

Abstract
Clostridioides difficile colonizes the intestines of susceptible individuals and releases toxins that mediate disease. To replicate and expand in the intestines, C. difficile ferments proline, and this activity is influenced by the availability of proline and trace nutrients. C. difficile must also compete with the commensal microbiota for these limited nutrients. The specific microbes present in the intestines that may shape the ability of C. difficile to benefit from proline fermentation are unknown. In this study we developed a panel of commensal Clostridia to test the hypothesis that the microbiota influences C. difficile growth through proline fermentation. The experimental panel of Clostridia was composed of murine and human isolates that ranged in their capacity to ferment proline in different media. Competition between wild type C. difficile and a mutant strain unable to ferment proline (prdB:CT) in the presence of these Clostridia revealed that bacteria closely related to Paraclostridium benzoelyticum and Paeniclostridium spp. decreased the benefit to C. difficile provided by proline fermentation. Conversely, Clostridium xylanolyticum drove C. difficile towards an increased reliance on proline fermentation for growth. Overall, the ability of C. difficile to benefit from proline fermentation is contextual and in part dependent on the microbiota.
1. Introduction
Clostridioides difficile is a Gram-positive, toxin producing bacterium that is responsible for nearly half a million intestinal infections in the United States annually [1]. In healthy individuals, the resident microbes in the intestines, or the microbiota, protect against C. difficile infection (CDI) through a process known as colonization resistance. Disruption of the microbiota, typically through the use of antibiotics, increases susceptibility to C. difficile colonization and disease that can result in severe colitis and death.
Several factors contribute to microbiota-mediated colonization resistance [2]. For instance, some primary bile salts, including cholate and taurocholate, can serve as germination factors for C. difficile spores, but are converted to secondary bile salts by the microbiota, thus limiting access to these required germinants [3,4]. Secondary bile salts then directly inhibit growth of vegetative C. difficile to further suppress pathogen growth [3]. The microbiota can also inhibit C. difficile growth through the production of antimicrobial bacteriocins and lantibiotics [5–7]. Finally, the microbiota can exclude C. difficile through nutrient competition, filling in nutrient niches that C. difficile requires for expansion during CDI [8].
While evidence supports the idea that disruption of the microbiota can open up nutrient niches for C. difficile [8,9], how competition for nutrients by the microbiota influences C. difficile metabolism is not well defined. Several nutrients increase in abundance following antibiotic treatment, including sugar alcohols, monosaccharides such as fructose and galactose, sialic acids, and amino acids [8,10]. These changes in nutrient abundance correlate with specific alterations in the microbiota [9,10], suggesting that specific taxa are responsible for driving changes in those nutrients under homeostatic conditions. One example of microbiota nutrient competition with C. difficile involves the release of sialic acids. Under homeostatic conditions, host-produced sialic acid is released and consumed by the microbiota. Following antibiotic treatment, concentrations of sialic acid increase and are then consumed by C. difficile to fuel pathogen expansion following initial colonization [11]. Sialic acid supplementation of human stool supernatants also leads to increased C. difficile growth in vitro [12], supporting the hypothesis that sialic acid consumption by the microbiota limits C. difficile growth on these nutrients.
C. difficile can metabolize a wide variety of nutrients, so sialic acid likely only represents one of many nutrient niches that open following disruption of the microbiota. Another nutrient that contributes to C. difficile expansion during CDI is the amino acid proline [13]. Proline abundance increases following antibiotic treatment [8], providing an early nutrient source for C. difficile, which can ferment proline for energy production and to maintain redox balance [14]. In human patients, proline fermentation is associated with CDI [15], and in mouse CDI models the ability to ferment proline provides C. difficile a fitness advantage [13]. Recently, we showed that C. difficile ferments proline during infection by measuring the byproduct of proline fermentation, 5-aminovalerate, in the stools of infected mice [13]. Interestingly, uninfected animals also had increasing 5-aminovalerate concentrations in their stools in the days following antibiotic treatment, signifying that other members of the microbiota ferment proline and could compete for proline against C. difficile. However, the specific taxa that influence the dependence of C. difficile on proline fermentation remain unknown.
In this study, we test the hypothesis that members of the Clostridia influence C. difficile proline fermentation. To this end, a representative panel of commensal Clostridia, consisting of human and animal isolates, was developed. This experimental panel was characterized by growth and proline fermentation capabilities in a variety of rich media. These bacteria were then cultured with C. difficile 027 ribotype [16] in vitro. While many members of the experimental panel had no noticeable effect on C. difficile proline fermentation, three Clostridia, Paraclostridium benzoelyticum, Paeniclostridium spp., and Clostridium xylanolyticum, altered the reliance of C. difficile on proline fermentation. These results suggest that the fitness advantage provided by the ability to ferment proline is partially dependent on the presence of particular members of the microbiota.
2. Results
2.1. Clostridia are predicted to compete with C. difficile for proline
A recent study by our group found that 5-aminovalerate, the byproduct of proline fermentation, increases in abundance by day 4 following cessation of antibiotic treatment of uninfected animals [13]. This implies that at least some members of the microbiota can ferment proline. To predict the taxa likely responsible for fermenting proline in the intestines, bacterial genomes were searched for proteins homologous to PrdB using AnnoTree [17]. PrdB is the catalytic subunit of the proline reductase responsible for reducing d-proline to 5-aminovalerate. Thus, bacterial genomes that encode PrdB likely have the capacity to ferment proline. A search of genomes in the Genome Taxonomy Database [18] revealed that several classes across the domain Bacteria encode for PrdB, including many that are not likely to reside in mammalian intestines (Fig. 1). However, amongst all genomes in our search the class Clostridia has the largest number of genomes with prdB (Fig. 1, Supplementary Table 1). The Clostridia belong to the Firmicutes phylum, which, along with the Bacteroidetes, form the dominant membership of bacteria in mammalian guts [19]. Clostridia as a class are widely diverse, but generally defined as Gram-positive, strictly anaerobic, and spore-forming. Most Clostridia have either neutral or beneficial roles in human health, though some members, including C. difficile, are pathogenic. Since a relatively large proportion of Clostridia were predicted to ferment proline based on the presence of PrdB, we hypothesized that some members of the Clostridia compete with C. difficile and influence C. difficile proline fermentation.
Fig. 1. Phylogenetic distribution of PrdB.
