

Abstract
N 6‐methyladenosine (m6A) modification of mRNA mediates diverse cellular and viral functions. Infection with Epstein–Barr virus (EBV) is causally associated with nasopharyngeal carcinoma (NPC), 10% of gastric carcinoma, and various B‐cell lymphomas, in which the viral latent and lytic phases both play vital roles. Here, we show that EBV transcripts exhibit differential m6A modification in human NPC biopsies, patient‐derived xenograft tissues, and cells at different EBV infection stages. m6A‐modified EBV transcripts are recognized and destabilized by the YTHDF1 protein, which leads to the m6A‐dependent suppression of EBV infection and replication. Mechanistically, YTHDF1 hastens viral RNA decapping and mediates RNA decay by recruiting RNA degradation complexes, including ZAP, DDX17, and DCP2, thereby post‐transcriptionally downregulating the expression of EBV genes. Taken together, our results reveal the critical roles of m6A modifications and their reader YTHDF1 in EBV replication. These findings contribute novel targets for the treatment of EBV‐associated cancers.
Keywords: Epstein–Barr virus, m6A modification, RNA decay, viral replication, YTHDF1
Subject Categories: Cancer; Microbiology, Virology & Host Pathogen Interaction; RNA Biology
The m6A reader YTHDF1 promotes viral RNA degradation by recruiting RNA decay complexes, including ZAP, DDX17, and DCP2. YTHDF1 suppresses EBV lytic replication to regulate EBV infection status.
Introduction
The N 6‐methyladenosine (m6A) RNA modification was discovered in the 1970s, and later studies revealed its presence across different regions of mRNA transcripts (Desrosiers et al, 1974; Wei et al, 1976). Notably, it is distinctly enriched near the stop codons, where the consensus motif G[G>A]m6AC[U>A>C] is found (Dominissini et al, 2012; Meyer et al, 2012). Several m6A‐associated proteins have been identified to date. Among them, the “writer” proteins comprise the enzyme METTL3 with its two co‐factors METTL14 and WTAP (Bokar et al, 1997; Liu et al, 2014; Ping et al, 2014; Schwartz et al, 2014), while the “eraser” proteins FTO and ALKBH5 act as RNA de‐methylases (Jia et al, 2011; Zheng et al, 2013). Finally, the so‐called “reader” proteins are mainly responsible for regulating RNA metabolism based on the m6A signal. The YTH domain‐containing proteins, including YTHDF1, YTHDF2, and YTHDF3, are the most studied “reader” proteins. YTHDF1 enhances the translation efficiency of m6A‐modified mRNAs (Wang et al, 2015b), whereas YTHDF2 mediates RNA degradation (Wang et al, 2014; Du et al, 2016). YTHDF3 can cooperate with either YTHDF1 or YTHDF2 to further influence RNA fate (Shi et al, 2017; Li et al, 2017a). Importantly, YTHDF1, YTHDF2, and YTHDF3 can function together to mediate the degradation of m6A‐mRNAs (Lasman et al, 2020; Zaccara & Jaffrey, 2020). However, other m6A readers can also regulate various RNA processes, including RNA splicing, RNA helicase activity, nuclear export, microRNA processing, or promoting RNA stability and translation (Alarcon et al, 2015; Liu et al, 2015; Xiao et al, 2016; Hsu et al, 2017; Wojtas et al, 2017; Huang et al, 2018).
The m6A modification was long known to be present in viral transcripts, such as those encoded by herpes simplex virus type 1 (HSV‐1), influenza A virus (IAV), simian virus 40 (SV40), Rous sarcoma virus, avian sarcoma virus, and adenovirus (Lavi & Shatkin, 1975; Krug et al, 1976; Sommer et al, 1976; Dimock & Stoltzfus, 1977; Moss et al, 1977; Chen‐Kiang et al, 1979; Kane & Beemon, 1985). Recently, the m6A RNA methylomes of RNA viruses have been sequenced, including human immunodeficiency virus (HIV), Zika virus (ZIKV), hepatitis C virus (HCV), enterovirus 71 (EV71), murine leukemia virus (MLV), human respiratory syncytial virus (RSV), and the plant virus alfalfa mosaic virus (Gokhale et al, 2016; Kennedy et al, 2016; Tirumuru et al, 2016; Lichinchi et al, 2016a; Lichinchi et al, 2016b; Courtney et al, 2017; Martinez‐Perez et al, 2017; Hao et al, 2018; Lu et al, 2018; Courtney et al, 2019; Xue et al, 2019). Moreover, the methylation pattern has also been elucidated in the transcripts of numerous DNA viruses, such as hepatitis B virus (HBV) and Kaposi’s sarcoma‐associated herpesvirus (KSHV) (Ye et al, 2017; Hesser et al, 2018; Imam et al, 2018; Tan et al, 2018; Baquero‐Perez et al, 2019). Crucially, the m6A machinery was reported to regulate viral replication and infectivity (Gonzales‐van Horn & Sarnow, 2017; Tan & Gao, 2018; Tirumuru & Wu, 2019).
Epstein–Barr virus (EBV), the first identified human oncovirus, is widely associated with B‐cell lymphoma, gastric cancer, and nasopharyngeal carcinoma (NPC) (Shi et al, 2016; Young et al, 2016; Liu et al, 2019). The ability of EBV to readily infect primary B cells in vitro and transform them into proliferating lymphoblastoid cells (LCLs) strongly supports its role in B‐cell malignancies (Young & Rickinson, 2004). Once inside infected cells, EBV can enter either the latent phase or the lytic phase, which have been categorized primarily based on the differential expression of viral proteins (Munz, 2019). In latency III, EBV expresses six EBV nuclear antigens (EBNA 1, 2, 3A, 3B, 3C, and ‐LP), three latent membrane proteins (LMP 1, 2A, and 2B) (Allday et al, 2015; Thorley‐Lawson, 2015). In latency IIa, EBV expresses EBNA1 and LMPs. Latently infected memory cells express no viral proteins (latency 0) (Babcock et al, 1998). During homeostatic proliferation, EBNA1 is transiently expressed in EBV‐infected memory B cells, and this pattern is called latency I (Hochberg et al, 2004). In latency IIb, EBV expresses six EBNAs but does not express LMPs (Doyle et al, 1993; Klein et al, 2013). EBV can be reactivated by external stimuli, such as 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) and sodium butyrate (NaB) (zur Hausen et al, 1978; Luka et al, 1979). During reactivation, the viral immediate‐early (IE) lytic genes BZLF1 and BRLF1 are expressed, which both play pivotal roles in virus reactivation and lytic replication (Countryman & Miller, 1985; Hardwick et al, 1988). In the lytic phase, EBV expresses its lytic genes to start the EBV lytic replication machinery to infectious virions and thereby infect more cells. Although most studies found that EBV exhibits a latency II pattern in NPC (Fahraeus et al, 1988), recent studies found that EBV genes of latency III pattern and lytic phase are also expressed in NPC, indicating a broad range of spontaneous lytic reactivation of EBV (Arvey et al, 2012; Tsai et al, 2013; Hu et al, 2016). Previous studies have found that DNA methylation, transcription factors, and miRNAs can regulate the expression of BZLF1 and BRLF1 to influence viral replication (Wille et al, 2013; Woellmer & Hammerschmidt, 2013; Wille et al, 2015; Li et al, 2016). Recently, the EBV epitranscriptome in EBV‐transformed LCLs and lymphoma cells was examined, and it is illustrated that m6A modification regulated EBV‐associated tumorigenesis (Lang et al, 2019). However, it remains unclear whether the EBV lifecycle is regulated by m6A modifications in NPC. In this study, we show that m6A modifications are present on numerous EBV transcripts. We also demonstrate that m6A‐binding protein YTHDF1 interacts with the RNA decay machinery to destabilize EBV transcripts, thereby suppressing the lytic replication of EBV.
Results
m6A modifications are widespread in EBV transcripts in NPC cells and primary cancer cells
To determine the presence of potential m6A modification sites in the EBV epitranscriptome, we performed methylated RNA immunoprecipitation and sequencing (MeRIP‐seq) (Dominissini et al, 2012, 2013; Meyer et al, 2012) on various cells infected with EBV in different life cycle and different biopsies. The examined samples included NPEC1‐Bmi1 cells and HK1 cells with acute EBV infection, C666 cells and CNE2EBV cells with latent EBV infection, CNE2EBV cells with lytic EBV reactivation, B cells with latent infection or lytic reactivation, one nontumor nasopharyngeal biopsy, two primary NPC biopsies, and two NPC patient‐derived xenograft (PDX) tissues.
The results revealed that m6A‐modified EBV transcripts were present in NPC cells, B cells, and tissue samples. These transcripts included EBNA1, the IE transcripts BZLF1 and BRLF1, and the lytic transcripts BHRF1, BXLF2, BNLF2a, and BSLF2/BMLF1 (Figs 1A and EV1). Notably, EBNA1 and BNLF2a were m6A‐modified in the cells under different EBV infection states and EBV‐infected tissues (Figs 1A and EV1). As expected, no EBV reads were found in the nontumor control biopsy (NPN in Fig EV1), whereas BZLF1, BSLF2/BMLF1, BNLF2a, and BHRF1 exhibited higher levels of m6A modifications in the PDX samples (PDX‐NPC04L and PDX‐NPC03W in Fig EV1). The m6A modifications on the transcripts of EBNA1, BZLF1, BRLF1, BHRF1, BSLF2/BMLF1, BNLF2a, and BXLF2 were confirmed by MeRIP‐qPCR in CNE2EBV cells with induced EBV reactivation (Appendix Fig S1). Additionally, there were differences in the expression levels and m6A levels of EBV transcripts among B cells, NPC cells, and tissue samples (Fig EV1). The m6A modification sites of EBV transcripts are summarized in Dataset EV1.
Figure 1. EBV transcripts are m6A modified.
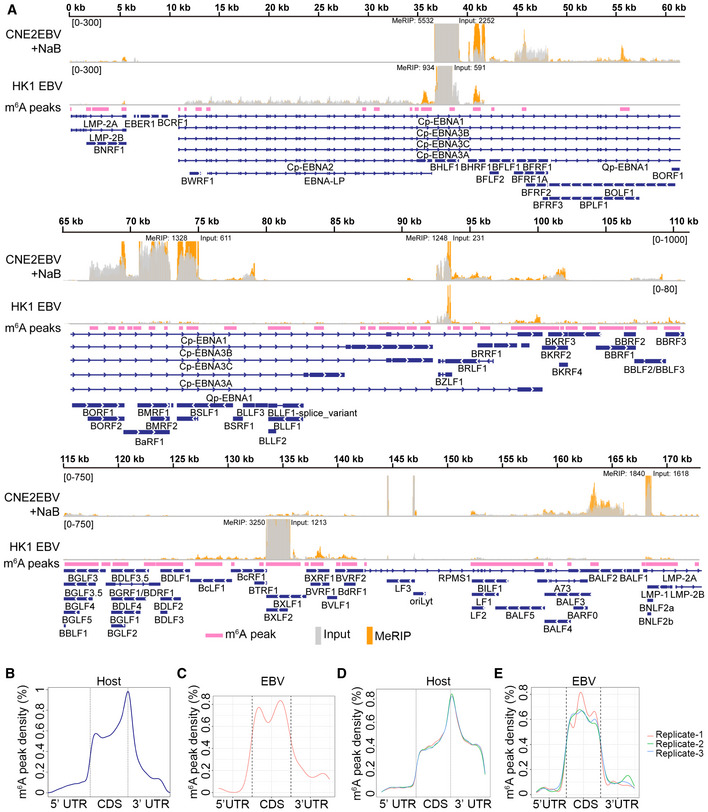
-
AMap showing the EBV epitranscriptome in CNE2EBV with induced EBV reactivation, or HK1 cells with acute EBV infection. CNE2EBV cells were treated with NaB (2 mM) for 24 h to reactivate EBV. For acute EBV infection, RNA of HK1 cells was harvested at 24 hpi. The m6A peaks, identified based on all MeRIP‐seq data using MACS2, are indicated as pink bars. The input and MeRIP coverage are indicated with gray and orange bars, respectively. The coverage is shown as a number if out of range.
-
B, CDistribution of the m6A peaks across host mRNAs (B) and EBV mRNAs (C) was analyzed based on the MeRIP‐seq data.
-
D, EDistribution of the m6A peaks across the host transcripts (D) and EBV transcripts (E) was analyzed based on three independent PA‐m6A‐seq experiments. PA‐m6A‐seq assays were performed in CNE2EBV cells following EBV reactivation, which was induced using TPA and NaB.
Figure EV1. The m6A modification landscape of EBV in NPC cells, B cells, and tissue samples.
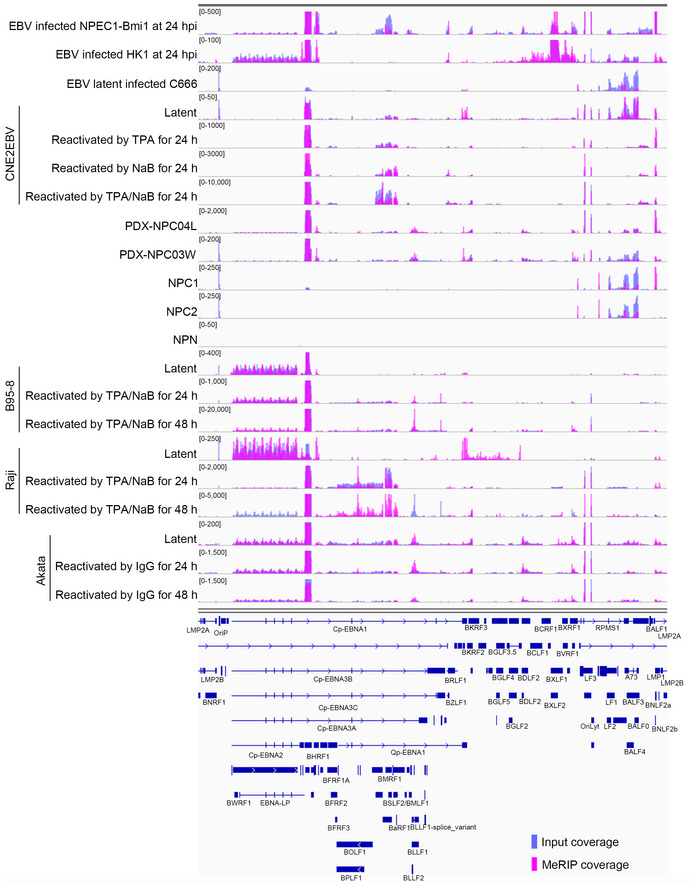
The m6A modification landscape of EBV in different cells with different stages of EBV infection is shown. The data presented from the top down are EBV acute infection in NPEC1‐Bmi1 sphere‐like cells; EBV acute infection in HK1 cells; EBV latent infection in C666 cells; CNE2EBV cells with latent infection, induced with TPA, induced with NaB, induced with both TPA and NaB; PDX‐NPC04L tissue; PDX‐NPC03W tissue; NPC1 tissue; NPC2 tissue; nontumor control biopsy (NPN); B95‐8 cells with latent EBV infection, reactivated with TPA and NaB at 24 h, or 48 h; Raji cells with EBV latent infection, reactivated with TPA and NaB at 24 h, or 48 h; Akata cells with EBV latent infection, reactivated with IgG at 24 h, or 48 h. The input and MeRIP coverage is indicated with blue and pink bars, respectively.
