

Abstract
Reduction of mitochondrial membrane potential (Δψm) is a hallmark of mitochondrial dysfunction. It activates adaptive responses in organisms from yeast to human to rewire metabolism, remove depolarized mitochondria, and degrade unimported precursor proteins. It remains unclear how cells maintain Δψm, which is critical for maintaining iron‐sulfur cluster (ISC) synthesis, an indispensable function of mitochondria. Here, we show that yeast oxidative phosphorylation mutants deficient in complex III, IV, V, and mtDNA, respectively, exhibit activated stress responses and progressive reduction of Δψm. Extensive omics analyses of these mutants show that these mutants progressively activate adaptive responses, including transcriptional downregulation of ATP synthase inhibitor Inh1 and OXPHOS subunits, Puf3‐mediated upregulation of import receptor Mia40 and global mitochondrial biogenesis, Snf1/AMPK‐mediated upregulation of glycolysis and repression of ribosome biogenesis, and transcriptional upregulation of cytoplasmic chaperones. These adaptations disinhibit mitochondrial ATP hydrolysis, remodel mitochondrial proteome, and optimize ATP supply to mitochondria to convergently maintain Δψm, ISC biosynthesis, and cell proliferation.
Keywords: mitochondrial membrane potential, mitochondrial stress responses, oxidative phosphorylation
Subject Categories: Membrane & Intracellular Transport, Metabolism
Oxidative phosphorylation deficiency causes mitochondrial membrane potential (Δψm) reduction and activates transcriptional and post‐transcriptional adaptions to maintain Δψm.

Introduction
Oxidative phosphorylation (OXPHOS) system consists of electron transport chain (ETC) complexes I–IV and the ATP synthase (complex V). ETC complexes oxidize reducing equivalents (NADH and FADH2) and transport electrons to oxygen. Energy released by electron transport pumps protons from matrix to inter‐membrane space (IMS) to generate ∆ψm, which powers ATP synthesis by complex V. OXPHOS‐linked mitochondrial diseases are caused by mutations of over 150 genes; these genes encode OXPHOS subunits, cofactor synthesis, OXPHOS complex assembly factors, and mitochondrial gene expression system (mitochondrial DNA maintenance, transcription, and translation; Gorman et al, 2016; Craven et al, 2017).
Reduction of ∆ψm is a common and dangerous situation associated with OXPHOS impairment because ∆ψm drives the import of carriers and pre‐sequence guided mitochondrial precursors, including enzymes for iron‐sulfur cluster (ISC) synthesis (Chacinska et al, 2009; Pfanner et al, 2019). ISCs are essential cofactors that are synthesized in mitochondrial matrix and required for house‐keeping enzymes such as DNA polymerases and ribosome recycling factor (Lill, 2009). Reduction of ∆ψm compromises ISC synthesis to cause secondary defects in extra‐mitochondrial ISC‐containing enzymes (Veatch et al, 2009).
Oxidative phosphorylation impairment and the associated reduction of ∆ψm activate stress responses in diverse organisms. Early studies in yeast highlight that OXPHOS impairment activates a retrograde signaling pathway, the RTG pathway, to rewire metabolism (to synthesize glutamate; Epstein et al, 2001; Liu & Butow, 2006). Recent studies in yeast show that ∆ψm reduction as well as other forms of mitochondrial stresses impairs the import of mitochondrial precursor proteins to cause mPOS (mitochondrial precursor over‐accumulation stress; Wang & Chen, 2015) and activate the UPRam (unfolded protein response activated by mistargeting of proteins; Wrobel et al, 2015), as well as the mitoCPR (mitochondrial compromised protein import response; Weidberg & Amon, 2018) to activate proteasome, inhibit cytoplasmic protein synthesis, and degrade precursors accumulated in the cytosol and clogged in the TOM import channel. In C. elegans, genetic and chemical disruption of OXPHOS decreases the mitochondrial import of transcription factor ATFS‐1, causing its nuclear translocation to upregulate glycolysis, repress the transcription of mitochondrial OXPHOS subunits, and promote mitochondrial biogenesis and quality control (Nargund et al, 2012; Nargund et al, 2015). In multi‐cellular organisms such as C. elegans, D. melanogaster, and human, ∆ψm reduction stabilizes PINK1 kinase on mitochondrial outer membrane (OM) to recruit ubiquitin E3 ligases Parkin to activate proteosomal degradation of mitochondrial proteins and mitophagy (Youle & Narendra, 2011). Recent studies showed that in mammalian cells, OXPHOS impairment activates inner membrane (IM) metalloprotease OMA1 to cleave DELE1, which then translocates to the cytosol to activate integrative stress response (Fessler et al, 2020; Guo et al, 2020).
These progresses are remarkable examples demonstrating how cells deal with the consequence of OXPHOS impairment and ∆ψm reduction by removing toxic precursor proteins and degrading dysfunctional mitochondria. But none has directly addressed how to maintain ∆ψm. Here, we have characterized the global adaptations of yeast OXPHOS mutants through multiple‐omics approaches. We report the surprising finding that accompanying increasing proteostatic stresses and reduction of ∆ψm, OXPHOS mutants progressively activate increasing number of adaptive responses. We report the key pathways and effectors of these adaptations and show they convergently maintain ∆ψm.
Results
Decreasing ∆ψm and proliferation rates among OXPHOS mutants
To model OXPHOS deficiency, we deleted complex III subunit Cor1 (cor1∆), complex IV subunit Cox9 (cox9∆), ATP synthase stalk subunit Atp7 (atp7∆), and mtDNA (ρ0). These cells were incapable of proliferation under respiratory growth condition (YPEG) (Appendix Fig S1A). Because some OXPHOS subunits are required for mtDNA maintenance (Merz & Westermann, 2009), we mated cor1∆, cox9∆, and atp7∆ cells with ρ0 cells to examine their mtDNA quality. The diploid cells had normal growth on YPEG plates (Appendix Fig S1A), indicating these mutants have intact mtDNA. For simplicity, we labeled cor1∆, cox9∆, atp7∆ as ∆III, ∆IV, and ∆V. Cells were cultured in synthetic medium with 2% glucose (SCD) to OD600 0.4–0.6, and RNAseq and proteomic analyses were performed within this OD range. At this condition, ρ0 cells consumed < 15% of total glucose (Appendix Fig S1B) and maintained exponential growth.
Although these mutants had common impairment of OXPHOS, we were surprised to find that comparing with wild‐type (WT) cells, ∆III and ∆IV cells had slightly decreased ∆ψm and proliferation rates, ∆V cells had lower and ρ0 cells had the lowest ∆ψm and proliferation rates (Fig 1A and B). Two factors may contribute to the increasing difficultness/stresses to maintain ∆ψm and proliferation among these mutants. First, mtDNA encodes subunits of complex III, IV, and V, and mtrRNAs, as well as mttRNAs and several other proteins. ρ0 cells have multiple disassembled OXPHOS complexes and disassembled mtRibosome, thus more proteostatic stresses as compared to other OXPHOS mutants. Second, the differential ∆ψm maintenance mechanisms among these mutants may cause extra stresses in ∆V and ρ0 cells (Appendix Fig S1C). In ∆III and ∆IV cells, ∆ψm is maintained by the reversal of complex V to hydrolyze ATP and export protons to IMS (Chinopoulos & Adam‐Vizi, 2010). In this way, ∆III and ∆IV cells hydrolyze 1 ATP to export 3.33 protons (yeast complex V contains a c10 ring) and 1 positive charge (exchange of ADP3‐ with ATP4‐). ρ0 cells lose mtDNA‐encoded Fo components of complex V and maintain ∆ψm through F1‐dependent ATP hydrolysis (Buchet & Godinot, 1998; Appleby et al, 1999). In this way, mitochondria import 1 ATP4‐ at the exchange of 1 ADP3‐ through the ATP/ADP antiporter Aac2, thus importing 1 negative charge at the cost of 1 ATP (Appendix Fig S1C). Theoretically, ∆V cells block proton flux into matrix and thus should have elevated ∆ψm. However, ∆V cells had intermediate ∆ψm between ∆III/IV and ρ0 cells (Fig 1A). We observed that ∆V cells had greatly reduced complex IV level (Appendix Fig S1D), similar to other mutants of complex V subunits (Hadikusumo et al, 1988; Marzuki et al, 1989; Paul et al, 1989; Spannagel et al, 1997; Marsy et al, 2008). This indicates that ∆ψm of ∆V cells is not maintained by the ETC. Interestingly, ∆ψm and the proliferation of ∆V cells were significantly impaired by deleting AAC2 (Appendix Fig S1E and F), indicating ∆ψm of ∆V cells is maintained by mechanism similar to that of ρ0 cells.
Figure 1. Yeast OXPHOS mutants have progressive reduction of ∆ψm and cell proliferation, and increasing activation of transcriptional responses.
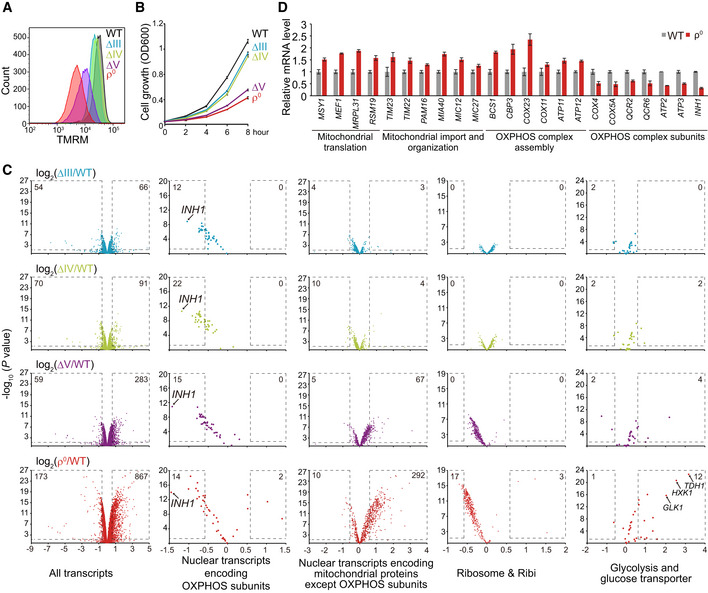
- Decreasing ∆ψm from ∆III to ρ0 cells. WT and mutant yeast cells were cultured to log phase and stained with TMRM for FACS analysis.
- Decreasing proliferation rates from ∆III to ρ0 cells. Cells were cultured from 0.05 OD600 in SCD. OD600 was measured every 2 h.
- Transcriptome analysis of OXPHOS mutants. WT and mutant yeast cells were cultured to log phase. RNAseq was performed with three biological replicates per genotype (Dataset EV1). All the transcripts and four categories of transcripts (gene lists in Table EV1B) are shown. Significantly changed genes (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed genes are indicated.
- qRT–PCR analysis of selected transcripts in ρ0 cells.
Data information: Data are mean ± SD from three biological replicates (B, D).
Source data are available online for this figure.
Progressive activation of transcription responses in OXPHOS mutants
We performed RNAseq analysis to understand how OXPHOS mutants respond to mitochondrial stresses (Fig 1C and Dataset EV1). mRNA level changes of selected transcripts in ρ0 cells were confirmed by qPCR analysis (Fig 1D). Apparently, there were increasing transcriptional changes from ∆III to ρ0 cells. In ρ0 cells, there were 173 downregulated transcripts and 867 upregulated transcripts reaching the significance threshold (P < 0.05, fold change > 1.5 or < 0.67). Gene ontology analysis (Ashburner et al, 2000) of transcripts with P < 0.05 in ρ0 cells showed that the three most downregulated biological processes all relate to ribosome biogenesis (Fig EV1A) and mainly consist of transcripts encoding ribosome subunits and ribosome processing and assembly (Ribi) factors. The most‐upregulated processes are mitochondrial translation, mitochondrial gene expression, and carbohydrate metabolic process (glucose transporters and glycolysis genes) (Fig EV1A). We further analyzed transcripts encoding mitochondrial proteins (gene list in Table EV1A) with P < 0.05 in ρ0 cells and showed the upregulation of mitochondrial gene expression, mitochondrial organization, and mitochondrial translation, and the downregulation of ATP metabolic process and oxidative phosphorylation (both consist of OXPHOS subunits) (Fig EV1B). We thus collected four gene lists for further analysis in all the OXPHOS mutants, these genes encode: nuclear‐encoded OXPHOS subunits, mitochondrial proteins other than OXPHOS subunits, ribosome subunits and Ribi factors, as well as glycolysis enzymes and glucose transporters (gene lists in Table EV1B).
Figure EV1. Transcriptome analysis of yeast OXPHOS mutants. Related to Fig 1 .
