

Abstract
Intracellular pH is a potent modulator of neuronal functions. By catalyzing (de)hydration of CO2, intracellular carbonic anhydrase (CAi) isoforms CA2 and CA7 contribute to neuronal pH buffering and dynamics. The presence of two highly active isoforms in neurons suggests that they may serve isozyme‐specific functions unrelated to CO2‐(de)hydration. Here, we show that CA7, unlike CA2, binds to filamentous actin, and its overexpression induces formation of thick actin bundles and membrane protrusions in fibroblasts. In CA7‐overexpressing neurons, CA7 is enriched in dendritic spines, which leads to aberrant spine morphology. We identified amino acids unique to CA7 that are required for direct actin interactions, promoting actin filament bundling and spine targeting. Disruption of CA7 expression in neocortical neurons leads to higher spine density due to increased proportion of small spines. Thus, our work demonstrates highly distinct subcellular expression patterns of CA7 and CA2, and a novel, structural role of CA7.
Keywords: actin cytoskeleton, carbonic anhydrase, dendritic spines, intracellular pH, multifunctional protein
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Neuroscience
Carbonic anhydrase CA7 is a pH‐regulatory molecule that is expressed in pyramidal neurons at the time of onset of dendritic spinogenesis. This study shows that CA7, but not CA2, binds and bundles actin filaments and has a morphogenetic role in cells.
Introduction
Intracellular carbonic anhydrases (CAi) are best known for their contribution to CO2/ ‐dependent buffering (βCO2) (Maren, 1967). There are five catalytically active CAi isoforms: CA1, CA2, CA3, CA7, and CA13 which differ in their tissue/cell‐type distribution, sensitivity to inhibitors as well as in their kinetic properties (Mboge et al, 2018). In the brain, CAi is abundant in choroid plexus epithelia (Halmi et al, 2006; Johansson et al, 2008) and glial cells (Cammer, 1984; Cammer & Tansey, 1988) but several studies have shown that CAis are present (Nogradi & Mihaly, 1990; Wang et al, 2002; Halmi et al, 2006; Kida et al, 2006) and accelerate the (de)hydration of CO2 to in neurons as well (Pasternack et al, 1993). Rat and mouse hippocampal neurons sequentially express two catalytically highly active CAi isoforms: Expression of CA7, the neuron‐specific isoform in the central nervous system, starts at postnatal day (P) 10–12, and it is the only cytoplasmic isoform until P18 when the expression of CA2 commences (Ruusuvuori et al, 2004, 2013). The presence of the highly mobile CO2/ buffer system has, in addition to βCO2, an important role in enhancing diffusion of acid–base species within the neuronal cytoplasm (Voipio, 1998; Geers & Gros, 2000). Notably, acts as a major carrier of current in GABAA receptors (Kaila & Voipio, 1987; Kaila et al, 1993), and the CA7‐driven replenishment of intraneuronal is required for the developmental onset of the paradoxical GABAergic excitation and network synchronization under conditions of prolonged activation of interneurons (Kaila et al, 1997; Ruusuvuori et al, 2004). Using intracellular pH (pHi) and patch‐clamp measurements, we showed that after P18 both isoforms work in parallel to catalyze the intraneuronal CO2 (de)hydration (Ruusuvuori et al, 2013). However, the co‐expression of two high‐activity isoforms in neurons may suggest that they serve isozyme‐specific functions unrelated to CO2‐(de)hydration.
The subcellular distribution of neuronal CAis is of great interest because in addition to the GABAergic current discussed above, glutamatergic signaling (Rose et al, 1999; Brini et al, 2017) evokes large, localized ionic fluctuations. Excitatory synapses are mostly formed on dendritic spines (Bourne & Harris, 2008), which undergo morphological and structural changes, upon synaptic activation (Yuste & Bonhoeffer, 2001). These structural changes are driven by fast disassembly of actin filaments, followed by re‐polymerization and re‐assembly of the filaments (Halpain, 2000). In rats and mice, a period of rapid synaptic formation and dendritic spine maturation from filopodia to mushroom spines (Dailey & Smith, 1996; Dunaevsky et al, 1999; Hotulainen et al, 2009) takes place at P10–P20, within a time‐window overlapping with the developmental upregulation of CA7. Based on this, we hypothesized that CA7 may play a specific role in dendritic spine development.
Here, we show that, in contrast to CA2, CA7 is highly compartmentalized within neurons and that it has a major influence on actin dynamics. Specifically, we demonstrate that CA7 directly binds and bundles actin in vitro and that it co‐localizes with filamentous actin (F‐actin) when overexpressed in fibroblasts and neurons. Further, we identify a novel role of CA7 in modulation of actin bundling that is not dependent on its catalytic activity. In CA7‐overexpressing neurons, CA7 is enriched in the actin‐dense dendritic spines whereas CA7 depletion increases overall spine density, with a shift to smaller spine heads. These results show that neuronal CA7 has tightly linked functions in both cellular ion homeostasis and cytoskeleton structure.
Results
CA7 binds to and bundles F‐actin
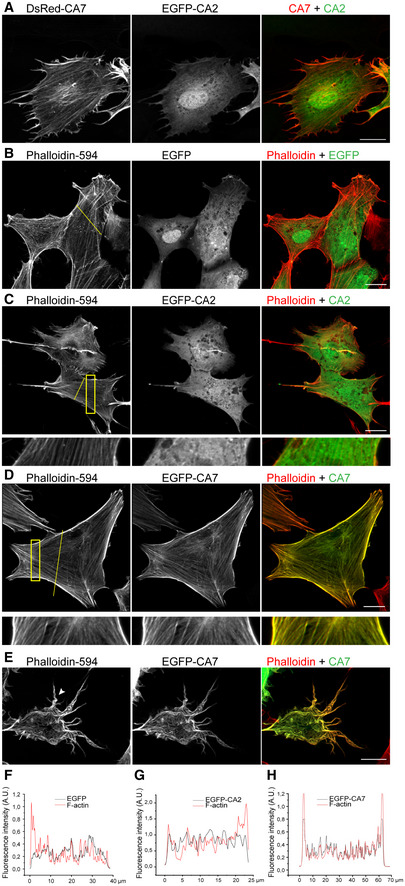
To study the subcellular localization of CA2 and CA7, we simultaneously overexpressed EGFP‐CA2 and DsRed‐CA7 fusion proteins in cultured NIH3T3 fibroblasts. The two isoforms showed strikingly different localization patterns. CA7 localized to cytosolic filamentous structures, whereas CA2 was homogenously distributed throughout the cytoplasm and nucleus (Fig 1A). This difference prompted us to test whether CA7 co‐localized with F‐actin. Experiments, in which we expressed EGFP, EGFP‐CA2, or EGFP‐CA7 in fibroblasts and stained F‐actin with phalloidin‐594 after fixation, showed no overlap of EGFP or EGFP‐CA2 with F‐actin (Fig 1B and C). In contrast, EGFP‐CA7 strongly co‐localized with F‐actin (Fig 1D). An exception to this were the outer edges of lamellipodia, mainly containing branched actin (Higgs, 2011), in which EGFP‐CA7 was not present (Fig EV1 and Movie EV1). Very high levels of CA7 overexpression also caused marked changes in actin structures and cellular morphology, including the formation of thick, curving cytosolic stress fibers and filopodia‐like plasmalemmal protrusions (Fig 1E). To compare the co‐localization of the overexpressed CA proteins with F‐actin in a quantitative manner, we measured the fluorescence intensities of EGFP and phalloidin‐594 from cross‐sectional cellular profiles (Fig 1F–H). These data, together with the correlation coefficients presented below in Fig 5, demonstrate that EGFP‐CA7, but not EGFP‐CA2, co‐localizes strongly with F‐actin.
Figure 1. Subcellular localization of CA7 and CA2 in fibroblasts.
-
ANIH3T3 fibroblasts co‐expressing DsRed‐CA7 and EGFP‐CA2 (n = 4 independent replicates).
-
B–ECo‐localization of EGFP and the two CA isoforms with filamentous actin studied in fibroblasts expressing (B) EGFP, (C) EGFP‐CA2, or (D, E) EGFP‐CA7 and stained with phalloidin‐594 to visualize F‐actin (n = 4, 10, and 8 independent replicates, respectively). A magnification of the area marked with the yellow rectangle in (C) and (D) shows the localization of EGFP‐CA2 and EGFP‐CA7 compared to phalloidin‐594. (E) EGFP‐CA7 caused a prominent overexpression phenotype with thick and curvy cytosolic actin bundles (arrow) and plasmalemmal protrusions (arrowhead).
-
F–HThe normalized fluorescence emission intensity profiles for F‐actin (red line) and (F) EGFP, (G) EGFP‐CA2, or (H) EGFP‐CA7 (black line).
Data information: For the plots, pixel intensities were measured through the cross‐section of the cell indicated by the yellow line in panels (B–D). Scale bar in (A–E) 20 µm.
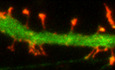
Figure EV1. CA7 co‐localizes with selective actin filaments.
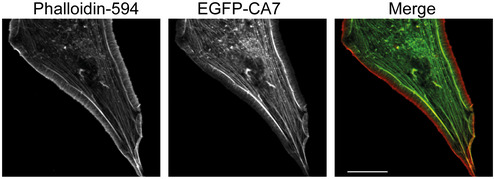
EGFP‐CA7 co‐localizes with subcellular F‐actin structures (visualized with Phalloidin‐594) except at the very edges of NIH3T3 fibroblast lamellipodia. Scale bar 20 µm.
Figure 5. Subcellular localization of the chimeric CA7 constructs in fibroblasts.
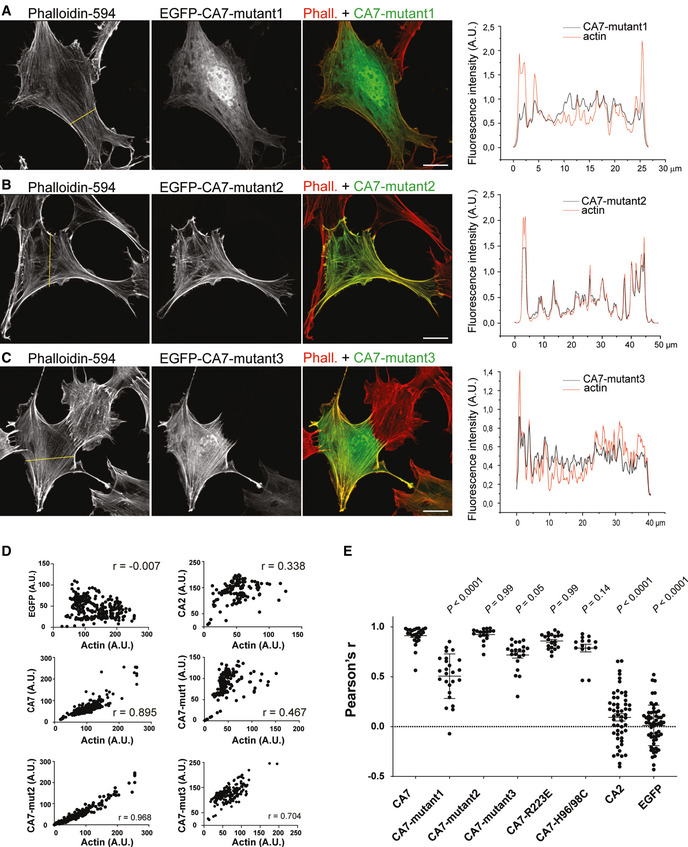
-
A–CNIH3T3 fibroblasts expressing EGFP‐CA7‐mutant1 (A), EGFP‐CA7‐mutant2 (B), and EGFP‐CA7‐mutant3 (C). F‐actin is visualized with Phalloidin‐594. In the right‐most panels of (A–C) are the normalized fluorescence intensity profiles of the mutated CA7 EGFP signal (black) and actin (red), and the yellow line in left‐most panels indicates the cross‐section from which the pixel intensities were measured. Scale bars 20 µm.
