

Abstract
Fusarium wilt (FW) disease of cotton, caused by the fungus Fusarium oxysporum f. sp. vasinfectum (Fov), causes severe losses in cotton production worldwide. Though significant advancements have been made in development of FW‐resistant Upland cotton (Gossypium hirsutum) in resistance screening programs, the precise resistance genes and the corresponding molecular mechanisms for resistance to Fov remain unclear. Herein it is reported that Fov7, a gene unlike canonical plant disease‐resistance (R) genes, putatively encoding a GLUTAMATE RECEPTOR‐LIKE (GLR) protein, confers resistance to Fov race 7 in Upland cotton. A single nucleotide polymorphism (SNP) (C/A) in GhGLR4.8, resulting in an amino acid change (L/I), is associated with Fov resistance. A PCR‐based DNA marker (GhGLR4.8SNP(A/C)) is developed and shown to cosegregate with the Fov resistance. CRISPR/Cas9‐mediated knockout of Fov7 results in cotton lines extremely susceptible to Fov race 7 with a loss of the ability to induce calcium influx in response to total secreted proteins (SEPs) of Fov. Furthermore, coinfiltration of SEPs with GhGLR4.8A results in a hypersensitive response. This first report of a GLR‐encoding gene that functions as an R gene provides a new insight into plant–pathogen interactions and a new handle to develop cotton cultivars with resistance to Fov race 7.
Keywords: disease‐resistant genes, Fusarium wilt, GLUTAMATE RECEPTOR‐LIKE genes, Gossypium hirsutum
Fusarium oxysporum f. sp. vasinfectum (Fov) is a widely distributed fungal pathogen seriously threatening cotton production. Herein, a GLUTAMATE RECEPTOR‐LIKE gene (GhGLR4.8) is identified as an atypical disease‐resistance gene specifying resistance to Fov race 7 through a genome‐wide association study in upland cotton, which provides a new insight into plant–pathogen interactions and facilitates the development of elite Fov‐resistant cultivars.
1. Introduction
Fusarium oxysporum is a soilborne fungal pathogen that infects more than 100 plant species and causes tremendous economic losses in numerous important crop plants, including cotton, tomato, banana, and melons.[ 1 ] Fusarium oxysporum f. sp. vasinfectum (Fov) causes Fusarium wilt (FW) of cotton, seriously threatening cotton production worldwide.[ 2 , 3 , 4 ] Fov is classified into eight races (1–8) based on pathogenicity to different cotton species and other crops.[ 4 ] In China, three races (races 3, 7, and 8) of Fov have been reported, and race 7 is identified as the most widely dispread race and possesses the highest virulence.[ 5 ] Chlamydospores produced by Fov can survive in the soil for long period in the absence of host plants, which makes Fov a difficult pathogen to be managed and eliminated in fields.[ 3 ]
Phenotypic and genetic analyses have revealed that one or two major R genes with complete to incomplete dominance, together with a few minor genes, contribute to Fov resistance. Three major resistance genes (FWR, Fov1, Fov4) and some quantitative trait loci (QTL) conferring resistance to Fov in different cotton species have been identified.[ 2 , 3 , 6 , 7 , 8 , 9 ] The resistance of Upland cotton to Fov race 7 is governed by a single dominant gene, FWR, and was mapped to chromosome 17 (D03).[ 2 ] Fov1 was mapped to chromosome 16 and confers resistance to Fov race 1 in G. barbadense Pima‐S7 and Pima 3‐79[ 7 ] while Fov4 was mapped to chromosome 14 and confers resistance to Fov race 4 in G. barbadense Pima‐S6.[ 8 ] GaGSTF9, a gene encoding the Phi class of glutathione S‐transferases and located on chromosome A11, may be a target for Fov resistance in G. arboretum.[ 10 ] Furthermore, GaGSTF9 was also identified as a candidate gene for Verticillium wilt resistance.[ 11 ] However, none of the Fov‐resistance genes has been characterized in Upland cotton, which accounts for more than 90% of cotton production worldwide. Whether the genes conferring resistance to Fov race 7 in G. hirsutum and in G. arboretum are the same is unclear.
2. Results
2.1. Genome‐Wide Association Study (GWAS) Analyses for Fusarium Wilt Resistance in Upland Cotton
Through direct screening from susceptible cultivars in disease nurseries, highly FW‐resistant germplasm has been identified, and has been widely deployed for breeding FW‐resistant Upland cotton.[ 9 ] To assess the favorable genetic variation during artificial selection over the past decades and finely map Fov‐resistance genes, a re‐sequenced population of 290 diverse Upland cotton accessions, collected from China, was employed to screen for Fov resistance. Among the population, 222 accessions were genotyped previously.[ 12 ] The resistance of the cotton population was screened for resistance in a disease nursery and the disease index (DI) to FW was investigated. The DI of the population ranged from 0 to 81.6, with 122 accessions susceptible to FW and 168 accessions resistant to FW (Figure S1 and Table S1, Supporting Information). In total, 2 719 708 high‐quality SNPs were identified, and population structure analysis was performed by using these SNPs. The result shows that the values of Evanno's ΔK present an obvious spike at K = 2 (Figure S2a, Supporting Information), which suggests that the population can be divided into two subpopulations (Figure S2b, Supporting Information). Principal component analysis (PCA) and neighbor‐joining analysis were also performed to further assess the genetic diversity of our new association panel and showed consistent results (Figure S2c,d, Supporting Information).
Candidate gene association analysis was performed to evaluate the association between the homologous genes of GaGSTF9 in G. hirsutum and Fov resistance. No SNPs in an upstream region of GhGSTF9 were associated with Fov resistance in G. hirsutum, differing from the result of GaGSTF9 (Figure S3, Supporting Information),[ 10 ] which suggested there may be a different mechanism for Fov resistance between G. hirsutum and G. arboretum.
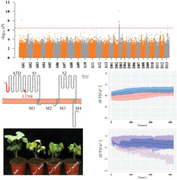
We performed GWAS with the Upland cotton population for Fov resistance using a mixed linear model approach.[ 13 ] The result revealed that a single region on chromosome D03 contains 15 SNPs that are significantly associated with Fov resistance, meeting a threshold for Bonferroni correction (P < 1/2 719 708, −log10 (P) > 6.43) (Figure 1a). The most significant SNP D03_2176763 with a −log10 (P) value of 10.106, accounted for 17.54% of the phenotypic variance (Table S2, Supporting Information). The Fov resistance in 194 accessions of this and previous population[12] had been identified through several previous studies and is indicated as HR, R, T, and S (Figure S4a and Table S3, Supporting Information).[ 14 ] We performed a case‐control association mapping of the 194 accessions, with 127 HR and R accessions as case and 67 T and S accessions as control. A continuous peak was observed on chromosome D03, and D03_2176763 exceeded the significant threshold, consistent with the GWAS result performed in 290 accessions (Figure S4b, Supporting Information). Both assays showed that FW resistance to race 7 in Upland cotton was conferred by a single major locus on chromosome 17 (D03), consistent with a previous study.[ 2 ] We therefore designated the locus for the resistance of Upland cotton to Fov race 7 as Fov7.
Figure 1.
GWAS analysis and identification of natural variation in GhGLR4.8 associated with Fusarium wilt resistance in cotton. a) Manhattan plot for the Fusarium wilt disease index. The Red solid line represents the Bonferroni‐adjusted significance threshold (−log10 (P) = 6.43). The most significant SNP (D03_2176763) is marked by the arrowhead. b) Regional Manhattan plot (from 1.97 to 2.37 Mb) for FW resistance on chromosome D03. The annotated genes are indicated by green boxes. c) Predicted structure of Gh_D03G0209. Red dots indicate signal peptide sequence. Four transmembrane domains are indicated as M1–M4. Two segments of LBD are indicated as S1 and S2. d) Gene structure display and DNA polymorphisms in the exon of GhGLR4.8. Blue‐shared regions indicate the most significant SNP. The numerical value indicates the number of different GhGLR4.8 haplotypes in 290 accessions. e) Comparison of the disease index (DI) between these haplotypes in the GWAS population. In box plots, center line indicates median, box limits denote upper and lower quartiles, and points indicate outliers. P‐value is calculated using two‐tailed Student's t‐test. f) Detection of amino acid substitution by four nonsynonymous SNPs in GhGLR4.8 in FW‐resistant (R1–R7) and FW‐susceptible (S1–S6) cotton cultivars through Sanger sequencing. S1: Emian 11, S2: Esha 28, S3: Ejing 92, S4: Xuzhou 142, S5: Xuzhou 209, S6: Xinluzao 4. R1: Yinshan 4, R2: Zhongmiansuo 12, R3: Jinmian 28, R4: Yumian 19, R5: Xinluzao 31, R6: Xinluzao 36, R7: Xinluzhong 14.
