

Abstract
The dynamic nature of histone post-translational modifications such as methylation or acetylation makes possible the alteration of disease associated epigenetic states through the manipulation of the associated epigenetic machinery. One approach is through small molecule perturbation. Chemical probes of epigenetic reader domains have been critical in improving our understanding of the biological consequences of modulating their targets, while also enabling the development of novel probe-based reagents. By appending a functional handle to a reader domain probe, a chemical toolbox of reagents can be created to facilitate chemiprecipitation of epigenetic complexes, evaluate probe selectivity, develop in vitro screening assays, visualize cellular target localization, enable target degradation and recruit epigenetic machinery to a site within the genome in a highly controlled fashion.
Keywords: : biotin, bivalent ligand, chemical probe, chemical tool, chromatin reader, epigenetics
Chemical probes have become instrumental tools for the biological community as they can be used to elucidate the roles of their molecular targets in biology and evaluate the therapeutic significance of a wide array of protein classes [1–3]. Such a chemical genetics approach allows for concrete conclusions to be drawn regarding the biological outcome of direct chemical inhibition of a specific domain within a target, in contrast to traditional genetic approaches such as shRNA which ablate the entire protein [4–6]. Specifically, the need for chemical probes for epigenetic protein targets has been fueled by the emergence of epigenetic proteins as potential therapeutic targets in a wide range of diseases [7,8]. A plethora of high-quality chemical probes have been described in recent years for all classes of epigenetic proteins – the readers, writers and erasers – and their utility in biological systems has been reviewed extensively [9–13]. We and others have focused on the development of chemical probes for epigenetic reader domains that bind acetyl-lysine (Kac) and methyl-lysine (Kme), and bromodomain antagonists are perhaps the poster child for the ‘probe-to-biology-to-drug’ revolution in chromatin regulation [9,14].
Despite remarkable successes with bromodomain ligands and other reader antagonists, the development and use of epigenetic reader chemical probes has also revealed some limitations. First, achieving complete selectivity for a member of an epigenetic reader family has proven challenging in some cases, which can make validation of a single target in disease difficult [1,5]. Chromatin reader families such as the bromo- and extra-terminal domains (BET) and chromodomains exemplify this challenge as homologs often exhibit high sequence similarity and conserved binding motifs [15–17]. While we have employed target class screening strategies involving both negative selections and competitor exchange kinetics in an attempt to circumvent this, exquisite selectivity often remains difficult [18]. Second, some families of Kme reader proteins bind their substrate via a surface groove binding mode, characterized by a wider, more accessible binding pocket and often an extensive hydrogen bond network between the reader domain and the peptide backbone of the endogenous substrate [19]. As a result, the development of peptidomimetic chemical probes which preserve these native interactions to gain high affinity is an attractive strategy in targeting these domains; however, peptidic ligands often suffer from poor cell permeability which can make achieving effective cellular concentrations difficult [1,18]. Finally, the noncatalytic nature of chromatin reader domains can make the evaluation of cellular inhibition via a chemical genetics approach less straightforward. Similarly, chromatin reader domains are often part of large multidomain proteins which participate in intricate protein complexes and localize to chromatin in a multivalent manner [20], and such multivalent-binding modes can make eliciting a biological response as a result of antagonism of a single reader domain difficult [21–24].
Although epigenetic reader probes often have these inherent limitations, they have enabled the community to investigate understudied targets from this protein family in a way that would have otherwise not been possible. Importantly, their utility can be greatly expanded through the creation of chemical tools, or chemical probes functionalized with a unique handle to enable applications beyond just target antagonism (Figure 1). This can allow probes to facilitate a greater breadth of biological experiments, as well as novel assays for the identification of new ligand starting points. Herein, we describe advancements in the use of chemical tools derived from chemical probes, with a focus on epigenetic reader probes. We will specifically concentrate on applications that have been uniquely facilitated by such tools to enable biological discovery.
Figure 1. . Functionalization of chemical probes enhances their utility.
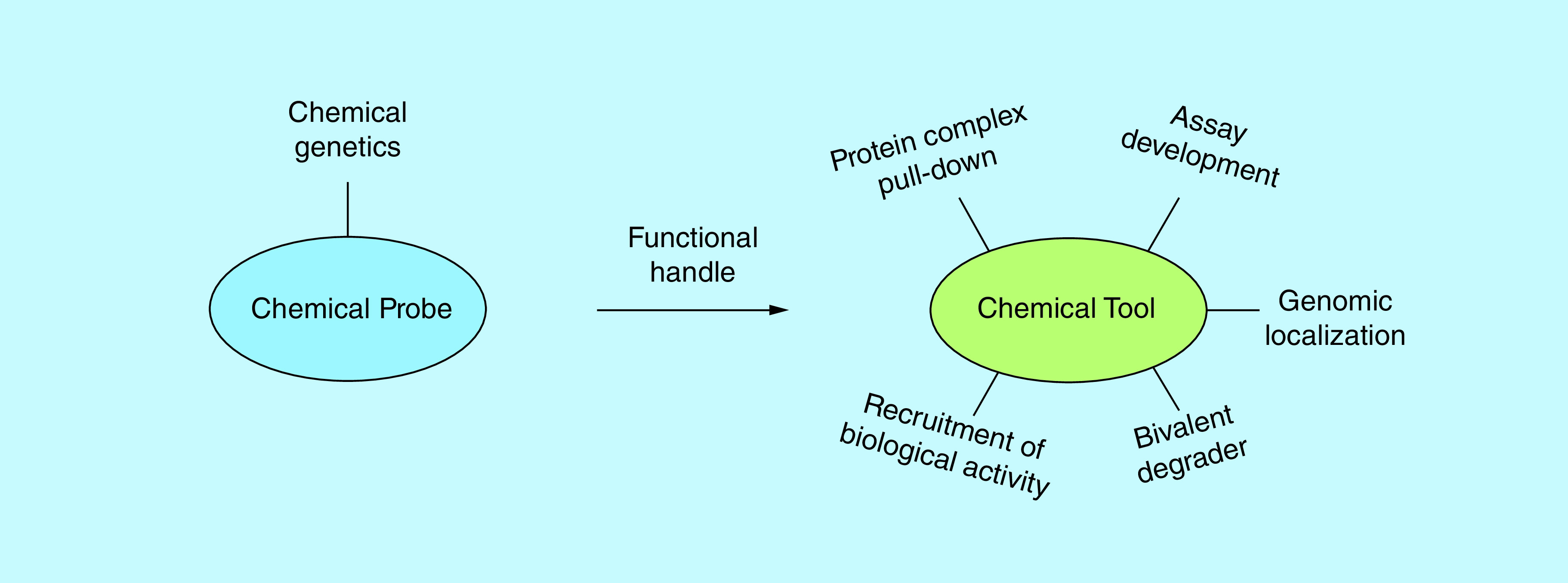
The addition of a variety of functional handles to previously characterized chemical probes allows for their use in a wide array of biological and biochemical platforms.
Biotin: the utility knife of chemical biology
One of the most ubiquitous handles in chemical biology is biotin. The small size of the compound paired with its high, covalent-like affinity (Kd = 10-14 M) and multivalent (4:1) binding with streptavidin makes it a powerful tool for affinity-based applications [25]. Biotin can be affixed to a probe via a variety of functional groups and often a polyethylene glycol (PEG) linker of varying lengths is included as a spacer between the biotin handle and the parent probe (Figure 2A). This linker generally allows the chemical probe to bind its target unencumbered by the biotin tag. As with most functional handles, the positioning of the PEG-biotin is critical to avoid diminishing target binding and often guided by extensive structure–activity relationships (SAR), x-ray or NMR structural data, or molecular modeling. In our laboratory, we have been able to take advantage of the surface-groove binding mode of our peptidomimetic chemical probes that target the Kme reader chromodomain family and affix PEG-biotin tags to the C-terminus which is largely solvent exposed. In the case of UNC3866, a chemical probe for the PRC1 chromodomains (dissociation constant (Kd) of ~100 nM for CBX4 and -7), only a two-fold reduction in binding potency for the target proteins is observed upon addition of PEG11-biotin [26].
Figure 2. . Applications of biotinylated reader domain chemical probes.
