

Abstract
Chiral 1,2-diamino compounds are important building blocks in organic chemistry for biological applications and as asymmetric inducers in stereoselective synthesis that are challenging to prepare in a straightforward and stereoselective manner. Herein, we disclose a cost-effective and readily available Cu-catalyzed system for the reductive coupling of a chiral allenamide with N-alkyl substituted aldimines to access chiral 1,2-diamino synthons as single stereoisomers in high yields. The method shows broad reaction scope and high diastereoselectivity and can be easily scaled using standard Schlenk techniques. Mechanistic investigations by density functional theory calculations identified the mechanism and origin of stereoselectivity. In particular, the addition to the imine was shown to be reversible, which has implications toward development of catalyst-controlled stereoselective variants of the identified reductive coupling of imines and allenamides.
Introduction
Chiral vicinal diamines are extremely valuable and important motifs in organic chemistry that are exploited by both nature and the pharmaceutical industry for their biological activities,1,2 and in stereoselective organic synthesis as powerful chiral inducers through application as organocatalysts,3 chiral ligands4 for transition metal catalyzed reactions, and as chiral auxiliaries.5 For example, a variety of biologically active pharmaceuticals and natural products are given in Figure 1 possessing either the chiral 1,2-diamino-fragment or its corresponding urea form.2 Representative therapeutics being developed for the treatment of important human diseases include antibiotics (penicillin,2e jogyamycin2j), anticancer compounds (cisplatin derivatives,6 LP992c), HIV protease inhibitors (NBD-110212d), NK1-antagonists7 (CP-99,994;2a Sch4250782g) for central-nervous-system (CNS) related diseases and rheumatoid arthritis, and influenza (tamiflu).2b
Figure 1.

Selected examples of chiral 1,2-diamine- and urea-derived biologically active molecules.
Due to the biological and synthetic value of chiral 1,2-diamines, stereoselective methods for their preparation are an important endeavor in organic chemistry.1a,1c,1d,8 Potential synthetic options to access the chiral vicinal diamine moiety can be envisioned to occur either by formation of the two C–N bonds starting from unsaturated hydrocarbons (2, Scheme 1A) or through direct C–C bond formation between C1 and C2 of the 1,2-diamine from two N-substituted reagents (Scheme 1B).1a,1c,1d,8 Using a C–N bond forming approach (Scheme 1A), diamination may be achieved by forming both C–N bonds at the same time,8,9 or sequentially through either aziridination10 followed by ring-opening with an amine nucleophile1a,1c,1d,11 or through aminohydroxylation12 followed by alcohol activation and amine substitution.1a−1d While direct catalytic 1,2-diamination of 2 represents an ideal strategy for diamine synthesis, the amino-groups added across the π-system are typically identical leading to the formation of diamines with identical substituents (i.e., R3 = R4 in 1),8,9 and a recent approach employing electrochemistry9b suffers from potentially forming high-energy/explosive diazocompounds13 en route to the desired diamines. Additionally, the aziridination/ring-opening strategy can suffer from poor stereoselectivity in the aziridination step and regiochemistry issues in the subsequent opening step, while the aminohydroxylation route requires regiocontrol in the aminohydroxylation step followed by additional transformations to convert 4 to the desired diamine. Alternatively, synthesis of 1 through C–C bond formation can be achieved through aza-pinacol coupling of two imines,14 nitro-Mannich,15 or glycine-Mannich16 reactions (Scheme 1B). Typical aza-pinacol coupling protocols only afford symmetrical diamines through homocoupling of a single imine; however, recent photoredox strategies14h−14l enabling the generation of α-aminoradicals17 from amines have enabled cross-selective coupling of imines and N-methylamines.14j−14l Furthermore, nucleophilic additions to imines using α-aminoanion derivatives18 from nitroalkanes (7)15 or protected glycines (8)16 offer another entry into the diamine core 1.
Scheme 1. Synthetic Strategies toward the Synthesis of 1,2-Diamines.

In regards to chiral amine synthesis, asymmetric allylation of imines using allyl organometallic nucleophiles (10) by direct addition or through catalyst control has been an area of intense research in organic chemistry (Scheme 2).19 The chiral allylamine products (11) are highly valuable in the context of the synthesis of complex amine-containing organic compounds because of the high versatility of the olefin functional group present within 11. Substituted allylorganometallic reagents (e.g., 12) allow for increased molecular complexity by introducing two stereocenters in the allyl addition reaction (e.g., 13, Scheme 2B). Therefore, we envisioned that use of an amino-substituted allyl reagent 12 in addition reactions with imine electrophiles would be a powerful strategy to prepare 1,2-diamines (13) with differential substitution patterns on nitrogen and containing an olefin motif for further functional group manipulations. Surprisingly, only a single example of such a strategy for the preparation of 1,2-diamines has been reported, which employs a lithiated derivative of 12 (M = Li) with chiral tert-butanesulfinimide derived aldimines affording products in moderate yields with mixtures of branched and linear allylation products.20 In contrast, amino-substituted allyl reagents 12 have been used in reactions employing carbonyl electrophiles to provide 1,2-aminoalcohols (16).21−23 Recently, the Krische22 group and our own lab23 have developed reductive coupling24,25 procedures for the catalytic generation of amino-substituted allyl reagents 12 and have studied their reactions with carbonyl electrophiles (Scheme 2C). These techniques represent orthogonal methodologies whereby the Krische22a system employs a chiral Ir-catalyst and processes aldehyde electrophiles using an achiral allenamide (15), while our work utilizes a Cu-catalyst and a chiral allenamide (15a) for reactions using ketone electrophiles.23 Based on our success in the stereoselective Cu-catalyzed reductive coupling of ketones and chiral allenamides to afford branched chiral 1,2-aminoalcohols 16(23a) or the corresponding linear products,23b and the lack of literature data for imine allylation reaction utilizing amino-substituted allylic nucleophiles, we began to investigate the reaction of allenamide 15a with imine electrophiles 9 for the stereoselective synthesis of chiral 1,2-diamine synthons 17 (Scheme 2D). The results of these studies leading to the identification of a practical and highly stereoselective synthesis of diamine synthons 17 using Cu-catalyzed reductive coupling are disclosed herein.
Scheme 2. Proposed Allylation Strategy toward the Synthesis of 1,2-Diamines.

Results and Discussion
Reaction Optimization
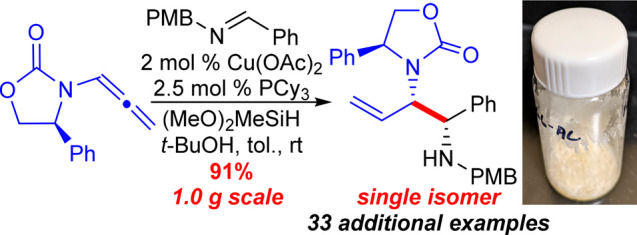
To investigate the proposed Cu-catalyzed reductive coupling of imines and allenamides, initial studies examined the ligand effect when employing DMB-protected imine 9a with chiral allenamide 15a in the reaction (Table 1). The phenyl-derived Evans oxazolidinone of allenamide 15a was specifically targeted due to its low-cost and high-availability,26 and because it allows for more deprotection options of the desired diamine products over other alkyl-substituted oxazolidinones (i.e., hydrogenolysis). The DMB-group of the aldimine was employed due to its acid lability to allow for chemoselective differentiation of the two amine protecting groups in the final products (18a/19a). Gratifyingly, a variety of phosphine ligands (entries 1–7) afforded urea product 19a presumably resulting from migration of the carbamate carbonyl (Scheme 3), whereas an N-heterocyclic carbene (NHC) ligand provided poor conversion (entry 8). In all cases, a single diastereomer of product was obtained as determined by 1H NMR spectroscopy of the unpurified reaction mixture. Notably, the bidentate phosphine dcpe that has been utilized previously in Cu-catalyzed reductive coupling of C-substituted allenes and imines25c afforded only a moderate yield with a substantial amount of unreacted imine (20%, entry 1). Monodentate phosphine ligands (entries 2–7) worked well with the exception of sterically demanding ligands that afforded poor conversion of the imine (entries 3, 4). Ultimately, the use of PCy3 as ligand afforded the highest yield of 19a in the reaction (entry 2). Use of solvents other than toluene in the reaction (entries 9–12) offered no improvements. Finally, addition of 2 equiv of t-BuOH to the reaction led to the exclusive formation of 18a in excellent yield and diastereoselectivity.
Table 1. Ligand Optimization for the Reductive Coupling Using 15aa.

| Entry | Ligand | %Yield 18ab | %Yield 19ab | % 9ab |
|---|---|---|---|---|
| 1 | dcpe | <5 | 58 | 20 |
| 2 | PCy3 | <5 | 86 | <5 |
| 3 | P(adam)3 | <5 | 28 | 31 |
| 4 | XPhos | <5 | 22 | 38 |
| 5 | P(NMe2)3 | <5 | 66 | <5 |
| 6 | P(OEt)3 | <5 | 60 | <5 |
| 7 | (PhO)2PNMe2 | <5 | 66 | <5 |
| 8 | SIMes | <5 | 8 | 67 |
| 9c | PCy3 | <5 | 51 | <5 |
| 10d | PCy3 | <5 | 54 | <5 |
| 11e | PCy3 | <5 | 52 | 21 |
| 12f | PCy3 | <5 | 31 | 14 |
| 13g | PCy3 | 90 | <5 | <5 |
129 mg (0.400 mmol) 9a, 96.6 mg (0.480 mmol) 15a, 5 mol % Cu(OAc)2, 6 mol % ligand, and 1.0 mL of toluene. A single diastereomer of product was obtained in all cases by analysis of the unpurified reaction mixture by 1H NMR spectroscopy. See the Supporting Information for further details.
Yield determined by 1H NMR spectroscopy on the unpurified reaction mixture using dimethylfumarate as analytical standard.
Reaction performed in MTBE.
Reaction performed in dioxane.
Reaction performed in CH2Cl2.
Reaction performed in THF.
Performed using 2.0 equiv of t-BuOH as additive. DMB = 2,4-dimethoxybenzyl.
Scheme 3. Proposed Reaction Catalytic Cycle.

An initial working hypothesis to understand the difference in product selectivity between the formation of urea 19a in the absence of t-BuOH versus the exclusive formation of diamino-derivative 18a when t-BuOH was used as an additive is given in Scheme 3. Regioselective hydrocupration of allenamide 15a by the LCuH23,25c catalyst 20 initially is expected to afford substituted linear allylcopper reagent 21 that may undergo E/Z isomerization through σ–π–σ equilibration prior to reaction with the imine electrophile. Then, diastereoselective reaction of intermediate 21 with imine 9a provides Cu-amide intermediate 22. To afford product 18a from 22, direct silylation of the amine by the silane must occur to regenerate the LCuH catalyst 20; however, this step is expected to be slow due to the weak strength of the N–Si bond (BDE ≈ 104 kcal/mol).27 Due to the strong basicity of the N-anion in 22, intramolecular attack of the oxazolidinone carbonyl may occur competitively to provide 23 containing an O–Cu bond that should more easily silylate due to the high bond strength of the O–Si bond (BDE ≈ 190 kcal/mol)28 affording urea 24 and regenerating the LCuH catalyst (20). Alternatively, when t-BuOH is present, protonation of the Cu–N bond of 22 by t-BuOH to afford product 18a directly and generate LCuOtBu is thermodynamically favorable based on the pKa values for a secondary amine (pyrrolidine: ∼44)29 vs t-BuOH (32) and supported by DFT calculations (vide infra).30 The LCu-OtBu intermediate can then undergo silylation to regenerate the LCuH catalyst 20. The role of alcohol additives to facilitate catalyst turnover by protonation of Cu–N intermediates has been documented previously.25c,31 Sterically hindered alcohols such as t-BuOH have been shown to be preferred since the rate of competitive protonation of the Cu–H catalyst is reduced with bulky alcohols.31
Next, the substrate scope of the Cu-catalyzed reductive coupling reaction using t-BuOH as additive to provide branched diamino-derived products 18 was examined (Scheme 4). In all cases, a single diastereomer (the (S,S,S)-diastereomer) of product was obtained as determined by analysis of the unpurified reaction mixture by 1H NMR spectroscopy. In general, a wide variety of imines could be employed in the reaction in good to excellent yields. Electron-deficient (18a – 18k) and electron-rich (18l–18o) aryl groups both performed well in the reaction. Heterocyclic imines (18k, 18s–18u) and C-substituted arenes (18p–18r) were also well tolerated. Finally, a sterically demanding imine (18m) or a m-NO2Ph group (18i) required heating at 65 °C to afford good reactivity. Use of an aliphatic aldimine (i.e., Ar1 = Me) did not provide any desired products.
Scheme 4. Imine Generality in the Cu-Catalyzed Reductive Coupling To Access 1,2-Diamino Synthons 18.

Conditions: 9 (0.400 mmol), 15a (96.6 mg, 0.48 mmol), Cu(OAc)2 (5 mol %), PCy3 (6.5 mol %), t-BuOH (76 μL, 0.80 mmol), Me(MeO)2SiH (99 μL, 0.80 mmol), and 1.0 mL of toluene, rt 24 h followed by treatment with NH4F/MeOH. See the Supporting Information for more details. A single diastereomer of product was obtained in all cases by analysis of the unpurified reaction mixture by 1H NMR spectroscopy. Yields represent isolated yield.
Reaction performed at 65 °C.
Isolated as an inseparable mixture of 18 and urea 19.
Initial analysis of the substrate scope for the urea-forming Cu-catalyzed reductive coupling reaction employing DMB-substituted imines in the absence of t-BuOH proved to be less general than the analogous reaction conducted with t-BuOH as the additive. In these problematic cases, a poor yield of desired product was obtained even at 65 °C; however, the imine remained while the allenamide had been consumed. As a result, the effect of the N-substituent of the imine electrophile was examined to improve the efficiency of the reaction to the desired product (Table 2). As an example, the 2-naphthyl N-DMB-imine (9pa) afforded a poor yield in the desired reaction (entry 1). A strong influence on reactivity and the electronic character of the aryl group (Ar2) of the imine was found (entries 1–5). Use of an electron-poor aryl group (entry 5) afforded the best reaction yield; however, a simple benzyl group also provided good reactivity (entry 3). As a result, for problematic DMB-derived imines, the reactivity can be improved by utilizing PMB, Bn, or p-CF3-benzyl as the N-substituent on the aldimine.
Table 2. Effect of Imine N-Substitution on Reactivitya.

| Entry | Ar2 | % yieldb |
|---|---|---|
| 1 | 2,4-dimethoxyphenyl (9pa) | 17 (19pa) |
| 2 | 4-methoxyphenyl (9pb) | 58 (19pb) |
| 3 | Phenyl (9pc) | 70 (19pc) |
| 4 | 4-fluorophenyl (9pd) | 71 (19pd) |
| 5 | 4-trifluoromethylphenyl (9pe) | 79 (19pe) |
Conditions: 9p (0.400 mmol), 106 mg (0.480 mmol) of 15a, 5 mol % Cu(OAc)2, 6 mol % PCy3, 99 μL (0.80 mmol) of (MeO)2MeSiH, and 1.0 mL of toluene. A single diastereomer of product was obtained in all cases by analysis of the unpurified reaction mixture by 1H NMR spectroscopy.
Yield determined by 1H NMR spectroscopy on the unpurified reaction mixture using dimethylfumarate as analytical standard.
Based on the results from Table 2, the substrate scope for the urea-forming Cu-catalyzed reductive coupling reaction in the absence of t-BuOH was investigated using this new knowledge (Table 3). DMB-substituted imines could be employed in good yields affording single diastereomers of product at room temperature when Ar1 was a simple phenyl group (19b), heterocyclic (19k, 19s, 19t), or substituted at the para-position with an electron-donating group (19l, 19n) or an electron-withdrawing group (19a). However, reactions employing imines containing halogenated arenes or more sterically demanding aryl groups were not successful utilizing the N-DMB derived imine and instead required heating and the use of either an N-Bn or an N-CH2-p-CF3Ph group on the aldimine (see 19de, 19ee, 19j and 19m, 19pe, respectively).
Table 3. Imine Generality in the Cu-Catalyzed Reductive Coupling To Access Chiral Ureasa.


Conditions: 9 (0.40 mmol), 15a (96.6 mg, 0.48 mmol), Cu(OAc)2 (5 mol %), PCy3 (6.5 mol %), Me(MeO)2SiH (99 μL, 0.80 mmol), and 1.0 mL of toluene, rt 24 h followed by treatment with NH4F/MeOH. See the Supporting Information for more details. A single diastereomer of product was obtained in all cases by analysis of the unpurified reaction mixture by 1H NMR spectroscopy. Yields represent isolated yield.
Reaction performed at 65 °C.
Isolated as an inseparable mixture of urea 19 and 18.
Stereochemical assignment of the products obtained in the Cu-catalyzed reductive coupling reaction as the (S,S,S)-diastereomer was determined unequivocally by X-ray crystallography (Scheme 5). While the branched products 18 were typically noncrystalline, formation of the HCl-salt of 18c afforded crystalline material whose structure was determined by single-crystal Xray analysis. Furthermore, conversion of products 18 to the urea 19 could also be achieved after isolation of 18 by subsequent treatment with n-BuLi (e.g., 18a → 19a). The urea product obtained from this sequence was identical to the material made from the reductive coupling reaction performed in the absence of t-BuOH by NMR spectroscopy confirming that the same stereoisomer of product was formed in both reductive coupling processes (i.e., with or without t-BuOH as additive).
Scheme 5. Stereochemistry Determination.

The synthetic utility of the reaction products obtained in the allenamide/imine reductive coupling reaction is highlighted in Scheme 6. The phenethyl group of urea 19a derived from the Evans oxazolidinone of the allenamide starting material could be cleaved in a three-step sequence consisting of alcohol activation (MsCl), base induced elimination, and enamide hydrolysis with aqueous acid to provide urea 25 in good overall yield without isolation of intermediates. Furthermore, synthon 27 is a viable intermediate to access chiral aminopiperidine 28 for the preparation of the potent NK-1 inhibitor compounds CP-99,994 and CP-122,721,2a,7 which could easily be accessed from reductive coupling product 18c (Scheme 6). The Cu-catalyzed reductive coupling was scaled to 1.0 g without the need for an inert atmosphere glovebox by performing the reaction on the “benchtop” using standard Schlenk techniques and preparing the (PCy3)Cu-catalyst by adding the PCy3 as a 20 wt % solution in toluene that is commercially available.32 The catalyst loading could be reduced to 2.0 mol % Cu providing 18c in good yield and excellent diastereocontrol in only 2 h of reaction time. Considering the low cost and high availability of the Cu-precatalyst,32 the ligand employed (PCy3),32 and the chiral allenamide 15a,26 along with the high catalytic activity of this system (2 mol % catalyst loading), the current method represents a highly practical and scalable method for the synthesis of diamino-synthons 18/19. Allylation of 18c was then carried out using allyl bromide, followed by ring-closing metathesis with the Hoveyda–Grubbs second generation catalyst to provide access to compound 27 as an orthogonally protected aminopiperidine derivative as a single stereoisomer.
Scheme 6. Synthetic Applications.

Mechanistic Modeling by DFT Analysis
To shed light onto the mechanism and origin of diastereoselectivity, we used dispersion-corrected DFT calculations (see Supporting Information for details). Specifically, we performed extensive conformational analysis on all intermediates and transition states using the B3LYP-D3 functional and a def2-SVP basis set33 with toluene as the solvent using the CPCM solvation model34 as implemented in Gaussian16. Further, to refine the energetics and compare methods, single-point calculations using the M06-L functional,35 as well as a larger basis set (def2-TZVPP) with B3LYP-D3, which yielded similar energetic profiles, were subsequently performed. For simplicity, only B3LYP-D3/def2-SVP optimization energetics will be discussed in the text. Structures were visualized using CYLview Version 1.0.561.36
As shown in Figure 2, initial investigations were conducted by first analyzing the hydrocupration of allenamide 15a with (PCy3)CuH as catalyst. Following coordination of the copper and allenamide π-bond, the energetically favored hydrocupration proceeds via TS–I-II (barrier of 10.4 kcal/mol with respect to separated 15a and LCuH structures) to form the branched allylcopper species II. Presumably this transition state benefits from lack of steric hindrance between the ligand and the chiral auxiliary, which were present in the alternative transition states. Specifically, alternate hydrocupration transition states leading to linear allylcopper species (TS–I-IIIcis and TS–I-IIItrans) were found to be much higher in energy by ∼3 kcal/mol for TS–I-IIItrans and by >7 kcal/mol for all other pathways and were therefore not productive.
Figure 2.

