

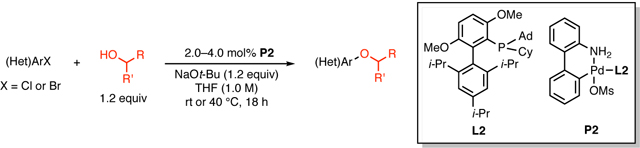
Abstract
An improved protocol for the Pd-catalyzed C–O cross-coupling of secondary alcohols is described. The use of biaryl phosphine L2 as the ligand was key to achieving efficient cross-coupling of (hetero)aryl chlorides with only a 20% molar excess of the alcohol. Additionally, we observed an unusual reactivity difference between an electron-rich aryl bromide and the analogous aryl chloride, and deuterium-labeling suggested that currently unidentified pathways for reduction play an important role in explaining this disparity.
Graphical Abstract
The synthesis of alkyl aryl ethers has seen significant advances in the past decade.1 Traditional approaches, such as the Williamson ether synthesis,2 the Mitsunobu reaction,3 and nucleophilic aromatic substitution,4 often require specific and limited classes of substrates to achieve efficient C–O bond formation. Transition-metal-catalyzed C–O cross-coupling reactions, including Pd,5 Cu6 and Ni7 catalysis, have also been improving to operate on an increasingly broad scope of (hetero)aryl halides and aliphatic alcohols. Alternative metal-free approaches involving sulfonate esters8a or diaryliodonium salts8b, 8c, 8d also show great promise.
The Pd-catalyzed O-arylation of aliphatic alcohols has been widely explored by our group and by others.5 As de-picted in Scheme 1, the slow rate of reductive elimination from intermediate [LnPdII(Ar)(alkoxide)] (IV) is generally believed to account for the diminished efficiency of C–O bond formation, compared to the analogous C–N cross-coupling processes.9 As a result, competitive β-hydride elimination can lead to the overall reduction of the aryl halide and the formation of undesired carbonyl side products. Between the two classes of aliphatic alcohols bearing β-hydrogens, secondary alcohols are typically considered more challenging coupling partners. Previously, the coupling reaction of primary alcohols has been shown to proceed with higher yields and less reduction of aryl halide, compared to that of the corresponding secondary alcohols under the same reaction conditions.5c, 5g, 5i, 5n The most recent report from our group on the coupling reaction of secondary alcohols introduced the use of a bulky biaryl phosphine ligand RockPhos (L1, Figure 1) to facilitate C–O bond formation.5i This method required the use of two equivalents of alcohol substrate at somewhat elevated temperature (90 °C) to provide moderate yields with a restricted scope of alcohol substrates (Scheme 2A).
Scheme 1.
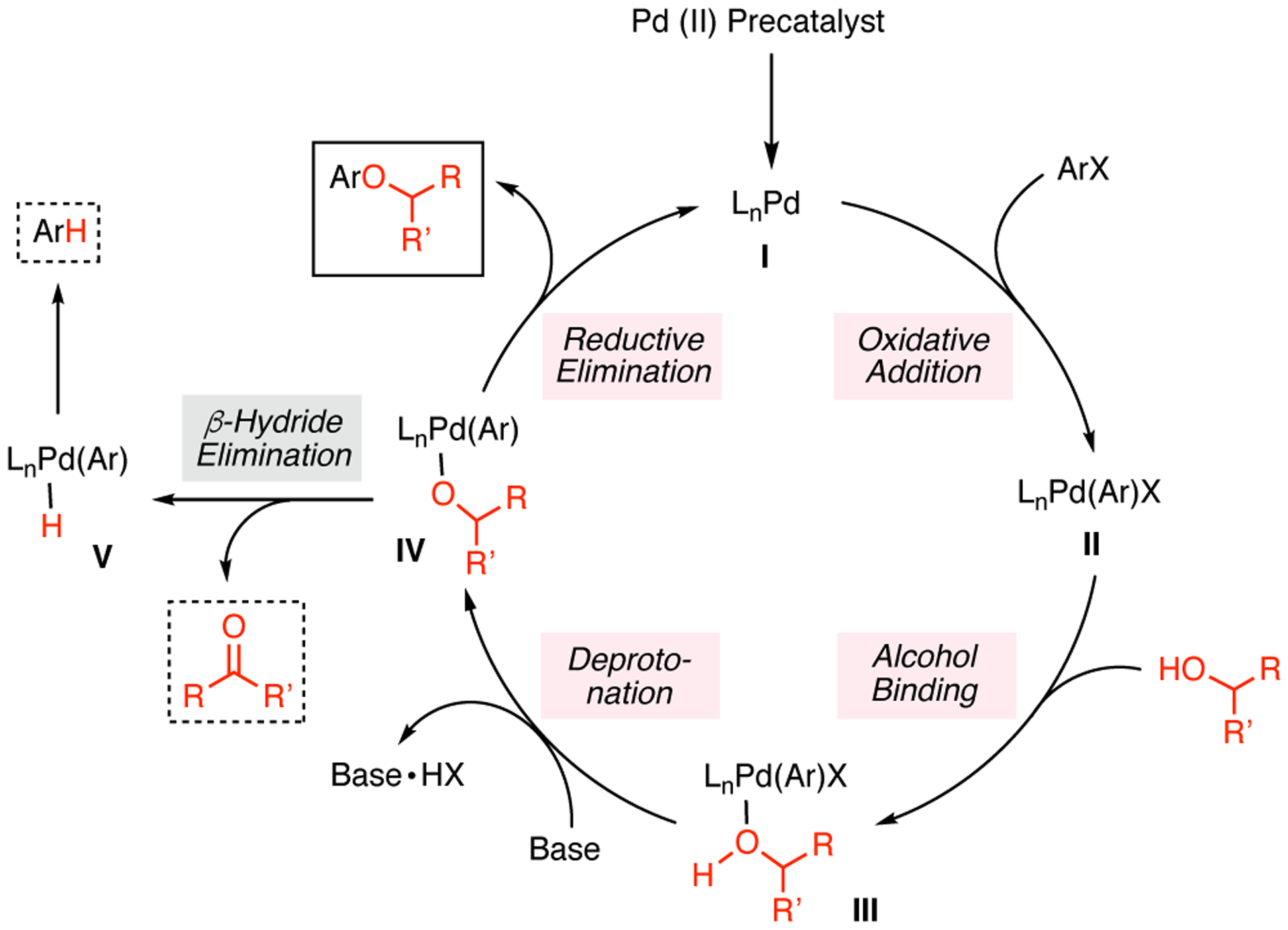
General catalytic cycle of Pd-catalyzed C–O cross-coupling of aryl halides with secondary alcohols.