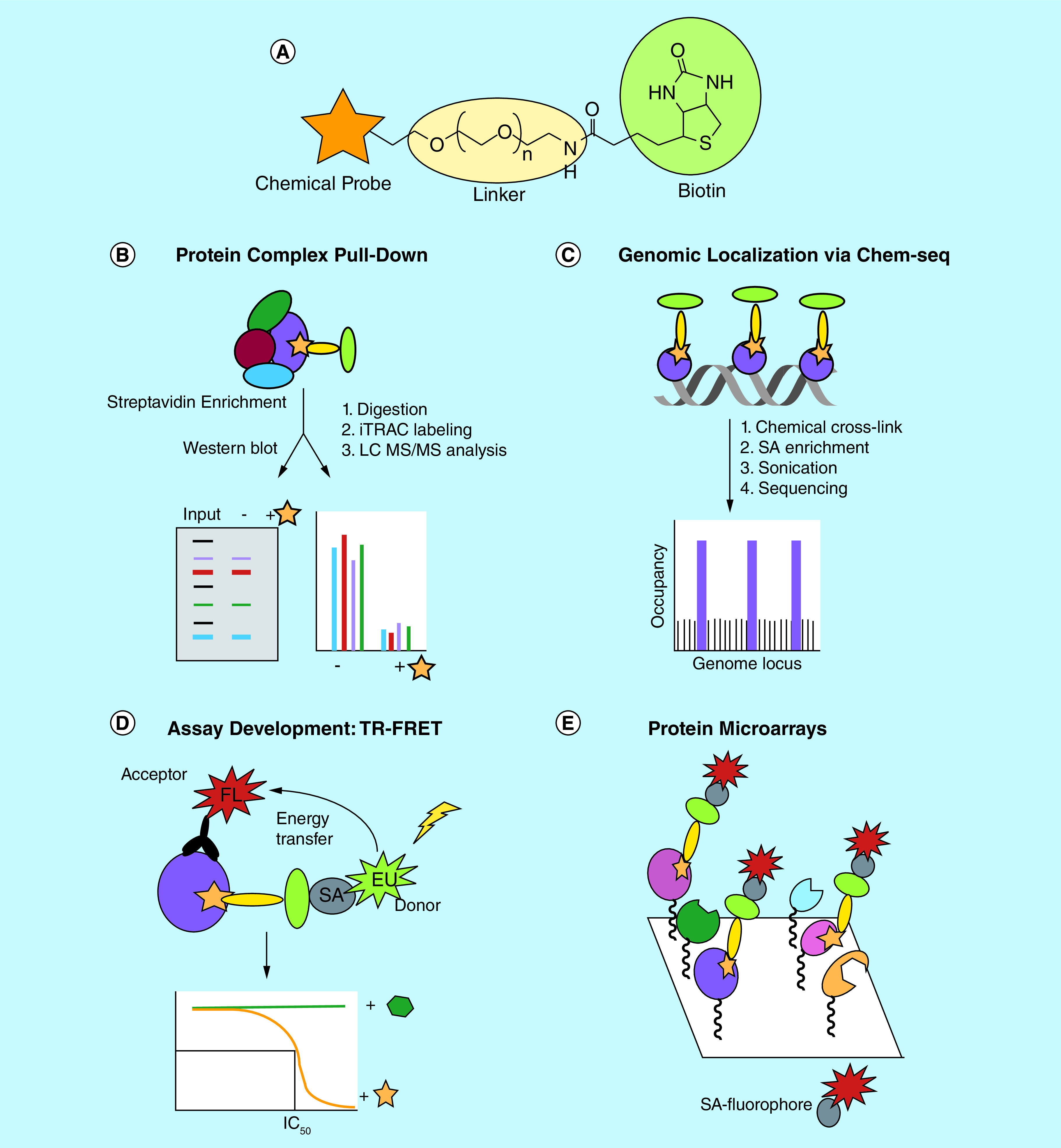
(A) The general structure of a chemical probe (orange star) tagged with biotin (green circle), with the two moieties connected via a PEG-linker (yellow oval). (B) Biotin tagged probes targeting reader domains are capable of pulling down intact protein complexes as determined by western blot or proteomics analysis. (C) The genomic localization of the probe and its target reader protein can be assessed via Chem-seq which couples chemical affinity capture via a biotinylated ligand with massively parallel DNA sequencing. (D) Biotinylated chemical tools can serve as tracer ligands in in vitro TR-FRET assays to screen for new reader domain ligands and evaluate ligand potency to establish SAR. (E) Protein microarrays can be used to assess chemical probe selectivity within the reader target class by incubation with a biotinylated chemical tool and visualization with fluorescent streptavidin.
FL: Fluorophore; PEG: Polyethylene glycol; SA: Streptavidin; SAR: Structure–activity relationships; TR-FRET: Time-resolved fluorescence energy transfer.
Chemiprecipitation-based applications
One of the most common applications of a biotinylated probe is its use as a chemiprecipitation reagent (Figure 2B). Such pull-down experiments from cell lysates have become a powerful method to confirm probe interaction with its target protein in a complex cellular environment. We and others have used this technique to demonstrate that epigenetic reader probes engage their endogenous, full length protein targets by either western blot analysis or proteomics, whereas biophysical assays are generally conducted with recombinant protein and isolated domains [15,26–28]. This method is especially powerful as extremely high potency is not necessarily required for effective chemiprecipitation. Furthermore, through addition of the parent probe as a soluble competitor to block pull-down of the target(s), the specific nature of the interaction can be further demonstrated [15,26,27]. In the case of UNC3866, whole cells were incubated with increasing concentrations of chemical probe prior to lysis and chemiprecipitation. The ability of UNC3866 to block chemiprecipitation of CBX7 with biotinylated UNC3866 additionally highlighted its ability to penetrate the cell membrane and engage endogenous CBX7 in whole cells [26].
Biotinylated chemical tools can also enable the pull-down of intact protein complexes of which the target is a member, confirming that the probe engages its target in the context of its protein partners. For example, biotinylated UNC3866 was shown to effectively chemiprecipitate core components of PRC1 (RING1B, BMI-1 and HPH2) through direct engagement with the CBX chromodomains [26]. Interestingly, although UNC3866 is capable of interacting with all five polycomb CBXs in vitro, biotinylated UNC3866 selectively pulls down endogenous CBX4, -7 and -8 and is unable to engage CBX2 or CBX6 in this context, perhaps due to the inaccessibility of the chromodomain-binding site within PRC1, suggestive of potential differences between the CBX homologs within a cellular environment. Ligands for EED, the Kme reader component of PRC2, including EED226 and UNC5114 have also been biotinylated and shown to pull-down all core PRC2 components including EED, EZH2 and SUZ12 from cell lysates by both western blot and chemoproteomics [27,29]. Importantly, the ability to chemiprecipitate protein complexes allows these chemical tools to be utilized in an unbiased fashion for the discovery of new protein partners of the target of interest via analysis by mass spectrometry [30,31]. Similarly, such an unbiased chemoproteomics approach can also be critical in broadly evaluating chemical probe selectivity as it enables unanticipated targets of the probe to be identified. Subsequent co-immunoprecipitation experiments can be performed to distinguish between proteins that are identified via chemoproteomics due to interaction with the primary target versus direct interaction with the biotinylated probe.
Chemical inhibitor-based ChIP
As epigenetic reader proteins continue to reveal their relevance in cancer and the potential benefits from small molecule intervention are becoming increasingly clear, it is important to understand how chemical probes and their targets interact genome-wide to gain better insight into global chromatin regulatory processes. Biotinylated tool compounds can serve as synthetic alternatives to antibodies to map the direct interactions of the chemical tool and its target(s) with chromatin throughout the genome (Figure 2C). Utilizing a biotinylated derivative of the BET bromodomain inhibitor JQ1, Anders et al. demonstrated genome-wide localization of the chemical tool and BRD4 uses a method that combines ligand affinity capture and massively parallel DNA sequencing known as Chem-seq [32,33]. In this experiment, live cells were incubated with biotin-JQ1 followed by chemical cross-linking of chromatin bound proteins to DNA and isolation and fragmentation of the DNA. DNA sequences associated with biotin-JQ1 were then enriched using streptavidin beads and identified via sequencing to be mapped to the genome. Importantly, comparison of these results with those generated via ChIP-seq with antibodies for BRD2, BRD3 and BRD4, the biological targets of JQ1, revealed that biotinylated JQ1 was localized to sites of actively transcribing genes that were also occupied by the BRD isoforms. It should be noted that there remains the possibility that biotinylated ligands targeting epigenetic reader domains will dislodge their protein target from chromatin, particularly if there are no other chromatin interacting domains to facilitate protein localization in the presence of the inhibitor. In this case, cells can be fixed prior to addition of the biotinylated chemical tool. This approach is also more amenable to biotinylated chemical tools that lack sufficient cellular permeability to achieve necessary intracellular concentrations. Tyler et al. implemented a similar strategy, and in order to circumvent potential permeability issues they synthesized JQ1 with a chemically reactive moiety to ‘build’ the biotinylated JQ1 tool molecule intracellularly via bioorthogonal chemical ligation by click chemistry [34]. Overall, this method highlights the potential for biotinylated chemical tools to enhance our understanding of probe-binding sites through the genome and the mechanisms by which probes alter gene expression programs.