Structures and relative free energies (in kcal/mol, with respect to separate LCuH catalyst and reactants) of possible hydrocupration pathways, optimized using B3LYP-D3/def2SVP-CPCM(toluene), M06-L/def2SVP-gas//B3LYP-D3/def2SVP-CPCM(toluene) (in parentheses), and B3LYP-D3/def2TZVPP-gas//B3LYP-D3/def2SVP-CPCM(toluene) {in braces}.
In turn, the branched allylcopper intermediate is expected to undergo isomerization to linear allylcopper species by σ–π–σ isomerization. Recently, Buchwald and co-workers reported branched-linear allylcopper isomerizations for a system with a bidentate phosphine ligand25c as well as with a CuH-catalyzed allylation of ketones and dienes.37 In our case, it was calculated that the branched allylcopper intermediate II can readily isomerize (barrier of only 6.9 and 10.0 kcal/mol via TS–II-IIIcisor TS–II-IIItrans, respectively) to form the nearly isoenergetic cis or trans linear allylcopper intermediates (IIIcis and IIItrans). Intermediate IIIcis was slightly favorable compared to intermediate IIItrans (by 0.6 kcal/mol), as was the cis isomerization transition state (TS–I-IIIcis was favored by 3.1 kcal/mol), presumably due to coordination between the oxazolidinone and the copper (Cu–O bond distance = 2.37 Å). However, upon coordination of the imine to the copper, the trans conformation (III′trans) becomes significantly more favored (as seen in Figure 3), likely due to unfavorable steric hindrance between the imine and the oxazolidinone in the III′cis conformation (see Supporting Information for further details).
Figure 3.

Structures and relative free energies (in kcal/mol, with respect to separate LCuH catalyst and reactants) for proposed mechanistic pathway, optimized using B3LYP-D3/def2SVP-CPCM(toluene), M06-L/def2SVP-gas//B3LYP-D3/def2SVP-CPCM(toluene) (in parentheses), and B3LYP-D3/def2TZVPP-gas//B3LYP-D3/def2SVP-CPCM(toluene) {in braces}. Optimized structures of transition states visualized with CYLview are shown (with PCy3 ligand faded out for clarity).
Next, we focused on the key C–C bond formation steps. As shown in Figure 3, after performing extensive conformational analysis on the subsequent diastereomeric C–C bond forming transition states with the allylcopper intermediates and the imine substrate (see Supporting Information for details), the most favorable pathway for diastereoselective C–C bond formation was identified to proceed from the trans linear intermediate III′trans through a Zimmerman–Traxler transition state TS-III′-IVtrans (S,S,S) (barrier of only 7.4 kcal/mol from complexed III′trans intermediate) to branched addition product IV(S,S,S). Further, in agreement with experiment, the competing diastereomeric transition state TS-III′-IVtrans (R,R,S) which would lead to the opposite diastereomer was determined to be much higher in energy. Notably, all C–C bond formation steps fromIII′transare reversible, as the branched addition products IV(S,S,S) and IV(R,R,S) were each uphill in energy (by ∼2 kcal/mol and ∼6 kcal/mol respectively), which can have implications for rational catalyst and reaction design (vide infra).
To gain insights into the origin of diastereoselectivity, we performed distortion–interaction and NCI analysis (Figure 4). Overall, comparing the structures of the lowest energy competing diastereomeric transition states TS-III′-IVtrans(S,S,S) and TS-III′-IVtrans(R,R,S) reveals that the structures of these transition states were remarkably similar, with key C–C and C–Cu bond distances differing by no more than 0.05 Å. However, the orientation of the chiral auxiliary is different, as the TS-III′-IVtrans(S,S,S) transition state has the oxazolidinone moiety of the enamide group of the substituted Cu(allyl) ligand in an s-trans conformation while the TS-III′-IVtrans(R,R,S) has this group in an s-cis conformation that, as shown in Figure 5, leads to a 2.2 kcal/mol energy destabilization. Furthermore, the ground state structures of chiral oxazolidinone-derived enamides are known to favor an s-trans conformation.38 In addition, distortion–interaction analysis39 (Figure 4a) showed that the distortion energy of the TS-III′-IVtrans(S,S,S) transition state was higher than that of the corresponding TS-III′-IVtrans(R,R,S) transition state (by 3.9 kcal/mol). However, the (S,S,S) system benefited from much stronger interaction energy (by 8.5 kcal/mol). Overall, this favorable interaction between the imine and allylcopper makes the TS-III′-IVtrans(S,S,S) the favorable diastereomeric transition state. Finally, noncovalent interaction (NCI) analysis (performed using Multiwfn40 software and visualized using VMD41 software) further supports the presence of favorable interactions in the TS-III′-IVtrans(S,S,S) transition state (Figure 4b). Specifically, in both transition states, there appeared to be favorable C–H···π interactions between the ligand and the benzyl group of the imine (highlighted inside the blue circle). However, comparing the areas in red circles, the TS-III′-IVtrans(S,S,S) system had stronger noncovalent interactions between the oxazolidinone group and the phenyl ring on the imine. This suggests that noncovalent interactions (i.e., between the oxazolidinone moiety and the protecting group) are critical for control of diastereoselectivity. Taken together, these results suggest that both the conformational preference for the s-trans geometry about the N-enamide group of the substituted Cu(allyl) ligand and favorable noncovalent interactions between the oxazolidinone group and the imine are the major contributing factors for diastereocontrol in these reactions.
Figure 4.

(A) Distortion–Interaction analysis of key diastereomeric C–C bond formation transition states. Electronic energies reported at B3LYP-D3/def2SVP-CPCM(toluene) level of theory. (B) Noncovalent Interaction analysis of key diastereomeric C–C bond formation transition states. Color code for the atoms is shown.
Figure 5.
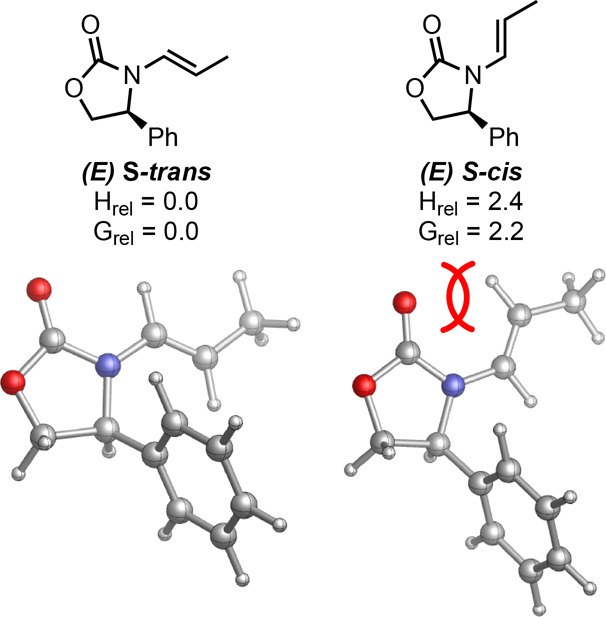
Energetic comparison of s-trans and s-cis conformations of an (E)-enamide system with chiral oxazolidinone, with steric hindrance causing allylic strain highlighted. Structures optimized using B3LYP-D3/def2SVP-CPCM(toluene) (Hrel and Grel shown in kcal/mol).
Following C–C bond formation, intermediate IV serves as a fork between two reaction pathways depending on whether or not there is t-BuOH present (as supported by experiments; vide supra). Specifically, in the presence of t-BuOH, the alcohol can act as a proton source to protonate the Cu–N bond and yield branched product 18, while the alkoxide binds to the copper. From this, the silane reagent can exchange hydride for the alkoxide group, reforming the catalyst. While this protonation of the amine moiety and concomitant release of the t-BuO-CuL was calculated to be initially energetically unfavorable (uphill by ∼4 kcal/mol), the exchange of hydride for the alkoxide was thermodynamically favorable (downhill by ∼10 kcal/mol), rendering this overall process energetically feasible. In the absence of t-BuOH, a thermodynamically favorable rearrangement of intermediate IV can yield the Cu-alkoxide urea intermediate V (∼7 kcal/mol exergonic), which can then readily undergo transmetalation with silane to reform the LCuH catalyst and furnish the silylated product of urea 19. This mechanistic model is consistent with experimental findings that the presence of t-BuOH has a profound effect on the product selectivity (but not diastereoselectivity) of the reaction toward either of the products (vide supra).
As previously noted, computational modeling of the imine addition predicts this step should be reversible. This phenomenon has important impacts for future developments of catalyst controlled enantioselective reactions utilizing a chiral catalyst in conjunction with an achiral allenamide. In this regard, reaction of achiral allenamide 15b with imine 9a using (S,S)-Ph-BPE as a chiral ligand was examined with and without t-BuOH as the additive (Scheme 7). Again, branched product 29 was formed as a single diastereomer when t-BuOH was present in the reaction, and urea 30 was formed as a single diastereomer in the absence of t-BuOH. Separate conversion of 29 to 30 using n-BuLi confirmed that the same relative stereochemistry was formed in both reactions. Importantly, 29 and 30 were formed in different enantiopurities (57:43 vs 80:20 er, respectively), supporting a reversible imine addition step in these reactions. For example, if imine addition were irreversible, reaction of 15b with a chiral catalyst to afford the analogous intermediate to 22 (Scheme 3) must be enantiodetermining and requires that urea product 30 formed from rearrangement of the intermediate 22 derivative to have identical enantiopurity to that of 29. However, when a chiral ligand is employed, rearrangement of the two enantiomers of intermediate 22 may occur at different rates because the transition states will be diastereomeric due to the chirality on the ligand. Therefore, the carbamate migration step may also be enantiodetermining if the imine addition step becomes reversible enabling different enantiomeric ratios to be obtained for a 29-selective vs a 30-selective process as was observed.
Scheme 7. Mechanistic Implications Relevant to Catalyst-Controlled Enantioinduction.