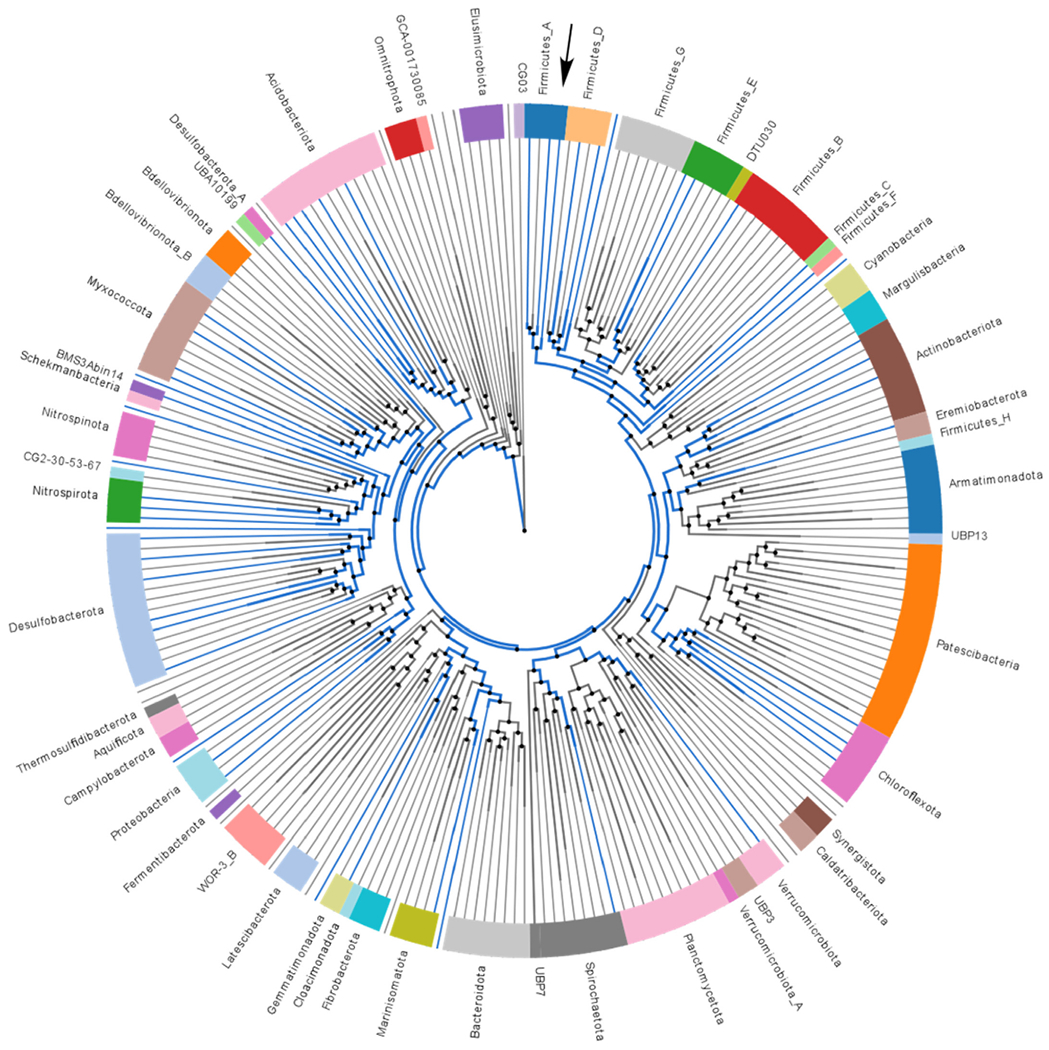
Phylogenetic tree displaying bacterial genomes grouped by phylum at the class taxonomic resolution. Lines indicate class level, with blue lines indicating a class containing at least one genome hit for a protein homologous to the catalytic subunit of proline reductase, PrdB. Black arrow indicates the class Clostridia. The colors on the outer wheel are only used to visually separate lineages. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.2. Development of a diverse experimental panel of clostridia
To test the prediction that Clostridia compete with C. difficile, an experimental panel of Clostridia isolates was developed from murine and human sources. Taking advantage of the heat-resistant spores produced by many Clostridia, feces taken from naïve mice were exposed to high heat to kill vegetative cells and leave only viable spores (Fig. 2A). Heat-treated feces were then plated on rich Eggerth-Gagnon (EG) agar anaerobically and incubated for several days at 37 °C. Colonies were initially chosen based on differences in colony morphologies and growing times and passaged at least twice to ensure pure cultures (Fig. 2A). Human isolates were taken from the American Type Culture Collection (ATCC) and the Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures and selected based on their importance in human health and relevance to CDI [20–26]. The human isolates consisted of C. scindens, Ruminococcus torques, Blautia obeum, and Dorea longicatena (Table 1).
Fig. 2. Diversity in an experimental panel of Clostridia.
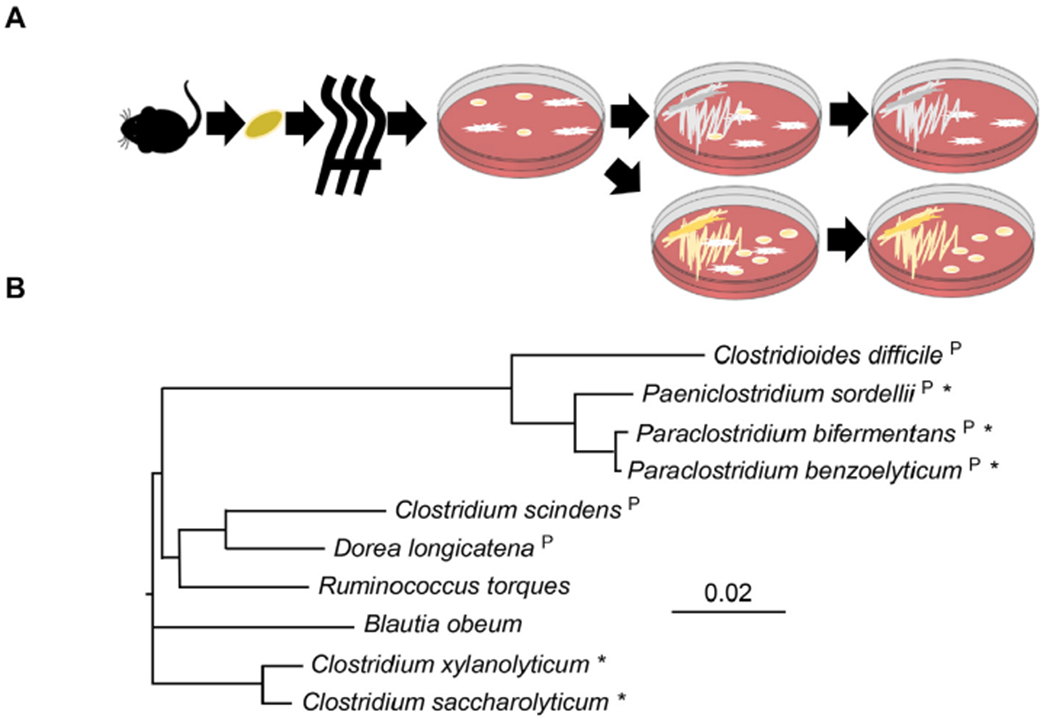
(A) Clostridia isolation strategy from mouse feces. Feces were collected fresh from mice and then heated to kill vegetative cells. Spores were plated onto non-selective Eggerth-Gagnon agar. Each isolate underwent at least two rounds of passage to ensure pure cultures. (B) Phylogenetic tree of bacteria in the panel (based on full-length 16S sequence of closest sequenced bacterium in the NCBI database). * indicates bacteria isolated using scheme in (A). All other bacteria are human isolates from ATCC or DSMZ (see Table 1). “P” indicates bacteria that are predicted to contain PrdB based on sequence homology to C. difficile PrdB.
