

Abstract
Microsatellite instability-high (MSI-H) tumors are characterized by high tumor mutation burden and responsiveness to checkpoint blockade. We identified tumor-specific frameshifts encoding multiple epitopes that originated from indel mutations shared among patients with MSI-H endometrial, colorectal and stomach cancers. Epitopes derived from these shared frameshifts have high population occurrence rates, wide presence in many tumor subclones and are predicted to bind to the most frequent MHC alleles in MSI-H patient cohorts. Neoantigens arising from these mutations are distinctly unlike self and viral antigens, signifying novel groups of potentially highly immunogenic tumor antigens. We further confirmed the immunogenicity of frameshift peptides in T cell stimulation experiments using blood mononuclear cells isolated from both healthy donors and MSI-H cancer patients. Our study uncovers the widespread occurrence and strong immunogenicity of tumor-specific antigens derived from shared frameshift mutations in MSI-H cancer and Lynch Syndrome patients, suitable for the design of common “off-the-shelf’ cancer vaccines.
Graphical Abstract
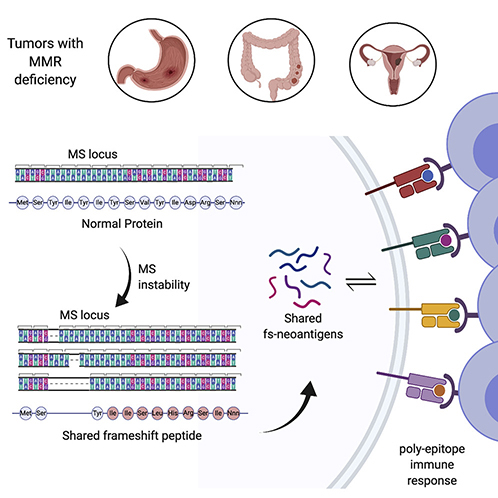
Introduction
Genetic alterations in tumor genomes that encode novel stretches of amino acids compared to normal cells are a potential source of immunogenic tumor-specific epitopes, commonly referred to as neoantigens. Total neoantigen burden, the sum of neoantigens predicted to be expressed by a tumor, has been demonstrated to be an independent proxy for response to immune checkpoint inhibitor therapy (Snyder et al., 2014; Rizvi et al., 2015; Van Allen et al., 2015; Samstein et al., 2019). However, determining neoepitopes in individual tumor samples remains fraught with uncertainties, such as the lack of congruence between neoantigen prediction pipelines. Microsatellite instability-high (MSI-H) tumors have high tumor burdens accompanied by effector T cell infiltration and are more responsive to checkpoint inhibitor therapy, making them suitable models to investigate neoantigen-based immune therapies (Mandal et al., 2019; Willis et al., 2019). The MSI-H tumor phenotype arises from defective DNA repair mechanisms due to a loss of mismatch repair (MMR) activity. MSI-H is typically characterized by the variation of DNA length in microsatellite loci – units of one to ten mono-, di-, tri-, or tetra-nucleotides repeated multiple times (Kim, Laird and Park, 2013). In healthy cells these unpaired nucleotides are recognized and excised by MMR, but in MSI-H tumors they remain unrepaired. Some of these microsatellite regions are located in coding regions, where their destabilization can cause frameshift (fs-) mutations that shift an open reading frame, thereby providing a substantial source of tumor-specific neoantigens (Turajlic et al., 2017; Mandal et al., 2019).
Inactivation of MMR genes plays a key role in the acquisition of the MSI-H phenotype in hypermutated tumors (Zighelboim et al., 2007; Walther et al., 2009; Ahmed et al., 2013; Diaz-Padilla et al., 2013). The sporadic form of MSI-H tumors occurs in 10–40% of colorectal and endometrial cancers and is mainly caused by biallelic hyperméthylation of the MLH1 promoter (Cunningham et al., 1998; Veigl et al., 1998). Lynch syndrome, sometimes referred to as hereditary nonpolyposis colorectal cancer (HNCC), is an inherited, autosomal-dominant disorder characterized by germline non-synonymous mutations in MMR genes. The majority of Lynch syndrome patients have germline mutations inMSH2 (~30%) and PMS2 (~70%) genes (Gatalica et al., 2016). Estimates suggest that as many as 1 in every 300 people may carry Lynch syndrome-associated germline alterations (Carethers et al., 2015; Chung and Rustgi, 2019; Cohen, Pritchard and Jarvik, 2019). Lynch syndrome patients have an 80% lifetime risk for developing colorectal or endometrial MSI-H cancers, accounting for 3–5% of all colorectal and endometrial cancers. The most common cancer associated with sporadic and hereditary-predisposed MSI-H type is colorectal cancer (80% of HNCC patients), followed by endometrial carcinoma (60% of HNCC patients). The MSI-H group accounts for up to 28.6% of low-grade and 54.3% of high-grade endometrioid cancers. Although less common, the MSI-H phenotype is also observed in other cancers such as bladder, gastric, ovarian, small bowel and renal due to a hereditary-predisposition (Vasen et al., 1996).
Most neoantigens that are predicted from non-synonymous point mutations are derived from patient-specific passenger mutations, as recurrent driver mutations infrequently generate immunogenic peptides (Marty et al., 2017). However, neoantigens expressed in the MSI-H phenotype are distinguished by unique features, namely, (1) a high mutational burden in well-defined, limited sequence spaces – namely microsatellite regions and (2) a restricted pattern of mutations due to nucleotide insertions or deletions (indels). This feature combination can induce a bottleneck, causing a high probability of shared indel mutations in protein-coding genes, leading to frameshift (fs-) peptides encoding multiple MHC-I-restricted epitopes (poly-epitope fs-peptide) likely to be common among multiple patients. Based on this premise, we investigated fs-mutations in the tumor genomes of MSI-H patients with colorectal, stomach and endometrial carcinomas and identified broadly shared, immunogenic, poly-epitope fs-peptides. Our study provides a foundation for the potential application of these shared epitopes in “off-the-shelf’ vaccines.
Results
MSI-H colorectal, stomach and endometrial cancers have a high fs-load
To interrogate the relationship between MSI status and fs-load, we utilized tumor whole exome sequencing (WES) data available at the Cancer Genome Atlas (TCGA) database. Although the majority of TCGA tumors are microsatellite stable or their status is unknown, ~20–30% of endometrial, colorectal and stomach adenocarcinomas (UCEC, COAD, STAD, respectively) are diagnosed as microsatellite unstable (MSI-H). In total, the MSI-H population in TCGA accounts for 338 patients (Figure 1A, B). Similar to previous studies (Mlecnik et al., 2016; Marty et al., 2017; Turajlic et al., 2017), we first annotated somatic mutation load on a pan-cancer scale and observed specific mutation frequencies vary across different tumor types (Figure S1A).
Figure 1.

Microsatellite instability in COAD, STAD and UCEC tumors documented in TCGA. Majority of MSI-H frameshifts are deletions. A. Quantification of patients with microsatellite instable (MSI) tumors by designation applied in TCGA. MSI-H is MSI-high, MSI-L is MSI-low, MSS is MS-stable, and Unknown - undetermined MS status. B. Table showing the fraction (absolute number) of patients with UCEC, COAD and STAD tumors identified as MSI-H, MSI-L, MSS or Unknown. C. Frameshift (fs-) load (Y-axis, log10) in different tumor types across TCGA. D. Segregation of fs-load by MSI designation in COAD, STAD and UCEC patients. E. Comparison of fs-load (Y-axis, log10) with insertion-deletion ratio (X-axis, log10) in COAD, STAD and UCEC patients. F. Schematic of the shared fs-peptide hypothesis. Examples of possible deletions within the MS locus of a protein coding gene that generates similar stretches of new amino acids. 0 – normal gene, 1–3 – deletions within MS locus.
However, the average frameshift (fs-) load, as determined by frameshift count per each patient, was selectively elevated in a subset of UCEC, COAD and STAD patients (Figure 1C). When we stratified UCEC, COAD and STAD patients according to MSI status, the majority of high fs-load patients overlapped with the MSI-H clinical biomarker, indicating a high specificity/selectivity of this biomarker in detecting indel-enriched tumor types (Figure 1D). Consistent with previous studies, most frameshifts stemmed from nucleotide deletions, as determined by correlating patients’ fs-load with insertion-to-deletion ratio (Figure 1E).