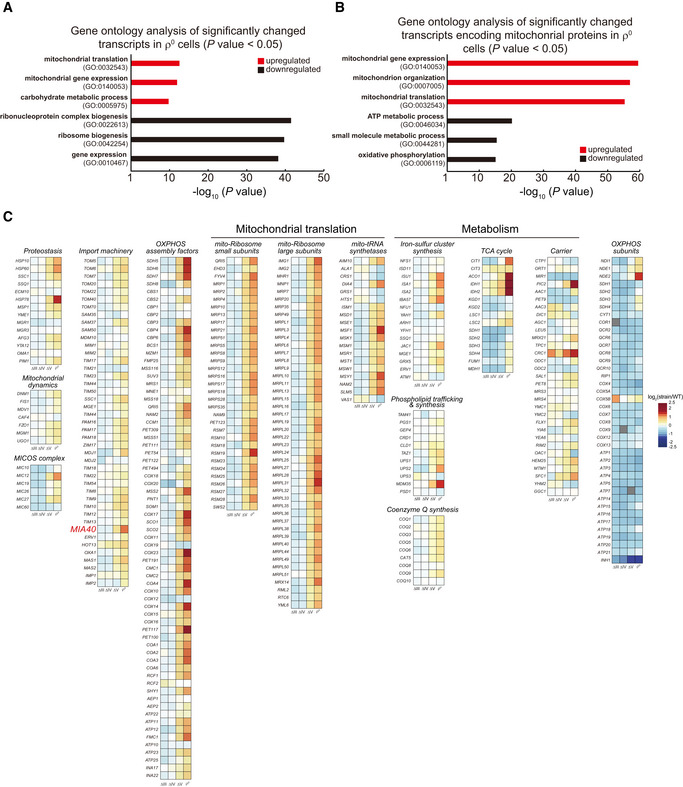
- Gene ontology analysis of significantly changed transcripts in ρ0 cells (P value < 0.05).
- Gene ontology analysis of significantly changed transcripts encoding mitochondrial proteins in ρ0 cells (P value < 0.05).
- Heat map plots of the mRNA level changes of genes encoding selected functional groups of mitochondrial proteins (gene list in Table EV1C) in the OXPHOS mutants. Gray boxes: deleted genes (COR1, COX9, and ATP7).
We observed interesting patterns of gene expression changes in these mutants (Fig 1C): (i) Nuclear transcripts encoding OXPHOS subunits were commonly downregulated in all the mutants. (ii) Nuclear transcripts encoding mitochondrial proteins except OXPHOS subunits were progressively upregulated in ∆V and ρ0 cells. We selected gene lists encoding several important pathways/functions of mitochondrial proteins (gene list in Table EV1C). The heat map plots of the mRNA level changes of these genes showed the increasing upregulation of mitochondrial proteostasis, import, organization, OXPHOS complex assembly, translation, and metabolism in ∆V and ρ0 cells (Fig EV1C). (iii) Transcripts encoding ribosomal subunits and Ribi factors were progressively downregulated in ∆V and ρ0 cells; (iv) Transcripts encoding glucose transporters and glycolysis enzymes were progressively upregulated in ∆V and ρ0 cells.
Proteome remodeling in OXPHOS mutants
To examine whether transcriptional changes remodel mitochondrial proteome, we performed stable isotope labeling using amino acids in cell culture (SILAC) analysis of ∆IV and ρ0 cells (Dataset EV2). Equal number of isotope‐labeled WT cells and isotope‐unlabeled mutant cells were mixed together. Mitochondrial fraction was purified for mass spectrometry analysis.
Our results showed that ∆IV cells had small changes of mitochondrial proteome with 17 downregulated proteins and 28 upregulated proteins reaching the significance threshold (P < 0.05, fold change > 1.5 or < 0.67). In contrast, 251 proteins were significantly upregulated and 84 proteins were significantly downregulated in ρ0 cells (Fig 2A). Integrating RNAseq and proteome results showed correlative changes of most mRNA and protein levels in ∆IV and ρ0 cells with two exceptions (Fig EV2). First, in ρ0 cells, some mtRibosome subunits were upregulated at both mRNA and protein levels, whereas the others were upregulated at mRNA but downregulated at protein levels. Because mtDNA encodes mtrRNAs, the scaffolds for mtRibosome assembly and function, we speculate the downregulated subunits may be destabilized by mtrRNA depletion. Second, in ρ0 cells, about half of the carriers showed discordant changes at mRNA and protein levels. The underlying reason remains unclear.
Figure 2. Remodeling of mitochondrial proteome and cytoplasmic ribosome in the OXPHOS mutants.
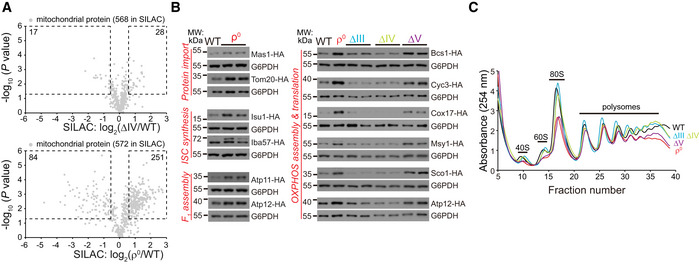
- SILAC analysis of mitochondrial proteome of ∆IV and ρ0 cells. Equal amount of WT cells labeled with heavy amino acids and unlabeled mutant cells were mixed to extract mitochondrial fraction for mass spectrometry analysis. Three biological replicates were prepared for each pair of strains (Dataset EV2). 568 high‐fidelity mitochondrial proteins (gene/protein list in Table EV1A) in ∆IV cells, and 572 such proteins in ρ0 cells were quantitatively measured. Significantly changed proteins (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed proteins are indicated.
- Western blot analysis of representative mitochondrial proteins endogenously tagged with HA. The selection markers were removed by the Cre‐loxP system. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Polysome fractionation analysis of OXPHOS mutants. Polysomes were fractionated by sucrose gradients (10%–50%), and the gradients were separated and measured at A254.
Source data are available online for this figure.
Figure EV2. Correlation between mRNA and protein level changes of selected mitochondrial proteins. Related to Fig 2.
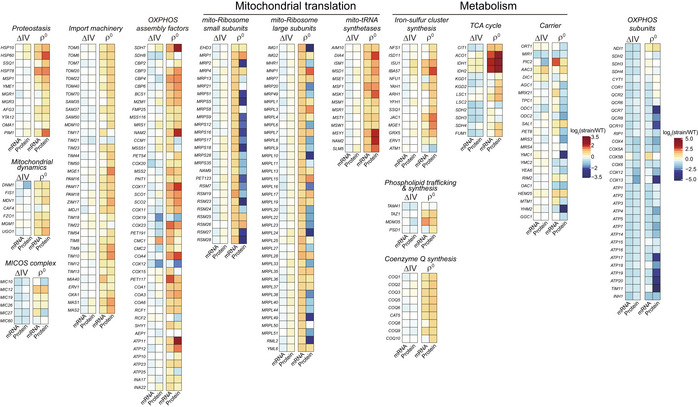
Heat map plots of the mRNA and protein level changes of genes encoding selected functional groups of mitochondrial proteins (gene list in Table EV1C) in the ∆IV and ρ0 cells.
We then tagged endogenous mitochondrial proteins with an HA tag and deleted selection marker with the Cre‐loxP system to preserve endogenous gene regulation. We confirmed the upregulation of mitochondrial import, ISC synthesis, and F1 assembly factors in ρ0 cells (Fig 2B). Importantly, the upregulation of mitochondrial proteins specifically occurred in ∆V and ρ0 cells but not in ∆III and ∆IV cells (Fig 2B), matching the RNAseq and SILAC results.
Ribosome biogenesis was progressively downregulated at transcript level (Fig 1C). Fractionation analysis showed that ∆III and ∆IV cells had similar polysome profiles as WT cells, but ∆V and ρ0 cells had significantly reduced ribosome contents, especially in the 40S, 60S, and 80S fractions (Fig 2C), correlating with transcript changes.
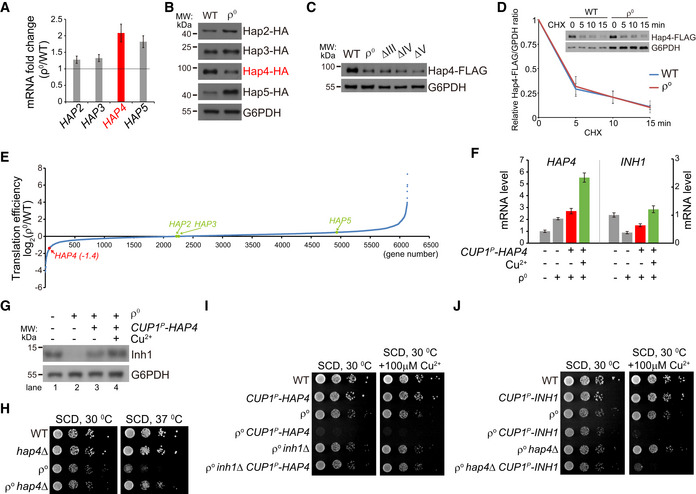
Transcriptional repression of Inh1, the ATP synthase inhibitor, maintains ∆ψm and proliferation of ρ0 cells
Downregulation of the transcript level of OXPHOS subunits happens in all the mutants (Fig 1C). We noticed the most downregulated one is Inh1 (Fig 1C), a conserved inhibitor of complex V (ATPIF1 in mouse and human) (Garcia‐Bermudez & Cuezva, 2016). Deletion of Inh1/ATPIF1 disinhibits the ATPase activity of F1 to maintain ∆ψm in both yeast and human cells (Clark‐Walker, 2007; Lefebvre et al, 2013; Chen et al, 2014; Martinez‐Reyes et al, 2016). But it is unclear whether Inh1/ATPIF1 is regulated by mitochondrial adaptive response.
We confirmed Inh1 downregulation at mRNA and protein levels in all the OXPHOS mutants (Fig 3A and B). Deleting Inh1 in ρ0 cells benefited cell proliferation at 37°C (Fig 3C). We then used copper‐inducible promoter to inducibly overexpress Inh1 (Fig 3D). Inh1 overexpression selectively inhibited ∆ψm and cell proliferation of ρ0 cells but had no effect in WT, ∆III, ∆IV, or ∆V cells (Fig 3E and F). To examine the detrimental effect of losing ∆ψm, we genetically tagged endogenous Nfs1 and Yah1, two ISC‐synthesis enzymes in mitochondrial matrix, and DNA polymerase Pol3, an essential nuclear ISC‐containing protein required for DNA replication (Lill, 2009), and examined their protein levels by Western blot. Inh1 overexpression in ρ0 cells greatly downregulated mitochondrial ISC‐synthesis enzymes Nfs1 and Yah1 and also downregulated nuclear ISC‐containing protein Pol3 (Fig 3G, lane 8 vs. 7). These results indicate ∆ψm reduction causes import defects to compromise ISC synthesis and destabilize ISC‐containing proteins. Taken together, OXPHOS mutants commonly downregulate Inh1 but this response is specifically critical for ρ0 cells to maintain ∆ψm and ISC synthesis, as well as cell proliferation.
Figure 3. Transcriptional repression of Inh1 maintains ∆ψm, ISC synthesis, and proliferation of ρ0 cells.

- qRT–PCR analysis of INH1 mRNA levels. Data are mean ± SD from three biological replicates.
- Western blot analysis of Inh1 protein levels. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Deletion of INH1 benefits the proliferation of ρ0 cells at 37°C. Serial dilutions (tenfold dilution) of the indicated strains were analyzed on SCD plates at 30°C and 37°C for 2 days.
- Western blot analysis of copper‐inducible overexpression of Inh1. A cassette expressing Inh1 under the control of CUP1 promoter was inserted into the HO locus. Inh1 overexpression was induced by 100 μM CuSO4 for 12 h.
- Overexpression of Inh1 decreases ∆ψm in ρ0 cells. Cells were cultured in SCD or SCD plus 100 μM CuSO4 for 12 h to mid‐log phase. Cells were then stained with 125 nM TMRM for FACS analysis.
- Overexpression of Inh1 represses the proliferation of ρ0 cells. Serial dilutions (tenfold dilution) of the indicated strains were analyzed on SCD or SCD plus 100 μM CuSO4 plates at 30°C for 2 days.
- Overexpression of Inh1 reduces the protein levels of mitochondrial ISC biosynthesis proteins (Nfs1 and Yah1) and nuclear ISC‐containing protein Pol3. Cells were cultured in SCD or SCD plus 100 μM CuSO4 for 12 h to mid‐log phase. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot. Nfs1 and Yah1 were endogenously tagged with FLAG, and Pol3 was endogenously tagged with HA.
- Cartoon illustration of the translational repression of Hap4 and the subsequent transcriptional downregulation of Inh1 to maintain ∆ψm in ρ0 cells.