-
DAnalysis of the mutated CA7 and F‐actin co‐localization in cultured fibroblasts. Scatterplots of fluorescent intensities per pixel (EGFP vs. Phalloidin‐594) along a cross‐section through a representative cell. Pearson’s correlation coefficient (r) for the analyzed cells is given in each panel.
-
EPearson’s correlation coefficient values calculated for the depicted constructs and compared to CA7. F‐actin had a strong positive correlation coefficient with EGFP‐CA7 (r = 0.91 ± 0.02, n = 26 cells). Neither EGFP alone (r = 0.01 ± 0.03, n = 56) nor EGFP‐CA2 (r = 0.09 ± 0.03, n = 53) co‐localized with F‐actin (P < 0.0001 for both constructs, when compared to CA7). From the five mutated CA7 constructs, EGFP‐CA7‐mutant1 (r = 0.51 ± 0.04, P < 0.0001, n = 25) and EGFP‐CA7‐mutant3 (r = 0.72 ± 0.03, P = 0.05, n = 21) co‐localized less with F‐actin actin when compared to CA7. The co‐localization of the other four mutated CA7 constructs, EGFP‐CA7‐mutant2 (r = 0.92 ± 0.02, P = 0.99, n = 18), EGFP‐CA7‐R223E (r = 0.86 ± 0.02, P = 0.99, n = 24), and EGFP‐CA7‐H96/98C (r = 0.79 ± 0.04, P = 0.14, n = 18) did not differ significantly from that of CA7. Data are shown as mean ± SEM. Data did not pass Shapiro–Wilk test for normality, statistical comparison against CA7 was done with Kruskal–Wallis test corrected for multiple comparisons.
Source data are available online for this figure.
Next, we examined whether there is a direct interaction between CA7 and F‐actin. For these experiments, we used purified recombinant mouse CA7 protein (mCA7). In an actin pull‐down assay, mCA7 co‐sedimented with F‐actin, demonstrating a direct interaction between the two proteins (Figs 2A and EV2A). Interestingly, pH modulated this interaction: at pH 6.5, mCA7 bound F‐actin with higher affinity than at pH 7.4 (P < 0.0001, two‐way ANOVA, three independent repeats for each pH and actin concentration) (Figs 2A and EV2A). CA2 did not bind F‐actin at either pH (Fig EV2B and C).
Figure 2. CA7 binds to filamentous actin and increases actin bundling.
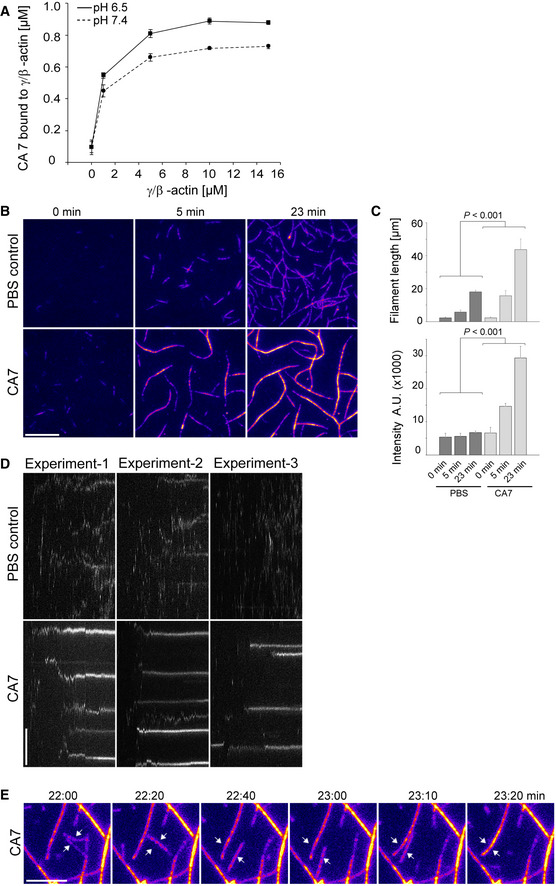
- Actin co‐sedimentation assay shows that CA7 binds to F‐actin. The binding is enhanced at more acidic pH (6.5 vs. 7.4). n = 3 independent replicates at each actin concentration, two‐way ANOVA, P < 0.001.
- Fluorescence time‐lapse images of F‐actin bundling in an in vitro bundling assay. A mixture of unlabeled and Rhodamine labeled non‐muscle actin was polymerized in the absence (PBS control, upper panel) or presence of mCA7 (lower panel). Numbers in images indicate the time after the onset of the experiment (0, 5, and 23 min). Intensity‐based fire‐coloring (Fiji) was used to visualize intensity changes. Scale bar 10 µm.
- Quantification of the mean increase in filament length (n = 10 filaments at each time point) and the mean relative fluorescence intensity values of cross‐sections for individual filaments /bundles (n = 30–31) in the absence and presence of mCA7 (1.12 µM). The data were analyzed using a general mixed model with time as a within‐unit factor and the presence of CA7 as a between‐unit factor. n = 3 independent repetitions, experiment repeats were included as a covariate and were non‐significant.
- Kymographs showing different time points in the line of interest (line width 1 µm) from the experiments analyzed in (C). Kymographs were generated with Fiji Multi Kymograph function. Total time is 159 frames = 26.5 min. Scale bar 5 μm (full height is 20 μm). F‐actin bundles can be detected as clear lines in kymographs.
- Fluorescence time‐lapse images of F‐actin bundling in an in vitro bundling assay. A mixture of unlabeled and Rhodamine labeled non‐muscle actin was polymerized in the presence of mCA7 (0.11 µM). Numbers above images indicate the time after the onset of the experiment (22–23 min 20 s). Intensity‐based fire‐coloring (Fiji) was used to visualize intensity changes. White arrows highlight the bundling filaments. Scale bar 5 μm. These data are from a representative video (n = 8 independent repeats).
Data information: Data are presented as mean ± SEM in (A) and mean ± SD in (C).
Source data are available online for this figure.
Figure EV2. CA7 directly interacts with F‐actin and enhances actin filament polymerization.
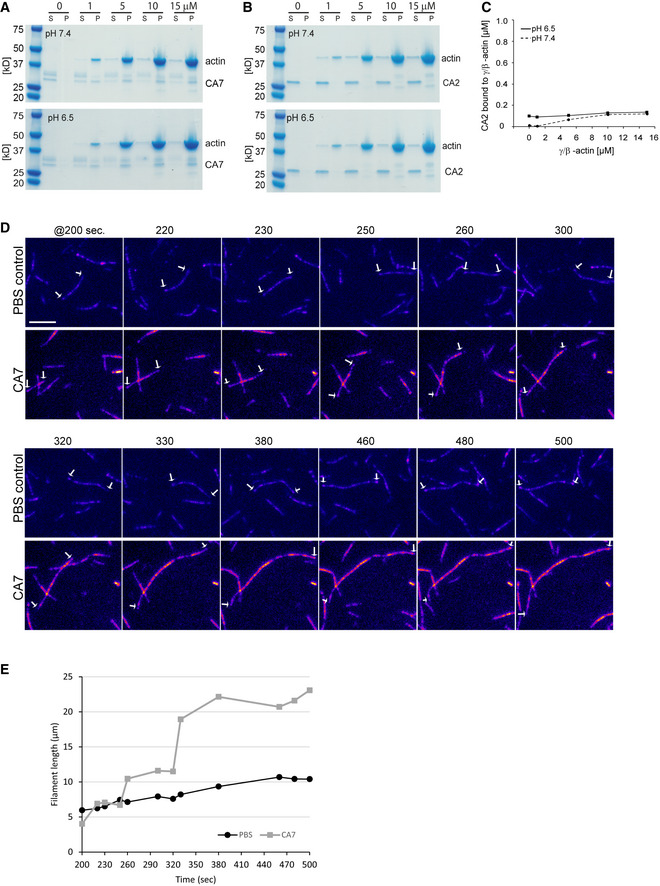
-
A, BActin co‐sedimentation assay was carried out at five different concentrations of β/γ‐actin and with 1 µM (A) CA7 or (B) CA2 at two different pH (7.4 or 6.5). After centrifugation, the supernatant (S) and pellet (P) fractions were separated and resolved by SDS‐PAGE. Staining the gels with Coomassie Blue showed that CA7 co‐sedimented in the pellets with actin, whereas CA2 was found only in the supernatant fraction. CA7; three repetitions and CA2; one repetition at each of the four actin concentrations/pH.
-
CAnalysis of the CA2 gels confirmed that the isoform does not interact with actin at either pH tested.
-
DIn the presence of mCA7, existing filaments assemble thus increasing elongation velocity in a stepwise manner. Fluorescence time‐lapse images of F‐actin bundling in the in vitro bundling assay. A mixture of unlabeled and Rhodamine labeled non‐muscle actin was polymerized in the absence (PBS control, upper row) or in the presence of mCA7 (1.12 µM, lower row). Numbers above images indicate the time after the onset of the experiment (images every 10 s) (First frame 200 s = 3 min 20 s, last frame 500 = 8 min 20 s). The filament/bundle ends that were followed over the experiment are indicated with white arrows. Intensity‐based fire‐coloring (Fiji) was used. Scale bar 5 μm.
-
EQuantification of the filament/bundle length for shown frames. Measured lengths were plotted to Excel x = time in seconds, y = length in μm.
As CA7‐overexpressing fibroblasts often showed abnormally thick actin structures, we set out to test whether CA7 modulates actin bundling. To this end, an in vitro actin‐bundling assay using total internal reflection fluorescence (TIRF) microscopy was used in the absence and presence of 1.12 μM mCA7 (Fig 2B, Movies [Link], [Link], [Link], [Link], [Link], [Link]). Both actin filament length and fluorescence intensity of their cross‐section (which takes into account the filament/bundle thickness) increased over time significantly in the presence of mCA7 when compared to the vehicle control (P < 0.001 for both length and intensity) (Fig 2C). Kymographs generated from the Movies [Link], [Link], [Link], [Link], [Link], [Link] showed that in the presence of mCA7, thick actin bundles appeared soon after starting the experiment (Fig 2D). Under control conditions, the intensity of filaments was constant throughout the experiment indicating that actin filaments became elongated as individual filaments (Fig 2B–D). In contrast, in the presence of mCA7, the intensity doubled or tripled in 5 min and was about 5‐fold brighter at the end point (after 23 min), suggesting that most of the actin is in bundles of 4–6 filaments (Fig 2B–D). In the presence of mCA7, the filaments started to elongate individually, but they soon bundled together (Fig 2B and D). For better visualization, we followed the process of bundling in a set of experiments at a lower concentration of mCA7 (0.11 µM) (Fig 2E). An analysis of actin filament elongation over time revealed further differences between the treatments (Fig EV2D and E). In PBS control experiments, filament length increased steadily (Fig EV2D, white arrows at the end of filaments; Fig EV2E, plot of filament length). In the presence of mCA7, filament/bundle elongation occurred stepwise, with a short filament added to the end (or close to end) of the original filament/bundle (Fig EV2D and E). Thus, under control conditions, actin polymerizes slowly to produce single filaments as shown in previous studies (Kuhn & Pollard, 2005). Notably, however, in the presence of mCA7, individual filaments form bundles of up to six filaments.
Based on the above results, we tested whether CA7‐mediated actin bundling stabilizes actin filaments. This was done by exposing transfected cells to latrunculin B, which sequesters free actin monomers and can thus be used as a robust method to quantify the rate of actin filament depolymerization (Fig 3). The experiments showed that in control cells expressing DsRed, actin filaments depolymerized within a few minutes (Fig 3A). In contrast, over 60% of the DsRed‐CA7 expressing cells had visible F‐actin structures even after 30 min of latrunculin B treatment (Fig 3B). These data demonstrate that CA7 directly binds to and bundles F‐actin, and that DsRed‐CA7 overexpression stabilizes existing actin filaments.
Figure 3. CA7 overexpressing NIH3T3 cells are resistant to latrunculin B treatment.