Further analysis indicated that distribution of the cellular m6A peaks occurred in the coding regions (CDS) and 3′ untranslated regions (UTR), with a sharp crest near the stop codon (Fig 1B), which was consistent with previous reports (Meyer et al, 2012; Dominissini et al, 2013). In contrast, the m6A peaks of EBV‐encoded messages might be mostly distributed along the entire CDS region (Fig 1C). Similarly, m6A sites in the transcripts of IAV, SV40, and KSHV are also enriched in the CDS (Courtney et al, 2017; Tsai et al, 2018; Baquero‐Perez et al, 2019). This phenomenon may either be virus‐specific or simply a statistical consequence of the relatively short non‐coding transcribed regions in highly compact viral genomes.
Additionally, a previously described photo cross‐linking‐assisted m6A sequencing technique (PA‐m6A‐seq) (Chen et al, 2015) was used to identify m6A residues in EBV transcripts. When the photoactivatable ribonucleoside 4‐thiouridine (4SU) is incorporated into mRNAs, it can be covalently cross‐linked with nearby aromatic amino acid residues in RNA‐binding proteins upon UV irradiation at 365 nm. The m6A sites can be precisely identified by scoring for thymidine (T) to cytidine (C) transitions in the sequencing data. PA‐m6A‐seq improves the accuracy of the methylation site assignments and provides a high‐resolution transcriptome‐wide m6A map. In this study, the addition of 200 µM 4SU had a modest effect on the cell viability at 20 h (Appendix Fig S2). CNE2EBV cells were treated with TPA (30 ng/ml) and NaB (2 mM) for 4 h to reactivate EBV and were then pulsed with 200 µM 4SU for 20 h. The total mRNA of these cells was incubated with m6A‐specific antibody and cross‐linked. The RNAs bound to the m6A antibody were sequenced. With PA‐m6A‐seq and bioinformatic analysis, a large number of m6A sites were identified in the viral transcripts (Fig EV2A). Consistent with our MeRIP‐seq results, the m6A sites of cellular mRNAs identified using PA‐m6A‐seq were enriched in the CDS and 3′ UTR, with a sharp crest near the stop codon (Fig 1D), while the m6A sites of the EBV mRNAs were dispersed throughout the entire CDS region (Fig 1E). The most consensus m6A motif, based on all PA‐m6A‐seq experimental replicates, was “GGAC” in the host genes and “GAC” in the EBV genes (Fig EV2B and C). These are both classical m6A motif (Dominissini et al, 2012; Meyer et al, 2012).
Figure EV2. Analysis of the EBV m6A epitranscriptome based on PA‐m6A‐seq data of CNE2EBV cells following EBV reactivation.
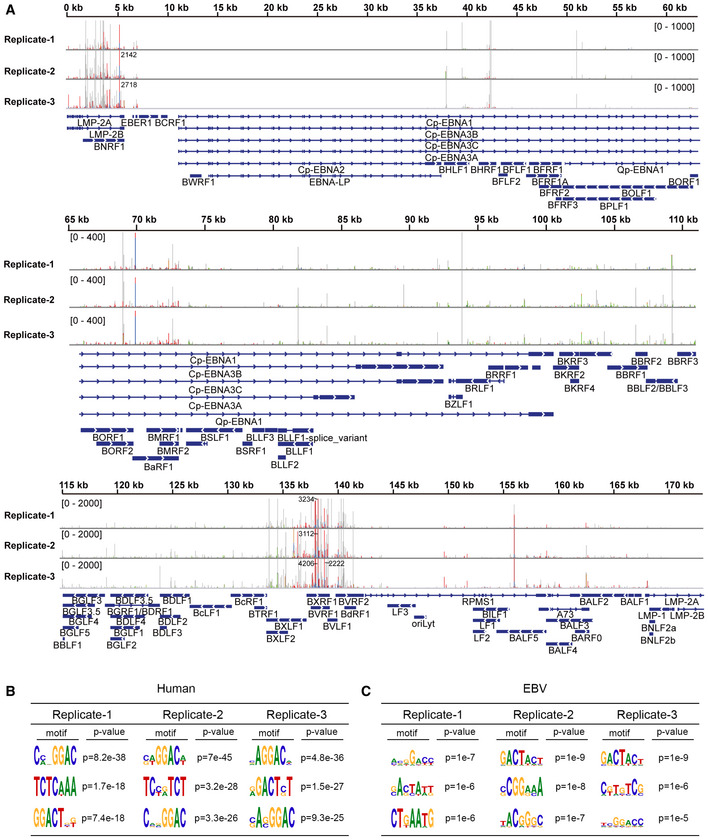
-
ASites of m6A modification in EBV transcripts were mapped by analyzing the PA‐m6A‐seq data from CNE2EBV cells following EBV reactivation. Three independent PA‐m6A‐seq experimental replicates are shown. CNE2EBV cells were induced with TPA (30 ng/ml) and NaB (2 mM) for 4 h to reactivate EBV and were then pulsed with 200 µM 4SU for 20 h. The 4SU‐based CLIP technique results in T‐to‐C mutations at cross‐linked 4SU residues that arise during reverse transcription. Red/blue bars indicate sites of T‐to‐C conversions. Green/brown bars indicate sites of A‐to‐G conversions. The coverage is shown as a number if out of range.
-
B, CMotif analysis to identify consensus sequences on host transcripts (B) and EBV transcripts (C) based on three independent PA‐m6A‐seq experiments. The top three motifs for each are shown.
Validation of m6A modifications of the BZLF1 mRNA
As an IE gene, BZLF1 plays critical roles in the reactivation and lytic replication of EBV (Countryman & Miller, 1985). m6A modification of BZLF1 mRNA was augmented not only in cells during acute infection and lytic reactivation but also in PDXs in vivo, compared to the modification levels detected in CNE2EBV cells with latent EBV infection (Fig 2A). The PA‐m6A‐seq data showed that BZLF1 mRNA had a potential m6A modification region harboring two potential m6A sites at bases 47 and 58, both positioned within a “GAC” motif (Fig 2A). Furthermore, EBV reactivation markedly increased the proportion of m6A‐modified BZLF1 transcripts in the CNE2EBV, Raji, and B95‐8 cells (Fig 2B). To determine whether the m6A levels at specific sites were induced by EBV reactivation, we used the single‐base elongation‐ and ligation‐based PCR method (SELECT) (Xiao et al, 2018). This method takes advantage of the fact that m6A inhibits both the single‐base elongation activity of the DNA polymerases and the nick ligation efficiency of ligases, which can be detected by qPCR. Within the m6A peaks of BZLF1 mRNA, two potential m6A sites are located at bases 47 and 58 (Fig 2A). Two “A” sites at bases 150 and 528, which are not associated with the "GAC" motifs, were selected as controls. The relative SELECT product abundance of the two potential m6A sites, normalized to the product abundance of negative control base 150, was markedly decreased after the induction of EBV reactivation (Fig 2C). As expected, EBV reactivation did not affect the relative SELECT product abundance of the negative control at base 528 (Fig 2C). Since less SELECT product abundance indicates a higher m6A modification level, the SELECT results revealed that EBV reactivation increased the m6A levels of the BZLF1 mRNA at bases 47 and 58 (Fig 2C). The MeRIP‐qPCR results revealed a prominent m6A enrichment in the BZLF1 mRNA in PDX tissues, compared with a nontumor control biopsy (Fig 2D).
Figure 2. The BZLF1 mRNA is m6A modified.
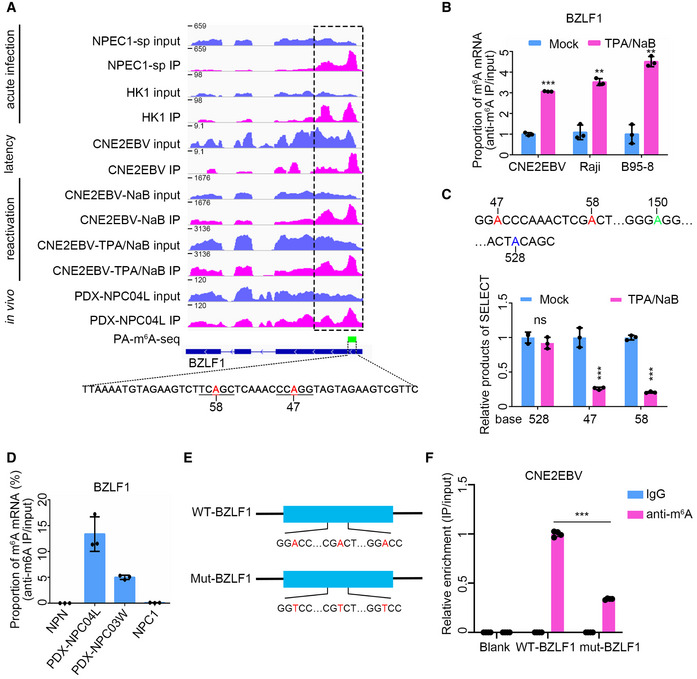
- m6A peak distribution of BZLF1 in different EBV infection stages was analyzed based on MeRIP‐seq data. The data presented from the top down are the EBV acute infected (24 hpi) NPEC1‐Bmi1 sphere‐like cells (NPEC‐sp), and HK1 cells; latently infected CNE2EBV cells; and CNE2EBV cells with reactivated EBV induced using NaB alone or TPA/NaB together. For acute EBV infection, RNA was harvested at 24 hpi. TPA (30 ng/ml) and NaB (2 mM) were used alone or together to treat the cells for 24 h to reactivate EBV. The PA‐m6A‐seq peak is indicated by the green box, and the potential m6A sites on the indicated sequence are shown in red. The region indicated by the dotted box was PCR‐amplified to construct the wild‐type BZLF11‐262nt plasmid.
- The BZLF1 m6A levels were measured using MeRIP‐qPCR in CNE2EBV, Raji, and B95‐8 cells with latent EBV infection or lytic reactivation. TPA (30 ng/ml) and NaB (2 mM) were used to treat the cells for 24 h to reactivate EBV. The cellular RNA was harvested for MeRIP‐qPCR assays. The fold enrichment was determined by calculating the 2−Δ C t of the MeRIP sample relative to the input sample. The mean value of the results in mock‐treated cells was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 compared to mock‐treated cells according to unpaired Student’s t‐test.
- The relative product abundance of SELECT at the predicted m6A sites in the latently infected or lytic reactivated CNE2EBV cells. TPA (30 ng/ml) and NaB (2 mM) were used to treat the CNE2EBV cells for 24 h to reactivate EBV. The mean value of results in mock‐treated cells was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ***P < 0.001 compared to the mock‐treated group according to unpaired Student’s t‐test.
- The validation of the BZLF1 m6A peaks by MeRIP‐qPCR in PDXs, NPC1, and NPN. One nontumor control biopsy (NPN), one NPC sample (NPC1), and two PDX samples (PDX‐NPC04L and PDX‐NPC03W) were used to perform the MeRIP‐qPCR assays. The fold enrichment was determined by calculating the 2−Δ C t of the MeRIP sample relative to the input sample. The data represent the means ± SD of n = 3 technical replicates.
- Construction of the WT‐BZLF1 and mut‐BZLF1 plasmids. The WT‐BZLF1 plasmid was constructed by inserting the BZLF11–262nt fragment (1–262 nt of the BZLF1 mRNA) into pcDNA3.1+ plasmid. The mut‐BZLF1 plasmid was constructed by introducing A>T mutations at the putative m6A sites of BZLF11–262nt.
- The IgG or anti‐m6A antibody enrichment of the WT‐BZLF1 and mut‐BZLF1 mRNAs was measured by RIP‐qPCR in the CNE2EBV cell lines. Blank indicates CNE2EBV cells without plasmids transfection. The fold enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. The mean value of the m6A enrichment level on WT‐BZLF1 mRNA was normalized to 1. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates. ***P < 0.001 according to unpaired Student’s t‐test.
To further validate the m6A modification sites of the BZLF1 mRNA, we constructed the wild‐type and mutant BZLF11–262nt plasmids by cloning a wild‐type 262 nt length fragment encompassing the m6A peak region in the BZLF1 mRNA identified according to the MeRIP‐seq data and a mutant fragment with A>T mutations at the potential m6A sites, respectively (Fig 2E). The plasmids were introduced into the CNE2EBV cells for MeRIP‐qPCR analysis, which exhibited a higher m6A level on the wild‐type BZLF11–262nt transcripts than the mutant control (Fig 2F), confirming the presence of m6A sites in the BZLF1 mRNA.
The m6A reader YTHDF1 represses EBV infection and lytic replication
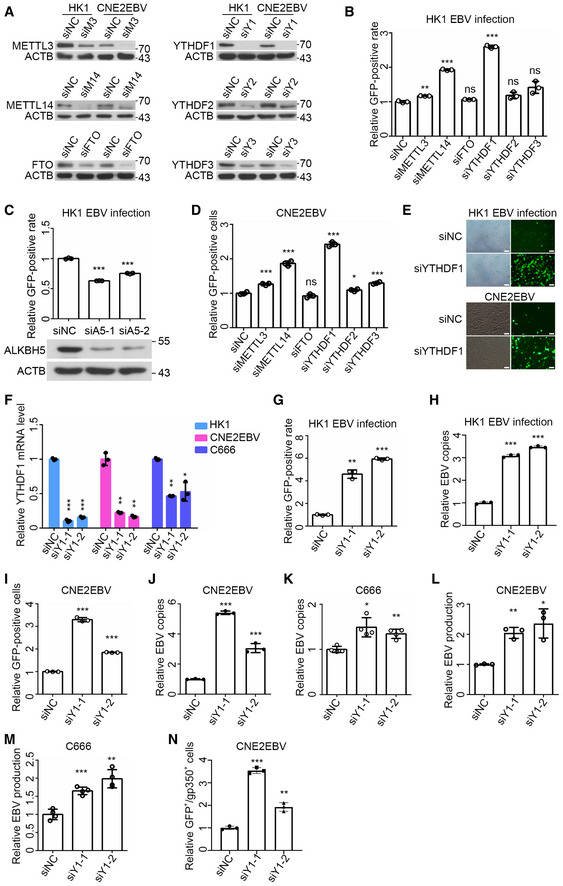
To investigate whether m6A modification influences EBV infection or replication, we used siRNA libraries to knock down m6A “writers”, “erasers”, and “readers” in the HK1 and CNE2EBV cells. Western blot analysis confirmed that the siRNAs could efficiently silence the targeted gene expression (Fig 3A). HK1 cells were infected with the EBV‐GFP virus and subjected to flow cytometry (FACS) analysis at 24 h post‐transfection (hpi). The FACS results showed that the knockdown of METTL3, METTL14, or YTHDF1 significantly increased the ratio of GFP‐positive HK1 cells (Fig 3B), while the ALKBH5 knockdown could reverse this phenotype (Fig 3C). CNE2EBV cells carried the recombinant EBV‐GFP virions. In the lytic replicative cycle, the number of EBV genome copies resulted in a heightened GFP fluorescence intensity, which could be determined by FACS. After siRNA transfection, CNE2EBV cells were treated with NaB (2 mM) for 24 h to reactivate EBV, followed by FACS to detect the GFP intensity. The results indicated that the knockdown of METTL3, METTL14, YTHDF1, YTHDF2, or YTHDF3 promoted the lytic replication of EBV in the CNE2EBV cells with induced EBV reactivation (Fig 3D). The effects of YTHDF1 knockdown on EBV infection and lytic replication were further confirmed using fluorescence imaging (Fig 3E).
Figure 3. Knockdown of YTHDF1 promotes EBV infection and replication.
-
AThe knockdown efficiency of METTL3, METTL14, FTO, YTHDF1, YTHDF2, and YTHDF3 in HK1 and CNE2EBV cells at 24 h after siRNA transfection was determined by Western blot analysis. ACTB was used as loading control.