Development of novel screening assays
While the applications discussed above largely focus on the use of biotinylated tools to improve biological understanding of their epigenetic reader targets, these reagents can also be extremely valuable for the development of novel assays. In contrast to the enzymatic writer and eraser target classes, the development of high-throughput in vitro assays for reader proteins is uniquely challenging in part because of their lack of catalytic activity which obviates substrate conversion assays. Assay development for epigenetic readers is also hindered by the relatively low affinity of reader domains for their cognate histone targets, with dissociation constants in the mM range for some proteins. This can make achieving a useful assay window and practical protein concentrations for high-throughput screening difficult [35]. Therefore, the use of functionalized chemical tools to improve existing assays and develop new assays to screen for novel hits compounds is of great significance in the field of epigenetics.
A recent paper from Rectenwald et al. describes a general platform for screening methyl-lysine reader domains using a Time-Resolved Fluorescence Energy Transfer assay (Figure 2D) [36]. This is a proximity-based assay that relies on the interaction between a biotinylated tracer ligand and 6X-His tagged protein to initiate energy transfer. The Time-Resolved Fluorescence Energy Transfer assays that were developed for the chromodomains of CBX2, CBX4, CBX7, CBX8 and CDYL2 made use of biotinylated UNC3866 as a potent bait ligand. In each case, the higher affinity of this inhibitor-based chemical tool for the chromodomain relative to biotinylated histone peptides (e.g., H3K9me3 and H3K27me3) allowed for a significant reduction in the amount of protein and other reagents required for the assay, while similarly increasing the assay throughput. A focused screen of CBX2 using this assay produced UNC3866 as a top hit as expected, and encouragingly, the IC50 value matched favorably with the previously reported Kd determined by isothermal titration calorimetry [26]. Therefore, a single biotinylated chemical tool enabled the development of a suite of robust assays to screen for novel chromodomain inhibitors in a target class fashion [18].
Protein microarray approaches
Protein microarrays can be useful tools in evaluating ligand selectivity or binding trends within a target class. Although rarely quantitative, they provide a rapid and inexpensive means to profile ligand binding across a large number of purified protein targets. Bedford et al. have developed extensive arrays for epigenetic readers, spanning more than 200 domains and numerous protein families in some cases [37,38]. Using GST fusion proteins immobilized on glass slides, binding to biotinylated ligands can be detected via incubation with fluorescently tagged streptavidin (Figure 2E). We and others have utilized this technology in conjunction with our biotinylated tool compounds to facilitate broad profiling of Kme reader chemical probes. For example, a microarray that contained close to 100 chromatin effector proteins was incubated with biotin-UNC3866 which yielded a preliminary qualitative assessment of the molecular targets of UNC3866 [26]. These results were then followed up with a quantitative analysis of the binding affinities by isothermal titration calorimetry.
A similar approach was taken to evaluate the selectivity of UNC1215, a Kme reader chemical probe that targets the MBT domains of L3MBTL3 [28]. In this case, despite validation in numerous orthogonal assays, binding between biotin-UNC1215 and L3MBTL3 was not observed on the array. A similar result was obtained with biotinylated H4K20me2, the endogenous substrate of L3MBTL3. This could be due to instability or improper folding of L3MBTL3 upon immobilization to the surface, inaccessibility of the protein binding site or protein precipitation from freeze/thaw cycles, among other things. Therefore, while this method is a powerful approach to examine ligand selectivity within the reader domain protein class using biotinylated tools, negative results can be difficult to interpret.
Protein microarrays can also be powerful target class screening tools for the discovery of inhibitors with unique selectivity profiles. Bae et al. utilized a microarray of 98 Kme reader proteins to screen a series of biotinylated UNC1215 analogs [39]. This ‘library-on-library’ screening approach led to the discovery of compounds with either increased or decreased specificity relative to UNC1215, and in one case, a compound was identified that interacted with the Tudor domain containing protein SPIN1. Follow-up structural studies facilitated the rational design of SPIN1 inhibitors with increased potency and selectivity. This study highlights the ability to use this platform and biotinylated chemical tools for Kme reader domains to ‘target-hop’ and identify ligands with unique selectivity profiles for further development into novel chemical probes.
Lighting up chemical probes with fluorophores
Fluorescent labeling of chemical probes creates powerful tools for a variety of applications in chemical biology and their uses have been extensively described [40–42]. From subcellular localization to assay development, fluorescent small molecules allow for a unique set of scientific inquires to be addressed with relative ease. Further, a wide array of fluorophores are available to develop such bioorthogonal tools which range in chemical structure, physiochemical properties and excitation and emission wavelengths in order to tailor the tool for a desired application [43]. For example, great consideration was taken in the selection of mero76, a cell penetrant long wavelength merocyanine dye, to functionalize UNC1215 [28,44]. The choice of this dye was driven by the desire to achieve a cell permeable and cell stable fluorescent tool to examine the colocalization of UNC1215 with GFP tagged L3MBTL3 and demonstrate cellular target engagement. Additionally, there is little spectral overlap between GFP and mero76 [40]. Using this fluorescent chemical tool, it was shown that mero76-UNC1215 colocalizes with full length L3MBTL3 in cells and that this interaction was compound dependent (Figure 3). Furthermore, the fact that mero76-UNC1215 did not displace GFP-L3MBTL3 from its nuclear loci revealed that the Kme-binding function of L3MBTL3 is likely not the only element contributing to the nuclear localization of L3MBTL3. This study represents an excellent example of utilizing a fluorescent chemical tool to better demonstrate cellular target engagement of the chemical probe. In a separate study, live cell imaging of BRD4 was achieved by functionalizing JQ1 with a dual tag containing a diazirine and a cyclopropane moiety, which could facilitate both chemical cross-linking with BRD4 via UV irradiation and fluorescent labeling of BRD4 using copper-free bioorthogonal chemistry in situ, respectively [45].
Figure 3. . Fluorescent chemical probes demonstrate cellular target engagement.