Figure 1.

Ligands and precatalysts employed in Pd-, Cu- and Ni-catalyzed C–O cross-coupling of secondary alcohols.
Scheme 2.
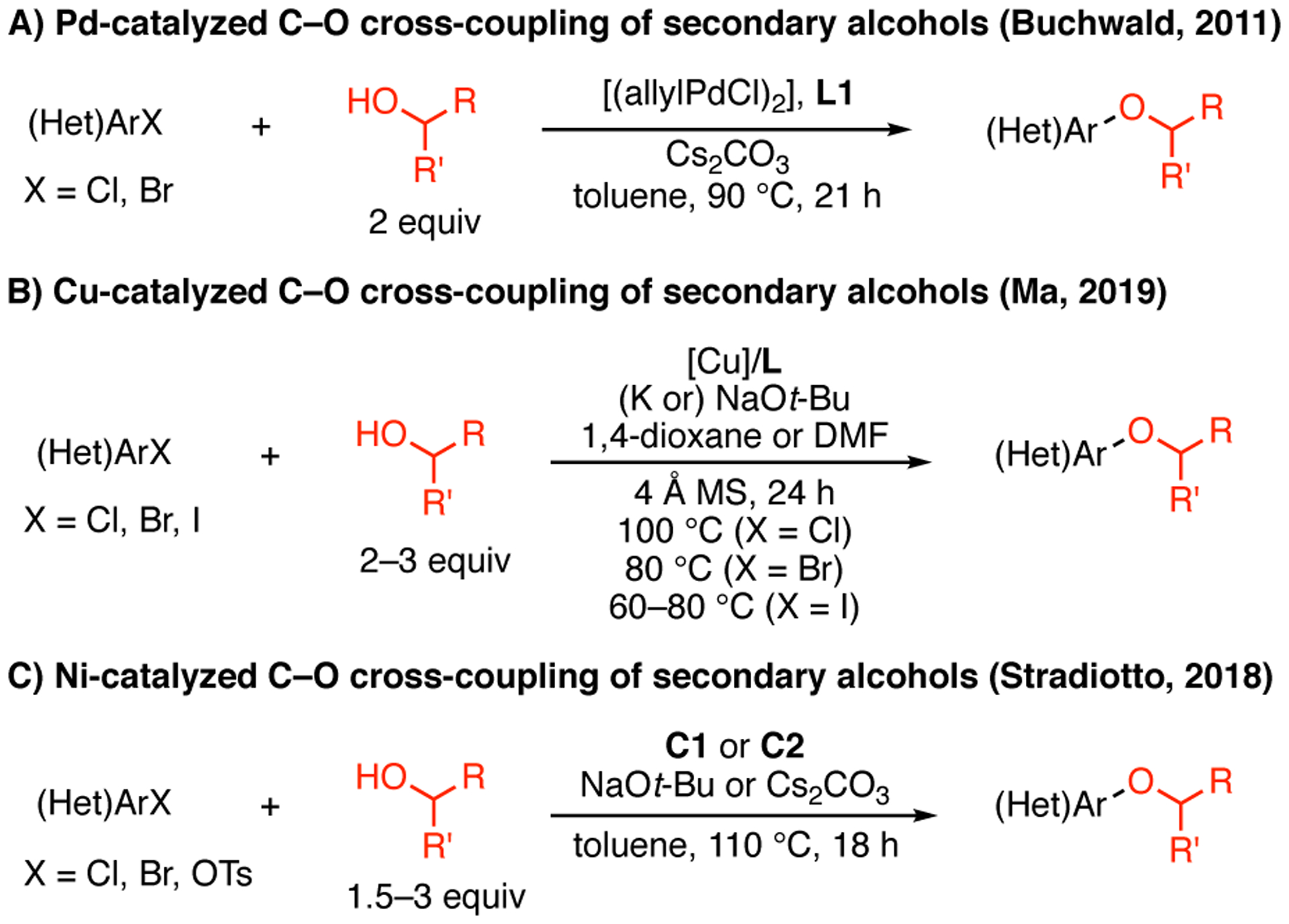
Literature precedents of transition-metalcatalyzed C–O cross-coupling of secondary alcohols.