Table 1.
Bacterial strains.
| Bacterial strain | Comments | Source |
|---|---|---|
| Clostridioides difficile ribotype 027 (CD196) | hypervirulent strain | [16] |
| C. difficile prdB::CT | 027 ribotype unable to ferment proline | [13] |
| Clostridium scindens | isolate from human feces | ATCC35704 |
| Ruminococcus torques | isolate from human feces | ATCC27756 |
| Blautia obeum | isolate from human feces | DSM25238 |
| Dorea longicatena | isolate from human feces | DSM13814 |
The murine isolates were broadly identified based on 16S and rpoB sequence identity to sequenced genomes, followed by metabolic profiling using API 20A metabolic strips (see Materials and Methods, Supplemental Table 2). Based on these characteristics, isolates were determined to be most closely related to P. bifermentans, C. xylanolyticum, P. benzoelyticum, and Paeniclostridium spp. To determine the relative diversity of the experimental Clostridia panel, full length 16S sequences from type strains most closely related to strains in the experimental panel were aligned. A phylogenetic tree based on these 16S sequences (Fig. 2B) revealed that the experimental panel contained a cluster of bacteria closely related to C. difficile (P. bifermentans, Paeniclostridium, and P. benzoelyticum) as well as lineages divergent from C. difficile. Basic morphology after Gram-staining also supports the conclusion that the experimental panel represents diverse members of Clostridia. For instance, while Clostridia are typically regarded as Gram-positive, C. saccharolyticum and C. xylanolyticum stain Gram-negative (Supplemental Fig. 1). The experimental panel also exhibited a wide range of bacterial shapes that include larger rods (P. benzoelyticum) and chains of coccobacilli (D. longicatena) (Supplemental Fig. 1). A BLAST search for PrdB in the experimental panel also revealed diversity in their predicted ability to ferment proline. Amongst the panel, P. bifermentans, P. benzoelyticum, Paeniclostridium spp., C. scindens, and D. longicatena contain proteins with high sequence identity to PrdB while C. saccharolyticum, C. xylanolyticum, R. torques, and B. obeum do not (Fig. 2B).
2.3. Clostridia growth characteristics and proline fermentation
To test the hypothesis that members of the Clostridia influence C. difficile proline fermentation, we first identified media that would allow growth of isolates and C. difficile to similar levels. Members of the experimental Clostridia panel were grown independently in tryptone yeast (TY) broth, brain-heart infusion broth supplemented with yeast extract (BHIS), or EG broth. TY is composed of trypsin-digested peptides and yeast extract and is thus high in amino acids and vitamins. BHIS also contains yeast extract with additional infusions of heart, brain, and dextrose. Similar to BHIS, EG broth is composed of yeast extract and dextrose, though it contains peptides from other sources and Tween-80. For each medium, bacteria were cultured for 16 h anaerobically, with growth measured via spectrometry at 630 nm (OD630). The OD630 was then compared to the density of wild type (WT) C. difficile and calculated as a percent of C. difficile growth in that same medium (Fig. 3). Additionally, the growth of a C. difficile mutant unable to ferment proline due to an interruption in prdB (prdB:CT) [13] was examined. In TY broth, only P. bifermentans, P. benzoelyticum, and Paeniclostridium spp. grew to a similar OD630 as C. difficile while D. longicatena grew to appreciable levels (>80%). The other bacteria in the panel grew very poorly (<30%) in TY broth. In BHIS broth, all bacteria exhibited moderate growth (30–80%), with P. bifermentans, P. benzoelyticum, Paeniclostridium spp., and C. xylanolyticum reaching a higher density than WT C. difficile. In EG broth, most bacteria from the panel grew to similar densities as C. difficile, though interestingly P. bifermentans exhibited only moderate growth in this medium. Overall, all members of the experimental Clostridia panel grew to similar densities as C. difficile in either BHIS or EG broth, which were used for subsequent experiments.
Fig. 3. Growth of Clostridia panel in different media.
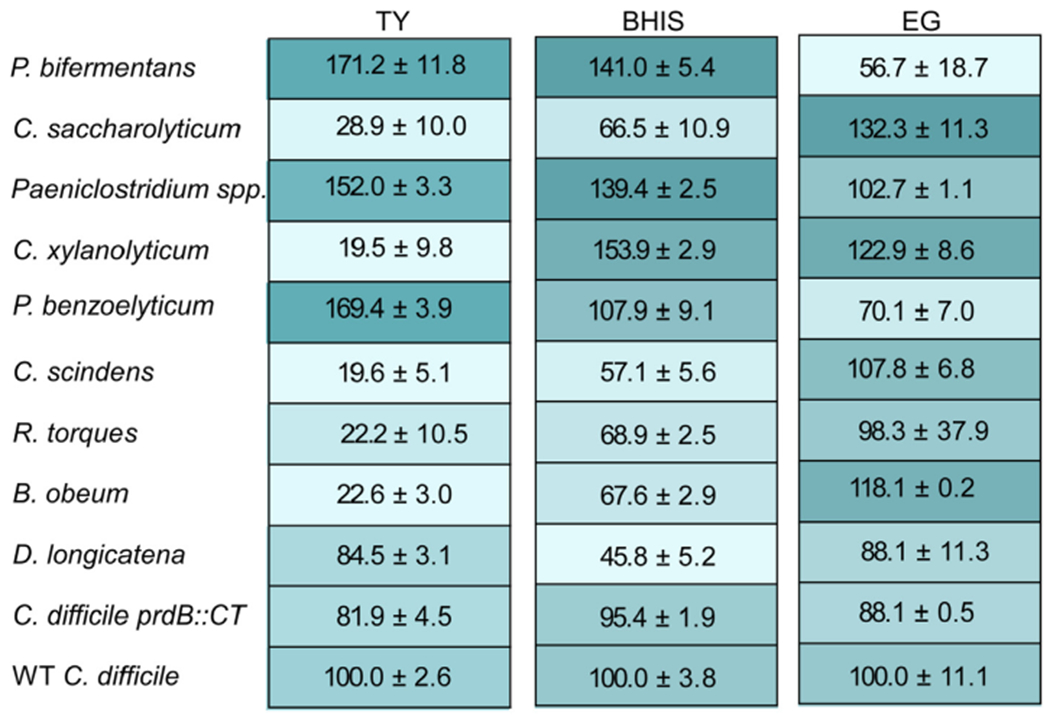
Average percent growth with 95% confidence interval of each bacterium relative to WT C. difficile for the indicated medium. Shading shows relative percent growth, with darker shading indicative of higher percent growth. Data represent growth after 16 h at 37 °C and at least 3 replicates.