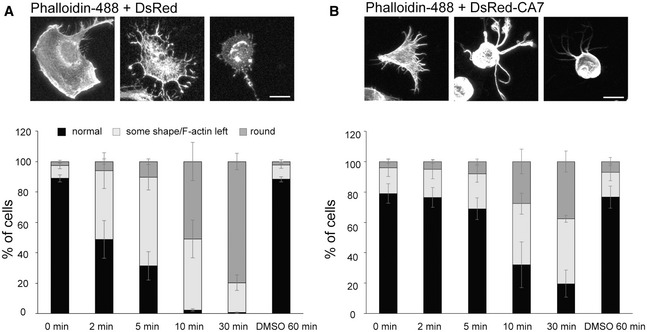
-
A, BNIH3T3 cells transfected with DsRed (A) or DsRed‐CA7 (B) were incubated in growth medium with 5 µM Latrunculin B for 0, 2, 5, 10, or 30 min, or with an equal amount of DMSO for 60 min. Analyses of experiments show that in cells transfected with DsRed‐CA7 F‐actin structures collapse more slowly (0 min: 83% “normal”; 2 min: 80%, P = 0.44; 5 min: 74%, P = 0.39; 10 min: 27%, P = 0.08; 30 min: 16%, P = 0.02; DMSO: 82%, P = 0.99; tested against 0 min with two‐way ANOVA, Dunnett’s multiple comparison test) than in the DsRed‐transfected ones (0 min: 89% “normal”; 2 min: 48%, P = 0.14; 5 min: 31%, P = 0.04; 10 min: 2%, P = 0.001; 30 min: 0.7%, P = 0.001; DMSO: 88%, P = 0.8; tested against 0 min with two‐way ANOVA, Dunnett’s multiple comparison test). For the analysis, cells were categorized to three groups as “normal”, “some shape/F‐actin left”, and “round”. The upper panel shows example images of the cells in all three categories for (A) DsRed‐ or (B) DsRed‐CA7 transfected cells (actin visualized with Phalloidin‐488).
Data information: One hundred cells per each time point from each experiment (n = 3) were counted and categorized. Scale bar 50 µm. Data are presented as mean ± SEM.
Source data are available online for this figure.
The CA7 surface motif DDERIH is crucial for actin binding
As no previously known actin‐binding domains (Paavilainen et al, 2004; Lee & Dominguez, 2010) are present in CA7, we sought to identify which CA7 sequence features contribute to actin binding. We first analyzed publicly available CA2 and CA7 protein structures (Eriksson et al, 1988; Di Fiore et al, 2010) to identify potential actin interaction surfaces. These comparisons revealed a structural motif at the CA7 protein surface that clearly differs from CA2 (Fig 4A and B). The amino acids 101–105, 113, 115, and 237–242 are distributed over the primary sequence of CA7, and form a continuous “ridge” at the protein surface (Fig 4B). Superimposing the CA7 α‐helix 6 on the twinfilin‐C/G‐actin structure (Paavilainen et al, 2007) suggested that CA7 α‐helix 6 including the amino acids 237–242 could also be a putative interaction site (Fig 4C) and that arginine 223 (R223) located to the helix might further contribute to actin binding as its positive charge is reversed in CA2 (Fig 4D). The alignment of the protein sequences of CA2 and CA7 confirmed that these amino acids are not conserved between the two isoforms (Fig 4D). In addition, a sequence comparison with the human cytosolic CA isoforms revealed that these motifs are unique to CA7 (Fig EV3).
Figure 4. Structure and sequence comparison of CA7 and CA2.
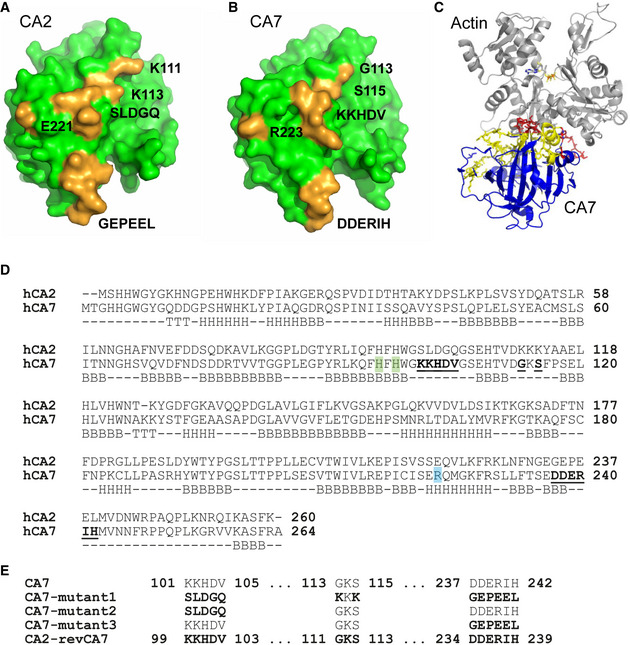
-
A, BThree‐dimensional representation of (A) CA2 and (B) CA7 structure. The amino acids 101–105 (SLDGQ in CA2 and KKHDV in CA7), 113, 115, and 237–242 (GEPEEL in CA2 and DDERIH in CA7) form a ridge at the protein surface (highlighted in yellow). R223 is located close to the ridge.
-
CSuperimposing the CA7 α‐helix‐6 (CA7 in blue, areas where mutations1–3 are located is in yellow, and the putative actin‐interacting CA7 helix in red) on the Twf‐C/G‐actin structure (gray) shows a sterically compatible structure.
-
DSequence alignment of human CA7 and CA2 protein sequences generated using the Clustal O (1.2.1) multiple sequence alignment. Residues forming a ridge at the CA7 protein surface in the CA7 3D structure are highlighted in the CA7 sequence (bold/underlined). R223 is marked in turquoise and H96 and H98 (mutated to gain a catalytically loss‐of‐function mutant) are highlighted in green. The row below sequence alignment indicates structural features of CA7 based on the UniProt database (B = beta strand, H = helix and, T = turn).
-
ESchematic representation of four mutants with a full (mutant1) or partial (mutant2 and mutant3) replacement of the amino acids encoding the ridge in CA7 by the corresponding CA2 sequence. In the reversed mutant (CA2‐revCA7), the amino acids replaced in mutant1 were introduced to CA2.
Data information: Panels (A–C) were prepared using PyMOL (PyMOL The PyMOL Molecular Graphics System, Version 1.4.1 Schrödinger, LLC.).
Figure EV3. The identified actin interaction sites are unique to CA7 among human cytosolic CAs.
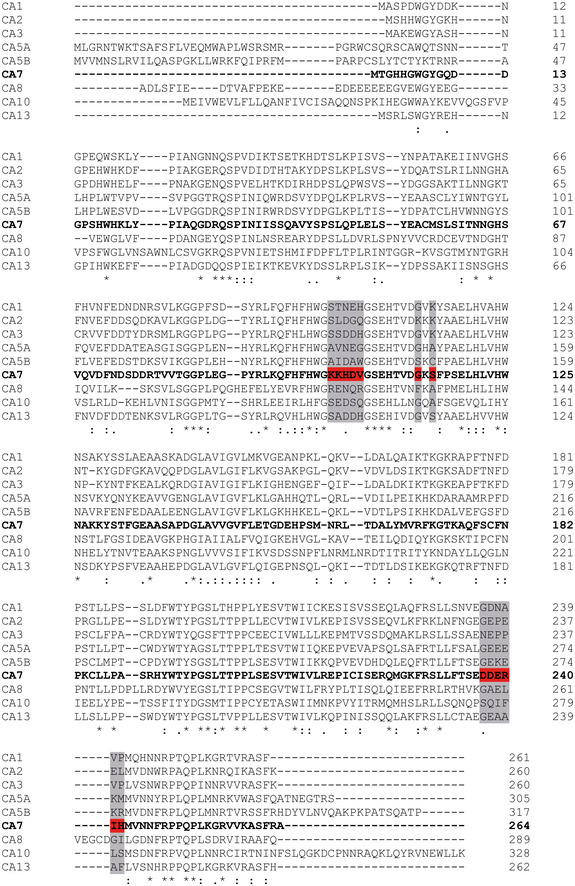
Sequence alignment of the catalytically active (CA1, CA2, CA3, CA5A, CA5B, CA7, and CA13) and catalytically inactive (CA8 and CA10) human cytosolic CA protein sequences generated using the Clustal O (1.2.1) multiple sequence alignment. The amino acids that were characterized as part of a putative actin‐binding site of CA7 (highlighted in red) are not conserved in the other cytosolic CA isoforms (highlighted in gray). An asterisk below the aligned sequences indicates fully conserved residues, a colon indicates residues with strongly similar properties (scoring > 0.5 in the Gonnet PAM 250 matrix) and a period indicates residues with weakly similar properties (scoring ≤ 0.5 in the Gonnet PAM 250 matrix).
To explore the putative CA7‐actin‐binding domains, we generated a set of CA7 mutants (EGFP‐CA7‐mutant1‐3) in which subsets of the amino acids mentioned above were replaced with the corresponding amino acid sequences of CA2 (Fig 4E). The mutants were expressed in NIH3T3 cells as EGFP‐fusion proteins and their subcellular distribution and co‐localization with F‐actin were examined after staining cells with phalloidin‐594 (Figs 5A–C and EV4). When amino acids forming the “ridge” (101–105, 113, 115, and 237–242) were replaced with the corresponding amino acids of CA2 (EGFP‐CA7‐mutant1; see Fig 4E), the strict co‐localization with F‐actin was abolished and the construct did not induce detectable changes in actin structure or cell morphology (Fig 5A). To characterize the interaction motif in more detail, we separately mutated amino acids 101–105 (KKHDV; EGFP‐CA7‐mutant2) or 237–242 (DDERIH; EGFP‐CA7‐mutant3). The localization of EGFP‐CA7‐mutant2, with only the KKHDV motif replaced by the corresponding CA2 sequence, was unchanged, when compared to EGFP‐CA7. EGFP‐CA7‐mutant2 co‐localized strongly with F‐actin (Fig 5B), and the overexpression phenotype with thick, curving cytosolic actin bundles and plasmalemmal protrusions (Fig EV4A) was similar to that seen with EGFP‐CA7. Interestingly, replacement of the DDERIH motif alone (EGFP‐CA7‐mutant3) reduced the co‐localization with F‐actin, and overexpression of the mutant did not visibly affect cellular actin structures (Fig 5C). We compared the co‐localization of the different EGFP‐tagged mutants with phalloidin‐594 by plotting the fluorescence intensities per pixel against each other (Figs 5A–C and EV4, EV5) and calculating Pearson’s correlation coefficient (Fig 5D and E). Comparison of the scatter plots from individual cells revealed that, firstly, F‐actin fluorescence was strongly correlated with that of CA7, but not with CA2. Secondly, the correlation coefficient of EGFP‐CA7‐mutant1 and EGFP‐CA7‐mutant3, but not that of EGFP‐CA7‐mutant2 was reduced when compared to CA7 (Fig 5E; note P = 0.05 for EGFP‐CA7‐mutant3). This indicates that the DDERIH motif forms an important part of the CA7‐actin interaction site.
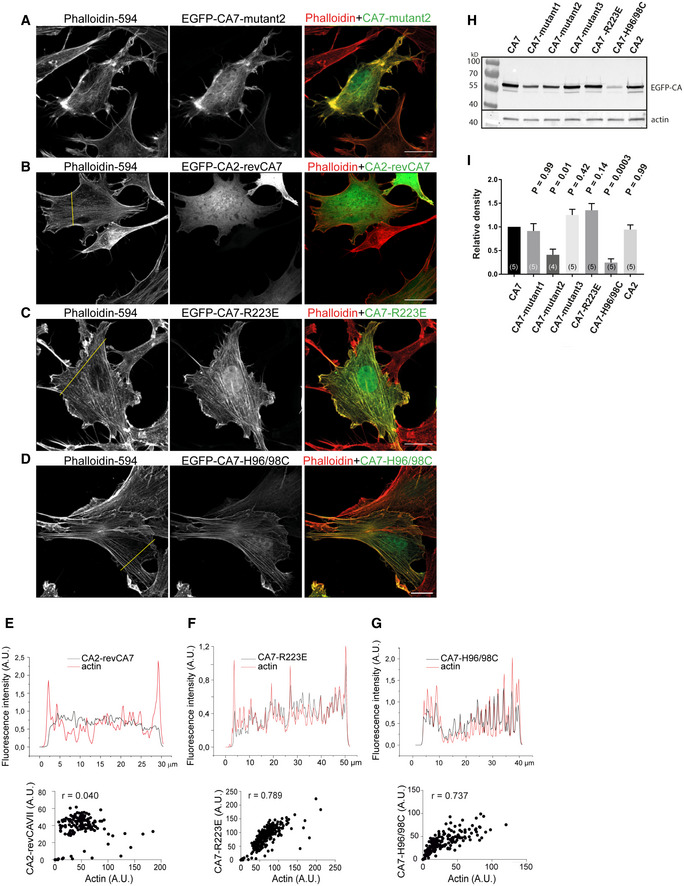
Figure EV4. Subcellular localization and expression levels of the chimeric CA fusion proteins EGFP‐CA7‐mutant2, EGFP‐CA2‐revCA7, EGFP‐CA7‐R223E, and EGFP‐CA7‐H96/98C in fibroblasts.