2.2. GhGLR4.8 Is Significantly Associated with Fusarium Wilt Resistance
FW‐resistance gene in Upland cotton was previously mapped on D03 with an interval genetic distance of 10.8 cM from JESPR304 and 5.7 cM from CIR035.[ 2 ] Sequence mapping placed the most significant SNP D03_2176763 at a distance of 540 158 and 70 570 bp from JESPR304 and CIR035, respectively (Figure S4c, Supporting Information). Linkage disequilibrium (LD) calculation showed that the LD decay distance for chromosome D03 in this 290 association panel is about 200 kb (Figure S5, Supporting Information), so we estimated a candidate region from 1.97 to 2.37 Mb (200 kb on either side of SNP D03_2176763). A total of 23 putative protein‐encoding genes were found in this 400 kb genomic region (Figure 1b; Table S4, Supporting Information). Although the candidate region on D03 contained 836 polymorphisms, only 17 polymorphisms were significantly associated with FW resistance (Figure 1b). Among them, 2 SNPs resulted in amino acid changes. D03_2125319, which is located in gene Gh_D03G0206, resulted in an amino acid change from phenylalanine to serine. Gh_D03G0206 is an ortholog of the Arabidopsis CYP83B1 encoding an oxime‐metabolizing enzyme in the glucosinolate biosynthetic pathway, a class of secondary metabolites found mainly in Brassicaceae.[ 15 ] SNP D03_2176763 is located in the second exon of Gh_D03G0209, which was annotated as a homologue of the Arabidopsis GLUTAMATE RECEPTOR‐LIKE 3.3 (GLR3.3)[ 16 ] playing important roles in the plant innate immune response.[ 17 , 18 ] To better understand GLR family members in cotton and possible function of Gh_D03G0209, genome‐wide identification and phylogenetic analysis of cotton GLRs were performed. A total of 36 genes harboring the essential domains of ionotropic glutamate receptors (iGluRs) were regarded as bona fide GLRs through Pfam prediction according to the newly assembled G. hirsutum accession Texas Marker‐1 (TM‐1) (Table S5, Supporting Information).[ 19 ] Phylogenetic analysis revealed that the 36 GLRs in cotton with 20 A. thaliana GLRs and three GLRs (OsGLR3.5,[ 20 ] SlGLR1.1, and SlGLR1.2[ 21 ]) could be divided into four clades (GLR1, GLR2, GLR3, and GLR4) (Figure S6, Supporting Information), Gh_D03G0209 corresponding to Ghir_D03G002390.1 was grouped into clade GLR4 with OsGLR3.5, SlGLR1.1 and SlGLR1.2 (Figure S6, Supporting Information). According to the arrangement of GLR subfamily on chromosome, Gh_D03G0209 was designated as GhGLR4.8. The predicted structure of the protein encoded by GhGLR4.8 is similar to iGluRs and AtGLRs,[ 22 , 23 ] with an extracellular amino‐terminal domain (ATD), two extracellular putative ligand‐banding domains (LBD) (S1 and S2), four transmembrane helices (M1–M4, 1 of which ‐M2‐ is not fully transmembrane), a cytoplasmic tail (carboxylterminal domain; CTD) and a signal peptide at the N terminus (Figure 1c). Sequence alignment of GhGLR4.8, GluA2 and AtGLRs revealed that the SNP D03_2176763 (C/A) lies in the predicted ATD domain causing an amino acid change from leucine (reference) to isoleucine (alternate) (Figure 1c; Figure S7, Supporting Information). Among the 290 members of the association panel, the disease index of accessions carrying the allele ‘AA’ was significantly lower than that of accessions carrying the allele ‘CC’ (Figure S8b, Supporting Information).
Further analysis showed that there are four nonsynonymous SNPs, including the SNP D03_2176763, within the coding region of Gh_D03G0209 (Figure 1d). Based on these four nonsynonymous SNPs, there are 12 haplotypes for Gh_D03G0209., i.e., haplotypes A‐E with the allele ‘AA’ of D03_2176763 and haplotypes F‐L with the allele ‘CC’ of D03_2176763 (Figure 1d). Varieties carrying haplotypes A‐E exhibited significantly lower disease index than haplotypes F‐L (Figure 1e). These results suggest that the two haplotypes based on the lead SNP D03_2176763 are associated with FW‐resistant and FW‐susceptible phenotypes in Upland cotton. Therefore, the gene Gh_D03G0209 (GhGLR4.8) is the most likely candidate gene for Fov7.
We then selected 6 highly susceptible and 7 highly resistant Upland cotton cultivars to sequence GhGLR4.8. The results revealed that all resistant varieties carry ‘A’ alleles (GhGLR4.8A) and all susceptible varieties carry ‘C’ alleles (GhGLR4.8C) at the position 2176763 of chromosome D03 (Figure 1f). The other three variations in GhGLR4.8 were not linked with Fov resistance (Figure S8a,c,d, Supporting Information) and did not differ consistently between resistant and susceptible cultivars (Figure 1f). Surprisingly, we found that no cotton in the wild group (wild cottons) was of the GhGLR4.8A genotype and a very low percentage of GhGLR4.8A genotype was identified in cottons from the ABI group (cottons from America, Brazil and India) (Figure S9, Supporting Information). On the contrary, the proportion of GhGLR4.8A genotype increased to 43% in cultivars from China (Figure S9, Supporting Information), suggesting evolution of GhGLR4.8 during cotton improvement. These results suggest that the SNP D03_2176763 is responsible for the variation of FW resistance in Upland cotton and GhGLR4.8A may be the causal resistance gene for Fov7.
2.3. GhGLR4.8A Confers Resistance to Fusarium Wilt in Upland Cotton
To investigate whether the expression levels of GhGLR4.8 are associated with FW resistance in the natural population, 9 lines each of FW‐resistant and FW‐susceptible varieties were selected to determine the expression levels of GhGLR4.8 by quantitative RT‐PCR (qRT‐PCR). No significant difference (Fold change < 2) was found in transcript level of GhGLR4.8 between susceptible and resistant varieties with or without Fov inoculation (Figure S10, Supporting Information), suggesting that GhGLR4.8‐mediated resistance to Fov is independent of its expression level. Within the FW resistance locus, there are a total of 23 genes (Table S4, Supporting Information). To determine the possible functions of these genes in FW resistance, virus‐induced gene silencing (VIGS) constructs were completed for all these 23 genes excluding Gh_D03G0224, the expression level of which was extremely low so we failed to amplify it. We assigned the rest 22 genes excluding Gh_D03G0209 to 3 groups with seven genes in each group. VIGS constructs for each gene in the individual group were mixed equally to generate multigene‐silenced cotton plants named TRV:groupI, TRV:groupII, and TRV:groupIII, respectively, according to our previous study.[ 24 ] The results of qRT‐PCR showed that the expression levels of these genes were successfully knocked down in seedlings 14 days after Agrobacterium infiltration (Figure S11b,c,d, Supporting Information). All three TRV:group cotton lines showed high resistance to Fov similar to the empty‐vector‐carrying cotton lines (TRV:00) (Figure S11a, Supporting Information). Meanwhile, GhGLR4.8 was knocked down in two highly resistant Upland cotton cultivars (YZ1 and Xinluzao 46) and one susceptible Upland cotton cultivar (Xinluzao 7) (Figure S12b, Supporting Information). The results showed that, compared with the TRV:00 plants, knockdown of GhGLR4.8A in YZ1 and Xinluzao 46 made the plants severely susceptible to Fov race 7, with severe leaf wilting and brown coloration in the vascular tissue and high disease severity (Figure S12a,c,d, Supporting Information). Furthermore, fungal biomass analysis showed that the amount of fungal DNA in TRV:GhGLR4.8A plants was significantly higher than that in TRV:00 plants (Figure S12e, Supporting Information). No significant difference in disease symptoms and fungal content was observed when GhGLR4.8C was knocked down in FW‐susceptible cotton cultivar Xinluzao 7 (Figure S12, Supporting Information). We also found that there was no significant effect on Verticillium wilt resistance when GhGLR4.8A was knocked down (Figure S13, Supporting Information). Taken together, these results suggested that GhGLR4.8A is the causal resistance gene for Fov7 specifying resistance to Fov race 7 in Upland cotton.
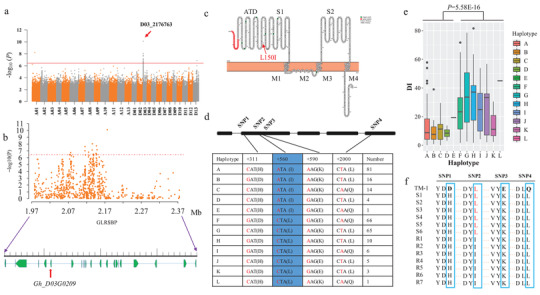
To further validate the role of Fov7 in resistance to Fov, we used CRISPR/Cas9 technology to knock out the Fov7 gene in Jin668 (J668), a highly resistant cultivar. A 20‐nt sequence in the Fov7 gene was chosen as the target site for Cas9 cleavage (Figure 2a) and generated multiple putative transgenic lines in the J668 background. The type of mutations were identified by Hi‐TOM.[ 25 ] Three lines (Fov7_KO#5, Fov7_KO#19, and Fov7_KO#20) were further verified through Sanger sequencing. The results showed seven‐base and single‐base deletions in Fov7 in Fov7_KO#5 and Fov7_KO#20 lines, respectively, while a two‐base insertion was observed in Fov7_KO#19 line, leading to a frameshift mutation (Figure 2a). After inoculation with Fov race 7, all three Fov7_KO lines exhibited extremely severe wilt symptoms, with obvious brown coloration of the vascular tissue, a high disease index and high levels of fungal DNA. In contrast, wild type plants were highly resistant to Fov, almost no brown coloration was observed and no fungal DNA was detected (Figure 2b–e). Additionally, Fov hyphae recovered from infected Fov7_KO lines grew well in culture, while no obvious Fov hyphae were recovered from infected wild type plants (Figure 2f). These results indicate that knocking out of Fov7 resulted in a loss of resistance to Fov (Figure 2).