Though tumor evolution is primarily regarded as driven by random mutational processes, there is accumulating evidence that some loci acquire mutations preferentially (Gerstung et al., 2017; Buljan, Blattmann and Aebersold, 2018; Iranzo, Martincorena and Koonin, 2018). Given the existing skewing in the underlying mutational process and the high occurrence of indel mutations in microsatellite regions within open reading frames (Cortes-Ciriano et al., 2017), we hypothesized MSI-H patients could share frameshift events. While missense somatic mutations share limited similarity across multiple tumors (Schumacher and Schreiber, 2015), we found fs-mutations in microsatellite (MS) unstable regions are likely to generate common fs-peptides when translated (Figure 1F). To gain an understanding of shared mutational events, we examined mutational load in cancer cell lines from the Cancer Cell Line Encyclopedia (Barretina et al., 2012; Ghandi et al., 2019) (CCLE) and found fs-mutations are shared among multiple cancer cell lines far more frequently than missense mutations. Intriguingly, cell lines derived from tumor types frequently displaying an MSI-H phenotype (stomach, colon and endometrial) tend to share fs-events more often than cell lines derived from other tissues (Figure S1B).
Colorectal, stomach and endometrial MSI-H adenocarcinomas are enriched in shared poly-epitope fs-peptides
To identify potentially immunogenic epitopes derived from fs-mutations, we developed an fs-neoantigen calling pipeline (see Methods). Using this pipeline, we analyzed the distribution of fs-mutations, fs-peptides, and corresponding fs-epitopes in MSI-H UCEC, COAD and STAD cohorts of TCGA patients (Figure 2A, Tables S1 and S2). We found many genes that were commonly mutated via indels in MS regions of all three tumor types. Up to 80% of MSI-H COAD and STAD patients shared genes commonly mutated in the form of MSI-derived indels (fs-genes), including ACVR2A, MIKI67, RPL22. Similarly, the top-listed shared frameshifted genes in >50% of MSI-H UCEC and COAD patients include CASP5, MUC6, KMT2C. As expected, the frequency of shared fs-peptides – mutation events derived from exactly the same fs-mutation – was lower. Only a few fs-peptides were shared among >40% of MSI-H UCEC (e.g. RPL22, SETD1B), 50% of MSI-H COAD patients (e.g. SGOL1, SEC31A, ACVR2A) or >50% of MSI-H STAD patients (e.g. RPL22, ACVR2A). Finally, the top-frequency of shared MHC class I epitopes was around 30% of MSI-H UCEC patients (e.g. OR7E24, MSYFPILFF epitope), around 60% of MSI-H COAD patients (e.g. SGOL1, LIWKRVFIL epitope) and around 50% MSI-H STAD patients (e.g. RNF43, RFFPITPPV epitope) (Figure 2A). Interestingly, the average frequency of shared fs-gene, fs-peptide and fs-neoepitope events in colon and stomach MSI-H tumors was twice as high compared to endometrial MSI-H tumors, possibly due to how different pathways of tumorigenesis and rates of cell growth may affect the rate of MS-related mutagenesis.
Figure 2.

Frequencies of shared fs-events and fs-peptides, and fs-epitope distribution in STAD, COAD and UCEC MSI-H tumors. A. Scatterplots of patient frequencies of frameshifted genes (LEFT), fs-peptides (CENTER) and fs-epitopes (RIGHT) in UCEC, STAD and COAD MSI-H tumors. B. Three scatterplots showing the selection criteria for identification of shared fs-peptides in MSI-H UCEC, COAD and STAD. Each dot represents a fs-peptide shared in at least 20% of patients in each cohort. Number of predicted 9-mer epitopes per peptide (X-axis) is plotted against the number of predicted interacting MHC alleles (Y-axis). Size of the circle represents the number of predicted pMHC interactions. Color of the dot reflects the somatic score of the fs-mutation. C. MHC-I epitope mapping of 46 shared fs-peptides from MSI-H UCEC, COAD and STAD tumors combined together. D. Quantification of MHC-I epitopes derived from 9 shared fs-peptides of MSI-H UCEC cohort shown per each patient (rows, left panel) or each MHC-I allele (rows, right panel).
To identify immunogenic fs-peptides with confidence, we developed a mutation ranking system based on the maximization of four parameters. First, we introduced a somatic score for each fs-mutation, where a higher score implies higher confidence that this mutation is truly somatic. We analyzed the distribution of fs-peptide lengths: on average, the length of MSI-H frameshift peptides was 20–30 amino acid (aa) residues, suggesting these peptides may encode multiple immunogenic epitopes per fs-mutation (Figure S2). Taking this into account, we maximized the number of putative neoepitopes per each fs-peptide: epitopes, predicted for each fs-peptide across all patients were pooled and the total number of unique epitopes was determined. Third, we grouped all MHC alleles predicted to bind those neoepitopes. Maximization of this parameter allowed us to pick poly-allelic fs-peptides, covering a diverse set of alleles in a population. Finally, to include the population MHC allele frequency parameter, we quantified the total amount of peptide-MHC (pMHC) interactions per frameshift (Figure 2B), together enabling the selection of fs-peptides that are likely immunogenic, encode poly-epitopes, bind a broad spectrum of MHC-I alleles, and are widely shared. Applying these selection parameters to fs-mutations shared by at least 20% of MSI-H patients, we identified 9, 37 and 23 shared fs-peptides that encode poly-epitopes in endometrial, colorectal and stomach MSI-H patients, respectively (Figures 2B, S2 and Table S2). Altogether, this fs-peptide set accounted for 46 unique peptide sequences with broad epitope mapping (Figure 2C), 5 of which, SLC35F5, SEC31A, TTK, SETD1B and RNF43, were shared among all MSI-H UCEC, COAD and STAD patients analyzed.
We next focused on frameshift-derived neoantigens from MSI-H endometrial carcinoma, as these have not been well characterized to date. To assess the distribution of shared fs-neoantigens in patients and determine the MHC class I alleles they bind to, we analyzed epitopes derived from the 9 MSI-H UCEC shared fs-peptides. The majority of the analyzed tumor specimens (>95% of the MSI-H UCEC patient cohort) potentially encoded neoepitopes derived from at least 2 fs-peptides (out of the 9). Importantly, the combination of neoepitope-yielding fs-peptides may vary depending on the patient. For example, SEC31A and ASTE1 fs-neoantigens were frequently found in the same patients together. Shared fs-neoantigens were predicted to bind to multiple frequently occurring MHC class I alleles (e.g. A0201, B0801, C0701/02), as well as a spectrum of less frequent ones (Figure 2D). Importantly, only the “mixture” of fs-epitopes derived from all 9 peptides has the potential to reach a good representation in all possible pMHC interactions per each MHC-I allele (Figure 3A). Together our data demonstrates the presence of shared poly-epitope fs-peptides across MSI-H UCEC, COAD and STAD patients, suggesting the possibility of developing an off-the-shelf MSI-H vaccine for these three tumor types.
Figure 3.

Genomic and expression properties of shared fs-mutations in MSI-H tumors. A. Friedman difference test, measuring the difference between pMHC interactions found in all MSI-H UCEC patients (ALL) and either pMHC interactions in each fs-peptide separately or combined together (POOL). Each bar represents the difference score per each MHC allele. B. Tumor allele frequency of nine shared frameshift mutations in normal (Top) and tumor (Bottom) tissues of MSI-H UCEC TCGA patients. C. Unsupervised hierarchical clustering of shared fs-mutation load (Left) and corresponding expression FPKM values of frameshifted genes (Right) in MSI-H UCEC (Top), COAD (Center) and STAD (Bottom) tumors. Patients plotted in columns; genes plotted in rows. D. Spearman correlation test between shared fs-mutation load and fs-gene expression. E. Normalized expression of nine shared fs-mutations in MSI-H UCEC patients. LEFT – ratio of indel to total read count spanning the microsatellite region in MSI-H and MSS patient cohorts of UCEC, STAD and COAD tumors. Statistical significance is derived from non-parametric Mann-Whitney two-tailed test. RIGHT – normalized frequency of fs-mutation in RNF43 within 100 nucleotide genomic loci: 50 nt upstream and 50 nt downstream of the shared frameshift in MSI-H and MSS RNAseq samples. F. LEFT - Clustering of MSI-H COAD patients with shared frameshift mutations from the CPTAC dataset. RIGHT – patients’ frequencies of all and shared frameshift mutations in the CPTAC dataset. G. MS/MS detection of predicted shared RNF43 frameshift in an MSI-H UCEC sample from the CPTAC UCEC dataset. MS/MS spectra of tryptic peptide (yellow fragment) derived from predicted fs-peptide (red sequence) is identified by Pepquery analysis (PMS p-value 0.00099).