-
A–G(A) Transfection with EGFP‐CA7‐mutant2 modified cellular F‐actin structures in a similar manner than CA7. (B) Introduction of KKHDV and DDERIH motifs to CA2 (EGFP‐CA2‐revCA7) did not affect the diffuse cytosolic localization of the isoform 2. (C) EGFP‐CA7‐R223E and (D) the catalytically loss‐of‐function mutant EGFP‐CA7‐H96/98C co‐localized with F‐actin. F‐actin is visualized with Phalloidin‐594 in (A–D). The normalized fluorescence emission intensity profiles of (E) EGFP‐CA2‐revCA7 (F) EGFP‐CA7‐R223E, and (G) EGFP‐CA7‐H96/98C (black lines) and F‐actin (red line). The yellow line in (B–D) indicates the cross‐section of the cell from which the pixel intensities were measured. Analysis of co‐localization is shown in lower panels of (E–G). Representative single‐cell pixel intensities of EGFP and phalloidin‐594 channels were plotted against each other and the Pearson’s correlation coefficient value (r) was calculated. n = 3–4 independent transfections/construct. Scale bars in (A–D) is 20 µm.
-
HA representative Western blot showing the expression levels of EGFP‐CA fusion proteins in 10 µg of lysate collected 24 h after transfection. The EGFP‐tagged CA fusion proteins are visible at approximately 60 kDa, and the β‐actin loading control is visible at 42 kDa.
-
IQuantification of the fusion proteins expression levels in NIH3T3 cells. Expression level of EGFP‐CA7 was set at 1 for each Western blot. n = 5 independent transfections for all fusion proteins except for CA7‐mutant2, for which n = 4 (data passed Shapiro–Wilk test for normality). Transfections’ efficacy of different mutant fusion proteins was compared to CA7 WT using one‐way ANOVA with Dunnett’s multiple comparisons test. Data are given as mean + SEM.
Source data are available online for this figure.
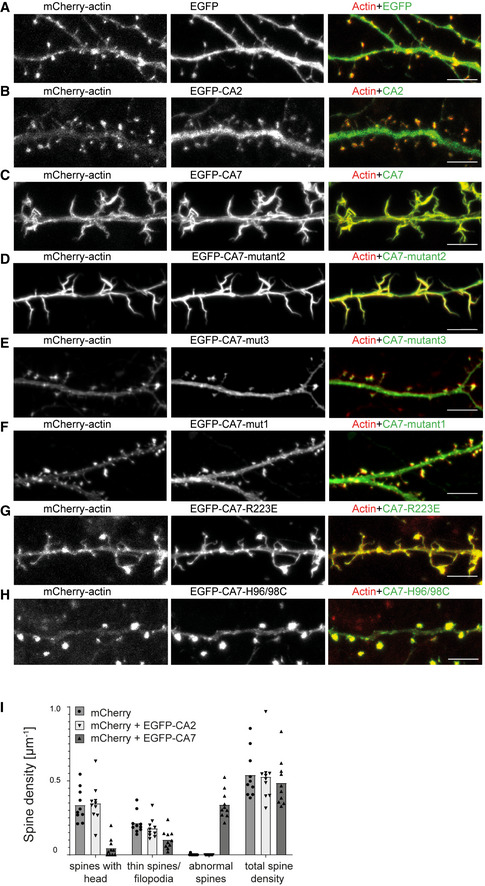
Figure EV5. Localization of WT and chimeric CA EGFP‐fusion proteins and their effect on dendritic spines in cultured hippocampal neurons.
-
A–H(A) Control experiment with neurons co‐expressing mCherry‐actin and EGFP. (B) Neuron transfected with mCherry‐actin and EGFP‐CA2. Compared to the spine‐targeted mCherry‐actin, CA2 localizes more diffusely along dendritic shafts and spines. Both (C) EGFP‐CA7 and (D) EGFP‐CA7‐mutant2 show a highly overlapping localization with mCherry‐actin and disruption of dendritic spine morphology. Spines were replaced by thick, filopodia‐like dendritic protrusions, which lack spine heads. The loss‐of‐function constructs (E) EGFP‐CA7‐mutant3 and (F) EGFP‐CA7‐mutant1 are more homogenously present in both dendrites and spines. (G) EGFP‐CA7‐R223E and (H) the catalytically inactive EGFP‐CA7‐H96/98C showed overlapping localization with mCherry‐actin. Scale bar 5 µm (A–C, G, H), 10 µm (D–F).
-
IEGFP‐CA7 expression disrupted normal spine morphology in cultured neurons. Control, only mCherry‐actin: spines with head 0.34 ± 0.04, thin spines/filopodia 0.21 ± 0.02, abnormal spines 0.00 ± 0.00, total 0.54 ± 0.05 spines/µm; n = 10 cells, 509 spines, 973 µm analyzed dendrite. EGFP‐CA2: spines with head 0.35 ± 0.04, thin spines/filopodia 0.17 ± 0.02, abnormal spines 0.00 ± 0.00, total 0.53 ± 0.06 spines/µm; n = 10 cells, 535 spines, 992 µm analyzed dendrite; EGFP‐CA7: spines with head 0.05 ± 0.02, thin spines /filopodia 0.10 ± 0.02, abnormal spines 0.34 ± 0.03, total 0.49 ± 0.05 spines/µm; n = 10 cells, 493 spines, 10,256 µm dendrite. Analyzed cells were pooled from two independent experiments. The bar diagrams show the mean.
Source data are available online for this figure.
Despite the changes seen with EGFP‐CA7‐mutant1 and EGFP‐CA7‐mutant3, an analogue mutant of CA2, containing all mutations of EGFP‐CA7‐mutant1 in a reversed manner (EGFP‐CA2‐revCA7, see Fig 4E), maintained the diffuse cytosolic localization pattern seen with CA2 (Fig EV4B and E).
When the positively charged arginine 223, which we hypothesized to play a role in CA7‐actin‐binding, was mutated to the negatively charged glutamic acid (EGFP‐CA7‐R223E), co‐localization with F‐actin was not affected (Figs 5E, and EV4C and F), but the generation of thick cytosolic actin bundles was reduced. Finally, to examine whether the catalytic activity of CA7 affects actin co‐localization, we expressed a catalytically inactive mutant, EGFP‐CA7‐H96/98C (Kiefer & Fierke, 1994), in NIH3T3 cells. This mutant retained its co‐localization with actin (Figs 5E, and EV4D and G), indicating that catalytic activity is dispensable for the interaction with actin. The different impact of the various mutants on actin cytoskeleton morphology did not seem to correlate with overall construct expression levels (Fig EV4H and I).
Together, these comparisons provide compelling evidence for structural motifs consisting of widely spread amino acids within the primary structure of CA7 and containing the DDERIH sequence as the CA7/actin‐binding site.
CA7 localizes to dendritic spines and its overexpression disrupts spine morphology
We moved on to study the subcellular localization of CA2 and CA7 in rat hippocampal neurons. The lack of a knockout (KO) controlled antibody, suitable for immunohistochemistry (IHC), hinders studies on endogenous CA7 (see Ruusuvuori et al, 2013). Hence, we examined the subcellular localization by simultaneously overexpressing DsRed‐CA7 and EGFP‐CA2 in neuronal cultures. While EGFP‐CA2 distributed homogenously along the somato‐dendritic axis, DsRed‐CA7 was prominent in the actin‐rich spines (Fig 6A). Co‐expression of mCherry‐actin and EGFP‐CA2 further confirmed that CA2, similar to EGFP alone, localizes diffusely to dendrites and spines and has no effect on the structure of spines (Fig EV5A and B). In contrast, the EGFP‐CA7 distribution overlapped with that of mCherry‐actin and, in line with the CA7‐dependent modulation of actin structures detected in fibroblasts, it had a marked effect on spine morphology: EGFP‐CA7‐overexpressing neurons had a high proportion of aberrant spines, i.e., thick, filopodia‐like dendritic protrusions with no clear spine head (Fig EV5C). The effects of EGFP‐CA2 and EGFP‐CA7 on spine density and structure in cultured neurons are summarized in Fig EV5I.
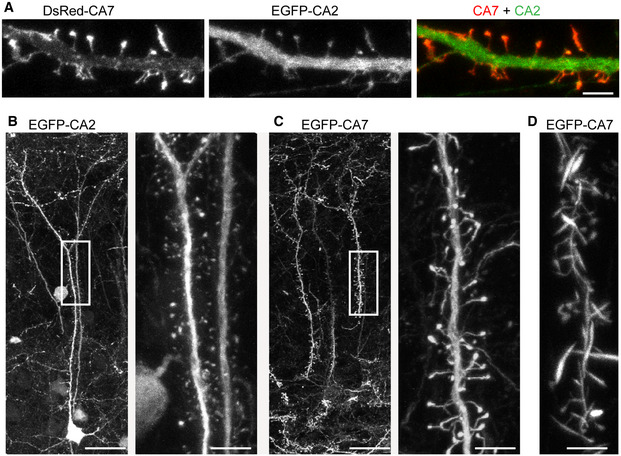
Figure 6. Localization of the overexpressed CA2 and CA7 in neurons.
-
AIsoform‐specific subcellular localization shown in cultured hippocampal neurons (DIV14) co‐expressing DsRed‐CA7 (left) and EGFP‐CA2 (middle).
-
B, CRepresentative confocal images of precocious in vivo expression of (B) EGFP‐CA2 and (C) EGFP‐CA7 in P40 mouse cortical layer 2/3 pyramidal neurons. Neurons were transfected at E14.5 with EGFP‐CA2 or EGFP‐CA7 using in utero electroporation and images were taken from fixed slices. Right panels in (B) and (C) show higher magnification of the primary apical dendrite marked with a box.
-
DThe strong expression of EGFP‐CA7 disrupted the normal spine morphology and induced the formation of thick, filopodia‐like protrusions.
Data information: n = 5 independent repeats for cultured neurons and two animals/construct in vivo. Scale bar in (A) 5 µm; (B and C): 5 µm, insets in (B, C) and panel (D): 25 µm.
Of the mutant proteins, EGFP‐CA7‐mutant2 co‐localized with mCherry‐actin and caused a similar change in spine morphology as CA7 (Fig EV5D). When the DDERIH motif was mutated individually (EGFP‐CA7‐mutant3), or together with the other amino acids forming the ridge (EGFP‐CA7‐mutant1), the subcellular distribution of the fusion proteins was homogenous along dendrites and dendritic spines, and overexpression of these constructs had no obvious effect on the spine morphology (Fig EV5E and F). EGFP‐CA7‐R223E and EGFP‐CA7‐H96/98C, both of which co‐localized with F‐actin in fibroblasts, showed strongly overlapping localization with mCherry‐actin in dendritic spines (Fig EV5G and H).