Conclusion
In conclusion, a highly stereoselective method for the reductive coupling of imines with a chiral allenamide was developed as a convenient strategy for the asymmetric synthesis of valuable 1,2-diamino synthons. The method employs readily available and cost-effective starting materials26 and catalyst (Cu(OAc)2/PCy3)32 and can be performed on the “bench-top” using standard Schlenk techniques without issues. Use of tert-butanol as an additive was shown to aid in the amine release and catalyst regeneration to avoid the formation of urea products that are exclusively obtained in the absence of this additive. The oxazolidinone moiety of the final products could be removed chemoselectively without disruption of the pendant terminal alkene, and an orthogonally protected chiral aminopiperidine derivative en route to important biologically active pharmaceuticals was demonstrated. Finally, mechanistic investigations by density functional theory calculations identified the mechanism for stereoselection in these processes as determined from the relative transition state barriers of N-substituted allylcopper complexes to the imine electrophile. This C–C bond forming addition step was shown to be reversible by calculation and was experimentally supported by the catalytic asymmetric reaction of a chiral catalyst with an achiral allenamide. These mechanistic insights are important for the development of future asymmetric catalyst-controlled procedures and are currently under further investigation in these laboratories.
Experimental Section
General
1H NMR spectra were recorded on Bruker 600 MHz spectrometers. Chemical shifts are reported in ppm from tetramethylsilane with the solvent resonance as the internal standard (CDCl3: 7.26 ppm). Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, h = hextet, hept = heptet, br = broad, m = multiplet), and coupling constants (Hz). 13C NMR spectra were recorded on a Bruker 600 MHz (151 MHz) instrument with complete proton decoupling. Chemical shifts are reported in ppm from tetramethylsilane with the solvent as the internal standard (CDCl3: 77.0 ppm). Liquid chromatography was performed using forced flow (flash chromatography) on silica gel purchased from Silicycle. Thin layer chromatography (TLC) was performed on glass-backed 250 μm silica gel F254 plates purchased from Silicycle. Visualization was achieved by using UV light, a 10% solution of phosphomolybdic acid in EtOH, or potassium permanganate in water followed by heating. HRMS was collected using a Jeol AccuTOF-DART mass spectrometer using DART source ionization. All reactions were conducted in oven or flame-dried glassware under an inert atmosphere of nitrogen or argon with magnetic stirring unless otherwise noted. Solvents were obtained from VWR as HPLC grade and transferred to septa sealed bottles, degassed by argon sparge, and analyzed by Karl Fischer titration to ensure water content was ≤600 ppm. Me(MeO)2SiH was purchased from Alfa Aesar and used as received. Allenamides 15 were prepared in one step as described in the literature.26 Aldehydes were purchased from Sigma-Aldrich, Combi-Blocks, TCI America, Alfa Aesar, or Oakwood Chemicals and used as received. Tricyclohexylphosphine and Cu(OAc)2 were purchased from the Strem Chemical Company and used as received. All other materials were purchased from VWR, Sigma-Aldrich, Combi-Blocks, or Alfa Aesar and used as received. Imines 9a,429b,439c,449ee,459h,469i,479j,489l,459m,479pb,499pc,50 and 9u(45) were synthesized as described in the literature.
General Procedure A for the Synthesis of Imines
A 25 mL round-bottom flask equipped with a magnetic stirring bar was charged with aldehyde (6.0 mmol, 1.0 equiv) and dichloromethane (8 mL). Anhydrous magnesium sulfate was added to this solution while stirring followed by 2,4-dimethoxy benzylamine (6.0 mmol, 1.0 equiv) dropwise. The reaction mixture was stirred at room temperature for 12 h under a nitrogen atmosphere. After the reaction is complete the crude reaction mixture was filtered through Celite to remove magnesium sulfate. The filtrate was concentrated in vacuo to yield the pure imine, which was stored under nitrogen in the fridge.
(E)-1-(4-Chlorophenyl)-N-(2,4-dimethoxybenzyl)methanimine (9d)
Following General Procedure A, 4-chloro benzaldehyde (0.84 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a pale-yellow solid (1.54 g, 89%). Mp −59.5–60.5 °C. 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 7.70 (d, J = 8.5 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 7.21–7.15 (m, 1H), 6.51–6.43 (m, 2H), 4.75 (s, 2H), 3.81 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 160.2, 158.3, 136.4, 134.9, 130.2, 129.4, 128.8, 119.6, 104.1, 98.5, 58.9, 55.4. HRMS (DART) m/z calcd for C16H17ClNO2 [M + H]+: 290.0948; Found [M + H]+: 290.0950.
(E)-1-(4-Chlorophenyl)-N-(4-(trifluoromethyl)benzyl)methanimine (9de)
Following General Procedure A, 4-chloro benzaldehyde (0.84 g, 6.0 mmol), 4-trifluoromethyl benzylamine (1.05 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as brown solid (1.5 g, 84%). Mp −39.7–41.3 °C. 1H NMR (600 MHz, CDCl3) δ 8.39 (s, 1H), 7.80 (dd, J = 8.5, 5.6 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.12 (t, J = 8.6 Hz, 2H), 4.85 (s, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.2, 143.3, 137.0, 134.4, 129.66 (C–F, 2J C–F = 33.22 Hz), 129.5, 129.45 (C–F, 2J C–F = 33.22 Hz), 129.23 (C–F, 2J C–F = 33.22 Hz), 128.9, 128.1, 126.97 (C–F, 1J C–F = 273.31 Hz), 125.49 (C–F, 3J C–F = 3.02 Hz), 125.46 (C–F, 3J C–F = 3.02 Hz), 125.44 (C–F, 3J C–F = 3.02 Hz), 125.41 (C–F, 3J C–F = 3.02 Hz), 125.17 (C–F, 1J C–F = 273.31 Hz), 123.36 (C–F, 1J C–F = 273.31 Hz), 121.56 (C–F, 1J C–F = 273.31 Hz), 64.3. 19F NMR (565 MHz, CDCl3) δ −108.88. HRMS (DART) m/z calcd for C15H12ClF3N [M + H]+: 298.0610; Found [M + H]+: 298.0640.
(E)-N-(2,4-Dimethoxybenzyl)-1-(4-fluorophenyl)methanimine (9e)
Following General Procedure A, 4-fluorobenzaldehyde (0.742 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a yellow solid (1.36 g, 84%). Mp −39.7–41.1 °C. 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1H), 7.76 (dd, J = 8.5, 5.7 Hz, 2H), 7.23–7.16 (m, 1H), 7.08 (t, J = 8.6 Hz, 2H), 6.52–6.45 (m, 2H), 4.76 (s, 2H), 3.81 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 165.06 (C–F, 1J C–F = 250.66 Hz), 163.40 (C–F, 1J C–F = 250.66 Hz), 160.23, 160.21, 158.32, 132.80 (C–F, 3J C–F = 3.02 Hz), 132.78 (C–F, 3J C–F = 3.02 Hz), 132.2, 130.15, 130.11, 130.06, 119.84, 115.66 (C–F, 2J C–F = 22.65 Hz), 115.51 (C–F, 2J C–F = 22.65 Hz), 104.1, 98.54, 58.85. 19F NMR (565 MHz, CDCl3) δ −109.87. HRMS (DART) m/z calcd for C16H17FNO2 [M + H]+: 274.1243; Found [M + H]+: 274.1269.
(E)-4-(2,4-Dimethoxybenzyl iminomethyl)benzonitrile (9f)
Following General Procedure A, 4-formyl benzonitrile (0.784 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a yellow solid (1.59 g, 95%). Mp −54.0–56.2 °C. 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 9.0 Hz, 1H), 6.51–6.44 (m, 2H), 4.80 (s, 2H), 3.80 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.4, 159.6, 158.4, 140.3, 132.3, 130.3, 128.6, 119.0, 118.6, 113.7, 104.2, 98.5, 59.1, 55.4. HRMS (DART) m/z calcd for C17H17N2O2 [M + H]+: 281.1290; Found [M + H]+: 281.1306.
Methyl (E)-4-(2,4-Dimethoxybenzyl iminomethyl)benzoate (9g)
Following General Procedure A, methyl-4-formyl benzoate (0.982 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a yellow solid (1.87 g, 100%). Mp −54.5–56.3 °C. 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 8.06 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 9.0 Hz, 1H), 6.48 (m, 2H), 4.79 (s, 2H), 3.91 (s, 3H), 3.79 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.7, 160.6, 160.2, 158.3, 140.3, 131.6, 130.2, 129.7, 128.0, 119.4, 104.1, 98.5, 59.1, 55.3, 55.3, 52.2. HRMS (DART) m/z calcd for C18H20NO4 [M + H]+: 314.1392; Found [M + H]+: 314.1422.
(E)-N-(2,4-Dimethoxybenzyl)-1-(pyridin-3-yl)methanimine (9k)
Following General Procedure A, 3-pyridinecarboxaldehyde (0.64 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a pale-yellow oil (1.51 g, 99%). 1H NMR (600 MHz, CDCl3) δ: 8.85 (s, 1H), 8.63 (d, J = 4.8 Hz, 1H), 8.34 (s, 1H), 8.14 (d, J = 7.9 Hz, 1H), 7.33–7.31 (dd, J = 7.9 Hz, 4.8 Hz, 1H), 7.19 (d, J = 8.9 Hz, 1H), 6.49–6.47 (m, 2H), 4.78 (s, 2H), 3.81 (s, 3H), 3.80 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.7, 158.3, 151.3, 150.2, 134.5, 131.9, 130.3, 123.6, 119.2, 104.1, 98.5, 59.1, 55.3. HRMS (DART) m/z calcd for C15H17N2O2 [M + H]+: 257.1290; Found [M + H]+: 257.1297.
(E)-1-(Benzo[d][1,3]dioxol-5-yl)-N-(2,4-dimethoxybenzyl)methanimine (9n)
Following General Procedure A, piperonal (0.89 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a pale-yellow solid (1.77 g, 99%). Mp −54.3–55.7 °C. 1H NMR (600 MHz, CDCl3) δ: 8.2 (s, 2H), 7.4 (s, 1H), 7.18 (d, J = 8.9 Hz, 1H), 7.12 (d, J = 7.9 Hz, 1H), 6.8 (d, J = 7.9 Hz, 1H), 6.47–6.46 (m, 2H), 5.9 (s, 2H), 4.71 (s, 2H), 3.808 (s, 3H), 3.802 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.8, 160.0, 158.2, 149.7, 148.2, 131.3, 130.0, 124.3, 120.1, 107.9, 106.7, 104.0, 101.4, 98.5, 58.6, 55.3. HRMS (DART) m/z calcd for C17H18NO4 [M + H]+: 300.1236; Found [M + H]+: 300.1253.
(E)-1-(2,3-Dihydrobenzofuran-5-yl)-N-(2,4-dimethoxybenzyl)methanimine (9o)
Following General Procedure A, 2,3-dihydrobenzofuran-5-carbaldehyde (0.886 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a pale-yellow oil (1.94 g, 72% purity, 78% yield). 1H NMR (600 MHz, CDCl3) δ 8.24 (s, 1H), 7.73 (s, 1H), 7.44 (d, J = 12 Hz, 1H), 7.19 (d, J = 8.9 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 6.49–6.43 (m, 2H), 4.71 (s, 2H), 4.60 (t, J = 8.7 Hz, 2H), 3.80 (s, 6H), 3.20 (t, J = 8.7 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 162.3, 161.3, 160.0, 158.2, 130.0, 129.8, 129.6, 127.7, 124.2, 120.3, 109.0, 104.0, 98.4, 71.7, 58.7, 55.3, 29.2. HRMS (DART) m/z calcd for C18H20NO3 [M + H]+: 298.1443; Found [M + H]+: 298.1466.
(E)-N-(2,4-Dimethoxybenzyl)-1-(naphthalen-2-yl)methanimine (9pa)
Following General Procedure A, 2-naphthaldehyde (0.937 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a white solid (1.78 g, 98%). Mp −91.5–93.9 °C. 1H NMR (600 MHz, CDCl3) δ: 8.48 (s, 1H), 8.05–8.03 (m, 2H), 7.89–7.87 (m, 1H), 7.85–7.83 (m, 2H), 7.51–7.49 (m, 2H), 7.24 (d, J = 8.9 Hz, 1H), 6.49–6.48 (m, 2H), 4.82 (s, 2H), 3.82 (s, 3H), 3.81 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ: 161.7, 160.1, 158.3, 134.6, 134.1, 133.1, 130.1, 129.9, 128.5, 128.3, 127.8, 127.0, 126.3, 124.0, 119.9, 104.9, 104.0, 98.5, 59.0, 55.4. HRMS (DART) m/z calcd for C20H20NO2 [M + H]+: 306.1494; Found [M + H]+: 306.1506.
(E)-N-(4-Fluorobenzyl)-1-(naphthalen-2-yl)methanimine (9pd)
Following General Procedure A, 2-naphthaldehyde (0.937 g, 6.0 mmol), 4-fluoro benzylamine (1.05 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as an off-white solid (1.28 g, 90% purity, 73% yield). Mp −87.7–88.9 °C. 1H NMR (600 MHz, CDCl3) δ 8.53 (s, 1H), 8.09 (s, 1H), 8.08 (d, J = 12.0 Hz, 1H), 7.92–7.86 (m, 3H), 7.54 (tdd, J = 8.0, 6.1, 3.3 Hz, 2H), 7.36 (dd, J = 8.4, 5.5 Hz, 2H), 7.07 (t, J = 8.7 Hz, 2H), 4.85 (s, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 162.8 (C–F, 1J C–F = 244.62 Hz), 162.1, 161.2 (C–F, 1J C–F = 244.62 Hz), 135.14 (C–F, 3J C–F = 3.02 Hz), 135.12 (C–F, 3J C–F = 3.02 Hz), 134.8, 133.7, 133.1, 130.2, 129.6, 129.5, 128.6, 128.5, 127.9, 127.2, 126.5, 123.9, 115.4 (C–F, 2J C–F = 21.14 Hz), 115.2 (C–F, 2J C–F = 21.14 Hz), 64.37. 19F NMR (565 MHz, CDCl3) δ – 115.92. HRMS (DART) m/z calcd for C18H15FN [M + H]+: 264.1189; Found [M + H]+: 264.1193.
(E)-1-(Naphthalen-2-yl)-N-(4-(trifluoromethyl)benzyl)methanimine (9pe)
Following General Procedure A, 2-naphthaldehyde (0.937 g, 6.0 mmol), 4-trifluoromethyl benzylamine (1.05 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as an off-white solid (1.71 g, 91%). Mp −105.9–107.9 °C. 1H NMR (600 MHz, CDCl3) δ 8.56 (s, 1H), 8.10 (s, 1H), 8.09 (d, J = 6 Hz, 1H), 7.95–7.85 (m, 3H), 7.64 (d, J = 8.0 Hz, 2H), 7.58–7.49 (m, 4H), 4.92 (s, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 162.7, 143.6, 134.9, 133.6, 133.1, 130.4, 129.61 (C–F, 2J C–F = 31.71 Hz), 129.40 (C–F, 2J C–F = 31.71 Hz), 129.18 (C–F, 2J C–F = 31.71 Hz), 129.18 (C–F, 2J C–F = 31.71 Hz), 128.69, 128.63, 128.1, 127.9, 127.3, 127.04 (C–F, 1J C–F = 271.8 Hz), 126.6, 125.50 (C–F, 3J C–F = 3.02 Hz), 125.47 (C–F, 3J C–F = 3.02 Hz), 125.45 (C–F, 3J C–F = 3.02 Hz), 125.42 (C–F, 3J C–F = 3.02 Hz), 125.24 (C–F, 1J C–F = 271.8 Hz), 123.8, 123.43 (C–F, 1J C–F = 271.8 Hz), 121.63 (C–F, 1J C–F = 271.8 Hz), 64.4. 19F NMR (565 MHz, CDCl3) δ −62.28. HRMS (DART) m/z calcd for C19H15F3N [M + H]+: 314.1157; Found [M + H]+: 314.1154.
(E)-1-([1,1′-Biphenyl]-4-yl)-N-(2,4-dimethoxybenzyl)methanimine (9q)
Following General Procedure A, 4-phenylbenzaldehyde (1.09 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a white solid (1.82 g, 94% purity, 86% yield). Mp −91.3–93.9 °C. 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.70–7.63 (m, 4H), 7.49 (t, J = 7.6 Hz, 2H), 7.40 (t, J = 6 Hz, 1H), 7.29–7.23 (m, 1H), 6.55–6.50 (m, 2H), 4.83 (s, 2H), 3.86 (s, 3H), 3.84 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.3, 160.1, 158.3, 143.2, 140.5, 135.4, 130.3, 130.1, 129.0, 128.8, 128.7, 128.5, 127.7, 127.4, 127.2, 127.1, 120.0, 104.1, 98.5, 59.0, 55.4. HRMS (DART) m/z calcd for C22H22NO2 [M + H]+: 332.1651; Found [M + H]+: 332.1667.
(E)-N-(2,4-Dimethoxybenzyl)-1-(p-tolyl)methanimine (9r)
Following General Procedure A, p-tolualdehyde (0.72 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a pale-yellow oil (1.28 g, 80%). 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 7.72 (d, J = 7.8 Hz, 2H), 7.27 (d, J = 7.4 Hz, 3H), 6.55–6.51 (m, 2H), 4.81 (s, 2H), 3.86 (s, 6H), 2.38 s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.7, 160.1, 158.2, 140.7, 133.8, 130.0, 129.8, 129.7, 129.2, 128.2, 120.1, 104.0, 98.5, 58.9, 55.3, 21.5. HRMS (DART) m/z calcd for C17H20NO2 [M + H]+: 270.1494; Found [M + H]+: 270.1495.
(E)-N-(2,4-Dimethoxybenzyl)-1-(furan-2-yl)methanimine (9s)
Following General Procedure A, furfural (0.57 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a brown oil (1.6 g, 91% purity, 99% yield). 1H NMR (600 MHz, CDCl3) δ: 8.08 (s, 1H), 7.49 (s, 1H), 7.18 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 3.4 Hz, 1H), 6.47–6.45 (m, 3H), 4.73 (s, 2H), 3.80 (s, 3H), 3.79 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ: 160.28, 158.46, 151.96, 150.13, 144.51, 130.69, 119.30, 113.59, 111.53, 104.07, 98.47, 58.89, 55.40, 55.33. HRMS (DART) m/z calcd for C14H16NO3 [M + H]+: 246.1130; Found [M + H]+: 246.1126.
(E)-N-(2,4-Dimethoxybenzyl)-1-(thiophen-2-yl)methanimine (9t)
Following General Procedure A, thiophene-2-carboxaldehyde (0.67 g, 6.0 mmol), 2,4-dimethoxybenzylamine (1.0 g, 6.0 mmol), magnesium sulfate (2.0 g), and dichloromethane (8 mL) were used. The title compound was obtained as a yellow solid (1.15 g, 74%). Mp −47.4–50.2 °C. 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 7.37 (d, J = 6 Hz, 1H), 7.29 (d, J = 3.6 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.06 (t, J = 6 Hz, 1H), 6.50–6.46 (m, 2H), 4.74 (s, 2H), 3.80 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.3, 154.8, 142.9, 130.3, 130.2, 128.6, 127.2, 119.6, 104.1, 98.4, 58.2, 55.4, 55.3. HRMS (DART) m/z calcd for C14H16NO2S [M + H]+: 262.0902; Found [M + H]+: 262.0915.
General Procedure B for the Synthesis of 18