Two recent reports from the groups of Ma6a and Stradiotto7a demonstrated great potential for utilizing Cu- and Ni-catalyzed methods, respectively, to prepare secondary alkyl aryl ethers. The use of two oxalic diamide ligands and sodium tert-butoxide enabled Cu-catalyzed C–O cross-coupling of secondary alcohols with aryl chlorides, bromides and iodides with excellent efficiency.6a Compared to that of more reactive aryl bromides and iodides, the coupling reaction of aryl chlorides still required higher catalyst loading (10 mol % Cu) as well as elevated temperature (100 °C). Ni-catalyzed C–O cross-coupling of secondary alcohols with aryl halides was achieved with the use of (Cy)PAd-DalPhos-based precatalysts,7a proving the feasibility of such transformation in the absence of photoredox catalysts.7b Although the Ni-catalyzed coupling of aryl chlorides with secondary alcohols was first achieved in this report, the method was limited to activated substrates and also required elevated temperature (110 °C). Additionally, all three metal-catalyzed methods necessitated the use of 100% or more molar excess of alcohols (i.e., more than two equivalents of alcohol, with two exceptions7a) to ensure successful C–O bond formation. Therefore, the development of a method that operates under mild conditions, preferably at room temperature, and that utilizes a smaller excess of alcohol (i.e., less than two equivalents of alcohol) for the coupling reactions of (hetero)aryl chlorides and secondary alcohols, remains a desirable goal.
We recently disclosed a catalyst system that employs one of two ligands for Pd-catalyzed C–O cross-coupling reactions of (hetero)aryl halides with primary alcohols.5b In particular, the use of a new (now commercially available, CAS No.: 2197989-24-3) hybrid biaryl phosphine ligand L2 (Figure 1) allowed the effective coupling of challenging electrophiles, including unactivated aryl chlorides (e.g., electron-rich (hetero)aryl chlorides). Herein, we report the Pd-catalyzed C–O cross-coupling of secondary alcohols, facilitated by the use of L2, with a diverse range of (hetero)aryl chlorides under improved reaction conditions: more than half of the reactions proceeded at room temperature, while only requiring a 20% molar excess of alcohols. Additionally, we present examples of the Oarylation of secondary alcohols with aryl bromides and the observation of an unusual difference in reactivity between an electron-rich aryl bromide and the corresponding aryl chloride.5b, 5i, 10
Following the conditions previously reported for the cross-coupling of primary alcohols,5b we employed palladacycle P2 (Figure 1) as the precatalyst and 1,4-dioxane as the solvent. Electron-rich aryl chloride 1, containing a para-morpholino substituent, and sec-butanol (s-BuOH) were chosen as model coupling partners. Lowering the number of equivalents of s-BuOH from 3 to 1.2 resulted in a decline in the efficiency of the reaction: the conversion of the starting aryl chloride 1 decreased by 50%, and the ratio of the reduction side product 4 to the coupling product 3 increased substantially (Table 1, entries 1–3). However, when the solvent was changed to THF, lowering the number of equivalents of s-BuOH to 1.2 had a negligible effect on the efficiency of the reaction (Table 1, entries 4–6). Therefore, THF was selected as an appropriate solvent for further exploration of the substrate scope.
Table 1.
Evaluation for Pd-catalyzed C–O cross-coupling of electron-rich aryl halides with different equivalents of s-BuOH in 1,4-dioxane and THF.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | X | solvent | equiv of s-BuOH | Conver version (%)b | yield of 3 (%)b | yield of 4 (%)b |
| 1 | Cl | dioxane | 3 | 100 | 94 | 8 |
| 2 | Cl | dioxane | 2 | 78 | 62 | 14 |
| 3 | Cl | dioxane | 1.2 | 50 | 22 | 18 |
| 4 | Cl | THF | 3 | 100 | 98 | 3 |
| 5 | Cl | THF | 2 | 100 | 97 | 4 |
| 6 | Cl | THF | 1.2 | 100 | 90 | 9 |
| 7 | Br | THF | 3 | 100 | 67 | 31 |
| 8 | Br | THF | 2 | 100 | 72 | 27 |
| 9 | Br | THF | 1.2 | 100 | 63 | 33 |
Reaction conditions: ArX (0.5 mmol), s-BuOH (x mmol), NaOt-Bu (0.6 mmol), P2 (2.0 mol %), solvent (0.5 mL, 1.0 M), rt, 18 h. dioxane = 1,4-dioxane. THF = tetrahydrofuran.
Determined by GC analysis using an internal standard. All yields presented are not normalized. In certain cases, the sum of yields being over 100% is a result of the error in the analytical method used.
Although it is widely accepted that aryl bromides exhibit higher reactivity in cross-coupling reactions than aryl chlorides,5b, 5i, 10 we observed an opposite trend between electron-rich aryl chloride 1 and aryl bromide 2. First, under the same set of reaction conditions (Table 1, entries 6 and 9), while the reaction of 1 provided a 90% yield of desired aryl ether 3, along with 9% reduction product 4, that of 2 only provided a 63% yield of 3, with a notable increase in the amount of 4 (33%) that was formed. Although this difference could be partially ameliorated by adjusting the quantity of s-BuOH to 2 equivalents, a further increase of the amount of alcohol utilized did not lead to an additional improvement in the yield of 3 (Table 1, entries 7 and 8).
A variety of (hetero)aryl chlorides and secondary alcohols were surveyed to examine the generality of this method (Scheme 3). The C–O cross-coupling reactions took place under mild conditions, using only a 20% molar excess of alcohols. Many traditionally-challenging substrates, including unactivated aryl chlorides (3, 5, 7) and fivemembered heterocycles (9, 10, 11) readily underwent C–O bond formation at room temperature. Various heterocycles, such as a quinoline (7), a pyridazine (8), a pyrazole (8), a thiadiazole (9), a benzisothiazole (10), a benzimidazole (11), a pyrazine (12), a quinazoline (13), a pyrazolopyrimidine (14), and a pyridine (15) were tolerated as structural components in the electrophiles. Functional groups such as an unprotected tertiary hydroxyl group (6), a carbamate group (7), and a lactone (14) were also compatible with the reaction conditions. While stericallyaccessible alcohols proved to be good coupling partners at room temperature, secondary alcohols with moderate steric encumbrance at either the α-carbon (12, 13) or the β-carbon (14, 15) required moderate heating (40 °C) to react with activated heteroaryl chlorides and afford corresponding heteroaryl ethers in ≥80% yields.