-
BThe EBV infection efficiency in HK1 cells transfected with the indicated siRNAs was analyzed by FACS. The cells were infected with EBV‐GFP virus at 24 h after siRNA transfection. The ratio of GFP‐positive cells was quantified by FACS at 24 hpi. The mean value of the GFP‐positive rates in the siNC‐transfected HK1 cells was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ns, **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
CThe ratio of GFP‐positive HK1 cells transfected with ALKBH5‐specific siRNAs (siA5‐1 and siA5‐2) or the siNC control was quantified using FACS. The experiments were performed similarly as described for (B) except for the different siRNA transfection. At 24 h post‐siRNA transfection, the protein level of ALKBH5 was determined by Western blot analysis. ACTB was used as loading control. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
DThe GFP‐positive cells of the EBV reactivated CNE2EBV cells transfected with indicated siRNAs. After 24 h of siRNA transfection, the cells were induced with NaB (2 mM) for 24 h. The GFP‐positive cells were quantified using FACS. The mean value of the GFP‐positive rates in the siNC transfected cells was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ns, *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
EThe GFP‐expressed cells were visualized by fluorescence microscopy after YTHDF1 knockdown in HK1 cells following EBV infection and CNE2EBV cells with induced EBV reactivation. The experiments were performed similarly as described for (B) in HK1 cells and (D) in CNE2EBV cells. Representative data from three independent experiments are shown. Scale bars: 50 μm.
-
FThe knockdown efficiency of YTHDF1 in HK1, CNE2EBV, and C666 cells, transfected with YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control for 24 h, was determined by qRT–PCR. The mRNA levels of YTHDF1 were normalized to the housekeeping gene ACTB. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
G, HThe HK1 cells were transfected with YTHDF1‐specific siRNA (siY1‐1 and siY1‐2) or the siNC control for 24 h, and then infected with EBV‐GFP virus. At 24 hpi, the ratio of GFP‐positive cells was quantified using FACS (G), and the number of EBV copies was quantified using qPCR (H). The EBV copies were determined using a specific primers targeting BamHI‐W fragment region of EBV, and GAPDH was used as a reference genome copy. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
I, JCNE2EBV cells were transfected with YTHDF1‐specific siRNA (siY1‐1 and siY1‐2) or the siNC control for 24 h, and then induced with NaB (2 mM). At 24 h post‐NaB‐induced EBV reactivation, the GFP‐positive cells (I) and the EBV copies (J) were quantified as described for (G and H). Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
KThe relative number of EBV copies in C666 cells transfected with the YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control was quantified using qPCR. At 24 h after siRNA transfection, the EBV copies were quantified as described for (H). The mean value of EBV copies in the siNC cells was normalized to 1. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates. *P < 0.05 and **P < 0.01 compared to siNC according to unpaired Student’s t‐test.
-
LThe relative production of EBV virions in CNE2EBV cells transfected with YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control following EBV reactivation was measured by qPCR. After 24 h of siRNA transfection, CNE2EBV cells were treated with NaB (2 mM) for 24 h, and the DNA from the cell‐free culture supernatant was harvested for qPCR. The number of EBV copies in the supernatant of the cells was quantified by qPCR using specific primers targeting BamHI‐W fragment region of EBV. The mean value of EBV copies in the siNC cells was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05 and **P < 0.01 compared to siNC according to unpaired Student’s t‐test.
-
MThe relative EBV production in C666 cells transfected with YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control was measured by qPCR. The DNA from the cell‐free culture supernatant was harvested at 24 h after siRNA transfection. The number of EBV copies in the supernatant of the cells was quantified by qPCR using specific primers targeting BamHI‐W fragment region of EBV. The mean value of EBV copies in the siNC cells was normalized to 1. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates. **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
-
NFACS analysis of gp350 expression in CNE2EBV cells transfected with YTHDF1‐specific siRNA (siY1‐1 and siY1‐2) or the siNC control following EBV reactivation. To measure gp350 expression in the reactivated CNE2EBV cells, cells were stained with anti‐gp350 mAb (72A1) followed by an Alexa Fluor 594‐conjugated secondary anti‐mouse antibody and analyzed by FACS. The quantification of the ratio of GFP‐positive and gp350‐positive (GFP+/gp350+) cells from three independent experiments was shown. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
Source data are available online for this figure.
To decipher the role of YTHDF1 in promoting EBV infection and lytic replication (Fig 3A–E), we further focused on the function and underlying mechanism of YTHDF1 in the modulation of the EBV lifecycles. qPCR was used to determine the number of EBV copies (Zhang et al, 2018b), and FACS was used to quantify the ratio of GFP‐positive cells, which revealed that YTHDF1 knockdown facilitated EBV infection and lytic replication in HK1 cells (Figs 3F–H and EV3A). It also promoted lytic replication in the CNE2EBV cells with induced EBV reactivation (Figs 3I and J, and EV3B). It did not affect the viability of either HK1 or CNE2EBV cells (Fig EV3C). Since C666 cells contained original‐infecting EBV without GFP, we used qPCR to quantify the endogenous EBV copies and found that YTHDF1 knockdown also promoted EBV replication in C666 cells (Fig 3K). Additionally, YTHDF1 knockdown also promoted EBV production in CNE2EBV cells with induced EBV reactivation and C666 cells (Fig 3L and M). Moreover, the FACS results revealed that YTHDF1 knockdown increased the ratio of the gp350‐positive cells in CNE2EBV cells with EBV‐induced reactivation (Figs 3N and EV3D). The immunofluorescence results indicated that YTHDF1 knockdown increased the expression of gB in CNE2EBV cells with EBV‐induced reactivation (Fig EV3E). Therefore, our results suggest that YTHDF1 knockdown promotes lytic replication and progeny production of EBV.
Figure EV3. FACS analysis and cell viability assay of HK1 and CNE2EBV cells with YTHDF1 knockdown.
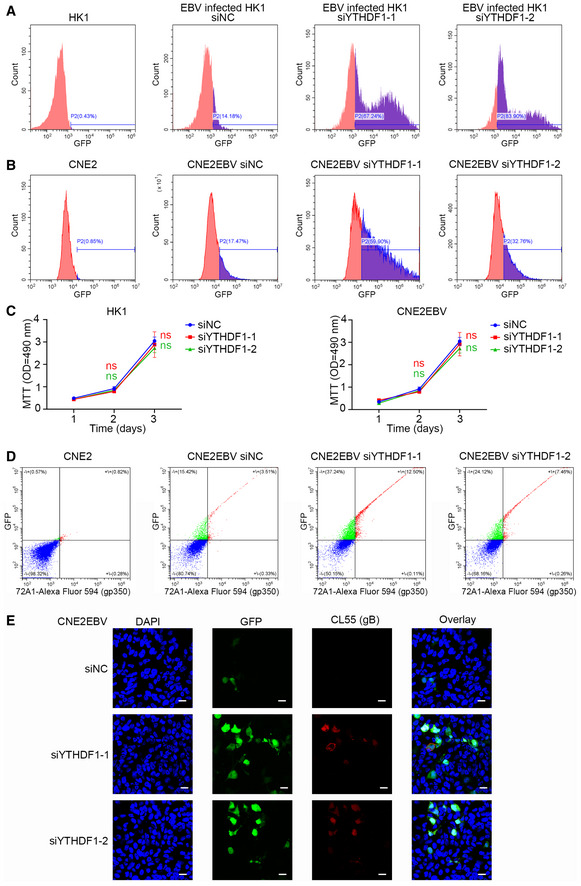
- The ratio of GFP‐positive HK1 cells transfected with YTHDF1‐specific siRNA (siYTHDF1‐1 and siYTHDF1‐2) or the siNC control was quantified using FACS. Representative data of Fig 3G from three independent experiments are shown.
- The GFP‐positive cells of CNE2EBV cells transfected with YTHDF1‐specific siRNA (siYTHDF1‐1 and siYTHDF1‐2) or the siNC control following EBV reactivation were measured using FACS. Representative data of Fig 3I from three independent experiments are shown.
- Growth curves of cells with YTHDF1 knockdown. MTT assays of CNE2EBV and HK1 cells with YTHDF1 knockdown. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates. ns compared to siNC according to unpaired Student’s t‐test.
- FACS analysis of gp350 expression in CNE2EBV cells transfected with YTHDF1‐specific siRNA (siYTHDF1‐1 and siYTHDF1‐2) or the siNC control following EBV reactivation. Representative data of Fig 3N from three independent experiments are shown.
- The ratio of GFP‐positive and gB‐expressing CNE2EBV transfected with YTHDF1‐specific siRNA (siYTHDF1‐1 and siYTHDF1‐2) or the siNC control following EBV reactivation was visualized by immunofluorescence. To measure gB expression in CNE2EBV cells following EBV reactivation, the cells were stained with anti‐gB mAb (CL55) followed by an Alexa Fluor 594‐conjugated secondary anti‐mouse antibody confocal laser scanning microscopy. Representative data from three independent experiments are shown. Scale bars: 20 μm.
YTHDF1 represses the expression of BZLF1 and BRLF1
Next, RNA‐seq was performed to determine the effect of YTHDF1 knockdown on the global EBV gene expression in acutely infected HK1 cells or CNE2EBV cells. The results indicated that the YTHDF1 knockdown upregulated most EBV genes’ expression, including BZLF1 and BRLF1 (Dataset EV2). Strikingly, we observed a remarkable elevation of the BZLF1 and BRLF1 transcript levels upon YTHDF1 knockdown in acutely infected HK1 cells, EBV latently infected or EBV lytic reactivated in CNE2EBV cells, as well as C666 cells (Fig 4A–C). BZLF1 protein level was also induced in the YTHDF1 knockdown cells (Fig 4D). Additionally, many cellular pathways related to viral replication were also affected by YTHDF1 knockdown (Appendix Fig S3A and B). On the other hand, EBV infection and lytic reactivation could in turn suppress YTHDF1 expression (Fig 4E and F), suggesting a positive feedback to facilitate EBV infection.
Figure 4. YTHDF1 knockdown promotes the expression of BZLF1 and BRLF1.
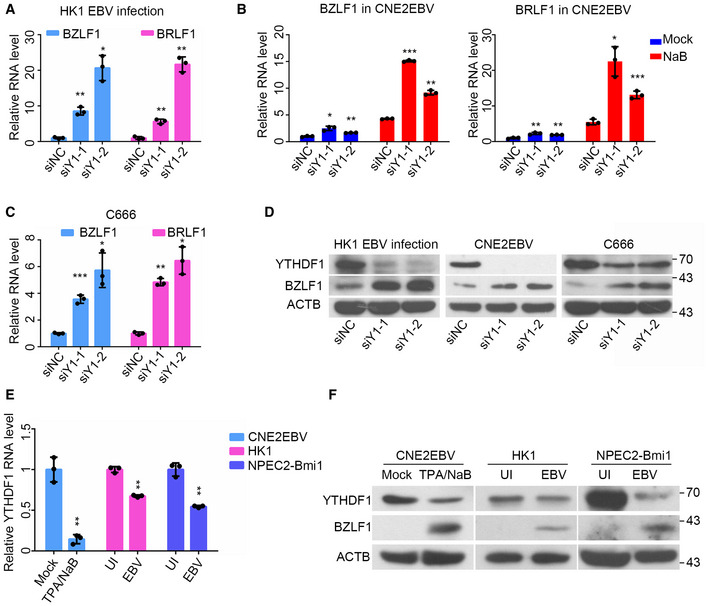
- The relative mRNA levels of BZLF1 and BRLF1 at 24 hpi following EBV infection of HK1 cells transfected with the YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control. After 24 h of siRNA transfection, HK1 cells were infected with EBV‐GFP and analyzed by qRT–PCR at 24 hpi. All the gene expression levels were normalized to the housekeeping gene ACTB. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05 and **P < 0.01 compared to siNC according to unpaired Student’s t‐test.
- The relative mRNA levels of BZLF1 and BRLF1 in the latently infected or reactivated (24 h post‐NaB treatment) CNE2EBV cells transfected with the YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control. After 24 h of siRNA transfection, CNE2EBV cells were treated with NaB (2 mM) or mock for 24 h, followed by qRT–PCR analysis. All the gene expression levels were normalized to the housekeeping gene ACTB. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The relative mRNA levels of BZLF1 and BRLF1 in C666 cells transfected with the YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control. The total RNA was harvested from the cells at 24 h after siRNA transfection. All gene expression levels were normalized to the housekeeping gene ACTB. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The protein levels of YTHDF1 and BZLF1 in the EBV‐infected (24 hpi) HK1 cells, CNE2EBV cells with induced EBV reactivation (24 h post‐NaB treatment) and C666 cells transfected with YTHDF1‐specific siRNA (siY1‐1 and siY1‐2) or the siNC control. The whole‐cell protein was extracted at same time as the RNA.
- The relative mRNA expression levels of YTHDF1 in CNE2EBV cells following EBV reactivation (24 h post‐NaB treatment), EBV‐infected HK1 or NPEC2‐Bmi1 cells at 24 hpi. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 according to unpaired Student’s t‐test.
- The protein level of YTHDF1, BZLF1, and ACTB in CNE2EBV cells following EBV reactivation (24 h post‐NaB treatment), EBV‐infected HK1, or NPEC2‐Bmi1 cells at 24 hpi. The whole‐cell protein was extracted at same time as the RNA.
Source data are available online for this figure.
Taken together, the data suggests that YTHDF1 knockdown promotes EBV infection and replication, at least partially through upregulating the expression of BZLF1 and BRLF1.
YTHDF1 decreases EBV transcript stability
Based on our observations that the knockdown of YTHDF1 could increase the expression of BZLF1 and BRLF1, as well as that both BZLF1 and BRLF1 transcripts were m6A‐modified, we further investigated whether YTHDF1 could bind to the m6A‐modified BZLF1 and BRLF1 mRNA. C666 cells and EBV‐reactivated CNE2EBV cells were subjected to RNA immunoprecipitation (RIP) assay using a YTHDF1‐specific antibody. Compared with the IgG control, YTHDF1 pull down yielded 19‐fold and 93‐fold enrichment of the BZLF1 and BRLF1 mRNAs in C666 cells and, likewise, 268‐fold and 252‐fold enrichment of BZLF1 and BRLF1 mRNAs in CNE2EBV cells with induced EBV reactivation (Fig 5A and B). When CNE2EBV cells were co‐transfected the myc‐YTHDF1 plasmid and the wild‐type or mutant BZLF11–262nt plasmid, the YTHDF1 pull down contained more wild‐type BZLF11–262nt mRNA than the mutant control (Fig 5C). It has been established that YTHDF1, YTHDF2, and YTHDF3 interact with one another (Shi et al, 2017). To investigate the potential joint roles of these proteins on EBV regulation, we knocked down either YTHDF2 or YTHDF3 and performed YTHDF1‐RIP in CNE2EBV cells with induced EBV reactivation. YTHDF2 or YTHDF3 depletion did not affect the binding of YTHDF1 to BZLF1 and BRLF1 mRNAs (Fig 5D). However, knockdown of the m6A writer METTL14 decreased the m6A level of BZLF1 and BRLF1 mRNAs and reduced the enrichment level of both transcripts by YTHDF1 (Fig 5E and F). Thus, we demonstrate that YTHDF1 interacts with the BZLF1 and BRLF1 transcripts, which is at least partially dependent on the m6A modifications.
Figure 5. Knockdown of YTHDF1 prolongs the half‐life of EBV transcripts.