Figure 2.
CRISPR/Cas9‐mediated knockout of Fov7 strongly suppresses resistance to Fov in Upland cotton. a) Identification of mutation type in knockout lines by PCR‐based sequencing. Three representative transgenic lines were generated in the J668 genetic background. The designations of d7, i2, and d1 denote a 7 bp deletion, a 2 bp insertion and a 1 bp deletion, respectively. b) Disease symptoms of J668 plants and three knockout transgenic lines at 20 days after inoculation with Fov. c) Vascular bundle coloration in longitudinal sections of inoculated J668 and transgenic stem. d) Disease index statistics of J668 and transgenic plants at 3 weeks after Fov inoculation. e) Relative content of Fov DNA in inoculated stem of J668 and transgenic plants. f) Fungal recovery assay of J668 and three knockout transgenic plants. Short sections cut from inoculated plants were incubated on potato dextrose agar (PDA) medium and the color of Fov mycelium is purple‐red. Data in d) and e) are presented as mean ± SD from three biological replicates.
2.4. Cosegregation between GhGLR4.8SNP(A/C) Marker and Fusarium Wilt Resistance
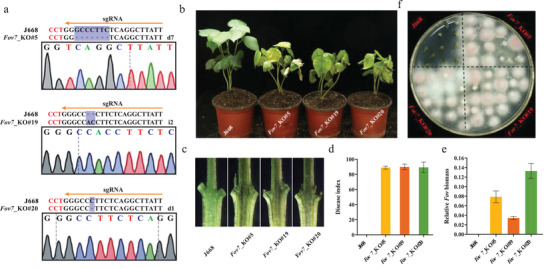
Based on the observation that resistant varieties carry the allele GhGLR4.8A and susceptible varieties carry the allele GhGLR4.8C, we designed a pair of primers with the polymorphism located at 3’ end of the forward primer. A gradient annealing temperature PCR was employed to find the optimal annealing temperature which can distinguish the DNA polymorphisms in GhGLR4.8 between resistant and susceptible varieties. Gel electrophoresis indicated that an obvious product with a size of 464 bp could be successfully amplified for GhGLR4.8A in four resistant varieties, whereas no clear product was detected for GhGLR4.8C in four susceptible varieties at the optimal annealing temperature (61–63.7 °C) (Figure 3a).
Figure 3.
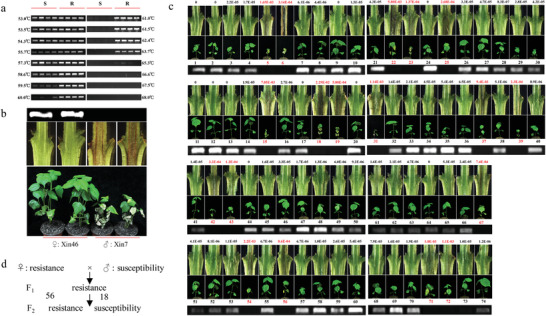
Cosegregation between the GhGLR4.8SNP(A/C)marker and FW resistance. a) Development of a PCR‐based DNA marker based on the nucleotide variation in GhGLR4.8 between resistant (R) and susceptible (S) cotton. Four susceptible cotton cultivars (Ejing 1, Jimian 8, Xinluzao 8, Emian11) and four resistant cotton cultivars (Yinshan 4, Zhongmiansuo 12, Jinmian 28, Yumian 11) were selected to explore suitable annealing temperature. Under annealing temperature of 61–63.7 °C, GhGLR4.8A and GhGLR4.8C genotype were distinguished based on the presence of a diagnostic band. b) Phenotype and genotype of Xinluzao 46 (Xin46) and Xinluzao 7 (Xin7). Xin46 served as female parent line and Xin7 serve as male line. c) Phenotype and genotype of F2 population derived from Xin46 × Xin7 cross. The resistance of F2 plants to Fov was reflected by disease symptom, vascular bundle coloration and the relative content of fungal DNA (indicated by values above the dissected stem). The numbers in the middle of seedlings and PCR band indicate individual F2 plants. The numbers and values of susceptible F2 individuals are marked in red. Resistance/susceptibility phenotype corresponded to the presence/absence of PCR bands. d) Calculation of the segregation ratio of resistant plants to susceptible plants (56R:18S, χ 2 = 0.037).
To further verify GhGLR4.8A is responsible for Fov resistance in cotton, an F2 segregation population was generated from a cross between the highly resistant cultivar Xinluzao 46 and the susceptible cultivar Xinluzao 7. First, the biparental lines were genotyped using the GhGLR4.8SNP(A/C) marker and phenotyped after inoculation with Fov. Xinluzao 7 was genotyped as GhGLR4.8C, and Xinluzao 46 was genotyped as GhGLR4.8A (Figure 3b). Then the F2 population comprising 74 individual plants were genotyped using the GhGLR4.8SNP(A/C) marker and the resistance to Fov was evaluated through pathogenicity tests. Among the 74 individual plants tested, 56 lines were genotyped as GhGLR4.8A and 18 lines were genotyped as GhGLR4.8C (Figure 3c). All the individuals genotyped as GhGLR4.8A were robust, exhibiting almost no brown coloration in the vascular tissues and an extremely low content of Fov in the stem after inoculation. While all the individuals genotyped as GhGLR4.8C exhibited severe leaf wilting, brown coloration in vascular tissues and a high content of Fov in the stem (Figure 3c). Based on the molecular identification and inoculation response, the segregation ratio of resistant plants to susceptible plants fit a 3:1 ratio (56R:18S, χ 2 = 0.037) (Figure 3d). A repeated experiment was conducted and similar results were obtained (Figure S14, Supporting Information). These results strongly implicate GhGLR4.8A as the causal resistance gene for Fov7, conferring resistance to Fov race 7 in Upland cotton.
2.5. GhGLR4.8A Triggers Immune Response and Induces Calcium Influx in Response to Fov
To examine whether Fov7 as a FW‐resistance gene could trigger plant immune response in responses to Fov, we performed Agrobacterium tumefaciens infiltration to transiently express GhGLR4.8A and GhGLR4.8C in Nicotiana benthamiana. Total secreted proteins (SEPs) of Fov were isolated and infiltrated in N. benthamiana 48 h after Agrobacterium infiltration. The results showed that coinfiltration of SEPs with GhGLR4.8A, but not with GhGLR4.8C, resulted in a hypersensitive response, which indicates that Fov could be recognized by GhGLR4.8A to trigger the plant immune response (Figure 4a).
Figure 4.
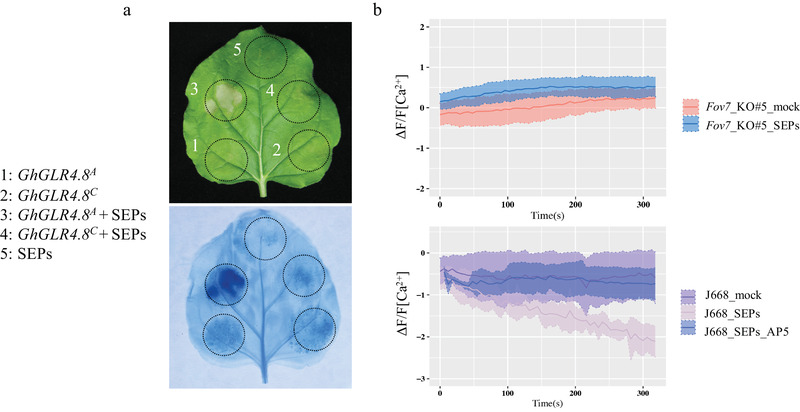
Total secreted proteins of Fov activate Ca2+ influx and trigger a hypersensitive response. a) Analysis of hypersensitive response triggered by coinfiltration of GhGLR4.8 and total Fov secreted proteins (SEPs) in Nicotiana benthamiana. GhGLR4.8A and GhGLR4.8C were expressed by Agrobacterium‐mediated transient transformation (Agro‐infiltration). For coinfiltration, tobacco leaves were infiltrated with SEPs 48 h after Agro‐infiltration. Cell death triggered by coinfiltration was visualized by trypan blue staining 60 h after Agro‐infiltration. Representative photographs are shown. b) Measurements of Ca2+ influx triggered by SEPs in cotton root meristem of Fov7 knockout and J668 seedlings by the scanning ion‐selective electrode method. AP5, an iGluRs antagonist. Error bars, mean ± SD, n = 4.
The initiation of innate immunity responses upon specific microbial epitopes, like flg22, elf18, chitin, and also wound signaling recognition, involve an apoplastic Ca2+ influx via GLR channels.[ 26 , 27 ] To verify the function of GhGLR4.8 in the regulation of Ca2+ influx following application of the SEPs of Fov, we performed scanning ion‐selective electrode assays to measure the calcium ion flux of cotton roots treated with SEPs. The results showed that the SEPs of Fov induced an increase in averaged Ca2+ influx in J668 carrying GhGLR4.8A. The iGluRs antagonist (2R)‐amino‐5‐phosphonopentanoate (AP5) significantly suppressed the SEPs‐induced response. The increase of [Ca2+]cyt influx observed in the GhGLR4.8A roots induced by SEPs was completely absent in the roots of Fov7‐knockout plants (Figure 4b).