Fs-peptides shared in MSI-H endometrial carcinoma are correctly translated and abundant
In addition to MHC-I binding affinity predictions, expression and abundance of neoantigens are also important correlates of immunogenicity. To better evaluate the immunogenic potential of fs-peptides, we analyzed tumor allele frequencies from the MSI-H UCEC patient cohort to estimate the abundance of the selected nine shared frameshifts. We compared corresponding fs-allele frequencies in normal and tumor samples and found that while they were almost non-detectable in normal tissues, in tumor biopsies their allele frequencies rose to 30–40% on average, suggesting these mutations were present in substantial fractions of the tumors (Figure 3B). The same conclusion was also made from orthogonal mutation recalling of selected WES matched tumor/normal datasets (Figures S2, S3 and Table S1). This suggests the combination of 9 fs-peptides has the potential to prime T cell responses that recognize the majority of the malignant cells in MSI-H UCEC tumors. The high mutation rates of MSI-H tumors might decrease the probability of shared fs-peptides being correctly translated. Therefore, we assessed the conditional probability of shared fs-peptides being correctly translated. For this purpose, we estimated all disruptive upstream and downstream mutation frequencies using TCGA MSI-H cohort and calculated their posterior probabilities. MSI-H UCEC shared fs-peptides had a high probability of being correctly translated, with a ~0.8 probability for TTK and RNF43 and >0.9 probability for the remaining frameshifts (Figure S2).
To determine whether genes encoding shared fs-peptides were expressed, we analyzed RNAseq from samples of MSI-H patients in TCGA and mass spectrometry (MS) data utilizing the COAD and UCEC datasets from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) (Zhang et al., 2014; Vasaikar et al., 2019; Dou et al., 2020). Unsupervised clustering of MSI-H patients was performed focusing on genes with predicted shared fs-events. We also plotted fragments per kilobase of transcript per million (FPKM) expression values of genes encoding shared fs-peptides and ranked them according to the previously obtained patient and gene rankings (Figure 3C). We observed no correlation between RNA expression and shared fs-load (Figure 3D), suggesting that frameshifted genes were not selectively epigenetically silenced in tumors. Considering the latter results, we analyzed the expression of nine shared fs-mutations in MSI-H UCEC patients. Of note, each fs-mutation was detected in a different fs-gene, except two, both of which occurred in one fs-gene, TTK. Therefore, formally we detected 8 uniquely mutated genes. Six of these fs-genes were expressed at the RNA level from matching tumor samples. To assess the expression level of fs-alleles, we compared the normalized read count containing an indel with the total amount of reads covering the targeted genomic loci utilizing RNAseq samples from MSI-H (n=270) and MSS (n=200) patients, used as controls. Basically, we performed variant allele frequency estimation from RNAseq samples with expression normalization. We detected significant and robust expression of fs-alleles in MSI-H patients compared to MSS patients (Figure 3E). Indel reads were also detected in RNAseq samples of MSS patients as well. We attribute this to either higher mutability of reverse-transcriptase applied during RNA sequencing protocols or imperfect MSI-H classification. Further confirming the expression of fs-peptides in tumors, we identified many shared fs-mutations in COAD and UCEC genomic samples collected through CPTAC. In addition to confirming the genomic presence of the underlying fs-mutation, predicted fs-peptides were therefore detected as protein as well (Figures 3F–G, S4 and Table S2). Taken together, we confirmed shared fs-mutations are not epigenetically silenced and have the potential of being correctly expressed within tumors.
Tumor fs-epitopes are more likely to be presented and are less similar to viral antigens than missense derived epitopes
We next examined the intrinsic properties of fs-derived epitopes compared to missense-derived epitopes and viral antigens. Tumor-derived neoantigens can have broad similarities to pathogen-derived (viral) antigens, and their expression may promote the response to checkpoint therapy (Luksza et al., 2017; Balachandran et al., 2017; Richman, Vonderheide and Rech, 2019; Zhang et al., 2019). To understand how fs-derived or missense-derived neoepitopes relate to viral antigens we compared both types of neoantigens to the viral antigens present in The Immune Epitope Database (IEDB). We first calculated the total number of neoantigens derived from missense and frameshift mutations of MSI-H patients. Even though the total frameshift and missense neoantigen loads were similar, the number of predicted MHC-I epitopes per mutation were different: 4 epitopes per one frameshift and 2 per one missense mutation on average (Figure S5). This observation is consistent with the idea that fs-mutations may be more immunogenic than missense mutations due to an increased probability of generating neoantigens. While many missense-derived epitopes are, by definition, one amino acid different from a self-peptide, the majority of fs-derived epitopes are unique, “non-self” peptide sequences and hence exhibit less similarity to the human proteome (Figure S5). This implies that fs-derived epitopes are unlikely to have been tolerized by the host immune system, and that the frameshift-specific T-cells will have little or minimal autoreactivity. We also compared these two epitope datasets with virus-derived antigens. At different blastp search stringency, the overall number of missense epitopes matched with viral epitopes was 3 times higher than matched fs-epitopes (Figure S5). We speculate this observation is due to the overall viral adaptation to the human proteome and host T-cell epitopes, as viruses mimic particular host functionalities in order to interact with the host cellular machinery as well as to escape host immune recognition. Therefore, fs-epitopes appear “less-self than either missense or virus-derived epitopes, and are likely of higher “quality” as a result (Balachandran et al., 2017).
Predicted shared poly-epitope fs-peptides are detected and presented on MHC class I of cancer cell lines from CCLE
To validate the presence of predicted shared fs-mutations in an external dataset, we queried cell lines in the Cancer Cell Line Encyclopedia (CCLE) (Figure 4 and Table S3). 34 of 46 shared poly-epitope fs-mutations were detected in multiple cancer cell lines derived from different tumor types. The number of detected shared fs-mutations, however, differed across cancer cell lines. Lines derived from intestine, endometrium, stomach and prostate cancers had 5–10 shared fs-mutations each, while hematopoietic, ovarian and lung cancer cell lines had 1–5 (Figure 4A). The presence of predicted shared fs-mutations in the last three tumor types suggests a broader occurrence of shared poly-epitope fs-peptides across tumors. Notably around 20, 40 and 60% of intestine, stomach and endometrial cell lines expressed shared fs-peptides, respectively (Figure 4B). Initially predicted in TCGA cohorts, fs-mutations expressing shared fs-peptides were also significantly shared in cancer cell lines compared to other fs-mutations derived from the same genes (Figure 4C). Allele coverage analysis indicated that predicted shared fs-mutations were present at 30–50% allele frequency on average in cancer lines (Figure 4D). RNAseq indicates gene expression patterns are unchanged upon acquiring shared frameshifts (Table S3), ruling out the possibility of epigenetic allele-specific silencing. Significantly, fs-epitopes derived from predicted shared mutations could be detected by tandem mass spectrometry (MS/MS) proteomic analysis of peptides eluted from MHC class I of HCT116, an MSI-H colorectal cancer cell line (Bassani-Sternberg et al., 2015) (Figure 4E, Table S2), establishing fs-mutations yield epitopes that can be processed and presented for recognition by the immune system. Finally, we used targeted PCR coupled with Sanger sequencing to verify the presence of selected shared indel sequences in CCLE cell lines. Our analysis showed indel mutations could be recalled with high specificity and sensitivity, reaching an AUC of 0.882 (Figures 4F, Data S1). Overall, the CCLE dataset analyses confirm the widespread occurrence of predicted shared fs-peptides in cancer cells as well as their presentability by MHC-I on the cell surface.
Figure 4.

Detection of shared fs-mutations in the cancer cell line encyclopedia (CCLE). A. Quantification of shared fs-mutations in cell lines per tissue of tumor origin and per each frameshifted gene. Histogram plot on the right shows the number of shared fs-mutations per cell line. Bar-plot is highlighted according to shared frameshift load: high (yellow) and low (grey). B. Absolute number of cell lines with detected shared fs-mutation compared to total number of cell lines in CCLE. Cell lines are sorted according to tissue origin. C. Distribution of all detected indels in genes with shared fs-mutations (34 genes in CCLE). Left – metagene, showing normalized frequency of all detected indels in 34 genes, around shared fs-mutation. Right– t-test of number of cell lines encoding shared fs-mutations versus all other fs-mutations, detected in selected 34 genes. D. Comparison of fs-allele frequency per cell line with fs-mutation frequency among cancer cell lines. E. MS/MS detection of fs-peptide epitopes eluted from MHC-I complexes of the HCT116 cell line. MS/MS spectra of an MHC class I epitope (dark orange) derived from shared SLC35G2 fs-peptide (light grey) is identified following Pepquery analysis (PMS p-value 0.001). Shared fs-mutation allele frequency is ~ 0.4 in HCT116 according to CCLE (bar plot). netMHC predictions of MHC class I allele affinities of MS/MS detected peptides using HCT116 MHC-alleles. Significant interactions with interaction thresholds of rank = 2.0 or KD < 500 nM are shown. F. ROC analysis of shared fs-mutation recall by Sanger sequencing in the selected cell lines (HCT116, LOVO, Hec59, HeclB). Indel calling by WES is highly specific and sensitive (91.2% and 85.2% respectively).