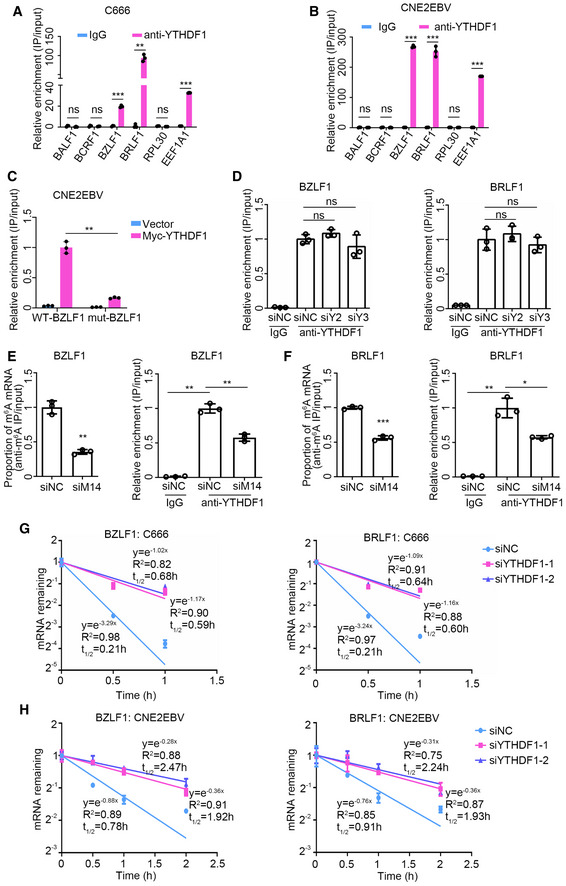
-
A, BThe relative YTHDF1‐RIP enrichment ratio of the indicated genes in C666 cells (A) and CNE2EBV cells with induced EBV reactivation (24 h post‐TPA/NaB treatment) (B). The EEF1A1 mRNA known to contain m6A sites and RPL30 mRNA containing no m6A modification were used as positive and negative controls, respectively. BALF1 and BCRF1 containing no m6A modification, identified in this study, were used as viral negative controls. The fold enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. The mean of the fold enrichment by IgG was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ns, **P < 0.01 and ***P < 0.001 compared to IgG according to unpaired Student’s t‐test.
-
CThe relative Myc‐RIP enrichment ratios of wild‐type and mutant BZLF11–262nt mRNA in CNE2EBV cells. CNE2EBV cells were co‐transfected with wild‐type or mutant BZLF11–262nt and myc‐YTHDF1 or vector control for 24 h. The fold Myc‐RIP enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. The mean of the fold Myc‐RIP enrichment level on WT‐BZLF1 mRNA was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 according to unpaired Student’s t‐test.
-
DThe relative enrichment of the BZLF1 and BRLF1 mRNA in CNE2EBV cells with transfected with siYTHDF2, siYTHDF3, or siNC siRNA following EBV reactivation. After 24 h of siRNA transfection, CNE2EBV cells were treated with TPA (30 ng/ml) and NaB (2 mM) for 24 h. The RIP assays were performed using YTHDF1‐specific antibody or IgG control in these cells. Fold enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ns, not significant according to unpaired Student’s t‐test.
-
E, FThe relative m6A and YTHDF1 enrichment of the BZLF1 (E) or BRLF1 (F) mRNAs measured in CNE2EBV cells transfected with the METTL14‐specific siRNAs (siM14) or siNC control following EBV reactivation. After 24 h of siRNA transfection, CNE2EBV cells were treated with TPA (30 ng/ml) and NaB (2 mM) for 24 h, followed by MeRIP and RIP assays. The fold enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 according to unpaired Student’s t‐test.
-
GResidual mRNA levels of BZLF1 and BRLF1 after termination of transcription via ActD treatment in C666 cells transfected with YTHDF1‐specific siRNAs or the siNC control. After 24 h of siRNA transfection, the C666 cells were treated with ActD and the mRNA levels analyzed by qRT–PCR. The relative mRNA level at 0 h after the ActD treatment was normalized to 1. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates.
-
HResidual mRNA levels of BZLF1 and BRLF1 after termination of transcription via ActD treatment in CNE2EBV cells transfected with YTHDF1‐specific siRNAs or the siNC control following EBV reactivation. After 24 h of siRNA transfection, CNE2EBV cells were induced with NaB (2 mM) for 24 h, treated with ActD, and analyzed by qRT–PCR. The relative mRNA level at 0 h after the ActD treatment was normalized to 1. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates.
To investigate whether YTHDF1 could affect the half‐lives of the EBV transcripts, we knocked down YTHDF1 in C666 cells and CNE2EBV cells with induced EBV reactivation. The cells were treated with the RNA polymerase inhibitor actinomycin D (ActD) to monitor the effects of YTHDF1 on the stability of the transcripts. Our results revealed that YTHDF1 knockdown prolonged the half‐lives of the BZLF1 and BRLF1 mRNAs (Fig 5G and H). These data suggest that YTHDF1 interacts with BZLF1 and BRLF1 mRNAs, thereby regulating their stability.
YTHDF1 interacts with the RNA degradation complex
While YTHDF1 was originally reported to promote RNA translation efficiency (Wang et al, 2015b), it was also reported to interact with the CCR4‐NOT complex to promote mRNA de‐adenylation and degradation (Du et al, 2016; Youn et al, 2018). To determine whether YTHDF1 inhibits EBV replication by interacting with the CCR4‐NOT complex, we knocked down the CCR4‐NOT complex components CNOT, CCR4, CAF1, or CCR4B in CNE2EBV cells. In contrast to YTHDF1 depletion, knockdown of any of these CCR4‐NOT complex components did not affect EBV replication (Appendix Fig S4A and B). To further explore the mechanism of the YTHDF1‐mediated decay of the EBV transcripts, we performed the YTHDF1 pull‐down assay in CNE2EBV cells and mass spectrometry (MS) analysis. The results revealed that YTHDF1 can interact with proteins from both translation complexes and RNA degradation complexes (Dataset EV3). Two components of the RNA degradation complex, ZAP (also named ZC3HAV1) and DDX17, were identified as interaction partner of YTHDF1 with high confidence (Fig 6A). Upon comparing the MS results of the YTHDF1 pull down in the CNE2EBV cells with the published YTHDF1 affinity purification/MS datasets from HeLa and HEK293 cells (Wang et al, 2015b; Youn et al, 2018), ZAP and DDX17 were also found to interact with the YTHDF1 in HEK293 cells (Fig EV4A). ZAP is an anti‐viral protein that inhibits the replication of HIV, HBV, and murine gammaherpesvirus 68 (Zhu et al, 2011; Xuan et al, 2012; Mao et al, 2013). ZAP can interact with the RNA degradation complex to enhance degradation of targeted mRNAs, and DDX17 can help to restructure the ZAP‐bound mRNAs for efficient degradation (Chen et al, 2008b). Using co‐IP, we further confirmed the YTHDF1 interaction with ZAP and DDX17 (Figs 6B and C, and EV4B and C).
Figure 6. YTHDF1 interacts with the RNA degradation complex.
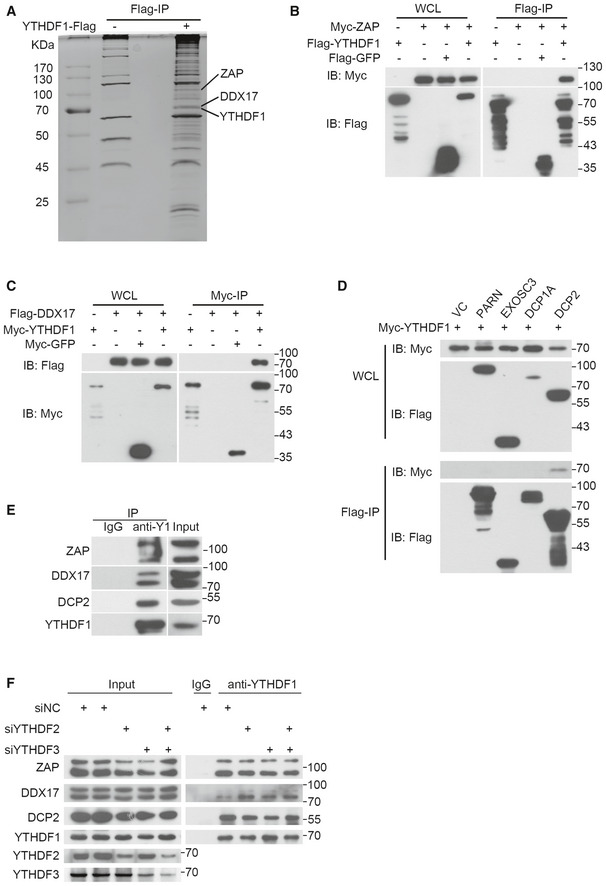
-
AThe interaction partners of YTHDF1 were identified by pull‐down assays and mass spectrometry. The Flag‐YTHDF1 pull‐down assay was performed in CNE2EBV cells transfected with a plasmid encoding Flag‐tagged YTHDF1. The proteins in the differentially abundant bands were identified by mass spectrometry.
-
B, CThe interactions between YTHDF1 and ZAP (B) or DDX17 (C) were measured by co‐IP in 293T cells. The results were detected using specific antibodies.
-
DThe interaction between YTHDF1 and DCP2 was detected by co‐IP in 293T cells.
-
EThe interactions between endogenous YTHDF1 and ZAP, DDX17, or DCP2 were detected by endogenous IP using a YTHDF1‐specific antibody in CNE2EBV cells.
-
FThe interactions between endogenous YTHDF1 and ZAP, DDX17, or DCP2 were detected by endogenous IP assays in CNE2EBV cells transfected with YTHDF2‐, YTHDF3‐specific siRNAs or the siNC control. YTHDF1, YTHDF2, YTHDF3, ZAP, DDX17, and DCP2 were detected in the immunoprecipitated complexes using specific antibodies, respectively.
Source data are available online for this figure.
Figure EV4. YTHDF1 interacts with ZAP, DDX17, and DCP2.
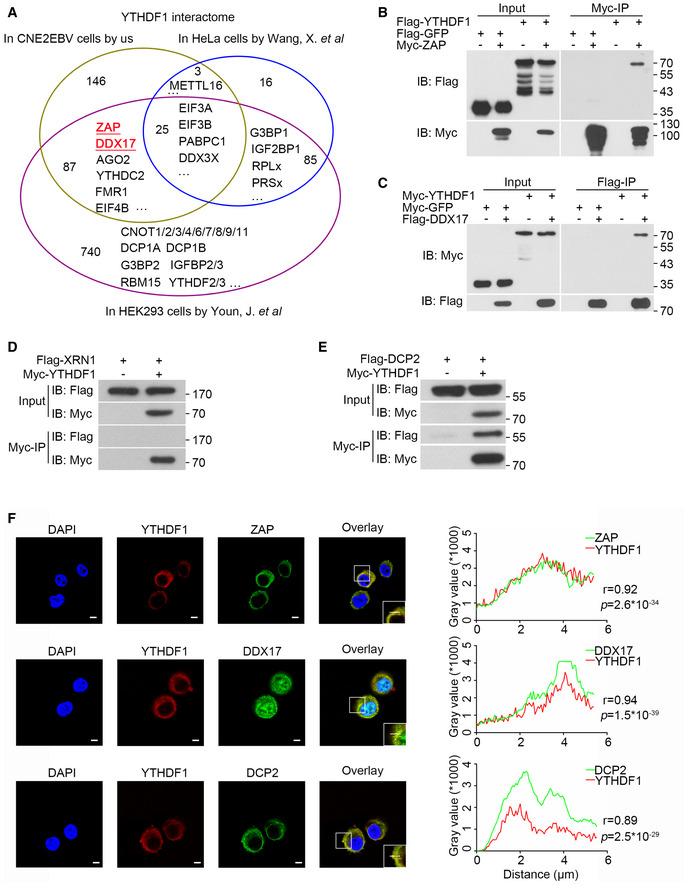
-
AVenn diagram of YTHDF1 interaction partners in CNE2EBV, HeLa, and HEK293 cells.
-
B, CThe interactions between YTHDF1 and ZAP (B) or DDX17 (C) were detected by co‐IP assays in 293T cells. The results were detected using the indicated antibodies.
-
DYTHDF1 did not interact with XRN1 according to the co‐IP assay in 293T cells.
-
EThe interaction of YTHDF1 with DCP2 detected by co‐IP assays in 293T cells.
-
FCo‐localization of the endogenous YTHDF1 with ZAP, DDX17, and DCP2 in HK1 cells detected by immunofluorescence. YTHDF1 was stained with the mouse monoclonal YTHDF1 antibody and an Alexa Fluor 594‐conjugated secondary antibody. ZAP, DDX17, and DCP2 were stained with the target‐specific rabbit primary antibodies and an Alexa Fluor 488‐conjugated secondary antibody. The nuclei were counterstained with DAPI (blue). The merged signal of YTHDF1 and ZAP, DDX17, or DCP2 is shown in yellow. Representative of three independent experiments are shown. Scale bars: 5 μm. The right panel shows the results of co‐localization analysis in the selected region analyzed under a confocal laser scanning microscope. r: correlation coefficients. The P‐values were calculated using Pearson’s correlation test.
Source data are available online for this figure.
ZAP can either recruit PARN and the 3′–5′ exosome complex to promote de‐adenylation‐dependent decay or recruit DCP1A, DCP2, and XRN1 to promote mRNA decapping (Zhu et al, 2011). However, we failed to detect any physical interaction between YTHDF1 and XRN1 (Fig EV4D), which was consistent with the findings of a previous report (Kretschmer et al, 2018). Further, 293T cells were co‐transfected with myc‐YTHDF1 and flag‐PARN, flag‐DCP1A, flag‐DCP2, or flag‐EXOSC3. The co‐IP results showed that only DCP2 interacted with YTHDF1 (Figs 6D and EV4E). Furthermore, IP of endogenous YTHDF1 antibody in the CNE2EBV cells using a specific antibody further demonstrated that YTHDF1 interacted with ZAP, DDX17, and DCP2 (Fig 6E). It has been implicated that YTHDF2 could interact with DCP2, while ZAP and DDX17 were identified as potential interaction partners of YTHDF2 and YTHDF3, whereas YTHDF1, YTHDF2, and YTHDF3 are known to interact with each other (Wang et al, 2014; Shi et al, 2017; Youn et al, 2018). To determine whether the interactions between YTHDF1 with ZAP, DDX17, or DCP2 are dependent on either YTHDF2 or YTHDF3, we performed IP of endogenous YTHDF1 in CNE2EBV cells following YTHDF2 and/or YTHDF3 knockdown. The results showed that knockdown of YTHDF2 and/or YTHDF3 did not reduce the interaction between YTHDF1 and ZAP, DDX17, or DCP2 (Fig 6F). Surprisingly, knockdown of either YTHDF2 and/or YTHDF3 augmented the interaction between YTHDF1 and DDX17, suggesting that YTHDF1, YTHDF2, and YTHDF3 might compete for binding to DDX17. Moreover, immunofluorescence staining indicated that YTHDF1 co‐localized with ZAP, DDX17, and DCP2 in the cytoplasm (Fig EV4F).
YTHDF1 destabilizes viral mRNAs by promoting decapping
To investigate the role of the YTHDF1‐associated protein complex in EBV infection and lytic replication, we used siRNAs to knock down ZAP, DDX17, or DCP2 in HK1, CNE2EBV, and C666 cells. At 24 h following siRNA transfection, the HK1 cells were infected with EBV‐GFP virus for 24 h, and the GFP‐positive cells were analyzed by FACS analysis. The results indicated that knockdown of these genes promoted EBV infection in HK1 cells (Fig 7A). On the other hand, CNE2EBV cells were induced by NaB for 24 h post‐siRNA transfection and subjected to FACS analysis. The results showed that knockdown of these genes promoted lytic replication of EBV in CNE2EBV cells with induced EBV reactivation (Fig 7B). At 24 h following siRNA transfection in C666 cells, the EBV copies were quantified by qPCR, which indicated that the knockdown of these genes increased viral replication (Fig 7C). Knockdown of ZAP, DDX17, or DCP2 increased the mRNA level of BZLF1 and BRLF1 in EBV‐infected HK1 cells (Appendix Fig S5A–C). Therefore, we conclude that knockdown of the YTHDF1‐associated proteins ZAP, DDX17, and DCP2 promotes EBV infection and replication in NPC cells.