Data information: The red highlight two cell lines for comparison (E, F).
Source data are available online for this figure.
Translational downregulation of HAP4 represses Inh1 transcription
To understand how Inh1 and OXPHOS subunits are downregulated at transcript level, we analyzed the HAP complex, consisting of Hap2‐5, which is responsible for the transcription of OXPHOS subunits (Boos et al, 2019; Mao & Chen, 2019). In ρ0 cells, most HAP subunits were upregulated at both mRNA and protein levels with one exception that Hap4 mRNA was increased twofold but its protein level was decreased (Fig EV3A and B). Downregulation of Hap4 protein was observed in all the OXPHOS mutants (Fig EV3C). In ρ0 cells, we did not detect change of Hap4 protein half‐life (Fig EV3D). We then performed the ribosome footprinting to measure genome‐wide changes of translation efficiency (Ingolia, 2010; Ingolia et al, 2012). ρ0 cells specifically downregulated the translation efficiency of Hap4 but not the other three HAP transcripts (Fig EV3E, Dataset EV3). The decrease of Hap4 translation efficiency exceeds the increase of its mRNA level, thus may cause a net reduction of Hap4 protein.
Figure EV3. Translational downregulation of Hap4 represses transcription of INH1. Related to Fig 3.
- qRT–PCR analysis of HAP complex components in ρ0 cells. Data are mean ± SD from three biological replicates. The horizontal line indicates fold change of 1.
- Western blot analysis of Hap proteins. Hap proteins were endogenously tagged with HA. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Western blot analysis of Hap4‐FLAG level in OXPHOS mutants. Hap4 was endogenously tagged with FLAG. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Protein half‐life of Hap4‐FLAG is not changed in ρ0 cells as compared to WT cells. Hap4 was endogenously tagged with FLAG. Cells were treated with cycloheximide (CHX) to block protein synthesis for the indicated time. Whole cell lysates were extracted for Western blot analysis. Protein samples of ρ0 cells were loaded twofold the amounts of samples of WT cells to equalize Hap4‐FLAG amount at 0 min of CHX treatment. Relative Hap4‐FLAG/G6PDH ratio at 0 min of CHX treatment was normalized as 1. Data are mean ± SD from three biological replicates.
- Genome‐wide translation efficiency changes in ρ0 cells versus WT cells (Dataset EV3). The translation efficiencies of Hap proteins are indicated.
- qRT–PCR analysis of HAP4 and INH1 mRNA levels upon Hap4 overexpression in the indicated strains. A cassette expressing Hap4 under the control of CUP1 promoter was inserted into the HO locus. Hap4 overexpression was induced by 100 μM CuSO4 for 12 h. Data are mean ± SD from three biological replicates.
- Western blot analysis of Inh1 level upon Hap4 overexpression in the indicated strains. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Deletion of HAP4 benefits the proliferation of ρ0 cells at 37°C. Serial dilutions (tenfold dilution) of the indicated strains were analyzed on SCD plates at 30°C and 37°C for 2 days.
- Overexpression of Hap4 inhibits the proliferation of ρ0 cells, but not the proliferation of ρ0 inh1∆ cells.
- Overexpression of Inh1 inhibits the proliferation of both ρ0 and ρ0 hap4∆ cells.
Data information: Serial dilutions (tenfold dilution) of the indicated strains were analyzed on SCD or SCD plus 100 μM CuSO4 plates at 30°C for 2 days (I, J).
Source data are available online for this figure.
To examine whether Hap4 level determines Inh1 transcription, we used the copper‐inducible CUP1 promoter to drive Hap4 expression in ρ0 cells. Without copper supplementation, we noticed the upregulation of HAP4 mRNA (Fig EV3F, left panel, highlighted in red) and the corresponding upregulation of INH1 mRNA (Fig EV3F, right panel, highlighted in red) and Inh1 protein (Fig EV3G, lane 3 vs. 2). This could be due to the tracing amount of copper in the synthetic medium. Copper supplementation further induced INH1 at mRNA (Fig EV3F, highlighted in green) and protein (Fig EV3G, lane 4 vs. 2) levels. Thus, Hap4 level is a key determinant of Inh1 transcription.
Functionally, deleting Hap4 benefited ρ0 cell proliferation at 37°C (Fig EV3H) and the leaky expression of Hap4 was sufficient to suppress ρ0 cell growth (Fig EV3I). Furthermore, deleting Inh1 completely abolished the growth suppression effect of Hap4 in ρ0 cells (Fig EV3I). Overexpression of Inh1 suppressed proliferation of ρ0 hap4∆ cells (Fig EV3J). These epistatic analyses demonstrate that Inh1 is the major downstream effector of Hap4. Taken together, ρ0 cells downregulate Hap4 translation to repress Inh1 transcription and thus to disinhibit the ATP hydrolysis activity of F1 component (Fig 3H).
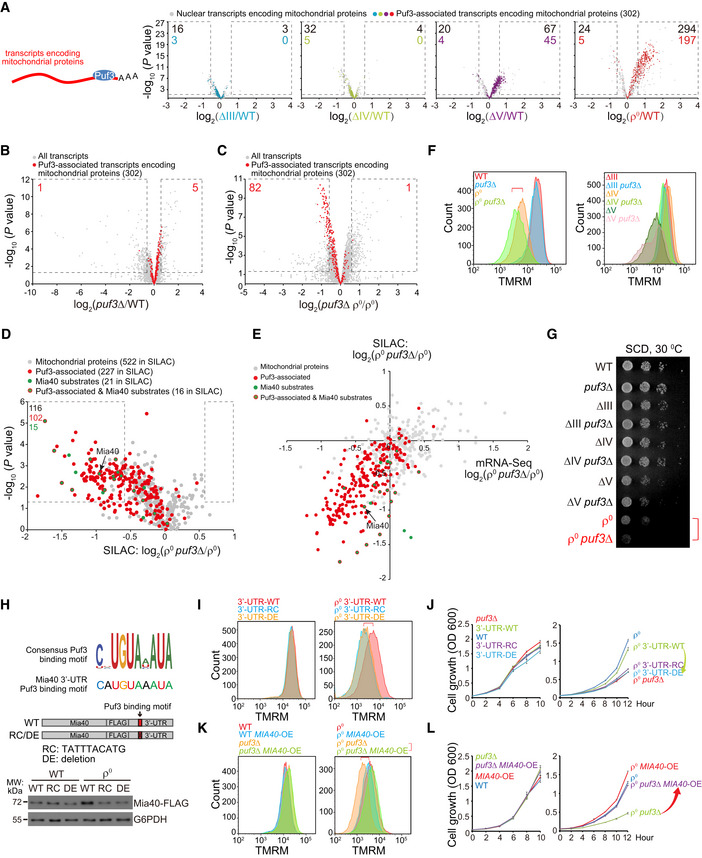
Puf3 promotes mitochondrial biogenesis to maintain ∆ψm and proliferation in ρ0 cells
Next, we focused on the global mitochondrial biogenesis response in ∆V and ρ0 cells. Puf3 is a cytoplasmic mRNA‐binding protein that mainly associates with mRNAs encoding mitochondrial proteins (Gerber et al, 2004; Lapointe et al, 2015; Lapointe et al, 2018). It promotes mRNA degradation under fermentative condition (Olivas & Parker, 2000; Houshmandi & Olivas, 2005) and gets phosphorylated to promote mRNA translation under respiratory growth condition (Lee & Tu, 2015). We combined two studies to generate a list of Puf3‐associated mRNAs (Table EV1D) (Gerber et al, 2004; Lapointe et al, 2015). We found that most‐upregulated transcripts encoding mitochondrial proteins in ∆V and ρ0 cells overlapped with transcripts known to associate with Puf3 (Fig 4A).
Figure 4. mRNA‐binding protein Puf3 upregulates Mia40 and global mitochondrial biogenesis to maintain ∆ψm .
-
AThe changes of Puf3‐associated transcripts encoding mitochondrial proteins in OXPHOS mutants. The RNAseq results in Fig 1C (Dataset EV1) are re‐analyzed. Transcripts encoding high‐fidelity mitochondrial proteins (gene list in Table EV1A) and the Puf3‐associated transcripts encoding mitochondrial proteins (gene list in Table EV1D) are shown. The significantly changed transcripts (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed transcripts are indicated.
-
B, CDeletion of PUF3 upregulates mitochondrial transcripts in WT cells (B), whereas downregulates mitochondrial transcripts in ρ0 cells (C). WT and mutant yeast cells were cultured to log phase. RNAseq was performed with three biological replicates per genotype (Dataset EV4). All the transcripts and the Puf3‐associated transcripts encoding mitochondrial proteins are shown. The significantly changed transcripts (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed transcripts are indicated.
-
DDeletion of PUF3 in ρ0 cells downregulates mitochondrial proteins. SILAC analysis of mitochondrial fractions from ρ0 and ρ0 puf3∆ cells are shown. Equal amount of ρ0 cells labeled with heavy amino acids and unlabeled ρ0 puf3∆ cells were mixed to extract mitochondrial fraction for mass spectrometry analysis. Three biological replicates were prepared for each pair of strains (Dataset EV5). Mitochondrial proteins encoded by Puf3‐associated mRNAs are highlighted in red. Mia40 substrates are highlighted in green (gene list in Table EV1E). Mia40 substrates encoded by Puf3‐associated transcripts are highlighted in green with red circle. The significantly changed proteins (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed proteins are indicated.
-
ECorrelation plots of fold changes (ρ0 puf3∆ versus ρ0) between mitochondrial proteins and their transcripts. Puf3‐associated transcripts encoding mitochondrial proteins are highlighted in red. Mia40 substrates are highlighted in green. Mia40 substrates encoded by Puf3‐associated transcripts are highlighted in green with red circle.
-
FDeletion of PUF3 decreases ∆ψm in ρ0 cells.
-
GDeletion of PUF3 decreases the proliferation of ρ0 cells. Serial dilutions (tenfold dilution) of the indicated strains were analyzed on SCD plates at 30°C for 2 days.
-
HSchematic illustration of the consensus Puf3‐binding motif (Lapointe et al, 2015) and the motif at Mia40 3’‐UTR. Mutating the Puf3‐binding motif of MIA40 mRNA abolishes Mia40 upregulation in ρ0 cells. Mia40 was endogenously tagged with FLAG, and its 3’‐UTR was edited as indicated. The selection markers were removed by the Cre‐loxP system. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot. RC: reverse complement. DE: deletion.
-
HSchematic illustration of the consensus Puf3‐binding motif (Lapointe et al, 2015) and the motif at Mia40 3’‐UTR. Mutating the Puf3‐binding motif of MIA40 mRNA abolishes Mia40 upregulation in ρ0 cells. Mia40 was endogenously tagged with FLAG, and its 3’‐UTR was edited as indicated. The selection markers were removed by the Cre‐loxP system. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot. RC: reverse complement. DE: deletion.
-
IMutating the Puf3‐binding motif of MIA40 mRNA decreases ∆ψm of ρ0 cells.
-
JMutating the Puf3‐binding motif of MIA40 mRNA impairs the proliferation of ρ0 cells. Cells were cultured from 0.05 OD in SCD, and OD600 was measured every 2 h.
-
KOverexpression of MIA40 rescues ∆ψm of ρ0 puf3∆ cells. Single copy plasmid expressing Mia40 driven by the MIA40 promoter was transformed into the indicated strains.
-
LOverexpression of MIA40 rescues the proliferation of ρ0 puf3∆ cells. Strains were prepared as in (K). Cells were cultured from 0.05 OD in SCD, and OD600 was measured every 2 h.
Data information: Cells were cultured to mid‐log phase and stained with 125 nM TMRM for FACS analysis (F, I, K). The red highlight two cell lines for comparison (F, G, I, K). Data are mean ± SD from three biological replicates (J, L).
Source data are available online for this figure.
To determine whether Puf3 is required for the upregulation of transcripts encoding mitochondrial proteins, we deleted PUF3 in both WT and ρ0 cells. RNAseq analysis (Dataset EV4) showed that deleting PUF3 modestly upregulated its associated transcripts in WT cells as previously reported (Olivas & Parker, 2000; Fig 4B), but significantly downregulated its associated transcripts (with 82 transcripts reaching the significance threshold) in ρ0 cells (Fig 4C), indicating Puf3 switches its role from promoting mRNA degradation in WT cells to mRNA stabilization in ρ0 cells.
Stable isotope labeling using amino acids in cell culture analysis of mitochondrial proteome showed that deleting PUF3 downregulated mitochondrial proteins in ρ0 cells with 116 proteins reaching significance threshold, among which 102 proteins are encoded by Puf3‐associated mRNAs (Fig 4D and Dataset EV5). Similar to ρ0 cells, ∆V cells also required Puf3 to upregulate mitochondrial mRNAs and proteins (Appendix Fig S2A and B).