Next, the ability to ferment proline was determined by measuring concentrations of 5-aminovalerate, the byproduct of proline fermentation. Bacteria from the panel were grown in either BHIS or EG broth for 6 or 8 h, respectively, and spent media were analyzed using liquid chromatography tandem mass spectrometry (LC-MS/MS). The resulting 5-aminovalerate concentrations were then normalized to the OD630 of the bacterial cultures to account for differences in growth rate. As expected, WT C. difficile fermented proline in BHIS broth (Fig. 4A). In accordance with the predictions based on the presence of PrdB, P. bifermentans, Paeniclostridium, P. benzoelyticum, C. scindens, and D. longicatena fermented proline while C. xylanolyticum, R. torques, and B. obeum did not. Surprisingly, despite not having a predicted PrdB homolog, C. saccharolyticum produced minor quantities of 5-aminovalerate.
Fig. 4. Clostridia influence C. difficile proline fermentation.
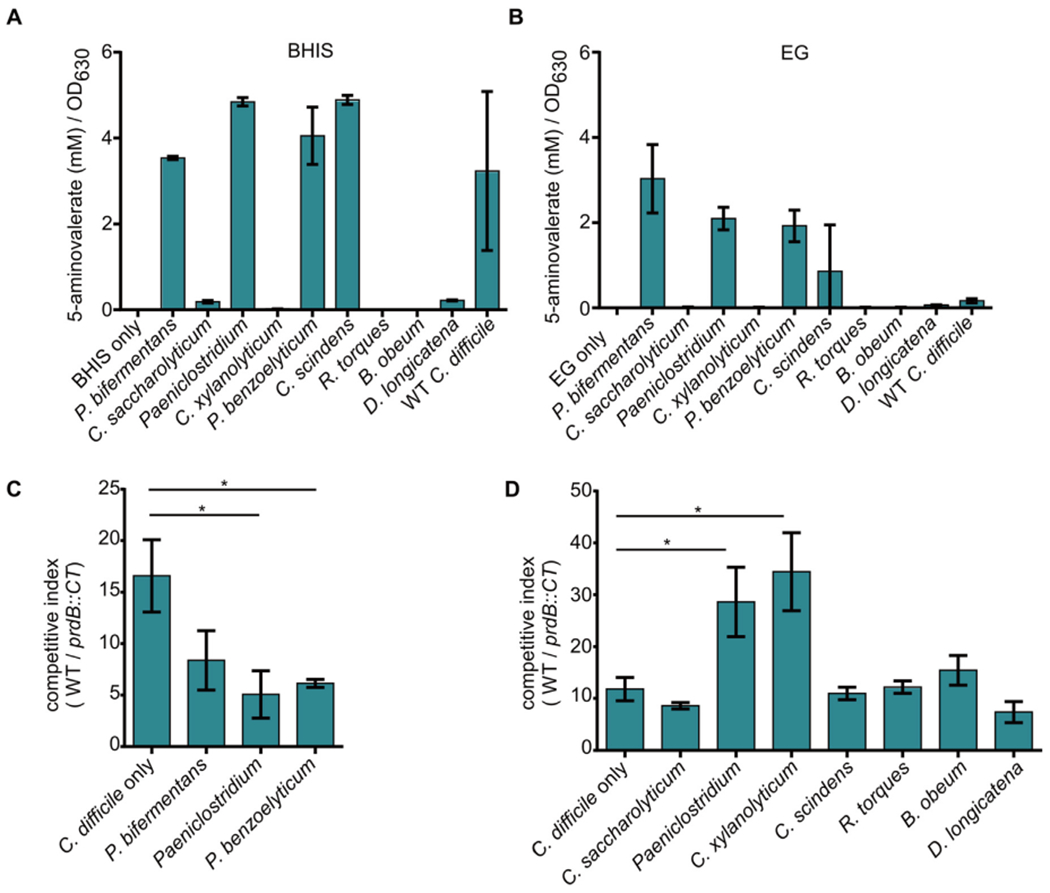
(A) 5-aminovalerate concentrations normalized to OD630 after 6 h of growth in BHIS broth. Error bars represent standard deviation. (B) 5-aminovalerate concentrations normalized to OD630 after 8 h of growth in EG broth. Error bars represent standard deviation. (C) Output ratio compared to input ratio of WT C. difficile compared to prdB::CT (competitive index) in BHIS broth supplemented with 30 mM l-proline grown alone or with the indicated bacterium. (D) Competitive index of WT C. difficile compared to prdB::CT in EG broth supplemented with 30 mM l-proline grown alone or with the indicated bacterium. * - p < 0.05, One-way ANOVA followed by Dunnett’s test for multiple comparisons.
In EG broth only a minor amount of 5-aminovalerate was detected in the WT C. difficile culture supernatant (Fig. 4B), indicating that proline fermentation is not a dominant form of metabolism in this medium for C. difficile. Conversely, P. bifermentans, P. benzoelyticum, Paeniclostridium spp., and C. scindens produced substantial 5-aminovalerate. Similar to C. difficile, D. longicatena did not produce high concentrations of 5-aminovalerate despite fermenting proline in BHIS broth (Fig. 4A). In EG broth, C. saccharolyticum also did not produce 5-aminovalerate. Together these data reveal that members of the experimental Clostridia panel have varying capacities to ferment proline that change depending on the nutrients available in the medium.
2.4. Clostridia alter C. difficile proline fermentation-dependent growth
C. difficile ferments proline in vitro and during infection, with the advantage provided by proline fermentation modified by the availabilities of multiple nutrients [13,27]. To test the hypothesis that commensal members of the Clostridia also influence C. difficile proline fermentation-dependent fitness, WT C. difficile and the prdB:CT mutant were competed in the absence and presence of members of the experimental Clostridia panel. In a defined medium where proline is not the limiting nutrient, both the WT and prdB:CT mutant grow similarly and show similar levels of sporulation after 12 h of incubation (Supplemental Fig. 2). In a medium where proline fermentation can provide a growth benefit, the ratio of WT and prdB:CT C. difficile (competitive index) provides a bioindicator of the relative fitness advantage afforded by proline fermentation. An increase in the ratio of recovered WT to prdB:CT indicates that the presence of the commensal Clostridia increases reliance on proline fermentation. A decrease in the competitive index indicates that C. difficile is benefiting less from the ability to ferment proline. To reduce artifacts from large differences in growth rates, competitions were performed in a medium where growth was roughly equivalent between C. difficile and the commensal Clostridia (Fig. 3).