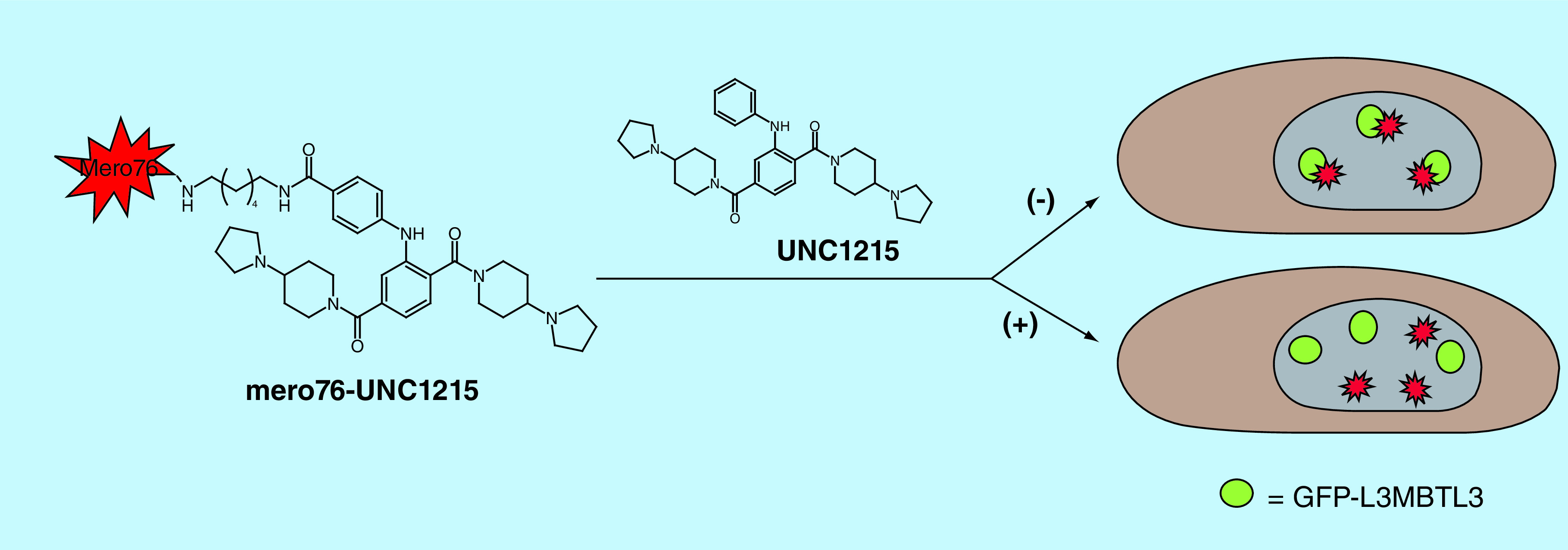
UNC1215 was tagged with the cell penetrant, long wavelength merocyanine dye mero76 and mero76-UNC1215 was shown to colocalize with GFP-L3MBTL3, indicating that UNC1215 engages its target in cells. Localization of mero76-UNC1215 was perturbed with the addition of untagged UNC1215.
Fluorescent conjugation can also be used to facilitate fluorescence-based assays and screening applications for epigenetic reader proteins in a similar fashion to the biotinylated tool compounds discussed above. Barnash et al. describe the development of a peptidomimetic combinatorial screening strategy to identify ligands for the chromodomain containing protein CDYL2 [15]. Here, hit compounds identified in the screen were synthesized with an alkyne moiety at the C-terminus for further modification through click chemistry to append fluorescein. The binding affinities of these ligands for a panel of seven closely related chromodomain containing proteins were then determined by fluorescence polarization to simultaneously evaluate potency and selectivity within the family [46]. Fluorescent analogs of reader inhibitors can also be utilized as tracer ligands in competition-based assays to screen for novel binders or evaluate SARs in a more high-throughput fashion. This was nicely demonstrated with a peptidic inhibitor of EED (compound 3) identified by combinatorial chemistry which was C-terminally labeled with fluorescein to enable a robust EED fluorescence polarization assay and establish SAR trends [27].
Targeted protein degradation with bivalent degraders
Epigenetic chemical probes can be powerful assets in elucidating the roles of their targets in chromatin biology; however, the noncatalytic and multivalent nature of reader proteins can often result in uncertainty as to whether or not potent reader domain ligands will elicit a strong biological response [9,28,47]. An emerging strategy in chemical biology is the use of bivalent degrader compounds to hijack the cell’s ubiquitin ligase and proteasome machinery to degrade a protein of interest (POI) rather than simply inhibit its function. This approach has been extensively reviewed elsewhere for a variety of protein classes and targets [48–54]. The basic architecture of a bivalent degrader involves three core components: a ligand that binds the POI targeted for degradation, a linker moiety and a ligand that recruits an E3 ubiquitin ligase. Unlike biotin and fluorophore handles, which can be appended to a ligand without too much concern for the length or composition of the linker connecting the two moieties, the two ligands of a bivalent degrader must be linked in a precise fashion to facilitate the formation of a ternary complex and subsequent ubiquitination of the POI.
Bivalent degraders are attractive for targeting chromatin reader proteins when high-quality ligands are sparse and inhibition alone does not result in a strong phenotype [10,50]. Further, the catalytic nature of bivalent degraders can circumvent potential limitations associated with weak-binding ligands, as achieving extremely high potency for Kme reader domains can be particularly challenging. Interestingly, bivalent degraders have also been shown to invoke highly selective degradation independent of ligand selectivity [50]. This is particularly relevant for chromatin reader proteins where exquisite target selectivity within a family of homologs can be hard to achieve with small molecules. For example, despite the immense investment in ligands for BET bromodomains in recent years, a high-quality chemical probe that selectively inhibits BRD2, BRD3 or BRD4 has yet to be realized. Despite this challenge, Zengerle et al. recently demonstrated the selective degradation of BRD4 with their bivalent degrader MZ1 which is composed of the BET chemical probe JQ1 linked to a recruiter of the von Hippel-Lindau (VHL) E3 ubiquitin ligase (Figure 4A). While MZ1 is capable of degrading all three BET bromodomains at high concentrations (1 μM), incubation with lower concentrations of MZ1 revealed selective degradation of BRD4 over BRD2 and BRD3 (Figure 4B). Remarkably, through the development of this JQ1-based bivalent chemical tool, one now has the ability to specifically interrogate BRD4-mediated biology in a way that was previously not possible [55]. Building on this success, Gadd et al. solved a structure of the BRD4-MZ1-VHL ternary complex and utilized structure-based design to rationally develop novel BRD4 degraders which exhibit enhanced efficacy and selectivity in cells [56]. Importantly, BET targeted chemical degraders have demonstrated improved efficacy over their chemical probe counterparts in various disease models suggesting a potential clinical advantage. For example, using the pan-BET degrader ARV-771, Raina et al. found that BET degradation resulted in dramatically improved efficacy in cellular models of castration-resistant prostate cancer and a castration-resistant prostate cancer mouse xenograph model as compared with BET inhibition alone [57].
Figure 4. . Bivalent degrader approach affords selective degradation of BRD4.
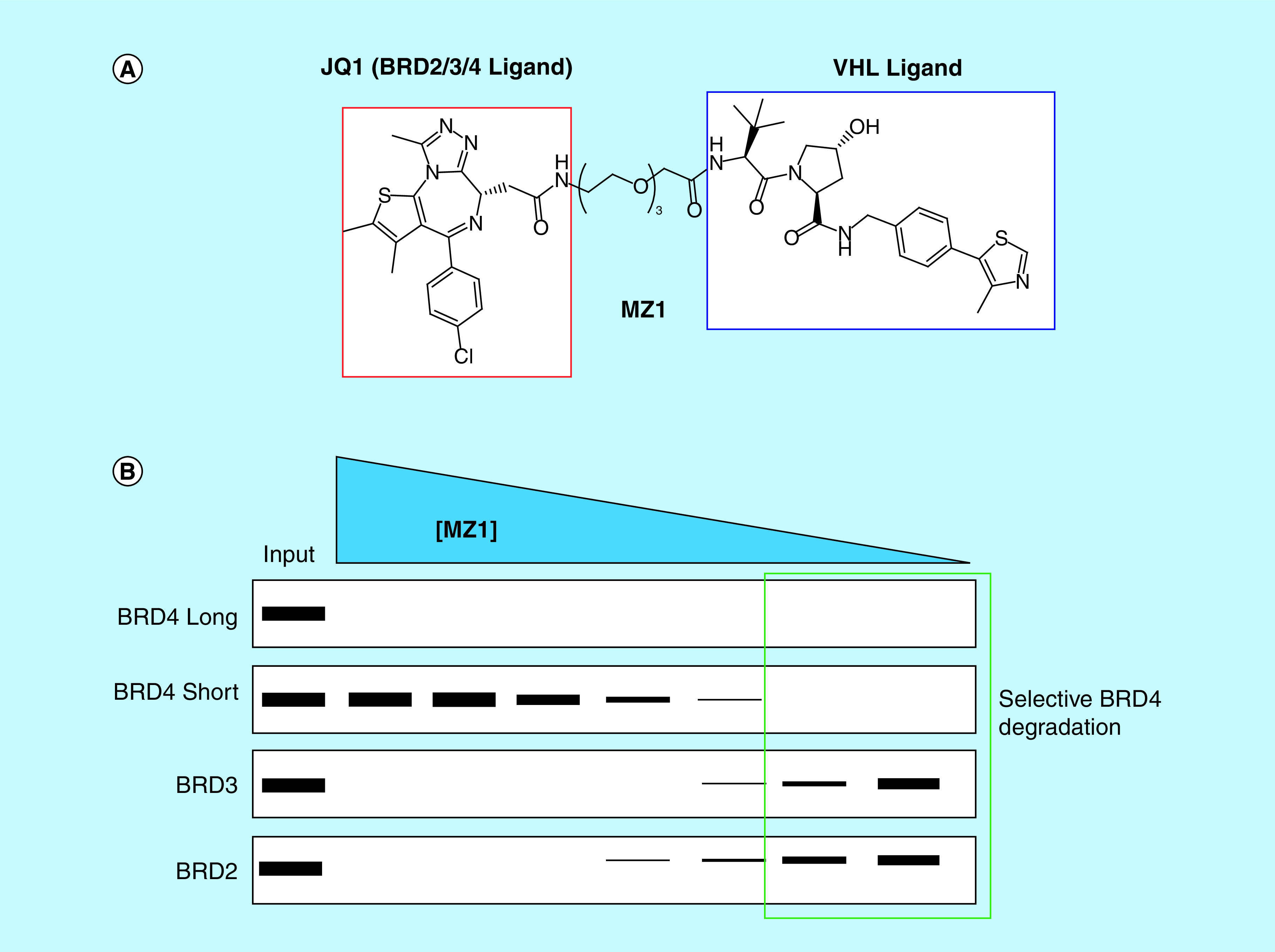
(A) Structure of the bivalent degrader, MZ1, which contains a bromo- and extra-terminal domain targeting ligand (JQ1) and a ligand for VHL. (B) MZ1 achieves selective degradation of BRD4 even though JQ1 is about equipotent for BRD2, BRD3 and BRD4.