Finally, to study the localization of CA7 and CA2 in neurons in vivo, we overexpressed EGFP‐CA7 or EGFP‐CA2 in cortical layer 2/3 pyramidal neurons using in utero electroporation and examined transfected neurons in fixed slices from P40 mice. Compared to EGFP‐CA2 (Fig 6B), EGFP‐CA7 localized strongly to dendritic spines (Fig 6C). The spine phenotype of CA7‐overexpressing neurons depended on the expression level. Strong CA7 overexpression induced the formation of abnormal, filopodia‐like dendritic protrusions that lacked a clear spine head (Fig 6D). This finding is in line with the observations made in cultured neurons (see Figs 6A and EV5C).
Genetic deletion of CA7 changes cortical layer 2/3 pyramidal neuron spine density and morphology in vivo
The subcellular localization of overexpressed CA7 and its interaction with the actin cytoskeleton raised the question whether genetic deletion have an effect on dendritic spines. For this, we did electrophysiological recordings and structural analysis of neurons using CA7 KO and wild‐type (WT) mice (Ruusuvuori et al, 2013). Since spines are the major site for excitatory synaptic input, we measured miniature excitatory post‐synaptic currents (mEPSCs) from WT and CA7 KO somatosensory cortex layer 2/3 pyramidal neurons (Fig 7A). Neither mEPSC amplitude (14.93 ± 1.16 pA vs. 14.71 ± 1.10 pA, P = 0.90) nor frequency (5.37 ± 1.62 Hz vs. 6.70 ± 2.24 Hz, P = 0.63) differed between the genotypes (n = 7 WT and n = 4 CA7 KO neurons from five WT and four CA7 KO mice). Structural analysis was done from Lucifer Yellow (LY) labeled somatosensory cortex layer 2/3 pyramidal neurons from P34–P37 WT and CA7 KO mice (Fig 7B) as described earlier (Fiumelli et al, 2013). Interestingly, constitutive genetic ablation of CA7 significantly changed dendritic architecture in vivo (Fig 7C). In CA7 KO neurons (n = 28 neurons, in total 8,279 spines analyzed from four animals), spine density on the second‐order apical and basal dendritic shafts increased by 38 ± 16% and by 42 ± 16%, respectively (for apical dendrites P = 0.000002, Mann–Whitney test and for basal dendrites P = 6.8 × 10−8, Student’s t‐test with Welch correction), in comparison to the WT neurons (n = 29 neurons for apical dendrites and 30 neurons for basal dendrites, in total 8,730 spines from two animals). The change in spine density was due to a specific increase in immature‐type of spines with small spine heads. The average spine head diameter in CA7 KO neurons was 0.37 ± 0.01 μm (n = 421 spines, from 15 neurons) and in WT neurons 0.48 ± 0.01 μm (n = 467 spines, from 15 neurons). Compared to WT, the distribution was shifted significantly toward smaller spine heads in the CA7 KO neurons (W = 134,540, P < 0.001, Wilcoxon rank‐sum test) (Fig 7D).
Figure 7. Layer 2/3 cortical pyramidal neurons in CA7 KO mice have high dendritic spine density and smaller spines but mEPSC frequency and amplitude are not affected.
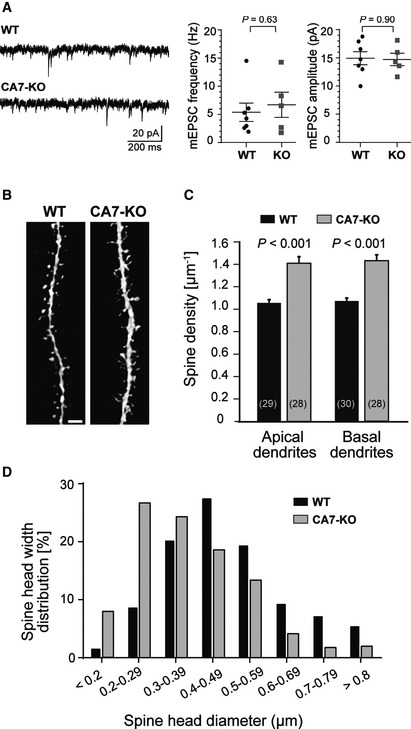
- Comparison of mEPSCs in cortical layer 2/3 pyramidal neurons from P30 to P40 WT and CA7 KO mice. Sample traces of mEPSC recordings from WT and CA7 KO neurons, low‐pass filtered at 1 kHz (left). The data are summarized in the bar diagrams (right). mEPSC frequency (P = 0.63) and amplitude (P = 0.90) were not significantly different between WT and CA7 KO and neurons (n = 7 and 5 neurons, respectively, Student’s independent samples t‐test).
- Representative confocal images of apical dendrites from Lucifer Yellow‐injected cortical layer 2/3 pyramidal neurons from WT and CA7 KO mice. The dendritic spine density and spine head size were examined in fixed slice preparations from P34 to P37 mice. Scale bar 2 µm.
- Summary of the spine density analysis done from the Lucifer Yellow‐injected neurons. Spine density was a higher in CA7 KO neurons both in apical and basal dendrites (n = 28 neurons for both) compared to WT (n = 29 neurons for apical and n = 30 for basal dendrite analysis) (P = 0.000002 for apical dendrites, analyzed with Mann–Whitney test, and P = 6.8 × 10−8 for basal dendrites, Student’s t‐test with Welch correction.) A total of 8279 spines were analyzed from four CA7 KO mice and 8730 spines from two WT control mice.
- The spine head width distribution differed significantly between the genotypes (n = 467 spines from WT and n = 421 spines from CA7 KO animals, 15 neurons analyzed from both genotypes, Wilcoxon rank‐sum test with continuity correction, W = 134,540, P < 0.001).
Data information: Data are given as mean ± SEM in (A) and as mean + SEM in (C).
Source data are available online for this figure.
Discussion
Our main finding is that the subcellular distribution of CA7 is dictated by its remarkably prominent binding to actin, in particular to F‐actin, which is a novel property with regard to all cytosolic CAs studied to date. Thus, in neurons, CA7 is expected to have a major influence on dendritic spine morphology and density as described below.
Expression of DsRed‐ and EGFP‐fusion constructs of CA7 and CA2 both in cultured fibroblasts and hippocampal neurons revealed a mutually exclusive subcellular localization. The diffuse cytoplasmic distribution of CA2 is well in line with previous results from non‐erythroid cells (Wang et al, 2002; Stridh et al, 2012; Al‐Samir et al, 2013) and fits with the idea that this ubiquitous and high‐activity isoform serves a housekeeping role in cytosolic pH buffering (see Introduction).
When Montgomery et al (1991) first characterized human CA7, they recognized several poorly conserved regions that were predicted to “be located toward the surface of the protein.” Our work shows that one of these regions, consisting of residues 232–248 encoded by exon 7, is critically involved in the CA7‐actin interaction. When we replaced amino acids 237–242 (DDERIH) with corresponding amino acids from CA2 either alone (EGFP‐CA7‐mutant3) or together with additional mutations (amino acids 101–105, 113, and 115; EGFP‐CA7‐mutant1), co‐localization of F‐actin and the mutated EGFP‐CA7 proteins decreased compared to EGFP‐CA7. Notably, replacing the corresponding sequence of CA2 by the CA7 amino acids forming this ridge (101–105, 113, 115, and 237–242) was not sufficient to induce actin binding by CA2, pointing to a complex three‐dimensional structure of the CA7 actin‐binding site.
Furthermore, while EGFP‐CA7 strongly co‐localized with F‐actin in fibroblast stress fibers, the edges of the highly dynamic lamellipodia were largely devoid of CA7. It should be noted here that in the lamellipodia of migrating fibroblasts, actin filaments are polymerized by Arp2/3 complex, resulting in strongly branched actin filament network at the leading edge. Our results thus demonstrate that CA7 preferably interacts with non‐branched actin filaments.
An important observation in this context is that the CA7‐actin interaction was enhanced at acidic pH (6.5 vs. 7.4). The pHi of the leading edge of migrating cell lamellipodia is known to be exceptionally high, primarily due to enhanced NHE1‐mediated acid‐extrusion, which in turn promotes cofilin activation and actin treadmilling (Grinstein et al, 1993; Martin et al, 2011). Thus, the alkaline microenvironment is expected to impair CA7–actin interaction and thereby serve as an additional factor governing CA7 localization as well as actin turnover.
We have made numerous, but unsuccessful attempts to create a specific CA7 antibody compatible in IHC using conventional immunization techniques. In addition, we have developed a llama VHH nanobody (cf. Paalanen et al, 2011) selective for CA7 in ELISA. Despite about a hundred of monoclonal lines of these VHH nanobodies, we have not succeeded in developing a CA7‐specific one that is suitable for IHC in brain tissue. In addition to our own CA7‐KO validated antibody that works in immunoblots (Ruusuvuori et al, 2013), we have also tested a number of commercially available antibodies, as well as those used in previous publications (Bootorabi et al, 2010; Del Giudice et al, 2013; Viikila et al, 2016). Unfortunately, these antibodies showed unspecific staining in our experimental settings when CA7‐KO mouse tissue was used as negative control. Hence, in the current study addressing the subcellular distribution of CA7, we were left with no other option than to express fluorescently tagged CA fusion proteins. We fully recognize the caveats in work conducted on fluorescently tagged proteins, especially those with DsRed, as discussed below.
The lack of CA7 in subcellular areas with branched actin is similar to the distribution of the actin‐binding protein drebrin‐A (Ludwig‐Peitsch, 2017). Drebrin‐A is known to stabilize actin filaments by binding along the actin double‐helix (Mikati et al, 2013). Interestingly, drebrin‐A localizes in the middle of the spine where the more stable actin filament pool (consisting of straight actin filaments) is located (Koganezawa et al, 2017). Because of the lack of a specific CA7 antibody, we are unable to study localization of endogenous CA7 in spines but, in light of its actin‐binding characteristics, it is possible that CA7 has a distribution within spines that is similar to drebrin‐A. This is an interesting question for future studies.
CA7 not only binds to F‐actin but it also increases actin bundling and changes actin dynamics in vitro, as visualized in our time‐lapse imaging experiments (Movies [Link], [Link], [Link], [Link], [Link], [Link]). While we cannot exclude the possibility that mCA7 enhances polymerization of individual filaments, the current TIRF imaging data suggest that the enhancement of filament/bundle elongation occurs through bundling of existing filaments/bundles together. The ability of CA7 to increase bundling by crosslinking actin filaments implies a bivalent binding mechanism. Conventional actin cross‐linkers, such as α‐actinin and fimbrin, most often work via homodimerization (Puius et al, 1998). It is important to note that we did all cell assays with DsRed‐ or EGFP‐tagged CA7 protein and sometimes these protein‐tags dimerize by themselves (Zhang et al, 2002). However, the TIRF assay, which showed clear crosslinking function for CA7, was done with CA7 purified with small, non‐dimerizing His‐tag. Thus, we are confident that the observed CA7 crosslinking stems from CA7 properties and not (only) DsRed or EGFP‐tag dimerization. In the future, it will be interesting to study whether CA7 affects actin filament turnover. The present latrunculin B treatment experiment (Fig 3) suggests that CA7 reduces the depolymerization rate of actin filaments, but direct measurement of the actin turnover rate by fluorescent recovery after photobleaching or photoactivation assays would give a more definitive answer.
Our bundling assay was done in the nominal absence of CO2/ , demonstrating that the CA7‐mediated enhancement of bundling is independent of the catalytic activity of CA7. This is well in line with our result that the enzymatically inactive CA7 H96/98C mutant maintains its co‐localization with F‐actin. Interestingly, deletion of the catalytically inactive CA‐related protein 8 has been shown to cause structural abnormalities in parallel fiber and Purkinje cell synapses (Hirasawa et al, 2007), further suggesting that enzymatic activity and morphogenetic functions of CAis are not based on shared molecular actions.