Deleting PUF3 had no effect on ∆ψm and cell proliferation in WT, ∆III, ∆IV, or ∆V cells, but decreased both ∆ψm and cell proliferation in ρ0 cells (Fig 4F and G). Thus, Puf3 promotes a global mitochondrial biogenesis response in ∆V and ρ0 cells, but is only required in ρ0 cells to maintain ∆ψm and cell proliferation.
Puf3 may promote mitochondrial biogenesis through mRNA stabilization and/or promoting mRNA translation. Ribosome footprinting (Dataset EV3) showed that there was no general upregulation of the translation efficiency of Puf3‐associated transcripts in ρ0 cells (Fig EV4A and B), and deleting PUF3 in ρ0 cells did not affect the translation efficiency of its associated transcripts (Fig EV4C and D). Polysome fractionation experiments also showed that Puf3 did not enter into the polysome fractions after mtDNA depletion (Fig EV4E). Thus, Puf3 has minimal effect on the translation efficiency of its associated mRNAs in ρ0 cells, which is mechanistically different from the glucose deprivation condition, in which Puf3 promotes translation of its associated mRNAs (Lee & Tu, 2015).
Figure EV4. Ribosome footprinting and polysome analysis. Related to Fig 4.
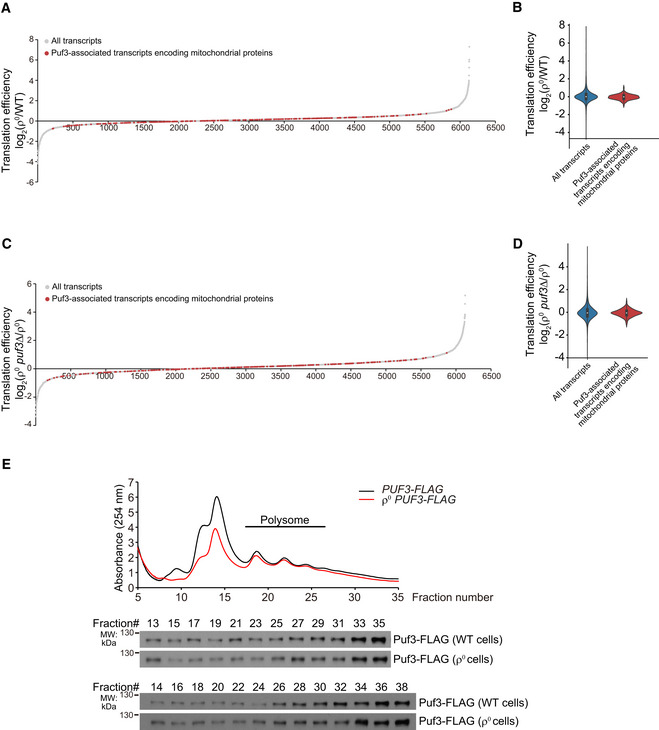
- Genome‐wide translation efficiency changes of ρ0 cells versus WT cells. The translation efficiencies of Puf3‐associated mitochondrial transcripts are highlighted in red. Translation efficiency was determined by ribosome footprinting (Dataset EV3).
- Violin plot of translation efficiency changes of ρ0 versus WT cells.
- Genome‐wide translation efficiency changes of ρ0 puf3∆ versus ρ0 cells. Translation efficiency was determined by ribosome footprinting (Dataset EV3).
- Violin plot of translation efficiency changes of ρ0 puf3∆ cells versus ρ0 cells.
- Polysome analysis of Puf3‐FLAG. Equal volume of lysates from the indicated fractions was loaded for Western blot analysis.
Source data are available online for this figure.
Mia40 is the critical mitochondrial protein upregulated by Puf3 to maintain ∆ψm and proliferation in ρ0 cells
It is intriguing that among the hundreds of mitochondrial mRNAs/proteins upregulated by Puf3, many of them, such as the mtRibosome subunits, seem to aggravate the folding stress inside mitochondrial matrix because many subunits cannot be properly assembled and are presumably degraded in ρ0 cells (Fig EV2). Similarly, upregulation of most OXPHOS assembly factors and mitochondrial tRNA synthetases in ρ0 cells seems to be a waste. Then, what factors upregulated by Puf3 help ∆ψm maintenance?
Because ∆ψm maintenance in ρ0 cells requires matrix F1 component and inner membrane (IM) carriers, enhancing the import pathway seems to be a meaningful response. In our RNAseq results, Mia40 was the most‐upregulated mitochondrial import factor in ρ0 cells (Fig EV1, highlighted in red in the “import machinery” category). Mia40 is an essential mitochondrial protein that mediates the import and oxidative folding of IMS proteins (Herrmann & Riemer, 2012; Peleh et al, 2016; Stojanovski et al, 2012). Its substrates include phospholipid carriers and small TIMs (Tim8, Tim9, Tim10, Tim12, and Tim13) critical for the biogenesis of OM β‐barrel proteins and IM carrier proteins (Koehler, 2004; Modjtahedi et al, 2016). Inactivation of Mia40 activates the UPRam (Wrobel et al, 2015). Thus, Mia40 has profound effect on mitochondrial proteome and phospholipids. MIA40 mRNA contains a Puf3‐binding site in its 3’‐UTR (Fig 4H). In our SILAC results, Mia40 and its substrates (gene list in Table EV1E) were among the significantly downregulated mitochondrial proteins upon PUF3 deletion in ρ0 cells (Fig 4, D and E, highlighted in green), supporting Mia40 as a substrate upregulated by Puf3. More importantly, many Mia40 substrates, including small TIMs, were downregulated more at protein levels as compared to mRNA levels in ρ0 puf3∆ cells (Fig 4E, highlighted in green), indicating Puf3 upregulates Mia40 to promote the import of Mia40 substrates in ρ0 cells.
We mutated the Puf3‐binding site in MIA40 mRNA 3’‐UTR by introducing reverse complement (RC) sequence or deletion (DE) (Fig 4H). Both manipulations did not affect the basal protein level of Mia40 in WT cells but selectively blocked Mia40 upregulation in ρ0 cells (Fig 4H). Abolishing Puf3 association with Mia40 mRNA (3’‐UTR‐RC and 3’‐UTR‐DE) had no effect on ∆ψm and the proliferation of WT cells, but decreased ∆ψm and the proliferation of ρ0 cells (Fig 4I and J). We further overexpressed Mia40 in ρ0 puf3∆ cells, in which hundreds of Puf3‐associated mitochondrial mRNAs remain downregulated. Strikingly, Mia40 overexpression rescued ∆ψm and the proliferation of ρ0 puf3∆ cells to the level of ρ0 cells (Fig 4K and L). These results strongly suggest Mia40 as the major effector upregulated by Puf3 to maintain ∆ψm and cell proliferation in ρ0 cells.
Puf3 hyper‐phosphorylation upregulates Mia40 and mitochondrial biogenesis
Then how does Puf3 switch from repressing to promoting mitochondrial biogenesis in ∆V and ρ0 cells? We generated PUF3‐FLAG knock‐in cells and observed that Puf3‐FLAG prepared from WT cells migrated as a slightly smeared band in Western blot and Puf3‐FLAG from ρ0 cells migrated as a wider and upshifted band (Fig 5A), indicating post‐translational modifications. Treatment with λ‐phosphatase collapsed Puf3‐FLAG from both WT and ρ0 cells into a single band with lower molecular weight (Fig 5A), indicating Puf3 is a phosphor protein in both WT and ρ0 cells. We thus run samples with the Phos‐tag gel, in which the migration of phosphor protein is impeded (Kinoshita et al, 2009). The result showed that Puf3 is a heavily phosphorylated protein in WT cells and gets hyper‐phosphorylated in ρ0 cells (Fig 5A). Furthermore, Puf3 hyper‐phosphorylation was induced by treating with complex V inhibitor oligomycin and in ∆V and ρ0 cells but not affected by genetic inactivation or chemical inhibition of complex III and complex IV (Fig 5B). Thus, Puf3 hyper‐phosphorylation correlates with the upregulation of mitochondrial biogenesis in ∆V and ρ0 cells.
Figure 5. Puf3 hyper‐phosphorylation upregulates Mia40 and mitochondrial biogenesis to maintain ∆ψm .
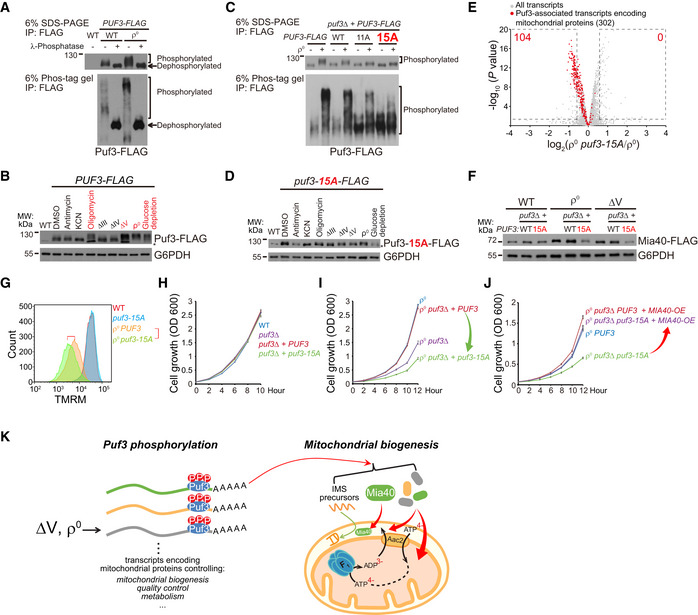
- Puf3 is phosphorylated in WT cells and is hyper‐phosphorylated in ρ0 cells. Puf3‐FLAG was immunoprecipitated from the indicated strains and treated with/without λ‐phosphatase, and analyzed by regular and Phos‐tag SDS–PAGE.
- Puf3 phosphorylation in response to OXPHOS inhibitors and in OXPHOS mutants. WT cells were treated with antimycin (10 µM), KCN (10 µM), or oligomycin (10 µM) for 3 h, or shifted from SCD to SCEG medium for 3 h. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot. Treatments and strains with Puf3 hyper‐phosphorylation was highlighted in red. Glucose depletion was used as a positive control. Asterisk indicates a non‐specific band.
- Puf3‐15A mutation inhibits Puf3 hyper‐phosphorylation in ρ0 cells. A cassette expressing WT or mutant Puf3‐FLAG (11A or 15A) under the control of PUF3 promoter was inserted into the HO site in puf3∆ cells. The 15A mutant that blocks Puf3 hyper‐phosphorylation is highlighted in red. Puf3‐FLAG was immunoprecipitated from the indicated strains and analyzed by regular and Phos‐tag SDS–PAGE.
- Puf3‐15A mutation inhibits Puf3 hyper‐phosphorylation upon oligomycin treatment and in ∆V and ρ0 cells. WT cells were treated with antimycin (10 µM), KCN (10 µM) or oligomycin (10 µM) for 3 h, or shifted from SCD to SCEG medium for 3 h. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot. Asterisk indicates a non‐specific band.
- Puf3‐15A mutation downregulates Puf3‐associated mitochondrial transcripts in ρ0 cells. ρ0 and ρ0 puf3‐15A cells were cultured to log phase. RNAseq was performed with three biological replicates per genotype (Dataset EV7). All the transcripts and the Puf3‐associated transcripts encoding mitochondrial proteins (gene lists in Table EV1D) are shown. The significantly changed transcripts (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed transcripts are indicated.
- Puf3‐15A mutation inhibits the upregulation of Mia40 in ∆V and ρ0 cells. Mia40 was endogenously tagged with FLAG. The selection markers were removed by the Cre‐loxP system. Whole cell lysates were extracted, and equal amount of proteins was loaded for Western blot.
- Puf3‐15A mutation decreases ∆ψm in ρ0 cells. Cells were cultured to mid‐log phase and stained with 125 nM TMRM for FACS analysis. The red highlight two cell lines for comparison.
- Puf3‐15A mutation has no effect on the proliferation of WT cells.
- Puf3‐15A mutation inhibits the proliferation of ρ0 cells.
- Mia40 overexpression rescues the proliferation defect of ρ0 puf3‐15A cells. Single copy plasmid expressing Mia40 driven by MIA40 promoter was transformed into the indicated strains.
- Cartoon illustration that Puf3 hyper‐phosphorylation in ∆V and ρ0 cells upregulates Puf3‐associated mitochondrial transcripts, especially Mia40, to enhance mitochondrial biogenesis and ∆ψm maintenance.
Data information: Cells were cultured from 0.05 OD in SCD, and OD600 was measured every 2 h; Data are mean ± SD from three biological replicates (H, I, J).
Source data are available online for this figure.