Detection of shared poly-epitope fs-peptides in MSI-H patients undergoing immunotherapy
MSI-H tumors are characterized by a high tumor mutational load and responsiveness to anti-programmed cell death 1 (PD-1)-based immune checkpoint inhibitor (ICI) immunotherapy (Le et al., 2015; Dudley et al., 2016). However, not all patients respond to therapy, suggesting additional differences between patients may underlie the lack of successful immunotherapy. We hypothesized that differential levels of fs-mutations translating into greater numbers of immunogenic peptides may in part account for the outcome of the immunotherapy. We first performed Cox regression and survival analyses of MSI-H UCEC patients in TCGA stratified by shared fs-load as high (top 50%) and low (bottom 50%), and analyzed tumor stage and patient age in the same strata to determine if fs-mutation numbers correlate with survival. Patient age and tumor stage were evenly represented in both fs-neoantigen high and fs-neoantigen low MSI-H cohorts. We did not detect any significant benefit in patients’ survival based on shared fs-neoantigen load in any MSI-H tumor types (Figure S6).
Next, we analyzed the distribution of 46 shared fs-mutations in cancer patients undergoing PD-1 blockade (12 MSI-H and 4 MSS patients, NCT01876511) (Le et al., 2015, 2017; Mandal et al., 2019). We confirmed the wide presence of shared mutations in tumor samples on a genomic level as well as high concordance with MS status (Figure 5A–B, Table S4). 70% of shared fs-mutations were present in > 20% of the MSI-H patients in the immunotherapy cohort (Figure 5C). We also analyzed the distribution of neoantigens derived from shared fs-peptides. Fs-neoantigens were widely present in MSI-H patients, whereas undetected in MSS patients (Figure 5D). The shared fs-neoantigen load was higher in MSI-H patients responding to PD-1 immunotherapy (Figure 5E), emphasizing the potential importance of fs-neoantigens in driving response to ICI. Though many antigen-independent mechanisms might underlie the poor response rates in a subset of those patients, a potential combination of PD-1 and shared MSI-H vaccine may therefore hold promise in improving outcomes of immunotherapy for non-responsive MSI-H patients.
Figure 5.

Detection of shared fs-neoantigens in MSI-H patients undergoing immunotherapy. A. Heatmap plots of shared fs-mutation allele frequency in normal (LEFT) and tumor (RIGHT) samples of patients undergoing PD-1 immunotherapy. MS status of each patient is highlighted in the far-right bar column. B. Distribution of shared fs-mutation frequencies in normal and tumor samples. The cutoff of 0.2 is suggested to filter somatic events. C. Distribution of population frequencies of 46 shared fs-mutations. 70% of shared fs-mutations are present in > 20% patients. D. Heatmap of shared fs-neoantigen load derived from fs-mutations with allele frequency > 0.2. MS status and objective clinical responses are shown in the right bar columns. E. Shared fs-neoantigen load in MSI-H patients classified by clinical objective response rate: CR/PR – complete and partial responses; SD/PD - stable and progressed disease. Statistical significance is determined by unpaired t-test (p-value < 0.049). Color code is the same as in D.
Poly-epitope fs-peptides shared in MSI-H UCEC patients are highly immunogenic
Our data has suggested that shared fs-mutations yield unique arrays of neoantigens that should be highly immunogenic. To assess the potential immunogenicity of the nine predicted fs-peptides identified from the MSI-H UCEC patient cohort (Figure 2D), we induced T cell responses against each neopeptide stretch using an immunogenicity assay designed to rapidly prime naïve T cells (Cimen Bozkus et al., 2019). We first designed long overlapping peptide (OLP) libraries spanning each fs-peptide (Table S2) to prime and expand T cells from 15 randomly picked healthy donors (HD). After expansion, the cells were stimulated with OLP pools and fs-peptide-specific T cell responses were evaluated by measuring IFN-γ production using ELISPOT (Figure 6A). Each fs-peptide was able to elicit T cell responses in a subset of subjects tested. Furthermore, some subjects had reactive T cells against multiple fs-peptides (Figure 6B–D). Importantly, when combined, the fs-peptide-specific T cells were significantly enriched across the subject cohort (Figure 6D), in agreement with our prediction that the combination of fs-epitopes derived from all 9 peptides has the best representation of all possible pMHC interactions per population (Figure 2D). We confirmed the fs-peptide-specific T cell responses in the same HD cohort by intracellular staining (ICS). Responses to fs-peptides were observed primarily in CD8+ T cells, indicating strong priming to these neoantigens (Figure 6E–G). In total, a majority of HD responded to at least one fs-peptide: IFN-γ+ reactive CD8 T cell population increased at least 2-fold compared to background in 11 patients out of 14 tested. Importantly, the reactive T cells produced TNF-α, in addition to IFN-γ, suggesting fs-peptide-specific T cells are polyfunctional (Figures 6E and S7). Additionally, we synthesized control peptides (15-aa) for each fs-peptide using the wild type sequence surrounding the fs-mutation site origin. Responses by HD T cells to stimulation with the WT OLP pool were not higher than the background (Figure 6H), suggesting that the observed T cell responses were specific to fs-peptides.
Figure 6.

Shared fs-peptides predicted from UCEC MSI-H tumors elicit T cell responses. A. T cell immunogenicity assay used to evaluate antigen-specific T cell responses. PBMCs from healthy donors (HD) were expanded in vitro following stimulation with fs-peptide OLPs as shown in Figure S10. Expanded T cells were re-stimulated with either the peptide pool they were expanded with or the control peptide pool MOG. Representative IFN-γ ELISPOT images for B. HD13 or C. for selected responsive HD. D. Summary of ELISPOT data, 5×104 cells/well (n=14). Statistical significance for MOG vs OLPs was evaluated by Wilcoxon signed-rank test. E. Representative flow cytometry plots demonstrating gating strategy. Summary of flow data (n=15) for IFN-γ in F. CD8 and G. CD4 T cell subsets. Statistical significance for DMSO vs OLPs was evaluated by Wilcoxon signed-rank test. **p=0.0032 for SLC22A9 and **0.0031 for CEFT. H. Frequency of IFN-γ or TNF-α producing CD8+ T cells upon stimulation with WT peptide pool. I. PBMCs from HD13 were stimulated and expanded with OLP pools for SLC35F5, SLC22A9 or RNF43. Expanded cells were re-stimulated either with pooled OLPs or the individual peptides constituting each peptide pool (detailed in Figure S8) or MOG. Frequencies of IFN-γ or TNF-α producing CD8+ T cells were measured by ICS in duplicates and average values are shown. J. PBMCs from MSI-H patients (Pt 1 and 3 with UCEC, Pt 2 with COAD) were stimulated and expanded with fs-peptide OLPs. After expansion, each group of cells was re-stimulated with the corresponding OLP pool or MOG. Frequencies of IFN-γ producing CD8+ T cells were measured by ICS, in duplicates. Average values are shown. Statistical significance for MOG vs OLPs was evaluated by unpaired t test for each patient. Pt 1 : SETD1B*: p=0.0118; Pt 2: SLC22A9_N***: p=0.0003, TTK*: p=0.0172, RNF43**: p=0.0031; Pt 3: SLC35F5**: p=0.0064, SLC22A9_C***: p=0.0008, OR7E24_C*: p=0.0167, RNF43*: p=0.0177. For all assays, stimulation with DMSO or MOG were used as negative controls and CEFT and/or PMA/Ionomycin were used as positive control. The spot numbers and % IFN-γ or IFN-γ/TNF-α values were calculated by subtracting the values obtained after MOG or DMSO stimulation from the values after peptide stimulation and negative values were set to zero.
Next, we investigated whether the shared fs-peptides can give rise to multiple immunogenic epitopes as suggested by our computational predictions. We selected a donor, HD13, that displayed CD8+ T cell effector responses upon stimulation with multiple fs-peptide OLP pools, namely SLC35F5, SLC22A9_C and RNF43 (Figure 6F) and deconvoluted each OLP pool by re-stimulating cells initially expanded with pooled peptides with the individual peptides constituting each pool (Figure S7). The data indicate that fs-peptides encode multiple MHC-I-restricted epitopes (Figure 6I). We also investigated whether the fs-peptide-specific T cell responses that were observed in the HD cohort correlated with the predicted high affinity epitope load. Epitope load was assessed by first determining the class I alleles of each subject by sequence-based MHC-I genotyping and enumerating the number of predicted epitopes with high binding affinity from a given fs-peptide to each subject’s genotype. We found no significant correlation between the predicted epitope load per patient and the in vitro measured response rate (Figure S7). This observation may suggest that it is not quantity but rather quality of the predicted antigens which is responsible for the T cell response rate (Balachandran et al., 2017; Luksza et al., 2017). Further inspection of the predicted antigens may help to understand the intrinsic properties underlying their exceptional T cell recognition.