The changes detected in the actin cytoskeleton of CA7‐overexpressing cells are consistent with the biochemical assay data and show that CA7 modulates higher‐order actin structures in the cytoplasm. Fibroblasts transfected with CA7 generated numerous filopodia‐like protrusions projecting from the cell surface and had thick, sometimes curving, cytosolic stress fibers. The phenotypic characteristics closely resemble those seen with the brain‐specific actin‐bundling protein drebrin‐A in both fibroblasts (Shirao et al, 1994) and in cultured neurons (Hayashi & Shirao, 1999; Mizui et al, 2005). Dendritic spine structure, just like fibroblast morphology, varied depending on the expression level of CA7. In neurons overexpressing high levels of CA7, in cell cultures or in vivo, dendritic spines eventually lose their morphological diversity (categorized as thin, stubby, and mushroom spines according to (Bourne & Harris, 2008; Hotulainen & Hoogenraad, 2010)) and turn into thick protrusions, which lack a clear spine head.
The excess of thin spines (spine head < 0.3 µm) in CA7 KO mouse cortical neurons in vivo suggests that the lack of CA7 either facilitates their formation, or prolongs transformation/maturation of spines. Our quantification of spine type was based on the head diameter (Bourne & Harris, 2008). It is well established that spines with small heads (i) are short‐lived and dynamic structures (Holtmaat et al, 2005); (ii) are more abundant in early development; and (iii) make only occasional contacts with presynaptic terminals (Berry & Nedivi, 2017). It is thus not surprising that the increased spine number in CA7 KO cortical neurons did not alter the basic pre‐ or post‐synaptic properties, measured as the frequency and amplitude of mEPSCs, respectively. It is tempting to suggest that the overproduction of small spines, actively searching for presynaptic partners (Ziv & Smith, 1996; Zuo et al, 2005), renders CA7 KO neurons in a more juvenile‐like morphological state.
The morphology of spines in CA7 KO neurons is different from WT and in particularly stark contrast to those observed upon CA7 overexpression. In the thick, filopodia‐like protrusions present in CA7 overexpressing neurons, CA7 promotes linear bundling of actin, thereby eliminating the dynamic spine head formed by a dense network of short, cross‐linked branched actin (Hotulainen et al, 2009; Hotulainen & Hoogenraad, 2010). The time‐lapse data on fibroblasts show that, when abundantly present, CA7‐dependent bundling of actin filaments immobilizes cell movements. Notably, reorganization of the spine actin cytoskeleton is a fundamental property of excitatory synaptic plasticity (Hlushchenko et al, 2016). Direct measurements of dendritic spine pH are scarce, but Diering et al (2011) have reported an NHE5‐dependent increase in spine pH, which develops over tens of minutes after chemical long‐term potentiation induction in cultured rat hippocampal neurons. This is in line with the idea that pH alterations associated with synaptic plasticity could impair the CA7‐actin interaction, thereby allowing modulation of post‐synaptic actin structures. Compared to the actions of other pH‐sensitive actin‐binding proteins, CA7 could thus counteract gelsolin‐dependent severing of F‐actin, which takes place upon acidification (Lagarrigue et al, 2003). Notably, in the central nervous system pronounced acidification of the intracellular compartment occurs under various pathophysiological conditions such as stroke (Pavlov et al, 2013) and seizures (Siesjö et al, 1993; Ziemann et al, 2008).
The actin‐associated localization of CA7 to dendritic spines (as suggested by our overexpression data) is an important observation. This is because synaptic activity is known to evoke long‐term structural changes in spine size and morphology (Sala & Segal, 2014), as well as large transient changes in ionic concentrations within the spine (Rose et al, 1999; Brini et al, 2017). The present study shows that CA7 is optimally localized to not only separately modulate F‐actin and pH, but also to provide a link between F‐actin dynamics and activity‐dependent pH transients within spines.
Materials and Methods
Animal experiment ethics
All experiments involving animals were conducted in accordance with the European Directive 2010/63/EU and were approved by the National Animal Ethics Committee of Finland or the Local Animal Ethics Committee, University of Helsinki. Animals were housed and bred in a conventional animal house. Both mice (C57BL/6 and ICR) and rats (Wistar) were group housed (weaned littermates of same sex in groups of 2–6) under a 12:12 h light:dark schedule (lights on at 06:00 h). Altromin food pellets and tap water were available ad libitum and animals had wood bedding/nesting material and wood blocks for enrichment. The CA7 KO mice (deletion of exons 5–7 of the Car7 gene) have been backcrossed to C57BL/6 and were used at P35–P40 for the mEPSC recordings and spine analyses.
Neuronal primary cultures, fibroblast cultures, and transfections
Hippocampal neuronal cultures were prepared as described previously (Bertling et al, 2012). Briefly, the hippocampi of embryonal day 17 Wistar rat fetuses of either sex were dissected, the meninges were removed, and the cells were dissociated with 0.05% papain and mechanical trituration. The cells were plated at a density of 100,000 cells/coverslip (diameter 13 mm), coated with Poly‐l‐Lysine (0.1 mg/ml; Sigma), in neurobasal medium (Gibco) supplemented with B‐27 (Invitrogen), l‐glutamine (Invitrogen), and penicillin‐streptomycin (Lonza). Transient transfections were performed after 13 days in vitro (DIV) using Lipofectamine 2000 (Invitrogen), as described earlier (Hotulainen et al, 2009). Prior to all experiments, we confirmed that cultures formed a dense network of neurons, ensuring the availability of a proper synaptic network. The neurons were imaged after fixation with 4% paraformaldehyde (PFA). Fibroblasts (NIH3T3, Sigma; Cat. No.: 93061524) were maintained in DMEM supplemented with 10% fetal bovine serum (Hyclone), 2 mM l‐glutamine (Invitrogen), and penicillin‐streptomycin (Lonza). Cells were transfected with Superfect (Qiagen) or Turbofect (Thermo Scientific)‐transfection reagent according to manufacturer’s instructions for 24 h and either imaged live or after fixation with 4% PFA.
Plasmid constructs
pEGFP‐N1 (EGFP) and mCherry‐C1 (mCherry) plasmids were purchased from Clontech Laboratories, Inc. Human GFP‐β‐actin (Dopie et al, 2012) and mCherry‐β‐actin plasmids were gifts from Maria Vartiainen (University of Helsinki, Finland) and Martin Bähler (University of Münster, Germany), respectively. Constructs containing full‐length CA2 and CA7 (human isoform 1) coding sequences were obtained from ImaGenes (human CA2 and human CA7 including start and stop codon; OCAAo5051H1054 and OCAAo5051E0588, respectively) and GeneCopoeia (human CA2 without stop codon and mouse CA7 including start and stop). Constructs encoding CA7‐R223E and CA7‐H96/98C were generated using site‐directed mutagenesis (Phusion high fidelity PCR, ThermoFisher) and the correct sequence of PCR amplified sequences was confirmed by full‐length sequencing of both strands (DNA Sequencing and Genomics Laboratory, Institute of Biotechnology, Helsinki). More complex mutants containing multiple nucleotide exchanges were commercially synthesized (GenScript). All coding sequences were either available as Gateway entry vectors or subcloned into pDONR or pENTR vectors using the Gateway technology (LifeTechnologies). Expression constructs encoding N‐ or C‐terminal fusion proteins of CA2 or CA7 and various reporter proteins (EGFP, DsRed, mCherry) were generated using the Gateway technology and appropriate destination vectors. To allow for stable expression in cell cultures and neurons in vivo, all destination vectors contained the modified CMV/chicken β‐actin promoter (CAG) (Niwa et al, 1991). The pCAG‐EGFP plasmid was a gift from Connie Cepko (Addgene plasmid # 11150) and served as a control (Matsuda & Cepko, 2004).
Expression level quantification in NIH3T3 cells
NIH3T3 cells were seeded on 24‐well plates with Poly‐l‐Lysine‐coated coverslips at a density of 60,000 cells/well and on 6‐well plates at a density of 300,000 cells/well. The following day, the cells were transfected with the CA7/2‐EGFP constructs using Turbofect transfection reagent (Thermo Scientific) according to the manufacturer’s instructions. For immunofluorescence, the cells were fixed 24 h after transfection with 4% PFA for 30 min. For western blotting, the cells were washed once with ice‐cold phosphate‐buffered saline (PBS) and collected in 150 µl radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (cOmplete, Roche) 24 h after transfections. 10 µg of the lysates was separated by 8–16% SDS‐PAGE (Bio‐Rad) and blots were probed with a 1:2,000 dilution of mouse anti‐GFP antibody (Clontech, Cat. No.: 632381), followed by 1:5,000 anti‐mouse Starbright Blue 700 (Bio‐Rad, Cat. No.: 12004158) (Fig EV4H) and 1:5,000 anti‐actin‐rhodamine (Bio‐Rad, Cat. No.: 12004164) as a loading control. The blots were imaged with ChemiDoc MP (Bio‐Rad). Quantification was done with ImageJ, and expression levels of the constructs were shown relative to the WT EGFP‐CA7 expression level, which was set at one. One lane was excluded because of a mistake in the cell transfection: The actin band was normal but there was no EGFP band on the lane.
Actin filament staining with phalloidin in fibroblasts
Fibroblasts were permeabilized with 0.1% Triton X‐100 in PBS. Filamentous actin was visualized with Phalloidin‐iFluor 488 (Abcam, cat:no ab176753), Alexa fluor 488‐, or Alexa fluor 594 –phalloidin (Invitrogen, Molecular probes cat:no A12379 and A12381), incubation for 30 min.
Actin filament visualization in cultured neurons
Neuronal cultures were co‐transfected with a mCherry‐actin and EGFP‐CA2/CA7 constructs on DIV13 and fixed with 4% PFA (30 min) 24 h after transfection.
Imaging
Fixed NIH3T3 fibroblasts were imaged under epifluorescence illumination using an upright Axio Imager.M2 microscope equipped with a 63× 1.4NA objective and with an Apotome 2 structured illumination slider (all from Zeiss). Images were acquired with a black and white CMOS camera (Hamamatsu ORCA Flash 4.0 V2) and ZEN 2 software (Zeiss). For quantification of co‐localization of the EGFP‐tagged proteins with 594‐Phalloidin in fibroblasts, we analyzed the pixel intensities along a virtual line across the cell, excluding the nucleus (ImageJ, http://imagej.nih.gov/ij). The placing of the line was done using the “actin channel”, and the experimenter was blinded to the transfections. We plotted the pixel intensities of both channels against each other and calculated the Pearson’s correlation coefficient (GraphPad Prism 7). Each individual experiment consists of 1–4 transfected wells. The person who analyzed the co‐localization and calculated Pearson’s coefficient values was blind to the transfection.
The dendritic spines of cultured hippocampal neurons were imaged using a Zeiss LSM 710 upright confocal microscope (63× 1.3NA objective) or an Axio Imager.M2 microscope (63× 1.4NA objective, Apotome 2 structured illumination slider). Image files were processed with ZEN 2012 (Carl Zeiss Microscopy GmbH), ImageJ 1.46r, and Photoshop CS4 (Adobe).
Spine analysis
For analysis of spine density and morphology in fixed rat cell cultures, serial image files corresponding to z‐stacks of 20–30 optical sections per dendritic segment were taken. Inclusion criteria: all healthy, pyramidal neuron looking cells which expressed moderate amount of mCherry and co‐expressed EGFP‐construct were imaged and included to analysis. We did not exclude any outliers.
NeuronStudio, a software package specifically designed for spine detection and analysis (Rodriguez et al, 2008), was used to analyze spine density. The detailed analysis of spine classes was performed as described in (Bertling et al, 2012). Classification of spines was done by using rules defined by (Rodriguez et al, 2008) and verified manually. For the plot, spines were divided to three groups: spines with head (Neurostudio: mushroom and stubby), filopodia/thin spines (Neurostudio: thin), and spines with abnormal morphology (Neurostudio: other), latter including all spines with branches, long thick protrusions or otherwise morphology not classified in any common spine classes. The mean value (+SEM) of separate images is shown.
Latrunculin B assay
For the Latrunculin B assay, fibroblasts were transfected with DsRed or DsRed‐CA7 24 h prior to treatment with 5 µM Latrunculin B in DMSO. Cells were fixed after 0, 2, 5, 10, and 30 min of Latrunculin B treatment and stained with phalloidin‐488. Control cells were treated with DMSO for 60 min and stained with phalloidin‐488. We made three independent replicates of such experiments. One hundred transfected cells from each time point from each experiment were categorized either as “normal”, “some shape/F‐actin left”, or “round” (example cells for the three categories are depicted in Fig 3A and B, upper panels). Inclusion criteria: all cells expressing DsRed (control) or DsRed‐CA7. We did not exclude any outliers.