Figure 7. YTHDF1 promotes viral mRNA decapping by recruiting the RNA degradation complex.
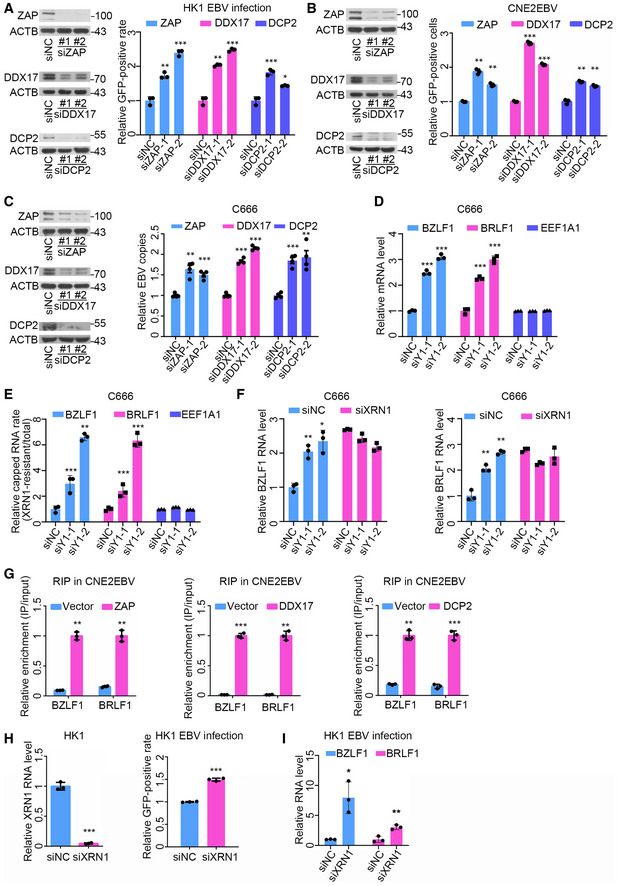
- The ratios of GFP‐positive HK1 cells transfected with the ZAP‐, DDX17‐, and DCP2‐specific siRNAs or the siNC control were quantified using FACS. At 24 h post‐siRNA transfection, the protein level of indicated genes was determined by Western blot analysis. ACTB was used as loading control. The experiments were performed similarly as described in Fig 3B, except the different siRNA transfection. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The GFP‐positive cells in CNE2EBV cells transfected with the ZAP‐, DDX17‐, and DCP2‐specific siRNAs or the control siRNA following EBV reactivation were quantified using FACS. At 24 h post‐siRNA transfection, the protein level of indicated genes was determined by Western blot analysis. ACTB was used as loading control. The experiments were performed similarly as described in Fig 3D, except the different siRNA transfection. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The relative number of EBV copies in C666 cells transfected with the ZAP‐, DDX17‐, and DCP2‐specific siRNAs or the siNC control was measured using qPCR. At 24 h post‐siRNA transfection, the protein level of indicated genes was determined by Western blot analysis. ACTB was used as loading control. The experiments were performed similarly as described in Fig 3K, except the different siRNA transfection. Experiments were independently repeated four times, and the results are represented as the means ± SD of n = 4 biological replicates. **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The relative mRNA levels of BZLF1 and BRLF1 in C666 cells transfected with the YTHDF1‐specific siRNAs (siY1‐1 and siY1‐2) or the siNC control. The experiments were performed similarly as described in Fig 4C. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The ratio of endogenous 5′‐capped BZLF1 and BRLF1 mRNAs in C666 cells at 24 h post‐transfection with the YTHDF1‐specific siRNAs or the siNC control. The ratios of endogenous 5′‐capped mRNAs were quantified by dividing the abundance of XRN1‐resistant transcripts with the total amount of transcripts. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The relative mRNA levels of BZLF1 and BRLF1 in C666 cells transfected with YTHDF1‐specific siRNAs and XRN1‐specific siRNAs. The C666 cells were co‐transfected with YTHDF1‐specific siRNAs and XRN1‐specific siRNAs for 24 h. The mRNA levels were determined using qPCR. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05 and **P < 0.01 compared to siNC according to unpaired Student’s t‐test.
- The relative enrichment of BZLF1 and BRLF1 mRNAs by ZAP, DDX17, or DCP2 in CNE2EBV cells following EBV reactivation. CNE2EBV cells were transfected with plasmids encoding myc‐ZAP, myc‐DDX17, myc‐DCP2, or vector control. At 24 h post‐transfection, the cells were treated with TPA (30 ng/ml) and NaB (2 mM) for 24 h. The RIP assays were performed using Myc‐beads. The fold enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 compared to Vector according to unpaired Student’s t‐test.
- Determination of the EBV infection efficiency in HK1 cells transfected with the XRN1‐specific siRNA or the siNC control. After 24 h of siRNA transfection, HK1 cells were infected with EBV and analyzed by FACS at 24 hpi. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. ***P < 0.001 compared to siNC according to unpaired Student’s t‐test.
- The relative mRNA levels of BZLF1 and BRLF1 in the EBV‐infected HK1 cells transfected with the XRN1‐specific siRNA (siXRN1) or the siNC control at 24 hpi. All gene expression levels were normalized to the housekeeping gene ACTB. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05 and **P < 0.01 compared to siNC according to unpaired Student’s t‐test.
Source data are available online for this figure.
DCP2 was recently identified as a crucial factor to degrade a subset of m6A‐modified RNAs via decapping (Luo et al, 2020). Since YTHDF1 could interact with DCP2, we hypothesized that YTHDF1 could recruit ZAP, DDX17, and DCP2 to the viral transcripts to promote RNA decay by decapping. XRN1 is a highly active 5′–3′ exoribonuclease, which requires 5′ monophosphate. XRN1 does not cleave RNAs that are capped at the 5′ ends. To determine whether the decapping efficiency could be affected by YTHDF1, we treated the total RNA extracted from the YTHDF1 knockdown and control cells with XRN1 enzyme to degrade the uncapped RNAs and measured the ratio of the capped RNAs to the total RNAs by qRT–PCR. The results showed that YTHDF1 knockdown significantly increased the mRNA level of BZLF1 and BRLF1, as well as the proportion of capped mRNAs of these transcripts in C666 cells (Fig 7D and E). Therefore, our data suggest that YTHDF1 destabilizes BZLF1 and BRLF1 transcripts, likely by promoting their decapping. Since the decapped RNA can be degraded by XRN1 protein, C666 cells were co‐transfected with YTHDF1‐specific and XRN1‐specific siRNAs. The qPCR results indicated that XRN1 knockdown increased the mRNA levels of BZLF1 and BRLF1, while YTHDF1 knockdown could not increase the mRNA levels of BZLF1 and BRLF1 in C666 cells following XRN1 knockdown (Fig 7F). Therefore, suppression of BZLF1 and BRLF1 expression by YTHDF1 is dependent on XRN1.
Further, RIP assays were performed to determine the interaction between ZAP, DDX17, and DCP2 with the viral mRNA in CNE2EBV cells with induced EBV reactivation. The results showed that ZAP, DDX17, and DCP2 could markedly enrich the mRNAs of BZLF1 and BRLF1 (Fig 7G). Next, CNE2EBV cells were co‐transfected with plasmids of either wild‐type BZLF11–262nt or mutant control and plasmids of either ZAP, DDX17, or DCP2. The RIP‐qPCR results showed that ZAP, DDX17, and DCP2 could enrich greater amounts of wild‐type BZLF11–262nt mRNAs than the mutant control mRNAs (Fig EV5A–C). Wild‐type BZLF11–262nt plasmid was co‐transfected with ZAP, DDX17, or DCP2 in the YTHDF1 knockdown CNE2EBV cells or control CNE2EBV cells. The RIP‐qPCR results showed that the interactions between the wild‐type BZLF11–262nt transcripts and ZAP, DDX17, or DCP2 proteins could be reduced by YTHDF1 knockdown in CNE2EBV cells (Fig EV5D–F). Therefore, the interaction of ZAP, DDX17, and DCP2 with m6A‐modified viral mRNA was partially dependent on YTHDF1. We then investigated the function of XRN1 in EBV infection and found that knockdown of XRN1 promoted EBV infection in HK1 cells and increased mRNA level of BZLF1 and BRLF1 (Fig 7H and I).
Figure EV5. The interactions of ZAP, DDX17, and DCP2 with m6A‐modified mRNAs are dependent on YTHDF1.
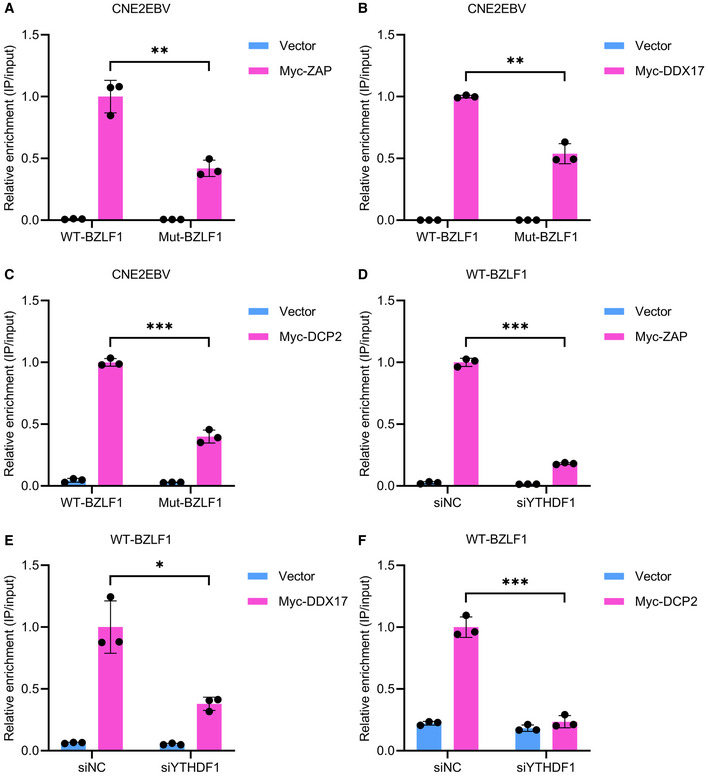
-
A–CThe relative RIP enrichment ratios of the wild‐type BZLF11–262nt (WT‐BZLF1) and mutant BZLF11–262nt (Mut‐BZLF1) mRNA following immunoprecipitation by ZAP (A), DDX17 (B), or DCP2 (C) in CNE2EBV cells transfected as indicated. The fold Myc‐RIP enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. **P < 0.01 and ***P < 0.001 according to unpaired Student’s t‐test.
-
D–FThe relative RIP enrichment of the wild‐type BZLF11–262nt mRNA following immunoprecipitation by ZAP (D), DDX17 (E), or DCP2 (F) in CNE2EBV cells transfected with the YTHDF1‐specific siRNAs (siYTHDF1) or the siNC control. The fold Myc‐RIP enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample. Experiments were independently repeated three times, and the results are represented as the means ± SD of n = 3 biological replicates. *P < 0.05 and ***P < 0.001 according to unpaired Student’s t‐test.
In conclusion, the m6A “reader” YTHDF1 recognizes the m6A modification sites in EBV transcripts, promotes m6A‐dependent RNA decay by recruiting the RNA degradation complexes components ZAP, DDX17, and DCP2, and thereby suppresses EBV infection and replication. Conversely, a low expression level of YTHDF1 protects the viral mRNA from RNA decay mediated by the RNA degradation complex.
Discussion
The m6A methylome has been identified in the epitranscriptomes of an increasing number of species. Recently, the architecture of the m6A methylome has been mapped for several viruses, including the RNA viruses HIV, ZIKV, HCV, IAV, EV71, MLV, RSV, and the plant virus alfalfa mosaic virus (Gokhale et al, 2016; Kennedy et al, 2016; Lichinchi et al, 2016a, 2016b; Tirumuru et al, 2016; Courtney et al, 2017; Martinez‐Perez et al, 2017; Hao et al, 2018; Lu et al, 2018; Courtney et al, 2019; Xue et al, 2019). Moreover, m6A modification sites have also been identified in the transcripts of DNA viruses, such as those encoded by HBV, SV40, and KSHV (Ye et al, 2017; Hesser et al, 2018; Imam et al, 2018; Tan et al, 2018; Tsai et al, 2018; Baquero‐Perez et al, 2019). Most importantly, KSHV, a γ‐herpesvirus related to EBV, was reported to be regulated by m6A modification. However, many differences exist between EBV and KSHV. For example, KSHV is mainly linked to Kaposi’s sarcoma and primary effusion lymphoma (Tan et al, 2018), whereas EBV is associated with NPC, 10% of gastric cancers, Burkitt’s lymphoma, and multiple other lymphomas. Therefore, it is worth investigating the m6A modification of EBV. A recent study showed that the m6A writer METTL14 plays a role in EBV‐associated tumorigenesis, and the authors also examined the viral epitranscriptome of EBV‐transformed LCLs and lymphoma cells (Lang et al, 2019). They also found that depletion of the m6A writer METTL14 could increase the protein level of BZLF1 and stabilize lytic EBV transcripts, including BRLF1. Here, the m6A modification landscape of EBV was profiled in NPC cells and tissues, and we demonstrated that the m6A reader YTHDF1 could bind to BZLF1 and BRLF1 transcripts, and promote their degradation to repress EBV infection and lytic replication. Together with the results from Lang et al, our data demonstrate that m6A modification can suppress lytic replication of EBV. Accordingly, EBV might co‐opt m6A modification in host cells to repress the lytic cycle reactivators BZLF1 and BRLF1, thereby suppressing lytic replication to maintain viral latency. However, m6A modification was found to mediate mRNA degradation of IFNB1 to promote virus infection and replication (Rubio et al, 2018; Winkler et al, 2018). Therefore, we speculate that there might be an equilibrium between the degradation of IFNB1 mRNA and the degradation of viral transcripts during the process of m6A‐mediated control of viral replication.
We observed that the m6A modification status varies largely depending on the EBV infection stage and EBV gene expression levels in B cells, NPC cells, and tissue samples. The m6A modification sites in host transcripts are enriched near the stop codon, resulting in a sharp peak of frequency. However, the m6A modification sites of EBV transcripts are generally located throughout the CDS region of the viral mRNA. EBV contains many overlapping transcripts, untranslated transcripts, unspliced coding transcripts et cetera. Consequently, the m6A peaks of EBV possibly cannot be accurately analyzed using short‐read MeRIP sequencing, which might affect the estimate that the m6A peaks belong to the corresponding overlapping viral genes. We found that EBNA1 and BNLF2a were m6A‐modified in almost all samples. Latent gene expression can be regulated by differential promoter usage (Tierney et al, 1994). Among them, EBNA1 transcription is mostly initiated from Wp/Cp in type III latency, from Qp in type I and IIa latency or Fp during the lytic cycle (Tierney et al, 1994). Therefore, our MeRIP‐seq results could not clearly define these promoters produced the identified EBNA1 transcripts. The lytic transcripts, including BNLF2a, are also expressed in latently infected cells or tissues, which might be due to a small number of cells undergoing spontaneous lytic reactivation. As BZLF1 and BRLF1 are the crucial factors regulating the lytic replication, we mainly focused on the effects of YTHDF1 on BZLF1 and BRLF1.