Finally, we investigated whether fs-peptide-specific T cell responses could be detected in MSI-H cancer patients. PBMCs from 3 patients, 2 with MSI-H UCEC and 1 with MSI-H COAD (Figure S7) were stimulated with fs-peptide OLP pools. Following T cell expansion in response to simulation with fs-peptides, we observed high frequencies of primarily fs-peptide-specific effector CD8+ T cell responses in all 3 MSI-H cancer patients that were monitored. (Figure 6J). To assess whether in vivo priming has occurred in MSI-H patients, we performed ex vivo T cell stimulation assays using PBMCs from 2 MSI-H patients, COAD and UCEC, and monitored IFN-γ formation by ELISPOT after 48 hours of stimulation with fs-peptide pools. Overall, we did not observe robust spontaneous responses against fs-peptides, except for SLC35F5 in Patient 3 (Figure S7). SLC35F5-specific responses were also observed in expanded cultures of T cells from Patient 3 (Figure 6J). These findings suggest either a lack of robust preexisting fs-peptide-specific T cell immunity in MSI-H patients or technical limitations in the detection of responses in non-expanded cells due to a low frequency of fs-peptide-specific T cells. Altogether, our data show that MSI-H patients have an increased frequency of high-quality T cell epitopes derived from shared fs-peptides, binding to a broad spectrum of MHC alleles, which are capable of inducing CD8+ T cell responses.
Discussion
In this study we evaluated MSI-H patients from TCGA for the presence of shared, immunogenic tumor-associated neoantigens. Our approach to detect neoantigens relies on two assumptions: (i) indel mutations occurring in frequently mutated microsatellite regions lead to identical fs-peptide extensions and (ii) these frequent, identical fs-peptide extensions encode poly-epitopes with broad MHC-I specificity. We confirmed the validity of our neoantigen selection approach by testing the immunogenicity of selected fs-peptides and finding selected peptides were highly immunogenic and generated strong CD8+ T cell responses in both healthy donors and MSI-H patients. Indeed, the ease with which we could prime CD8+ T cells from healthy donors (HD) suggests that these epitopes are particularly immunogenic. This observation is exceptional since studies have reported that the majority of predicted missense-derived neoantigens preferentially elicit CD4+ T cell responses, even when the vaccines were designed based on the predictions for MHC-I affinity (Ott et al., 2017; Keskin et al., 2018; Cimen Bozkus et al., 2019).
We found fs-peptide-specific T cells to primarily elicit CD8+ responses. This could be because the fs-peptides we tested were selected based on their predicted high-affinity binding to multiple MHC-I molecules. Alternatively, we characterized fs-peptide-specific T cell responses by priming HD T cells in vitro, where the dynamics of T cell priming and induction are likely to be significantly different than in the context of cancer, where immunosuppressive mechanisms are at play. Although in MSI-H patients fs-peptides again mainly induced CD8+ T cells, this observation is limited by sample size. Another limitation is that we have not directly evaluated the tumor cell killing capacity of fs-peptide-specific T cells due to the inaccessibility of autologous or MHC-I-matched tumor cell lines with confirmed expression of fs-mutations. Further studies will be required to fully determine any potential bias exhibited by T cell subsets against fs-peptides, the cytotoxicity of fs-peptide-specific T cells and the prevalence of preexisting memory T cells against fs-peptides in MSI-H cancer patients.
From a tumor evolution perspective, multiple tumor-intrinsic mechanisms to avoid immune responses against immunogenic fs-epitopes exist, including the upregulation of checkpoint molecules to evade the development of antitumor T cell responses (Le et al., 2015; Gatalica et al., 2016; Mlecnik et al., 2016; Mittica et al., 2017). The blockade of this mechanism has been proven to be effective in improving response rates in a range of MSI-H tumors in multiple clinical trials (Le et al., 2015, 2017). However, several other immune resistance mechanisms are described, including downregulation of MHC alleles and/or β-microglobulin expression; inactivation/loss of antigen processing and/or interferon-γ-response pathway genes; and disruption of immunogenic neoantigens by acquired mutations (Gao et al., 2016; Roh et al., 2017; Sharma et al., 2017). We examined this latter possibility with respect to mutational escape of shared poly-epitope fs-peptides. In certain cases, we observed mutations that were potentially disruptive to the predicted neoantigens (Figure S2).
Of note, shared fs-mutation load was not predictive for patients’ survival across all tested tumors, (Figure S6). Although shared fs-neoantigen load was marginally higher in responders to anti-PD-1 immunotherapy, the shared fs-neoantigens were present in the non-responsive group as well (Figure 5E). Many antigen-independent mechanisms might underlie the poor response rates in a subset of those patients and a potential combination of PD-1 blockade and a shared fs-neoantigen vaccine may therefore hold promise in improving outcomes of immunotherapy for non-responsive MSI-H patients. Another important observation is the high-occurrence of predicted shared fs-peptides in genomic samples of independently collected cohorts and datasets, including CPTAC, an immunotherapy cohort and cancer cell lines (Figures 3, 4, 5). Finding shared fs-deletions in RNAseq and fs-peptides in MS/MS samples suggests that fs-neoantigens are present both at transcriptional and protein levels. Similar to our discovery, a few published reports identified the same shared fs-deletions and characterized their potential biological role (Giannakis et al., 2014; Tu et al., 2019). The retained expression of fs-derived neoepitopes may be due to the fact that their RNA can exhibit a high rate of turnover and processivity within cancer cells that is otherwise not deleterious. Indeed, mRNAs that encode frameshift mutations may be rapidly degraded through the nonsense-mediated decay (NMD) pathway, which is accompanied by nascent peptide decay on the 80S ribosome (Frischmeyer et al., 2002; Conti and Izaurralde, 2005; Isken and Maquat, 2008; Schweingruber et al., 2013; Kurosaki, Popp and Maquat, 2019). While the expression of fs-genes may be downregulated at the RNA level, the translated product is destabilized and quickly processed by proteasome producing short, presentable peptides at a higher rate (Buchwald et al., 2010; Apcher et al., 2011).
Finally, we investigated the qualities of fs-mutations. As mutation derived neoantigens are highly similar to self-peptides, previous reports used similarity to immunogenic viral epitopes or dissimilarity from self to assess whether a distribution of neoantigens is predictive of outcome to checkpoint blockade immunotherapy or long-term survival (Balachandran et al., 2017; Luksza et al., 2017; Richman, Vonderheide and Rech, 2019; Zhang et al., 2019). To determine whether similar principles applied to fs-neoantigens, we investigated similarities between fs-neoantigens, viral epitopes and missense neoantigens. We found missense-derived neoantigens to be 3 times more similar to viral epitopes than fs-neoantigens. We attributed this to host-virus co-evolution and viral mimicry of host function. The fs-mutations are therefore even “further from self’ than viral antigens (Figure S5). Taken together, we conclude that frameshifts represent a unique and intrinsically different sequence space of high-quality antigens with a great potential for discovering immunogenic epitopes which can be targeted by immune therapies.
A recent study investigated the presence and sequences of fs-derived neoepitopes in TCGA and arrived at similar conclusions (Koster and Plasterk, 2019), and a few previous reports have also investigated the immunogenicity of unique fs-mutations but on a significantly smaller scale (Woerner et al., 2003; Schwitalle et al., 2008; Garbe, Maletzki and Linnebacher, 2011; Maletzki et al., 2013; Wagner, Mullins S and Linnebacher, 2018). Furthermore, the data presented here provides a set of preselected fs-mutations for developing targeted sequencing panels for diagnostic purposes. The usage of targeted sequencing panels for diagnostics have already proven essential for developing actionable treatments, particularly in the selection of targeted regimens. We believe the same paradigm will become useful for precision immunotherapies, with physicians being able to select the ideal individualized cancer vaccine formulations based on the results of targeted sequencing panels. Our work also revealed the possibility of designing common cancer vaccines in specific tumor subtypes with broad MHC-I specificity. By applying such tailored vaccines for MSI-H endometrial, colorectal and stomach carcinomas, one can potentially achieve immunological responses against existing neoplasms or develop preventive memory T cell responses in high-risk patient populations, like those with Lynch syndrome.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for the resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nina Bhardwaj (nina.bhardwaj@mssm.edu)
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
Source data for TCGA part (Figures 1–3, S1, S2) is available at GDC data commons and generated by the TCGA Research Network (TCGA, January 2018, https://www.cancer.gov/tcga). Data for CPTAC analysis part (Figures 3, S4, Table S2) is available at the Clinical Proteomic Tumor Analysis Consortium, (https://proteomics.cancer.gov/data-portal). Both datasets used by this study (prospective colon and endometrial cancer samples) are published elsewhere (Vasaikar et al., 2019; Dou et al., 2020). Source data for CCLE analysis part (Figure 4) is available at the Cancer Cell Line Encyclopedia, Broad Institute (https://portals.broadinstitute.org/ccle ) and published elsewhere (Barretina et al., 2012). Source data for MHC-I peptide elution analysis from HCT116 cell line (Figure 4) is available at Proteomics Identification Database (PRIDE) and published elsewhere (Bassani-Sternberg et al., 2015). Source data for viral versus tumor epitope comparisons (Figure S6) is available at Immune Epitope Database (IEDB) at NIAID. Source data for MSI-H immunotherapy cohort (NCT01876511) is available upon request from Timothy Chan and published elsewhere (Le et al., 2015; Mandal et al., 2019).
