesized by IDT (Belgium) and Microsynth AG (Switzerland). Two technical replicates for each biological replicate were assessed. The mRNA expression level for each gene of interest was determined relative to a normalization factor (i.e., the average of the reference genes), using the method (Fernández et al, 2018).
Disease gene databases
We used DECIPHER (Database of genomic variation and phenotype in humans using Ensembl resources) to explore clinical phenotypes in male cases of 17p13.1 de novo microdeletion and microduplication (decipher.sanger.ac.uk) (Firth et al, 2009). We also consulted the CNV database of the center of Medical Genetics in Antwerp (Belgium) for additional cases, one female case with microduplication was identified (internal reference no. 119193, information kindly shared by Prof. Frank Kooy). Additionally, the Open Targets Platform was used as a secondary independent exploratory analysis for FXR2 disease association (www.targetvalidation.org; Carvalho‐Silva et al, 2019). Only sources from nervous system disorders that are not related to cancer or peripheral disorders were considered. The disease association score ranges from 0 to 1 with scores close to 1 corresponding to strong disease association and scores close to 0 corresponding to no evidence supporting an association. 1,089 Candidate genes of epilepsy were compiled based on the recent review on epilepsy‐associated genes, CT database (www.ctdbase.org; keywords used “Disease=name:Epilepsy” & “Gene‐disease association=curated”) and the EpilepsyGene database (wzgenomics.cn/EpilepsyGene) (Ran et al, 2015; Guo et al, 2017; Wang et al, 2017). The list of 122 genes associated with SE was taken from Bhatnagar and Shorvon (2015). The intersection between candidate FXR2 mRNAs and epilepsy gene was further analyzed for protein‐protein interactions using the String database (http://string‐db.org).
Microelectrode array recordings in vitro
To investigate neuronal excitability, we used the in vitro MEA recordings. WT and Fxr2 KO mice were anesthetized with isoflurane, the brain was quickly removed and transferred into bubbled ice‐cold 95% O2/5% CO2 equilibrated artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl, 2.5 KCl, 1 Na2HPO4, 26 NaHCO3, 11 glucose, 1.2 MgCl2, 0.6 CaCl2 (pH 7.3). Horizontal brain slices cut with an angle of 15° containing the hippocampus (350 μm) were prepared using a vibratome (Leica Biosystem) and the dissected hippocampus was incubated for 30 min at RT in standard aCSF. Slices were afterward transferred into magnesium‐free aCSF containing 100 μM picrotoxin (a GABAA receptor antagonist) for at least 1 h at RT before recording. A single hippocampal slice was placed in a submerged chamber (MultiChannel System, Reutlingen, Germany) and the CA3‐CA1 circuit was carefully positioned on top of an 8 × 8 electrode grid layout (60 TiN electrodes, electrode distance 200 μm, electrode diameter 30 μm). The slices were held in place with a heavy platinum anchor and perfused with 0‐Mg picrotoxin aCSF at a rate of 2 ml/min at RT. After 30 min of stabilization, hippocampal activity was sampled at 10 kHz and recorded with a MEA2100‐acquisition System for approximately 30 min (MultiChannel System, Reutlingen, Germany). Data were acquired with the MultiChannel Experimenter 2.8 (MultiChannel System, Reutlingen, Germany) and extracted for analysis using the MultiChannel Analyzer 2.6 (MultiChannel System, Reutlingen, Germany). Bursts were analyzed following a low‐pass 100 Hz Bessel filter, and the threshold was set at −5.5 standard deviations of noise amplitude. Active electrodes were defined as electrodes with at least one spike. In total, 16 Fxr2 KO and 9 WT hippocampal slices were taken from 8 Fxr2 KO and 6 WT male mice of 8–12 weeks old. Data formatting, burst detection and rasterplot generation were performed with a custom‐written script in R (https://github.com/adrianclo/meaR).