To a 20 mL crimp cap vial with a stir bar in an Ar filled glovebox were charged Cu(OAc)2 (3.6 mg, 20 μmol) and PCy3 (7.3 mg, 26 μmol) followed by toluene (1.0 mL) and tert-butanol (76.5 μL, 2 equiv). The mixture was stirred for 5 min. Allenamide 15a (96.6 mg, 480 μmol) followed by imine (400 μmol) was then charged, and the vial was sealed with a crimp-cap septum and removed from the glovebox. Dimethoxymethylsilane (0.099 mL, 2 equiv) was then charged to the reaction mixture (Caution:dimethoxymethylsilane should be handled in a well-ventilated fume hood because it is known to cause blindness. Syringes were quenched with 2 M NaOH, gas evolution! prior to disposal). The mixture was then stirred at rt for 24 h. The reaction was quenched by addition of 200 mg of NH4F and 2.5 mL of MeOH followed by agitation at rt for 30 min. A 10 mL volumen of 5% NaHCO3 was then added to the mixture followed by extraction with DCM (2 × 5 mL). The combined organics were dried with Na2SO4, filtered, and concentrated in vacuo. Crude product was purified by flash chromatography on silica gel to afford the desired product.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(4-(trifluoromethyl)phenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18a)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 180 mg (85%) of 18a as a white foam as a single diastereomer. Rf = 0.43 (50% EtOAC/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.50 (d, J = 8.0 Hz, 1H), 7.36 (dd, J = 5.1, 1.8 Hz, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.21–7.17 (m, 1H), 6.99 (d, J = 6 Hz, 1H), 6.49 (s, 1H), 6.47 (d, J = 8.3 Hz, 1H), 5.15–5.09 (dt, J = 18 Hz, 12 Hz, 1H), 4.73 (d, J = 12 Hz, 1H), 4.70 (d, J = 17.1 Hz, 1H), 4.61 (t, J = 8.2 Hz, 1H), 4.52 (t, J = 12 Hz, 1H), 4.17 (d, J = 6 Hz, 1H), 4.11 (t, J = 8.0 Hz, 1H), 3.93 (t, J = 9.6 Hz, 1H), 3.83 (s, 6H). 3.66 (d, J = 12 Hz, 1H), 3.36 (d, J = 18 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.8, 158.3, 145.2, 138.2, 132.6, 130.5, 129.94 (C–F, 2J C–F = 31.71 Hz), 129.73 (C–F, 2J C–F = 31.71 Hz), 129.51 (C–F, 2J C–F = 31.71 Hz), 129.29 (C–F, 2J C–F = 31.71 Hz), 129.17, 129.11, 128.8, 127.8, 126.90 (C–F, 1J C–F = 271.8 Hz), 125.12 (C–F, 3J C–F = 4.53 Hz), 125.09 (C–F, 2J C–F = 4.53 Hz), 123.29 (C–F, 1J C–F = 271.8 Hz), 121.49 (C–F, 1J C–F = 271.8 Hz), 120.5, 119.6, 103.6, 98.6, 70.2, 63.4, 61.2, 59.3, 55.4, 55.2, 46.0. 19F NMR (565 MHz, CDCl3) δ – 62.36. HRMS (DART) m/z calcd for C29H30F3N2O4 [M + H]+: 527.2158; Found [M + H]+: 527.2153.
(S)-3-((1S,2S)-1-((2,4-dimethoxybenzyl)amino)-1-phenylbut-3-en-2-yl)-4-phenyloxazolidin-2-one (18b)
According to General Procedure B, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 147 mg (80%) of 18b as a colorless foam as a single diastereomer. Rf = 0.35 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.32 (dd, J = 4.5, 2.3 Hz, 3H), 7.25–7.22 (d, J = 6 Hz, 2H), 7.22–7.19 (d, J = 6 Hz, 1H), 7.18–7.16 (d, J = 12 Hz, 2H), 7.15–7.14 (m, 2H), 7.03 (d, J = 8.0 Hz, 1H), 6.48 (s, 1H), 6.46 (d, J = 8.3 Hz, 1H), 5.05–4.97 (dt, J = 18 Hz, 12 Hz, 1H), 4.70 (d, J = 17.0 Hz, 1H), 4.64 (d, J = 10.2 Hz, 1H), 4.59 (t, J = 6 Hz, 1H), 4.46 (t, J = 8.6 Hz, 1H), 4.12–4.04 (dt, J = 18 Hz, 6 Hz, 2H), 3.99 (d, J = 10.0 Hz, 1H), 3.81 (s, 6H), 3.67 (d, J = 13.4 Hz, 1H), 3.34 (d, J = 13.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.6, 140.6, 138.9, 133.3, 130.6, 128.98, 128.96, 128.4, 128.2, 127.8, 127.4, 120.9, 119.0, 103.6, 98.6, 70.2, 63.2, 61.5, 58.9, 55.4, 55.2, 45.8. HRMS (DART) m/z calcd for C28H31N2O4 [M + H]+: 459.2284; Found [M + H]+: 459.2300.
(S)-3-((1S,2S)-1-((4-Methoxybenzyl)amino)-1-phenylbut-3-en-2-yl)-4-phenyloxazolidin-2-one (18c)
According to General Procedure B, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 153 mg (89%) of 18c as a white solid as a single diastereomer. Mp 101–104 °C. Rf = 0.41 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.37–7.33 (m, 3H), 7.31 (m, 2H), 7.27 (m, 3H), 7.20 (d, J = 6 Hz, 2H), 7.18–7.14 (m, 2H), 6.96 (d, J = 12 Hz, 2H), 5.10 (dt, J = 16.5, 9.6 Hz, 1H), 4.76–4.69 (m, 2H), 4.66 (t, J = 8.4 Hz, 1H), 4.60 (t, J = 12 Hz, 1H), 4.18 (t, J = 12 Hz, 1H), 4.12 (t, J = 12 Hz, 1H), 4.02 (d, J = 10.2 Hz, 1H), 3.86 (s, 3H), 3.64 (d, J = 13.0 Hz, 1H), 3.41 (d, J = 12.9 Hz, 1H), 1.91 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 159.1, 158.7, 140.5, 138.5, 133.2, 132.5, 129.8, 128.9, 128.9, 128.4, 128.2, 127.8, 127.5, 119.2, 113.7, 70.3, 63.2, 61.5, 60.4, 59.0, 55.3, 49.9, 21.0, 14.2. HRMS (DART) m/z calcd for C27H29N2O3 [M + H]+: 429.2178; Found [M + H]+: 429.2196.
(S)-3-((1S,2S)-1-(4-Chlorophenyl)-1-((2,4-dimethoxybenzyl)amino)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18d)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 146 mg (74%) of 18d as a colorless foam and a single diastereomer. Rf = 0.33 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.36–7.33 (m, 3H), 7.23 (d, J = 8.2 Hz, 2H), 7.20–7.16 (m, 2H), 7.12 (d, J = 8.1 Hz, 2H), 6.99 (d, J = 8.1 Hz, 1H), 6.49 (s, 1H), 6.47 (d, J = 8.2 Hz, 1H), 5.11–5.03 (dt, J = 18 Hz, 12 Hz, 1H), 4.72 (s, 1H), 4.69 (d, J = 7.9 Hz, 1H), 4.60 (t, J = 8.1 Hz, 1H), 4.49 (t, J = 8.6 Hz, 1H), 4.10 (t, J = 7.9 Hz, 1H), 4.05 (d, J = 10.1 Hz, 1H), 3.95 (t, J = 9.6 Hz, 1H), 3.83 (s, 6H), 3.66 (d, J = 13.4 Hz, 1H), 3.33 (d, J = 13.4 Hz, 1H), 2.08 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.8, 158.4, 139.3, 138.4, 133.0, 132.8, 130.6, 129.8, 129.10, 129.07, 128.4, 127.8, 120.6, 119.4, 103.6, 98.6, 70.2, 63.4, 60.8, 59.2, 55.4, 55.2, 45.9. HRMS (DART) m/z calcd for C28H30ClN2O4 [M + H]+: 493.1894; Found [M + H]+: 493.1929.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(4-fluorophenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18e)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 147 mg (77%) of 18e as a colorless foam and a single diastereomer. Rf = 0.35 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.37–7.32 (m, 3H), 7.20–7.12 (m, 4H), 7.00 (d, J = 8.1 Hz, 1H), 6.94 (t, J = 8.6 Hz, 2H), 6.50 (s, 1H), 6.48 (d, J = 6 Hz, 1H), 5.06 (dt, J = 18.2, 9.5 Hz, 1H), 4.70 (d, J = 16.6 Hz, 2H), 4.60 (t, J = 8.1 Hz, 1H), 4.49 (t, J = 8.6 Hz, 1H), 4.10 (t, J = 7.9 Hz, 1H), 4.05 (d, J = 10.1 Hz, 1H), 3.96 (t, J = 9.6 Hz, 1H), 3.83 (s, 6H), 3.66 (d, J = 13.4 Hz, 1H), 3.34 (d, J = 13.5 Hz, 1H), 2.10 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 162.93 (C–F, 1J C–F = 244.62 Hz), 161.31 (C–F, 1J C–F = 244.62 Hz), 160.2, 158.8, 158.4, 138.5, 136.4, 133.0, 130.5, 129.9, 129.8, 129.07 (C–F, 3J C–F = 4.53 Hz), 129.05 (C–F, 3J C–F = 4.53 Hz), 127.8, 120.7, 119.2, 115.13 (C–F, 2J C–F = 21.14 Hz), 114.99 (C–F, 2J C–F = 21.14 Hz), 103.6, 98.6, 70.2, 63.5, 60.7, 59.1, 55.4, 55.2, 45.9. 19F NMR (565 MHz, CDCl3) δ −115.14. HRMS (DART) m/z calcd for C28H30FN2O4 [M + H]+: 477.2190; Found [M + H]+: 477.2204.
4-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-2-((S)-2-oxo-4-phenyloxazolidin-3-yl)but-3-en-1-yl)benzonitrile (18f)
According to General Procedure B, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 157 mg (81%) of 18f as a colorless foam and a single diastereomer. Rf = 0.36 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.54 (d, J = 7.9 Hz, 2H), 7.39–7.34 (m, 3H), 7.30 (d, J = 7.9 Hz, 2H), 7.22–7.18 (m, 2H), 6.95 (d, J = 8.1 Hz, 1H), 6.49 (s, 1H), 6.46 (d, J = 8.1 Hz, 1H), 5.14 (dt, J = 16.9, 9.8 Hz, 1H), 4.73 (d, J = 10.1 Hz, 1H), 4.66 (d, J = 17.0 Hz, 1H), 4.59 (t, J = 8.2 Hz, 1H), 4.51 (t, J = 8.6 Hz, 1H), 4.20 (d, J = 10.0 Hz, 1H), 4.12 (t, J = 8.1 Hz, 1H), 3.82 (s, 7H), 3.63 (d, J = 13.5 Hz, 1H), 3.32 (d, J = 13.5 Hz, 1H), 2.20 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.4, 158.7, 158.2, 146.8, 137.9, 132.0, 130.5, 129.3, 129.2, 129.1, 127.8, 120.3, 119.9, 118.8, 111.2, 103.7, 98.6, 70.2, 63.4, 61.3, 59.4, 55.4, 55.2, 46.2. HRMS (DART) m/z calcd for C29H30N3O4 [M + H]+: 484.2236; Found [M + H]+: 484.2255.
Methyl 4-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-2-((S)-2-oxo-4-phenyloxazolidin-3-yl)but-3-en-1-yl)benzoate (18g)
According to General Procedure B, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 137 mg (66%) of 18g as a colorless foam and a single diastereomer. Rf = 0.34 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.93 (d, J = 8.0 Hz, 2H), 7.35 (dd, J = 4.6, 1.6 Hz, 3H), 7.29–7.24 (m, 3H), 7.21–7.15 (m, 2H), 6.99 (d, J = 8.1 Hz, 1H), 6.49 (s, 1H), 6.47 (d, J = 8.2 Hz, 1H), 5.07 (dt, J = 14.2, 9.5 Hz, 1H), 4.67 (d, J = 16.5 Hz, 2H), 4.61 (t, J = 8.1 Hz, 1H), 4.49 (t, J = 8.6 Hz, 1H), 4.11 (m, 2H), 4.01 (t, J = 9.6 Hz, 1H), 3.89 (s, 3H), 3.82 (s, 6H), 3.66 (d, J = 13.4 Hz, 1H), 3.33 (d, J = 13.4 Hz, 1H), 2.18 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.9, 160.3, 158.8, 158.4, 146.4, 138.4, 132.7, 130.6, 129.5, 129.4, 129.1, 129.0, 128.5, 127.8, 120.6, 119.5, 103.6, 98.6, 70.2, 63.3, 61.3, 59.1, 55.4, 55.2, 52.0, 46.0. HRMS (DART) m/z calcd for C30H33N2O6 [M + H]+: 517.2339; Found [M + H]+: 517.2378.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(4-nitrophenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18h)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 106 mg (53%) of 18h as a pale-yellow foam and a single diastereomer. Rf = 0.39 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.13–8.07 (m, 2H), 7.40–7.32 (m, 5H), 7.21 (dd, J = 6.3, 2.4 Hz, 2H), 6.95 (d, J = 8.1 Hz, 1H), 6.49 (s, 1H), 6.46 (d, J = 8.1 Hz, 1H), 5.17 (dt, J = 16.9, 9.8 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.66 (d, J = 17.0 Hz, 1H), 4.61 (t, J = 8.3 Hz, 1H), 4.52 (t, J = 8.6 Hz, 1H), 4.27 (d, J = 10.0 Hz, 1H), 4.16–4.10 (m, 1H), 3.82 (s, 6H), 3.64 (d, J = 13.5 Hz, 1H), 3.33 (d, J = 13.5 Hz, 1H), 2.28 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.4, 158.7, 158.2, 147.4, 137.9, 132.2, 130.5, 129.4, 129.3, 129.2, 127.8, 123.4, 120.2, 120.0, 103.7, 98.7, 70.2, 63.5, 61.1, 59.5, 55.4, 55.2, 46.2. HRMS (DART) m/z calcd for C28H30N3O6 [M + H]+: 504.2135; Found [M + H]+: 504.2119.
(S)-3-((1S,2S)-1-(Benzylamino)-1-(3-nitrophenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18i)
The reaction was set up according to General Procedure B and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 88 mg (50%) of 18i as a pale-yellow foam and a single diastereomer and as a 91:9 mixture of the branched 18i to the rearranged product 19i. Rf = 0.71 (20% EtOAc/DCM). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 6.0 Hz, 1H), 7.91 (s, 1H), 7.47 (d, J = 7.7 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.28–7.16 (m, 9H), 7.05 (d, J = 6.7 Hz, 2H), 5.16 (dt, J = 17.0, 9.7 Hz, 1H), 4.69 (d, J = 10.1 Hz, 1H), 4.59 (d, J = 17.0 Hz, 1H), 4.53 (t, J = 8.4 Hz, 1H), 4.47 (t, J = 8.6 Hz, 1H), 4.22 (d, J = 10.0 Hz, 1H), 4.02 (t, J = 8.2 Hz, 1H), 3.77 (t, J = 9.8 Hz, 1H), 3.56 (d, J = 13.3 Hz, 1H), 3.36 (d, J = 13.2 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 158.7, 148.3, 137.5, 134.3, 132.1, 129.4, 129.3, 129.2, 128.49, 128.47, 127.7, 127.3, 123.3, 122.8, 120.3, 70.2, 63.5, 61.5, 59.6, 50.9. HRMS (DART) m/z calcd for C26H26N3O4 [M + H]+: 444.1923; Found [M + H]+: 444.1952.
(S)-3-((1S,2S)-1-(Benzylamino)-1-(3-bromophenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18j)
The reaction was set up according to General Procedure B and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (2.5% E.A. in DCM) to provide 173 mg (82%) of 18j as a pale-yellow foam and a single diastereomer. Rf = 0.53 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.41–7.29 (m, 10H), 7.16 (t, J = 7.7 Hz, 1H), 7.14–7.09 (m, 3H), 5.14 (dt, J = 16.8, 9.5 Hz, 1H), 4.75 (dd, J = 18 Hz, 12 Hz, 2H), 4.60 (t, J = 8.3 Hz, 1H), 4.55 (t, J = 8.5 Hz, 1H), 4.09 (t, J = 8.0 Hz, 1H), 4.04 (d, J = 10.2 Hz, 1H), 3.99 (t, J = 9.5 Hz, 1H), 3.68 (d, J = 13.2 Hz, 1H), 3.45 (d, J = 13.2 Hz, 1H), 1.96 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 159.0, 143.1, 140.1, 138.1, 132.6, 131.2, 130.7, 130.0, 129.1, 129.0, 128.5, 128.4, 127.8, 127.2, 126.8, 122.6, 119.7, 70.2, 63.3, 61.4, 59.3, 50.7. HRMS (DART) m/z calcd for C26H26BrN2O2 [M + H]+: 477.1178; Found [M + H]+: 477.1182.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(pyridin-3-yl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18k)
According to General Procedure B, the product was purified by silica gel chromatography (40% E.A. in DCM) to provide 143 mg (78%) of 18k as a pale-yellow foam as a single diastereomer. Rf = 0.1 (60% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.40 (d, J = 73.8 Hz, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.38–7.31 (m, 3H), 7.23–7.14 (m, 3H), 6.99 (d, J = 8.1 Hz, 1H), 6.49 (s, 1H), 6.47 (d, J = 8.1 Hz, 1H), 5.09 (dt, J = 17.0, 9.8 Hz, 1H), 4.72 (d, J = 10.2 Hz, 1H), 4.68 (d, J = 17.1 Hz, 1H), 4.57 (t, J = 8.1 Hz, 1H), 4.50 (t, J = 8.6 Hz, 1H), 4.16–4.08 (m, 2H), 3.93 (t, J = 9.7 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.67 (d, J = 13.5 Hz, 1H), 3.35 (d, J = 13.5 Hz, 1H), 2.24 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.4, 158.8, 158.3, 150.5, 149.0, 138.1, 136.3, 135.6, 132.5, 130.6, 129.2, 129.1, 127.8, 123.5, 120.3, 120.0, 103.7, 98.7, 70.2, 63.4, 59.2, 59.0, 55.4, 55.2, 46.0. HRMS (DART) m/z calcd for C27H30N3O4 [M + H]+: 460.2236; Found [M + H]+: 460.2264.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(4-methoxyphenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18l)
According to General Procedure B, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 143 mg (73%) of 18l as a colorless foam and a single diastereomer. Rf = 0.25 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.33 (dd, J = 4.4, 2.3 Hz, 3H), 7.16 (dd, J = 5.9, 2.7 Hz, 2H), 7.10 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.1 Hz, 1H), 6.83–6.78 (m, 2H), 6.50 (s, 1H), 6.49–6.45 (d, J = 12 Hz, 1H), 5.06–4.98 (dt, J = 18 Hz, 12 Hz, 1H), 4.73 (d, J = 17.0 Hz, 1H), 4.67 (d, J = 10.1 Hz, 1H), 4.59 (t, J = 7.9 Hz, 1H), 4.47 (t, J = 12 Hz, 1H), 4.11–4.04 (m, 2H), 3.97 (d, J = 10.0 Hz, 1H), 3.83 (s, 6H), 3.78 (s, 3H), 3.68 (d, J = 13.5 Hz, 1H), 3.35 (d, J = 13.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.21, 158.9, 158.8, 158.5, 138.9, 133.4, 132.5, 130.6, 129.4, 128.96, 128.94, 127.8, 121.0, 118.9, 113.5, 103.6, 98.6, 70.2, 63.3, 60.8, 58.9, 55.4, 55.2, 55.1, 45.7. HRMS (DART) m/z calcd for C29H33N2O5 [M + H]+: 489.2389; Found [M + H]+: 489.2386.
(S)-3-((1S,2S)-1-(Benzylamino)-1-(2-methoxyphenyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18m)
The reaction was set up according to General Procedure B and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (10% E.A. in DCM) to provide 127 mg (74%) of 18m as a colorless foam as a single diastereomer and as a 95:5 mixture of the branched 18m to rearranged product 19m. Rf = 0.36 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.27–7.20 (m, 4H), 7.20–7.11 (m, 5H), 7.10 (t, J = 6 Hz, 1H), 6.92 (d, J = 6 Hz, 3H), 6.76 (t, J = 7.4 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 4.76 (dt, J = 18.1, 9.4 Hz, 1H), 4.61 (d, J = 16.8 Hz, 1H), 4.48 (t, J = 8.3 Hz, 2H), 4.42 (d, J = 10.1 Hz, 1H), 4.38 (t, J = 8.7 Hz, 1H), 4.08–3.95 (m, 1H), 3.92 (t, J = 7.9 Hz, 1H), 3.60 (d, J = 8.2 Hz, 1H), 3.59 (s, 3H), 3.29 (d, J = 13.0 Hz, 1H), 2.11 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 159.4, 157.9, 140.8, 139.3, 134.3, 129.0, 128.68, 128.65, 128.49, 128.45, 128.2, 128.0, 127.0, 120.4, 118.0, 110.6, 70.2, 58.3, 55.1, 50.7. HRMS (DART) m/z calcd for C27H29N2O3 [M + H]+: 429.2178; Found [M + H]+: 429.2194.
(S)-3-((1S,2S)-1-(Benzo[d][1,3]dioxol-5-yl)-1-((2,4-dimethoxybenzyl)amino)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18n)
According to General Procedure B, the product was purified by silica gel chromatography (15% E.A. in DCM) to provide 163 mg (81%) of 18n as a colorless foam and a single diastereomer. Rf = 0.26 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.37–7.31 (m, 3H), 7.19–7.11 (m, 2H), 7.04 (d, J = 8.1 Hz, 1H), 6.72 (s, 1H), 6.69 (d, J = 7.9 Hz, 1H), 6.62 (d, J = 7.9 Hz, 1H), 6.49 (s, 1H), 6.48 (d, J = 7.3 Hz, 1H), 5.92 (s, 2H), 5.05 (dt, J = 17.3, 8.6 Hz, 1H), 4.75 (d, J = 17.0 Hz, 1H), 4.71 (d, J = 10.2 Hz, 1H), 4.58 (t, J = 8.0 Hz, 1H), 4.47 (t, J = 8.6 Hz, 1H), 4.08 (t, J = 7.8 Hz, 1H), 3.97 (m, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.70 (d, J = 12.0 Hz, 1H), 3.36 (d, J = 13.4 Hz, 1H), 2.02 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.4, 147.7, 146.8, 138.6, 134.6, 133.2, 130.6, 129.0, 127.8, 122.0, 120.9, 119.0, 108.1, 107.7, 103.6, 100.9, 98.6, 70.2, 63.4, 61.1, 59.0, 55.4, 55.2, 45.8. HRMS (DART) m/z calcd for C29H31N2O6 [M + H]+: 503.2182; Found [M + H]+: 503.2200.
(S)-3-((1S,2S)-1-(2,3-Dihydrobenzofuran-5-yl)-1-((2,4-dimethoxybenzyl)amino)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18o)