Custom computer code and pipelines are either described in the Method Detail section or available at GitHub: https://github.com/VladimirRoudko/shared_frameshift_neoantigen/. Alternatively, the code is available upon request by the first author (V.R).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human healthy donor PBMC samples were purchased from New York Blood Center (https://nybloodcenter.org). Human MSI-H cancer patients’ PBMCs were obtained under consent form linked to IRB-19–02392. Sex, gender and age of healthy donors’ samples is unavailable as it is an anonymous donation. Sex, gender and age of consented cancer patients are provided in Figure S7. Conditions of in vitro studies conducted with primary PBMC cultures are specified in Methods section. Conditions and maintenance of cancer cell lines (Figure 4) are described in Data S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using statistical tools available in PRISM8. Non-parametric Mann-Whitney two-tailed test was used to infer the statistical significance of indel allele expression in MSI-H cohorts (Figure 3), and shared fs-peptide enrichment in the immunotherapy cohort and CCLE dataset (Figures 4 and 5). Statistical significance of MS/MS identifications by Pepquery (Figures 3, 4 and S4) was set to the default approach (see Methods section for details). Standard T-test and Wilcoxon sign-ranked test were used to infer statistical significance for T cell responses (Figure 6). In all performed tests, significance was defined by p-value set to 0.05. Survival and hazard ratio analyses were performed using the survival package in R.
METHOD DETAIL
Computational analysis of TCGA
Tumor-associated antigens were predicted using somatic mutation datasets, called by the internal mutation pipelines of The Cancer Genome Atlas (TCGA 2018 version). Therefore, the results obtained in this paper are in part based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Called somatic missense and frameshift mutations by Mutect, Somatic Sniper, Varscan and Muse were combined together (union) per each patient. For somatic missense mutations, corresponding 17-amino acid residue-length normal peptides, surrounding a mutation site, were converted to tumor-specific peptides and used for MHC-I epitope prediction. In the case of frameshift mutations, the tumor specific peptide was called as follows: the major mRNA isoform corresponding to the frameshift mutation, translated starting with the “−8” aminoacid residue position from the mutation site until the stop codon within the new open reading frame as defined by the frameshift mutation. Predicted frameshift peptides were used for MHC-I epitope prediction by NetMHC v4.0 (Andreatta and Nielsen, 2016; Nielsen and Andreatta, 2016). A rank score threshold (< 2.0%) was used to filter the predicted binders. MHC allele types for >5000 patients from TCGA were from a previously published paper (Charoentong et al., 2017). Collected epitope data was analyzed using statistical packages Prism and R. To characterize mutation expression at the RNA level, hg19-aligned RNAseq bam files were downloaded from GDC (https://gdc.cancer.gov). Obtained .bam files were processed with samtools to extract RNAseq reads, covering 250 nt genomic loci around shared fs-mutation (samtools view -b -L {target.region.bed} {input.bam} > {output.bam}). To count indel events in extracted RNAseq bam files, we applied samtools mpileup (samtools mpileup -uf {reference.fasta} {input.bam} | bcftools view -l {target.region.bed} - | grep “INDEL” > {output.vcf}. Finally, the obtained data was processed with custom scripts and analyzed in PRISM8. To recall mutations with orthogonal approaches we applied bamreadcount. Sequencing read coverages for genomic loci of 46 shared frameshift mutations were downloaded from GDC using standard curl request. Obtained sequencing files were analyzed with bamreadcount (bam-readcount -q 10 -b 10 -d 100000 -l {target.region.bed} -f {reference.fasta} {input.bam} | grep “chr” > {output.file}. Readcounts were processed using custom python scripts to extract read coverages and qualities of targeted mutation sites. To assess the probability of fs-peptides being correctly translated, we deconvoluted the conditional probability shown in Figure S2F the following way: PccFM|cfm = 1 - [ (Pupm | cfm + PdPM | cfm) x 3/4 × 3/27 + (PdPM | cfm x 2/3 + PdFM | cfm x 1/3) x Sk=Σk=1n Cnk x (7/lenFM)k + (PuFM | cfm + PdFM | cfm) x 2/3 ] x ½, where lenFM – is the length of the frameshift.
Peptide comparison with virus epitope databases
The collection of viral MHC-I epitopes was downloaded from the IEDB database and preformatted for BLAST usage (makeblastdb -in iedb.fasta -parse_seqids -dbtype prot). Predicted frameshift and missense T-cell epitopes from MSI-H patients were compared with IEDB epitopes using blastp (blastp -db {iedb.fasta} -query {input.frameshift.fasta} -outfmt “6 qseqid sseqid pident ppos positive mismatch gapopen length qlen slen qstart qend sstart send qseq sseq evalue bitscore” -word_size 3 -gapopen 32767 -gapextend 32767 -evalue 1 - max_hsps_per_subject 1 -matrix BLOSUM62 -max_target_seqs 10000000 -out {output.file}. To compare predicted epitopes with the human proteome, we used gap aware last-align. First, we preformatted the human proteome (December 2016 version, Ensembl) using lastdb -p human.proteome human.proteome.fasta. Then we used lastal to compare epitopes with this database (lastal -f MAF -r 2 -q 1 -m 100000000 -a 100000 -d 15 -l 4 -k 1 -j1 -P 10 human.proteome {input.frameshifts.fasta} > {output.fasta}. Finally, the obtained results were processed with custom scripts (bash, python) and analyzed in PRISM8.
Computational genomic analysis of CCLE, CPTAC and MSI-H immunotherapy cohorts
CCLE: Somatic mutation data and normalized RNAseq expression values for genes with shared fs-mutations were obtained from https://portals.broadinstitute.org/ccle. CPTAC: Somatic mutation data and the MS-status of patients was obtained from published studies (Vasaikar et al., 2019; Dou et al., 2020). Immunotherapy cohort: Somatic mutation data, MS-status and matching normal and tumor WES datasets were generously provided by T. Chan from published studies (Le et al., 2015; Mandal et al., 2019). To match obtained WES datasets with patient clinical responses, we performed independent MHC-typing with Optitype (Schubert et al., 2014) and compared it to originally published data. The data was statistically analyzed in PRISM8.
Pepquery analysis of CPTAC and HCT116 MHC-I peptidome proteomic datasets
MS/MS datasets were downloaded from PRIDE (https://www.ebi.ac.uk/pride/archive/ ) or CPTAC endometrial and colon studies (https://proteomics.cancer.gov/data-portal ). Retrieved data was analyzed using the standalone version of Pepquery v.1.4.1 (Wen, Wang and Zhang, 2019; Wen et al., 2020) (http://www.pepquery.org). Briefly, raw MS/MS spectra was converted to MGF format using msconvert (http://proteowizard.sourceforge.net/tools.shtml), which was then supplied to stand-alone Pepquery (Wen, Wang and Zhang, 2019). In the case of analysis of the HCT116 MHC-I MS/MS dataset (Bassani-Sternberg et al., 2015), predicted fs peptides were computationally sliced into overlapping 8-, 9-, 10- and 11-mer epitopes. The produced list of epitopes was submitted to pepquery analysis (pepquery -o “pep” -varMod 75,117 -e 0 -t 1 -tol 10 -tolu ppm -itol 0.05 -prefix “pep” -ms “${input.ms.file}” -pep “${input.frameshift.peptide}” -db “${reference.proteome}” -n 1000 -m 1 -maxLength 11 -minLength 8 -um -hc FALSE -cpu 30 To analyze whole cell MS/MS spectra of CPTAC datasets, pepquery command line was configured accordingly to reflect the MS/MS experimental settings (java -Xmx10G -jar pepquery-1.4.1.jar -o “pep” -fixMod 6 -varMod 117 -tol 10 -tolu ppm -itol 0.05 -prefix “pep” -t 1 -ms “${input.ms.file}” -i “${input.ms.file}” -db “${reference.proteome}” -n 1000 -m 1 - maxLength 50 -minLength 5 -um -hc FALSE -cpu 10 was applied for “VU” files, and java - Xmx10G -jar pepquery-1.4.1.jar -o “pep” -fixMod 6,62,108 -varMod 117 -tol 10 -tolu ppm -itol 0.05 -prefix “pep” -t 1 -ms “${input.ms.file}” -i “${input.ms.file}” -db “${reference.proteome}” -n 1000 -m 1 -maxLength 50 -minLength 5 -um -hc FALSE -cpu 10 for “PNNL” files). Obtained results are listed in Table S2.