Statistical analysis
Data are expressed as means ± SEM and analyzed with GraphPad Prism (version 8) and R (version 3.6.1). Differences between two groups were analyzed with two‐tailed Student's t‐test. We used two‐way ANOVA when two factors were analyzed simultaneously in the study (i.e., genotype and treatment). We used Repeated Measures ANOVA when a time component was involved. Tukey HSD or Bonferroni were used for post hoc comparisons. Relative frequency of observations was analyzed with the chi‐square test. Cumulative frequencies were analyzed with the Kolmogorov–Smirnov test. The significance of overlapping genes between the different datasets analyzed was calculated using Fisher's exact test considering the following variables: (i) total population size corresponding to all the immunoprecipitated mRNAs (N) = 14,809; (ii) FXR2P targets = 488. Differences of P < 0.05 were considered significant.
Author contributions
ACL and TA designed the experiments. ACL performed and analyzed the seizure experiments, the Western Blots and DECIPHER. NR performed the RIP‐seq experiment and qPCR validation. ACL and NR compiled the epilepsy risk gene list. DG performed and analyzed the qPCRs and Western Blots. MLH performed and analyzed the electrophysiology experiment. R scripts for MEA experiments by ACL. LT analyzed the RIP‐seq data. AB and MS shared preliminary data on the seizure phenotype in the Fxr2 KO. ACL, TA and CB wrote the manuscript. CB supervised the project. All co‐authors have read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
We are thankful to Annick Crevoisier and Eef Lemmens for excellent administrative support and to Jonathan Royaert, Karin Jonkers, Joanna Viguiè, Mélanie Reinero, and Christiane Devenoges for technical assistance with the mouse colonies. We thank Dr. Frank Kooy for sharing details on a female microduplication patient, Dr. Mark Fiers for preliminary analyses and Dr. Esperanza Fernández for initial discussions. We are grateful to Dr. Ron Stoop for critical reading of the manuscript and Dr. Rob Willemsen for providing the original breeders for the Fxr2 KO mouse strain. This work was supported by SNSF 310030‐182651, SNSF NCCR Synapsy 51NF40‐158776 and Etat de Vaud. NR was supported by the European BrainTrain initiative (FP7‐People‐ITN‐2008‐238055). Initial support was provided by VIB.
EMBO Reports (2021) 22: e51404.
Contributor Information
Tilmann Achsel, Email: tilmann.achsel@unil.ch.
Claudia Bagni, Email: claudia.bagni@unil.ch, Email: claudia.bagni@uniroma2.it.
Data availability
Data generated by this study are not deposited in public databases and are available upon request from the corresponding authors.
References
- Achsel T, Bagni C (2016) Cooperativity in RNA–protein interactions: the complex is more than the sum of its partners. Curr Opin Neurobiol 39: 146–151 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Zukin RS (2019) A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 101: 1070–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belligni EF, Di Gregorio E, Biamino E, Calcia A, Molinatto C, Talarico F, Ferrero GB, Brusco A, Silengo MC (2012) 790 Kb microduplication in chromosome band 17p13.1 associated with intellectual disability, afebrile seizures, dysmorphic features, diabetes, and hypothyroidism. Eur J Med Genet 55: 222–224 [DOI] [PubMed] [Google Scholar]
- Berry‐Kravis E (2002) Epilepsy in fragile X syndrome. Dev Med Child Neurol 44: 724–728 [DOI] [PubMed] [Google Scholar]
- Berry‐Kravis E, Raspa M, Loggin‐Hester L, Bishop E, Holiday D, Bailey DB (2010) Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil 115: 461–472 [DOI] [PubMed] [Google Scholar]
- Bertaso F, Roussignol G, Worley P, Bockaert J, Fagni L, Ango F (2010) Homer1a‐dependent crosstalk between NMDA and metabotropic glutamate receptors in mouse neurons. PLoS One 5: e9755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar M, Shorvon S (2015) Genetic mutations associated with status epilepticus. Epilepsy Behav 49: 104–110 [DOI] [PubMed] [Google Scholar]