2.6. Knockout of GhGLR4.8 Impairs Cotton Cell Wall Fortification in Response to Fov
To identify the signaling pathways regulated by GhGLR4.8 in response to pathogen, a dual RNA sequencing (RNA‐seq) was carried out to analyze the transcriptome profile of both plant and pathogen genes in Fov‐infected hypocotyls of J668 and Fov7_KO#5 at 5 days after inoculation (dpi) and 10 dpi. The trimmed reads from samples of Fov‐infected Fov7_KO#5 hypocotyls and in vitro‐grown Fov were mapped to the genome of Fov race 4.[ 28 ] It was found that 0.08–0.68% of the total reads derived from Fov at 5–10 dpi, yielding uniquely aligned read counts from 20799 to 236807 (Table S6, Supporting Information). The varied fungal mRNA reads over the time course revealed hyphal proliferation within host xylem vessels. Of all the differentially expressed fungal genes (DEFGs), 1297 and 2759 host‐induced genes were identified at 5 dpi and 10 dpi, respectively (Figure S15a,b; Table S7, Supporting Information). Gene ontology (GO) enrichment analysis showed that the categories of carbohydrate metabolic process was highly enriched among host‐induced genes, most of which (70/93) were glycoside hydrolase (Figure S15c and Table S7, Supporting Information), one of the families of cell wall‐degrading enzymes (CWDEs).
Of all the differentially expressed plant genes (DEPGs) in hypocotyls of Fov7_KO#5 compared with J668, 161 and 2336 genes were upregulated, while 1298 and 3313 genes were downregulated at 5 dpi and 10 dpi, respectively (Figure 5a,b; Table S8, Supporting Information). More DEPGs were also identified in Fov7_KO#5 than that in J668 at 10 dpi versus 5 dpi (Figure S16a,b and Table S9, Supporting Information), highlighting a stronger response in Fov7_KO#5 to Fov versus J668. Consistent with the increased secretion of glycoside hydrolases during Fov colonization, host genes involved in cell wall fortification were highly enriched among downregulated genes, revealing that knockout of GhGLR4.8A impaired the process of cell wall fortification in response to Fov (Figure 5c; Table S8, Supporting Information). Surprisingly, genes related to plant defense were highly enriched among upregulated genes (Figure 5d; Table S8, Supporting Information). Moreover, enhanced plant defense‐related responses and reduced cell wall fortifications were observed in Fov7_KO#5 at 10 dpi versus 5 dpi (Figure S16c and Table S9, Supporting Information), while weakened plant defense‐related responses were observed in J668 at 10 dpi versus 5 dpi (Figure S16d and Table S9, Supporting Information). These results suggests that loss‐of‐function of GhGLR4.8A impairs cell wall fortification during cotton‐Fov interaction and defense‐related response is enhanced by more colonization of Fov in Fov7_KO#5 xylem vessels.
Figure 5.
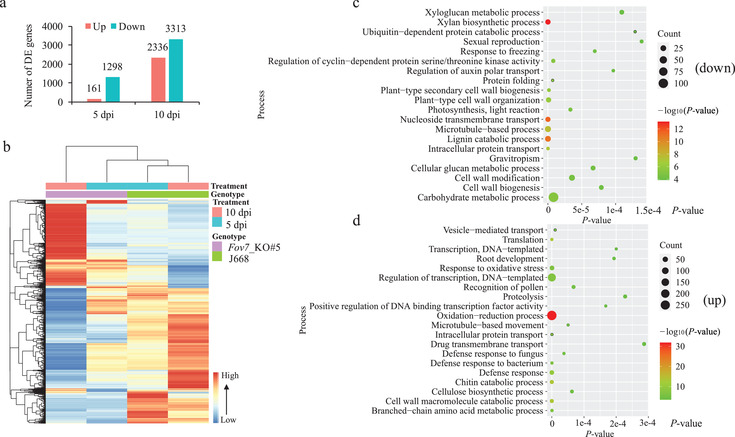
Transcriptome profiles of genes in hypocotyls of J668 and Fov7_KO#5 at various time points after infection with Fov. a) Number of differentially expressed plant genes (DEPGs) (P < 0.05, |log2(FC)| > 2) in hypocotyls of Fov‐infected Fov7_KO#5 versus J668 at different time points after inoculation. Fold change is calculated by inoculated Fov7_KO#5/J668. dpi, days post inoculation. b) Heat map of DEPGs in hypocotyls of Fov7_KO#5 versus J668 at different time points after inoculation. c,d) Gene ontology (GO) enrichment analysis of all c) down‐regulated or all d) up‐regulated genes. TOP 20 significantly enriched biological process GO terms are show. Three biological replicates were included for each treatment.
3. Discussion
Employment of disease‐resistance genes are considered as one of the most efficient strategies to control plant disease.[ 29 ] Significant achievement has been made in breeding for FW resistance through traditional phenotype evaluation under field conditions. However, the evolution of new and highly virulent Fov races have emerged as one of major threats to cotton production.[ 9 ] The identification and characterization of disease resistance genes is indispensable for both understanding resistance mechanism and efficient crop genetic improvement.
Herein, we identified the FW‐resistance gene, Fov7, conferring resistance against Fov race 7 in Upland cotton and developed a PCR‐based DNA markers associated with Fov resistance. Fov7 is located on chromosome 17 (D03), consistent with previous molecular mapping with F2 populations[ 2 , 30 ] but different from the chromosomal location observed in G. arboretum.[ 10 ] Different location of QTLs for Fov resistance among interspecific and intraspecific populations suggested a different genetic basis of Fov resistance in G. hirsutum and G. barbadense.[ 30 ] Our results showed that Fov7 varies greatly in different species, especially in G. barbadense (Figure S17, Supporting Information). Unlike G. hirsutum, most G. barbadense cultivars are susceptible to Fov race 7 in China.[ 31 ] Thus, Fov7 can be employed to G. barbadense breeding through marker assisted selection.
Fov7 encodes a GLUTAMATE RECEPTOR‐LIKE (GLR) protein, which is distinct from typical race‐specific disease resistance proteins. The tomato‐Fol pathosystem is a well‐established model system to study plant‐F. oxysporum interactions.[ 32 ] R genes in tomato conferring resistance to different Fol races have been characterized.[ 32 , 33 , 34 , 35 ] The I gene for Fol race 1 encodes a membrane anchored leucine‐rich repeat receptor‐like protein (LRR‐RLP);[ 35 ] the I‐2 gene for Fol race 2 encodes a coiled‐coil nucleotide‐binding leucine‐rich repeat (CC‐NB‐LRR) protein.[ 36 ] The I‐3 and I‐7 genes for Fol race 3 encode an S‐receptor‐like kinase (SRLK)[ 37 ] and leucine‐rich repeat receptor‐like protein (LRR‐RLP),[ 32 ] respectively. In our study, GhGLR4.8A was identified as a major R gene specifying resistance to Fov in Upland cotton, suggesting the molecular basis of cotton against Fov was distinct from tomato against Fol, which provides a new insight into plant‐F. oxysporum interactions. An increasing number of atypical resistance genes have been identified. Examples include Lr34 and Lr67 against wheat rust diseases,[ 38 , 39 ] Fhb1 and Fhb7 against Fusarium head blight in wheat,[ 40 , 41 ] and ZmFBL41 against banded leaf and sheath blight in maize.[ 42 ] Like ZmFBL41, the expression of GhGLR4.8 showed no difference between resistant and susceptible cotton cultivars (Figure S10, Supporting Information). Most of the known atypical disease resistance genes confer resistance to multiple pathogens. However, Fov7 just confers resistance to Fov but not to V. dahliae (Figure S13, Supporting Information), which causes another disastrous fungal disease that threatens cotton production worldwide.
Glutamate receptors are best known for their role as neurotransmitters mediating excitatory synaptic transmission in vertebrate brain.[ 43 ] Most recently, GLR‐3 was identified as a cold receptor in the peripheral sensory neuron ASER, suggesting the multifunction of GLRs acting as both chemical receptor and thermal receptor.[ 44 ] Plant GLRs are homologues of mammalian iGluRs[ 45 ] and have evolved many plant‐specific physiological functions, such as in sperm chemotaxis and transcriptional regulation,[ 46 ] pollen tube morphogenesis,[ 47 ] leaf‐to‐leaf wound signaling,[ 27 , 48 ] and in the plant defense response.[ 17 , 18 ] A total of 20 GLRs have been identified in A. thaliana and were grouped into three clades.[ 49 ] Moreover, a fourth clade of GLRs has been found in some monocotyledons and dicotyledons.[ 50 , 51 ] Phylogenetic analysis showed that cotton GLRs were divided into four subfamilies and GhGLR4.8 was grouped into clade GLR4 (Figure S6, Supporting Information), suggesting evolutionary expansion of GLR gene family in cotton. GLR3 subfamily was the most studied plant GLRs, AtGLR3.4 and the related AtGLR3.2 were primarily expressed in the phloem of roots,[ 52 ] AtGLR3.3 and AtGLR3.6 were localized to the phloem and xylem parenchyma in leaves, respectively,[ 18 ] and pear GLR4 was preferentially expressed in phloem.[ 51 ] GhGLR4.8A was identified as Fov7 mediating the resistance to Fov, a xylem‐colonizing fungus, suggesting that acquired resistance to Fov might be related to a different response between GhGLR4.8A and GhGLR4.8C vascular cells. The plant cell wall serves as both mechanical and defensive barrier to restrict the invading of pathogens, alterations of plant cell wall have been demonstrated to have a significant impact on disease resistance.[ 53 ] To gain access to the cell of plants, pathogens produce an array of cell wall‐degrading enzymes (CWDEs) to break down the barrier.[ 54 ] Our results showed that knockout of GhGLR4.8A impaired the process of cell wall fortification in response to Fov (Figure 5c; Table S8, Supporting Information), consistent with the highly enriched categories of carbohydrate metabolic process among host‐induced genes (Figure S15c and Table S7, Supporting Information).