VHL: von Hippel-Lindau.
Chemical probes targeting other bromodomains have also been successfully tagged to facilitate target degradation. Bassi et al. demonstrated that while small molecule inhibition of the bromodomains of PCAF/GCN5 using the potent binder GSK4027 (IC50 = 50 nM) showed little effect on modulating cytokine production, a PCAF/GCN5 chemical degrader (GSK983) effectively modulated the expression of multiple inflammatory cytokines mirroring PCAF-deficient immune cells [58]. In another study, ligands for the bromodomains of the SMARCA2 and SMARCA4 ATPases enabled the development of potent degraders of these BAF complex subunits. Encouragingly, degradation of SMARCA2 and SMARCA4 with ACBI1 led to pronounced antiproliferative effects and cell death across multiple cancer cell lines, suggesting that this may be a viable strategy to treat tumors sensitive to the loss of SMARCA2 and -4 [59].
Bromodomain containing proteins have been more widely targeted using this chemical degradation strategy largely due to the increased availability of bromodomain chemical probes and clinical candidates in some cases [14,54,60–64]; however, this approach could be equally powerful in unlocking the therapeutic potential of other classes of chromatin readers such as Kme reader proteins. We recently reported UNC6852, a first-in-class chemical degrader of PRC2 which is composed of the potent EED ligand EED226 linked to the VHL ligand VH032 (Figure 5A) [65]. UNC6852 is capable of selectively degrading EED as well as the other core PRC2 components SUZ12 and EZH2, as confirmed by proteomics analysis (Figure 5B). Further, UNC6852 results in a decrease in H3K27me3 which is installed primarily by PRC2 and reduced cell proliferation in DB cells harboring an EZH2Y641N gain of function mutation. This study provides confirmation that Kme reader proteins can be successfully targeted through a bivalent degrader approach and suggests that this strategy can be extended to other members within this target class.
Figure 5. . Bivalent degrader targeting EED leads to polycomb repressive complex 2 degradation.
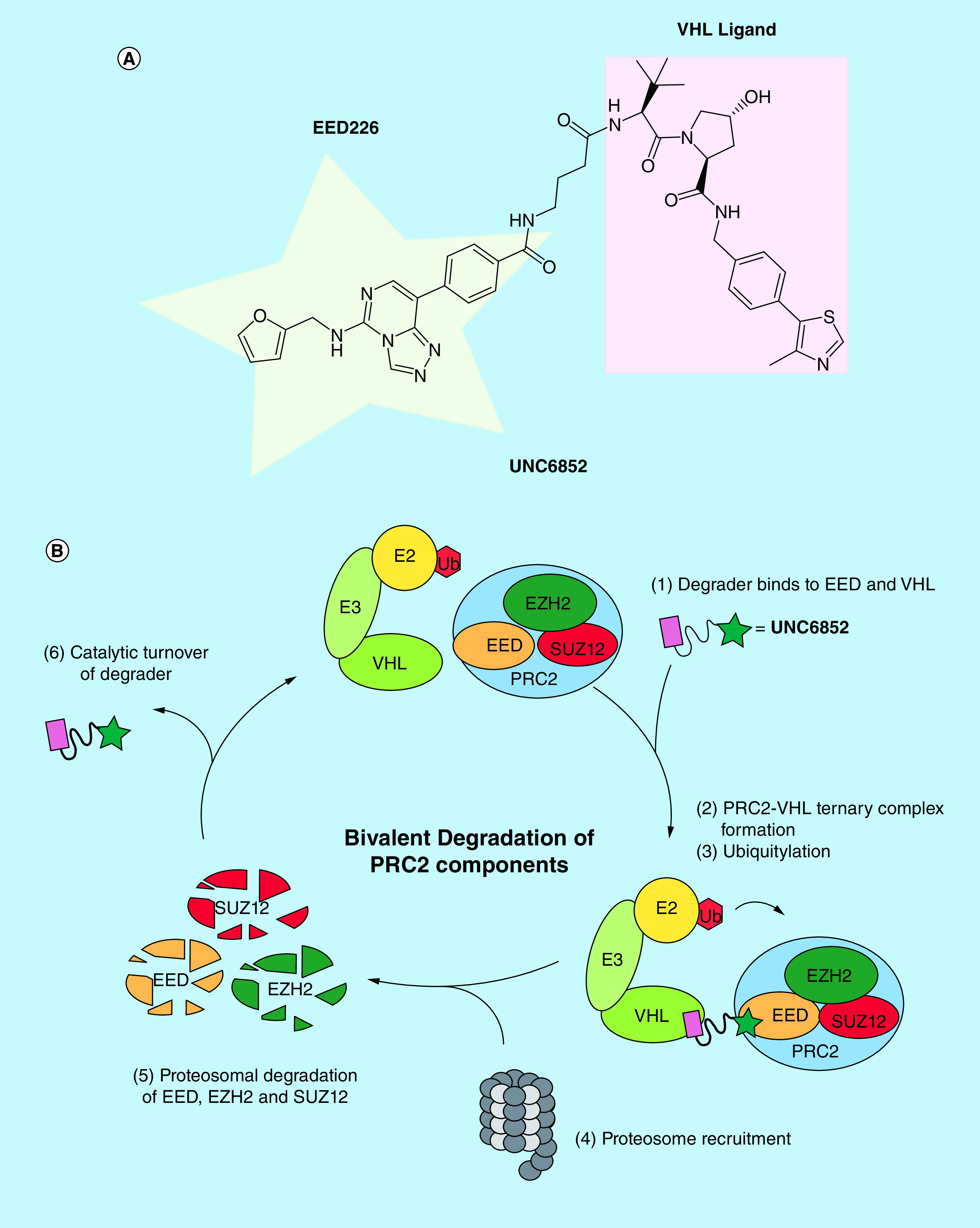
(A) Structure of UNC6852 which is composed of the EED ligand, EED226, tethered via an alkyl linker to a VHL ligand. (B) In the catalytic cycle of UNC6852, a ternary complex is first formed between PRC2, UNC6852 and VHL. Subsequent PRC2 ubiquitylation marks the complex for degradation and it is recruited to the proteasome, resulting in degradation of the core PRC2 components (EZH2, EED and SUZ12).
VHL: von Hippel-Lindau.