Scheme 3. Pd-catalyzed C–O cross-coupling of (hetero)aryl chlorides with secondary alcohols.a.
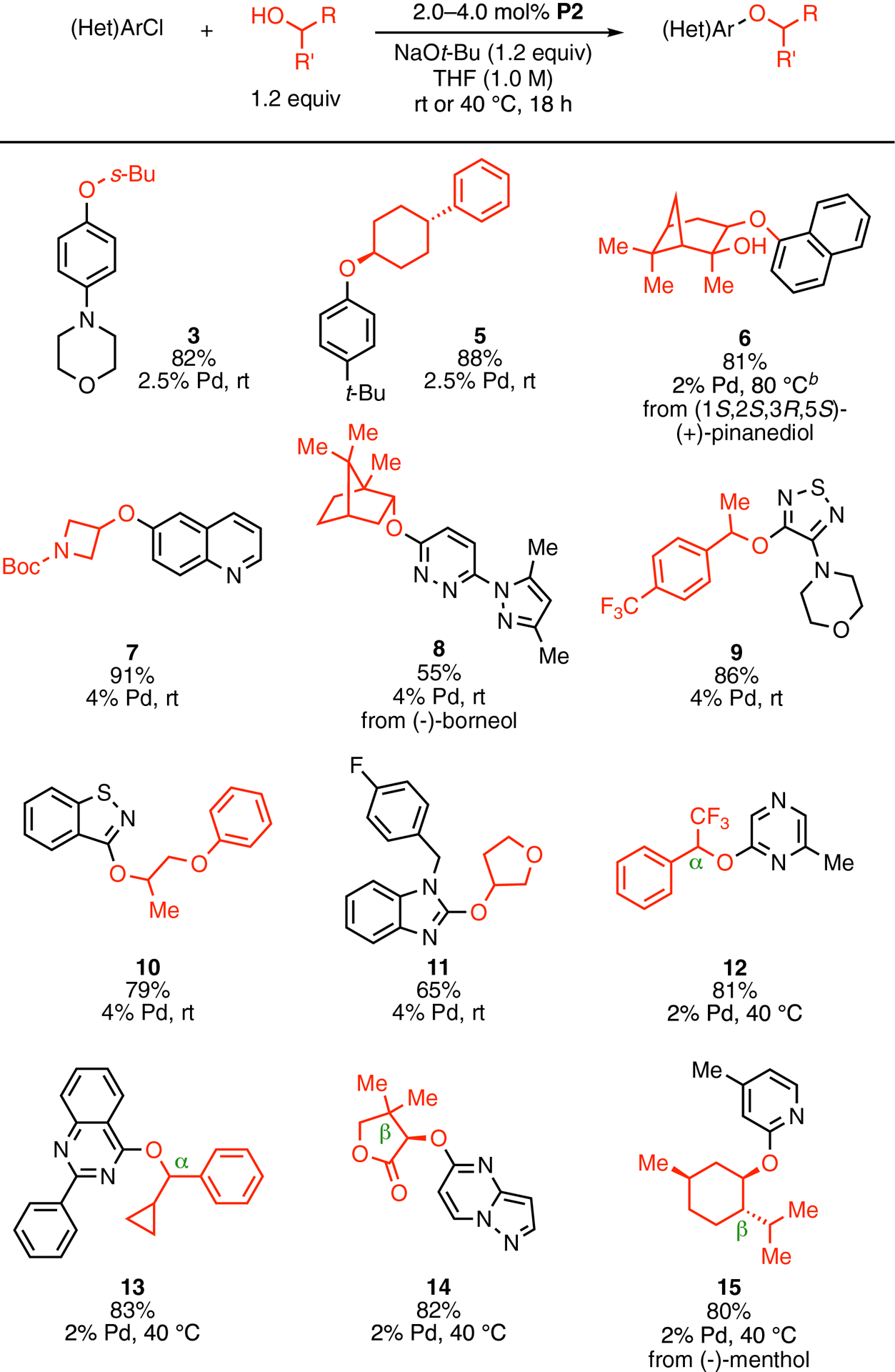
aReaction conditions: ArCl (1.0 mmol), alcohol (1.2 mmol), NaOt-Bu (1.2 mmol), P2 (2.0–4.0 mol %), THF (1.0 mL, 1.0 M), rt–40 °C, 18 h. Isolated yields represent the average result of two runs. bTHF (7.0 mL) and higher temperature were used due to the poor solubility of the combination of aryl chloride, alcohol and NaOt-Bu under standard reaction conditions.
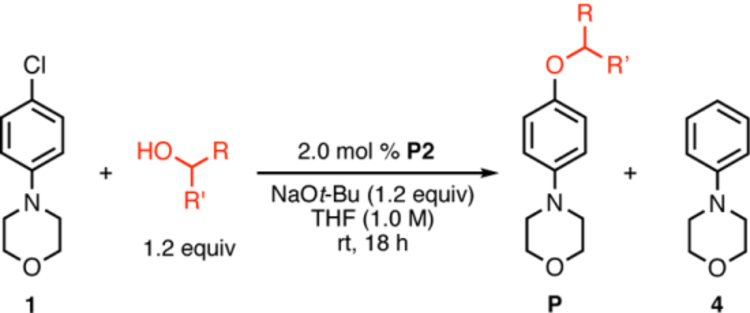
The coupling reactions between electron-rich aryl chlorides and more sterically-demanding nucleophiles, however, remained challenging, as demonstrated in the reactions of aryl chloride 1 (Table 2). As the steric congestion around the α-carbon of the alcohol increased (Table 2, entries 1 and 2), both the conversion and the yield of desired product decreased by approximately 40%, while a small increase in reduction product 4 was observed. We hypothesize that increasing the steric bulk around the α-carbon could negatively impact the binding tendency of alcohol nucleophiles to the oxidative-addition complex II (Scheme 1), thus accounting for the less efficient C–O bond formation. Although heteroaryl ethers 9, 12, and 13 were prepared and isolated in >80% yields, benzylic alcohols proved to be more difficult coupling partners for electron-rich aryl chloride 1 (Table 2, entries 3 and 4). The steric environment of the benzylic carbon also played an important role in the coupling process, as a change from a methyl to an ethyl group led to a 60% decrease in conversion, and only a trace amount of desired product was detected (Table 2, entries 3 and 4).