The m6A reader YTHDF1 has been reported to increase RNA translation efficiency. YTHDF1 was recently found to promote mRNA de‐adenylation, suggesting a role of YTHDF1 in RNA decay (Wang et al, 2015b; Du et al, 2016; Zaccara & Jaffrey, 2020). Here, we found that YTHDF1 destabilized EBV transcripts stability in EBV‐infected cells by promoting the RNA decapping. Mechanistically, YTHDF1 can interact with the RNA degradation proteins ZAP, DDX17, and DCP2. Previous studies have indicated that ZAP and DDX17 can recruit RNA degradation complexes to regulate the stability of various viral transcripts (Guo et al, 2007; Chen et al, 2008b; Zhu et al, 2011; Todorova et al, 2015). Therefore, we speculate that ZAP and DDX17 can help YTHDF1 to mediate the destabilization of EBV transcripts. We also found that the knockdown of the YTHDF1 interaction partners ZAP, DDX17, and DCP2 promoted EBV infection and replication. Since DCP2 is an RNA‐decapping enzyme, we focused on the role of YTHDF1 in RNA decapping. A recent study identified DCP2 as a crucial factor for the degradation of a subset of m6A‐modified RNAs via decapping (Luo et al, 2020). Here, we revealed that YTHDF1 could promote mRNA decapping of the endogenous BZLF1 and BRLF1 transcripts. This mechanism was different from that of YTHDF2, which was reported to promote RNA decay through mRNA de‐adenylation instead of mRNA decapping (Du et al, 2016).
Previous studies have found that YTHDF2 not only destabilizes targeted RNAs, but also facilitates translation in cells under heat shock stress (Wang et al, 2014; Zhou et al, 2015). Under conditions of starvation or viral infection, translation could be compromised and decapping could be stimulated (Parker & Sheth, 2007). Although YTHDF1 mostly promotes translational efficiency of m6A‐modified mRNAs, we reveal that YTHDF1 recruits ZAP, DDX17, and DCP2 to degrade the m6A‐modified viral transcripts in EBV‐infected cells. We speculate that a balance is maintained between the roles of YTHDF1 in translation and mRNA decay. EBV infection stress might influence the function of YTHDF1. Our findings suggest that during EBV infection in host cells, YTHDF1 promotes viral RNA decay by inducing RNA decapping.
In conclusion, many EBV transcripts are m6A‐modified, including BZLF1 and BRLF1. The m6A reader YTHDF1 represses EBV infection and replication by destabilizing the transcripts of BZLF1 and BRLF1. YTHDF1 promotes the RNA decapping efficiency of the targeted viral RNA by interacting with the essential components of the RNA decay complex, including ZAP, DDX17, and DCP2. Moreover, the interactions between the RNA decay complex and the m6A‐modified viral mRNA are dependent on YTHDF1. The treatment of EBV‐associated cancers remains a substantial challenge. Further investigation is warranted to determine whether m6A modification plays a pivotal role in the progression of NPC and other EBV‐related cancers, with the hope of uncovering novel therapeutic targets.
Materials and Methods
Reagents
The antibodies used in this study are listed below in the format of name (application, catalogue, supplier): anti‐YTHDF1 (WB, ab99080, Abcam); anti‐DCP2 (WB, A302‐597A, Thermo); anti‐DDX17 (WB, ab180190, Abcam); anti‐ZAP (WB, PA5‐31650, Invitrogen); anti‐BZLF1 (WB, sc53904, Santa Cruz); anti‐METTL3 (WB, ab195352, Abcam); anti‐METTL14 (WB, ab98166, Abcam); anti‐FTO (WB, ab124892, Abcam); anti‐YTHDF2 (WB, ab170118, Abcam); anti‐YTHDF3 (WB, ab103328, Abcam); anti‐ACTB (WB, 66009‐1‐Ig, ProteinTech); anti‐Myc‐tag (WB, 16286‐1‐AP, ProteinTech); and anti‐Flag‐tag (WB, 66008‐1‐Ig, ProteinTech). The anti‐gB monoclonal antibody (CL55) was a kind gift from Professor Richard Longnecker (Northwestern University, USA); horseradish peroxidase (HRP)‐conjugated goat anti‐mouse (WB, #31460, Invitrogen); HRP‐conjugated goat anti‐rabbit (WB, #61‐6520, Invitrogen); anti‐YTHDF1 (RIP, 17479‐1‐AP, ProteinTech); anti‐YTHDF1 (IF, 66745‐1‐Ig, ProteinTech); goat anti‐rabbit Alexa 488 (IF, A11008, Thermo); and goat anti‐mouse Alexa 594 (IF, A11005, Thermo). Other regents like DAPI (D9542), phorbol 12‐myristate 13‐acetate (TPA) (P8139), sodium butyrate (NaB) (B5887), ActD (A9415), and TRIzol (T924) are obtained from Sigma‐Aldrich (USA).
Patients and tissue samples
NPC tissues and PDX samples were obtained from the Sun Yat‐sen University Cancer Center (Guangzhou, China). Samples were collected after written informed consent forms were obtained from the patients, and all related procedures were performed with the approval of the Internal Review and Ethics Board of Sun Yat‐sen University Cancer Center (Approval No.: GZR2018‐140). The PDX mouse model experiments were reviewed and approved by the Ethics Board and the Clinical Research Committee of Sun Yat‐sen University Cancer Center (Approval No.: L102012018220M). The NPC PDX tissues were EBER positive, CK14 positive, and CD19 negative.
Cell culture
HEK293 cells (ATCC, CRL‐1573) and 293T cells (ATCC, CRL‐3216) were purchased from ATCC. Bmi1‐immortalized primary NPECs (NPEC1‐Bmi1 and NPEC2‐Bmi1) were established in our laboratory and were grown in serum‐free keratinocyte medium (Life Technology, USA) (Song et al, 2006; Xiong et al, 2015). C666 cells were a kind gift from Sai Wah Tsao (University of Hong Kong, China) (Cheung et al, 1999). Raji and B95‐8 cells were a kind gift from Professor Chao‐Nan Qian (Sun Yat‐sen University, China) (Yang et al, 2013). EBV‐GFP‐positive AKATA cells were a kind gift from Professor Maria G. Masucci (Karolinska Institute, Sweden). HK1 and CNE2 cells were a kind gift from Professor Quentin Liu (Sun Yat‐sen University, China) (Yan et al, 2014). CNE2EBV cells, EBV‐EGFP‐infected CNE2 cells, were cultured in the presence of G418 (500 μg/ml). HK1, CNE2, CNE2EBV, C666, Raji, and B95‐8 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, GIBCO). AKATA cells were grown in RPMI 1640 medium supplemented with 5% FBS. HEK293 cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS. All cells were maintained in a humidified atmosphere comprising 5% CO2 at 37°C. All cells were tested to ensure that they are free from Mycoplasma infection.
Plasmids
YTHDF1‐pENTER plasmid was purchased from Vigene Biosciences (Shandong, China). Plasmids encoding myc‐tagged YTHDF1 and ZAP were constructed by the respective cDNAs into the pcDNA6‐MYC plasmid. The plasmids encoding flag‐EXOSC3 was constructed by incorporating EXOSC3‐flag into the pcDNA3.1+ plasmid. The plasmids of PEYFP‐EXOSC3, PCMV‐HA‐flag‐DDX17, PCMV‐HA‐flag‐DCP2, and PCMV‐HA‐flag‐PARN were a generous gift from Professor Guangxia Gao (Chinese Academy of Sciences, China). Myc‐tagged DDX17 and DCP2 were PCR‐amplified and then inserted into the pcDNA6‐MYC plasmid.
The plasmids of WT‐BZLF1 and mut‐BZLF1 were constructed by inserting the wild‐type BZLF11–262nt sequence (5′‐ATTGCACCTTGCCGGCCACCTTTGCTATCTTTGCTGAAGATGATGGACCCAAACTCGACTTCTGAAGATGTAAAATTTACACCTGACCCATACCAGGTGCCTTTTGTACAAGCTTTTGACCAAGCTACCAGAGTCTATCAGGACCTGGGAGGGCCATCACAAGCTCCTTTGCCTTGTGTGCTGTGGCCGGTGCTGCCAGAGCCTCTGCCACAAGGCCAGCTCACTGCCTATCATGTTTCAGCCGCACCAACTGGGTCGTGGT‐3′) or the mutant sequence (5′‐ATTGCACCTTGCCGGCCACCTTTGCTATCTTTGCTGAAGATGATGGTCCCAATCTCGTCTTCTGAAGATGTAAAATTTACACCTGTCCCATACCAGGTGCCTTTTGTACAAGCTTTTGTCCAAGCTACCAGAGTCTATCAGGTCCTGGGAGGGCCATCACAAGCTCCTTTGCCTTGTGTGCTGTGGCCGGTGCTGCCAGAGCCTCTGCCACAAGGCCAGCTCACTGCCTATCATGTTTCAGCCGCACCAACTGGGTCGTGGT‐3′) into the pcDNA3.1+ plasmid, respectively.
Virus preparation and infection
The EBV‐GFP (strain Akata) was prepared in AKATA cells as described previously (Kanda et al, 2004; Wang et al, 2015a; Zhang et al, 2018a). Briefly, EBV was produced in AKATA cells by reactivation with 0.8% (v/v) of a goat anti‐human IgG (Huayang Zhenglong Biochem Lab, China). The MOI of the EBV suspension was determined by detecting the BamHI‐W fragment region of the EBV genome using TaqMan real‐time PCR. A calibration curve was constructed using the DNA extracted from the EBV‐positive Namalwa cell line, which contains two integrated viral genomes per cell, as a standard. 5 × 104 HK1 cells were seeded in 24‐well plates, infected with serial dilutions of EBV, and analyzed by FACS at 24 hpi to evaluate the infection efficiency of the newly purified EBV. The percentage of infected HK1 cells was 1–10% at the MOI of 250 or 500, about 10–20% at the MOI of 1,000, and about 30% at the MOI of 2,000 (Appendix Fig S6). EBV infection of HK1 cells was mediated in a virus titer‐dependent manner. We performed the EBV infection assays at an MOI of 1,000 to investigate the function of the genes. Briefly, HK1 cells were exposed to EBV at an MOI of 1,000 for 3 h at 37°C, and the unbound virus was removed by washing twice with Hanks’ solution. The infected cells were cultured in fresh medium for 24 h, followed by the determination of the ratio of GFP‐positive cells via flow cytometry (Beckman Coulter FC500) and fluorescence microscopy.
Induction of the EBV lytic cycle
Raji and B95‐8 cells were seeded at a density of 5 × 105 cells per ml, and 2× 106 CNE2EBV cells were plated on 10 cm dishes before lytic induction. After 24 h, the cells were treated with TPA (30 ng/ml) and/or NaB (2 mM) and were incubated for 0, 24, and 48 h, respectively. Following the induction of EBV lytic cycle, the cells were harvested, and the cellular RNA was extracted using TRIzol reagent.
siRNA transfection
A total of 1 × 105 cells per well were seeded into 12‐well plates for 12 h and transfected with the indicated siRNA duplexes using RNAiMAX (Invitrogen) according to the manufacturer’s instructions. The cells were used for further experiments 24 h after transfection. The siRNA pools targeting METTL3, METTL14, WTAP, FTO, YTHDF1, YTHDF2, YTHDF3, and XRN1, as well as control siRNA (siNC), were synthesized by RIBOBIO (Guangzhou, China). The single siRNA duplexes used in this study are listed in Appendix Table S1.
Immunoprecipitation and mass spectrometry
The transfected cells were lysed in IP lysis buffer [50 mM Tris–HCl, pH 7.4; 150 mM NaCl; 5 mM EDTA; 0.5% NP‐40 containing 1 mM phenylmethylsulfonyl fluoride, and Roche complete protease inhibitor cocktail (04693159001, Roche)] for 20 min at 4°C. The supernatants were collected by centrifugation at 12,000 g for 20 min at 4°C, leaving behind 10% as the input.
For co‐IP with overexpressed flag‐ or the myc‐tagged proteins, the supernatant was incubated with 25 μl Anti‐c‐Myc Agarose Affinity Gel (A7470, Sigma) or Anti‐FLAG‐M2 Affinity Gel (A2220, Sigma) for 2 h at 4°C.
For endogenous IP, the supernatant was incubated with 2 μg YTHDF1‐specific antibody (17479‐1‐AP, ProteinTech) or rabbit IgG for 2 h at 4°C. The mixture was then incubated with 25 μl Pierce Protein A/G Agarose (20421, Thermo Scientific) for 2 h at 4°C.
After washing three times with lysis buffer to remove the unbound proteins, the sample was resuspended in 2× SDS sample loading buffer and denatured for 5 min at 98°C. The complex was then analyzed by Western blot analysis with the indicated antibodies.
For Flag‐YTHDF1 pull down in CNE2EBV cells, the cells were transfected with the plasmid encoding Flag‐YTHDF1 or empty vector for 24 h. The cells were then lysed in IP lysis buffer and the supernatant was incubated with 25 μl Anti‐FLAG‐M2 Affinity Gel (A2220, Sigma) for 2 h at 4°C. After washing three times with lysis buffer, the samples were suspended in 2× SDS sample loading buffer, denatured for 5 min at 98°C, and separated by SDS–PAGE. The differential bands were isolated and analyzed by mass spectrometry. The proteins identified in the Flag‐YTHDF1 samples are summarized in Dataset EV3.
Genome annotation
The AKATA EBV genome (GenBank: KC207813.1) and annotation files were provided by Dr. Lin Zhen (Tulane University, USA). Mapping and annotatoion were performed using the UCSC hg38 human reference genomes and UCSC calJac3 marmosets reference genomes.
MeRIP‐seq and analysis
The total RNA was isolated using the TRIzol and was incubated with DNase I at 37°C for 30 min. The RNA integrity was validated using an Agilent 2100 Bioanalyzer to ensure that all RNA integrity numbers were > 9.0. The mRNA was purified from the total RNA using the GenElute™ mRNA Miniprep Kit (MRN10, Sigma). The mRNA was further fragmented using the Ambion RNA Fragmentation Reagents (AM8740, Life Technologies) and purified via ethanol precipitation. The fragmented mRNA was analyzed on an Agilent 2100 Bioanalyzer to ensure that the distribution of the RNA fragment sizes was around ~ 150 nt. Then, 1% of the fragmented RNA was retained as input for RNA‐seq. For MeRIP, the fragmented RNA was denatured for 5 min at 70°C, chilled on ice for 3 min, and incubated with the anti‐m6A antibody (202003, Synaptic Systems) conjugated to Protein A/G Dynabeads (88803, Thermo Fisher Scientific) in 500 μl of MeRIP Buffer (50 mM Tris–HCl, 750 mM NaCl and 0.5% NP‐40) overnight at 4°C. The unbound RNA was removed using a magnetic separator, and the beads were washed three times with MeRIP buffer. The MeRIP RNA was collected by adding TRIzol to the beads. Finally, the MeRIP RNA samples were analyzed on an Agilent 2100 Bioanalyzer to verify the RNA quality. The sequencing libraries (input RNA‐seq and MeRIP‐seq) were prepared using the Illumina TruSeq Stranded mRNA Kits, beginning the protocol at the “elute prime‐fragment” step. This step was slightly modified to only prime the samples by heating at 80°C for 2 min. The samples were sequenced on an Illumina HiSeq 2500 with a single read of 50 bp.