According to General Procedure B, the product was purified by silica gel chromatography (15% E.A. in DCM) to provide 151 mg (75%) of 18o as a colorless foam and a single diastereomer. Rf = 0.18 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.36–7.30 (m, 3H), 7.19–7.13 (m, 2H), 7.07–7.03 (m, 2H), 6.86 (d, J = 8.1 Hz, 1H), 6.65 (d, J = 8.1 Hz, 1H), 6.52–6.45 (m, 2H), 5.00 (dt, J = 16.9, 9.6 Hz, 1H), 4.75 (d, J = 17.0 Hz, 1H), 4.67 (d, J = 10.2 Hz, 1H), 4.58 (t, J = 7.9 Hz, 1H), 4.53 (t, J = 8.7 Hz, 2H), 4.47 (t, J = 8.6 Hz, 1H), 4.08 (t, J = 8.0 Hz, 2H), 3.92 (d, J = 10.1 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H) 3.69 (d, J = 13.4 Hz, 1H), 3.36 (d, J = 13.4 Hz, 1H), 3.15 (t, J = 8.7 Hz, 2H), 2.06 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 159.4, 158.8, 138.9, 133.5, 132.5, 130.6, 128.96, 128.93, 128.4, 127.8, 127.1, 124.4, 121.0, 118.9, 108.6, 103.6, 98.6, 71.2, 70.2, 63.3, 61.0, 58.8, 55.4, 55.2, 45.7, 29.7. HRMS (DART) m/z calcd for C30H33N2O5 [M + H]+: 501.2389; Found [M + H]+: 501.2426.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(naphthalen-2-yl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18p)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 151 mg (74%) of 18p as a colorless foam as a single diastereomer. Rf = 0.30 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.83–7.75 (m, 3H), 7.63 (s, 1H), 7.45 (tdd, J = 7.7, 5.9, 3.2 Hz, 2H), 7.36 (dt, J = 6.4, 2.7 Hz, 4H), 7.23–7.19 (m, 2H), 7.06 (d, J = 7.9 Hz, 1H), 6.49 (m, 2H), 5.10 (dt, J = 17.4, 8.8 Hz, 1H), 4.71 (d, J = 17.0 Hz, 1H), 4.66 (t, J = 8.0 Hz, 1H), 4.62 (d, J = 10.2 Hz, 1H), 4.51 (t, J = 8.6 Hz, 1H), 4.22 (d, J = 7.4 Hz, 2H), 4.11 (t, J = 7.6 Hz, 1H), 3.83 (s, 6H), 3.70 (d, J = 13.4 Hz, 1H), 3.40 (d, J = 13.4 Hz, 1H), 2.23 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.6, 138.8, 138.1, 133.22, 133.21, 133.1, 130.6, 129.0, 128.1, 127.88, 127.80, 127.7, 125.9, 125.8, 125.7, 120.9, 119.2, 103.6, 98.6, 70.3, 63.2, 61.7, 59.1, 55.5, 55.3, 46.0. HRMS (DART) m/z calcd for C32H33N2O4 [M + H]+: 509.2440; Found [M + H]+: 509.2411.
(S)-3-((1S,2S)-1-([1,1′-Biphenyl]-4-yl)-1-((2,4-dimethoxybenzyl)amino)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18q)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 144 mg (67%) of 18q as a colorless foam and a single diastereomer. Rf = 0.26 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.59 (d, J = 7.9 Hz, 2H), 7.51 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.37–7.30 (m, 4H), 7.26 (d, J = 6.0 Hz, 3H), 7.21–7.16 (m, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.52–6.47 (m, 2H), 5.08 (dt, J = 18.1, 9.6 Hz, 1H), 4.76 (d, J = 17.0 Hz, 1H), 4.70 (d, J = 10.2 Hz, 1H), 4.63 (t, J = 8.0 Hz, 1H), 4.50 (t, J = 8.6 Hz, 1H), 4.17–4.05 (m, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.73 (d, J = 13.5 Hz, 1H), 3.42 (d, J = 13.4 Hz, 1H), 2.07 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.6, 140.7, 140.1, 139.8, 138.8, 133.2, 130.6, 129.0, 128.9, 128.8, 128.7, 127.8, 127.2, 126.9, 126.8, 121.0, 119.2, 103.6, 98.6, 70.3, 63.2, 61.3, 59.0, 55.4, 55.2, 45.9. HRMS (DART) m/z calcd for C34H35N2O4 [M + H]+: 535.2597; Found [M + H]+: 535.2631.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(p-tolyl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18r)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 143 mg (76%) of 18r as a colorless foam and a single diastereomer. Rf = 0.29 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.33 (dd, J = 4.6, 2.3 Hz, 3H), 7.18–7.14 (m, 2H), 7.08 (s, 4H), 7.05 (d, J = 8.1 Hz, 1H), 6.51–6.46 (m, 2H), 5.00 (dt, J = 18.1, 9.6 Hz, 1H), 4.74 (d, J = 17.0 Hz, 1H), 4.66 (d, J = 10.2 Hz, 1H), 4.61 (t, J = 7.9 Hz, 1H), 4.47 (t, J = 8.6 Hz, 1H), 4.13 (t, J = 9.6 Hz, 1H), 4.08 (t, J = 6 Hz, 1H), 3.95 (d, J = 9.9 Hz, 1H), 3.83 (s, 6H), 3.68 (d, J = 13.4 Hz, 1H), 3.35 (d, J = 13.3 Hz, 1H), 2.31 (s, 3H), 2.02 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.6, 139.0, 137.5, 137.0, 133.50, 130.6, 128.96, 128.94, 128.91, 128.3, 127.8, 121.0, 118.9, 103.6, 98.6, 70.3, 63.1, 61.2, 58.8, 55.4, 55.2, 45.7, 21.1. HRMS (DART) m/z calcd for C29H33N2O4 [M + H]+: 473.2440; Found [M + H]+: 473.2459.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(furan-2-yl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18s)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 154 mg (86%) of 18s as a colorless foam and as a single diastereomer. Rf = 0.33 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.32 (s, 4H), 7.20–7.14 (m, 2H), 7.11 (d, J = 7.9 Hz, 1H), 6.48 (dd, J = 7.9, 1.4 Hz, 2H), 6.26 (d, J = 1.7 Hz, 1H), 6.16 (d, J = 3.2 Hz, 1H), 5.18–5.07 (dt, J = 18 Hz, 12 Hz, 1H), 4.89 (d, J = 17.0 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.60 (t, J = 8.1 Hz, 1H), 4.49 (t, J = 12 Hz, 1H), 4.32 (t, J = 9.6 Hz, 1H), 4.12 (d, J = 10.2 Hz, 1H), 4.07 (t, J = 6 Hz, 1H), 3.82 (s, 6H), 3.73 (d, J = 13.2 Hz, 1H), 3.47 (d, J = 13.1 Hz, 1H), 1.91 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.2, 158.8, 158.7, 153.1, 141.9, 138.9, 133.0, 130.8, 128.9, 128.9, 127.7, 120.6, 118.8, 109.8, 108.7, 103.7, 98.6, 70.3, 60.8, 60.4. 58.8, 55.4, 55.2, 45.8. HRMS (DART) m/z calcd for C26H29N2O5 [M + H]+: 449.2076; Found [M + H]+: 449.2068.
(S)-3-((1S,2S)-1-((2,4-Dimethoxybenzyl)amino)-1-(thiophen-2-yl)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18t)
According to General Procedure B, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 137 mg (74%) of 18t as a pale-yellow foam and as a single diastereomer. Rf = 0.33 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.36–7.31 (m, 3H), 7.21 (d, J = 5.1 Hz, 1H), 7.18–7.13 (m, 2H), 7.07 (d, J = 8.1 Hz, 1H), 6.90 (ddd, J = 4.8, 3.5, 1.1 Hz, 1H), 6.85 (d, J = 3.4 Hz, 1H), 6.50 (s, 1H), 6.48 (d, J = 7.9 Hz, 1H), 5.15 (dt, J = 16.2, 9.6 Hz, 1H), 4.84 (d, J = 17.0 Hz, 1H), 4.77 (d, J = 10.2 Hz, 1H), 4.56 (dd, J = 8.6, 7.2 Hz, 1H), 4.47 (td, J = 8.6, 1.1 Hz, 1H), 4.39 (d, J = 9.6 Hz, 1H), 4.08 (ddd, J = 11.6, 6.7, 2.8 Hz, 2H), 3.82 (s, 3H), 3.83 (s, 3H), 3.80 (d, J = 13.4 Hz, 1H), 3.48 (d, J = 13.4 Hz, 1H), 2.11 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.8, 158.3, 145.5, 138.6, 132.8, 130.8, 129.0, 127.7, 126.2, 126.0, 124.8, 120.6, 119.3, 103.6, 98.6, 70.3, 63.5, 60.4, 59.0, 57.2, 55.4, 55.2, 46.0. HRMS (DART) m/z calcd for C26H29N2O4S [M + H]+: 465.1848; Found [M + H]+: 465.1852.
(S)-3-((1S,2S)-1-(5-Bromothiophen-2-yl)-1-((2,4-dimethoxybenzyl)amino)but-3-en-2-yl)-4-phenyloxazolidin-2-one (18u)
According to General Procedure B, the product was purified by silica gel chromatography (3% E.A. in DCM) to provide 172 mg (79%) of 18u as a pale-yellow foam as a single diastereomer and a 95:5 mixture of the branched to rearranged product. Rf = 0.38 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.35 (dd, J = 4.2, 2.5 Hz, 3H), 7.17 (dd, J = 6.0, 2.6 Hz, 2H), 7.04 (d, J = 8.1 Hz, 1H), 6.84 (d, J = 6 Hz, 1H), 6.59 (d, J = 6 Hz, 1H), 6.50 (s, 1H), 6.48 (d, J = 12 Hz, 1H), 5.24 (dt, J = 16.7, 9.5 Hz, 1H), 4.89–4.83 (m, 2H), 4.54 (t, J = 8.0 Hz, 1H), 4.50 (t, J = 12 Hz, 1H), 4.39 (d, J = 9.6 Hz, 1H), 4.11 (t, J = 12 Hz, 1H), 3.84 (s, 3H), 3.83 (s, 3H), 3.80 (d, J = 13.5 Hz, 1H), 3.51 (d, J = 13.4 Hz, 1H), 2.24 (s, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.8, 158.0, 147.7, 138.0, 132.2, 130.7, 129.2, 129.1, 129.1, 127.7, 126.4, 120.3, 119.8, 111.6, 103.6, 98.6, 70.2, 63.6, 59.4, 57.7, 55.4, 55.2, 46.1. HRMS (DART) m/z calcd for C26H28BrN2O4S [M + H]+: 543.0953; Found [M + H]+: 543.0949.
General Procedure C for the Synthesis of 19
To a 20 mL crimp cap vial with a stir bar in an Ar filled glovebox were charged Cu(OAc)2 (3.6 mg, 20 μmol) and PCy3 (7.3 mg, 26 μmol) followed by toluene (1.0 mL), and the mixture was stirred for 5 min. Allenamide 15a (96.6 mg, 480 μmol) followed by imine (400 μmol) was then charged, and the vial was sealed with a crimp-cap septum and removed from the glovebox. Dimethoxymethylsilane (0.099 mL, 2 equiv) was then charged to the reaction mixture (Caution:dimethoxymethylsilane should be handled in a well-ventilated fume hood because it is known to cause blindness. Syringes were quenched with 2 M NaOH, gas evolution! prior to disposal). The mixture was then stirred at rt for 24 h. The reaction was quenched by addition of 200 mg of NH4F and 2.5 mL of MeOH followed by agitation at rt for 30 min. A 10 mL volume of 5% NaHCO3 was then added to the mixture followed by extraction with DCM (2 × 5 mL). The combined organics were dried with Na2SO4, filtered, and concentrated in vacuo. Crude product was purified by flash chromatography on silica gel to afford the desired product 19.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-(4-(trifluoromethyl)phenyl)-4-vinylimidazolidin-2-one (19a)
According to General Procedure C, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 198 mg (94%) of 19a as a colorless foam as a single diastereomer. Rf = 0.36 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.53 (d, J = 8.0 Hz, 2H), 7.35–7.30 (m, 2H), 7.26 (m, 4H), 7.18 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 8.2 Hz, 1H), 6.41 (d, J = 6.0 Hz, 1H), 6.35 (d, J = 2.4 Hz, 1H), 5.59 (ddd, J = 17.0, 9.3, 8.7, 1.0 Hz, 1H), 5.20 (d, J = 10.1 Hz, 1H), 4.90 (t, J = 7.0 Hz, 1H), 4.85 (d, J = 17.1 Hz, 1H), 4.78 (d, J = 14.7 Hz, 1H), 4.32 (m, 1H), 4.25 (dd, J = 7.9, 3.4 Hz, 1H), 4.06–4.03 (m, 1H), 4.01 (d, J = 7.9 Hz, 1H), 3.86 (d, J = 14.7 Hz, 1H), 3.79 (s, 3H), 3.59 (s, 3H), 3.39 (t, J = 8.3 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.1, 160.7, 158.6, 143.0, 137.6, 134.8, 131.7, 130.55 (C–F, 2J C–F = 31.71 Hz), 130.39 (C–F, 2J C–F = 31.71 Hz), 130.18 (C–F, 2J C–F = 31.71 Hz), 129.96 (C–F, 2J C–F = 31.71 Hz), 128.7, 127.8, 127.6, 127.4, 126.7 (C–F, 1J C–F = 271.8 Hz), 125.51 (C–F, 3J C–F = 3.02 Hz), 125.48 (C–F, 3J C–F = 3.02 Hz), 124.89 (C–F, 1J C–F = 271.8 Hz), 123.09 (C–F, 1J C–F = 271.8 Hz), 121.4, 121.29 (C–F, 2J C–F = 271.8 Hz), 116.2, 104.2, 98.1, 66.2, 64.9, 63.3, 61.9, 55.3, 54.9, 40.8. 19F NMR (565 MHz, CDCl3) δ −62.54. HRMS (DART) m/z calcd for C29H30F3N2O4 [M + H]+: 527.2158; Found [M + H]+: 527.2173.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-phenyl-4-vinylimidazolidin-2-one (19b)
According to General Procedure C, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 158 mg (86%) of 19b as a colorless foam as a single diastereomer. Rf = 0.31 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.33 (t, J = 7.5 Hz, 3H), 7.30–7.24 (m, 7H), 7.07 (d, J = 6.0 Hz, 2H), 7.03 (d, J = 8.2 Hz, 1H), 6.40 (dd, J = 8.2, 2.4 Hz, 1H), 6.38 (d, J = 2.4 Hz, 1H), 5.61 (ddd, J = 17.0, 10.1, 8.7 Hz, 1H), 5.18 (d, J = 10.1 Hz, 1H), 5.12 (t, J = 7.8 Hz, 1H), 4.86 (d, J = 17.0 Hz, 1H), 4.81 (d, J = 14.8 Hz, 1H), 4.34–4.24 (m, 2H), 4.04 (m, 1H), 3.98 (d, J = 7.7 Hz, 1H), 3.83 (d, J = 6.6 Hz, 2H), 3.80 (s, 3H), 3.63 (s, 3H), 3.46 (t, J = 8.2 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.1, 160.5, 158.7, 138.8, 137.8, 135.2, 131.5, 128.6, 128.5, 128.0, 127.6, 127.1, 120.8, 116.6, 104.0, 98.1, 66.4, 65.1, 63.6, 61.9, 55.3, 55.0, 40.6. HRMS (DART) m/z calcd for C28H31N2O4 [M + H]+: 459.2284; Found [M + H]+: 459.2304.
(4S,5S)-4-(4-Chlorophenyl)-1-((S)-2-hydroxy-1-phenylethyl)-3-(4-(trifluoromethyl)benzyl)-5-vinylimidazolidin-2-one (19de)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 143 mg (71%) of 19de as a colorless foam as a single diastereomer. Rf = 0.46 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.9 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.24 (dd, J = 8.5, 6.5 Hz, 5H), 7.19 (d, J = 7.9 Hz, 2H), 6.98 (d, J = 8.1 Hz, 2H), 5.59 (ddd, J = 17.1, 10.1, 8.7 Hz, 1H), 5.17 (d, J = 10.1 Hz, 1H), 4.89 (d, J = 15.1 Hz, 1H), 4.85 (d, J = 17.0 Hz, 1H), 4.65 (s, 1H), 4.29 (m, 2H), 4.06 (m, 1H), 3.90 (d, J = 7.9 Hz, 1H), 3.70 (d, J = 15.1 Hz, 1H), 3.48 (t, J = 8.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.7, 140.1, 137.4, 135.8, 134.6, 134.3, 130.33 (C–F, 2J C–F = 31.71 Hz), 130.11 (C–F, 2J C–F = 31.71 Hz), 129.90 (C–F, 2J C–F = 31.71 Hz), 129.68 (C–F, 2J C–F = 31.71 Hz), 129.2, 128.8, 128.79, 128.73, 127.9, 127.6, 126.77 (C–F, 1J C–F = 273.31 Hz), 125.72 (C–F, 3J C–F = 3.02 Hz), 125.69 (C–F, 3J C–F = 3.02 Hz), 125.67 (C–F, 3J C–F = 3.02 Hz), 125.64 (C–F, 3J C–F = 3.02 Hz), 124.97 (C–F, 1J C–F = 273.31 Hz), 123.16 (C–F, 1J C–F = 273.31 Hz), 121.7, 121.36 (C–F, 1J C–F = 273.31 Hz), 66.4, 64.6, 63.2, 61.8, 45.4. 19F NMR (565 MHz, CDCl3) δ −62.50. HRMS (DART) m/z calcd for C27H25ClF3N2O2 [M + H]+: 501.1557; Found [M + H]+: 501.1583.
(4S,5S)-4-(4-Fluorophenyl)-1-((S)-2-hydroxy-1-phenylethyl)-3-(4-(trifluoromethyl)benzyl)-5-vinylimidazolidin-2-one (19ee)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 149 mg (74%) of 19ee as a colorless foam as a single diastereomer. Rf = 0.40 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.45 (d, J = 7.9 Hz, 2H), 7.25 (t, J = 7.7 Hz, 2H), 7.20–7.14 (m, 3H), 7.11 (d, J = 7.9 Hz, 2H), 6.94 (dd, J = 8.4, 5.3 Hz, 2H), 6.88 (t, J = 8.5 Hz, 2H), 5.53–5.45 (ddd, J = 17.0, 10.1, 8.7 Hz, 1H), 5.10 (d, J = 10.1 Hz, 1H), 4.79 (t, J = 15.6 Hz, 2H), 4.58 (t, J = 6.5 Hz, 1H), 4.24–4.19 (m, 2H), 4.01–3.95 (m, 1H), 3.84 (d, J = 8.0 Hz, 1H), 3.64 (d, J = 15.1 Hz, 1H), 3.41 (t, J = 8.3 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 163.63 (C–F, 1J C–F = 249.15 Hz), 161.98 (C–F, 1J C–F = 249.15 Hz), 160.7, 140.2, 137.4, 134.4, 133.01 (C–F, 3J C–F = 3.02 Hz), 132.99 (C–F, 3J C–F = 3.02 Hz), 130.31 (C–F, 2J C–F = 31.71 Hz), 130.09 (C–F, 2J C–F = 31.71 Hz), 129.88 (C–F, 2J C–F = 31.71 Hz), 129.66 (C–F, 2J C–F = 31.71 Hz), 129.1, 129.0, 128.8, 128.7, 127.9, 127.6, 126.78 (C–F, 1J C–F = 273.31 Hz), 125.69 (C–F, 3J C–F = 3.02 Hz), 125.67 (C–F, 3J C–F = 3.02 Hz), 125.64 (C–F, 3J C–F = 3.02 Hz), 125.61 (C–F, 3J C–F = 3.02 Hz), 124.97 (C–F, 1J C–F = 273.31 Hz), 123.16 (C–F, 1J C–F = 273.31 Hz), 121.6, 121.35 (C–F, 1J C–F = 273.31 Hz), 116.09 (C–F, 2J C–F = 21.14 Hz), 115.95 (C–F, 2J C–F = 21.14 Hz), 66.6, 64.7, 63.2, 61.9, 45.3. 19F NMR (565 MHz, CDCl3) δ −62.52, −112.82. HRMS (DART) m/z calcd for C27H25F4N2O2 [M + H]+: 485.1852; Found [M + H]+: 485.1861.
(4S,5S)-1-Benzyl-5-(3-bromophenyl)-3-((S)-2-hydroxy-1-phenylethyl)-4-vinylimidazolidin-2-one (19j)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 131 mg (69%) of 19j as a colorless foam as a single diastereomer. Rf = 0.46 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.42 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 7.5 Hz, 2H), 7.34–7.24 (m, 8H), 7.17 (t, J = 7.8 Hz, 1H), 7.11 (d, J = 6.6 Hz, 2H), 7.01 (d, J = 7.6 Hz, 1H), 5.59 (ddd, J = 17.0, 10.0, 8.7 Hz, 1H), 5.21 (d, J = 10.1 Hz, 1H), 4.98 (d, J = 14.9 Hz, 1H), 4.91 (d, J = 17.0 Hz, 1H), 4.82 (t, J = 7.0 Hz, 1H), 4.38–4.27 (m, 2H), 4.08–4.05 (m, 1H), 3.94 (d, J = 7.5 Hz, 1H), 3.64 (d, J = 14.9 Hz, 1H), 3.48 (t, J = 8.1 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.6, 140.3, 137.5, 135.9, 134.5, 131.6, 130.4, 130.2, 128.8, 128.7, 128.6, 127.9, 127.7, 127.6, 125.9, 123.0, 121.4, 66.2, 64.8, 62.8, 62.0, 45.7. HRMS (DART) m/z calcd for C26H26BrN2O2 [M + H]+: 477.1178; Found [M + H]+: 477.1207.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-(pyridin-3-yl)-4-vinylimidazolidin-2-one (19k)
According to General Procedure C, the product was purified by silica gel chromatography (50% E.A. in DCM) to provide 183 mg (99%) of 19k as a pale-yellow foam as a single diastereomer and as a 86:14 mixture of the rearranged 19k to branched product 18k. Rf = 0.10 (60% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 8.50 (s, 1H), 8.26 (s, 1H), 7.44 (d, J = 6.0 Hz, 1H), 7.36–7.30 (m, 3H), 7.28–7.24 (m, 4H), 7.06 (d, J = 8.2 Hz, 1H), 6.40 (dd, J = 8.3, 2.4 Hz, 1H), 6.35 (d, J = 2.4 Hz, 1H), 5.58 (ddd, J = 17.1, 10.0, 8.7 Hz, 1H), 5.20 (d, J = 10.1 Hz, 1H), 4.90 (t, J = 6.9 Hz, 1H), 4.85 (d, J = 17.1 Hz, 1H), 4.76 (d, J = 14.6 Hz, 1H), 4.34–4.29 (m, 1H), 4.26 (dd, J = 7.8, 3.3 Hz, 1H), 4.07–4.02 (m, 1H), 3.97 (d, J = 8.1 Hz, 1H), 3.86 (dd, J = 18.0 6.0 Hz 2H), 3.79 (s, 3H), 3.60 (s, 3H), 3.43 (t, J = 8.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.1, 160.7, 158.6, 149.6, 149.1, 137.6, 134.5, 131.8, 129.1, 128.7, 127.85, 127.82, 127.6, 121.6, 116.1, 104.2, 98.1, 66.3, 64.8, 61.8, 61.5, 55.3, 55.0, 40.7. HRMS (DART) m/z calcd for C27H30N3O4 [M + H]+: 460.2236; Found [M + H]+: 460.2247.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-(4-methoxyphenyl)-4-vinylimidazolidin-2-one (19l)
According to General Procedure C, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 184 mg (94%) of 19l as a colorless foam as a single diastereomer. Rf = 0.29 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.35 (t, J = 7.7 Hz, 2H), 7.31–7.25 (m, 3H), 7.02 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 6.0 Hz, 2H), 6.42–6.40 (m, 2H), 5.64–5.56 (ddd, J = 17.0, 9.3, 8.6 Hz, 1H), 5.18 (d, J = 10.3 Hz, 1H), 5.15 (t, J = 7.1 Hz, 1H), 4.87 (d, J = 17.0 Hz, 1H), 4.79 (d, J = 14.8 Hz, 1H), 4.35–4.24 (m, 2H), 4.07–4.04 (m, 1H), 3.94 (d, J = 7.8 Hz, 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.67 (s, 3H), 3.46 (t, J = 8.2 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.0, 160.5, 159.4, 158.7, 137.9, 135.3, 131.4, 130.6, 128.6, 128.4, 127.7, 127.6, 120.8, 116.7, 113.9, 104.0, 98.1, 66.6, 65.1, 63.1, 61.8, 55.3, 55.2, 55.1, 40.5. HRMS (DART) m/z calcd for C29H33N2O5 [M + H]+: 489.2389; Found [M + H]+: 489.2387.