Table 2.
Alcohol evaluation for Pd-catalyzed C–O cross-coupling of electron-rich aryl chloride 1.a
 | ||||
|---|---|---|---|---|
| entry | alcohol | Conversion (%)b | yield of P (%)c | yield of 4 (%)c |
| 1 | 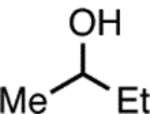 |
100 | 90 | 9 |
| 2 |  |
68 | 53 | 20 |
| 3 | 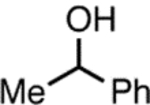 |
100 | 64 | 29 |
| 4 |  |
40 | 5 | 15 |
Reaction conditions: ArCl (0.5 mmol), alcohol (0.6 mmol), NaOt-Bu (0.6 mmol), P2 (2.0 mol %), THF (0.5 mL, 1.0 M), rt, 18 h.
Determined by GC using an internal standard.
Determined by 1H NMR using an internal standard.
As we continued to examine the scope of C–O cross-coupling reactions of aryl bromides, we noticed that the difference in reaction efficiency between the cross-coupling of aryl bromides and aryl chlorides was greatest for highly electron-rich substrates, such as 2 vs 1 (to prepare 3 in Schemes 4 and 3, respectively). In contrast, for weakly electron-rich, electron-neutral and electrondeficient aryl bromides (to prepare 5, 16 and 17, respectively), the cross-coupling reactions proceeded with comparable levels of efficiency (>80% yield, Scheme 4).
Scheme 4. Pd-catalyzed C–O cross-coupling of aryl bromides with secondary alcohols.a.

aReaction conditions: ArBr (1.0 mmol), alcohol (2.0 mmol), NaOt-Bu (1.2 mmol), P2 (2.0–2.5 mol %), THF (1.0 mL, 1.0 M), rt, 18 h. Isolated yields represent the average result of two runs. b1.5 equiv of alcohol. cTHF (5.0 mL) and higher temperature were used due to the poor solubility of the combination of aryl chloride, alcohol and NaOt-Bu under standard reaction conditions.
To gain an understanding of the difference in reactivity between aryl halides 1 and 2, we performed experiments to examine the cause of increased reduction in the cross-coupling reaction of aryl bromide 2. In order to ascertain whether reduction product 4 resulted solely from β-hydride elimination, we prepared α-deutero-alcohol 18-d (98% d1, see Supporting Information) and examined its reaction with aryl bromide 2. When 2 and protio-alcohol 18 were subjected to the standard cross-coupling conditions, the desired product 19 and the reduction product 4 were observed in 47% and 54% yield, respectively as determined by 1H NMR analysis, while ketone 20 was formed in 31% yield, as judged by GC analysis (Scheme 5A). Ketone 20 is believed to result from β-hydride elimination from the intermediate [LnPdII(Ar)(alkoxide)] (IV, Scheme 1), and should theoretically be formed in a 1:1 ratio with 4. Therefore, these results indicate that β-hydride elimination only accounts for a fraction of the formation of 4. When 2 and 18-d were subjected to the cross-coupling conditions, the desired product 19-d was formed in 65% yield and contained the same amount of deuterium (98%) as in 18-d (estimated by 1H NMR analysis, Scheme 5B). Reduction product 4-d was formed in 36% yield (by 1H NMR analysis) and was 80% d1 (by HRMS analysis). Ketone 20 was detected in 20% yield by GC analysis. Taken together, these experiments demonstrate that not all reduction side product arises from β-hydride (deuteride) elimination, and some stems from (as yet) unidentified processes. It is conceivable that the reduction byproduct may arise from protodemetalation of the oxidative addition complex [LnPdII(Ar)X] (II, Scheme 1). Similar findings have been reported by Hartwig, in his studies of the Pd-catalyzed amination of aryl bromides in the presence of bidentate ligands.11
Scheme 5. Pd-catalyzed C–O cross-coupling of aryl bromide 2 with A) cyclohexanol 18, and B) deuterocyclohexanol 18-d.a.