Previous studies suggested GLRs act as amino acid‐gated Ca2+ channels to perceive changes in apoplastic amino acid concentrations in the regulation of plant defense responses.[ 17 , 18 ] Thus far, 12 proteinogenic amino acids, and also GSH, have been identified as GLRs agonists.[ 17 ] Moreover, Glu is indicated as a new DAMP (damage‐associated molecular pattern) activating GLR ion channels, eliciting defense signal propagation through altered [Ca2+]cyt.[ 18 ] Pull‐down experiments suggested that Fol Six4 (Avr1) can interact with glutamate decarboxylase,[ 34 ] an enzyme catalyze glutamate to gamma‐aminobutyric acid. We showed that SEPs of Fov could activate Ca2+ influx and trigger a hypersensitive response in a GLR‐dependent manner (Figure 4), suggesting that Fov7 may directly or indirectly interact with Fov effectors. There is evidence that some atypical resistance proteins could not only bind small molecules but also bind proteinaceous ligands.[ 55 ] For example, WAKs respond to changes during pathogen attack through binding with oligogalacturonides, which are regarded as a DAMP.[ 56 , 57 ] Recently, a study reported that the wheat Stb6 gene, encoding a conserved wall associated receptor kinase (WAK)‐like protein, detects the presence of its matching Avr effectors (AvrStb6) to control qualitative pathogen resistance in a gene‐for‐gene manner.[ 55 ] The direct or indirect interaction between Fov7 and the corresponding effectors of Fov race 7 requires further exploration.
4. Conclusions
In the present study, an atypical disease‐resistance gene specifying resistance to Fov race 7 was identified in Upland cotton through integrating a genome‐wide association study with gene function analyses. We found that a point mutation in the exon of GhGLR4.8 was associated with field‐evolved resistance of Upland cotton to Fusarium wilt. CRISPR/Cas9 editing of the gene validated its function and a PCR‐based DNA marker for this polymorphism was developed and subsequently shown to cosegregate with Fov resistance in an F2 segregation population. Furthermore, the resistant genotype of GhGLR4.8 (GhGLR4.8A) triggered immune response and induced calcium ion influx in response to SEPs of Fov, indicating that some unknown Fov secreted protein has the potential to activate GLR‐mediated ion channel, and RNA‐seq analyses revealed that knockout of GhGLR4.8A impaired cotton cell wall fortification in response to Fov. We demonstrate that GhGLR4.8 acts as an atypical R gene specifying resistance to Fov race 7, providing new insights into the interaction between plants and F. oxysporum, and the basis of GhGLR4.8 marker‐assisted selection to develop elite Fusarium wilt‐resistant cultivars.
5. Experimental Section
Plant Materials and Field Assays
The cotton varieties used in this study were inbred cultivars of Upland cotton and derived from China. A total of 290 cotton accessions were collected to perform GWAS for Fusarium wilt resistance, of which 222 accessions were selected from the previously described 352 resequenced accessions.[ 12 ] The population was grown in a heavily Fov‐infected fields at Kuche, Xinjiang, China. The F2 population resulted from the cross Xinluzao 46 × Xinluzao7, two cultivars widely planted in Xinjiang province. Xinluzao 46 is a highly resistant cultivar and Xinluzao7 exhibits susceptibly to Fov. The cross and subsequent self‐pollinated of F1 were both conducted in greenhouse at Huazhong Agricultural University.
The disease severity of the cotton populations was scored by the Fusarium wilt disease grade (0, 1, 2, 3, 4) and disease index (DI). Evaluation of disease grade and DI followed the technical specifications for evaluating resistance of cotton to diseases and insect pests‐part 4: Fusarium wilt (GB/T 22101.4‐2009). The resistance of cotton were classified into five levels based on the relative disease index (RDI), where RDI = 0 indicates immunity (I), 0 < RDI ≤ 5 indicates high resistance (HR), 5 < RDI ≤ 10 indicates resistance (R), 10 < RDI ≤ 20 indicates tolerance (T) and RDI > 20 indicates susceptibly (S). The resistance levels of the 352 cotton accessions were also searched from a monographs named ‘cotton varieties and genealogy in China’,[ 14 ] and found that a number of 194 cultivars have been identified through national varieties certification under fields to acquire its resistance. In total, there were 53 HR varieties, 74 R varieties, 22 T varieties, and 45 S varieties.
Variation Calling, Population Structure and Linkage Disequilibrium (LD) Analysis
Paired‐end resequencing reads were mapped to the TM‐1 genome[16] and SNP calling were performed as previously described.[12] STRUCTURE (version 2.3.4) was used to analyze the population structure of the 290 cotton accessions.[ 58 ] TASSEL (version 5.0) was used to perform PCA.[ 59 ] LD was calculated by Plink (version 1.07) software.[ 60 ]
Genome‐Wide Association Studies for Fusarium Wilt Resistance
A total of 2 719 708 high‐quality SNPs (MAF > 0.05) were used to perform GWAS for Fusarium wilt resistance measured by DI in 290 accessions. The compressed mixed linear model (P + G + Q + K) of TASSEL (version 5.0) was used to perform association analysis.[ 13 , 59 ] The threshold of significant association was set as P = 1/N (N indicates the number of SNPs). For the 194‐accessions panel, Fusarium wilt resistance was measured by resistance grade. A total of 2 143 700 high‐quality SNPs (MAF > 0.05) were used to perform a case‐control association using Plink (version 1.07) software.[ 60 ] HR and R were taken as case, T and S were taken as control.
Identification of GLR Gene Family Members in Upland Cotton
Hidden Markov model (HMM) was used to identify cotton GLR candidate genes. The protein sequence of TM‐1 genome[19] was download from cottongen (https://www.cottongen.org), and the HMM files of PF00060 (ligand‐gated ion channel) and PF00497 (solute binding protein) were download from Pfam database (http://pfam.xfam.org). Hmmer3.2 was used to search the protein sequence to identify GLR candidate genes,[ 61 ] and the genes with PF00060 and PF00497 domains were considered as bona fide GLRs.
Phylogenetic Analysis, Structure Predication, and Sequence Alignment
Construction of phylogenetic tree was performed using the neighborjoining (NJ) method using MEGA6.0 software.[ 62 ] Protein feature visualization was performed using Protter software,[ 63 ] and transmembrane predictions were performed using TMHMM. Protein sequence alignments were performed with DNAMAN software (Lynnon Biosoft). The figures of alignments were prepared with ESPript (http://espript.ibcp.fr). Amino acids sequences were obtained from The Arabidopsis Information Resource (https://www.arabidopsis.org) for ATGLRs, NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein) for GluA2 and cottongen for GhGLRs. The LBD boundaries for GhGLR4.8 were predicted using the alignment of GhGLR4.8 and AtGLRs as guidelines. The ATD boundaries for GhGLR4.8 were predicted using the alignment of GhGLR4.8 and GluA2 as guidelines.
Fov Inoculation
Fov race 7 isolate F17 was cultured in potato lactose broth (PLB, 200 g potato and 20 g lactose per 1 liter) for 3–4 days. The concentration of spores was adjusted to 1 × 107 conidia per mL for inoculation. Cotton seedlings prepared to inoculate were cultivated in Hoagland's solution in a controlled environment chamber under a 16 h light/8 h dark cycle at 25 °C. Roots of the prepared cotton plants were dipped into the spore suspension for 30 min then transplanted into sterilized soil for growing and observing the symptom of Fusarium wilt.
Qualitative and Quantitative Detection of Fov in Stem of Infected Cotton Plants
Qualitative detection of Fov was monitored by cotton dissection and a fungal recovery assay. The fungal recovery assay followed previous methods described by Fradin.[ 64 ] Briefly, first internode of seedlings was cutoff, after surface sterilized, cut them into short slices and incubated on PDA solid medium at 25 °C. Fungal colonization was evaluated by brown coloration in vascular tissue and DNA content of Fov in stem at 20 days after inoculation. Cotyledon nodes were dissected to longitudinal cross‐section and the brown coloration was observed under a stereoscopic microscope (MZFLIII; Leica, Wetzlar, Germany). Quantitative detection of Fov in cotton was conducted by qRT‐PCR. The amplification of the Fov specific gene was compared to that of the cotton UB7 to quantify fungal DNA levels according to the method of Abd‐Elsalam et al.[ 65 ]
VIGS Analysis
TRV vector construct and Agrobacterium tumefaciens infiltration were conducted as previously described but some modifications.[ 66 ] A 300–500 bp length specific coding sequence of GhGLR4.8 and other 22 genes were selected as targets for insertion into the TRV:00 vector then amplified from the cDNA of G. hirsutum Xinluzao 46. Primer pairs are listed in Table S10 in the Supporting Information. Two restriction endonucleases, BamHI and KpnI, were used to digest TRV:00 plasmid. After purification, the PCR products were fused to the linearized vectors through In‐Fusion Enzyme (Clonetch). The constructs were transformed into A. tumefaciens GV3101 by electroporation. For suppression of only one gene, TRV:GhGLR4.8 Agro‐infiltration was prepared as described by Gao et al.[ 66 ] For cosuppression of two or more genes together, TRV constructs of the other 21 genes were classified into three groups, and each group comprised 7 TRV constructs which were mixed in equal volumes for Agro‐infiltration according to Miao et al.[ 24 ] These TRV vectors were then Agro‐infiltrated into the cotyledons of ten‐day‐old seedlings of G. hirsutum cv. Xinluzao 46, G. hirsutum cv. Xinluzao 7 and G. hirsutum cv. YZ1 as described by Gao et al.[ 66 ] Infiltrated seedlings were grown at 25 °C in a controlled environment chamber with a 16 h light/8 h dark photoperiod cycle. About two weeks after infiltration, qRT‐PCR was performed to detect the expression of each of these genes in leaves to evaluate whether these genes were efficiently knocked down. The primers used for qRT‐PCR are listed in Table S10 in the Supporting Information.