Read mapping and m6A peak calling were performed as described previously (Dominissini et al, 2013; Li et al, 2017b). Briefly, the reads from the mRNA input (RNA‐seq) and MeRIP‐seq libraries were aligned to the hg38 human reference genome or the calJac3 marmoset reference genome with the AKATA EBV genome (GenBank: KC207813.1) using STAR (Dobin et al, 2013). We deleted the reads that could be mapped to both the EBV genome and the host genome. MACS2 (Zhang et al, 2008) was used for m6A peak calling in the MeRIP‐seq data, and further analyses were carried out using all peaks detected in at least two replicates. Since some EBV transcripts overlapped on the same locus, the m6A peaks in these regions were annotated: using BEDtools (Quinlan, 2014) to calculate the RPKM normalized reads count of every overlapped EBV transcript and the m6A peak regions from the input RNA‐seq data, respectively. A Pearson correlation coefficient was calculated based on the RPKM of the m6A peak, and the one with a higher Pearson correlation coefficient was annotated on the transcript. The m6A peak regions of EBV are shown in Dataset EV1. The RNA‐seq reads were normalized using the RSEM method (Li & Dewey, 2011). The reads of EBV and host genome are listed in Dataset EV4.
PA‐m6A‐Seq and analysis
PA‐m6A‐Seq was performed as described previously (Chen et al, 2015). Briefly, the CNE2EBV cells were induced with TPA and NaB for 4 h and then pulsed with 200 µM 4‐thiouridine (4SU) (T4509, Sigma) for 20 h. The RNA was isolated using TRIzol, and mRNA was purified using GenElute™ mRNA Miniprep Kit. Then, 10 µg of oligo‐dT purified mRNA was incubated with 7.5 µg of m6A polyclonal antibody in 500 μl of MeRIP Buffer (50 mM Tris–HCl, 750 mM NaCl, 0.5% NP‐40, proteinase inhibitor, and RNasin) for 2 h at 4°C. After incubation, the sample was irradiated three times with 0.15 J of at UV light 365 nm. The cross‐linked RNA was digested using 0.1 U/μl RNase T1 for 15 min at 22°C. Then, 100 μl pre‐blocked Protein A/G Dynabeads was incubated with the reaction mixture for 1 h at 4°C. The beads were washed three times with IP wash buffer (50 mM HEPES‐KOH, pH 7.5, 300 mM KCl, 0.05% NP‐40, proteinase inhibitor, and RNasin) and incubated with RNase T1 at a final concentration of 15 U/μl for 15 min at 22°C. After washing the beads three times with high salt wash buffer (50 mM HEPES‐KOH, pH 7.5, 500 mM KCl, 0.05% NP‐40, proteinase inhibitor, and RNasin), the beads–RNA mixture was resuspended in 100 μl of 1× NEB T4 polynucleotide kinase buffer and 10% (v/v) NEB T4 PNK. Then, the samples were incubated at 37°C for 20 min on a shaker incubator at maximum speed. ATP was added to a final concentration of 100 μM. The RNA fragments were then washed and digested with proteinase K to remove covalently bound peptides. Finally, the RNA was extracted using TRIzol. The PA‐m6A‐Seq libraries were prepared as described in the NEB protocol (E7300L, NEB). The samples were sequenced with a single read of 75 bp.
The PA‐m6A‐Seq data were analyzed as previously reported (Chen et al, 2015). The reads from the PA‐m6A‐Seq libraries were aligned to the hg38 human reference genome with the AKATA EBV genome (GenBank: KC207813.1) using STAR (Dobin et al, 2013), allowing up to 1 mismatch. The reads that could be mapped to both the EBV genome and the human genome were deleted. Only reads containing mutations were considered. Characteristic T>C mutations resulting from 4SU incorporation and cross‐linking were present among the aligned reads. Additionally, A>G mutations were also present, corresponding to negative‐strand alignments. All data were processed using in‐house Perl scripts and SAMtools (Li et al, 2009). The regional distribution of m6A modifications was plotted using the Guitar package (Cui et al, 2016). Motif analysis of m6A peaks was performed using the MEME package (Bailey et al, 2009).
Quantitative PCR with reverse transcription (qRT–PCR)
The total RNA was isolated using TRIzol Reagent. For gene expression analyses, 1 μg of RNA was reverse‐transcribed using the GoScript Reverse Transcription System in a 20 μl reaction mixture. The mRNA level was determined by qRT–PCR using the SYBR Green I Master mix and analyzed on a Roche LightCycler 480 instrument. All gene expression levels were normalized to that of the housekeeping genes ACTB or GAPDH. The primers are listed in Dataset EV5.
Quantification of mRNA methylation using MeRIP‐qPCR
We carried out MeRIP enrichment followed by qRT–PCR to quantify the changes in the m6A modification of the indicated genes of interest. Briefly, after MeRIP, all of the MeRIP RNA and input RNA samples were subjected to qRT–PCR. The fold enrichment was determined by calculating the 2−Δ C t of the MeRIP sample relative to the input sample. The primers are listed in Dataset EV5.
Determination of site‐specific m6A modification levels using SELECT
SELECT was performed as described previously (Xiao et al, 2018). Briefly, the total RNA was mixed with 40 nM Up Primer, 40 nM Down Primer, and 5 µM dTTP in 17 μl 1× CutSmart Buffer (NEB). The RNA and primers were annealed using a temperature gradient of 90°C for 1 min, 80°C for 1 min, 70°C for 1 min, 60°C for 1 min, 50°C for 1 min, and then 40°C for 6 min. The mixture was then mixed with 0.01 U Bst 2.0 DNA Polymerase, 0.5 U SplintR Ligase, and 10 nM ATP to a final volume of 20 μl. The final reaction mixture was incubated at 40°C for 20 min, 80°C for 20 min, and then kept at 4°C. Finally, qPCR was used to analyze the relative product abundance. The primers are listed in Dataset EV5.
Measurement of mRNA stability
C666 cells were transfected with YTHDF1‐specific siRNA or control siRNA for 24 h and then treated with ActD (5 μg/ml). RNA was collected at 0, 0.5, and 1 h after treatment and examined by qRT–PCR. Similarly, CNE2EBV cells were transfected with YTHDF1‐specific siRNA or control siRNA for 24 h and then induced with NaB (2 mM) for 24 h. RNA was collected at 0, 0.5, 1, and 2 h following ActD treatment and examined by qRT–PCR.
The turnover rates and half‐lives of mRNAs were estimated according to previously published papers (Chen et al, 2008a; Huang et al, 2018). After ActD treatment, the change in mRNA concentration at a given time (dC/dt) is proportional to the constant of the mRNA decay rate (K decay) and the mRNA concentration (C) according to the following equation:
The mRNA degradation rate (K decay) was estimated by:
To calculate the mRNA half‐life (t 1/2), when 50% of the mRNA decayed (that is, C/C 0 = 1/2), we used the equation:
where:
RIP‐qPCR
The RIP assay was performed using Magna RIP Quad RNA‐Binding Protein Immunoprecipitation Kit (Millipore, CA, USA, 17–704).
For the endogenous YTHDF1‐RIP, C666 cells were plated into a 10‐cm dish and allowed to grow for 24 h. Then, the cells were lysed and the lysate was incubated with anti‐YTHDF1 antibody or with a rabbit IgG antibody conjugated to Protein A/G Dynabeads in 1 ml RIP buffer overnight at 4°C. The beads were then washed five times with wash buffer and the RNA was collected by adding TRIzol to the beads.
Similarly, CNE2EBV cells were plated onto a 10‐cm dish and induced by TPA (30 ng/ml) and NaB (2 mM) for 24 h. Following EBV reactivation, CNE2EBV cells were lysed with lysis buffer and the RNA was collected as described above.
For the Myc‐RIP, CNE2EBV cells were transfected with the Myc‐ZAP, DDX17, or DCP2 and pcDNA6‐MYC plasmids. At 24 h post‐transfection, CNE2EBV cells were induced with TPA (30 ng/ml) and NaB (2 mM) for 24 h. The cells were then lysed and the cell lysates were incubated with rabbit anti‐c‐Myc Agarose Affinity Gel overnight at 4°C. The beads were then washed five times with wash buffer. For Myc‐DCP2 RIP assays, the beads were washed additional three times with high salt wash buffer (50 mM Tris–HCl, pH 7.4; 500 mM NaCl; 0.5% NP‐40; 1 mM EDTA). The RNA was collected by adding the TRIzol Reagent to the beads.
For the Myc‐RIP of wild‐type or mutant BZLF11–262nt mRNA in CNE2EBV cells, the cells were co‐transfected with the indicated plasmids and/or siRNAs for 24 h. The cells were then lysed and the cell lysates were incubated with rabbit anti‐c‐Myc Agarose Affinity Gel overnight at 4°C. Then, the beads were washed five times with wash buffer and the RNA was collected by adding TRIzol to the beads.
Finally, the input samples (1%) and all the IP RNA samples were subjected to qRT–PCR. The relative RIP enrichment was determined by calculating the 2−Δ C t of the RIP sample relative to the input sample.
Immunofluorescence analysis and confocal microscopy
For the co‐localization assays, the HK1 cells were seeded into 24‐well plates and allowed to grow for 24 h. Then, the cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X‐100 in PBS on ice, and blocked with 5% BSA in PBS. The slides were stained with specific antibodies, washed three times with PBS, stained with conjugated Alexa Fluor secondary antibodies, the nuclei were counterstained with DAPI, and the samples were mounted using ProLong Gold.
To detect the expression of gB, CNE2EBV cells were seeded into 24‐well plates and allowed to grow for 12 h. The cells were then transfected with YTHDF1‐specific siRNAs or siNC control for 24 h, followed by EBV reactivation with NaB for 24 h. The reactivated cells were then fixed with 4% paraformaldehyde in PBS and blocked with 5% BSA in PBS. The slides were stained with anti‐gB mAb (CL55), washed three times with PBS, stained with Alexa Fluor 594‐conjugated secondary anti‐mouse antibody, the nuclei were counterstained with DAPI, and the samples were mounted using ProLong Gold.
The fluorescence images were recorded using an FV1000 confocal laser scanning microscope (OLYMPUS, Japan).
Measurement of gp350 by flow cytometry
To measure of the expression of gp350 in the reactivated CNE2EBV cells, the cells were seeded into 12‐well plates and allowed to grow for 12 h. The cells were transfected with YTHDF1‐specific siRNAs or siNC control for 24 h, followed by EBV reactivation with NaB for 24 h. The cells were stained with anti‐gp350 mAb (72A1, (ATCC cell line ID: HB168)) followed by staining with Alexa Fluor 594‐conjugated secondary anti‐mouse antibody, washed three times with PBS, and analyzed by flow cytometry (Beckman Coulter FC500).
mRNA decapping assay
C666 cells were plated into 6‐well plates for 24 h and transfected with YTHDF1‐specific or control siRNAs. After 24 h, RNA was extracted from the transfected cells, and the presence of the 5′‐cap was determined as described previously (Du et al, 2016). Briefly, the cytoplasmic RNA samples (3 µg) were treated with three units of 5′‐phosphate‐dependent exonuclease XRN1 (New England BioLabs, USA) for 2 h at 37°C according to the manufacturer’s protocol. The treated RNAs were then purified overnight by ethanol precipitation and subjected to qPCR along with the total RNA sample. Finally, the ratios of capped RNA were calculated by dividing the RNA expression in XRN1‐treated samples by that in total RNA samples.
Western blot analysis
Western blot analysis was performed as described previously (Zhang et al, 2018a). Briefly, the cells were lysed in RIPA buffer containing a protease inhibitor mixture (Roche), and the proteins were resolved using SDS–PAGE, transferred onto a polyvinylidene difluoride (PVDF) membrane, and incubated with the indicated primary antibodies. The PVDF membrane was then incubated with goat anti‐mouse or goat anti‐rabbit HRP‐conjugated secondary antibodies at 1:3,000 dilution. The membrane was washed and visualized using enhanced chemiluminescence reagent (ECL, Millipore).
Statistical analyses
Data were analyzed by two‐tailed unpaired Student’s t‐test in R (v.3.3.0) and GraphPadPrism 8 (GraphPad Software Inc., La Jolla, CA). All results were expressed as the mean ± SD from at least three independent experiments. Differences with P‐values < 0.05 were considered statistically significant. In all results, ns denotes “not significant”, * denotes P < 0.05, ** denotes P < 0.01, and *** denotes P < 0.001.
Author contributions
Conception and experiment design, supervision, and manuscript writing: M‐SZ and ZZ; Performance and analysis of the key experiments and manuscript writing: T‐LX; Bioinformatics analysis: ZZ, XL, and YY; Experiments: XW, Y‐JZ, HZ, WC, M‐LC, YL, AZ, D‐LD, Q‐YZ, LY, S‐QC, and Z‐WX; Assistance in editing this manuscript: GR; Suggestions for this work: H‐BW, QZ, JZ, HH, DL, Y‐XZ, and JW; Revising the manuscript: HH, BZ, and BEG; Research design and data interpretation: BZ and JC; Reading and approval of the final version of the manuscript: All authors.
Conflict of interest
J.C. is a scientific founder of the Genovel Biotech Corp.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
M.‐S.Z. was supported by the National Key R&D Program (2017YFA0505600, 2017YFC0908503, and 2016YFA0502100), the National Natural Science Foundation of China (81830090, 81520108022), the Guangzhou Science Technology and Innovation Commission (201607020038), Natural Science Foundation of Guang Dong Province (No. 2017A030312003), and the Guangdong Province key research and development program (2019B020226002). L.Y. was supported by the National Natural Science Foundation of China (81802775). H.‐B.W. was supported by the National Natural Science Foundation of China (81672703). S.C. was supported by the National Natural Science Foundation of China (81702719). B.E.G. was supported by a Burroughs Wellcome Career Award in Medical Sciences. J.C. is a Leukemia & Lymphoma Society (LLS) Scholar and was supported by the National Institutes of Health (NIH) R01 Grants CA214965 and CA236399.
EMBO Reports (2021) 22: e50128.
Contributor Information
Zhixiang Zuo, Email: zuozhx@sysucc.org.cn.
Mu‐Sheng Zeng, Email: zengmsh@sysucc.org.cn.
Data availability
The key raw data have been uploaded onto the Research Data Deposit public platform (www.researchdata.org.cn), with the approval RDD number RDDB2021001069. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the BIG Data Center at the Beijing Institute of Genomics, Chinese Academy of Science, under Project accession number PRJCA004279 (accessible at http://bigd.big.ac.cn/gsa‐human).