- Blüthgen N, van Bentum M, Merz B, Kuhl D, Hermey G (2017) Profiling the MAPK/ERK dependent and independent activity regulated transcriptional programs in the murine hippocampus in vivo . Sci Rep 7: 45101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontekoe CJ, McIlwain KL, Nieuwenhuizen IM, Yuva‐Paylor LA, Nellis A, Willemsen R, Fang Z, Kirkpatrick L, Bakker CE, McAninch R et al (2002) Knockout mouse model for Fxr2: a model for mental retardation. Hum Mol Genet 11: 487–498 [DOI] [PubMed] [Google Scholar]
- Bregola G, Zucchini S, Rodi D, Binaschi A, D'Addario C, Landuzzi D, Reinscheid R, Candeletti S, Romualdi P, Simonato M (2002) Involvement of the neuropeptide nociceptin/orphanin FQ in kainate seizures. J Neurosci 22: 10030–10038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho‐Silva D, Pierleoni A, Pignatelli M, Ong CK, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E et al (2019) Open Targets Platform: new developments and updates two years on. Nucleic Acids Res 47: D1056–D1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro S, Paratore S, Fradale F, de Vrij FMSS, Willemsen R, Oostra BA (2008) Genes and pathways differentially expressed in the brains of Fxr2 knockout mice. Neurobiol Dis 32: 510–520 [DOI] [PubMed] [Google Scholar]
- Chapman AG (2000) Glutamate and epilepsy. J Nutr 130(4S Suppl): 1043S–1045S [DOI] [PubMed] [Google Scholar]
- Chen B, Feng B, Tang Y, You Y, Wang Y, Hou W, Hu W, Chen Z (2016a) Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time‐window. Exp Neurol 283: 29–38 [DOI] [PubMed] [Google Scholar]
- Chen JWY, Naylor DE, Wasterlain CG (2007) Advances in the pathophysiology of status epilepticus. Acta Neurol Scand 115: 7–15 [DOI] [PubMed] [Google Scholar]
- Chen X, Dong G, Zheng C, Wang H, Yun W, Zhou X (2016b) A reduced susceptibility to chemoconvulsant stimulation in adenylyl cyclase 8 knockout mice. Epilepsy Res 119: 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Chang YW, Huang YS (2019) Dysregulated translation in neurodevelopmental disorders: an overview of autism‐risk genes involved in translation. Dev Neurobiol 79: 60–74 [DOI] [PubMed] [Google Scholar]
- Clasadonte J, Morel L, Barrios‐Camacho CM, Chiang MSR, Zhang J, Iyer L, Haydon PG, Yang Y (2016) Molecular analysis of acute and chronic reactive astrocytes in the pilocarpine model of temporal lobe epilepsy. Neurobiol Dis 91: 315–325 [DOI] [PubMed] [Google Scholar]
- Coeytaux A, Jallon P, Galobardes B, Morabia A (2000) Incidence of status epilepticus in French‐speaking Switzerland: EPISTAR. Neurology 55: 693–697 [DOI] [PubMed] [Google Scholar]
- Conlon EG, Manley JL (2017) RNA‐binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev 31: 1509–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covolan L, Mello LEAM (2000) Temporal profile of neuronal injury following pilocarpine or kainic acid‐induced status epilepticus. Epilepsy Res 39: 133–152 [DOI] [PubMed] [Google Scholar]
- Curia G, Gualtieri F, Bartolomeo R, Vezzali R, Biagini G (2013) Resilience to audiogenic seizures is associated with p‐ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front Cell Neurosci 7: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RSG, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172: 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH (2011) Perturbing PSD‐95 interactions with NR2B‐subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther 19: 1780–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146: 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O, Darnell RB (2009) Discrimination of common and unique RNA‐binding activities among Fragile X mental retardation protein paralogs. Hum Mol Genet 18: 3164–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Wallace GC, Holmes C, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK et al (2012) Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220: 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, O'Brien TJ, Jette N, Scheffer IE, de Curtis M, Perucca P (2018) Epilepsy. Nat Rev Dis Prim 4: 18024 [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C et al (2008) X‐linked protocadherin 19 mutations cause female‐limited epilepsy and cognitive impairment. Nat Genet 40: 776–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonaventura C, Labate A, Maschio M, Meletti S, Russi E (2017) AMPA receptors and perampanel behind selected epilepsies: current evidence and future perspectives. Expert Opin Pharmacother 18: 1751–1764 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorofeeva NA, Grigorieva YS, Nikitina LS, Lavrova EA, Nasluzova EV, Glazova MV, Chernigovskaya EV (2017) Effects of ERK1/2 kinases inactivation on the nigrostriatal system of Krushinsky‐Molodkina rats genetically prone to audiogenic seizures. Neurol Res 39: 918–925 [DOI] [PubMed] [Google Scholar]