CRISPR/Cas9 Gene Editing and Cotton Transformation
CRISPR/Cas9‐mediated knockout of cotton GhGLR4.8 were carried out as previously described.[ 67 ] A 20 bp specific targeting sequences for GhGLR4.8 named sgRNA1 was designed based on homology searches against the cotton genome.[ 16 ] A fused gRNA‐tRNA‐sgRNA1 was generated using pGTR plasmid as template. pRGEB32‐GhU6.9, a modified vector using cotton endogenous promoter U6.9 to induce the transcription of gRNA, was digested by Endonuclease BsaI. The fused plasmid containing sgRNA1 was ligated to linear pRGEB32‐GhU6.9 by ClonExpress II One Step Cloning Kit (Vazyme), generating GhGLR4.8 CRISPR/Cas9 constructs. The CRISPR/Cas9 plasmid was transformed into A. tumefaciens strain GV3101. Hypocotyls of cotton cultivar G. hirsutum cv. J668 were used as explants for genetic transformation of cotton according to previous research.[ 68 ] After obtaining regenerated plants, the leaf DNA of these transformants was extracted and taken as template for PCR amplification using a primer flanking the target site. The PCR products were sequenced to detect the targeted mutation.
Extraction of Total Secreted Protein
Extraction of Fov secreted proteins were performed as reported for Verticillium dahliae with modification.[ 69 ] The seedlings of Xinluzao 7 were grown in sterilized MS medium. The Fov strain F17 was cultured at 25 °C for 4 days in Czapek liquid medium supplemented with the root sections of seven‐day‐old Xinluzao 7 seedlings. The culture was initially filtered by double gauze then centrifuged at 5000 g for 15 min. The culture supernatants were further filtered by passage through a 0.22‐µm filter (Millipore Express PES Membrane). To obtain a high concentration of total secreted proteins, the fungal filtrate was further passed through an Amicon Ultra‐15 3 kDa Centrifugal Filter Devices (Millipore). The volume was concentrated to 1 mL, 14 mL 1× PBS was added and centrifuged again and repeated three times, converting the solution buffer of secreted proteins from Czapek liquid medium to 1× PBS. Finally, the filtrate was concentrated to 500 µL.
Agrobacterium‐Mediated Gene Expression in N. benthamiana
The DNA sequences of GhGLR4.8A and GhGLR4.8C were amplified from Yinshan 4 and Xinluzao 8, respectively, and cloned into pGWB417 using the Gateway system. Transient expression in N. benthamiana leaves was performed as previously described.[ 70 ] A. tumefaciens GV3101 cultures containing the GhGLR4.8 gene were infiltrated at an OD600 of 0.2. Total secreted proteins (SEPs) of Fov were infiltrated in N. benthamiana 48 h after Agrobacterium infiltration. SEPs used here was not concentrated through ultra‐filtration, but was the culture supernatants of Fov cultured at 25 °C for 4 days in potato lactose broth (PLB, 200 g potato and 20 g lactose per 1 liter) supplemented with the root sections of seven‐day‐old Xinluzao 7 seedlings. Trypan blue was used to stain leaves for cell death 60 h after Agrobacterium infiltration. Experiments were replicated biologically three times with five plants each repeat.
Measurement of Extracellular Ca2+ Influx
Seeds of J668 and one transgenic line (Fov7_KO#5) were sterilized and planted in MS medium. One week later, the seedlings were gently removed and transferred into Hoagland's solution one day prior the measurement. Roots were cutoff and balanced in 5 mL testing buffer for 30 min (0.1 × 10 −3 m KCl, 0.1 × 10 −3 m CaCl2, 0.1 × 10 −3 m MgCl2, 0.5 × 10 −3 m NaCl, 0.3 × 10 −3 m MES, 0.2 × 10 −3 m Na2SO4, pH 6.0). Then, The Ca2+ flux in the meristem of the roots was measured by Xu‐Yue Science & Technology Co. (www.xuyue.net) using Non‐invasive Micro‐test Technology (NMT) as described previously.[ 71 ] There were at least four seedlings for each treatment. Pharmacological treatments were done by adding 200 µL 1× PBS or concentrated Fov total secreted proteins (SEPs) to testing buffer and gently pipetting the solution. Addition of 1× PBS was set as control. J668 seedlings were treated with an iGluRs antagonists (AP 50 µm) 1 h before the SEPs treatment. After 1× PBS and SEPs were added, the Ca2+ flux was measured immediately. Ion influx (pmol cm−2 s−1) was calculated by fractional flux changes (ΔF/F) using the equation ΔF/F = (F − F 0)/F 0 to correct background intensity values,[ 72 ] where F 0 denotes the average ion flux of baseline and F denotes the ion flux at every 6 s interval.
RNA‐Seq Analysis
For RNA‐seq, two‐week‐old seedlings of J668 and Fov7_KO#5 were inoculated with Fov and hypocotyls were harvested in a time course at 5, 10 d post inoculation (dpi). 10 plants were pooled at each time points. Fov was cultured in PLB for 4 days, then the spores were collected. Three biological replicates were included for each treatment. RNA was extracted using PureLink RNA Mini Kit (ambion) from three biological replicates of samples collected at each time point of Fov‐inoculated J668 or Fov7_KO#5 and from Fov spores. Library preparation and Illumina sequencing was performed at Novogene from three biological repeats of samples. Raw reads were trimmed by fastp to get clean reads.[ 73 ] The trimmed reads of J668 and Fov7_KO#5 were uniquely mapped to the TM‐1 genome[ 74 ] with HISAT2[ 75 ] using the –dta option, the trimmed reads of Fov7_KO#5 were also uniquely mapped to the Fov race 4 genome[ 28 ] with Fov spores cultured in vitro as control. Since lacking of gene prediction and annotation of Fov race 4 genome, MAKER pipeline[ 76 ] was used to predict protein‐coding genes of Fov race 4, protein domains and Gene Ontology (GO) terms for each gene were annotated using InterProScan.[ 77 ] Read counts for each gene model was counted using Stringtie and a Python script (http://ccb.jhu.edu/software/stringtie/dl/prepDE.py).[ 78 ] R package DEseq2[ 79 ] was used to determine differentially expressed genes and genes with less than 10 counts across all samples were excluded. Gene Ontology (GO) analysis was performed using a custom PERL script. Heat map was created using R package pheatmap.
Statistical Analysis
Data for relative expression levels, disease index and calcium influxes were presented as mean ± SD. Two‐tailed Student's t‐tests was performed to compare the disease index between the haplotypes of GhGLR4.8 in the GWAS population. significance was defined as P ≤ 0.05. Statistical analysis was carried out using GraphPad Prism software.
Data Availability
The raw sequencing data of the 222 accessions genotyped previously are available in the NCBI Sequence Read Archive (SRA) under accession number SRP080913,[ 12 ] and other 68 accessions are available in the NCBI BioProject under accession number PRJNA579217. The RNA‐seq data for Fov cultured in vitro, Fov‐inoculated J668 and Fov7_KO#5 have been deposited in the NCBI BioProject under accession number PRJNA667289.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
L.Z. and X.Z. conceived and designed the project. L.Z. designed the experiment. J.M., W.S., X.N., C.Y., J.K., and A.A. prepared the 290 cotton samples and investigated the disease severity in field. S.L. performed data analysis. S.L., X.Z., S.X., T.Q., T.W., Y.W., and Z.Z. performed experiments. J.L. offered resequencing data of a part of cotton accessions. S.L. wrote the manuscript draft. L.Z., S.J.K., K.L., and X.Z. revised it.
Supporting information
Supporting Information
Supplemental Table 1
Supplemental Table 3
Supplemental Table 7
Supplemental Table 8
Supplemental Table 9
Supplemental Table 10
Acknowledgements
This work was supported by funding from National Natural Science Foundation of China (U1703231), National Key Research and Development Project of China (2018YFD0100403), and the International cooperative project from Ministry of Science and Technology from China (KY201702009).