In BHIS broth, C. difficile was grown either alone or with P. bifermentans, Paeniclostridium spp, or P. benzoelyticum. Following 16 h of incubation, cultures were plated onto cycloserine-cefoxitin-fructose agar (CCFA) to select for only C. difficile or BHIS agar with lincomycin to select for only the lincomycin-resistant prdB:CT mutant. The germinant taurocholate was not added to the CCFA or BHIS agar so that only vegetative C. difficile, and not spores, were enumerated. When grown alone, WT C. difficile was recovered in approximately 15-fold higher levels than the prdB:CT mutant. When P. bifermentans, Paeniclostridium spp, or P. benzoelyticum was present in the media, the total C. difficile levels were significantly lower compared to when C. difficile was grown alone (Supplemental Fig. 3A), which was expected as total nutrients in batch culture are shared amongst multiple bacterial species. However, the competitive index for WT C. difficile and prdB:CT was significantly lower only when Paeniclostridium spp. and P benzoelyticum were present in the medium. This suggests that Paeniclostridium spp. and P. benzoelyticum lower the fitness advantage provided by proline fermentation, possibly through competition for proline.
In EG broth, C. difficile was grown either alone or with C. saccharolyticum, Paeniclostridium spp., C. xylanolyticum, C. scindens, R. torques, B. obeum, and D. longicatena. When competed alone, WT C. difficile was recovered at 10-fold higher levels than the prdB:CT mutant. The lower competitive index in EG broth compared to BHIS broth is consistent with a lower reliance on proline fermentation in this medium (Fig. 4B). Incubation with C. saccharolyticum, C. scindens, R. torques, B. obeum, and D. longicatena did not significantly alter the competitive index compared to C. difficile grown alone. However, growth with Paeniclostridium spp. and C. xylanolyticum resulted in a significant increase in the competitive index, indicating that proline fermentation was more beneficial to C. difficile in the presence of these bacteria. Incubation with members of the experimental Clostridia panel had varying effects on overall C. difficile growth (Supplemental Fig. 3B), though these changes did not correlate with differences in the competitive index. Overall, these data reveal that diverse Clostridia isolates can impact the fitness advantage provided by C. difficile proline fermentation, either driving C. difficile towards increased reliance on proline fermentation or decreasing the growth benefits of proline fermentation.
3. Discussion
Susceptibility to CDI is largely dependent on the structure of the gut microbiota, with a diverse microbiota conferring protection against C. difficile colonization [28]. The microbiota are also responsible for clearance of C. difficile in animal models of CDI and are likely determining factors in recurrent infections [29]. While there are multiple examples of how C. difficile can interact with the microbiota [28], the specific taxa that influence critical metabolic processes in C. difficile are largely unknown. Proline fermentation is a key metabolic strategy in C. difficile that allows for the expansion of the pathogen during CDI. Following antibiotic treatment, proline availability increases in the intestines [8], which can increase transcription of proline reductase [27], the enzyme responsible for reducing d-proline to 5-aminovalerate using electrons from an oxidized amino acid donor. Accessibility of proline and other nutrients including zinc and selenium all influence C. difficile proline reductase transcription or reduction [13]. Yet, it is unclear if the microbiota can also alter the fitness benefit provided by proline fermentation.
Proline reductase is encoded by the prd operon, with prdB encoding the selenocysteine-containing catalytic subunit of the enzyme. We reasoned that bacteria encoding homologs to PrdB would likely be able to ferment proline and serve as candidate competitors for this amino acid that could alter the ability of C. difficile to ferment proline. Based on their predicted ability to ferment proline and large representation in the gut microbial community, a panel of commensal Clostridia was developed that included murine isolates described here and human isolates. The murine isolates, identified based on 16S and rpoB sequencing and on metabolic characteristics, were most closely related to P. bifermentans, Paeniclostridium spp, P. benzoelyticum, C. xylanolyticum, and C. saccharolyticum. P. bifermentans [30] is a commensal bacterium that can neverthless exacerbate inflammation in a murine ulcerative colitis model [31] and is rarely associated with human infections [32]. Paeniclostridium as a genus includes P. sordellii (formerly Clostridium sordellii) and P. ghonii; here, despite 16S sequence similarity, we could not categorize our isolate as belonging to either of the described species largely due to the lack of indole production from l-tryptophan (Supplementary Table 2). The type strain for P. benzoelyticum was recently isolated from marine sediment [30], while C. xylanolyticum was described after isolation from wood chips [33]. This suggests the isolates described in this manuscript either represent novel strains associated with mammals or that these species are ubiquitous in the gut and the environment. Along with C. saccharolyticum [34], roles for P. benzoelyticum and C. xylanolyticum in the intestines have not been elucidated. The human isolates in the experimental Clostridia panel (B. obeum, C. scindens, D. longicatena) were chosen based on potential interactions with C. difficile in the gut and association with human disease [3,20–26,35,36]. Overall the bacteria chosen for the experimental panel represent a diverse cohort (Fig. 2B).
To test how these Clostridia may alter C. difficile metabolism, it was necessary to identify a medium that allowed for adequate growth of the isolates to levels comparable to C. difficile. TY, BHIS, and EG broth were chosen as they are rich media known to support the growth of an array of microbes. C. difficile and closely related bacteria (Fig. 2B) grew well with TY, though the majority of other isolates exhibited poor growth. This result was expected, as C. difficile, presumably along with closely related bacteria, relies on the abundant amino acids in TY as energy sources. Perhaps due to the increased diversity of nutrient sources and easily accessible carbohydrate dextrose, BHIS supported growth of all bacteria in the panel to levels closer to C. difficile, particularly C. xylanolyticum. With the exception of P. bifermentans, EG broth supported the growth of all isolates to levels very similar to C. difficile. Thus the diversity of the experimental panel was clear in their capacities to grow in different nutrient environments.