- Egbenya DL, Hussain S, Lai YC, Xia J, Anderson AE, Davanger S (2018) Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol Cell Neurosci 92: 93–103 [DOI] [PubMed] [Google Scholar]
- Falcón‐Moya R, Sihra TS, Rodríguez‐Moreno A (2018) Kainate receptors: role in epilepsy. Front Mol Neurosci 11: 217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández E, Gennaro E, Pirozzi F, Baldo C, Forzano F, Turolla L, Faravelli F, Gastaldo D, Coviello D, Grasso M et al (2018) FXS‐like phenotype in two unrelated patients carrying a methylated premutation of the FMR1 gene. Front Genet 9: 442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, Li KW, Rajan N, De Rubeis S, Fiers M, Smit AB, Achsel T, Bagni C (2015) FXR2P exerts a positive translational control and is required for the activity‐dependent increase of PSD95 expression. J Neurosci 35: 9402–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández E, Rajan N, Bagni C (2013) The FMRP regulon: from targets to disease convergence. Front Neurosci 7: 191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C (2007) The fragile X mental retardation protein‐RNP granules show an mGluR‐dependent localization in the post‐synaptic spines. Mol Cell Neurosci 34: 343–354 [DOI] [PubMed] [Google Scholar]
- Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, Pringsheim T, Lorenzetti DL, Jetté N (2017) Prevalence and incidence of epilepsy: a systematic review and meta‐analysis of international studies. Neurology 88: 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van VS, Moreau Y, Pettett RM, Carter NP (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet 84: 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follwaczny P, Schieweck R, Riedemann T, Demleitner A, Straub T, Klemm AH, Bilban M, Sutor B, Popper B, Kiebler MA (2017) Pumilio2‐deficient mice show a predisposition for epilepsy. Dis Model Mech 10: 1333–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca A, Aalbers M, Frigerio F, Fiordaliso F, Salio M, Gobbi M, Cagnotto A, Gardoni F, Battaglia GS, Hoogland G et al (2011) Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis 43: 507–515 [DOI] [PubMed] [Google Scholar]
- Gafurov B, Bausch SB (2013) GABAergic transmission facilitates ictogenesis and synchrony between CA3, hilus, and dentate gyrus in slices from epileptic rats. J Neurophysiol 110: 441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano L, Palestra F, Giuffrida Maria G, Molinaro A, Iodice A, Bernardini L, La Boria P, Accorsi P, Novelli A (2014) 17p13.1 microdeletion: genetic and clinical findings in a new patient with epilepsy and comparison with literature. Am J Med Genet Part A 164: 225–230 [DOI] [PubMed] [Google Scholar]
- Glazova MV, Nikitina LS, Hudik KA, Kirillova OD, Dorofeeva NA, Korotkov AA, Chernigovskaya EV (2015) Inhibition of ERK1/2 signaling prevents epileptiform behavior in rats prone to audiogenic seizures. J Neurochem 132: 218–229 [DOI] [PubMed] [Google Scholar]
- Gong XW, Li JB, Lu QC, Liang PJ, Zhang PM (2014) Effective connectivity of hippocampal neural network and its alteration in Mg2+‐free epilepsy model. PLoS One 9: e92961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Polich ED, Su J, Gao Y, Christopher DM, Allan AM, Wang M, Wang F, Wang G, Zhao X (2015) Fragile X proteins FMRP and FXR2P control synaptic GluA1 expression and neuronal maturation via distinct mechanisms. Cell Rep 11: 1651–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Shang D‐M, Cao J‐H, Feng K, He Y‐C, Jiang Y, Wang S, Gao Y‐F (2017) Identifying and analyzing novel epilepsy‐related genes using random walk with restart algorithm. Biomed Res Int 2017: 6132436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X‐PP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO (2004) Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43: 31–42 [DOI] [PubMed] [Google Scholar]