(4S,5S)-1-Benzyl-3-((S)-2-hydroxy-1-phenylethyl)-5-(2-methoxyphenyl)-4-vinylimidazolidin-2-one (19m)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (10% E.A. in DCM) to provide 118 mg (69%) of 19m as a colorless foam as a single diastereomer. Rf = 0.37 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.26 (tdd, J = 14.3, 11.1, 7.6 Hz, 10H), 7.13 (d, J = 6.9 Hz, 3H), 6.94 (t, J = 7.5 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 5.68 (ddd, J = 16.8, 10.0, 8.3 Hz, 1H), 5.20–5.14 (m, 2H), 4.94 (dd, J = 16.1, 12.9 Hz, 2H), 4.50 (m, 1H), 4.33–4.27 (m, 2H), 4.06–4.00 (m, 1H), 3.70 (d, J = 15.0 Hz, 1H), 3.61 (s, 3H), 3.59 (m, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.7, 157.5, 138.0, 136.8, 135.6, 129.3, 128.5, 128.4, 127.6, 127.5, 127.3, 120.6, 119.7, 110.9, 65.2, 61.9, 55.1, 45.6. HRMS (DART) m/z calcd for C27H29N2O3 [M + H]+: 429.2178; Found [M + H]+: 429.2195.
(4S,5S)-4-(Benzo[d][1,3]dioxol-5-yl)-3-(2,4-dimethoxybenzyl)-1-((S)-2-hydroxy-1-phenylethyl)-5-vinylimidazolidin-2-one (19n)
According to General Procedure C, the product was purified by silica gel chromatography (10% E.A. in DCM) to provide 197 mg (98%) of 19n as a colorless foam as a single diastereomer. Rf = 0.26 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.36–7.32 (m, 2H), 7.29–7.25 (m, 4H), 7.03 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 7.9 Hz, 1H), 6.57 (s, 1H), 6.49 (d, J = 7.9 Hz, 1H), 6.42–6.38 (m, 2H), 5.95–5.91 (m, 2H), 5.61–5.53 (ddd, J = 17.1, 9.3, 8.5 Hz, 1H), 5.18 (d, J = 10.1 Hz, 1H), 5.06 (d, J = 7.9 Hz, 1H), 4.88 (d, J = 17.0 Hz, 1H), 4.77 (d, J = 14.8 Hz, 1H), 4.33–4.22 (m, 2H), 4.03 (m, 1H), 3.89 (d, J = 7.7 Hz, 1H), 3.83 (d, J = 14.8 Hz, 1H), 3.80 (s, 3H), 3.68 (s, 3H), 3.42 (t, J = 8.2 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.9, 160.5, 158.7, 147.4, 137.8, 135.2, 132.6, 131.4, 128.6, 127.6, 120.9, 120.8, 116.6, 108.0, 107.1, 104.0, 101.1, 98.1, 66.5, 65.1, 63.5, 61.8, 55.3, 55.1, 40.6. HRMS (DART) m/z calcd for C29H31N2O6 [M + H]+: 503.2182; Found [M + H]+: 503.2211.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-5-(furan-2-yl)-3-((S)-2-hydroxy-1-phenylethyl)-4-vinylimidazolidin-2-one (19s)
According to General Procedure C, the product was purified by silica gel chromatography (5% E.A. in DCM) to provide 149 mg (83%) of 19s as a colorless foam as a single diastereomer. Rf = 0.28 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.34–7.28 (m, 3H), 7.27–7.21 (m, 4H), 7.06 (d, J = 8.0 Hz, 1H), 6.41–6.37 (m, 2H), 6.26–6.23 (m, 1H), 6.08 (d, J = 3.2 Hz, 1H), 5.61 (ddd, J = 17.1, 10.1, 8.7 Hz, 1H), 5.16 (d, J = 10.1 Hz, 1H), 5.08–5.05 (m, 1H), 4.98 (d, J = 18.0 Hz, 1H), 4.71 (d, J = 15.0 Hz, 1H), 4.30–4.23 (m, 2H), 4.08 (d, J = 6.7 Hz, 1H), 3.98–3.94 (m, 1H), 3.85 (d, J = 15.0 Hz, 1H), 3.77 (s, 3H), 3.74–3.69 (m, 4H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.5, 160.2, 158.6, 151.0, 142.8, 137.7, 135.1, 131.0, 128.6, 127.6, 127.6, 120.6, 116.8, 110.2, 108.7, 104.0, 98.3, 65.2, 62.7, 62.0, 57.1, 55.3, 55.2, 40.7. HRMS (DART) m/z calcd for C26H29N2O5 [M + H]+: 449.2076; Found [M + H]+: 449.2066.
(4S,5S)-1-(2,4-Dimethoxybenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-(thiophen-2-yl)-4-vinylimidazolidin-2-one (19t)
According to General Procedure C, the product was purified by silica gel chromatography (3% E.A. in DCM) to provide 184 mg (99%) of 19t as a colorless foam as a single diastereomer and as a 92:8 mixture of the rearranged 19t to the branched product 18t. Rf = 0.34 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.37–7.32 (m, 2H), 7.30–7.25 (m, 4H), 7.24 (d, J = 5.1 Hz, 1H), 7.08–7.04 (m, 1H), 6.92 (t, J = 6.0 Hz, 1H), 6.76 (d, J = 3.5 Hz, 1H), 6.43 (m, 2H), 5.63–5.54 (ddd, J = 17.0, 10.1, 8.6 Hz, 1H), 5.21 (d, J = 10.1 Hz, 1H), 5.06 (t, J = 7.0 Hz, 1H), 4.98 (d, J = 18.0 Hz, 1H), 4.82 (d, J = 14.8 Hz, 1H), 4.35–4.25 (m, 3H), 4.01 (m, 1H), 3.94 (d, J = 14.9 Hz, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.59 (d, J = 8.7 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.6, 160.4, 158.7, 142.9, 137.6, 135.0, 131.4, 128.7, 127.7, 127.6, 126.6, 125.8, 125.7, 121.1, 116.5, 104.0, 98.2, 66.7, 65.1, 62.0, 59.2, 55.3, 55.1, 40.8. HRMS (DART) m/z calcd for C26H29N2O4S [M + H]+: 465.1848; Found [M + H]+: 465.1881.
(4S,5S)-1-((S)-2-Hydroxy-1-phenylethyl)-4-(naphthalen-2-yl)-3-(4-(trifluoromethyl)benzyl)-5-vinylimidazolidin-2-one (19pe)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 130 mg (79%) of 19pe as a colorless foam as a single diastereomer. Rf = 0.43 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.59–7.56 (m, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.52–7.49 (m, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.27–7.22 (m, 3H), 7.11 (t, J = 7.6 Hz, 2H), 7.08–7.02 (m, 3H), 7.01–6.95 (m, 4H), 5.46–5.38 (ddd, J = 17.2, 10.0, 8.6 Hz, 1H), 4.96 (d, J = 10.1 Hz, 1H), 4.72 (d, J = 15.1 Hz, 1H), 4.62 (d, J = 17.0 Hz, 1H), 4.13–4.09 (m, 2H), 3.90 (d, J = 7.8 Hz, 1H), 3.86 (d, J = 8.9 Hz, 1H), 3.53 (d, J = 15.2 Hz, 1H), 3.42 (t, J = 8.2 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.8, 140.4, 137.5, 134.64, 134.62, 133.4, 133.1, 130.2 (C–F, 2J C–F = 31.71 Hz), 130.01 (C–F, 2J C–F = 31.71 Hz), 129.80 (C–F, 2J C–F = 31.71 Hz), 129.58 (C–F, 2J C–F = 31.71 Hz), 129.2, 128.9, 128.7, 127.9, 127.86, 127.81, 127.7, 127.1, 126.82 (C–F, 1J C–F = 273.31 Hz), 126.68, 126.62, 125.66 (C–F, 3J C–F = 3.02 Hz), 125.63 (C–F, 3J C–F = 3.02 Hz), 125.61 (C–F, 3J C–F = 3.02 Hz), 125.58 (C–F, 3J C–F = 3.02 Hz), 125.02 (C–F, 1J C–F = 273.31 Hz), 124.2, 123.21 (C–F, 1J C–F = 273.31 Hz), 121.5, 121.41 (C–F, 1J C–F = 273.31 Hz), 66.3, 64.8, 64.0, 62.0, 45.4. 19F NMR (565 MHz, CDCl3) δ −62.47. HRMS (DART) m/z calcd for C31H28F3N2O2 [M + H]+: 517.2103; Found [M + H]+: 517.2121.
(4S,5S)-1-Benzyl-3-((S)-2-hydroxy-1-phenylethyl)-5-(naphthalen-2-yl)-4-vinylimidazolidin-2-one (19pc)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 125 mg (70%) of 19pc as a colorless foam as a single diastereomer. Rf = 0.60 (20% EtOAc/DCM). 1H NMR (600 MHz, CDCl3) δ 7.81–7.71 (m, 3H), 7.49 (s, 1H), 7.48–7.43 (m, 2H), 7.34–7.19 (m, 9H), 7.10 (d, J = 12.0 Hz, 2H), 5.62 (ddd, J = 17.0, 10.1, 8.7 Hz, 1H), 5.15 (d, J = 10.1 Hz, 1H), 5.00 (d, J = 14.9 Hz, 1H), 4.82 (d, J = 17.0 Hz, 1H), 4.35–4.27 (m, 2H), 4.14 (d, J = 7.8 Hz, 1H), 4.08 (dd, J = 11.2, 2.5 Hz, 1H), 3.63–3.58 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.9, 137.7, 136.2, 135.1, 134.9, 133.3, 133.1, 129.0, 128.74, 128.70, 128.6, 127.9, 127.8, 127.79, 127.74, 127.6, 127.1, 126.5, 126.4, 124.3, 121.3, 66.3, 64.9, 63.5, 62.0, 45.6. HRMS (DART) m/z calcd for C30H29N2O2 [M + H]+: 449.2229; Found [M + H]+: 449.2259.
(4S,5S)-1-(4-Fluorobenzyl)-3-((S)-2-hydroxy-1-phenylethyl)-5-(naphthalen-2-yl)-4-vinylimidazolidin-2-one (19pd)
The reaction was set up according to general procedure C and stirred at 65 °C for 24 h. The product was purified by silica gel chromatography (5% E.A. in DCM) to provide 132 mg (71%) of 19pd as a colorless foam as a single diastereomer. Rf = 0.62 (20% EtOAc/DCM). 1H NMR (600 MHz, CDCl3) δ 7.85–7.75 (m, 3H), 7.54–7.48 (m, 3H), 7.36 (t, J = 7.6 Hz, 2H), 7.33–7.26 (m, 3H), 7.25–7.22 (m, 1H), 7.09 (dd, J = 8.4, 5.5 Hz, 2H), 6.98 (t, J = 8.6 Hz, 2H), 5.69 (ddd, J = 17.0, 10.2, 8.6 Hz, 1H), 5.20 (d, J = 10.1 Hz, 1H), 4.96 (d, J = 14.9 Hz, 1H), 4.91 (t, J = 6.1 Hz, 1H), 4.86 (d, J = 17.0 Hz, 1H), 4.39–4.30 (m, 2H), 4.15–4.08 (m, 2H), 3.68–3.61 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3) δ 163.12 (C–F, 1J C–F = 246.13 Hz), 161.49 (C–F, 1J C–F = 246.13 Hz), 160.8, 137.6, 134.8, 134.7, 133.3, 133.1, 130.46, 132.02 (C–F, 3J C–F = 4.53 Hz), 131.99 (C–F, 3J C–F = 4.53 Hz), 130.46, 130.41, 129.1, 128.7, 127.87, 127.80, 127.7, 127.1, 126.6, 126.5, 124.3, 121.4, 115.60 (C–F, 2J C–F = 21.14 Hz), 115.46 (C–F, 2J C–F = 21.14 Hz), 66.3, 64.9, 63.6, 62.0, 45.0. 19F NMR (565 MHz, CDCl3) δ – 114.61. HRMS (DART) m/z calcd for C30H28FN2O2 [M + H]+: 467.2135; Found [M + H]+: 467.2105.
Synthesis of 29 from Achiral Allenamide 15b
To a 20 mL crimp cap vial with a stir bar in an Ar filled glovebox were charged Cu(OAc)2 (1.8 mg, 10 μmol) and Ph-BPE (5.1 mg, 10 μmol) followed by toluene (2.5 mL) and tert-butanol (26.3 μL, 275 μmol). The mixture was stirred for 5 min. Allenamide 15b (37.5 mg, 300 μmol) followed by imine 9a (250 μmol) was then charged, and the vial was sealed with a crimp-cap septum and removed from the glovebox. Dimethoxymethylsilane (0.061 mL, 2 equiv) was charged to the reaction mixture, and the reaction mixture was stirred at rt for 24 h. The reaction was quenched by addition of 100 mg of NH4F and 1.5 mL of MeOH followed by agitation at rt for 30 min. A 5 mL volume of 5% NaHCO3 was then added to the mixture followed by extraction with DCM (2 × 3 mL). The combined organics were dried with Na2SO4, filtered, and concentrated in vacuo. Crude product was purified by flash chromatography on silica gel (25% EtOAc/hexanes) to afford 78 mg (69%) of 29 as a white solid as a single diastereomer and as a 57:43 mixture of enantiomers as determined via chiral HPLC analysis (Chiracel AD-3 85:15 heptane/isopropanol 1.50 mL/min, 254 nm). Rf = 0.45 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.60 (d, J = 7.8 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 6.91 (d, J = 8.2 Hz, 1H), 6.44 (d, J = 2.3 Hz, 1H), 6.40 (dd, J = 8.1, 2.4 Hz, 1H), 5.47 (ddd, J = 17.2, 10.5, 6.9 Hz, 1H), 5.05 (d, J = 10.6 Hz, 1H), 4.99 (d, J = 17.2 Hz, 1H), 4.46 (t, J = 6.0 Hz, 1H), 4.29 (q, J = 12.0 Hz, 1H), 4.21 (q, J = 12.0 Hz, 1H), 3.79 (s, 6H), 3.75 (d, J = 9.6 Hz, 1H), 3.71 (d, J = 13.7 Hz, 1H), 3.42 (q, J = 8.1 Hz, 1H), 3.25 (d, J = 13.7 Hz, 1H), 3.16 (td, J = 8.7, 6.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 160.3, 158.7, 158.6, 144.8, 131.4, 130.7, 130.6, 130.2 (q, J = 31.71 Hz), 128.9, 128.7, 125.3 (q, J = 3.02 Hz), 119.7, 103.6, 98.6, 98.6, 62.2, 62.1, 61.7, 60.9, 55.3, 55.2, 46.1, 40.8. 19F NMR (565 MHz, CDCl3) δ −62.36. HRMS (DART) m/z calcd for C23H26F3N2O4 [M + H]+: 451.1845; Found [M + H]+: 451.1881.
Synthesis of 30 from Achiral Allenamide 15b
To a 20 mL crimp cap vial with a stir bar in an Ar filled glovebox were charged Cu(OAc)2 (1.8 mg, 10 μmol) and Ph-BPE (5.1 mg, 10 μmol) followed by toluene (0.5 mL). The mixture was stirred for 5 min. Allenamide 15b (37.5 mg, 300 μmol) followed by imine 9a (250 μmol) was then charged, and the vial was sealed with a crimp-cap septum and removed from the glovebox. Dimethoxymethylsilane (0.061 mL, 2 equiv) was then charged to the reaction mixture. The mixture was then stirred at rt for 24 h. The reaction was quenched by addition of 100 mg of NH4F and 1.5 mL of MeOH followed by agitation at rt for 30 min. A 5 mL volume of 5% NaHCO3 was then added to the mixture followed by extraction with DCM (2 × 3 mL). The combined organics were dried with Na2SO4, filtered, and concentrated in vacuo. Crude product was purified by flash chromatography on silica gel (50% EtOAc/hexanes) to afford 68 mg (60%) of 30 as a colorless liquid and as an 80:20 mixture of enantiomers as determined via chiral HPLC analysis (Chiracel AD-3 90:10 heptane/isopropanol 1.00 mL/min, 220 nm). Rf = 0.28 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 8.3 Hz, 1H), 6.37 (dd, J = 8.3, 2.4 Hz, 1H), 6.32 (d, J = 2.4 Hz, 1H), 5.62 (ddd, J = 17.1, 10.1, 8.6 Hz, 1H), 5.22 (d, J = 10.1 Hz, 1H), 5.04 (d, J = 17.0 Hz, 1H), 4.70 (d, J = 14.7 Hz, 1H), 4.01 (d, J = 8.2 Hz, 1H), 3.86 (d, J = 14.7 Hz, 1H), 3.78 (s, 3H), 3.76–3.68 (m, 2H), 3.63 (t, J = 8.4 Hz, 1H), 3.55 (s, 3H), 3.36–3.31 (m, 1H), 3.26–3.22 (m, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 162.3, 160.6, 158.6, 142.7, 135.0, 131.7, 130.6 (q, J = 31.71 Hz), 127.6, 126.7 (q, J = 273.31 Hz), 125.5 (q, J = 3.02 Hz), 121.2, 116.2, 104.1, 98.0, 68.2, 63.8, 62.2, 55.3, 54.8, 46.1, 40.8. 19F NMR (565 MHz, CDCl3) δ −62.50. HRMS (DART) m/z calcd for C23H26F3N2O4 [M + H]+: 451.1845; Found [M + H]+: 451.1882.
Synthesis of 18c on 1.0 g Scale
To a 20 mL crimp cap vial was charged Cu(OAc)2 (16.1 mg, 88.9 μmol), and the vial was sealed with a crimp-cap septum. The vial was evacuated and backfilled with nitrogen 3 times and then charged with toluene (5 mL), 20% PCy3 solution in toluene (194 μL, 111 μmol), and tert-butanol (0.85 mL, 8.89 mmol), and the mixture was allowed to stir at rt for 10 min until all the Cu(OAc)2 dissolved. A 50 mL two-neck round-bottom flask was then charged with imine 9c (1.0 g, 4.44 mmol) and allene 15a (1.07 g, 5.33 mmol), and the flask was evacuated and backfilled with nitrogen 3 times. The flask was then charged with toluene (5 mL). The imine/allene flask was then charged with the catalyst solution. Dimethoxymethyl silane (1.1 mL, 8.89 mmol) was charged to the reaction mixture (caution:dimethoxymethylsilane should be handled in a well-ventilated fume hood because it is known to cause blindness. Syringes were quenched with 2 M NaOH, gas evolution! prior to disposal), and the reaction was allowed to stir at rt for 2 h. A 50 mL round-bottom flask was charged with NH4F (2 g) and MeOH (20 mL), and the reaction mixture was transferred via pipet to this flask and allowed to stir at rt for 30 min. The volatiles were concentrated in vacuo, and 50 mL of 5% NaHCO3 solution were added to the flask. The mixture was extracted with CH2Cl2 (2 × 20 mL), and the combined organics were dried over Na2SO4 and concentrated in vacuo. The crude residue was purified by silica gel chromatography (5% EtOAc/DCM) to afford 1.655 g (91%) of 18c as a white solid as a single diastereomer.
Synthesis of 25
To a solution of 207.5 mg (393 μmol) of 19a in 2 mL of CH2Cl2 at 0 °C were charged 66 μL (473 μmol) of triethylamine followed by dropwise addition of 30.5 μL (393 μmol) of MsCl. The mixture was stirred for 30 min at 0 °C, and then 4 mL of 10% NH4Cl were added. The mixture was extracted with CH2Cl2 (3 × 5 mL). The combined organics were dried with anhydrous Na2SO4 and filtered, and the volatiles were removed in vacuo. The crude residue was then dissolved in 2 mL of THF and cooled to 0 °C. A 1.0 M concentration of potassium tert-butoxide (433 μL, 433 μmol) in THF was then added, and the mixture was warmed to room temperature and stirred for 30 min. To the mixture was added 5 mL of 10% brine followed by extraction with CH2Cl2 (3 × 5 mL). The combined organics were dried with anhydrous Na2SO4 and filtered, and the volatiles were removed in vacuo. The crude residue was then dissolved in 4 mL of THF in a crimp cap vial. To the solution were then added 788 μL (3.94 mmol) of 5.0 M aqueous H2SO4. The vial was purged with argon, sealed, and immersed in an oil bath at 50 °C. After 3 h the reaction mixture was cooled to room temperature, and 15 mL of saturated aqueous NaHCO3 were added. The mixture was extracted with CH2Cl2 (3 × 5 mL). The combined organics were dried with anhydrous Na2SO4 and filtered, and the volatiles were removed in vacuo. The crude residue was purified by flash chromatography (20% EtOAc/DCM) to afford 112 mg (70%) of 25 as a colorless foam. Rf = 0.22 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 8.3 Hz, 1H), 6.37 (d, J = 8.3 Hz, 1H), 6.31 (d, J = 2.4 Hz, 1H), 5.74 (ddd, J = 17.2, 10.2, 7.2 Hz, 1H), 5.41 (s, 1H), 5.12 (d, J = 10.2 Hz, 1H), 5.10 (d, J = 12 Hz, 1H), 4.67 (d, J = 14.8 Hz, 1H), 4.06 (d, J = 7.5 Hz, 1H), 3.86–3.82 (m, 2H), 3.76 (s, 3H), 3.54 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 161.6, 160.5, 158.5, 143.2, 136.1, 131.4, 130.6 (q, J = 33.22 Hz), 127.5, 126.7 (q, J = 271.8 Hz), 125.5 (q, J = 3.02 Hz), 118.0, 116.5, 104.1, 97.9, 65.5, 62.1, 55.3, 54.9, 39.8. 19F NMR (565 MHz, CDCl3) δ −62.49. HRMS (DART) m/z calcd for C21H22F3N2O3 [M + H]+: 407.1583; Found [M + H]+: 407.1600.
Synthesis of 19a from 18a
To a solution of 100 mg (190 μmol) of 18a in 1.0 mL of THF at −10 °C were added 114 μL (285 μmol) of 2.5 M solution of nBuLi in hexanes. The reaction mixture was allowed to warm to room temperature and stirred for 1 h. To the mixture were added 2 mL of saturated NH4Cl, and the mixture was extracted with CH2Cl2 (3 × 3 mL). The combined organics were dried with anhydrous Na2SO4 and filtered, and the volatiles were removed in vacuo. The crude residue was purified by flash chromatography (5% EtOAc/DCM) to afford 94 mg (94%) of 19a as a colorless foam.
Synthesis of 26