We performed SILAC analysis of Puf3 phosphorylation (Dataset EV6). We reached 70% peptide coverage and identified 43 phosphorylation sites in the N‐terminal unstructured region of Puf3 (Fig EV5A). Phosphorylation of 34 sites could be reliably quantified (Fig EV5B and Dataset EV6). We progressively mutated the sites with enhanced phosphorylation to alanine and reconstituted the mutant PUF3‐FLAG into puf3∆ cells. We found that mutation of 15 sites (Puf3‐15A, sites highlighted in red in Fig EV5B) abolished most of the Puf3 hyper‐phosphorylation in ρ0 cells without affecting the basal Puf3 phosphorylation in WT cells (Fig 5C). Puf3 hyper‐phosphorylation upon oligomycin treatment and in ∆V cells were also inhibited by the 15A mutation (Fig 5D).
Figure EV5. SILAC analysis of Puf3 phosphorylation. Related to Fig 5.
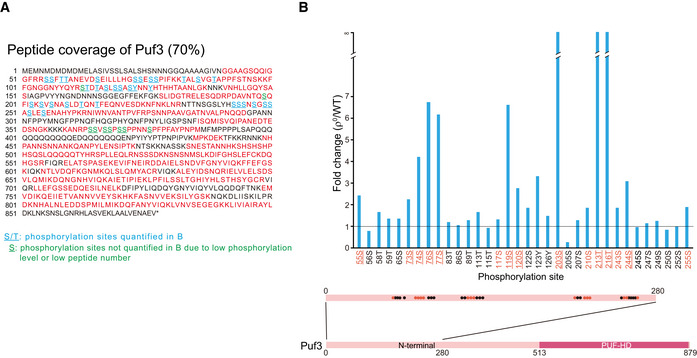
- Peptide coverage map of Puf3. Peptide sequences covered by mass spectrometry are shown in red. Underlined residues are phosphorylated. Blue underlined residues: phosphorylation sites quantitatively analyzed in Fig EV5B. Green underlined residues: phosphorylation sites not quantitatively analyzed due to low phosphorylation level or low total peptide number. Mass spectrometry data are shown in Dataset EV6.
- Quantitative analysis of Puf3 site‐specific phosphorylation (ρ0 versus WT). The localization of phosphorylation sites at the N terminus of Puf3 is illustrated. Residues mutated in Puf3‐15A are highlighted in red. Residues mutated in Puf3‐11A are highlighted in red and underlined. The horizontal line indicates fold change of 1.
Puf3‐associated mitochondrial transcripts were decreased by the Puf3‐15A mutation in ρ0 cells (Fig 5E, and Dataset EV7). Importantly, Puf3‐15A mutation did not affect the protein level of Mia40 in WT cells but selectively prevented Mia40 upregulation in ∆V and ρ0 cells (Fig 5F). Accordingly, Puf3‐15A mutation did not affect ∆ψm and cell proliferation in WT cells but decreased both ∆ψm and cell proliferation in ρ0 cells (Fig 5G–I). Mia40 overexpression rescued the proliferation defect of ρ0 puf3‐15A cells (Fig 5J). Taken together these findings, Puf3 gets hyper‐phosphorylated in ∆V and ρ0 cells to upregulate mitochondrial biogenesis, particularly Mia40 (Fig 5K). This response is critical for ρ0 cells to maintain ∆ψm and cell proliferation.
Snf1/AMPK represses ribosome biogenesis and upregulates glycolysis to maintain ∆ψm and proliferation in ∆V and ρ0 cells
∆V and ρ0 cells consume glycolytic ATP in an expensive way to maintain ∆ψm. Transcriptional upregulation of glucose transporters and glycolysis genes and repression of ribosome biogenesis genes in these two mutants (Fig 1C) seem to enhance glycolytic ATP production and reduce ATP expenditure. Ribosomes compose approximately 30% of cellular mass and ribosome biogenesis consumes about 80% of newly synthesized nucleotides (Lunt & Vander Heiden, 2011). In yeast, about 60% of total transcription is to transcribe rRNA and 50% of RNA polymerase II transcription and 90% of mRNA splicing are devoted to ribosomal proteins (Warner, 1999).
We found that the stress‐responsive kinase Snf1, the yeast AMPK (Hedbacker & Carlson, 2008), mediates these two changes. snf1∆ cells were inviable after mtDNA depletion. Therefore, ρ0 snf1∆ cells were generated by acute mtDNA depletion through EB treatment. RNAseq (Dataset EV8) showed that deleting SNF1 downregulated transcripts encoding glucose transporter and glycolysis genes in both WT and ρ0 cells, and specifically upregulated ribosome and Ribi transcripts in ρ0 cells (Fig 6A, highlighted in red). Polysome fractionation experiment showed that deleting SNF1 in WT cells upregulated monosome and polysomes (Fig 6B, solid red line vs. solid black line), indicating Snf1 represses ribosome biogenesis through post‐transcriptional mechanisms in WT cells. SNF1 deletion in ρ0 cells significantly upregulated 40S, 60S, and 80S ribosomes but had minimal effect on polysomes (Fig 6B, dashed red line vs. dashed black line). The production of excessive ribosomes in ρ0 snf1∆ cells is apparently uneconomic for energy metabolism. Accordingly, SNF1 deletion reduced ∆ψm and impaired cell proliferation in ∆V and ρ0 cells but not in WT, ∆III, or ∆IV cells (Fig 6C and D).
Figure 6. Snf1/AMPK upregulates glycolysis and represses ribosome biogenesis to maintain ∆ψm .

- Snf1 maintains glycolysis and glucose transporter gene expression in both WT and ρ0 cells, and represses ribosome and Ribi gene expression in ρ0 cells. ρ0 snf1∆ cells were prepared by acute EtBr (25 µg/ml) treatment of snf1∆ cells in SCD for 2 days. RNAseq was performed with three biological replicates per genotype (Dataset EV8). All the transcripts and two categories of transcripts (gene lists in Table EV1B) are shown. Significantly changed genes (P value < 0.05; fold change > 1.5 or < 0.67) are highlighted with dashed lines; numbers of significantly changed genes are indicated.
- Snf1 represses ribosome biogenesis in WT and ρ0 cells. Polysomes of the indicated strains were fractionated by sucrose gradients (10%–50%), and the gradients were separated and measured at A254.
- Deletion of SNF1 decreases ∆ψm in both ∆V and ρ0 cells.
- Deletion of SNF1 decreases the proliferation of both ∆V and ρ0 cells. Serial dilutions of the indicated strains were analyzed on SCD plates at 30°C for 2 days.
- Deletion of MIG1 in ρ0 snf1∆ cells rescues ∆ψm.
- Deletion of MIG1 in ρ0 snf1∆ cells partially rescues cell proliferation. A plasmid overexpressing MIG1 under the control of MIG1 promoter was transformed into the indicated strains. Serial dilutions (tenfold dilution) were analyzed on SCD plates at 30°C for 2 days. V: empty vector.
- Combining MIG1 deletion and NOG2 overexpression in ρ0 snf1∆ cells fully rescues cell proliferation. A plasmid overexpressing NOG2 under the control of TEF1 promoter was transformed into the indicated strains. Serial dilutions (tenfold dilution) were analyzed on SCD plates at 30°C for 2 days. V: empty vector.
- Cartoon illustration that Snf1 signaling in ∆V and ρ0 cells upregulates glycolysis and represses ribosome biogenesis to optimize energy metabolism.
Data information: Cells were cultured to mid‐log phase and stained with 125 nM TMRM for FACS analysis (C, E). The red highlight two cell lines for comparison (C, D, E).
To determine the contributions of Snf1’s two downstream pathways, we tried to upregulate glycolysis and repress ribosome biogenesis in ρ0 snf1∆ cells. Snf1 phosphorylates transcription repressor Mig1 and induces its nuclear export to turn on gene transcription (Treitel et al, 1998; Hedbacker & Carlson, 2008). RNAseq showed that deleting MIG1 in ρ0 snf1∆ cells upregulated glucose transporter and glycolysis genes and modestly repressed ribosome and Ribi genes (Appendix Fig S3A and Dataset EV9). Functionally, deleting MIG1 in ρ0 snf1∆ cells partially restored ∆ψm and proliferation (Fig 6E and F). Nog2 is a GTPase that binds to and inhibits the nuclear export of pre‐60S subunit and its overexpression inhibits ribosome biogenesis (Matsuo et al, 2014; Wang & Chen, 2015). We overexpressed Nog2 in ρ0 snf1∆ cells and observed no rescuing effect (Fig 6G). However, Nog2 overexpression in ρ0 snf1∆ mig1∆ cells fully restored cell proliferation to the level of ρ0 cells (Fig 6G). These results suggest glycolysis upregulation plays a major role and ribosome biogenesis repression plays a minor role downstream of Snf1 activation. Activation of both pathways fully mimics the effect of Snf1 activation (Fig 6H).
Three adaptive responses, namely Inh1 downregulation, Puf3‐mediated mitochondrial biogenesis, and Snf1 activation, are activated in ∆V but only Snf1 is required in ∆V cells. We examined if Snf1 and the other two adaptive responses have synergistic effect in ∆V cells. In ∆V snf1∆ cells, deleting either one or both of the responses exacerbated proliferation (Appendix Fig S3B, highlighted in red). As control, deleting all the three responses in WT cells had no effect on proliferation (Appendix Fig S3C).
Upregulation of cytosolic chaperones maintains ∆ψm
We observed that accompanying increasing stresses in WT, ∆III, ∆IV, ∆V, and ρ0 cells, there was gradual induction of chaperones, ubiquitin‐proteasome system, and autophagy genes (gene lists in Table EV1F). These quality control/degradation genes were further induced in Inh1‐overexpressing ρ0 cells (Fig 7A and Dataset EV10). These genes are most likely induced in response to the accumulation of mitochondrial precursor proteins under import arrest (Wrobel et al, 2015; Boos et al, 2019). Proteasome activation has been shown to alleviate proteostatic stress caused by precursor accumulation (Wrobel et al, 2015).
Figure 7. Upregulation of chaperones maintains ∆ψm .

- Summary of the growth results of the indicated strains. −: no effect on growth. +: growth inhibition. ++: strong growth inhibition. Genes that are preferentially required in ρ0 puf3∆ cells are highlighted in red.
- Deletion of HSC82 selectively impairs the proliferation of ρ0 puf3∆ cells. Cells were cultured from 0.05 OD in SCD, and OD600 was measured every 2 h. Data are mean ± SD from three biological replicates.
- Deletion of HSC82 selectively decreases ∆ψm in ρ0 puf3∆ cells. Cells were cultured to mid‐log phase and stained with 125 nM TMRM for FACS analysis. The red highlight two cell lines for comparison.
- Cartoon illustration of the progressive activation of adaptive responses to convergently maintain ∆ψm in OXPHOS mutants.
Source data are available online for this figure.
To understand whether chaperones and autophagy have a role under low ∆ψm condition, we selected a representative group of genes and deleted them in WT, ρ0, and ρ0 puf3∆ cells. These genes include chaperones Hsp70 and Hsp90, disaggregase Hsp104, antiaggregase Hsp26 and Hsp42, membrane chaperone Hsp12 (Verghese et al, 2012), autophagy gene Atg8, and mitophagy receptors Atg32 and Atg33 (Okamoto et al, 2009; Kanki et al, 2009a; Kanki et al, 2009b). As summarized in Fig 7B, disrupting autophagy, mitophagy, disaggregase, and antiaggregase individually or in combination had no effect on cell proliferation. Interestingly, deleting HSC82, the dominant isoform of Hsp90, impaired growth selectively in ρ0 puf3∆ cells, but not in WT and ρ0 cells (Fig 7B and C). Similarly, ρ0 puf3∆ cells relied more on Hsp70 family members Ssa1 and Ssa2 to maintain cell proliferation as compared to WT and ρ0 cells (Fig 7B). Deleting HSC82 in ρ0 puf3∆ cells decreased ∆ψm (Fig 7D), indicating Hsp90 activity becomes critical for ∆ψm maintenance under very low ∆ψm condition.