Liu S., Zhang X., Xiao S., Ma J., Shi W., Qin T., Xi H., Nie X., You C., Xu Z., Wang T., Wang Y., Zhang Z., Li J., Kong J., Aierxi A., Yu Y., Lindsey K., Klosterman S. J., Zhang X., Zhu L., A Single‐Nucleotide Mutation in a GLUTAMATE RECEPTOR‐LIKE Gene Confers Resistance to Fusarium Wilt in Gossypium hirsutum . Adv. Sci. 2021, 8, 2002723. 10.1002/advs.202002723
References
- 1. Michielse C. B., Rep M., Mol. Plant Pathol. 2009, 10, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang P., Su L., Qin L., Hu B., Guo W., Zhang T., Theor. Appl. Genet. 2009, 119, 733. [DOI] [PubMed] [Google Scholar]
- 3. Ulloa M., Hutmacher R. B., Davis R. M., Wright S. D., Marsh B., J. Cotton Sci. 2006, 10, 114. [Google Scholar]
- 4. Davis R. M., Colyer P. D., Rothrock C. S., Kochman J. K., Plant Dis. 2006, 90, 692. [DOI] [PubMed] [Google Scholar]
- 5. Sun W., Jian G., Chen Q., Sci. Agric. Sin. 1999, 32, 51. [Google Scholar]
- 6. Wang C., Roberts P. A., Phytopathology 2006, 96, 727. [DOI] [PubMed] [Google Scholar]
- 7. Ulloa M., Congli W., Hutmacher R. B., Wright S. D., Michael D. R., Saski C. A., Roberts P. A., Mol. Genet. Genomics 2011, 286, 21. [DOI] [PubMed] [Google Scholar]
- 8. Ulloa M., Hutmacher R. B., Roberts P. A., Wright S. D., Nichols R. L., Davis R. M., Theor. Appl. Genet. 2013, 126, 1405. [DOI] [PubMed] [Google Scholar]
- 9. Zhang J., Sanogo S., Ma Z., Qu Y., Crop Sci. 2015, 55, 2435. [Google Scholar]
- 10. Du X., Huang G., He S., Yang Z., Sun G., Ma X., Li N., Zhang X., Sun J., Liu M., Jia Y., Pan Z., Gong W., Liu Z., Zhu H., Ma L., Liu F., Yang D., Wang F., Fan W., Gong Q., Peng Z., Wang L., Wang X., Xu S., Shang H., Lu C., Zheng H., Huang S., Lin T., Zhu Y., Li F., Nat. Genet. 2018, 50, 796. [DOI] [PubMed] [Google Scholar]
- 11. Gong Q., Yang Z., Chen E., Sun G., He S., Butt H. I., Zhang C., Zhang X., Yang Z., Du X., Li F., Plant Cell Physiol. 2018, 59, 275. [DOI] [PubMed] [Google Scholar]
- 12. Wang M., Tu L., Lin M., Lin Z., Wang P., Yang Q., Ye Z., Shen C., Li J., Zhang L., Zhou X., Nie X., Li Z., Guo K., Ma Y., Huang C., Jin S., Zhu L., Yang X., Min L., Yuan D., Zhang Q., Lindsey K., Zhang X., Nat. Genet. 2017, 49, 579. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z., Ersoz E., Lai C.‐Q., Todhunter R. J., Tiwari H. K., Gore M. A., Bradbury P. J., Yu J., Arnett D. K., Ordovas J. M., Buckler E. S., Nat. Genet. 2010, 42, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Z., Cotton Varieties and Their Genealogy in China, China Agriculture Press, Beijing, China, 1996. [Google Scholar]
- 15. Sánchez‐Pujante P. J., Borja‐Martínez M., Pedreño M. Á., Almagro L., Planta 2017, 246, 19. [DOI] [PubMed] [Google Scholar]
- 16. Zhang T., Hu Y., Jiang W., Fang L., Guan X., Chen J., Zhang J., Saski C. A., Scheffler B. E., Stelly D. M., Hulse‐Kemp A. M., Wan Q., Liu B., Liu C., Wang S., Pan M., Wang Y., Wang D., Ye W., Chang L., Zhang W., Song Q., Kirkbride R. C., Chen X., Dennis E., Llewellyn D. J., Peterson D. G., Thaxton P., Jones D. C., Wang Q., Xu X., Zhang H., Wu H., Zhou L., Mei G., Chen S., Tian Y., Xiang D., Li X., Ding J., Zuo Q., Tao L., Liu Y., Li J., Lin Y., Hui Y., Cao Z., Cai C., Zhu X., Jiang Z., Zhou B., Guo W., Li R., Chen Z. J., Nat. Biotechnol. 2015, 33, 531. [DOI] [PubMed] [Google Scholar]
- 17. Forde B. G., Roberts M. R., F1000Prime Rep. 2014, 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Toyota M., Spencer D., Sawai‐Toyota S., Wang J., Tong Z., Koo A. J., Howe G. A., Gilroy S., Science 2018, 361, 1112. [DOI] [PubMed] [Google Scholar]
- 19. Wang M., Tu L., Yuan D., Zhu C. S., Li J., Liu F., Pei L., Wang P., Zhao G., Ye Z., Huang H., Yan F., Ma Y., Zhang L., Liu M., You J., Yang Y., Liu Z., Huang F., Li B., Qiu P., Zhang Q., Zhu L., Jin S., Yang X., Min L., Li G., Chen L. L., Zheng H., Lindsey K., Lin Z., Udall J. A., Zhang X., Nat. Genet. 2019, 51, 224. [DOI] [PubMed] [Google Scholar]
- 20. Ni J., Yu Z., Du G., Zhang Y., Taylor J. L., Shen C., Xu J., Liu X., Wang Y., Wu Y., Rice 2016, 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aouini A., Matsukura C., Ezura H., Asamizu E., Gene 2012, 493, 36. [DOI] [PubMed] [Google Scholar]
- 22. Wudick M. M., Michard E., Oliveira Nunes C., Feijo J. A., J. Exp. Bot. 2018, 69, 4151. [DOI] [PubMed] [Google Scholar]
- 23. Alfieri A., Doccula F. G., Pederzoli R., Grenzi M., Bonza M. C., Luoni L., Candeo A., Romano Armada N., Barbiroli A., Valentini G., Schneider T. R., Bassi A., Bolognesi M., Nardini M., Costa A., Proc. Natl. Acad. Sci. USA 2020, 117, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miao Y., Xu L., He X., Zhang L., Shaban M., Zhang X., Zhu L., Plant J. 2019, 98, 329. [DOI] [PubMed] [Google Scholar]
- 25. Liu Q., Wang C., Jiao X., Zhang H., Song L., Li Y., Gao C., Wang K., Sci. China: Life Sci. 2019, 62, 1. [DOI] [PubMed] [Google Scholar]
- 26. Kwaaitaal M., Huisman R., Maintz J., Reinstadler A., Panstruga R., Biochem. J. 2011, 440, 355. [DOI] [PubMed] [Google Scholar]
- 27. Mousavi S. A., Chauvin A., Pascaud F., Kellenberger S., Farmer E. E., Nature 2013, 500, 422. [DOI] [PubMed] [Google Scholar]
- 28. Seo S., Pokhrel A., Coleman J. J., Mol. Plant‐Microbe Interact. 2020, 33, 138. [DOI] [PubMed] [Google Scholar]
- 29. Michelmore R. W., Christopoulou M., Caldwell K. S., Annu. Rev. Phytopathol. 2013, 51, 291.23682913 [Google Scholar]
- 30. Wang P., Shi L., Su L., Hu B., Agric. Sci. China 2010, 9, 1799. [Google Scholar]
- 31. Zhu Y. J., Zhang X. Y., Jin‐Rong L. I., Han L., Zhang W., Xinjiang Agric. Sci. 2010, 47, 268. [Google Scholar]
- 32. Gonzalez‐Cendales Y., Catanzariti A. M., Baker B., McGrath D. J., Jones D. A., Mol. Plant Pathol. 2016, 17, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Wit P. J. G. M., New Phytol. 2016, 212, 805. [DOI] [PubMed] [Google Scholar]
- 34. de Sain M., Rep M., Int. J. Mol. Sci. 2015, 16, 23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Catanzariti A. M., Do H. T., Bru P., de Sain M., Thatcher L. F., Rep M., Jones D. A., Plant J. 2017, 89, 1195. [DOI] [PubMed] [Google Scholar]
- 36. Simons G., Groenendijk J., Wijbrandi J., Reijans M., Vos P., Plant Cell 1998, 10, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Catanzariti A. M., Lim G. T., Jones D. A., New Phytol. 2015, 207, 106. [DOI] [PubMed] [Google Scholar]
- 38. Krattinger S. G., Lagudah E. S., Spielmeyer W., Singh R. P., Huerta‐Espino J., McFadden H., Bossolini E., Selter L. L., Keller B., Science 2009, 323, 1360. [DOI] [PubMed] [Google Scholar]
- 39. Moore J. W., Herrera‐Foessel S., Lan C., Schnippenkoetter W., Ayliffe M., Huerta‐Espino J., Lillemo M., Viccars L., Milne R., Periyannan S., Kong X., Spielmeyer W., Talbot M., Bariana H., Patrick J. W., Dodds P., Singh R., Lagudah E., Nat. Genet. 2015, 47, 1494. [DOI] [PubMed] [Google Scholar]
- 40. Li G., Zhou J., Jia H., Gao Z., Fan M., Luo Y., Zhao P., Xue S., Li N., Yuan Y., Ma S., Kong Z., Jia L., An X., Jiang G., Liu W., Cao W., Zhang R., Fan J., Xu X., Liu Y., Kong Q., Zheng S., Wang Y., Qin B., Cao S., Ding Y., Shi J., Yan H., Wang X., Ran C., Ma Z., Nat. Genet. 2019, 51, 1106. [DOI] [PubMed] [Google Scholar]