A crimp cap vial was charged with 100 mg (233 μmol) of 18c, CH3CN (1 mL), K2CO3 (161 mg, 1.17 mmol), TBAI (17.2 mg, 46.7 μmol), and allyl bromide (101 μL, 1.17 mmol). The mixture was heated at 85 °C for 18 h. The reaction was quenched with 5 mL of water, and the mixture was extracted with MTBE (3 × 3 mL). The combined organics were dried with anhydrous Na2SO4 and filtered, and the volatiles were removed in vacuo. The crude residue was purified by flash chromatography (20% EtOAc/hexanes) to afford 90 mg (82%) of 26 as a yellow solid. Rf = 0.60 (50% EtOAc/hexanes). 1H NMR (600 MHz, CDCl3) δ 7.38–7.31 (m, 4H), 7.29–7.25 (m, 3H), 7.25–7.20 (m, 3H), 7.18 (d, J = 6.0 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 6.93–6.88 (m, 2H), 5.95 (dtd, J = 17.2, 9.5, 4.0 Hz, 1H), 5.25 (d, J = 10.1 Hz, 1H), 5.20 (d, J = 17.2 Hz, 1H), 5.12–5.05 (m, 1H), 4.88–4.82 (m, 1H), 4.61–4.52 (m, 2H), 4.52–4.45 (m, 2H), 4.08 (dd, J = 7.4, 5.3 Hz, 1H), 3.87 (d, J = 11.7 Hz, 1H), 3.85 (s, 3H), 3.82 (d, J = 13.3 Hz, 1H), 3.66–3.61 (m, 1H), 2.96 (d, J = 13.2 Hz, 1H), 2.56 (dd, J = 13.3, 9.1 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 159.0, 158.7, 140.1, 137.1, 134.7, 131.8, 131.0, 130.0, 128.7, 128.6, 128.0, 127.8, 127.6, 119.2, 118.0, 113.8, 70.4, 62.4, 56.9, 56.7, 55.4, 52.9, 52.5. HRMS (DART) m/z calcd for C30H33N2O3 [M + H]+: 469.2491; Found [M + H]+: 469.2504.
Synthesis of 27
To a 20 mL crimp cap vial with a stir bar in an Ar filled glovebox were added 48 mg (0.10 mmol) of 26 followed by 2 mL of toluene and 3.2 mg (5.1 μmol) of a Hoveyda–Grubbs II catalyst. The vial was sealed and removed from the glovebox. The solution was heated at 90 °C for 12 h. The reaction mixture was concentrated, and the crude residue was purified by flash chromatography (50% EtOAc/hexanes) to afford 35 mg (78%) of 27 as a colorless foam. 1H NMR (600 MHz, CDCl3) δ 7.44 (d, J = 7.3 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.35–7.31 (m, 1H), 7.28–7.21 (m, 6H), 7.15 (d, J = 6.0 Hz, 2H), 6.88 (d, J = 7.9 Hz, 2H), 5.41–5.37 (m, 1H), 5.20 (dd, J = 8.7, 2.8 Hz, 1H), 5.15–5.09 (m, 1H), 4.72–4.68 (m, 1H), 4.34 (t, J = 6.0 Hz, 1H), 3.99 (d, J = 4.4 Hz, 1H), 3.97 (s, 1H), 3.96–3.93 (m, 1H), 3.80 (s, 3H), 3.24 (dt, J = 17.9, 2.5 Hz, 1H), 2.95 (d, J = 13.2 Hz, 1H), 2.76 (dd, J = 18.0, 2.8 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ 158.7, 157.9, 142.8, 138.2, 130.7, 129.5, 128.8, 128.7, 128.6, 128.5, 128.1, 128.0, 126.4, 123.2, 113.8, 70.7, 67.8, 59.4, 59.3, 55.3, 53.5, 51.6. HRMS (DART) m/z calcd for C28H29N2O3 [M + H]+: 441.2178; Found [M + H]+: 441.2205.
Acknowledgments
Startup funding from the Virginia Commonwealth University and the Bill and Melinda Gates Foundation (The Medicines for All Institute, grant number OPP#1176590) is gratefully acknowledged. We thank Dr. Faik Musayev (VCU) for X-ray crystallographic analysis that was partially supported by Structural Biology resources provided by NIH Shared Instrumentation Grant S10OD021756 (MKS) and Virginia General Assembly Higher Education Equipment Trust Fund (HEETF) to VCU. M.D.E. acknowledges the NSF for a summer REU fellowship (CHE-1851916). We thank Dr. Joseph Turner (VCU) for assistance in collecting HRMS data. O.G. is grateful to the University of Maryland College Park for start-up funds, the NSF (CAREER 1751568) and by the NIGMS of the NIH (R35GM137797), and computational resources from UMD Deepthought2 and MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c02971.
Computational details, intermediate and transition state coordinates, copies of 1H and 13C NMR spectra of all new compounds, and X-ray diffraction analysis data of 18c·HCl (PDF)
Accession Codes
CCDC 2045972 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Reviews; a Lucet D.; Le Gall T.; Mioskowski C. The Chemistry of Vicinal Diamines. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. . [DOI] [PubMed] [Google Scholar]; b Kotti S. R. S. S.; Timmons C.; Li G. Vicinal Diamino Functionalities as Privileged Structural Elements in Biologically Active Compounds and Exploitation of Their Synthetic Chemisty. Chem. Biol. Drug Des. 2006, 67, 101–114. 10.1111/j.1747-0285.2006.00347.x. [DOI] [PubMed] [Google Scholar]; c Viso A.; Fernandez de la Pradilla R.; Tortosa M.; Garcia A.; Flores A. Update 1 of: α,β-Diamino Acids: Biological Significance and Synthetic Approaches. Chem. Rev. 2011, 111, PR1–PR42. [DOI] [PubMed] [Google Scholar]; d Bergmeier S. C. The Synthesis of Vicinal Amino Alcohols. Tetrahedron 2000, 56, 2561–2576. 10.1016/S0040-4020(00)00149-6. [DOI] [Google Scholar]
- Selected examples:; a Desai M. C.; Lefkowitz S. L.; Thadeio P. F.; Longo K. P.; Snider R. M. Discovery of a Potent Substance P Antagonist: Recognition of the Key Molecular Determinant. J. Med. Chem. 1992, 35, 4911–4913. 10.1021/jm00104a018. [DOI] [PubMed] [Google Scholar]; b Farina V.; Brown J. D. Tamiflu: The Supply Problem. Angew. Chem., Int. Ed. 2006, 45, 7330–7334. 10.1002/anie.200602623. [DOI] [PubMed] [Google Scholar]; c Clark P. G. K.; Vieira L. C. C.; Tallant C.; Fedorov O.; Singleton D. C.; Rogers C. M.; Monteiro O.; Bennett J. M.; Baronio R.; Muller S.; Daniels D. L.; Mendez J.; Knapp S.; Brennan P. E.; Dixon D. J. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Curreli F.; Kwon Y. D.; Zhang H.; Scacalossi D.; Belov D. S.; Tikhonov A. A.; Andreev I. A.; Altieri A.; Kurkin A. V.; Kwong P. D.; Debnath A. K. Structure-Based Design of a Small Molecule CD4-Antagonis with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. 10.1021/acs.jmedchem.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kamath A.; Ojima I. Advances in the Chemistry of b-Lactam and Its Medicinal Applications. Tetrahedron 2012, 68, 10640–10664. 10.1016/j.tet.2012.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; f D’Ambrosio M.; Guerriero A.; Debitus C.; Ribes O.; Pusset J.; Leroy S.; Pietra F. Agelastatin A, a New Skeleton Cytotoxic Alkaloid of the Oroidin Family. Isolation from the Axinellid Sponge Agelas dendromorpha of the Coral Sea. J. Chem. Soc., Chem. Commun. 1993, 1305–1306. 10.1039/c39930001305. [DOI] [Google Scholar]; g Reichard G. A.; Stengone C.; Paliwal S.; Mergelsberg I.; Majmundar S.; Wang C.; Tiberi R.; McPhail A. T.; Piwinski J. J.; Shih B.-Y. Asymmetric Synthesis of 4,4-Disubstituted-2-Imidazoli-dinones: Potent NK1 Antagonists. Org. Lett. 2003, 5, 4249–4251. 10.1021/ol030104p. [DOI] [PubMed] [Google Scholar]; h De Clercq P. J. Biotin: A Timeless Challenge for Total Synthesis. Chem. Rev. 1997, 97, 1755–1792. 10.1021/cr950073e. [DOI] [PubMed] [Google Scholar]; i Welin E. R.; Ngamnithiporn A.; Klatte M.; Lapointe G.; Pototschnig G. M.; McDermott M. S. J.; Conklin D.; Gilmore C. D.; Tadross P. M.; Haley C. K.; Negoro K.; Glibstrup E.; Grunanger C. U.; Allan K. M.; Virgil S. C.; Slamon D. J.; Stoltz B. M. Concise Total Syntheses of (−)-Jorunnamycin A and (−)-Jorumycin Enabled by Asymmetric Catalysis. Science 2019, 363, 270–275. 10.1126/science.aav3421. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Iwatsuki M.; Nishihara-Tsukashima A.; Ishiyama A.; Namatame M.; Watanabe Y.; Handasah S.; Pranamuda H.; Marwoto B.; Matsumoto A.; Takahashi Y.; Otoguro K.; Omura S. Jogyamycin, a New Antiprotozoal Aminocyclopentitol Antibiotic, Produced by Streptomyces Sp. a-WM-JG-16.2. J. Antibiot. 2012, 65 (3), 169–171. 10.1038/ja.2011.136. [DOI] [PubMed] [Google Scholar]; k Nishimura S.; Matsunaga S.; Shibazaki M.; Suzuki K.; Furihata K.; van Soest R. W. M.; Fusetani N. Massadine, a Novel Geranylgeranyltransferase Type I Inhibitor from the Marine Sponge Stylissa aff. Org. Lett. 2003, 5, 2255–2257. 10.1021/ol034564u. [DOI] [PubMed] [Google Scholar]
- a Doyle A. G.; Jacobsen E. N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; b Taylor M. S.; Jacobsen E. N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- a Bennani Y. L.; Hanessian S. trans-1,2-Diaminocyclohexane Derivatives as Chiral Reagents, Scaffolds, and Ligands for Catalysis: Applications in Asymmetric Synthesis and Molecular Recognition. Chem. Rev. 1997, 97, 3161–3196. 10.1021/cr9407577. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Machacek M. R.; Aponick A. Predicting Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: a Working Model. Acc. Chem. Res. 2006, 39, 747–760. 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]; c Surry D. S.; Buchwald S. L. Diamine Ligands in Copper-Catalyzed Reactions. Chem. Sci. 2010, 1, 13–31. 10.1039/c0sc00107d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Review:; a Kizirian J.-C. Chiral Tertiary Diamines in Asymmetric Synthesis. Chem. Rev. 2008, 108, 140–205. 10.1021/cr040107v. [DOI] [PubMed] [Google Scholar]
- a Dufrasne F.; Galanski M. The Relation Between Stereochemistry and Biological Activity of Platinum (II) Complexes Chelated with Chiral Diamine Ligands: an Intricate Problem. Curr. Pharm. Des. 2007, 13, 2781–2794. 10.2174/138161207781757060. [DOI] [PubMed] [Google Scholar]; b Apps M. G.; Choi E. H. Y.; Wheate N. J. The State-of-Play and Future of Platinum Drugs. Endocr.-Relat. Cancer 2015, 22, R219. 10.1530/ERC-15-0237. [DOI] [PubMed] [Google Scholar]
- Review:Huang S.-C.; Korlipara V. L. Neurokinin-1 Receptor Antagonists: a Comprehensive Patent Survey. Expert Opin. Ther. Pat. 2010, 20, 1019–1045. 10.1517/13543776.2010.495121. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Zhu Y.; Cornwall R. G.; Du H.; Zhao B.; Shi Y. Catalytic Diamination of Olefins via N-N Bond Activation. Acc. Chem. Res. 2014, 47, 3665–3678. 10.1021/ar500344t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cardona F.; Goti A. Metal-Catalysed 1,2-Diamination Reaction. Nat. Chem. 2009, 1, 269–275. 10.1038/nchem.256. [DOI] [PubMed] [Google Scholar]
- Selected examples of catalytic diamination:; a Muniz K.; Barreiro L.; Romero R. M.; Martinez C. Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc. 2017, 139, 4354–4357. 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]; b Fu N.; Sauer G. S.; Saha A.; Loo A.; Lin S. Metal-Catalyzed Electrochemical Diazidation of Alkenes. Science 2017, 357, 545–579. [DOI] [PubMed] [Google Scholar]; c Yuan Y.-A.; Lu D.-F.; Chen Y.-R.; Xu H. Iron-Catalyzed Direct Diaxidation for a Broad Range of Olefins. Angew. Chem., Int. Ed. 2016, 55, 534–538. 10.1002/anie.201507550. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sequeira F. C.; Turnpenny B. W.; Chemler S. R. Copper-Promoted and coper-Catalyzed Intermolecular Alkene Diamination. Angew. Chem., Int. Ed. 2010, 49, 6365–6368. 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Li G.; Wei H.-X.; Kim S. H.; Carducci M. D. A Novel Electrophilic Diamination Reactionof Alknes. Angew. Chem., Int. Ed. 2001, 40, 4277–4280. . [DOI] [PubMed] [Google Scholar]; f de Haro T.; Nevado C. Flexible Gold-Catalyzed Regioselective Oxidative Difunctionalization of Unactivated Alkenes. Angew. Chem., Int. Ed. 2011, 50, 906–910. 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]; g Muniz K.; Streuff J.; Hovelmann C. H.; Nunez A. Exploring the Nickel-Catalyzed Oxidation of Alkenes: a Diamination by Sulfamide Transfer. Angew. Chem., Int. Ed. 2007, 46, 7125–7127. 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]; h Turnpenny B. W.; Chemler S. R. Copper-Catalyzed Alkene Diamination: Synthesis of Chiral 2-Aminomethyl Indolines and Pyrrolidines. Chem. Sci. 2014, 5, 1786–1793. 10.1039/C4SC00237G. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Zhao B.; Du H.; Cui S.; Shi Y. Synthetic and Mechanistic Studies on Pd(0)-Catalyzed Diamination of Conjugated Dienes. J. Am. Chem. Soc. 2010, 132, 3523–3532. 10.1021/ja909459h. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Du H.; Zhao B.; Shi Y. Catalytic Asymmetric Allylic and Homoallylic Diamination of Terminal Olefins via Formal C-H Activation. J. Am. Chem. Soc. 2008, 130, 8590–8591. 10.1021/ja8027394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews:; a Zhu Y.; Wang Q.; Cornwall R. G.; Shi Y. Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev. 2014, 114, 8199–8256. 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]; b Damiano C.; Intrieri D.; Gallo E. Aziridination of Alkenes Promoted by Iron or Ruthenium Complexes. Inorg. Chim. Acta 2018, 470, 51–67. 10.1016/j.ica.2017.06.032. [DOI] [Google Scholar]; c Degennaro L.; Trinchera P.; Luisi R. Recent Advances in the Stereoselective Synthesis of Aziridines. Chem. Rev. 2014, 114, 7881–7929. 10.1021/cr400553c. [DOI] [PubMed] [Google Scholar]
- Chai Z.; Yang P. -JH.; Zhang H.; Wang S.; Yang G. Synthesis of Chiral Vicinal Diamines by Silver(I)-Catalyzed Enantioselective Aminolysis of N-Tosylaziridines. Angew. Chem., Int. Ed. 2017, 56, 650–654. 10.1002/anie.201610693. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Bodkin J. A.; McLeod M. D. The Sharpless Asymmetric Aminohydroxylation. J. Chem. Soc. Perkin Trans. 1 2002, 2733–2746. 10.1039/b111276g. [DOI] [Google Scholar]; b Heravi M. M.; Lashaki T. B.; Fattahi B.; Zadsirjan V. Application of Asymmetric Sharpless Aminohydroxylation in the Total Synthesis of Natural Products and Some Synthetic Complex Bio-active Molecules. RSC Adv. 2018, 8, 6634. 10.1039/C7RA12625E. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selected examples:; c Li G.; Angert H. H.; Sharpless K. B. N-Halocarbamate Salts Lead to More Efficient Catalytic Asymmetric Aminohydroxylation. Angew. Chem., Int. Ed. Engl. 1996, 35, 2813–2817. 10.1002/anie.199628131. [DOI] [Google Scholar]; d Williamson K. S.; Yoon T. P. Iron-Catalyzed Aminohydroxylation of Olefins. J. Am. Chem. Soc. 2010, 132, 4570–4571. 10.1021/ja1013536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keicher T.; Lobbecke S.. Organic Azides: Syntheses and Applications. In Organic Azides: Syntheses and Applications; Bräse S., Banert K., Eds.; John Wiley & Sons Ltd: West Sussex, 2010; pp 3–28. [Google Scholar]
- Selected examples of Aza-pinacol:; a Betschart C.; Seebach D. Preparation of 1,2-Diarylethylenediamines by Aminative Reductive Coupling of Aromatic Aldehydes with Low-Valent Titanium Reagents. Helv. Chim. Acta 1987, 70, 2215–2231. 10.1002/hlca.19870700826. [DOI] [Google Scholar]; b Periasamy M.; Reddy M. R.; Kanth J. V. B. Low Valent Titanium Induced One Pot Syntheses of Imidazolines. Tetrahedron Lett. 1996, 37, 4767–4770. 10.1016/0040-4039(96)00930-6. [DOI] [Google Scholar]; c Roskamp E. J.; Pedersen S. F. Convenient Routes to Vicinal Diamines. Coupling of Nitriles of N-(Trimethylsilyl)imines Promoted by NbCl4(THF)2. J. Am. Chem. Soc. 1987, 109, 3152–3154. 10.1021/ja00244a052. [DOI] [Google Scholar]; d Shimizu M.; Iida T.; Fujisawa T. Highly Enantioselective Iminio Pinacol Coupling Leading to the Synthesis of 1,2-Diphenylethyenediamine Derivatives. Chem. Lett. 1995, 24, 609–610. 10.1246/cl.1995.609. [DOI] [Google Scholar]; e Zhong Y.-W.; Izumi K.; Xu M.-H.; Lin G.-Q. Highly Diastereoselective and Enantioselective Synthesis of Enantiopure C2-Symmetrical Vicinal Diamines by Reductive Homocoupling of Chiral N-tert-Butanesulfinyl Imines. Org. Lett. 2004, 6, 4747–4750. 10.1021/ol0479803. [DOI] [PubMed] [Google Scholar]; f Zhong Y.-W.; Xu M.-H.; Lin G.-Q. Samarium Diiodide-Induced Asymmetric Syunthesis of Optically Pure Unsymmetrical Vicinal Diamines by Reductive Cross-Coupling of Nitrones with N-tert-Butanesulfinyl Imines. Org. Lett. 2004, 6, 3953–3956. 10.1021/ol048444d. [DOI] [PubMed] [Google Scholar]; g Zhou M.; Li K.; Chen D.; Xu R.; Xu G.; Tang W. Enantioselective Reductive Coupling of Imines Templated by Chiral Diboron. J. Am. Chem. Soc. 2020, 142, 10337–10342. 10.1021/jacs.0c04558. [DOI] [PubMed] [Google Scholar]; h Okamoto S.; Ariki R.; Tsujioka H.; Sudo A. A Metal-Free Approach to 1,2-Diamines via Visible Light-Driven Reductive Coupling of Imines with Perylene as a Photoredox Catalyst. J. Org. Chem. 2017, 82, 9731–9736. 10.1021/acs.joc.7b01838. [DOI] [PubMed] [Google Scholar]; i Nakajima M.; Fava E.; Loescher S.; Jiang Z.; Rueping M. Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]; j Fava E.; Millet A.; Nakajima M.; Loescher S.; Rueping M. Reductive Umpolung of Carbonyl Derivatives with Visible-Light Photoredox Catalysis: Direct Access to Vicinal Diamines and Amino Alcohols via α-Amino Radicals and Ketyl Radicals. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. 10.1002/anie.201511235. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Uraguchi D.; Kinoshita N.; Kizu T.; Ooi T. Synergistic Catalysis of Ionic Bronsyted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]; l Kizu T.; Uraguchi D.; Ooi T. Independence from the Sequence of Single-Electron Transfer of Photoredox Process in Redox-Neutral Asymmetric Bond-Forming Reaction. J. Org. Chem. 2016, 81, 6953–6958. 10.1021/acs.joc.6b00445. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Westermann B. Asymmetric Catalytic Aza-Henry Reactions Leading to 1,2-Diamines and 1,2-Diaminocarboxylic acids. Angew. Chem., Int. Ed. 2003, 42, 151–153. 10.1002/anie.200390071. [DOI] [PubMed] [Google Scholar]; b Phillips A. M. F.; Guedes da Silva M. F. C.; Pombeiro A. J. L. The stereoselective Nitro-Mannich Reaction in the Synthesis of Active Pharmaceutical Ingredients and Other Biologically Active Compounds. Front. Chem. 2020, 8, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noble A.; Anderson J. C. Nitro-Mannich Reaction. Chem. Rev. 2013, 113, 2887–2939. 10.1021/cr300272t. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Arrayas R. G.; Carretero J. C. Catalytic Asymmetric Direct Mannich Reaction: a Powerful Tool for the Synthesis of a,b-Diamino Acids. Chem. Soc. Rev. 2009, 38, 1940–1948. 10.1039/b820303b. [DOI] [PubMed] [Google Scholar]; b Saranya S.; Harry N. A.; Krishnan K. K.; Anilkumar G. Recent Developments and Perspectives in the Asymmetric Mannich Reaction. Asian J. Org. Chem. 2018, 7, 613–633. 10.1002/ajoc.201700679. [DOI] [Google Scholar]