CA7 biochemistry: mCA7 production, pull‐down assay, and bundling (in vitro TIRF) assay
Recombinant mCA7 was produced by Icosagen (Tartu, Estonia) in CHOEBNALT85 cells. The protein was produced with a secretion signal peptide and a C‐terminal His‐tag for purification. CA7 was purified from the cell culture supernatant by Ni‐affinity chromatography. The concentration of the purified protein was 1.5 mg/ml, and the purity was estimated to be > 90%. Actin co‐sedimentation assay was carried out in 20 mM Hepes pH 7.4/6.5, in the presence of 0.2 mM DTT. mCA7 was stored in PBS but the buffer was changed to Hepes (pH 7.4/6.5) before the experiment. Lyophilized powder of CA2 (Sigma) was reconstituted in MilliQ and diluted in Hepes‐buffer pH 7.4/6.5 to 33 μM. ZnCl2 (1 μM) was added to CA7/CA2 1 h before incubation with actin. β/γ‐G‐actin (0, 1, 5, 10 and 15 μM) was pre‐polymerized in Hepes‐buffer pH 7.4/6.5 by addition of 1/10 of 10×‐initiation mixture (1 M KCl, 10 mM EGTA, 50 mM MgCl2, 2.5 mM ATP, and 20 mM Hepes pH 7.4/6.5) for 30 min at room temperature. CA7 or CA2 (1 μM) was added to polymerized actin, gently mixed, and incubated for another 30 min at room temperature. Actin filaments were sedimented by centrifugation for 30 min at 20°C in a Beckman Optima MAX Ultracentrifuge at 353,160 × g in a TLA100 rotor. Equal proportions of supernatants and pellets were run on 13.5% SDS‐polyacrylamide gels, which were stained with Coomassie Blue. The intensities of β/γ‐actin and CA7/CA2 bands were quantified with QuantityOne program (Bio‐Rad), analyzed, and plotted as CA7/CA2 bound to actin (μM, CA7/CA2 in pellet) against actin. The mCA7‐actin co‐sedimentation assay was repeated three times for each pH value, and averaged curves were presented (± SEM). All experiments were included in the analysis.
In vitro TIRF imaging was performed as previously described (Suarez et al, 2011) but the muscle actin was substituted with non‐muscle actin (Cytoskeleton), prepared according to the manufacturer's instructions, and non‐muscle Rhodamine actin (Cytoskeleton) was used for labeling the filaments. A mixture of 0.5 µM unlabeled and 0.05 µM Rhodamine labeled non‐muscle actin was polymerized in the presence of 1.12 or 0.11 µM mCA7 or with an identical volume of PBS as a control in the nominal absence of CO2/ (three independent repeats for both treatments). Images were captured with Nikon Eclipse Ti‐E N‐STORM microscope, equipped with Andor iXon + 885 EMCCD camera, and 100× Apo TIRF oil objective (NA 1.49), a 150 mW 561 nm laser line was used for visualization of Rhodamine actin. Actin filament polymerization was followed (images every 10 s) until the imaging field was full with filaments (typically around 30–40 min). Bundling was quantified by measuring the mean relative fluorescence intensity of a cross‐section for an individual filament (n = 10–11 filaments per repeat, ImageJ) and actin fiber length (n = 3–4 per repeat) at three time points (0, 5, and 23 min). The analyzed filaments were chosen randomly and all experiments were included in the analysis. The data were analyzed using a general mixed model with time as a within‐unit factor and the presence of CA7 as a between‐unit factor, experiment repeats were included as a covariate. Kymographs were created from the same videos with Fiji. The highest density of filaments/bundles was visually estimated from the end of the video (time) focusing on an area in the middle. We used “Multi Kymograph” function with line width 1 µm. For Fig EV2E, filament/bundle length was measured manually with line function with Fiji. If filament was not straight, more than one line was used (max 4) and measurements were summed together.
In utero electroporation and slice preparation
The rat in utero protocol described by Fiumelli et al (2013) was used with some modifications. Timed‐pregnant ICR mice with E14.5 embryos were given Temgesic (0.05–0.1 mg/kg, s.c.) and anesthetized with isoflurane (4.2% induction, 2.5% during surgery). All embryos were injected with 1.25 µl plasmid DNA solution (3–4 µg/µl EGFP‐CA7 or EGFP‐CA2 construct in 0.9% NaCl and 0.1% Fast Green). Electroporation was done with 5 mm diameter circular electrodes (Sonidel Limited) with five pulses (40–45 V, 50 ms duration at 100 ms intervals), delivered with a square‐wave generator (CUY21vivo SC, Sonidel Limited). Detection of EGFP‐CA2 and EGFP‐CA7 was done on 50 µm coronal cryosections from fixed brains (P40 mice of both sex were transcardially perfused under terminal anesthesia with 4% PFA, over‐night postfixation in 4% PFA) with a Zeiss LSM 710 upright confocal microscope.
Cortical layer 2/3 spine analysis and mEPSC recordings
Post hoc iontophoretic injection of Lucifer Yellow
Male WT and CA7 KO mice were terminally anesthetized at P34–P37 by an intraperitoneal injection of pentobarbital (100 mg/kg) and perfused transcardially first with saline, followed by 4% PFA and 0.125% glutaraldehyde solution (pH 7.4). Brains were removed and postfixed for 2 h in 4% PFA. Coronal sections of 200 µm thickness were cut on a vibratome in ice‐cold PBS (pH 7.4). Coronal sections were pre‐stained for 10 min with methylene blue, which allows the visualization of neuronal somata, mounted into an injection chamber, and placed on the fixed stage of a Zeiss microscope equipped with a micromanipulator. Layer 2/3 pyramidal neurons were loaded iontophoretically with a 0.4% Lucifer yellow solution (Sigma‐Aldrich, St. Louis, MO) using sharp micropipettes and a negative current of 70 nA until the dendrites were fluorescing brightly. For each animal, neurons were labeled from 2 to 3 slices.
Immunohistochemistry
The Lucifer Yellow‐injected slices were preincubated for 1 h in a PBS solution containing sucrose (5%), bovine serum albumin (2%), Triton X‐100 (1%), and sodium azide (0.1%), followed by 48 h at room temperature with the anti‐LY antibody (rabbit IgG, Cat. No. A5750, Invitrogen, Carlsbad, CA; 1:4,000 dilution). Slices were then rinsed in PBS solution and incubated for an additional 24 h with Alexa conjugated secondary antibodies (Alexa Fluor® 568, Invitrogen; 1:1,000). After mounting, the slices were coverslipped using Immumount (Thermo Scientific, Pittsburgh, PA) and stored at +4°C until analysis.
Confocal laser scanning microscopy and image analysis
Second‐order dendrites were imaged for spine analysis using LSM700 confocal microscope and 63× oil‐immersion objective. Spine analysis was performed on acquired stacks of images using a homemade plug‐in written for OsiriX software (Pixmeo, Geneva, Switzerland). This plug‐in allows precise spine quantification, individual tagging, and measurement in 3D by scrolling through the z‐axis. We defined spines as structures emerging from the dendrites that were longer than 0.4 µm and for which we could distinguish an enlargement at the tip (spine head). Spines head diameters were measured at their largest width in xy‐axis on the z‐image corresponding to the central axis of the spine head. The difference in spine head width distribution between WT and CA7 KO mice was analyzed using a Wilcoxon rank‐sum test with continuity correction. Note that for illustration purposes, images presented in figures are maximum intensity projections of z‐stacks with volume rendering, further processed with a Gaussian blur filter.
Inclusion criteria: Layer 2/3 pyramidal neurons in the somatosensory cortex with Lucifer Yellow (LY)‐filled dendritic tree. Exclusion criteria: LY‐filled neurons outside layer 2/3 and neurons where LY‐injections failed (leading to partially filled dendritic arbor). We did not exclude any outliers. All data were counted and the person who analyzed spine density/spine head size was blind to the genotype. Data were first tested for normality using D’Agostino and Pearson normality test. Since the data set for WT apical dendrites did not pass the test, we used non‐parametric Mann–Whitney test for testing the difference in apical dendrites. In basal dendrites, both data sets passed the normality test, and Student’s t‐test was used.
mEPSC recordings and analysis
Male CA7 KO and WT mice (P30–P40) were anesthetized with halothane and decapitated. Acute coronal brain slices (400 µm) were cut using Campden vibratome (Campden Instruments 7000 SMZ‐2) in ice‐cold (< 4°C) cutting solution containing (in mM) 87 NaCl, 2.5 KCl, 0.5 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 7 MgCl2 and 50 sucrose, equilibrated with 95% O2 and 95% CO2 to pH 7.4. Before starting experiments, slices were let to recover for 1 h at +34°C in standard solution containing (mM): 124 NaCl, 3 KCl, 2 CaCl2, 25 NaHCO3, 1.1 NaH2PO4, 1.3 MgSO4, and 10 D‐glucose, (300 ± 5 mOsm). Whole‐cell voltage‐clamp recordings from layer 2/3 somatosensory cortex pyramidal neurons were obtained with a HEKA EPC‐10 amplifier with 20 kHz sampling rate and 4 kHz low‐pass filter. Slices were perfused with standard solution (see above, perfusion 3.5 ml/min), and all measurements were done in the presence of 100 µM picrotoxin and 0.5 µM TTX. Temperature in the recording chamber was 32 ± 1°C. The cells were clamped to −65 mV. Borosilicate patch pipette resistances ranged from 3 to 5.5 MΩ when filled with pipette solution containing (mM) 140 CsMs, 2 MgCl2, and 10 HEPES (liquid junction potential of 13 mV was taken into account). Only cells with a resting membrane potential below −55 mV and stable holding current were included in the analysis. Series resistance was compensated and recordings with unstable series resistance (change > 30%) were excluded from the analysis. The person who did and analyzed the mEPSC experiments was blind to the genotype. Events were manually detected with minianalysis software (Synaptosoft) after 1,000 Hz low‐pass filtering and with threshold set to 4× RMS noise.
Author contributions
PB, KK, PH, and ER conceptualized the study. PB, MB, and VP designed constructs. EB, PB, PS, MS, PU, VOP, LV, KK, PH, and ER designed the experiments. EB, PB, PS, EK, GG, MAV, MS, IS, PU, and VOP collected data. EB, PS, EK, GG, MS, IS, PU, MB, VOP, and LV analyzed data. PB, KK, PS, VOP, and PH reviewed and edited the paper. ER wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We wish to acknowledge Alexander Alafuzoff for his indispensable help with statistical analysis; Merle Kampura for her expertise in performing in utero electroporation and handling cell cultures; and Mikko Liljeström and the Biomedicum Imaging Unit (BIU), Helsinki, for kind help and technical assistance with the in vitro TIRF imaging experiments. We acknowledge the use of the INSTRUCT‐HiLIFE protein crystallization core facility, University of Helsinki, member of Biocenter Finland and Instruct‐FI for assistance in the SEC‐MALLS experiments. This work was supported by grants from the Academy of Finland (to K.K. SA 294375, SA 319237 and SA 276576, to P.H. SA 266351, and to V.O.P SA 289737 and SA 314672), the Jane and Aatos Erkko Foundation (to K.K.), the University of Helsinki (to P.H. the three‐year research grant), and the Sigrid Juselius Foundation (to V.O.P).
EMBO Reports (2021) 22: e50145.
Data availability
The data supporting the findings of this study are included in the supplemental material. Additional data are available from the corresponding author on request. No data are deposited in databases.