- Hellier JL, White A, Williams PA, Edward Dudek F, Staley KJ (2009) NMDA receptor‐mediated long‐term alterations in epileptiform activity in experimental chronic epilepsy. Neuropharmacology 56: 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T (2018) A brave new world of RNA‐binding proteins. Nat Rev Mol Cell Biol 19: 327–341 [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA (1998) Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology 50: 735–741 [DOI] [PubMed] [Google Scholar]
- Janumpalli S, Butler LS, MacMillan LB, Limbird LE, McNamara JO (1998) A point mutation (D79N) of the alpha2A adrenergic receptor abolishes the antiepileptogenic action of endogenous norepinephrine. J Neurosci 18: 2004–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jie X, Li C, Wang H, Zhang Q, Han Z (2018) Therapeutic effects of scoparone on pilocarpine (Pilo)‐induced seizures in mice. Biomed Pharmacother 97: 1501–1513 [DOI] [PubMed] [Google Scholar]
- Kazim SF, Chuang SC, Zhao W, Wong RK, Bianchi R, Iqbal K (2017) Early‐onset network hyperexcitability in presymptomatic Alzheimer’s disease transgenic mice is suppressed by passive immunization with anti‐human APP/Aβ antibody and by mGluR5 blockade. Front Aging Neurosci 9: 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E, Schaeffer SM, Dhamne SC, Lipton JO, Lindemann L, Honer M, Jaeschke G, Super CE, Lammers SH, Modi ME et al (2018) MGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology 43: 1457–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoike Y, Shimojima K, Liang JS, Fujii H, Maegaki Y, Osawa M, Fujii S, Higashinakagawa T, Yamamoto T (2010) A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J Hum Genet 55: 155–162 [DOI] [PubMed] [Google Scholar]
- Korotkov AA, Glazova MV, Nikitina LS, Dorofeeva NA, Kirillova OD, Chernigovskaya EV (2017) The role of ERK1/2 kinases in the molecular mechanisms of regulation of glutamatergic and GABAergic neurons during the development of convulsive seizures in Krushinskii‐Molodkina rats. Neurosci Behav Physiol 47: 311–320 [Google Scholar]
- Kuroda Y, Ohashi I, Tominaga M, Saito T, Nagai J‐I, Ida K, Naruto T, Masuno M, Kurosawa K (2014) De novo duplication of 17p13.1‐p13.2 in a patient with intellectual disability and obesity. Am J Med Genet Part A 164: 1550–1554 [DOI] [PubMed] [Google Scholar]
- Lähteinen S, Pitkänen A, Saarelainen T, Nissinen J, Koponen E, Castrén E (2002) Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci 15: 721–734 [DOI] [PubMed] [Google Scholar]
- Lam J, DuBois JM, Rowley J, González‐Otárula KA, Soucy JP, Massarweh G, Hall JA, Guiot MC, Rosa‐Neto P, Kobayashi E (2019) In vivo metabotropic glutamate receptor type 5 abnormalities localize the epileptogenic zone in mesial temporal lobe epilepsy. Ann Neurol 85: 218–228 [DOI] [PubMed] [Google Scholar]
- Larson J, Munkácsy E (2015) Theta‐burst LTP. Brain Res 1621: 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque M, Avoli M (2013) The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev 37: 2887–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque M, Avoli M, Bernard C (2016) Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J Neurosci Methods 260: 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Otsu Y, Murphy TH, Raymond LA (2003) Developmental decrease in NMDA receptor desensitization associated with shift to synapse and interaction with postsynaptic density‐95. J Neurosci 23: 11244–11254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Jacobi AA, Anderson SA, Lynch DR (2016) D‐serine and serine racemase are associated with PSD‐95 and glutamatergic synapse stability. Front Cell Neurosci 10: 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini I, Ivanovski I, Iodice A, Rosato S, Pollazzon M, Mussini M, Belligni EF, Coutton C, Marinelli M, Barbieri V et al (2016) Endocrinological abnormalities are a main feature of 17p13.1 microduplication syndrome: a new case and literature review. Mol Syndromol 7: 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord MC, Lorenzana A, Bloom CS, Chancer ZO, Schauwecker PE (2008) Effect of age on kainate‐induced seizure severity and cell death. Neuroscience 154: 1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Ceja L, García‐Barba C (2017) The glutamate receptor antagonists CNQX and MPEP decrease fast ripple events in rats treated with kainic acid. Neurosci Lett 655: 137–142 [DOI] [PubMed] [Google Scholar]