Directing biological activity: genomic targeting of epigenetic reader probes
When tagged with the appropriate machinery, the utility of epigenetic reader probes can be further expanded through their ability to recruit endogenous chromatin modifying complexes to targeted loci in order to regulate the expression of target genes (Figure 6A). Such applications of epigenetic bioengineering at the interface of chemistry and biology which combine the flexibility of pharmacologic manipulation with the specificity of DNA-binding elements are of great interest [66,67]. Additionally, they can provide attractive alternatives to using genetic fusion proteins for genomic localization. In recent years, numerous research groups have taken complimentary approaches to achieve genomic targeting of epigenetic reader probes, often in a temporal fashion, in turn hijacking the cells natural machinery, manipulating the epigenetic landscape and allowing for loci-specific alterations of gene expression [68]. Importantly, these approaches may inform new therapeutic strategies to target a variety of disease states caused by transcriptional dysfunction.
Figure 6. . Recruitment of reader proteins to specific genomic loci to control gene expression.
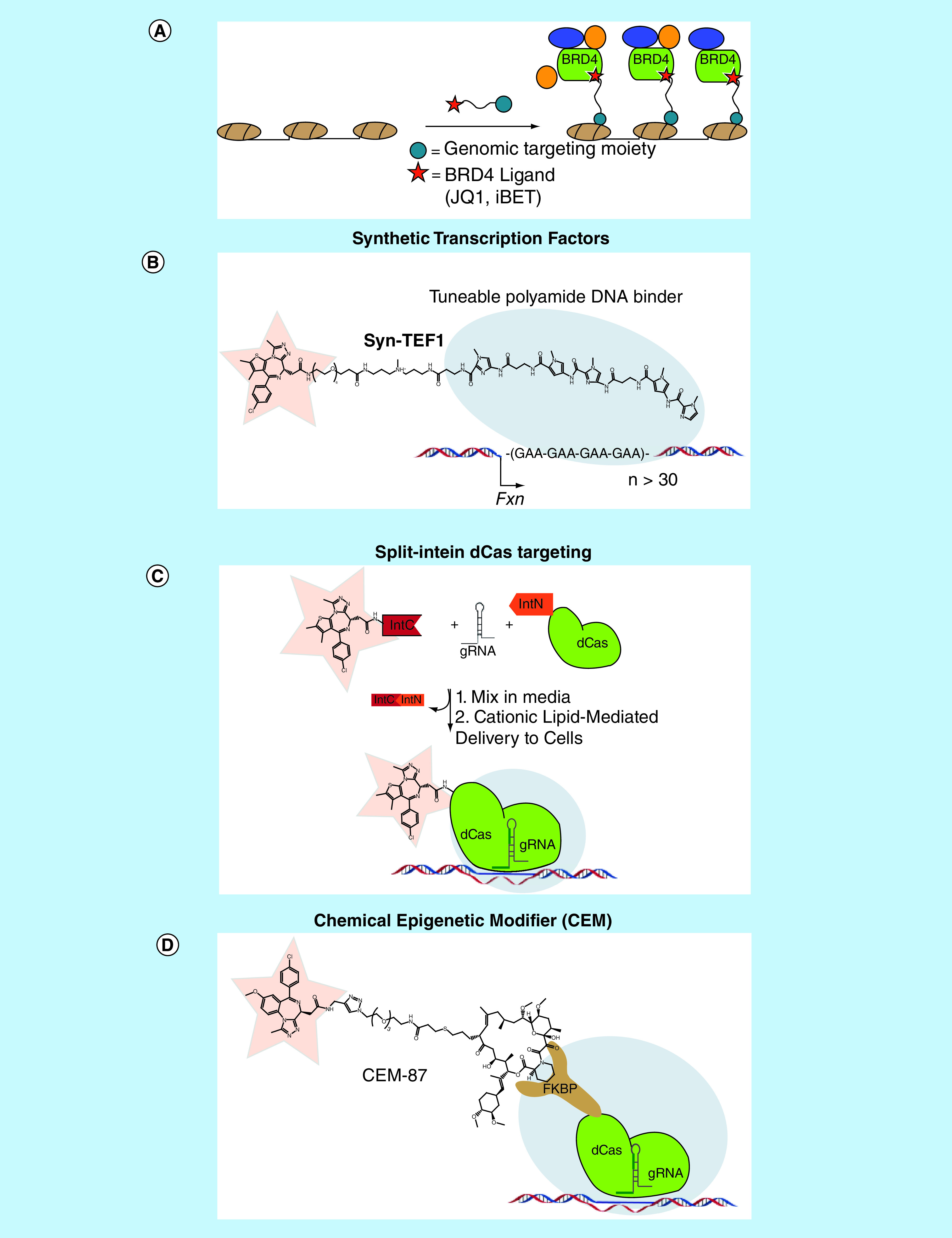
(A) General strategy of using bivalent molecules containing a BET bromodomain ligand and a genomic targeting moiety to site specifically recruit BRD4 to perturb gene expression. (B) Syn-TEF1 recruits BRD4 to the FXN gene by localizing to GAA repeats via its polyamide DNA-binding moiety, in turn activating FXN expression. (C) Mixing JQ1 fused to IntC with a guide RNA and dCas-IntN in cell media results in an assembled dCas-JQ1: guide RNA complex which can then be delivered to cells via cationic lipid-mediated delivery and localize to its target DNA sequence. (D) A dCas9-FKBP fusion protein targets CEM-87 to a gene of interest, which induces transcription at that loci through the recruitment of BRD4.
CEM: Chemical epigenetic modifier
Synthetic transcription factors
Natural transcription factors typically bind near a gene and recruit associated transcriptional machinery to nearby promoters through unique protein–protein interactions resulting in increased gene expression [69]. The genomic ‘address’ of transcriptional regulation is largely determined by the DNA-binding domain of the transcription factor [69,70]. The use of synthetic transcription elongation factors (Syn-TEFs) to control cell fate is an emerging technology [66,71]. Syn-TEFs can be designed to specifically regulate the genomic location and timing of transcription, allowing for a level of control not achievable by simple overexpression of transcription factors [66,72]. Syn-TEFs are bivalent molecules composed of a DNA-binding domain that can be tailored for a specific promoter sequence and a small molecule ligand, tethered by a linker region [70]. Pyrrole- and imidazole-based polyamides have emerged as a unique class of synthetic DNA binders as they can be programmed to bind specific DNA sequences, thus allowing for localization of Syn-TEFs and transcription elongation at targeted loci [70–75].
A landmark example from the Ansari laboratory demonstrated the ability to localize a Syn-TEF and induce transcription in a heterochromatic environment (Figure 6B) [67]. Specifically, they utilized a Syn-TEF with a DNA-binding domain that targeted a GAA repeat to enable transcription of the (FXN) gene, which is held in a heterochromatic state in Friedreich's ataxia (FRDA), a terminal neurological disorder [76]. The DNA-binding domain was tethered to JQ1 to localize BRD4 to the FXN gene, as BRD4 binds to areas of active transcription and engages P-TEFb to stimulate transcription through phosphorylation of Pol II [77,78]. Upon treatment of FRDA cells with the JQ1-derived Syn-TEF, FXN expression was selectively activated in a dose-dependent manner without altering BRD4 occupancy at endogenous loci and global transcriptome profiles. Furthermore, these results were recapitulated in primary cells derived from FRDA patients and in mice transplanted with human cells carrying an FXN-Luc gene. Overall, Syn-TEFs are powerful chemical biology tools enabled by high-quality chemical probes, particularly epigenetic reader probes such as JQ1 which can recruit transcriptional machinery to chromatin in a precise fashion to alter transcription.
dCas9 targeting systems
The development of technologies utilizing the DNA-binding protein Cas9 and its mutant analog which lacks endonuclease activity (dCas9) have similarly allowed for genomic-specific alterations in gene expression [68,79–85]. Recently, the Muir laboratory utilized a unique split intein-mediated protein trans-splicing technique to site specifically link dCas9 with chemical probes to recruit chromatin-binding proteins at specific genetic elements (Figure 6C) [84,86]. This method has the distinct advantage over standard bioconjugation techniques in that it can be performed in cell media allowing for delivery of the reaction product directly to cells. They utilized the epigenetic reader probes JQ1 and UNC3866 as synthetic cargo to recruit their target reader domains (BET bromodomains and PRC1 chromodomains, respectively) to specific genomic loci and in turn harness endogenous cellular transcriptional machinery. A two-hybrid luciferase-based assay under the control of a Gal4 promoter revealed that dCas9-JQ1 could localize to its target Gal4 sequence and initiate transcription through the recruitment of endogenous BET proteins. Additionally, they were able to demonstrate genomic localization of endogenous BRD4 by ChIP-qPCR. Similarly, dCas9-UNC3866 was capable of selectively recruiting PRC1 machinery to target sites. This method can be easily expanded to include a variety of chemical cargos to generate a unique set of probe-based tools to study nuclear processes and highlights the ability to direct an epigenetic chemical probe to a specific region of chromatin using the dCas9-targeting vehicle.