Discussion
Progressive activation of mitochondrial stress responses in response to increasing stresses among OXPHOS mutants
Through extensive omics analyses and characterizations of pathways and effectors, we summarize the global stress responses in OXPHOS mutants and show how these responses maintain ∆ψm (Fig 7E). Briefly, in ∆III/∆IV cells, a common response, Inh1 downregulation, is activated to release mitochondrial ATP hydrolysis activity. In ∆IV and ρ0 cells with increased stresses, two additional responses are induced. One response is the activation of Snf1/AMPK to upregulate glycolysis and repress ribosome biogenesis. This response enhances ATP supply and reduces ATP expenditure, thus optimizing ATP supply to mitochondria. The other response is the phosphorylation of Puf3 to upregulate global mitochondrial biogenesis. We identified Mia40, the import receptor of IMS proteins, as the key factor upregulated by Puf3. The exact mechanism of ∆ψm maintenance by Mia40 remains unclear, but may link with its broad effect on mitochondrial proteome and lipid metabolism. Finally, when ∆ψm gets further decreased in ρ0 cells, cells upregulate chaperones and degradation pathways. Chaperones bind precursor proteins and may escort substrates to import or degradation pathways. The requirement of Hsp90 to maintain ∆ψm in ρ0 puf3∆ cells (Fig. 7D) indicates its role in mitochondrial protein import. These diverse cytoplasmic and mitochondrial responses convergently maintain ∆ψm (Fig 7E). The conserved effectors mechanistically connect mitochondrial stress responses with mitochondrial repair/compensation, filling a major gap in our understanding of mitochondrial adaptive responses.
Caveats and remaining questions of the current study are also evident. First, what aspects of mitochondrial dysfunction are being sensed and how the initial signals are activated remain unclear. Reduction of ∆ψm is an attractive inducer, but may not be the only one, especially in ρ0 cells with extensive proteostatic stresses. Second, whether these diverse pathways are activated by a common upstream molecule or by different sensor molecules are unclear. Third, other types of changes, e.g., protein translocation from mitochondria to other subcellular locations cannot be revealed by our approach. Future studies are thus warranted.
Evolutionary conservation of the stress responses and implication for mitochondrial diseases
The yeast strategy to cope with OXPHOS dysfunction provides a unique angle to consider human mitochondrial diseases. First, our results highlight the importance of removing Inh1/ATPIF1 to maintain ∆ψm. ATPIF1 knockout increases ∆ψm in OXPHOS‐defective mammalian cell lines(Lefebvre et al, 2013; Chen et al, 2014). Whether ATPIF1 knockout can have beneficiary effects in mitochondrial diseases remains to be determined. Second, yeast activates Snf1 to upregulate glycolytic ATP supply and suppress ATP expenditure processes, thus channeling glycolytic ATP to mitochondria; yeast also activates mitochondrial biogenesis through Puf3 hyper‐phosphorylation. Interestingly, mammalian AMPK upregulates glycolysis, represses ATP consumption processes, and also promotes mitochondrial biogenesis (Herzig & Shaw, 2018). Thus, AMPK activation in mammalian cells achieves two major adaptations in yeast, and AMPK is an attractive target for mitochondrial diseases. AMPK activation, glycolysis upregulation, and mitochondrial biogenesis have been documented in mouse and C. elegans OXPHOS disease models (Gioran et al, 2019; Haynes et al, 2007; Kuhl et al, 2017; Nargund et al, 2015; Viscomi et al, 2011; Wredenberg et al, 2002). Importantly, pharmacological activation of AMPK by AICAR partially corrects COX deficiency (Viscomi et al, 2011). New generation of AMPK agonists with better pharmacological properties as compared to AICAR may provide a better therapeutic opportunity (Cokorinos et al, 2017; Myers et al, 2017).
Materials and Methods
Reagents or resources used in this paper are listed in Appendix Table S1.
Yeast strains and media
The yeast strains used in this study are listed in Table EV2. All the strains were derived from the wild‐type strain W303‐1A. To generate stable ρ0 cell lines, corresponding WT cells were treated with 25 µg/ml EtBr in SCD for 2 days and then plated on YPD plates. Single clones were picked up after 2–3 days. The ρ0 clones were streaked on YPEG plates to verify the absence of respiratory growth and were confirmed by DAPI staining and Western blot detecting mtDNA‐encoded Cox2.
Gene deletions were performed by PCR‐based homologous recombination to replace the target gene with appropriate selection marker (Longtine et al, 1998; Gueldener et al, 2002). C‐terminal tagging was similarly performed by PCR‐based homologous recombination to replace the endogenous stop codon with cassettes containing appropriate tags and selection markers. To preserve the intact 3’‐UTR for Puf3 regulation, C‐terminal tagging of Puf3‐associated genes was performed by using plasmids pUG27‐CYC1t‐3 × HA or pUG27‐CYC1t‐3 × FLAG containing tags and floxed selection markers, and then, the selection markers were removed by plasmid pSH47 expressing Cre recombinase (Gueldener et al, 2002). MIA40 3’‐UTR mutants were generated by replacing MIA40 3’‐UTR ~150 bp with mutated sequences in which the Puf3‐binding motif (CATGTAAATA) was deleted or reverse complemented. INH1 or HAP4 overexpression strains were generated by introducing a cassette expressing INH1 or HAP4 under the control of CUP1 promoter into the HO locus. PUF3‐15A strain was generated by introducing a cassette expressing PUF3‐15A under the control of endogenous PUF3 promoter (1 kb upstream of ORF) into the HO locus after disrupting endogenous PUF3.
Media used in this study included SCD (0.67% yeast nitrogen base without amino acids, 0.079% complete supplement mixture, and 2% glucose), and SCEG (0.67% yeast nitrogen base without amino acids, 0.079% complete supplement mixture, 3% ethanol, and 3% glycerol), YPD (1% yeast extract, 2% peptone, and 2% glucose), YPEG (1% yeast extract, 2% peptone, 3% ethanol, and 3% glycerol). Strains were grown at 30°C if not indicated.
Plasmids
The plasmids pUG27‐CYCt‐3 × HA or pUG27‐CYCt‐3 × FLAG were generated as follows: CYC1 terminator was inserted into pUG27 (Gueldener et al, 2002) at BglII site by Gibson assembly (Gibson et al, 2009) to generate pUG27‐CYCt; a 3 × HA or 3 × FLAG tag was inserted into pUG27‐CYCt at HindIII‐SalI sites.
Plasmid overexpressing MIA40 was generated as follows: a cassette including MIA40 promoter (1 kb upstream of ORF), ORF, and downstream 1 kb was inserted into p417‐TEF1 at SacI‐BamHI sites (TEF1 promoter was removed by restriction enzyme digestion; Wu & Tu, 2011).
Plasmid expressing PUF3‐15A mutant was made as follows: a fragment including PUF3 promoter (1 kb upstream of ORF), ORF, a 3 × FLAG tag and downstream 1 kb was generated by overlap extension PCR, and inserted into HO‐natNT2 (KanMX4 in HO‐KanMX4 was replaced by natNT2) at SalI‐BglII sites (Voth et al, 2001). Mutations of PUF3 phosphorylation sites were introduced by QuikChange site‐directed mutagenesis (Agilent Technologies).
Plasmid expressing MIG1 was generated as follows: a cassette including MIG1 promoter (1 kb upstream of ORF), ORF, and downstream 1 kb was inserted into pRS416 at KpnI‐SacI sites(Sikorski & Hieter, 1989).
Plasmid expressing NOG2 was generated by inserting NOG2 ORF into p417‐TEF1‐natNT2 (KanMX4 in p417‐TEF1 was replaced by natNT2) at EcoRI‐SalI sites.
Growth test
Growth curves were performed by measuring absorbance at 600 nm (A600) in biological triplicates. Strains were cultured from 0.05 OD, and A600 was measured every 2 or 3 h for 12 h at the indicated temperature.
For drop dilution assays, strains were grown in SCD media to mid‐log phase (OD ~0.4–0.6), washed with ddH2O, and then diluted to 10−1, 10−2, 10−3, and 10−4 OD. Drop dilutions on the indicated plates and incubated at the indicated temperatures for 2 days.
Mitochondrial membrane potential measurement
Strains were cultured to mid‐log phase in SCD and washed once with washing buffer (10 mM HEPES, pH 7.4, 5% glucose). 0.2 OD cells were stained for 20 min in 1 ml staining buffer (10 mM HEPES, pH 7.4, 5% glucose, 125 nM TMRM). The florescence of 20,000 cells was analyzed by BD FACSAria II (BD Biosciences), and results were plotted with FlowJo V10. Mitochondrial localization of TMRM signal was confirmed by microscope.
Glucose consumption assay
Glucose concentration was measured by Glucose Assay Kit (Sigma). Strains were cultured from 0.05 OD, and media were collected at 0.4, 0.8, and 1.5 OD. The media were diluted 40 times and measured following the manufacturer’s instruction. Glucose concentration of the original SCD media was set as 100%.
Total RNA isolation
Strains were grown to mid‐log phase in SCD, and 5–10 OD cells were collected, snap‐frozen in liquid nitrogen, and stored at −80°C. Total RNA was isolated using hot phenol method as described previously (Lyne et al, 2003). Cells were resuspended in 600 µl TES buffer (10 mM Tris–HCl, pH 7.5, 10 mM EDTA, pH 8, 0.5% SDS), and immediately added with 600 µl acidic phenol‐chloroform‐isoamylalcohol (25:24:1). Samples were incubated at 65°C for 1 h and mixed thoroughly every 10 min. RNA was extracted with acidic phenol‐chloroform‐isoamylalcohol (25:24:1, two times) and chloroform‐isoamylalcohol (24:1, one time). Then, RNA was precipitated by adding 1 volume isopropanol and 0.1 volume 3 M NaAc (pH 5.2), and dissolved in RNase‐free water. RNA integrity was assessed by 1% agarose gel electrophoresis.
qRT–PCR
Total RNA was digested with DNase I (NEB) and extracted again. Complementary DNA was synthesized with 1 µg RNA by PrimeScript™ RT Reagent Kit (Takara). qRT–PCR was performed by using CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad) in biological triplicates. PCRs were prepared with TB Green Premix Ex Taq (Takara). Primers used for qPCR were listed in Table EV3, and ACT1 was selected as reference gene for normalization. Data were analyzed by 2‐∆∆Ct method, and arithmetic mean ± SD were shown in plots.
RNAseq and data processing
mRNA was purified from total RNA by Oligo d(T)25 Magnetic beads (NEB). mRNA was fragmented by incubating with magnesium ion at 94°C for 5 min. Reverse transcription was performed by SuperScript II Reverse Transcriptase (Invitrogen). Complementary DNA was synthesized by DNA polymerase Ⅰ (Takara) and Random primers (Takara). Sequencing libraries were prepared by NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB). The quality of libraries was assessed by 2100 Bioanalyzer Instrument (Agilent) and Qubit (Thermo Fisher Scientific). Libraries were quantified by KAPA Library Quantification Kit (Roche) and sequenced by Hiseq2500 (Illumina). Three biological replicates were prepared for each strain.
All the raw data were assessed by FastQC and showed good quality. Alignment to S. cerevisiae genome (R‐64‐1‐1) was performed by STAR (2.6.1a) with default parameters. Output BAM files were indexed using SAMtools (1.9). Reads counts for each gene were quantified to the Ensembl R64 GTF annotation file by Rsubread package (1.34.7) with default parameters. Normalization to RPKM (reads per kilobase of exon model per million mapped reads) and differential expression analysis was executed by edgeR (3.26.8). TMM (trimmed mean of M‐values) normalization was applied to dataset to account for compositional difference between the libraries. Then the common dispersion and tagwise dispersions were estimated, and the quasi‐likelihood F‐tests was performed. All the non‐coding RNAs and mtDNA‐encoded mRNAs were removed from results due to lack of polyadenylation.
Gene function classification
Gene functional categories were generated according to literatures or Saccharomyces Genome Database (SGD) and listed in Table EV1. High‐fidelity nuclear‐encoded mitochondrial gene list was selected from gene ontology mitochondrion (GO: 0005739) category after manual checking of the SGD database for protein annotation and localization. OXPHOS subunits, glycolysis genes, and glucose transporter genes are from SGD. Gene list of cytosolic ribosome subunits is from Steffen et al, (2012) and gene list of ribosome processing and assembly (Ribi) is from Woolford and Baserga (2013). Gene list of Puf3‐associated transcripts was combined from Gerber et al, 2004; Lapointe et al, (2015), and gene list of Puf3‐associated transcripts encoding mitochondrial proteins was selected according to high‐fidelity mitochondrial gene list. Gene list of Mia40 substrates is from Herrmann & Riemer, 2012; Mordas and Tokatlidis (2015). Gene list of “Proteostasis” is from Quiros et al, (2015). Gene list of “Import machinery” is from Schmidt et al, (2010). Gene list of “OXPHOS assembly factors” was combined from Ndi et al, (2018), Ruhle and Leister (2015), and Soto et al, (2012). Gene list of “mitoRibosome” was combined from Amunts et al, (2014) and Desai et al, (2017). Gene list of “Iron‐sulfur cluster synthesis” is from Lill (2009). Gene list of “Phospholipid trafficking and synthesis” is from Tatsuta et al (2014). Gene list of “Coenzyme Q” is from Stefely and Pagliarini (2017). Gene list of “Carriers” is from Ogunbona and Claypool (2019). Gene list of “cytoplasmic chaperone” is from Verghese et al, (2012). Gene list of proteasome subunits is from Boos et al, (2019), and gene list of Cdc48 cofactors is from Wu et al, (2016). Gene list of “Autophagy” is from Farre and Subramani (2016), Reggiori and Klionsky (2013), and Farre and Subramani (2016).