References
- Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF (2015) HNRNPA2B1 is a mediator of m(6)A‐dependent nuclear RNA processing events. Cell 162: 1299–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allday MJ, Bazot Q, White RE (2015) The EBNA3 family: two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr Top Microbiol Immunol 391: 61–117 [DOI] [PubMed] [Google Scholar]
- Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM (2012) An atlas of the Epstein‐Barr virus transcriptome and epigenome reveals host‐virus regulatory interactions. Cell Host Microbe 12: 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Decker LL, Volk M, Thorley‐Lawson DA (1998) EBV persistence in memory B cells in vivo . Immunity 9: 395–404 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero‐Perez B, Antanaviciute A, Yonchev ID, Carr IM, Wilson SA, Whitehouse A (2019) The Tudor SND1 protein is an m(6)A RNA reader essential for replication of Kaposi's sarcoma‐associated herpesvirus. Elife 8: e47261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM (1997) Purification and cDNA cloning of the AdoMet‐binding subunit of the human mRNA (N6‐adenosine)‐methyltransferase. RNA 3: 1233–1247 [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ezzeddine N, Shyu AB (2008a) Messenger RNA half‐life measurements in mammalian cells. Methods Enzymol 448: 335–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Guo X, Lv F, Xu Y, Gao G (2008b) p72 DEAD box RNA helicase is required for optimal function of the zinc‐finger antiviral protein. Proc Natl Acad Sci USA 105: 4352–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T et al (2015) High‐resolution N(6) ‐methyladenosine (m(6) A) map using photo‐crosslinking‐assisted m(6) A sequencing. Angew Chem 54: 1587–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen‐Kiang S, Nevins JR, Darnell JE Jr (1979) N‐6‐methyl‐adenosine in adenovirus type 2 nuclear RNA is conserved in the formation of messenger RNA. J Mol Biol 135: 733–752 [DOI] [PubMed] [Google Scholar]
- Cheung ST, Huang DP, Hui AB, Lo KW, Ko CW, Tsang YS, Wong N, Whitney BM, Lee JC (1999) Nasopharyngeal carcinoma cell line (C666–1) consistently harbouring Epstein‐Barr virus. Int J Cancer 83: 121–126 [DOI] [PubMed] [Google Scholar]
- Countryman J, Miller G (1985) Activation of expression of latent Epstein‐Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci USA 82: 4085–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney DG, Kennedy EM, Dumm RE, Bogerd HP, Tsai K, Heaton NS, Cullen BR (2017) Epitranscriptomic enhancement of influenza A virus gene expression and replication. Cell Host Microbe 22: 377–386.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney DG, Chalem A, Bogerd HP, Law BA, Kennedy EM, Holley CL, Cullen BR (2019) Extensive epitranscriptomic methylation of A and C residues on murine leukemia virus transcripts enhances viral gene expression. MBio 10: e01209‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Wei Z, Zhang L, Liu H, Sun L, Zhang SW, Huang Y, Meng J (2016) Guitar: an R/bioconductor package for gene annotation guided transcriptomic analysis of RNA‐related genomic features. Biomed Res Int 2016: 8367534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA 71: 3971–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimock K, Stoltzfus CM (1977) Sequence specificity of internal methylation in B77 avian sarcoma virus RNA subunits. Biochemistry 16: 471–478 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–206 [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Salmon‐Divon M, Amariglio N, Rechavi G (2013) Transcriptome‐wide mapping of N(6)‐methyladenosine by m(6)A‐seq based on immunocapturing and massively parallel sequencing. Nat Protoc 8: 176–189 [DOI] [PubMed] [Google Scholar]
- Doyle MG, Catovsky D, Crawford DH (1993) Infection of leukaemic B lymphocytes by Epstein Barr virus. Leukemia 7: 1858–1864 [PubMed] [Google Scholar]
- Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L (2016) YTHDF2 destabilizes m(6)A‐containing RNA through direct recruitment of the CCR4‐NOT deadenylase complex. Nat Commun 7: 12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahraeus R, Fu HL, Ernberg I, Finke J, Rowe M, Klein G, Falk K, Nilsson E, Yadav M, Busson P et al (1988) Expression of Epstein‐Barr virus‐encoded proteins in nasopharyngeal carcinoma. Int J Cancer 42: 329–338 [DOI] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre AB, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, Quicke KM, Vazquez C, Willer J et al (2016) N6‐methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales‐van Horn SR, Sarnow P (2017) Making the mark: the role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe 21: 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ma J, Sun J, Gao G (2007) The zinc‐finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci USA 104: 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H, Hao S, Chen H, Chen Z, Zhang Y, Wang J, Wang H, Zhang B, Qiu J, Deng F et al (2018) N6‐methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res 47: 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Lieberman PM, Hayward SD (1988) A new Epstein‐Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J Virol 62: 2274–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesser CR, Karijolich J, Dominissini D, He C, Glaunsinger BA (2018) N6‐methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi's sarcoma‐associated herpesvirus infection. PLoS Pathog 14: e1006995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley‐Lawson DA (2004) Demonstration of the Burkitt's lymphoma Epstein‐Barr virus phenotype in dividing latently infected memory cells in vivo . Proc Natl Acad Sci USA 101: 239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J et al (2017) Ythdc2 is an N(6)‐methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 27: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Lin Z, Wu Y, Dong J, Zhao B, Cheng Y, Huang P, Xu L, Xia T, Xiong D et al (2016) Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired‐end transcriptome sequencing. Front Med 10: 61–75 [DOI] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL et al (2018) Recognition of RNA N(6)‐methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20: 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY, Kim SJ, Mason CE, Horner SM, Siddiqui A (2018) N6‐methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc Natl Acad Sci USA 115: 8829–8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG et al (2011) N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K (2004) Production of high‐titer Epstein‐Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J Virol 78: 7004–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SE, Beemon K (1985) Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol 5: 2298–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, Marshall JB, Poling BC, Tsai K, Gokhale NS, Horner SM et al (2016) Posttranscriptional m(6)A editing of HIV‐1 mRNAs enhances viral gene expression. Cell Host Microbe 19: 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E, Nagy N, Rasul AE (2013) EBV genome carrying B lymphocytes that express the nuclear protein EBNA‐2 but not LMP‐1: Type IIb latency. Oncoimmunology 2: e23035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer J, Rao H, Hackert P, Sloan KE, Hobartner C, Bohnsack MT (2018) The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5'‐3' exoribonuclease XRN1. RNA 24: 1339–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, Morgan MA, Shatkin AJ (1976) Influenza viral mRNA contains internal N6‐methyladenosine and 5'‐terminal 7‐methylguanosine in cap structures. J Virol 20: 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Singh RK, Pei Y, Zhang S, Sun K, Robertson ES (2019) EBV epitranscriptome reprogramming by METTL14 is critical for viral‐associated tumorigenesis. PLoS Pathog 15: e1007796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera‐Castrejon A, Schneir D, Bayerl J, Mizrahi O, Peles S, Tawil S et al (2020) Context‐dependent functional compensation between Ythdf m(6)A reader proteins. Genes Dev 34: 1373–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi S, Shatkin AJ (1975) Methylated simian virus 40‐specific RNA from nuclei and cytoplasm of infected BSC‐1 cells. Proc Natl Acad Sci USA 72: 2012–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liu S, Hu J, Luo X, Li N, M.Bode A, Cao Y (2016) Epstein‐Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int J Biol Sci 12: 1309–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X et al (2017a) Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res 27: 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C et al (2017b) FTO plays an oncogenic role in acute myeloid leukemia as a N6‐methyladenosine RNA demethylase. Cancer Cell 31: 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, Rana TM (2016a) Dynamics of the human and viral m(6)A RNA methylomes during HIV‐1 infection of T cells. Nat Microbiol 1: 16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, Rana TM (2016b) Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe 20: 666–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X et al (2014) A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10: 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T (2015) N6‐methyladenosine‐dependent RNA structural switches regulate RNA–protein interactions. Nature 518: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Sun XS, Li XY, Tang LQ, Chen QY, Lin HX, Liang YJ, Yan JJ, Lin C, Guo SS et al (2019) The diagnostic and prognostic values of plasma Epstein‐Barr virus DNA for residual cervical lymphadenopathy in nasopharyngeal carcinoma patients: a retrospective study. Cancer Commun 39: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Tirumuru N, St Gelais C, Koneru PC, Liu C, Kvaratskhelia M, He C, Wu L (2018) N(6)‐methyladenosine‐binding proteins suppress HIV‐1 infectivity and viral production. J Biol Chem 293: 12992–13005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luka J, Kallin B, Klein G (1979) Induction of the Epstein‐Barr virus (EBV) cycle in latently infected cells by n‐butyrate. Virology 94: 228–231 [DOI] [PubMed] [Google Scholar]
- Luo Y, Schofield J, Simon MD, Slavoff SA (2020) Global profiling of cellular substrates of human Dcp2. Biochemistry 59: 4176–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, Cuconati A, Block TM, Guo JT, Guo H (2013) Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog 9: e1003494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Perez M, Aparicio F, Lopez‐Gresa MP, Belles JM, Sanchez‐Navarro JA, Pallas V (2017) Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc Natl Acad Sci USA 114: 10755–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149: 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B, Gershowitz A, Stringer JR, Holland LE, Wagner EK (1977) 5'‐Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J Virol 23: 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz C (2019) Latency and lytic replication in Epstein‐Barr virus‐associated oncogenesis. Nat Rev Microbiol 17: 691–700 [DOI] [PubMed] [Google Scholar]
- Parker R, Sheth U (2007) P bodies and the control of mRNA translation and degradation. Mol Cell 25: 635–646 [DOI] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6‐methyladenosine methyltransferase. Cell Res 24: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014) BEDTools: the swiss‐army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I (2018) RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev 32: 1472–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter‐Ovanesyan D, Habib N, Cacchiarelli D et al (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep 8: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Peng SL, Yang LF, Chen X, Tao YG, Cao Y (2016) Co‐infection of Epstein‐Barr virus and human papillomavirus in human tumorigenesis. Chin J Cancer 35: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C (2017) YTHDF3 facilitates translation and decay of N6‐methyladenosine‐modified RNA. Cell Res 27: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Salditt‐Georgieff M, Bachenheimer S, Darnell JE, Furuichi Y, Morgan M, Shatkin AJ (1976) The methylation of adenovirus‐specific nuclear and cytoplasmic RNA. Nucleic Acids Res 3: 749–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LB, Zeng MS, Liao WT, Zhang L, Mo HY, Liu WL, Shao JY, Wu QL, Li MZ, Xia YF et al (2006) Bmi‐1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Can Res 66: 6225–6232 [DOI] [PubMed] [Google Scholar]
- Tan B, Gao SJ (2018) The RNA epitranscriptome of DNA viruses. J Virol 92: e00696‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Liu H, Zhang S, da Silva SR, Zhang L, Meng J, Cui X, Yuan H, Sorel O, Zhang SW et al (2018) Viral and cellular N(6)‐methyladenosine and N(6),2'‐O‐dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat Microbiol 3: 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley‐Lawson DA (2015) EBV persistence‐introducing the virus. Curr Top Microbiol Immunol 390: 151–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney RJ, Steven N, Young LS, Rickinson AB (1994) Epstein‐Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J Virol 68: 7374–7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L (2016) N6‐methyladenosine of HIV‐1 RNA regulates viral infection and HIV‐1 Gag protein expression. Elife 5: e15528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N, Wu L (2019) HIV‐1 envelope proteins up‐regulate N (6)‐methyladenosine levels of cellular RNA independently of viral replication. J Biol Chem 294: 3249–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova T, Bock FJ, Chang P (2015) Poly(ADP‐ribose) polymerase‐13 and RNA regulation in immunity and cancer. Trends Mol Med 21: 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R et al (2013) Spontaneous lytic replication and epitheliotropism define an Epstein‐Barr virus strain found in carcinomas. Cell Rep 5: 458–470 [DOI] [PubMed] [Google Scholar]
- Tsai K, Courtney DG, Cullen BR (2018) Addition of m6A to SV40 late mRNAs enhances viral structural gene expression and replication. PLoS Pathog 14: e1006919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G et al (2014) N6‐methyladenosine‐dependent regulation of messenger RNA stability. Nature 505: 117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Zhang H, Zhang JP, Li Y, Zhao B, Feng GK, Du Y, Xiong D, Zhong Q, Liu WL et al (2015a) Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun 6: 6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C (2015b) N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B (1976) 5'‐Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry 15: 397–401 [DOI] [PubMed] [Google Scholar]
- Wille CK, Nawandar DM, Panfil AR, Ko MM, Hagemeier SR, Kenney SC (2013) Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein‐Barr virus lytic gene expression and viral replication. J Virol 87: 935–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille CK, Nawandar DM, Henning AN, Ma S, Oetting KM, Lee D, Lambert P, Johannsen EC, Kenney SC (2015) 5‐hydroxymethylation of the EBV genome regulates the latent to lytic switch. Proc Natl Acad Sci USA 112: E7257–7265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai‐Schmiedel J, Friedman N, Le‐Trilling VTK et al (2018) m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol 20: 173–182 [DOI] [PubMed] [Google Scholar]
- Woellmer A, Hammerschmidt W (2013) Epstein‐Barr virus and host cell methylation: regulation of latency, replication and virus reactivation. Curr Opin Virol 3: 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS (2017) Regulation of m(6)A transcripts by the 3'–>5' RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell 68: 374–387 e312 [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY et al (2016) Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Wang Y, Tang Q, Wei L, Zhang X, Jia G (2018) An Elongation‐ and ligation‐based qPCR amplification method for the radiolabeling‐free detection of locus‐specific N(6) ‐methyladenosine modification. Angew Chem 57: 15995–16000 [DOI] [PubMed] [Google Scholar]
- Xiong D, Du Y, Wang HB, Zhao B, Zhang H, Li Y, Hu LJ, Cao JY, Zhong Q, Liu WL et al (2015) Nonmuscle myosin heavy chain IIA mediates Epstein‐Barr virus infection of nasopharyngeal epithelial cells. Proc Natl Acad Sci USA 112: 11036–11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y, Liu L, Shen S, Deng H, Gao G (2012) Zinc finger antiviral protein inhibits murine gammaherpesvirus 68 M2 expression and regulates viral latency in cultured cells. J Virol 86: 12431–12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Zhao BS, Zhang Z, Lu M, Harder O, Chen P, Lu Z, Li A, Ma Y, Xu Y et al (2019) Viral N6‐methyladenosine upregulates replication and pathogenesis of human respiratory syncytial virus. Nat Commun 10: 4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Zhang Y, He B, Xiang J, Wang ZF, Zheng FM, Xu J, Chen MY, Zhu YL, Wen HJ et al (2014) IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat Commun 5: 3661 [DOI] [PubMed] [Google Scholar]
- Yang HJ, Huang TJ, Yang CF, Peng LX, Liu RY, Yang GD, Chu QQ, Huang JL, Liu N, Huang HB et al (2013) Comprehensive profiling of Epstein‐Barr virus‐encoded miRNA species associated with specific latency types in tumor cells. Virol J 10: 314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen ER, Nilsen TW (2017) Kaposi's sarcoma‐associated herpesvirus utilizes and manipulates RNA N6‐adenosine methylation to promote lytic replication. J Virol 91: e00466‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM et al (2018) High‐density proximity mapping reveals the subcellular organization of mRNA‐associated granules and bodies. Mol Cell 69: 517–532.e11 [DOI] [PubMed] [Google Scholar]
- Young LS, Rickinson AB (2004) Epstein‐Barr virus: 40 years on. Nat Rev Cancer 4: 757–768 [DOI] [PubMed] [Google Scholar]
- Young LS, Yap LF, Murray PG (2016) Epstein‐Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16: 789–802 [DOI] [PubMed] [Google Scholar]
- Zaccara S, Jaffrey SR (2020) A unified model for the function of YTHDF proteins in regulating m6A‐modified mRNA. Cell 181: 1582–1595.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, Dong XD, Li SB, Du Y, Xiong D et al (2018a) Ephrin receptor A2 is an epithelial cell receptor for Epstein‐Barr virus entry. Nat Microbiol 3: 1–8 [DOI] [PubMed] [Google Scholar]
- Zhang PF, Zheng XH, Li XZ, Tian T, Zhang SD, Hu YZ, Jia WH (2018b) Nasopharyngeal brushing: a convenient and feasible sampling method for nucleic acid‐based nasopharyngeal carcinoma research. Cancer Commun 38: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB (2015) Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526: 591–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Chen G, Lv F, Wang X, Ji X, Xu Y, Sun J, Wu L, Zheng YT, Gao G (2011) Zinc‐finger antiviral protein inhibits HIV‐1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc Natl Acad Sci USA 108: 15834–15839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H, O'Neill FJ, Freese UK, Hecker E (1978) Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272: 373–375 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The key raw data have been uploaded onto the Research Data Deposit public platform (www.researchdata.org.cn), with the approval RDD number RDDB2021001069. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the BIG Data Center at the Beijing Institute of Genomics, Chinese Academy of Science, under Project accession number PRJCA004279 (accessible at http://bigd.big.ac.cn/gsa‐human).