Chemical epigenetic modifiers
A similar system exploiting the power of dCas9 was recently developed by the Hathaway laboratory which utilizes chemical epigenetic modifiers (CEM). CEMs are gene-regulating bifunctional molecules consisting of an epigenetic probe that can recruit chromatin machinery tethered to a ligand that binds to a DNA-protein anchor, which in this case is FK506, a potent ligand for FKBP (Figure 6D). Through the use of a dCas9-FKBP fusion protein, the CEMs can be targeted to any genomic location [83]. CEMs are highly tunable reagents and have the added advantage of being cell permeable via passive diffusion. CEMs have recently been developed incorporating a variety of bromodomain chemical probes including those for the BET bromodomains, CBP/p300 and BRPF1, all of which are associated with euchromatin and active transcription [87]. In each case, a significant increase in gene activation was seen upon CEM addition in a dose-dependent and gene-specific manner. Specifically, all CEMs were capable of inducing expression of endogenous target gene MYOD1 at nanomolar concentrations, with the BET bromodomain recruiting CEM (CEM87) showing the greatest activity. This highlights the value of being able to easily utilize a variety of CEMs to tailor the expression of endogenous genes. Additionally, ChIP-seq analysis for BRD4 at the MYOD1 gene post CEM87 treatment showed genomic localization consistent with MYOD1 target sites.
Conclusion & future perspective
The development of a high-quality chemical probe for an epigenetic reader domain is an immense accomplishment in its own right; however, it is clear that the innovation need not stop there. While epigenetic reader probes have the potential to aid in validating new targets, increase biological understanding of these targets, and translate into compounds of therapeutic value, their utility can be greatly expanded through the generation of tools based on the probe. The diverse handles that can be appended to a probe to create bivalent molecules are not limited to those discussed herein; yet, is remarkable how much can be accomplished through the addition of a simple biotin tag. Furthermore, recent chemical genetic approaches facilitated by probe-based protein degraders and epigenetic bioengineering strategies using probes to tightly regulate gene expression patterns truly epitomize chemical biology and highlight the importance of this field. Notably, the development of such epigenetic tool compounds to enable further biological discovery has largely been facilitated thanks to the disclosure of chemical probes to the scientific community. By continuing to make novel, first-in-class probes publicly available, we can undoubtedly depend on the creativity of the chemical biology community to develop innovative tools and probe-based applications to further promote scientific discovery.
Executive summary.
High-quality potent and selective chemical probes of epigenetic reader domains have proven to be critical in improving our understanding of the biological consequences of modulating their targets and validating new epigenetic targets for drug discovery.
The utility of chemical probes can be greatly expanded through the creation of chemical tools, or chemical probes functionalized with a unique handle to allow for applications beyond just target inhibition and enable the development of novel probe-based technologies.
Biotinylation of reader probes can facilitate the chemiprecipitation of epigenetic protein complexes, evaluation of probe selectivity within the proteome, development of new in vitro screening assays and mapping of genome-wide interactions between the probes and their reader targets, among other things.
The conjugation of fluorophores to reader probes can similarly aid in assay development, while also allowing for fluorescent imaging to visualize probe localization within the cell and cellular target engagement.
The use of bivalent degraders, which tether a probe to an E3 ligase recruiting moiety, are a promising strategy in targeting chromatin reader proteins when high-quality ligands are sparse and inhibition alone does not result in a strong phenotype.
Epigenetic bioengineering strategies in which genomic targeting moieties are appended to reader chemical probes to recruit biological activity to specific genomic loci can tightly regulate gene expression patterns.
Footnotes
Financial & competing interests disclosure
LI James gratefully acknowledges the National Institute on Drug Abuse, National Institutes of Health (NIH; grant R61DA047023), the National Cancer Institute, (NIH; grant R01CA242305), and the University Cancer Research Fund, University of North Carolina at Chapel Hill for support. JM Waybright acknowledges the National Cancer Institute for a training grant (grant T32CA217824). The authors have no other relevant affiliations of financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Arrowsmith CH, Audia JE, Austin C et al. The promise and peril of chemical probes. Nat. Chem. Biol. 11(8), 536–541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garbaccio RM, Parmee ER. The impact of chemical probes in drug discovery: a pharmaceutical industry perspective. Cell Chem. Biol. 23(1), 10–17 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Frye SV. The art of the chemical probe. Nat. Chem. Biol. 6(3), 159–161 (2010). [DOI] [PubMed] [Google Scholar]; • Editorial explaining the criteria that defines a chemical probe.
- 4.Schreiber SL. Small molecules: the missing link in the central dogma. Nat. Chem. Biol. 1(2), 64–66 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Runcie AC, Chan KH, Zengerle M, Ciulli A. Chemical genetics approaches for selective intervention in epigenetics. Curr. Opin. Chem. Biol. 33, 186–194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawasumi M, Nghiem P. Chemical genetics: elucidating biological systems with small-molecule compounds. J. Invest. Dermatol. 127(7), 1577–1584 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11(5), 384–400 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat. Chem. Biol. 8(5), 417–427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James LI, Frye SV. Chemical probes for methyl lysine reader domains. Curr. Opin. Chem. Biol. 33, 135–141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A concise review of currently available chemical probes for methyl-lysine reader domains.
- 10.Ackloo S, Brown PJ, Muller S. Chemical probes targeting epigenetic proteins: applications beyond oncology. Epigenetics 12(5), 378–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 17(4), 268 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Milosevich N, Hof F. Chemical inhibitors of epigenetic methyllysine reader proteins. Biochemistry 55(11), 1570–1583 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 17(3), 160–183 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Noguchi-Yachide T. BET bromodomain as a target of epigenetic therapy. Chem. Pharm. Bull. 64(6), 540–547 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Barnash KD, Lamb KN, Stuckey JI et al. Chromodomain ligand optimization via target-class directed combinatorial repurposing. ACS Chem. Biol. 11(9), 2475–2483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19(12), 1218–1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamps JJ, Huang J, Poater J et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 6, 8911 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnash KD, James LI, Frye SV. Target class drug discovery. Nat. Chem. Biol. 13(10), 1053–1056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14(11), 1025–1040 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8(12), 983–994 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothbart SB, Dickson BM, Ong MS et al. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 27(11), 1288–1298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savitsky P, Krojer T, Fujisawa T et al. Multivalent histone and DNA engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-rich chromatin. Cell Rep. 17(10), 2724–2737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suh JL, Watts B, Stuckey JI et al. Quantitative characterization of bivalent probes for a dual bromodomain protein, transcription initiation factor TFIID subunit 1. Biochemistry 57(14), 2140–2149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vangamudi B, Paul TA, Shah PK et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 75(18), 3865–3878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dundas CM, Demonte D, Park S. Streptavidin-biotin technology: improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 97(21), 9343–9353 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Stuckey JI, Dickson BM, Cheng N et al. A cellular chemical probe targeting the chromodomains of polycomb repressive complex 1. Nat. Chem. Biol. 12(3), 180–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnash KD, The J, Norris-Drouin JL et al. Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS Comb. Sci. 19(3), 161–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James LI, Barsyte-Lovejoy D, Zhong N et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 9(3), 184–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi W, Zhao K, Gu J et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 13(4), 381–388 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Drewes G, Knapp S. Chemoproteomics and chemical probes for target discovery. Trends Biotechnol. 36(12), 1275–1286 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Moellering RE, Cravatt BF. How chemoproteomics can enable drug discovery and development. Chem. Biol. 19(1), 11–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders L, Guenther MG, Qi J et al. Genome-wide localization of small molecules. Nat. Biotechnol. 32(1), 92–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Development of Chem-seq which is a method that couples ligand-affinity capture and massively parallel DNA sequencing to identify the sites bound by small chemical molecules throughout the human genome.