Indel recall using targeted genomic DNA PCR assay and Sanger sequencing
To validate CCLE-derived indel frequencies using an orthogonal experimental approach, we designed targeted high-fidelity PCR assay using a set of loci-specific primers (Data S1). Obtained PCRs were purified and subjected to Sanger sequencing (Genscript). Obtained sequences were aligned to human reference genome using Clustal Omega multiple sequence alignment tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). Finally, alignments were analyzed for the presence of indels in MS regions. Recalled CCLE indels (Table S3) were used for ROC analysis (http://www.rad.jhmi.edu/jeng/javarad/roc/JROCFITi.html).
Patient samples
The use of patient-derived specimens was approved by the Institutional Review Boards at Mount Sinai Hospital (IRB-19–02392) and all patients provided written informed consent before the initiation of any study procedures. All patients analyzed in this study were diagnosed with cancer and demonstrated loss of expression of one or more MMR proteins by immunohistochemistry. Patient blood was collected by the clinical personnel and MNCs were isolated by density gradient centrifugation using Ficoll-Paque™ Plus (GE Healthcare). Only freshly isolated patient PBMCs were used in immunogenicity assays. Therefore, assays were performed once for each patient. Healthy donor specimens were procured from New York Blood Center as a leukopak and MNCs were isolated by density gradient centrifugation using Ficoll-Paque™ Plus (GE Healthcare). PBMCs were cryopreserved in human serum containing 10% DMSO. HD PBMCs were used after thawing.
Peptide synthesis
Custom peptide libraries for WT and mutated peptides were chemically synthesized by GenScript (USA/China). Each peptide had >85% purity as determined by high performance liquid chromatography. MOG and CEFT peptide pools were commercially available at JPT Peptide Technologies (Germany). Each peptide was resuspended in DMSO and used at a final concentration of 1 μg/mL. Sequences of mutated peptides are shown in Table S2 and the WT sequences are as follows: for SLC35F5 GKLTATQVAKISFFF, for SEC31A QAVQSQGFINYCQKK, for SLC22A9 LEILKSTMKKELEAA, for TTK ESHNSSS SKTFEKKR and YSGGESHNSSSSKTF, for SETD1B MENSHPPHHHHQQPP, for OR7E24 MSYFPILFFFFLKRC, for RNF43 KSSLSARHPQRKRRG and for ASTE1 AEIFLPKGRSN SKKK.
T-cell immunogenicity assays
6×105 healthy donor PBMCs were cultured in X-VIVO15 media (LONZA) with cytokines promoting dendritic cell (DC) differentiation, GM-CSF (SANOFI, 1000 IU/mL), IL-4 (R&D Systems, 500 IU/mL) and Flt3L (R&D Systems, 50 ng/mL) overnight in U-bottom 96-well plates at 105 cells/well. After 24 hours, cells were stimulated with peptide pools (each peptide at 1 μg/mL) in the presence of adjuvants promoting DC maturation, LPS (Invivogen, 0.1 μg/mL), R848 (Invivogen, 10 μM) and IL-1β (R&D Systems 10 ng/mL), in X-VIVO15. Stimulation with DMSO (vehicle) and MOG pool (JPT, 1 μg/mL) were used as negative controls and CEFT pool (JPT, 1 μg/mL) were used as positive controls. Next day, cells were fed with IL-2 (R&D Systems, 10 IU/mL) and IL-7 (R&D Systems, 10 ng/mL) in RPMI media (Gibco) containing 10% human serum. Cells were fed every 2–3 days. IL-2 and IL-7 were not added at the last feeding. After 10 days of culture, cells were harvested and re-stimulated with peptides (1 μg/mL) in the presence of anti-CD28 (BD Biosciences, 0.5 mg/mL) and anti-CD49d (BD Biosciences, 0.5 mg/mL) antibodies. Where indicated, cells were stimulated with PMA (Sigma-Aldrich, 50 ng/mL) and ionomycin (Sigma-Aldrich, 1 μg/mL), as positive control. IFN-γ formation was measured by flow cytometry or ELISPOT. For flow cytometry, 1 hour after re-stimulation with peptides, cells were added BD GolgiStop™, containing monensin and BD GolgiPlug™, containing brefeldin A according to manufacturer’s suggestion. IFN-γ production was measured 8–12-hours after the addition of protein transport inhibitors by intracellular staining using BD Cytofix/Cytoperm™ reagents according to manufacturer’s protocol. A combination of the following antibodies was used: for surface staining; CD3 (Clone: OKT3 or SK7, FITC), CD4 (Clone: RPA-T4, BV785 or PerCP-Cy5.5) and CD8a (Clone: RPA-T8, APC) and for intracellular staining IFN-γ (Clone: B27, PE), TNF-α (Clone: Mab11, PE/Cy7) and IL-2 (Clone: MQ1–17H12, PerCP-Cy5.5). All antibodies were purchased via BioLegend. LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit by Thermo Fischer Scientific was used for live and dead cell discrimination. Data was acquired using the BD Fortessa or Canto and the data was analyzed on FlowJo V10 (TreeStar). For ELISPOT analysis, cells were stimulated in plates with mixed cellular ester membrane that were coated with anti-IFN-γ antibody (Mabtech, Clone 1-D1k, 4 μg/mL) and blocked by incubating with 10% human serum containing media at 37°C for at least 1 h prior to addition of cells. Cells were seeded in duplicates at either 5×104 or 105 per well for analysis of expanded cells and at 2.5×105 cell per well for ex vivo analysis and stimulated as detailed above. Plates were processed for IFN-γ detection after 48-hours of culture. Plates were first incubated with biotinylated anti-IFN-γ antibody (clone 7-B6–1 by Mabtech, used at 0.2 μg/mL) for 2 h at 37°C, then 1 h at room temperature with streptavidin-AP conjugate (Roche, used at 0.75 U/mL) and lastly with the SigmaFast BCIP/NBT substrate for 15 minutes at room temperature. Plates were washed 6x with PBS containing 0.05% Tween-20 and 3x with water in between each step. Plates were scanned and analyzed by ImmunoSpot software.
Supplementary Material
Figure S1. Mutational analysis of TCGA cancers and cancer cell lines from the CCLE, Related to Figure 1. A. Mutational load (insertion, deletion, somatic variant count or missense mutation) per patient across different tumor types in TCGA as quantified from WES. COAD, STAD and UCEC tumor types are shown in red. B. Comparison of frequencies of shared somatic indels and missense point mutations in human cancer cell lines from CCLE. LEFT – number of cancer cell lines sharing the same fs-mutation or missense point mutation from all CCLE (fs-all and ms-all, respectively) or from stomach, colon and endometrium cell lines (fs-S/C/E and ms-S/C/E respectively). Statistical significance determined by non-parametric Mann-Whitney, one-tailed test. RIGHT – absolute number of shared mutations from all CCLE (fs-all and ms-all respectively) or stomach, colon and endometrium cell lines (fs-S/C/E and ms-S/C/E respectively).