- Review:Leitch J. A.; Rossolini T.; Rogova T.; Maitland J. A. P.; Dixon D. J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. 10.1021/acscatal.9b05011. [DOI] [Google Scholar]
- For examples of diamine synthesis employing 2-aza-allylanions, see:; a Reddy L. R.; Kotturi S.; Shenoy R.; Nalivela K. S.; Patel C.; Raval P.; Zalavadiya V. Umpolung Synthesis of Vicinal Diamines: Diastereoselective Addition of 2-Azaallyl Anions to Davis-Ellman’s Imines. Org. Lett. 2018, 20, 5423–5426. 10.1021/acs.orglett.8b02331. [DOI] [PubMed] [Google Scholar]; b Wu L.; Xie C.; Mei H.; Dai Y.; Han J.; Soloshonok V. A.; Pan Y. Synthesis of Trifluoromethyl-Containing Vicinal Diamines by Asymmetric Decarboxylative Mannich Addition Reactions. J. Org. Chem. 2015, 80, 3187–3194. 10.1021/acs.joc.5b00124. [DOI] [PubMed] [Google Scholar]; c Shao X.; Li K.; Malcolmson S. J. Enantioselective Synthesis of anti-1,2-Diamines by Cu-Catalyzed Reductive Couplings of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc. 2018, 140, 7083–7087. 10.1021/jacs.8b04750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews:; a Yus M.; González-Gómez J. C.; Foubelo F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev. 2011, 111 (12), 7774–7854. 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]; b Yus M.; González-Gómez J. C.; Foubelo F. Diastereoselective Allylation of Carbonyl Compounds and Imines: Application to the Synthesis of Natural Products. Chem. Rev. 2013, 113 (7), 5595–5698. 10.1021/cr400008h. [DOI] [PubMed] [Google Scholar]; c Huo H. – X.; Duvall J. R.; Huang M. – Y.; Hong R. Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronoates. Org. Chem. Front. 2014, 1, 303–320. 10.1039/C3QO00081H. [DOI] [Google Scholar]; d Friestad G. K.; Mathies A. K. Recent Developments in Asymmetric Catalytic Addition to C=N Bonds. Tetrahedron 2007, 63, 2541–2569. 10.1016/j.tet.2006.11.076. [DOI] [Google Scholar]; e Ding H.; Friestad G. K. Asymmetric Addition of Allylic Nucleophiles to Imino Compounds. Synthesis 2005, 2005, 2815–2829. 10.1055/s-2005-872181. [DOI] [Google Scholar]; f Kobayashi S.; Mori Y.; Fossey J. S.; Salter M. M. Catalytic Enantioselective Formation of C-C Bonds by Addition to Imines and Hydrazones: a Ten Year Update. Chem. Rev. 2011, 111, 2626–2704. 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]; g Yamamoto Y.; Asao N. Selective Reactions Using Allylic Metals. Chem. Rev. 1993, 93, 2207–2293. 10.1021/cr00022a010. [DOI] [Google Scholar]
- Uphade M. B.; Reddy A. A.; Khandare S. P.; Prasad K. R. Stereoselective Addition of a Lithium Anion of 1,1-Diphenyl-2-aza-pentadiene to Sulfinimines: Application to the Synthesis of (−)-Epiquinamide. Org. Lett. 2019, 21, 9109–9113. 10.1021/acs.orglett.9b03507. [DOI] [PubMed] [Google Scholar]
- Amino-substituted allyl reagents in aldehyde allylation:; a Barrett A. G. M.; Seefeld M. A. B-[(E)-3-(Diphenylamino)allyl]diisopinocampheylborane: An Excellent Reagent for the Stereoselective Synthesis of anti-β-Diphenylamino Alcohols. J. Chem. Soc., Chem. Commun. 1993, 339–341. 10.1039/C39930000339. [DOI] [Google Scholar]; b Barrett A. G. M.; Seefeld M. A. The Use of B-[(E)-3-(Diphenylamino)allyl]diisopinocompheylborane as a Reagent for the Stereoselective Synthesis of anti-β-Diphenylamino Alcohols and trans-1-Diphenylamino-2-(1-hydroxylalkyl)-cyclopropanes. Tetrahedron 1993, 49, 7857–7870. 10.1016/S0040-4020(01)88011-X. [DOI] [Google Scholar]; c Barrett A. G. M.; Seefeld M. A.; Williams D. J. Covenient Asymmetric Synthesis of anti-β-Amino Alcohols: An X-ray Crystallographic Study of (4R)-2,2-Dimethyl-4-[(2S)-(diphenylmethyleneamino)-(1S)-hydroxy-3-buten-1-yl]-1,3-dioxolane. J. Chem. Soc., Chem. Commun. 1994, 1053–1054. 10.1039/C39940001053. [DOI] [Google Scholar]; d Trost B. M.; Cregg J. J.; Quach N. Isomerization of N-Allyl Amides to Form Geometrically Defined Di-, Tri0,m and Tetrasubstituted Enamides. J. Am. Chem. Soc. 2017, 139, 5133–5139. 10.1021/jacs.7b00564. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Takenouchi Y.; Ito H. Copper(I)-Catalyzed Boryl Substitution of Allyl Aminals: Selective Synthesis of Linear γ-AminoallylBoronates. Synthesis 2017, 49, 4738–4744. 10.1055/s-0036-1588802. [DOI] [Google Scholar]; f Hoffmann R. W.; Bruckner D.; Gerusz V. Domino-Hydroformylation-Allylboration-Hydroformylation for the Synthesis of trans-2-Alkylpiperidin-3-ols. Heterocycles 2000, 52, 121–124. 10.3987/COM-99-S39. [DOI] [Google Scholar]; g Clement H. A.; Hall D. G. Synthesis of α-Hydroxyalkyl Dehydroazepanes via Catalytic Enantioselective Borylative Migration of an Enol Nonaflate. Tetrahedron Lett. 2018, 59, 4334–4339. 10.1016/j.tetlet.2018.10.053. [DOI] [Google Scholar]; h Hayama K.; Kojima R.; Kubota K.; Ito H. Synthesis of Chrial N-Heterocyclic Allylboronates via the Enantioselective Borylative Dearomatization of Pyrroles. Org. Lett. 2020, 22, 739–744. 10.1021/acs.orglett.9b04581. [DOI] [PubMed] [Google Scholar]; i Panda S.; Coffin A.; Nguyen Q. N.; Tantillo D. J.; Ready J. M. Synthesis and Utility of Dihydropyridine Boronic Esters. Angew. Chem., Int. Ed. 2016, 55, 2205–2209. 10.1002/anie.201510027. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Kadota I.; Kawada M.; Saya S.; Yamamoto Y. Novel Route to the Synthesis of Hydroxylated Piperidine and Pyrrolidine Derivatives via the Intramolecular Reaction of γ-Aminoallylstannane with Aldehyde. Tetrahedron Lett. 1996, 37, 2109–2112. 10.1016/0040-4039(96)00205-5. [DOI] [Google Scholar]; k Kadota I.; Kawada M.; Muramatsu Y.; Yamamoto Y. Asymmetric Total Syntheses of Hydroxylated Piperidine Alkaloids via the Intramolecular Reaction of γ-Aminoallylstannane with Aldehyde. Tetrahedron: Asymmetry 1997, 8, 3887–3893. 10.1016/S0957-4166(97)00563-6. [DOI] [Google Scholar]
- a Spielmann K.; Xiang M.; Schwartz L. A.; Krische M. J. Direct Conversion of Primary Alcohols to 1,2-Amino Alcohols: Enantioselective Iridium-Catalyzed Carbonyl Reductive Coupling of Phthalimido-Allene via Hydrogen Auto-Transfer. J. Am. Chem. Soc. 2019, 141, 14136–14141. 10.1021/jacs.9b08715. [DOI] [PMC free article] [PubMed] [Google Scholar]; For nonenantioselective variants, see:; b Skucas E.; Zbieg J. R.; Krische M. J. anti-Aminoallylation of Aldehydes via Ruthenium-Catalyzed Transfer Hydrogenative Coupling of Sulfonamido Allenes: 1,2-Aminoalcohols. J. Am. Chem. Soc. 2009, 131, 5054–5055. 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zbieg J. R.; Mcinturff E. L.; Krische M. J. Allenamide Hydro-Hydroxyalkylation: 1,2-Amino Alcohols via Ruthenium-Catalyzed Carbonyl anti-Aminoallylation. Org. Lett. 2010, 12, 2514–2516. 10.1021/ol1007235. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhang W.; Chen W.; Xiao H.; Krische M. J. Carbonyl anti-(α-Amino)allylation via Ruthenium Catalyzed Hydrogen Autotransfer: Use of an Acetylenic Pyrrole as an Allylmetal Pronucleophile. Org. Lett. 2017, 19, 4876–4879. 10.1021/acs.orglett.7b02336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gargaro S. L.; Klake R. K.; Burns K. L.; Elele S. O.; Gentry S. L.; Sieber J. D. Access to a Catalytically Generated Umpolung Reagent through the Use of Cu-Catalyzed Reductive Coupling of Ketones and Allenes for the Synthesis of Chiral Vicinal Aminoalcohol Synthons. Org. Lett. 2019, 21, 9753–9758. 10.1021/acs.orglett.9b03937. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Klake R. K.; Gargaro S. L.; Gentry S. L.; Elele S. O.; Sieber J. D. Development of a Strategy for Linear-Selective Cu-Catalyzed Reductive Coupling of Ketones and Allenes for the Synthesis of Chiral γ-Hydroxyaldehyde Equivalents. Org. Lett. 2019, 21, 7992–7998. 10.1021/acs.orglett.9b02973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews; a Nguyen K. D.; Park B. Y.; Luong T.; Sato H.; Garza V. J.; Krische M. J. Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hassan A.; Krische M. J. Unlocking Hydrogenation for C-C Bond Formation: A Brief Overview of Enantioselective Methods. Org. Process Res. Dev. 2011, 15, 1236–1242. 10.1021/op200195m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Han S. B.; Kim I. S.; Krische M. J. Enantioselective Iridium-Catalyzed Carbonyl Allylation from the Alcohol Oxidation Level via Transfer Hydrogenation: Minimizing Pre-activation for Synthetic Efficiency. Chem. Commun. 2009, 7278–7287. 10.1039/b917243m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Holmes M.; Schwartz L. A.; Krische M. J. Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052. 10.1021/acs.chemrev.8b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Agrawal T.; Sieber J. D. Recent Developments in C-C Bond Formation Using Catalytic Reductive Coupling Strategies. Synthesis 2020, 52, 2623–2638. 10.1055/s-0040-1707128. [DOI] [Google Scholar]
- For examples of imine allylation by reductive coupling, see:; a Oda S.; Sam B.; Krische M. J. Hydroaminomethylation Beyond Carbonylation: Allene-Imine Reductive Coupling by Ruthenium-Catalyzed Transfer Hydrognation. Angew. Chem., Int. Ed. 2015, 54, 8525–8528. 10.1002/anie.201503250. [DOI] [PubMed] [Google Scholar]; b Chen T.-Y.; Tsutsumi R.; Montgomery T. P.; Volchkov I.; Krische M. J. Ruthenium Catalyzed C-C Coupling of Amino Alcohols with Dienes via Transfer Hydrogenation: Redox-Triggered Imine Addition and Related Hydroaminoalkylations. J. Am. Chem. Soc. 2015, 137, 1798–1801. 10.1021/ja5130258. [DOI] [PubMed] [Google Scholar]; c Liu T. Y.; Yang Y.; Buchwald S. L. Regiodivergent and Diastereoselective CuH-Catalyzed Allylation of Imines with Terminal Allenes. Angew. Chem., Int. Ed. 2016, 55, 14077–14080. 10.1002/anie.201608446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both the (R)- and (S)-enantiomers of 4-phenyl-2-oxazolidinone (Evans’ oxazolidinone) are commercially available at a cost of ∼$0.25/gram on 1 kg scale (Combi-Blocks/Oakwood Chemical, Nov. 16, 2020). The allenamide is made in one step from 4-phenyl-2-oxazolidinone; see:Bousfield T. W.; Kimber M. C. A simple one-pot preparation of N-allenyl amides, ureas, carbamates, and sulfonamides using a DMSO/tBuOK protocol. Tetrahedron Lett. 2015, 56, 350–352. 10.1016/j.tetlet.2014.11.093. [DOI] [Google Scholar]
- Cottrell T. L.The Strengths of Chemical Bonds, 2d ed.; Butterworth: London, 1958; p 216. [Google Scholar]
- Benson S. Bond Energies. J. Chem. Educ. 1965, 42, 502–518. 10.1021/ed042p502. [DOI] [Google Scholar]
- Bordwell F. G.; Drucker G. E.; Fried H. E. Acidities of Carbon and Nitrogen Acids: the Aromaticity of the Cyclopentadienyl Anion. J. Org. Chem. 1981, 46, 632–635. 10.1021/jo00316a032. [DOI] [Google Scholar]
- Olmstead W. N.; Margolin Z.; Bordwell F. G. Acidities of Water and Simple Alcohols in Dimethyl Sulfoxide Solution. J. Org. Chem. 1980, 45, 3295–3299. 10.1021/jo01304a032. [DOI] [Google Scholar]
- a Ascic E.; Buchwald S. L. Highly Diastereo- and Enantioselective CuH-Catalyzed Synthesis of 2,3-Disubstituted Indolines. J. Am. Chem. Soc. 2015, 137, 4666–4669. 10.1021/jacs.5b02316. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang S.; del Pozo J.; Romiti F.; Mu Y.; Torker S.; Hoveyda A. H. Delayed Catalyst Function Enables Direct Enantioselective Conversion of Nitriles to NH2-amines. Science 2019, 364, 45–51. 10.1126/science.aaw4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalyst prices from Strem Chemical Company (Nov. 16, 2020): (a) Cu(OAc)2 $1.48/g at 100 g scale; (b) 20 wt% PCy3 in toluene 0.50 $/g at 500 g scale.
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Takano Y.; Houk K. N. Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules. J. Chem. Theory Comput. 2005, 1, 70–77. 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Jin X.; Yu H. S.; Truhlar D. G.; He X. Revised M06-L Functional for Improved Accuracy on Chemical Reaction Barrier Heights, Noncovalent Interactions, and Solid-State Physics. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8487–8492. 10.1073/pnas.1705670114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legault C. Y.CYLview, rev 1.0.561; Universite′de Sherbrooke, 2009; http://www.cylview.org. [Google Scholar]
- Li C.; Liu R. Y.; Jesikiewicz L. T.; Yang Y.; Liu P.; Buchwald S. L. CuH-Catalyzed Enantioselective Ketone Allylation with 1,3-Dienes: Scope, mechanism, and applications. J. Am. Chem. Soc. 2019, 141, 5062–5070. 10.1021/jacs.9b01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xiong H.; Hsung R. P.; Shen L.; Hahn J. M. Chiral Enamide. Part 1: Epoxidations of Chiral Enamides. A Viable Approach to Chiral Nitrogen Stabilized Oxyallyl Cations in [4 + 3] cycloadditions. Tetrahedron Lett. 2002, 43, 4449–4453. 10.1016/S0040-4039(02)00849-3. [DOI] [Google Scholar]; b Song Z.; Lu T.; Hsung R. P.; Al-Rashid Z. F.; Ko C.; Tang Y. Stereoselective Simmons-Smith Cyclopropanation of Chiral Enamides. Angew. Chem., Int. Ed. 2007, 46, 4069–4072. 10.1002/anie.200700681. [DOI] [PubMed] [Google Scholar]; c Lu T.; Song Z.; Hsung R. P. A Mutually, p-Facial Selective Cyclopropanation of Chiral Enamides Using Dirhodium(II) Carbenoids. Org. Lett. 2008, 10, 541–544. 10.1021/ol702824s. [DOI] [PubMed] [Google Scholar]; d Xu Y.-S.; Tang Y.; Feng H.-J.; Liu J.-T.; Hsung R. P. A Highly Regio- and Stereoselective Synthesis of α-Fluorinated Imides via Fluorination of Chiral Enamides. Org. Lett. 2015, 17, 572–575. 10.1021/ol503591d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on Distortion–Interaction analysis, seeBickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD - Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Liu R. Y.; Yang Y.; Buchwald S. L. Regiodivergent and Diastereoselective CuH-Catalyzed Allylation of Imines with Terminal Allenes. Angew. Chem., Int. Ed. 2016, 55, 14077–14080. 10.1002/anie.201608446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B.; Arndtsen B. A. Palladium-Catalyzed Stille-Type Coupling of N-Acyl Iminium Ions with Distannanes: A Multicomponent Synthesis of α-Amidostannanes. ACS Catal. 2014, 4, 843–846. 10.1021/cs401164z. [DOI] [Google Scholar]
- Koch V.; Lorion M. M.; Barde E.; Braese S.; Cossy J. Cobalt-Catalyzed α-Arylation of Substituted α-Halogeno β-Lactams. Org. Lett. 2019, 21, 6241–6244. 10.1021/acs.orglett.9b02122. [DOI] [PubMed] [Google Scholar]
- Schaufelberger F.; Hu L.; Ramstrom O. trans-Symmetric Dynamic Covalent Systems: Connected Transamination and Transimination Reactions. Chem. - Eur. J. 2015, 21, 9776–9783. 10.1002/chem.201500520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigg R.; McMeekin P.; Sridharan V. X = Y-ZH systems as potential 1,3-dipoles. Proton Sponge Effects on the 1,2-Prototropic Formation of Azomethine Ylides from Arylidene Benzylamines. Tetrahedron 1995, 51, 13331–13346. 10.1016/0040-4020(95)00846-Z. [DOI] [Google Scholar]
- Lee B.; Lee K. H.; Lim B. W.; Cho J.; Nam W.; Hur N. H. Direct Synthesis of Imines via Solid State Reactions of Carbamates with Aldehydes. Adv. Synth. Catal. 2013, 355, 389–394. [Google Scholar]
- Achar T. K.; Maiti S.; Mal P. IBX Works Efficiently Under Solvent Free Conditions in Ball Milling. RSC Adv. 2014, 4, 12834–12839. 10.1039/C4RA00415A. [DOI] [Google Scholar]
- Green J. C.; Zanghi J. M.; Meek S. J. Diastereo- and Enantioselective Synthesis of Homoallylic Amines Bearing Quaternary Carbon Centers. J. Am. Chem. Soc. 2020, 142, 1704–1709. 10.1021/jacs.9b11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly J. D.; Jacobsen E. N. Thiourea-Catalyzed Enantioselective Hydrophosphonylation of Imines: Practical Access to Enantiomerically Enriched α-Amino Phosphonic Acids. J. Am. Chem. Soc. 2004, 126, 4102–4103. 10.1021/ja0494398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.