- 33.Filippakopoulos P, Qi J, Picaud S et al. Selective inhibition of BET bromodomains. Nature 468(7327), 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Initial discovery of BET inhibitor JQ1 which is used in a variety of applications discussed in this review.
- 34.Tyler DS, Vappiani J, Caneque T et al. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science 356(6345), 1397–1401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herold JM, Ingerman LA, Gao C, Frye SV. Drug discovery toward antagonists of methyl-lysine binding proteins. Curr. Chem. Genomics 5, 51–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rectenwald JM, Hardy PB, Norris-Drouin JL et al. A general TR-FRET assay platform for high-throughput screening and characterizing inhibitors of methyl-lysine reader proteins. SLAS Discov. 24(6), 693–700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shanle EK, Shinsky SA, Bridgers JB et al. Histone peptide microarray screen of chromo and Tudor domains defines new histone lysine methylation interactions. Epigenetics Chromatin 10, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Espejo A, Cote J, Bednarek A, Richard S, Bedford MT. A protein-domain microarray identifies novel protein-protein interactions. Biochem. J. 367(Pt 3), 697–702 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bae N, Viviano M, Su X et al. Developing spindlin1 small-molecule inhibitors by using protein microarrays. Nat. Chem. Biol. 13(7), 750–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lavis LD, Raines RT. Bright ideas for chemical biology. ACS Chem. Biol. 3(3), 142–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao M, Yu F, Lv C, Choo J, Chen L. Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chem. Soc. Rev. 46(8), 2237–2271 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Allali-Hassani A, Wasney GA, Siarheyeva A, Hajian T, Arrowsmith CH, Vedadi M. Fluorescence-based methods for screening writers and readers of histone methyl marks. J. Biomol. Screen. 17(1), 71–84 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Lavis LD, Raines RT. Bright building blocks for chemical biology. ACS Chem. Biol. 9(4), 855–866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacNevin CJ, Watanabe T, Weitzman M et al. Membrane-permeant, environment-sensitive dyes generate biosensors within living cells. J. Am. Chem. Soc. 41(18), 7275–7282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Wang D, Li L et al. “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J. Am. Chem. Soc. 136(28), 9990–9998 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Moerke NJ. Fluorescence polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding. Curr. Protoc. Chem. Biol. 1(1), 1–15 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Jani KS, Jain SU, Ge EJ et al. Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc. Natl Acad. Sci. USA 116(17), 8295–8300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol. Ther. 174, 138–144 (2017). [DOI] [PubMed] [Google Scholar]; • Detailed review on the development and use of bivalent degraders.
- 49.Bondeson DP, Crews CM. Targeted protein degradation by small molecules. Annu. Rev. Pharmacol. Toxicol. 57, 107–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu S, Cui D, Chen X, Xiong X, Zhao Y. PROTACs: an emerging targeting technique for protein degradation in drug discovery. BioEssays 40(4), e1700247 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheepstra M, Hekking KFW, van Hijfte L, Folmer RHA. Bivalent ligands for protein degradation in drug discovery. Comput. Struct. Biotechnol. J. 17, 160–176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An S, Fu L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36, 553–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang CY, Qin C, Bai L, Wang S. Small-molecule PROTAC degraders of the bromodomain and extra terminal (BET) proteins – a review. Drug Discov. Today Technol. 31, 43–51 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10(8), 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Details the discovery and application of the selective BRD4 degrader MZ1.
- 56.Gadd MS, Testa A, Lucas X et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13(5), 514–521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Elegant use of structure-based-design for the development of more selective and effective BET bivalent degraders.
- 57.Raina K, Lu J, Qian Y et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA 113(26), 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bassi ZI, Fillmore MC, Miah AH et al. Modulating PCAF/GCN5 immune cell function through a PROTAC approach. ACS Chem. Biol. 13(10), 2862–2867 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Farnaby W, Koegl M, Roy MJ et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 15(7), 672–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Henssen A, Thor T, Odersky A et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 4(11), 2080–2095 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fong CY, Gilan O, Lam EY et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525(7570), 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ocana A, Nieto-Jimenez C, Pandiella A. BET inhibitors as novel therapeutic agents in breast cancer. Oncotarget 8(41), 71285–71291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghoshal A, Yugandhar D, Srivastava AK. BET inhibitors in cancer therapeutics: a patent review. Expert Opin. Ther. Pat. 26(4), 505–522 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Markowski MC, De Marzo AM, Antonarakis ES. BET inhibitors in metastatic prostate cancer: therapeutic implications and rational drug combinations. Expert Opin. Investig. Drugs 26(12), 1391–1397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potjewyd F, Turner A-MT, Beri J et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol. 27(1), 47–56.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eguchi A, Lee GO, Wan F, Erwin GS, Ansari AZ. Controlling gene networks and cell fate with precision-targeted DNA-binding proteins and small-molecule-based genome readers. Biochem. J. 462(3), 397–413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Erwin GS, Grieshop MP, Ali A et al. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 358(6370), 1617–1622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Landmark paper utilizing synthetic transcription factors to induce transcription at disease relevant loci via BET recruitment.
- 68.Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13(2), 127–137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morimoto RI. Transcription factors: positive and negative regulators of cell growth and disease. Curr. Opin. Cell Biol. 4(3), 480–487 (1992). [DOI] [PubMed] [Google Scholar]
- 70.Arora PS, Ansari AZ, Best TP, Ptashne M, Dervan PB. Design of artificial transcriptional activators with rigid poly-L-proline linkers. J. Am. Chem. Soc. 124(44), 13067–13071 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Khalil AS, Lu TK, Bashor CJ et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell 150(3), 647–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mapp AK, Ansari AZ, Ptashne M, Dervan PB. Activation of gene expression by small molecule transcription factors. Proc. Natl Acad. Sci. USA 97(8), 3930–3935 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carlson CD, Warren CL, Hauschild KE et al. Specificity landscapes of DNA binding molecules elucidate biological function. Proc. Natl Acad. Sci. USA 107(10), 4544–4549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dervan PB. Molecular recognition of DNA by small molecules. Bioorg. Med. Chem. 9(9), 2215–2235 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Erwin GS, Grieshop MP, Bhimsaria D et al. Synthetic genome readers target clustered binding sites across diverse chromatin states. Proc. Natl Acad. Sci. USA 113(47), E7418–E7427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pandolfo M. Friedreich's ataxia: clinical aspects and pathogenesis. Semin. Neurol. 19(3), 311–321 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19(4), 523–534 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Yang Z, Yik JH, Chen R et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19(4), 535–545 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Braun SMG, Kirkland JG, Chory EJ, Husmann D, Calarco JP, Crabtree GR. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 8(1), 560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao D, Liang FS. Chemical inducible dCas9-guided editing of H3K27 acetylation in mammalian cells. Methods Mol. Biol. 1767, 429–445 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Ma D, Peng S, Xie Z. Integration and exchange of split dCas9 domains for transcriptional controls in mammalian cells. Nat. Commun. 7, 13056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shrimp JH, Grose C, Widmeyer SRT, Thorpe AL, Jadhav A, Meier JL. Chemical control of a CRISPR-Cas9 acetyltransferase. ACS Chem. Biol. 13(2), 455–460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Butler KV, Chiarella AM, Jin J, Hathaway NA. Targeted gene repression using novel bifunctional molecules to harness endogenous histone deacetylation activity. ACS Synth. Biol. 7(1), 38–45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liszczak GP, Brown ZZ, Kim SH, Oslund RC, David Y, Muir TW. Genomic targeting of epigenetic probes using a chemically tailored Cas9 system. Proc. Natl Acad. Sci. USA 114(4), 681–686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hilton IB, D'Ippolito AM, Vockley CM et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33(5), 510–517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ludwig C, Pfeiff M, Linne U, Mootz HD. Ligation of a synthetic peptide to the N terminus of a recombinant protein using semisynthetic protein trans-splicing. Angew. Chem. Int. Ed. Engl. 45(31), 5218–5221 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Chiarella AM, Butler KV, Gryder BE et al. Dose-dependent activation of gene expression is achieved using CRISPR and small molecules that recruit endogenous chromatin machinery. Nat. Biotechnol. (2019) (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates the utility of chemical epigenetic modifiers in the regulation of gene expression.