C. difficile was competed against an isogenic strain with an interruption in prdB in the presence of a single member of the Clostridia panel. The relative ratio of WT and prdB:CT C. difficile allowed us to distinguish between growth differences due to competition for proline in proline fermentation versus proline in peptide synthesis. However, this approach does not directly assess other forms of nutrient competition that are not dependent on proline fermentation. In BHIS broth, Paeniclostridium spp. and P. benzoelyticum reduced the ratio of WT and prdB:CT, suggesting that these bacteria limit the benefit provided by proline fermentation. As Paeniclostridium spp. and P. benzoelyticum both ferment proline to a high degree in BHIS (Fig. 4A), a possible explanation for the results is that these bacteria are better able to acquire proline through an unknown mechanism. This would limit proline available for C. difficile fermentation and drive C. difficile towards an alternate means of energy production. Unexpectedly, Paeniclostridium spp. enhances the benefit provided by proline fermentation in EG broth, leading to increased recovery of WT over the prdB :CT mutant (Fig. 4D). As Paeniclostridium spp. produces lower levels of 5-aminovalerate in EG broth compared to BHIS, it is conceivable that in EG broth Paeniclostridium spp. shifts its metabolism towards energy-producing pathways that do not rely on proline fermentation. This might then increase proline availability for C. difficile. C. xylanolyticum also increased the competitive advantage of C. difficile proline fermentation in EG broth (Fig. 4D). C. xylanolyticum does not ferment proline (Fig. 4B) and thus relies on alternate means of producing energy. This undefined metabolic pathway may consume nutrients that otherwise could be used by C. difficile for energy production or redox balance, therefore increasing the reliance of C. difficile on proline fermentation. The mechanisms describing how Paeniclostridium spp., P. benzoelyticum, and C. xylanolyticum influence C. difficile proline fermentation await further testing. Furthermore, members of the experimental Clostridia panel likely affect C. difficile through mechanisms independent of proline competition (Supplemental Fig. 3). One such mechanism could be alterations in C. difficile sporulation, though based on similar numbers of spores between the strains in defined medium (Supplemental Fig. 2) we do not anticipate changes in sporulation would significantly impact the relative ratios of the wt and mutant strains. More extensive studies exploring how commensal microbes alter C. difficile metabolism, growth, and sporulation may highlight novel microbe-microbe interactions. Lastly, these experiments were performed in batch culture, with limited nutrients and a build-up of metabolic waste products. How C. difficile proline fermentation is altered under continuous flow conditions or in the gut environment in the presence of a complex microbiota will be interesting areas of future study.
C. difficile ferments proline in multiple media in vitro and at different stages of infection in both mice and humans [13,15]. However, the benefit provided by proline fermentation is highly context dependent and regulated by the availabilities of proline, zinc, and selenium. This study suggests that the presence of certain members of the microbiota also shape the metabolism of C. difficile. Understanding the environment that pushes C. difficile towards proline fermentation, including nutrient availability, host response, and presence or absence of specific commensal bacteria, may aid in the rationale design of therapeutics that take advantage of this metabolic process.
4. Materials and methods
4.1. Bacterial strains and culture conditions
For information on the C. difficile, ATCC, and DSMZ strains used in the study, see Table 1. For information on isolates in the experimental panel, see “Clostridia isolation” section. Unless otherwise stated, C. difficile was routinely cultured in brain-heart infusion media (BHI) supplemented with yeast extract (BHIS) (52 g/L BHI, 5 g/L yeast extract, 0.03% l-cysteine). As indicated, members of the experimental Clostridia panel were cultured either in BHIS or in Eggerth-Gagnon (EG) media (2.8 g/L Lab Lemco powder, 10 g/L proteose peptone no.3, 5 g/L yeast extract, 4 g/L Na2HPO4,1.5 g/L d-glucose, 0.2 g/L l-cystine, 0.5 g/L l-cysteine, 0.5 g/L Tween 80). EG agar was supplemented with 0.5 g/L soluble starch, 50 mL defibrinated sheep blood, and 16 g/L agar. When appropriate, lincomycin (Linc) was added at 20 μg/mL. C. difficile defined medium (CDDM) was made as previously described [37] with the exception of adding a minimal amount of proline (0.3 mg/mL). All bacteria in this study were cultured anaerobically in an atmosphere of 5% CO2, 5% H2, and 90% N2.
4.2. Clostridia isolation and identification
Fecal pellets from WT C57BL/6J mice (Jackson Labs) were collected and immediately placed into 1 mL phosphate-buffered saline (PBS). Pellets were homogenized then incubated at 65 °C for 20 min to kill vegetative bacteria. Homogenates were then moved into an anaerobic chamber, further diluted with PBS, and plated onto EG agar plates. Plates were incubated at 37 °C for 1–4 days. Colonies with different morphologies were chosen and struck onto fresh EG agar plates. Following incubation at 37 °C and growth, single colonies were selected and struck out on a fresh EG agar plate at least one more time to ensure pure cultures. Single colonies were then used to inoculate EG broth, incubated at 37 °C, then stored at −80 °C in 30% glycerol. Using this strategy, 5 isolates were chosen for further experimentation. These were isolates m31, m33, m34, m36, and m38.
To identify bacterial isolates, each isolate above was first cultured on EG agar plates, then a single colony was chosen to inoculate EG broth. After 24 h of growth, 5 μL of each culture was added to 40 μL HotShot Lysis buffer (25 mM NaOH, 0.2 mM EDTA, pH = 12) and incubated at 95 °C for 30 min. Forty μL of HotShot Neutralization buffer (40 mM Tris-HCl, pH = 5) was then added to the reaction. PCR was then performed to amplify part of the 16S gene using degenerative primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) [38]. PCR reactions were then run on an agarose gel and bands were extracted, then sequenced. Performing a nucleotide BLAST search against the returned sequenced resulted in multiple matches for the isolates. To further identify the isolates, degenerate primers to Clostridial rpoB (FW 5′- CCNAAYCTDATHGARRTHCA-3’; RV 5′-CADTCTDGAHAGACC-3′) were designed. Following PCR amplification, isolation of DNA from an agarose gel, and sequencing, isolate m31 was the closest match to Paraclostridium bifermentans and m33 was the closest match to Clostridium saccharolyticum.
To further characterize isolates, metabolic testing was performed using API 20A test strips (BioMerieux). Isolates m31, m34, m36, and m38 were tested by first growing overnight on EG agar. Single colonies were then selected to inoculate the experimental media as per manufacturer instructions. Results of the metabolic assay were analyzed against the BioMerieux database and published literature [30]. Isolate m36 was identified as C. xylanolyticum and m38 was identified as P. benzoelyticum. Sequences for isolate m34 matched most closely to those of Paeniclostridium sordellii, however the metabolic profile did not match this bacterium. Therefore, here we refer to m34 as Paeniclostridium spp.