Protein isolation and Western blot
5–10 OD yeast cell pellets were resuspended in 300 μl yeast lysis buffer (50 mM NaCl, 50 mM NaF, 100 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 10% glycerol, 14 mM 2‐mercaptoethanol, 2 mM PMSF, 5 μM pepstatin A, and 10 μM leupeptin). After adding ∼100 μl of glass beads, cells were lysed via three rounds of bead‐beating (40 s of beating followed by 1 min of cooling on ice), and centrifuged at 18,407 g for 10 min at 4°C to collect the supernatants. Proteins were separated by 4–12% ‐gradient gel and then transferred onto nitrocellulose membrane (GE healthcare). Immunodetection was performed by Western Lightning Plus‐ECL (PerkinElmer).
SILAC analysis of mitochondrial proteome
Strains used for SILAC mass spectrometry were in lys2∆ arg4∆ CAN1 + background. Strains were cultured to mid‐log phase in SCD (0.67% yeast nitrogen base without amino acids, 0.079% complete supplement mixture lacking arginine and lysine, 2% glucose) supplemented with 50 mg/l [13C6/15N4] L‐arginine and 50 mg/l [13C6/15N2] L‐lysine (Cambridge Isotope Laboratory) or non‐labeled L‐arginine and L‐lysine. The average incorporation efficiency of labeled amino acids was detected by LC‐MS/MS, reaching 98.0%.
Three biological replicates were prepared for each strain. Equal amounts of heavy‐isotope and light‐isotope labeled cells (~150 OD) were harvested and mixed together. Mitochondria were isolated following a previously described method with some modifications (Diekert et al, 2001). Briefly, cells were washed once with water, resuspended in TD buffer (10 mM DTT and 100 mM Tris‐SO4, pH 9.4) and incubated for 15 min at 30°C. Cells were then pelleted and washed once with SP buffer (1.2 M sorbitol and 20 mM potassium phosphate, pH 7.4) and treated with Zymolyase 100T (MP Biomedical) for 40 min at 30°C to generate spheroplasts. After two times of washes with SP buffer, the spheroplasts were resuspended in SHE buffer (0.6 M sorbitol, 20 mM HEPES‐KOH, pH 7.4, 1 mM EGTA, pH 8.0, and 2 mM MgCl2) supplemented with protease inhibitors and BSA, and homogenized by a French press (EmulsiFlex‐C3, AVESTIN Inc.) at pressures in the range of 1,000–1,500 psi. The mitochondria‐enriched fraction was obtained by differential centrifugation, snap‐frozen by liquid nitrogen, and stored at −80°C until use.
Mitochondria pellets were lysed in 100 µl 1% SDS lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% SDS, EDTA‐free protease inhibitor cocktail (Roche)) and boiled at 98°C for 10 min. After centrifugation at 18,407 g, 4°C for 10 min, the supernatant was collected and mitochondrial proteins were precipitated overnight at −20°C by adding 8 volumes of ice‐cold acetone. The protein pellets were redissolved in 8 M urea in 50 mM ammonium bicarbonate (pH 8.0). Aliquot of 100 μg protein was reduced in 2 mM DTT at 56°C for 30 min followed by alkylation in 5 mM iodoacetamide at dark for 1 h. Around 2.5 μg of sequencing grade modified trypsin was added to each protein solution and incubated at 37 °C overnight. The resulting tryptic peptides were desalted and fractionated into eight fractions with Thermo Scientific™ Pierce™ High pH Reversed‐Phase Peptide Fractionation Kit (Thermo Fisher Scientific).
Each fraction of peptides was separated by a home‐made analytical capillary column (50 μm × 15 cm) packed with 5 μm spherical C18 reversed‐phase material (YMC). A Waters nanoAcquity UPLC system was used to generate the following HPLC gradient: 0–30% B in 75 min, 30–70% B in 15 min, 70–90% B in 5 min (A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile). The eluted peptides were sprayed into a Q Exactive mass spectrometer (Thermo Fisher Scientific) equipped with a nano‐ESI ion source. The mass spectrometer was operated in data‐dependent mode with one MS scan followed by ten HCD (High‐energy Collisional Dissociation) scans for each cycle.
Data were analyzed using MaxQuant (1.6.17.0) and searched against UniProt_Scerevisiae protein database. The search parameters were set as follows: variable modification included “Oxidation (M)”, “Acetyl (Protein N‐term” and “Carbamidomethyl (C)”; digestion enzyme was chosen as “Trypsin/P”, and 2 missed cleavage sites were allowed; protein quantification was based on at least one unique peptide with minimal length of 7 amino acids; protein identification was based on at least one peptide, and false discovery rate at peptide spectrum level and protein level was 0.01; “First search peptide tolerance” was 20 ppm, and “Main search peptide tolerance” was 4.5 ppm. Other parameters were set by default if not indicated. More than 3000 proteins were identified in each experiment, and only high‐fidelity mitochondrial proteins with 3 quantification results were kept for analysis. The ratios of proteins were log2‐transformed and loaded into Perseus (1.6.14.0) to calculate P value (two‐tailed t‐test). Protein groups with P < 0.05, and fold change > 1.5 or < 0.67 were considered as significantly changed in ∆IV or ρ0 cells.
Puf3 protein immunoprecipitation, phosphatase treatment, and Phos‐tag gel analysis
Yeast cells were cultured in SCD to mid‐log phase, and 50 OD cells were harvested. Cell pellets were washed once with 5% TCA, flash frozen in liquid nitrogen, and stored at −80°C until use. Cells were resuspended in 1 volume 1 M Tris–HCl (pH 8.0) plus 2 volumes SDS buffer (100 mM Tris–HCl, pH 7.5, 300 mM NaCl, 4% SDS, 8% BME), and lysed by six rounds of bead beating (20 s beating followed by 1 min of cooling on ice). The cell lysates were diluted with 20 volumes Triton buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton), and incubated with 8–10 µl anti‐FLAG agarose beads (Sigma) at 4°C for 5–6 h. The beads were washed three times with Triton buffer for 10 min and eluted with FLAG peptides (2 mg/ml) overnight at 4°C.
For phosphatase treatment, the beads with immunoprecipitates were treated with 400 U Lambda phosphatase (NEB) at 30°C for 20 min. The reaction was stopped by adding 4× sample buffer (0.2 M Tris–HCl, pH 6.8, 8% SDS, 28% glycerol, 8% BME, 0.08% Bromophenol Blue) and heated at 98°C for 10 min.
Puf3‐FLAG immunoprecipitates were loaded onto 6% Phos‐tag gel, run at 80 V for 1 h, 120 V for 30 min, then at 150 V till the leading front dye run out the gel. Gel was rinsed once with ddH2O, washed three times with transfer buffer (25 mM Tris, 192 mM Glycine) supplemented with 10 mM EDTA for 8 min, and then washed once with transfer buffer for 8 min. Transfer proteins to PVDF membrane at 140 V, 300 mA for 3 h at 4°C.
SILAC analysis of Puf3 phosphorylation sites
PUF3‐FLAG and PUF3‐FLAG ρ0 cells were cultured to mid‐log phase in SCD supplemented with labeled or unlabeled arginine and lysine. About 350 OD cells were harvested for each strain and mixed together. The protein extraction and beads incubation were performed as described above. After that, anti‐FLAG agarose beads were washed four times with yeast lysis buffer and eluted overnight with FLAG peptides at 4°C. The sample was separated by 4–12% gradient gel and stained with coomassie blue. Protein bands on the gradient gel were de‐stained and then reduced in 10 mM DTT at 56°C for 30 min followed by alkylation in 55 mM iodoacetamide at dark for 1 h. The protein bands were isolated and in‐gel digested with sequencing grade trypsin (10 ng/μl trypsin, 50 mM ammonium bicarbonate, pH 8.0) overnight at 37°C. Peptides were extracted with 5% formic acid/50% acetonitrile and 0.1% formic acid/75% acetonitrile sequentially and then concentrated to ~20 μl. The extracted peptides were separated by an analytical capillary column (50 μm × 15 cm) packed with 5 μm spherical C18 reversed‐phase material (YMC). A Waters nanoAcquity UPLC system (Waters) was used to generate the following HPLC gradient: 0–30% B in 40 min, 30–70% B in 15 min (A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile). The eluted peptides were sprayed into a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nano‐ESI ion source. The mass spectrometer was operated in data‐dependent mode with one MS scan followed by four CID (Collision‐Induced Dissociation) and four HCD (High‐energy Collisional Dissociation) MS/MS scans for each cycle. Database searches were performed on an in‐house Mascot server (Matrix Science Ltd.) against yeast Puf3 protein sequence. SILAC quantification of the phosphorylated peptides was performed by Mascot Distiller. The search parameters are 7 ppm mass tolerance for precursor ions; 0.5 Da mass tolerance for product ions; three missed cleavage sites were allowed for trypsin digestion; and the following variable modifications were included: oxidation on methionine, carbamidomethylation on cysteine, phosphorylation on serine, threonine, and tyrosine. The tandem mass spectra of matched phosphorylated peptides were manually checked for their validity. Phosphorylation level of each specific amino acid sites was quantified manually.
Ribosome footprinting and data analysis
Ribosome footprinting was performed as previously described (Ingolia, 2010; Ingolia et al, 2012). Briefly, yeast cells were cultured to mid‐log phase in SCD, treated with 100 μg/ml cycloheximide for 2 min. 500 OD cells were harvested by vacuum filtration and snap‐frozen in polysome lysis buffer (20 mM Tris–HCl, pH 8, 140 mM KCl, 1.5 mM MgCl2, 100 μg/ml cycloheximide, and 1% (v/v) Triton). 250 μl aliquot of cell lysate was digested by 7.5 μl RNase Ⅰ (Ambion) at room temperature for 1 h, and digestion was stopped by adding 5 μl SUPERase•In (Ambion). The samples were loaded on sucrose gradients (10%–50%) and centrifuged for 3 h at 209,230 g, 4°C with SW41 Ti rotor. The gradients were separated by Piston Gradient Fractionator™ (Biocomp), and A254 values of gradients were continually monitored by TriaxTM Flow Cell (Biocomp). Fractions representing the monosome peak were collected. RNA was isolated by TRIzol reagent (Invitrogen) and separated by 15% polyacrylamide TBE‐urea gel. 26‐nt to 34‐nt region demarcated by the marker oligos was carefully excised. After gel extraction, rRNA was removed by Ribo‐Zero rRNA Removal Kit (Epicentre). The RNA fragments were end‐repaired by T4 Polynucleotide Kinase (NEB) and ligated with Universal miRNA Cloning Linker (NEB) by T4 RNA Ligase 2 (NEB). Reverse transcription was performed with SuperScript III (Invitrogen), and then, the products were circularized by CircLigase (Epicentre). The libraries were amplified by Phusion High‐Fidelity DNA Polymerase (NEB) and sequenced by Hiseq2500 (Illumina). Alignment to yeast genome (R‐64‐1‐1) was performed by using TopHat (2.1.1) with library‐type set as fr‐secondstrand. RPKM was calculated by Rsubread package (1.34.7) and edgeR (3.26.8).
Polysome fractionation analysis
Polysome fractionation analysis was performed as previously described (Lee & Tu, 2015). Yeast cells were cultured to mid‐log phase in SCD, treated with 100 µg/ml cycloheximide for 15 min, and 75 OD cells were harvested. Cell pellets were resuspended in 500 μl lysis buffer (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 30 mM MgCl2, 200 μg/ml cycloheximide, 200 mg/ml heparin, 0.1% BME, EDTA‐free protease inhibitor cocktail (Roche), 1 mM EDTA, 1 mM PMSF, 5 mM pepstatin A, 10 mM leupeptin, 0.2 mM Na‐orthovanadate, 10 mM β‐glycerolphosphate, 10 mM NaF, and 20 U/ml SUPERase•In) and mixed with 250 μl glass beads. The cells were lysed by six rounds of bead beating (20 s beating followed by 1 min of cooling on ice). The supernatant was collected after centrifugation at 18,407 g, 4°C for 10 min. 200 μl cell lysate was loaded onto sucrose gradients (10–50%), and fractionated by centrifugation at 209,230 g, 4°C for 3 h with SW41 Ti rotor. The gradients were separated and monitored by Piston Gradient Fractionator™ and TriaxTM Flow Cell. For Puf3 polysome analysis, fractions representing the polysome peak were collected. Proteins were precipitated by 20% TCA and boiled at 98°C for 10 min with 50 µl 4× sample buffer.