- Medvedev A, MacKenzie L, Hiscock JJ, Willoughby JO (2000) Kainic acid induces distinct types of epileptiform discharge with differential involvement of hippocampus and neocortex. Brain Res Bull 52: 89–98 [DOI] [PubMed] [Google Scholar]
- Mooneyham KA, Holden KR, Cathey S, Dwivedi A, Dupont BR, Lyons MJ (2014) Neurodevelopmental delays and macrocephaly in 17p13.1 microduplication syndrome. Am J Med Genet Part A 164: 2887–2891 [DOI] [PubMed] [Google Scholar]
- Nadler JV, Evenson DA, Smith ME (1981) Evidence from lesion studies for epileptogenic and non‐epileptogenic neurotoxic interactions between kainic acid and excitatory innervation. Brain Res 205: 405–410 [DOI] [PubMed] [Google Scholar]
- Nateri AS, Raivich G, Gebhardt C, Da Costa C, Naumann H, Vreugdenhil M, Makwana M, Brandner S, Adams RH, Jefferys JGR et al (2007) ERK activation causes epilepsy by stimulating NMDA receptor activity. EMBO J 26: 4891–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, Wasterlain CG (2013) Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis 54: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newland CF, Cull‐Candy SG (1992) On the mechanism of action of picrotoxin on GABA receptor channels in dissociated sympathetic neurones of the rat. J Physiol 447: 191–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R (2006) Picrotoxin‐like channel blockers of GABAA receptors. Proc Natl Acad Sci USA 103: 6081–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kosti I, Ares M, Cline M, Mandel‐Gutfreund Y (2014) RBPmap: a web server for mapping binding sites of RNA‐binding proteins. Nucleic Acids Res 42: W361–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernice HF, Schieweck R, Kiebler MA, Popper B (2016) mTOR and MAPK: from localized translation control to epilepsy. BMC Neurosci 17: 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Löscher W, Vezzani A, Becker AJ, Simonato M, Lukasiuk K, Gröhn O, Bankstahl JP, Friedman A, Aronica E et al (2016) Advances in the development of biomarkers for epilepsy. Lancet Neurol 15: 843–856. [DOI] [PubMed] [Google Scholar]
- Pugliatti M, Beghi E, Forsgren L, Ekman M, Sobocki P (2007) Estimating the cost of epilepsy in Europe: a review with economic modeling. Epilepsia 48: 2224–2233 [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972) Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281–294 [DOI] [PubMed] [Google Scholar]
- Ran X, Li J, Shao Q, Chen H, Lin Z, Sun ZS, Wu J (2015) EpilepsyGene: a genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res 43: D893–D899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A et al (2013) A compendium of RNA‐binding motifs for decoding gene regulation. Nature 499: 172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE (2010) Neuroprotection by glutamate receptor antagonists against seizure‐induced excitotoxic cell death in the aging brain. Exp Neurol 224: 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluth‐Bolard C, Sanlaville D, Labalme A, Till M, Morin I, Touraine R, Edery P (2010) 17p13.1 microdeletion involving the TP53 gene in a boy presenting with mental retardation but no tumor. Am J Med Genet Part A 152: 1278–1282 [DOI] [PubMed] [Google Scholar]
- Sethna F, Feng W, Ding Q, Robison AJ, Feng Y, Wang H (2017) Enhanced expression of ADCY1 underlies aberrant neuronal signalling and behaviour in a syndromic autism model. Nat Commun 8: e14359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna F, Moon C, Wang H (2014) From FMRP function to potential therapies for fragile X syndrome. Neurochem Res 39: 1016–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Puttachary S, Thippeswamy A, Kanthasamy AG, Thippeswamy T (2018) Status epilepticus: behavioral and electroencephalography seizure correlates in kainate experimental models. Front Neurol 9: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skliris A, Papadaki O, Kafasla P, Karakasiliotis I, Hazapis O, Reczko M, Grammenoudi S, Bauer J, Kontoyiannis DL (2015) Neuroprotection requires the functions of the RNA‐binding protein HuR. Cell Death Differ 22: 703–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Serysheva E, Yuva‐Paylor LA, Oostra BA, Nelson DL, Paylor R (2006) Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X‐related proteins. Hum Mol Genet 15: 1984–1994 [DOI] [PubMed] [Google Scholar]