4.3. Bioinformatics
To examine the phylogenetic distribution of PrdB, Annotree [17] was used (accessed and created 10/2019) with the following parameters: KEGG ID K10794, % identity = 30, E value = 0.00001, % subject alignment = 70, % query alignment = 70. To determine the relatedness of the experimental Clostridia panel, full length 16S sequences were used. For C. difficile and the ATCC and DSMZ strains, 16S sequences were found through the sequenced strains in NCBI. For the bacteria isolated from this study, the 16S sequences from the closest sequenced bacterium found in the NCBI database were used. In the case of the isolated Paeniclostridium spp, the 16S sequence for P. sordellii was used. 16S sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) following by tree generation using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
4.4. Growth assay
Bacteria in the experimental panel, including WT C. difficile and the prdB::CT mutant, were struck out onto EG agar plates and incubated for 24 h at 37 °C. Bacteria were then scraped into 1 mL PBS. Bacterial density was normalized based on optical density (OD) at 630 nm, then 200 μL of fresh media in 96-well plates was inoculated with a 1:20 dilution of the normalized bacterial samples. Samples were incubated for 16 h at 37 °C, then OD630 was measured. The final values were calculated by dividing each sample OD in their respective medium by the average OD630 of WT C. difficile in the same medium. Each bacterium in the experimental panel was grown in biological triplicates in each medium. The media for the growth assay were tryptone-yeast broth (TY) (30 g/L tryptone, 20 g/L yeast extract), BHIS broth, and EG broth.
To determine growth rates and enumerate spores, either WT or prdB:CT were grown in CDDM. Growth was measured via spectrometry at OD600. Vegetative cells and spores were enumerated by plating bacteria incubated after 12 h onto BHIS agar plus 0.1% taurocholate. Cultures plated directly onto the agar led to colonies arising from both vegetative cells and spores. To specifically enumerate spores, samples were heated at 65 °C for 20 min, then plated onto solid media.
4.5. 5-Aminovalerate measurements
To determine 5-aminovalerate concentrations in spent media, bacteria in the experimental panel were struck out onto EG agar plates and incubated overnight. Bacteria from plates were then used to inoculate fresh BHIS or EG broth and incubated overnight. To begin the experiment, fresh BHIS or EG broth was inoculated with a 1:100 dilution of the overnight cultures and incubated for 6 h (BHIS) or 8 h (EG) at 37 °C. Following incubation, the OD630 of the cultures were measured and 1 mL of culture was removed, then centrifuged at high speed. 500 μL of the supernatant was then filtered using a 0.22 μm column filter. The flow through was collected and stored at −20 °C until further analysis. To determine 5-aminovalerate concentrations using liquid chromatography-mass spectrometry, samples were processed as previously described [13].
4.6. Competition assay
Bacteria in the experimental panel were first struck out onto EG agar plates and incubated overnight. These bacteria were then used to inoculate 5 mL of either BHIS or EG broth, depending on the medium for that competition. Following 24 h of incubation, the cultures were diluted to a concentration of 1 × 107 CFU/mL. For the control WT C. difficile versus prdB::CT alone, the two strains were mixed in a 1:1 ratio (total concentration 1 × 107 CFU/mL). For microbiota-C. difficile competitions, the 1:1 ratio of WT C. difficile and prdB::CT was mixed in an equal 1:1 ratio with an individual Clostridial member of the experimental panel (total concentration 1 × 107 CFU/mL; 2:1:1 ratio of microbiota: WT C. difficile: prdB::CT). The experimental media were prepared by supplementing 2 mL (one well of a 96-deep well plate) of either BHIS or EG broth with a final concentration of 30 mM l-proline and incubating anaerobically for 24 h. To begin the experiment, 1 × 104 CFU were added into the experimental media. Bacteria were incubated for 16 h, then diluted in PBS and plated onto cycloserine cefoxitin fructose agar (CCFA)(selects for only C. difficile) and BHIS + Linc (selects for only C. difficile prdB::CT). The competitive index was calculated by dividing the output ratio of WT and prdB::CT to the input ratio. Each competition was performed at least 3 independent times.
Supplementary Material
Acknowledgements
We thank the members of the Skaar laboratory for their critical and careful review of this manuscript.
Funding
This research is supported in part by the Ernest W. Goodpasture Chair in Pathology (E.P∙S.), and the Vanderbilt Digestive Diseases Research Center NIH grand DK058404 (E.P∙S.). C.A.L. is supported by the Childhood Infections Research Program training grant (NIH T32AI095202), the Burroughs Wellcome Fund Postdoctoral Enrichment Program, and the Simons Foundation through the Jane Coffin Childs Memorial Fund for Medical Research. T.M. and K.N. were supported by the Aspirnaut program through Vanderbilt University Medical Center. W.N.B. is supported by the American Heart Association Postdoctoral Fellowship. The funders had no role in the study design, data collections and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.anaerobe.2020.102210.
References
- [1].Lessa FC, Winston LG, McDonald LC, Difficil EIPC, Burden of Clostridium difficile infection in the United States REPLY, N. Engl. J. Med. 372 (24) (2015) 2369–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lewis BB, Pamer EG, Microbiota-based therapies for Clostridium difficile and antibiotic-resistant enteric infections, Annu. Rev. Microbiol. 71 (2017) 157–178, 10.1146/annurev-micro-090816-093549. [DOI] [PubMed] [Google Scholar]
- [3].Buffie CG, Bucci V, Stein RR, McKenney PT, Ling LL, Gobourne A, et al. , Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile, 205-U7, Nature 517 (7533) (2015), 10.1038/nature13828.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Studer N, Deshamais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, et al. , Functional intestinal bile acid 7 alpha-dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model, ARTN 191, Front. Cell. Infect. Mi. 6 (2016), 10.3389/fcimb.2016.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kang JD, Myers CJ, Harris SC, Kakiyama G, Lee IK, Yun BS, et al. , Bile acid 7 alpha-dehydroxylating gut bacteria secrete antibiotics that inhibit Clostridium difficile: role of secondary bile acids, 27-+. Cell. Chem. Biol. 26 (1) (2019) 10.1016/j.chembiol.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rea MC, Clayton E, O’Connor PM, Shanahan F, Kiely B, Ross RP, et al. , Antimicrobial activity of lacticin 3147 against clinical Clostridium difficile strains, J. Med. Microbiol. 56 (7) (2007) 940–946, 10.1099/jmm.0.47085-0. [DOI] [PubMed] [Google Scholar]