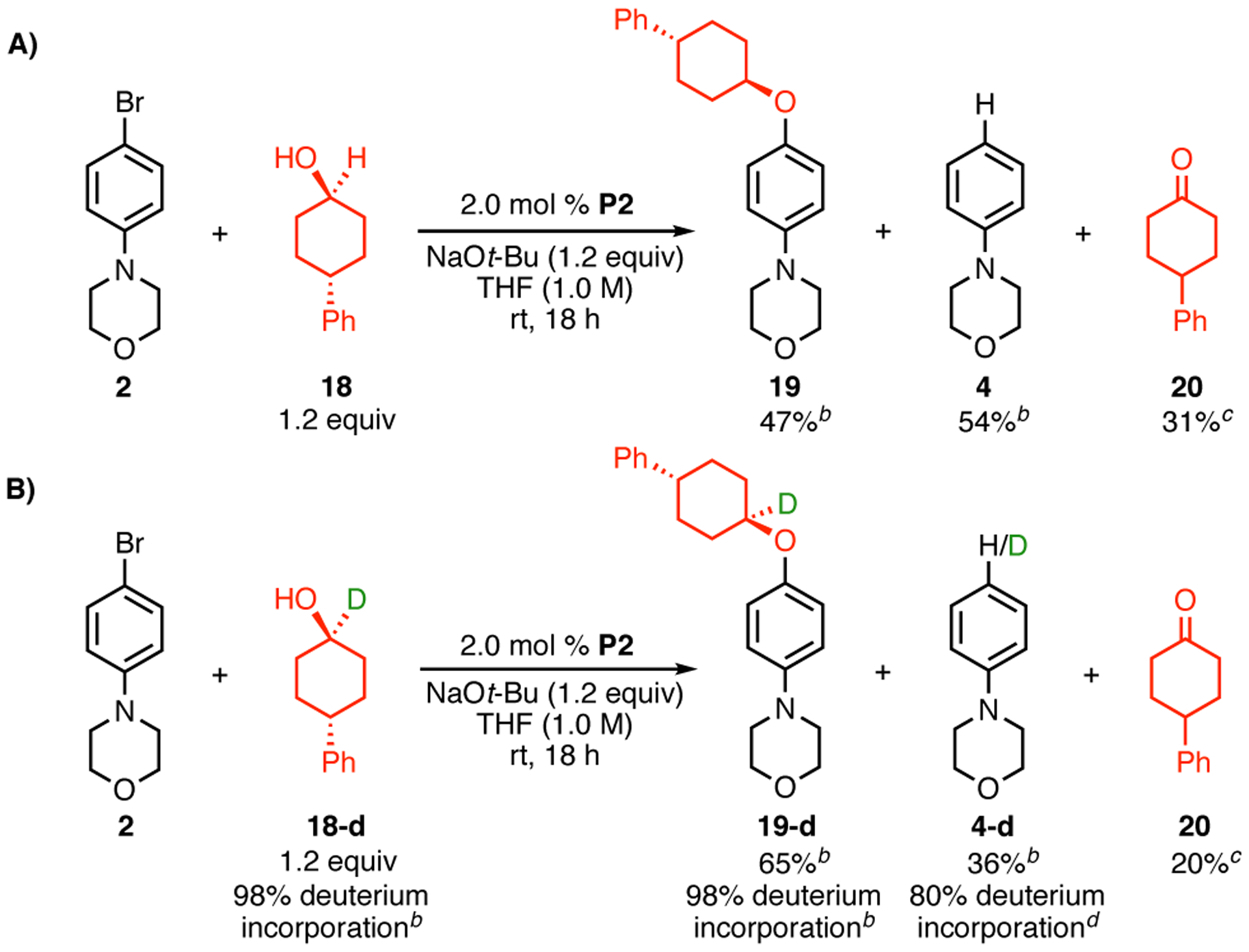
aReaction conditions: 2 (0.5 mmol), 18 or 18-d (0.6 mmol), NaOt-Bu (0.6 mmol), P2 (2.0 mol %), THF (0.5 mL, 1.0 M), rt, 18 h. bDetermined by 1H NMR analysis using an internal standard. cDetermined by GC analysis using an internal standard. dDetermined by HRMS analysis.
In conclusion, we have developed a significantly improved procedure for the Pd-catalyzed C–O cross-coupling of secondary alcohols. This protocol employs a previouslydisclosed hybrid biaryl phosphine ligand L2, while using THF as the reaction solvent in lieu of 1,4-dioxane as in our previous report. A variety of (hetero)aryl ethers were obtained in higher yields from the corresponding (hetero)aryl halides under more user-friendly reaction conditions than in our earlier method. For instance, a 20% molar excess of alcohols sufficed to allow the cross-coupling reaction of (hetero)aryl chlorides at room temperature or 40 °C. An interesting but unconventional reactivity difference between an electron-rich aryl bromide 2 and chloride 1 was discovered. A deuterium-labeling study suggested the possibility of as yet unidentified pathways responsible for the greater reduction of aryl bromide 2, indicating the need for further studies to establish a better detailed understanding of the mechanism of the Pd-catalyzed C-O coupling under these conditions.
Supplementary Material
ACKNOWLEDGMENT
Research reported in this publication was supported by the National Institutes of Health (R35-GM122483) and Arnold and Mabel Beckman Foundation for a postdoctoral fellowship to A.W.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We acknowledge MilliporeSigma (formerly Aldrich) for the generous donation of 1-adamantylzinc bromide solution, and Ryan King (MIT) for one batch of 2-iodo-2’,4’,6’-triisopropyl-3,6-dimethoxybiphenyl used in this work. We thank Dr. Jeffrey Yang (MIT) and Dr. Bryan Ingoglia (MIT) for helpful discussions. We thank Dr. Scott McCann (MIT), Dr. Richard Liu (MIT), and Dr. Christine Nguyen (MIT) for their assistance with the preparation of this manuscript.