Author contributions
Conception of the project, study supervision, and manuscript writing: HJ. Most of the experiments, data analysis, figure preparation, and manuscript editing: SiL. SNF1‐related experiments and data analysis: ShL. Puf3 phosphorylation analysis: BH. Generation of part of the strains: LaL. MS experiments and data analysis: LiL and SC. RNAseq and data analysis: JW and TC.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank Dr. Lilin Du and Dr. Wenhui Li, and Lijing Pan for technical assistance and thank Dr. Xi Wu for discussion and suggestion. The research is supported by Beijing Municipal Science and Technology Commission.
EMBO Reports (2021) 22: e51606.
Data availability
The deep‐sequencing raw data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al, 2002) and are accessible through GEO Series accession number GSE162197 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162197). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD022805 (http://www.ebi.ac.uk/pride/archive/projects/PXD022805).
References
- Amunts A, Brown A, Bai XC, Llacer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SHW, Ramakrishnan V (2014) Structure of the yeast mitochondrial large ribosomal subunit. Science 343: 1485–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby RD, Porteous WK, Hughes G, James AM, Shannon D, Wei YH, Murphy MP (1999) Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur J Biochem 262: 108–116 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos F, Kramer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zoller E, van der Laan M et al (2019) Mitochondrial protein‐induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol 21: 442–451 [DOI] [PubMed] [Google Scholar]
- Buchet K, Godinot C (1998) Functional F1‐ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA‐depleted rho degrees cells. J Biol Chem 273: 22983–22989 [DOI] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Birsoy K, Mihaylova MM, Snitkin H, Stasinski I, Yucel B, Bayraktar EC, Carette JE, Clish CB, Brummelkamp TR et al (2014) Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep 7: 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Adam‐Vizi V (2010) Mitochondria as ATP consumers in cellular pathology. Biochem Biophys Acta 1802: 221–227 [DOI] [PubMed] [Google Scholar]
- Clark‐Walker GD (2007) The F1‐ATPase inhibitor Inh1 (IF1) affects suppression of mtDNA loss‐lethality in Kluyveromyces lactis . FEMS Yeast Res 7: 665–674 [DOI] [PubMed] [Google Scholar]
- Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjobsted R, Jorgensen NO, Tran JL, Jatkar A, Cialdea K, Esquejo RM et al (2017) Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non‐human primates and mice. Cell Metab 25: 1147–1159. [DOI] [PubMed] [Google Scholar]
- Craven L, Alston CL, Taylor RW, Turnbull DM (2017) Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet 18: 257–275 [DOI] [PubMed] [Google Scholar]
- Desai N, Brown A, Amunts A, Ramakrishnan V (2017) The structure of the yeast mitochondrial ribosome. Science 355: 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekert K, de Kroon AI, Kispal G, Lill R (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol 65: 37–51 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CB, Waddle JA, Wt H, Dave V, Thornton J, Macatee TL, Garner HR, Butow RA (2001) Genome‐wide responses to mitochondrial dysfunction. Mol Biol Cell 12: 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 17: 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer‐Bender MF, Hanf M, Philippou‐Massier J, Krebs S, Zischka H, Jae LT (2020) A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579: 433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Bermudez J, Cuezva JM (2016) The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochem Biophys Acta 1857: 1167–1182 [DOI] [PubMed] [Google Scholar]
- Gerber AP, Herschlag D, Brown PO (2004) Extensive association of functionally and cytotopically related mRNAs with Puf family RNA‐binding proteins in yeast. PLoS Biol 2: E79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Gioran A, Piazzesi A, Bertan F, Schroer J, Wischhof L, Nicotera P, Bano D (2019) Multi‐omics identify xanthine as a pro‐survival metabolite for nematodes with mitochondrial dysfunction. EMBO J 38: e99558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM (2016) Mitochondrial diseases. Nat Rev Dis Primers 2: 16080 [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre‐mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M (2020) Mitochondrial stress is relayed to the cytosol by an OMA1‐DELE1‐HRI pathway. Nature 579: 427–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadikusumo RG, Meltzer S, Choo WM, Jean‐Francois MJ, Linnane AW, Marzuki S (1988) The definition of mitochondrial H+ ATPase assembly defects in mit‐ mutants of Saccharomyces cerevisiae with a monoclonal antibody to the enzyme complex as an assembly probe. Biochem Biophys Acta 933: 212–222 [DOI] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans . Dev Cell 13: 467–480 [DOI] [PubMed] [Google Scholar]
- Hedbacker K, Carlson M (2008) SNF1/AMPK pathways in yeast. Front Biosci 13: 2408–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J (2012) Mitochondrial disulfide relay: redox‐regulated protein import into the intermembrane space. J Biol Chem 287: 4426–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houshmandi SS, Olivas WM (2005) Yeast Puf3 mutants reveal the complexity of Puf‐RNA binding and identify a loop required for regulation of mRNA decay. RNA 11: 1655–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT (2010) Genome‐wide translational profiling by ribosome footprinting. Methods Enzymol 470: 119–142 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome‐protected mRNA fragments. Nat Protoc 7: 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch‐Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL et al (2009a) A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 20: 4730–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009b) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17: 98–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita‐Kikuta E, Koike T (2009) Separation and detection of large phosphoproteins using Phos‐tag SDS‐PAGE. Nat Protoc 4: 1513–1521 [DOI] [PubMed] [Google Scholar]
- Koehler CM (2004) The small Tim proteins and the twin Cx3C motif. Trends Biochem Sci 29: 1–4 [DOI] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, Larsson NG (2017) Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6: e30952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe CP, Stefely JA, Jochem A, Hutchins PD, Wilson GM, Kwiecien NW, Coon JJ, Wickens M, Pagliarini DJ (2018) Multi‐omics reveal specific targets of the RNA‐binding protein Puf3p and its orchestration of mitochondrial biogenesis. Cell systems 6: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe CP, Wilinski D, Saunders HA, Wickens M (2015) Protein‐RNA networks revealed through covalent RNA marks. Nat Methods 12: 1163–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CD, Tu BP (2015) Glucose‐regulated phosphorylation of the PUF protein Puf3 regulates the translational fate of its bound mRNAs and association with RNA granules. Cell Rep 11: 1638–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Du Q, Baird S, Ng AC, Nascimento M, Campanella M, McBride HM, Screaton RA (2013) Genome‐wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy 9: 1770–1779 [DOI] [PubMed] [Google Scholar]
- Lill R (2009) Function and biogenesis of iron‐sulphur proteins. Nature 460: 831–838 [DOI] [PubMed] [Google Scholar]
- Liu Z, Butow RA (2006) Mitochondrial retrograde signaling. Annu Rev Genet 40: 159–185 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464 [DOI] [PubMed] [Google Scholar]
- Lyne R, Burns G, Mata J, Penkett CJ, Rustici G, Chen D, Langford C, Vetrie D, Bahler J (2003) Whole‐genome microarrays of fission yeast: characteristics, accuracy, reproducibility, and processing of array data. BMC Genom 4: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Chen C (2019) The hap complex in yeasts: structure, assembly mode, and gene regulation. Front Microbiol 10: 1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsy S, Frachon P, Dujardin G, Lombes A, Lemaire C (2008) Respiratory mutations lead to different pleiotropic effects on OXPHOS complexes in yeast and in human cells. FEBS Lett 582: 3489–3493 [DOI] [PubMed] [Google Scholar]
- Martinez‐Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R et al (2016) TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell 61: 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzuki S, Watkins LC, Choo WM (1989) Mitochondrial H+‐ATPase in mutants of Saccharomyces cerevisiae with defective subunit 8 of the enzyme complex. Biochem Biophys Acta 975: 222–230 [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Granneman S, Thoms M, Manikas RG, Tollervey D, Hurt E (2014) Coupled GTPase and remodelling ATPase activities form a checkpoint for ribosome export. Nature 505: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz S, Westermann B (2009) Genome‐wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol 10: R95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modjtahedi N, Tokatlidis K, Dessen P, Kroemer G (2016) Mitochondrial proteins containing coiled‐coil‐helix‐coiled‐coil‐helix (CHCH) domains in health and disease. Trends Biochem Sci 41: 245–260 [DOI] [PubMed] [Google Scholar]
- Mordas A, Tokatlidis K (2015) The MIA pathway: a key regulator of mitochondrial oxidative protein folding and biogenesis. Acc Chem Res 48: 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D et al (2017) Systemic pan‐AMPK activator MK‐8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357: 507–511 [DOI] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS‐1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM (2012) Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 337: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndi M, Marin‐Buera L, Salvatori R, Singh AP, Ott M (2018) Biogenesis of the bc1 complex of the mitochondrial respiratory chain. J Mol Biol 430: 3892–3905 [DOI] [PubMed] [Google Scholar]
- Ogunbona OB, Claypool SM (2019) Emerging roles in the biogenesis of cytochrome c oxidase for members of the mitochondrial carrier family. Front Cell Dev Biol 7: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kondo‐Okamoto N, Ohsumi Y (2009) Mitochondria‐anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17: 87–97 [DOI] [PubMed] [Google Scholar]
- Olivas W, Parker R (2000) The Puf3 protein is a transcript‐specific regulator of mRNA degradation in yeast. The EMBO Journal 19: 6602–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MF, Velours J, Arselin de Chateaubodeau G, Aigle M, Guerin B (1989) The role of subunit 4, a nuclear‐encoded protein of the F0 sector of yeast mitochondrial ATP synthase, in the assembly of the whole complex. Eur J Biochem 185: 163–171 [DOI] [PubMed] [Google Scholar]
- Peleh V, Cordat E, Herrmann JM (2016) Mia40 is a trans‐site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. eLife 5, e16177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N, Warscheid B, Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20: 267–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Langer T, Lopez‐Otin C (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16: 345–359 [DOI] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ (2013) Autophagic processes in yeast: mechanism, machinery and regulation. Genetics 194: 341–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhle T, Leister D (2015) Assembly of F1F0‐ATP synthases. Biochem Biophys Acta 1847: 849–860 [DOI] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11: 655–667 [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto IC, Fontanesi F, Liu J, Barrientos A (2012) Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochem Biophys Acta 1817: 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spannagel C, Vaillier J, Arselin G, Graves PV, Velours J (1997) The subunit f of mitochondrial yeast ATP synthase–characterization of the protein and disruption of the structural gene ATP17. Eur J Biochem 247: 1111–1117 [DOI] [PubMed] [Google Scholar]
- Stefely JA, Pagliarini DJ (2017) Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci 42: 824–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, McCormick MA, Pham KM, MacKay VL, Delaney JR, Murakami CJ, Kaeberlein M, Kennedy BK (2012) Ribosome deficiency protects against ER stress in Saccharomyces cerevisiae . Genetics 191: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Bragoszewski P, Chacinska A (2012) The MIA pathway: a tight bond between protein transport and oxidative folding in mitochondria. Biochem Biophys Acta 1823: 1142–1150 [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Scharwey M, Langer T (2014) Mitochondrial lipid trafficking. Trends Cell Biol 24: 44–52 [DOI] [PubMed] [Google Scholar]
- Treitel MA, Kuchin S, Carlson M (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae . Mol Cell Biol 18: 6273–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch JR, McMurray MA, Nelson ZW, Gottschling DE (2009) Mitochondrial dysfunction leads to nuclear genome instability via an iron‐sulfur cluster defect. Cell 137: 1247–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese J, Abrams J, Wang Y, Morano KA (2012) Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol Mol Biol Rev 76: 115–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC‐1alpha axis. Cell Metab 14: 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Richards JD, Shaw JM, Stillman DJ (2001) Yeast vectors for integration at the HO locus. Nucleic Acids Res 29: E59–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen XJ (2015) A cytosolic network suppressing mitochondria‐mediated proteostatic stress and cell death. Nature 524: 481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440 [DOI] [PubMed] [Google Scholar]
- Weidberg H, Amon A (2018) MitoCPR‐A surveillance pathway that protects mitochondria in response to protein import stress. Science 360: eaan4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ (2013) Ribosome biogenesis in the yeast Saccharomyces cerevisiae . Genetics 195: 643–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG (2002) Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 99: 15066–15071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S et al (2015) Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524: 485–488 [DOI] [PubMed] [Google Scholar]
- Wu X, Li L, Jiang H (2016) Doa1 targets ubiquitinated substrates for mitochondria‐associated degradation. J Cell Biol 213: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Tu BP (2011) Selective regulation of autophagy by the Iml1‐Npr2‐Npr3 complex in the absence of nitrogen starvation. Mol Biol Cell 22: 4124–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
The deep‐sequencing raw data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al, 2002) and are accessible through GEO Series accession number GSE162197 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162197). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD022805 (http://www.ebi.ac.uk/pride/archive/projects/PXD022805).