References
- Al‐Samir S, Papadopoulos S, Scheibe RJ, Meissner JD, Cartron JP, Sly WS, Alper SL, Gros G, Endeward V (2013) Activity and distribution of intracellular carbonic anhydrase II and their effects on the transport activity of anion exchanger AE1/SLC4A1. J Physiol 591: 4963–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96: 43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertling E, Ludwig A, Koskinen M, Hotulainen P (2012) Methods for three‐dimensional analysis of dendritic spine dynamics. Methods Enzymol 506: 391–406 [DOI] [PubMed] [Google Scholar]
- Bootorabi F, Janis J, Smith E, Waheed A, Kukkurainen S, Hytonen V, Valjakka J, Supuran CT, Vullo D, Sly WS et al (2010) Analysis of a shortened form of human carbonic anhydrase VII expressed in vitro compared to the full‐length enzyme. Biochimie 92: 1072–1080 [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31: 47–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Carafoli E, Cali T (2017) The plasma membrane calcium pumps: focus on the role in (neuro)pathology. Biochem Biophys Res Commun 483: 1116–1124 [DOI] [PubMed] [Google Scholar]
- Cammer W (1984) Carbonic anhydrase in oligodendrocytes and myelin in the central nervous system. Ann N Y Acad Sci 429: 494–497 [DOI] [PubMed] [Google Scholar]
- Cammer W, Tansey FA (1988) Carbonic anhydrase immunostaining in astrocytes in the rat cerebral cortex. J Neurochem 50: 319–322 [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ (1996) The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16: 2983–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Giudice R, Monti DM, Truppo E, Arciello A, Supuran CT, De Simone G, Monti SM (2013) Human carbonic anhydrase VII protects cells from oxidative damage. Biol Chem 394: 1343–1348 [DOI] [PubMed] [Google Scholar]
- Di Fiore A, Truppo E, Supuran CT, Alterio V, Dathan N, Bootorabi F, Parkkila S, Monti SM, De SG (2010) Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg Med Chem Lett 20: 5023–5026 [DOI] [PubMed] [Google Scholar]
- Diering GH, Mills F, Bamji SX, Numata M (2011) Regulation of dendritic spine growth through activity‐dependent recruitment of the brain‐enriched Na+/H+ exchanger NHE5. Mol Biol Cell 22: 2246–2257 21551074 [Google Scholar]
- Dopie J, Skarp K‒P, Kaisa Rajakyl__ E, Tanhuanpää K, Vartiainen MK (2012) Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA 109: E544–E552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R (1999) Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA 96: 13438–13443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson AE, Jones TA, Liljas A (1988) Refined structure of human carbonic anhydrase II at 2.0 Å resolution. Proteins 4: 274–282 [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem BJ, Dayer AG, Kaila K, Martin JL, Vutskits L (2013) An ion transport‐independent role for the cation‐chloride cotransporter KCC2 in dendritic spinogenesis in vivo . Cereb Cortex 23: 378–388 [DOI] [PubMed] [Google Scholar]
- Geers C, Gros G (2000) Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev 80: 681–715 [DOI] [PubMed] [Google Scholar]
- Grinstein S, Woodside M, Waddell TK, Downey GP, Orlowski J, Pouyssegur J, Wong DCP, Foskett JK (1993) Focal localization of the Nhe‐1 isoform of the Na+/H+ antiport – assessment of effects on intracellular Ph. EMBO J 12: 5209–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmi P, Parkkila S, Honkaniemi J (2006) Expression of carbonic anhydrases II, IV, VII, VIII and XII in rat brain after kainic acid induced status epilepticus. Neurochem Int 48: 24–30 [DOI] [PubMed] [Google Scholar]
- Halpain S (2000) Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci 23: 141–146 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Shirao T (1999) Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci 19: 3918–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN (2011) Discussing the morphology of actin filaments in lamellipodia. Trends Cell Biol 21: 2–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa M, Xu X, Trask RB, Maddatu TP, Johnson BA, Naggert JK, Nishina PM, Ikeda A (2007) Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol Cell Neurosci 35: 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlushchenko I, Koskinen M, Hotulainen P (2016) Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton 73: 435–441 [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K (2005) Transient and persistent dendritic spines in the neocortex in vivo . Neuron 45: 279–291 [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, Tanhuanpaa K, Faix J, Rivera C, Lappalainen P (2009) Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol 185: 323–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC (2010) Actin in dendritic spines: connecting dynamics to function. J. Cell Biol 189: 619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson PA, Dziegielewska KM, Liddelow SA, Saunders NR (2008) The blood‐CSF barrier explained: when development is not immaturity. BioEssays 30: 237–248 [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J (1987) Postsynaptic fall in intracellular pH induced by GABA‐activated bicarbonate conductance. Nature 330: 163–165 [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA (1993) The role of bicarbonate in GABAA receptor‐mediated IPSPs of rat neocortical neurones. J Physiol 464: 273–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J (1997) Long‐lasting GABA‐mediated depolarization evoked by high‐ frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network‐driven, bicarbonate‐dependent K+ transient. J Neurosci 17: 7662–7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida E, Palminiello S, Golabek AA, Walus M, Wierzba‐Bobrowicz T, Rabe A, Albertini G, Wisniewski KE (2006) Carbonic anhydrase II in the developing and adult human brain. J Neuropathol Exp Neurol 65: 664–674 [DOI] [PubMed] [Google Scholar]
- Kiefer LL, Fierke CA (1994) Functional characterization of human carbonic anhydrase II variants with altered zinc binding sites. Biochemistry 33: 15233–15240 [DOI] [PubMed] [Google Scholar]
- Koganezawa N, Hanamura K, Sekino Y, Shirao T (2017) The role of drebrin in dendritic spines. Mol Cell Neurosci 84: 85–92 [DOI] [PubMed] [Google Scholar]
- Kuhn JR, Pollard TD (2005) Real‐time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys J 88: 1387–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarrigue E, Ternent D, Maciver SK, Fattoum A, Benyamin Y, Roustan C (2003) The activation of gelsolin by low pH: the calcium latch is sensitive to calcium but not pH. Eur J Biochem 270: 4105–4112 [DOI] [PubMed] [Google Scholar]
- Lee SH, Dominguez R (2010) Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 29: 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig‐Peitsch WK (2017) Juxtanuclear Drebrin‐Enriched zone. Drebrin: From Structure and Function to Physiological and Pathological Roles 1006: 329–336 [DOI] [PubMed] [Google Scholar]
- Maren TH (1967) Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 47: 595–781 [DOI] [PubMed] [Google Scholar]
- Martin C, Pedersen SF, Schwab A, Stock C (2011) Intracellular pH gradients in migrating cells. Am J Physiol Cell Physiol 300: C490–C495 [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro . Proc Natl Acad Sci USA 101: 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mboge MY, Mahon BP, McKenna R, Frost SC (2018) Carbonic anhydrases: role in pH control and cancer. Metabolites 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikati MA, Grintsevich EE, Reisler E (2013) Drebrin‐induced stabilization of actin filaments. J Biol Chem 288: 19926–19938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizui T, Takahashi H, Sekino Y, Shirao T (2005) Overexpression of drebrin A in immature neurons induces the accumulation of F‐actin and PSD‐95 into dendritic filopodia, and the formation of large abnormal protrusions. Mol Cell Neurosci 30: 149–157 [DOI] [PubMed] [Google Scholar]
- Montgomery JC, Venta PJ, Eddy RL, Fukushima YS, Shows TB, Tashian RE (1991) Characterization of the human gene for a newly discovered carbonic anhydrase, CA VII, and its localization to chromosome 16. Genomics 11: 835–848 [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 108: 193–199 [DOI] [PubMed] [Google Scholar]
- Nogradi A, Mihaly A (1990) Light microscopic histochemistry of the postnatal‐development and localization of carbonic anhydrase activity in glial and neuronal cell‐types of the rat central nervous system. Histochemistry 94: 441–447 [DOI] [PubMed] [Google Scholar]
- Paalanen MM, Ekokoski E, El KM, Tuominen RK, Verrips CT, Boonstra J, Blanchetot C (2011) The development of activating and inhibiting camelid VHH domains against human protein kinase C epsilon. Eur J Pharm Sci 42: 332–339 [DOI] [PubMed] [Google Scholar]
- Paavilainen VO, Bertling E, Falck S, Lappalainen P (2004) Regulation of cytoskeletal dynamics by actin‐monomer‐binding proteins. Trends Cell Biol 14: 386–394 [DOI] [PubMed] [Google Scholar]
- Paavilainen VO, Hellman M, Helfer E, Bovellan M, Annila A, Carlier MF, Permi P, Lappalainen P (2007) Structural basis and evolutionary origin of actin filament capping by twinfilin. Proc Natl Acad Sci USA 104: 3113–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack M, Voipio J, Kaila K (1993) Intracellular carbonic anhydrase activity and its role in GABA‐ induced acidosis in isolated rat hippocampal pyramidal neurones. Acta Physiol Scand 148: 229–231 [DOI] [PubMed] [Google Scholar]
- Pavlov I, Kaila K, Kullmann DM, Miles R (2013) Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J Physiol 591: 765–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puius YA, Mahoney NM, Almo SC (1998) The modular structure of actin‐regulatory proteins. Curr Opin Cell Biol 10: 23–34 [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008) Automated three‐dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3: e1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Kovalchuk Y, Eilers J, Konnerth A (1999) Two‐photon Na+ imaging in spines and fine dendrites of central neurons. Pflugers Arch 439: 201–207 [DOI] [PubMed] [Google Scholar]
- Ruusuvuori E, Li H, Huttu K, Palva JM, Smirnov S, Rivera C, Kaila K, Voipio J (2004) Carbonic anhydrase isoform VII acts as a molecular switch in the development of synchronous gamma‐frequency firing of hippocampal CA1 pyramidal cells. J Neurosci 24: 2699–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruusuvuori E, Huebner AK, Kirilkin I, Yukin A, Blaesse P, Helmy MM, Kang HJ, Muayed M, Hennings JC, Voipio J et al (2013) Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J 32: 2275–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Segal M (2014) Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94: 141–188 [DOI] [PubMed] [Google Scholar]
- Shirao T, Hayashi K, Ishikawa R, Isa K, Asada H, Ikeda K, Uyemura K (1994) Formation of thick, curving bundles of actin by drebrin A expressed in fibroblasts. Exp Cell Res 215: 145–153 [DOI] [PubMed] [Google Scholar]
- Siesjö BK, Katsura K, Mellergård P, Ekholm A, Lundgren J, Smith ML (1993) Acidosis‐related brain damage. Prog Brain Res 96: 23–48 [PubMed] [Google Scholar]
- Stridh MH, Alt MD, Wittmann S, Heidtmann H, Aggarwal M, Riederer B, Seidler U, Wennemuth G, McKenna R, Deitmer JW et al (2012) Lactate flux in astrocytes is enhanced by a non‐catalytic action of carbonic anhydrase II. J. Physiol 590: 2333–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C, Roland J, Boujemaa‐Paterski R, Kang H, McCullough BR, Reymann AC, Guerin C, Martiel JL, De la Cruz EM, Blanchoin L (2011) Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr Biol 21: 862–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viikila P, Kivela AJ, Mustonen H, Koskensalo S, Waheed A, Sly WS, Pastorek J, Pastorekova S, Parkkila S, Haglund C (2016) Carbonic anhydrase enzymes II, VII, IX and XII in colorectal carcinomas. World J Gastroentero 22: 8168–8177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voipio J (1998) Diffusion and buffering aspects of H+, HCO3‐, and CO2 movements in brain tissue, In pH and brain function, Kaila K, Ransom BR (eds.), pp 45–66. Hoboken, NJ: Wiley‐Liss; [Google Scholar]
- Wang WG, Bradley SR, Richerson GB (2002) Quantification of the response of rat medullary raphe neurones to independent changes in pH(O) and P‐CO2. J Physiol 540: 951–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001) Morphological changes in dendritic spines associated with long‐term synaptic plasticity. Annu Rev Neurosci 24: 1071–1089 [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY (2002) Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol 3: 906–918 [DOI] [PubMed] [Google Scholar]
- Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA, Welsh MJ, Wemmie JA (2008) Seizure termination by acidosis depends on ASIC1a. Nat Neurosci 11: 816–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ (1996) Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17: 91–102 [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB (2005) Development of long‐term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46: 181–189 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
The data supporting the findings of this study are included in the supplemental material. Additional data are available from the corresponding author on request. No data are deposited in databases.