- 41. Wang H., Sun S., Ge W., Zhao L., Hou B., Wang K., Lyu Z., Chen L., Xu S., Guo J., Li M., Su P., Li X., Wang G., Bo C., Fang X., Zhuang W., Cheng X., Wu J., Dong L., Chen W., Li W., Xiao G., Zhao J., Hao Y., Xu Y., Gao Y., Liu W., Liu Y., Yin H., Li J., Li X., Zhao Y., Wang X., Ni F., Ma X., Li A., Xu S. S., Bai G., Nevo E., Gao C., Ohm H., Kong L., Science 2020, 368, 844. [DOI] [PubMed] [Google Scholar]
- 42. Li N., Lin B., Wang H., Li X., Yang F., Ding X., Yan J., Chu Z., Nat. Genet. 2019, 51, 1540. [DOI] [PubMed] [Google Scholar]
- 43. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R., Sibley D., Pharmacol. Rev. 2010, 62, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gong J., Liu J., Ronan E. A., He F., Cai W., Fatima M., Zhang W., Lee H., Li Z., Kim G.‐H., Pipe K. P., Duan B., Liu J., Xu X. Z. S., Cell 2019, 178, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lam H.‐M., Chiu J., Hsieh M.‐H., Meisel L., Oliveira I. C., Shin M., Coruzzi G., Nature 1998, 396, 125. [DOI] [PubMed] [Google Scholar]
- 46. Ortiz‐Ramirez C., Michard E., Simon A. A., Damineli D. S. C., Hernandez‐Coronado M., Becker J. D., Feijo J. A., Nature 2017, 549, 91. [DOI] [PubMed] [Google Scholar]
- 47. Erwan M., Lima P. T., Filipe B., Ana Catarina S., Maria Teresa P., Carvalho J. O. E., Matthew G., Lai‐Hua L., Gerhard O., Feijó J. A., Science 2011, 332, 434.21415319 [Google Scholar]
- 48. Nguyen C. T., Kurenda A., Stolz S., Chételat A., Farmer E. E., Proc. Natl. Acad. Sci. USA 2018, 115, 10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiu J. C., Brenner E. D., DeSalle R., Nitabach M. N., Holmes T. C., Coruzzi G. M., Mol. Biol. Evol. 2002, 19, 1066. [DOI] [PubMed] [Google Scholar]
- 50. De Bortoli S., Teardo E., Szabo I., Morosinotto T., Alboresi A., Biophys. Chem. 2016, 218, 14. [DOI] [PubMed] [Google Scholar]
- 51. Chen J., Jing Y., Zhang X., Li L., Wang P., Zhang S., Zhou H., Wu J., Sci. Rep. 2016, 6, 32013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vincill E. D., Clarin A. E., Molenda J. N., Spalding E. P., Plant Cell 2013, 25, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bacete L., Melida H., Miedes E., Molina A., Plant J. 2018, 93, 614. [DOI] [PubMed] [Google Scholar]
- 54. Kubicek C. P., Starr T. L., Glass N. L., Annu. Rev. Phytopathol. 2014, 52, 427. [DOI] [PubMed] [Google Scholar]
- 55. Saintenac C., Lee W.‐S., Cambon F., Rudd J. J., King R. C., Marande W., Powers S. J., Bergès H., Phillips A. L., Uauy C., Hammond‐Kosack K. E., Langin T., Kanyuka K., Nat. Genet. 2018, 50, 368. [DOI] [PubMed] [Google Scholar]
- 56. Alexandre B., Francesca S., Alberto M., Felice C., Giulia D. L., Proc. Natl. Acad. Sci. USA 2010, 107, 9452.20439716 [Google Scholar]
- 57. Ferrari S., Savatin D. V., Sicilia F., Gramegna G., Cervone F., Lorenzo G. D., Front. Plant Sci. 2013, 4, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Evanno G., Regnaut S., Goudet J., Mol. Ecol. 2005, 14, 2611. [DOI] [PubMed] [Google Scholar]
- 59. Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y., Buckler E. S., Bioinformatics 2007, 23, 2633. [DOI] [PubMed] [Google Scholar]
- 60. Purcell S., Neale B., Todd‐Brown K., Thomas L., Ferreira M. A., Bender D., Maller J., Sklar P., de Bakker P. I., Daly M. J., Sham P. C., Am. J. Hum. Genet. 2007, 81, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Finn R. D., Clements J., Eddy S. R., Nucleic Acids Res. 2011, 39, W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tamura K., Stecher G., Peterson D., Filipski A., Kumar S., Mol. Biol. Evol. 2013, 30, 2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Omasits U., Ahrens C. H., Muller S., Wollscheid B., Bioinformatics 2014, 30, 884. [DOI] [PubMed] [Google Scholar]
- 64. Fradin E. F., Zhang Z., Juarez Ayala J. C., Castroverde C. D., Nazar R. N., Robb J., Liu C. M., Thomma B. P., Plant Physiol. 2009, 150, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abd‐Elsalam K. A., Asran‐Amal A., Schnieder F., Migheli Q., Verreet J. A., J Plant Dis Prot. 2006, 113, 14. [Google Scholar]
- 66. Gao W., Long L., Zhu L. F., Xu L., Gao W. H., Sun L. Q., Liu L. L., Zhang X. L., Mol. Cell. Proteomics 2013, 12, 3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang P., Zhang J., Sun L., Ma Y., Xu J., Liang S., Deng J., Tan J., Zhang Q., Tu L., Daniell H., Jin S., Zhang X., Plant Biotechnol. J. 2018, 16, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li J., Wang M., Li Y., Zhang Q., Lindsey K., Daniell H., Jin S., Zhang X., Plant Biotechnol. J. 2019, 17, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang L., Ni H., Du X., Wang S., Ma X. W., Nurnberger T., Guo H. S., Hua C., New Phytol. 2017, 215, 368. [DOI] [PubMed] [Google Scholar]
- 70. Ma L., Lukasik E., Gawehns F., Takken F. L. W., Methods Mol. Biol. 2012, 835, 61. [DOI] [PubMed] [Google Scholar]
- 71. Shabala L., Ross T., McMeekin T., Shabala S., FEMS Microbiol. Rev. 2006, 30, 472. [DOI] [PubMed] [Google Scholar]
- 72. Keinath N. F., Waadt R., Brugman R., Schroeder J. I., Grossmann G., Schumacher K., Krebs M., Mol. Plant 2015, 8, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen S., Zhou Y., Chen Y., Gu J., Bioinformatics 2018, 34, i884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu Y., Chen J., Fang L., Zhang Z., Ma W., Niu Y., Ju L., Deng J., Zhao T., Lian J., Baruch K., Fang D., Liu X., Ruan Y. L., Rahman M. U., Han J., Wang K., Wang Q., Wu H., Mei G., Zang Y., Han Z., Xu C., Shen W., Yang D., Si Z., Dai F., Zou L., Huang F., Bai Y., Zhang Y., Brodt A., Ben‐Hamo H., Zhu X., Zhou B., Guan X., Zhu S., Chen X., Zhang T., Nat. Genet. 2019, 51, 739. [DOI] [PubMed] [Google Scholar]
- 75. Kim D., Langmead B., Salzberg S. L., Nat. Methods 2015, 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Campbell M. S., Holt C., Moore B., Yandell M., Curr. Protoc. Bioinf. 2014, 48, 4.11.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hunter S., Jones P., Mitchell A., Apweiler R., Attwood T. K., Bateman A., Bernard T., Binns D., Bork P., Burge S., de Castro E., Coggill P., Corbett M., Das U., Daugherty L., Duquenne L., Finn R. D., Fraser M., Gough J., Haft D., Hulo N., Kahn D., Kelly E., Letunic I., Lonsdale D., Lopez R., Madera M., Maslen J., McAnulla C., McDowall J., McMenamin C., Mi H., Mutowo‐Muellenet P., Mulder N., Natale D., Orengo C., Pesseat S., Punta M., Quinn A. F., Rivoire C., Sangrador‐Vegas A., Selengut J. D., Sigrist C. J., Scheremetjew M., Tate J., Thimmajanarthanan M., Thomas P. D., Wu C. H., Yeats C., Yong S. Y., Nucleic Acids Res. 2012, 40, D306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pertea M., Pertea G. M., Antonescu C. M., Chang T.‐C., Mendell J. T., Salzberg S. L., Nat. Biotechnol. 2015, 33, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Love M. I., Huber W., Anders S., Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Table 1
Supplemental Table 3
Supplemental Table 7
Supplemental Table 8
Supplemental Table 9
Supplemental Table 10
Data Availability Statement
The raw sequencing data of the 222 accessions genotyped previously are available in the NCBI Sequence Read Archive (SRA) under accession number SRP080913,[ 12 ] and other 68 accessions are available in the NCBI BioProject under accession number PRJNA579217. The RNA‐seq data for Fov cultured in vitro, Fov‐inoculated J668 and Fov7_KO#5 have been deposited in the NCBI BioProject under accession number PRJNA667289.