Footnotes
The authors declare the following competing financial interest(s): MIT has or has filed patents on ligands/precatalysts that are described in the paper from which S.L.B. and former coworkers receive royalty payments.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data for new compounds (PDF)
REFERENCES
- 1.Mandal S; Mandal S; Ghosh SK; Sar P; Ghosh A; Saha R; Saha B, A review on the advancement of ether synthesis from organic solvent to water. RSC Adv. 2016, 6, 69605–69614. [Google Scholar]
- 2.(a) Wang Z, Williamson Ether Synthesis. In Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc.: John Wiley & Sons, Inc.: Hoboken, 2010; pp 3026–3030; [Google Scholar]; (b) Fuhrmann E; Talbiersky J, Synthesis of Alkyl Aryl Ethers by Catalytic Williamson Ether Synthesis with Weak Alkylation Agents. Org. Process Res. Dev 2005, 9, 206–211. [Google Scholar]
- 3.(a) Fletcher S, The Mitsunobu reaction in the 21st century. Org. Chem. Front 2015, 2, 739–752; [Google Scholar]; (b) Swamy KCK; Kumar NNB; Balaraman E; Kumar KVPP, Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev 2009, 109, 2551–2651; [DOI] [PubMed] [Google Scholar]; (c) Manivel P; Rai NP; Jayashankara VP; Arunachalam PN, Base catalyzed Mitsunobu reactions as a tool for the synthesis of aryl sec-alkyl ethers. Tetrahedron Lett. 2007, 48, 2701–2705. [Google Scholar]
- 4.(a) Caron S; Ghosh A, Nucleophilic Aromatic Substitution. In Practical Synthetic Organic Chemistry, John Wiley & Sons, Inc.: Hoboken, 2011; pp 237–253; [Google Scholar]; (b) Henderson AS; Medina S; Bower JF; Galan MC, Nucleophilic Aromatic Substitution (SNAr) as an Approach to Challenging Carbohydrate–Aryl Ethers. Org. Lett 2015, 17, 4846–4849. [DOI] [PubMed] [Google Scholar]
- 5.(a) Laffoon SD; Chan VS; Fickes MG; Kotecki B; Ickes AR; Henle J; Napolitano JG; Franczyk TS; Dunn TB; Barnes DM; Haight AR; Henry RF; Shekhar S, Pd-Catalyzed Cross-Coupling Reactions Promoted by Biaryl Phosphorinane Ligands. ACS Catal. 2019, 9, 11691–11708; [Google Scholar]; (b) Zhang H; Ruiz-Castillo P; Buchwald SL, Palladium-Catalyzed C–O Cross-Coupling of Primary Alcohols. Org. Lett 2018, 20, 1580–1583; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sawatzky RS; Hargreaves BKV; Stradiotto M, A Comparative Ancillary Ligand Survey in Palladium-Catalyzed C–O Cross-Coupling of Primary and Secondary Aliphatic Alcohols. Eur. J. Org. Chem 2016, 2016, 2444–2449; [Google Scholar]; (d) Rangarajan TM; Brahma R; Ayushee; Prasad AK; Verma AK; Singh RP, Mild and efficient palladium/BrettPhos-catalyzed methoxylation and deuteriomethoxylation of activated aryl bromides. Tetrahedron Lett. 2015, 56, 2234–2237; [Google Scholar]; (e) Rangarajan TM; Singh R; Brahma R; Devi K; Singh RP; Singh RP; Prasad AK, BrettPhos Ligand Supported Palladium-Catalyzed C-O Bond Formation through an Electronic Pathway of Reductive Elimination: Fluoroalkoxylation of Activated Aryl Halides. Chem. Eur. J 2014, 20, 14218–14225; [DOI] [PubMed] [Google Scholar]; (f) Cheung CW; Buchwald SL, Mild and General Palladium-Catalyzed Synthesis of Methyl Aryl Ethers Enabled by the Use of a Palladacycle Precatalyst. Org. Lett 2013, 15, 3998–4001; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Maligres PE; Li J; Krska SW; Schreier JD; Raheem IT, C-O Cross-Coupling of Activated Aryl and Heteroaryl Halides with Aliphatic Alcohols. Angew. Chem. Int. Ed 2012, 51, 9071–9074; [DOI] [PubMed] [Google Scholar]; (h) Gowrisankar S; Neumann H; Beller M, A Convenient and Practical Synthesis of Anisoles and Deuterated Anisoles by Palladium-Catalyzed Coupling Reactions of Aryl Bromides and Chlorides. Chem. Eur. J 2012, 18, 24982502; [DOI] [PubMed] [Google Scholar]; (i) Wu X; Fors BP; Buchwald SL, A Single Phosphine Ligand Allows Palladium-Catalyzed Intermolecular C-O Bond Formation with Secondary and Primary Alcohols. Angew. Chem. Int. Ed 2011, 50, 9943–9947; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Enthaler S; Company A, Palladium-catalysed hydroxylation and alkoxylation. Chem. Soc. Rev 2011, 40, 4912–4924; [DOI] [PubMed] [Google Scholar]; (k) Gowrisankar S; Sergeev AG; Anbarasan P; Spannenberg A; Neumann H; Beller M, A General and Efficient Catalyst for Palladium-Catalyzed C−O Coupling Reactions of Aryl Halides with Primary Alcohols. J. Am. Chem. Soc 2010, 132, 11592–11598; [DOI] [PubMed] [Google Scholar]; (l) Milton EJ; Fuentes JA; Clarke ML, Palladium-catalysed synthesis of aryl-alkyl ethers using alkoxysilanes as nucleophiles. Org. Biomol. Chem 2009, 7, 26452648; [DOI] [PubMed] [Google Scholar]; (m) Withbroe GJ; Singer RA; Sieser JE, Streamlined Synthesis of the Bippyphos Family of Ligands and Cross-Coupling Applications. Org. Process Res. Dev 2008, 12, 480–489; [Google Scholar]; (n) Vorogushin AV; Huang X; Buchwald SL, Use of Tunable Ligands Allows for Intermolecular Pd-Catalyzed C−O Bond Formation. J. Am. Chem. Soc 2005, 127, 8146–8149. [DOI] [PubMed] [Google Scholar]