- [7].Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, et al. , Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile, P. Natl. Acad. Sci. USA 107 (20) (2010) 9352–9357, 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jenior ML, Leslie JL, Young VB, Schloss PD, Clostridium difficile colonizes alternative nutrient niches during infection across distinct murine gut microbiomes, UNSP e00063-17, mSystems 2 (4) (2017), 10.1128/mSystems.00063-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jenior ML, Leslie JL, Young VB, Schloss PD, Clostridium difficile alters the structure and metabolism of distinct cecal microbiomes during initial infection to promote sustained colonization, ARTN e00261-18, mSphere 3 (3) (2018), 10.1128/mSphere.00261-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, et al. , Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection, ARTN 3114, Nat. Commun. 5 (2014), 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, et al. , Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens, 96-+, Nature 502 (7469) (2013) 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wilson KH, Perini F, Role of competition for nutrients in suppression of clostridium-difficile by the colonic microflora, Infect. Immun. 56 (10) (1988) 2610–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lopez CA, Beavers WN, Weiss A, Knippel RJ, Zackular JP, Chazin W, et al. , The immune protein calprotectin impacts Clostridioides difficile metabolism through zinc limitation, mBio 10 (6) (2019), 10.1128/mBio.02289-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jackson S, Calos M, Myers A, Self WT, Analysis of proline reduction in the nosocomial pathogen Clostridium difficile, J. Bacteriol. 188 (24) (2006) 8487–8495, 10.1128/Jb.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Battaglioli EJ, Hale VL,Chen J, Jeraldo P, Ruiz-Mojica C, Schmidt BA, et al. , Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea, ARTN eaam7019, Sci. Transl. Med. 10 (464) (2018), 10.1126/scitranslmed.aam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, et al. , Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium, Epub 2009/09/29, Genome Biol. 10 (9) (2009) R102, 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mendler K, Chen H, Parks DH, Lobb B, Hug LA, Doxey AC, AnnoTree: visualization and exploration of a functionally annotated microbial tree of life, Nucleic Acids Res. 47 (9) (2019) 4442–4448, 10.1093/nar/gkz246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. , A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life, Nat. Biotechnol. 36 (10) (2018) 996, 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- [19].Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, Knight R, The human microbiome in evolution, BMC Biol. 15 (1) (2017) 127, 10.1186/s12915-017-0454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mullish BH, McDonald JAK, Pechlivanis A, Allegretti JR, Kao D, Barker GF, et al. , Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection, Gut 68 (10) (2019) 1791–1800, 10.1136/gutjnl-2018-317842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Takahashi K, Nishida A, Fujimoto T, Fujii M, Shioya M, Imaeda H, et al. , Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in crohn’s disease, Digestion 93 (1) (2016) 59–65, 10.1159/000441768. [DOI] [PubMed] [Google Scholar]
- [22].Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, et al. , Potential roles of gut microbiome and metabolites in modulating ALS in mice, Nature 572 (7770) (2019) 474–480, 10.1038/s41586-019-1443-5. [DOI] [PubMed] [Google Scholar]
- [23].Rajilic-Stojanovic M, de Vos WM, The first 1000 cultured species of the human gastrointestinal microbiota, FEMS Microbiol. Rev. 38 (5) (2014) 996–1047, 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Petrov VA, Saltykova IV, Zhukova IA, Alifirova VM, Zhukova NG, Dorofeeva YB, et al. , Analysis of gut microbiota in patients with Parkinson’s disease, Bull. Exp. Biol. Med. 162 (6) (2017) 734–737, 10.1007/s10517-017-3700-7. [DOI] [PubMed] [Google Scholar]
- [25].Kamo T, Akazawa H, Suda W, Saga-Kamo A, Shimizu Y, Yagi H, et al. , Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure, PloS One 12 (3) (2017), e0174099, 10.1371/journal.pone.0174099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mondot S, Lepage P, Seksik P, Allez M, Treton X, Bouhnik Y, et al. , Structural robustness of the gut mucosal microbiota is associated with Crohn’s disease remission after surgery, Gut 65 (6) (2016) 954–962, 10.1136/gutjnl-2015-309184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bouillaut L, Self WT, Sonenshein AL, Proline-dependent regulation of Clostridium difficile stickland metabolism, J. Bacteriol. 195 (4) (2013) 844–854, 10.1128/Jb.01492-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Theriot CM, Young VB, Interactions between the gastrointestinal microbiome and Clostridium difficile, Annu. Rev. Microbiol. 69 (2015) 445–461, 10.1146/annurev-micro-091014-104115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leslie JL, Vendrov KC, Jenior ML, Young VB, The gut microbiota is associated with clearance of Clostridium difficile infection independent of adaptive immunity, ARTN e00698-18, mSphere 4 (1) (2019), 10.1128/mSphereDirect.00698-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jyothsna TSS, Tushar L, Sasikala C, Ramana CV, Paraclostridium benzoelyticum gen. nov. sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii (vol 66, pg 1268, 2016), Int.J. Syst. Evol. Microbiol. 66 (2016) 2459, 10.1099/ijsem.0.001144. [DOI] [PubMed] [Google Scholar]
- [31].Kutsuna R, Tomida J, Morita Y, Kawamura Y, Paraclostridium bifermentans exacerbates pathosis in a mouse model of ulcerative colitis, ARTN e0197668, PloS One 13 (5) (2018), 10.1371/journal.pone.0197668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hale A, Kirby JE, Albrecht M, Fatal spontaneous Clostridium bifermentans necrotizing endometritis: a case report and literature review of the pathogen, Open Forum. Infect. Di. 3 (2) (2016), 10.1093/ofid/ofw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rogers GM, Baecker AAW, Clostridium-xylanolyticum sp-nov, an anaerobic xylanolytic bacterium from decayed pinus-patula wood chips, Int. J. Syst. Bacteriol. 41 (1) (1991) 140–143, 10.1099/00207713-41-1-140. [DOI] [Google Scholar]
- [34].Murray WD, Khan AW, Ethanol-production by a newly isolated anaerobe, clostridium-saccharolyticum - effects of culture-medium and growth-conditions, Can. J. Microbiol. 29 (3) (1983) 342–347, 10.1139/m83-057. [DOI] [Google Scholar]
- [35].Amrane S, Bachar D, Lagier JC, Raoult D, Clostridium scindens is present in the gut microbiota during Clostridium difficile infection: a metagenomic and culturomic analysis, J. Clin. Microbiol. 56 (5) (2018), 10.1128/JCM.01663-17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [36].Louis P, Hold GL, Flint HJ, The gut microbiota, bacterial metabolites and colorectal cancer, Nat. Rev. Microbiol. 12 (10) (2014) 661–672, 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- [37].Karasawa T, Ikoma S, Yamakawa K, Nakamura S, A defined growth medium for Clostridium difficile, Epub 1995/02/01, Microbiology 141 (Pt 2) (1995) 371–375, 10.1099/13500872-141-2-371. [DOI] [PubMed] [Google Scholar]
- [38].Weisburg WG, Barns SM, Pelletier DA, Lane DJ, 16S ribosomal DNA amplification for phylogenetic study, J. Bacteriol. 173 (2) (1991) 697–703, 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.