- Tamanini F, Van Unen L, Bakker C, Sacchi N, Galjaard H, Oostra BA, Hoogeveen AT (1999) Oligomerization properties of fragile‐X mental‐retardation protein (FMRP) and the fragile‐X‐related proteins FXR1P and FXR2P. Biochem J 343(Pt 3): 517–523 [PMC free article] [PubMed] [Google Scholar]
- Thomas RH, Berkovic SF (2014) The hidden genetics of epilepsy ‐ a clinically important new paradigm. Nat Rev Neurol 10: 283–292 [DOI] [PubMed] [Google Scholar]
- Toth M (2001) RNA binding proteins in epilepsy. Gene Func Dis 2: 95–98 [Google Scholar]
- Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH (2015) A definition and classification of status epilepticus ‐ report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 56: 1515–1523 [DOI] [PubMed] [Google Scholar]
- Trinka E, Kälviäinen R (2017) 25 years of advances in the definition, classification and treatment of status epilepticus. Seizure 44: 65–73 [DOI] [PubMed] [Google Scholar]
- Tu J, Xiao B, Naisbitt S, Yuan J, Petralia R, Brakeman P, Doan A, Aakalu V, Lanahan A, Sheng M et al (1999) Coupling of mGluR/Homer and PSD‐95 complexes by the Shank family of postsynaptic density proteins. Neuron 23: 583–592 [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA (1989) Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse 3: 154–171 [DOI] [PubMed] [Google Scholar]
- Vincent P, Mulle C (2009) Kainate receptors in epilepsy and excitotoxicity. Neuroscience 158: 309–323 [DOI] [PubMed] [Google Scholar]
- Vuong CK, Wei W, Lee JA, Lin CH, Damianov A, de la Torre‐Ubieta L, Halabi R, Otis KO, Martin KC, O’Dell TJ et al (2018) Rbfox1 regulates synaptic transmission through the inhibitory neuron‐specific vSNARE Vamp1. Neuron 98: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Mahaffey CL, Sun W, Yang Y, Chao HT, Frankel WN (2011) Etiology of a genetically complex seizure disorder in Celf4 mutant mice. Genes, Brain Behav 10: 765–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lin ZJ, Liu L, Xu HQ, Shi YW, Yi YH, He N, Liao WP (2017) Epilepsy‐associated genes. Seizure 44: 11–20 [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Naylor DE, Liu H, Niquet J, Baldwin R (2013) Trafficking of NMDA receptors during status epilepticus: Therapeutic implications. Epilepsia 54: 78–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Incontro S, Nicoll RA, Roche KW (2016) PSD‐95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc Natl Acad Sci USA 113: E4736–4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mahaffey CL, Bérubé N, Maddatu TP, Cox GA, Frankel WN (2007) Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet 3: e124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeesman S, Kjaergaard S, Hove HD, Kirchhoff M, Stevens JM, Nowaczyk MJMJM (2012) Microdeletion in distal 17p13.1: A recognizable phenotype with microcephaly, distinctive facial features, and intellectual disability. Am J Med Genet Part A 158: 1832–1836 [DOI] [PubMed] [Google Scholar]
- Zhand A, Sayad A, Ghafouri‐Fard S, Arsang‐Jang S, Mazdeh M, Taheri M (2018) Expression analysis of GRIN2B, BDNF, and IL‐1β genes in the whole blood of epileptic patients. Neurol Sci 39: 1945–1953 [DOI] [PubMed] [Google Scholar]
- Zhang J, Hou L, Klann E, Nelson DL (2009) Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol 101: 2572–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, O'Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G (1995) The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J 14: 5358–5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, Meng H, Tang B, Li BM, Liu XR et al (2018) Novel mutations and phenotypes of epilepsy‐associated genes in epileptic encephalopathies. Genes, Brain Behav 17: e12456 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Data Availability Statement
Data generated by this study are not deposited in public databases and are available upon request from the corresponding authors.