- 6.(a) Chen Z; Jiang Y; Zhang L; Guo Y; Ma D, Oxalic Diamides and tert-Butoxide: Two Types of Ligands Enabling Practical Access to Alkyl Aryl Ethers via Cu-Catalyzed Coupling Reaction. J. Am. Chem. Soc 2019, 141, 3541–3549; [DOI] [PubMed] [Google Scholar]; (b) Bhunia S; Pawar GG; Kumar SV; Jiang Y; Ma D, Selected Copper-Based Reactions for C−N, C−O, C−S, and C−C Bond Formation. Angew. Chem. Int. Ed 2017, 56, 16136–16179; [DOI] [PubMed] [Google Scholar]; (c) Lam PYS, Chapter 7 Chan-Lam Coupling Reaction: Copper-promoted C-Element Bond Oxidative Coupling Reaction with Boronic Acids. In Synthetic Methods in Drug Discovery: Volume 1, The Royal Society of Chemistry: 2016; Vol. 1, pp 242–273; [Google Scholar]; (d) Lin H; Sun D, Recent Synthetic Developments and Applications of the Ullmann Reaction. A Review. Org. Prep. Proced. Int 2013, 45, 341–394; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Niu J; Zhou H; Li Z; Xu J; Hu S, An Efficient Ullmann-Type C−O Bond Formation Catalyzed by an Air-Stable Copper(I)−Bipyridyl Complex. J. Org. Chem 2008, 73, 7814–7817; [DOI] [PubMed] [Google Scholar]; (f) Altman RA; Shafir A; Choi A; Lichtor PA; Buchwald SL, An Improved CuBased Catalyst System for the Reactions of Alcohols with Aryl Halides. J. Org. Chem 2008, 73, 284–286; [DOI] [PubMed] [Google Scholar]; (g) Zhang H; Ma D; Cao W, N,N-Dimethylglycine-Promoted Ullmann-Type Coupling Reactions of Aryl Iodides with Aliphatic Alcohols. Synlett 2007, 2007, 0243–0246; [Google Scholar]; (h) Wolter M; Nordmann G; Job GE; Buchwald SL, Copper-Catalyzed Coupling of Aryl Iodides with Aliphatic Alcohols. Org. Lett 2002, 4, 973–976. [DOI] [PubMed] [Google Scholar]
- 7.(a) MacQueen PM; Tassone JP; Diaz C; Stradiotto M, Exploiting Ancillary Ligation To Enable Nickel-Catalyzed C–O Cross-Couplings of Aryl Electrophiles with Aliphatic Alcohols. J. Am. Chem. Soc 2018, 140, 5023–5027; [DOI] [PubMed] [Google Scholar]; (b) Terrett JA; Cuthbertson JD; Shurtleff VW; MacMillan DWC, Switching on elusive organometallic mechanisms with photoredox catalysis. Nature 2015, 524, 330–334; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mann G; Hartwig JF, Nickel- vs Palladium-Catalyzed Synthesis of Protected Phenols from Aryl Halides. J. Org. Chem 1997, 62, 5413–5418. [Google Scholar]
- 8.(a) Sach NW; Richter DT; Cripps S; Tran-Dubé M; Zhu H; Huang B; Cui J; Sutton SC, Synthesis of Aryl Ethers via a Sulfonyl Transfer Reaction. Org. Lett 2012, 14, 3886–3889; [DOI] [PubMed] [Google Scholar]; (b) Lindstedt E; Stridfeldt E; Olofsson B, Mild Synthesis of Sterically Congested Alkyl Aryl Ethers. Org. Lett 2016, 18, 42344237. [DOI] [PubMed] [Google Scholar]; (c) Sundalam SK; Stuart DR, Base Mediated Synthesis of Alkyl-aryl Ethers from the Reaction of Aliphatic Alcohols and Unsymmetric Diaryliodonium Salts. J. Org. Chem 2015, 80, 64566466. [DOI] [PubMed] [Google Scholar]; (d) Lindstedt E; Ghosh R; Olofsson B, Metal-Free Synthesis of Aryl Ethers in Water. Org. Lett 2013, 15, 6070–6073. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hartwig JF, Electronic Effects on Reductive Elimination To Form Carbon−Carbon and Carbon−Heteroatom Bonds from Palladium(II) Complexes. Inorg. Chem 2007, 46, 1936–1947; [DOI] [PubMed] [Google Scholar]; (b) Widenhoefer RA; Buchwald SL, Electronic Dependence of C−O Reductive Elimination from Palladium (Aryl)neopentoxide Complexes. J. Am. Chem. Soc 1998, 120, 6504–6511. [Google Scholar]
- 10.(a) Ingoglia BT; Wagen CC; Buchwald SL, Biaryl monophosphine ligands in palladium-catalyzed C–N coupling: An updated User’s guide. Tetrahedron 2019, 75, 4199–4211; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ruiz-Castillo P; Blackmond DG; Buchwald SL, Rational Ligand Design for the Arylation of Hindered Primary Amines Guided by Reaction Progress Kinetic Analysis. J. Am. Chem. Soc 2015, 137, 3085–3092; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barrios-Landeros F; Carrow BP; Hartwig JF, Effect of Ligand Steric Properties and Halide Identity on the Mechanism for Oxidative Addition of Haloarenes to Trialkylphosphine Pd(0) Complexes. J. Am. Chem. Soc 2009, 131,8141–8154; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Littke AF; Fu GC, Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed 2002, 41, 4176–4211; [DOI] [PubMed] [Google Scholar]; (e) Grushin VV; Alper H, Transformations of Chloroarenes, Catalyzed by Transition-Metal Complexes. Chem. Rev 1994, 94, 1047–1062. [Google Scholar]
- 11.Hamann BC; Hartwig JF, Systematic Variation of Bidentate Ligands Used in Aryl Halide Amination. Unexpected Effects of Steric, Electronic, and Geometric Perturbations. J. Am. Chem. Soc 1998, 120, 3694–3703. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.