Figure S2. Properties of shared frameshift peptides, related to Figure 2. A. Distribution of fs-peptide frequency per 10% somatic score bins. Somatic score is defined as a ratio of the number of times each frameshift mutation has passed the filters of somatic mutation callers to the total number that each frameshift mutation has been called. Black lines indicate the mean frequency. B. Distribution of fs-peptide length across all predicted fs-peptides in MSI-H tumors. C. Combined criteria applied to select shared fs-mutation events in MSI-H UCEC, COAD and STAD tumors. Selection function is a multiplication of somatic score, number of pMHC interactions (pMHC) and average fs-epitope frequency (AEF). Fs-mutations shared by at least 20% of patients are ranked according to the selection function. Cutoff value is shown by the red line. D. Estimation of “translatability” and correctness of 9 shared fs-peptides in MSI-H UCEC. In the reduced approximation, any upstream mutation (upstream fs-mutation uFM or upstream point mutation uPM) may alter the fs-peptide if uFM appears somewhere between a start codon and a apredicted shared fs-mutation or if uPM leads to an abortive stop codon; alternatively, any downstream mutation (downstream fs-mutation dFM or downstream point mutation dPM) may alter the shared fs-peptide if dFM occurs between the shared mutation and a novel stop codon, defined by the frame of fs-peptide or if dPM occurs within the predicted T cell epitope or leads to an abortive stop codon. E. Quantification of detrimental events around shared fs-mutations. F. Probabilistic function to estimate conditional probability of fs-peptide being correctly translated given that this fs-mutation has occurred; table showing estimated posterior probabilities for each nine shared fs-peptide. G. Allele frequencies of 46 shared deletion events in MSI-H and MSS TCGA samples determined by bam-read count analysis workflow. Mutations are classified as one-, two-, three- or four-nucleotide deletion events. Majority of shared frameshifts are one-nucleotide deletions, with statistically significant higher allele frequency in tumor samples. H. Shared tumor allele frequency in MS locus of RNF43 gene. T – Tumor; TN – Tissue Normal; BN – Blood Normal. MSIH and MSS stands for MS unstable and stable tumors respectively.
Figure S3. Recall of 46 shared fs-mutations by bamreadcount analysis pipeline, related to Figures 2 and 3. A. Heatmap of 46 shared fs-mutation allele frequencies in MSI-H tumor (LEFT) and normal (RIGHT) WES samples from TCGA. B. Box-plot representation of allele frequencies per each fs-mutation in MSI-H UCEC and COAD/STAD tumor and normal samples. C. Classification of shared fs-mutations by allele frequency bins, as defined by conventional thresholds. Majority of shared fs-mutations are subclonal in MSI-H tumors (VAF < 0.25), however nearly every patient has at least one shared fs-mutation with intermediate-clonal range allele frequency.
Figure S4. Genomic and proteomic detection of shared fs-mutations in CPTAC colon and endometrial cancers, related to Figure 3. A. Heatmap of shared fs-mutations identified in the CPTAC MSI-H UCEC cohort. B. Distribution of population frequencies of all and shared fs-mutations in COAD and UCEC CPTAC cohorts. C. Representative MS/MS spectra of tryptic peptides derived from predicted shared fs-peptides identified by Pepquery analysis in whole cell MS proteomic datasets in CPTAC.
Figure S5. Comparison of missense epitopes and fs-epitopes in MSI-H patients with the human proteome and annotated viral epitopes (IEDB), related to Figure 3. A. Quantification of epitopes per missense or frameshift mutation. On average, frameshift mutations generate 4 epitopes, while missense about 2. B. Mapping missense and fs-epitopes back to the human proteome (ensemble, 2016). “Epitope load” indicates the total amount of predicted neoantigens from missense and frameshift mutations. “Similar match” shows the number of epitopes 8 amino acids in length successfully aligned to the human proteome, allowing 1 mismatch. “Perfect match” indicates the number of 9 amino acid long epitopes perfectly matched to the human proteome with no mismatches. C. BLAST comparison of missense epitopes and fs-epitopes with viral epitopes from IEDB, allowing different number of mismatches (marked as “miss” on the x axis). Missense-derived epitopes are more similar to viral epitopes then frameshift-derived on average.
Figure S6. Survival analysis and other confounding parameter distributions of MSI-H patients with high and low shared frameshift load, related to Figure 5. A. Kaplan-Meier plot of survival analysis and Cox’s proportional hazards model of MSI-H UCEC, COAD and STAD patients, classified by shared fs-epitope load (HIGH vs LOW). B. LEFT - distribution of patients’ age in all, shared fs-epitope high and shared fs-epitope low MSI-H UCEC, COAD and STAD cohorts. RIGHT – patients’ frequency (y axis) across tumor stages (x axis) in the same cohorts.
Figure S7. Fs-peptide-specific T cells T cell immunity, related to Figure 6. PBMCs from healthy donors (HD) were expanded in vitro following stimulation with fs-peptide OLPs. DMSO was used as vehicle control. A. Representative flow cytometry overlay plots for IFN-γ (y axis) and TNF-α (x axis) production by CD8+ T cells. Cytokine production upon OLP or DMSO stimulation is shown in red or blue, respectively. B. Frequencies of CD8+ T cells producing IFN-γ or TNF-α in response to each fs-peptide OLP pool were plotted for each subject. Green denotes HD producing only TNF-α, blue denotes HD producing only IFN-γ and yellow denotes HD producing both IFN-γ and TNF-α. % IFN-γ values were calculated by subtracting the values obtained after DMSO stimulation from the values after OLP pool stimulation and negative values were set to zero. C. Comparison of predicted total epitope load and experimental response rate. Summary of log2 transformed data for IFN-γ /TNF-α response by CD8+ T cells against fs-peptides and the quantification (log2 transformed) of pMHC interactions for each subject per fs-peptide. Interactions were counted when IC50<500 nM or percentile rank<2. Note, T cell response rates do not correlate with pMHC counts (Pearson correlation coefficient r ~ 0.181). D. Demographics and clinical characteristics of MSI-H patients. Patient data depicted above corresponds to measurements at the time samples were obtained and experiments were run. F: female, NED: no evidence of cancer. Reference values for the measurements indicated in the chart are as follows: HGB: 12-16, HCT: 34-47, WBC: 4.5-11, neutrophil: 40-78, lymphocyte: 15-50, monocyte 2-11. E. Spontaneous T cell immunity against fs-peptides in MSI-H cancer patients. PBMCs from MSI-H patients (Pt 2 with COAD and Pt 3 with UCEC) were seeded at 2.5×105 cells per well and stimulated ex vivo with fs-peptide OLP pools, CEFT pool or the control peptide pool MOG. Stimulation with PMA and ionomycin was performed as positive control. IFN-γ formation was measured by ELISPOT after 48 hours of culture. The spot numbers were calculated by subtracting the number of spots formed after MOG stimulation from the number of spots formed after OLP pool stimulation and negative values were set to zero. Statistical significance was calculated by comparing MOG vs the corresponding test group via t test.
Table S1. Aggregation of indel calling in TCGA COAD, STAD and UCEC samples by TCGA calling pipelines (Mutect, Varscan, Muse, Somatic Sniper) and orthogonal validation with bam-readcount approach. Related to Figures 1 and 2.
Table S2. Frameshift neoantigen prediction using NetMHC.v.4.0. Patient frequencies of fs-mutations, fs-peptides and neoantigens in TCGA cohorts. Selection of 46 shared fs-peptides. Identification of shared fs-peptides in whole tumor MS/MS datasets by Pepquery. Analysis of MHC-I elutions of MSI-H cancer cell lines using Pepquery approach. Overlapping peptide design to test immunogenicity of fs-peptides in vitro. Related to Figures 2, 3 and 6.
Table S3. Analysis of shared fs-mutations in Cancer Cell Line Encyclopedia (CCLE) database. Related to Figure 4.
Table S4. Analysis of shared events in MSI-H immunotherapy cohort generously provided by T. Chan from published studies (Le et al., 2015; Mandal et al., 2019). Related to Figure 5.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| a-CD4, BV785 or PerCP-Cy5.5 | BioLegend | Clone: RPA-T8 |
| a-CD8a, APC | BioLegend | Clone: RPA-T4 |
| IFN-γ, PE | BioLegend | Clone: B27 |
| TNF-α, PE/Cy7 | BioLegend | Clone: Mab11 |
| IL-2, PerCP-Cy5.5 | BioLegend | Clone: MQ1–17H12 |
| a-IFN-γ, ELISPOT | Mabtech | Clone 1-D1k |
| a-IFN-γ, biotinilated | Mabtech | clone 7-B6–1 |
| streptavidin-AP conjugate | Sigma Aldrich | 11089161001 |
| a-CD28 | BD Biosciences | clone CD28.2, 556620 |
| a-CD49d | BD Biosciences | clone L25, 340976 |
| a-CD3, FITC | BioLegend | Clone: OKT3 or SK7 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Thermo Fischer Scientific | NA |
| Biological Samples | ||
| Peripheral Blood Mononuclear Cells (PBMC) | Human healthy donor provided by New York Blood Center; | NA |
| Peripheral Blood Mononuclear Cells (PBMC) | Cancer patients, under IRB-19–02392 | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| custom peptide libraries provided in the Figure S10 | Genscript, https://www.genscript.com | NA |
| MOG peptide pool | JPT Peptide Technologies, https://www.jpt.com | PM-MOG |
| CEFT peptide pool | JPT Peptide Technologies, https://www.jpt.com | PM-CEFT |
| GM-CSF | SANOFI | NA |
| IL4 | R&D Systems | 204-IL-010 |
| Flt3L | R&D Systems | 308-FKE-010 |
| LPS | Invivogen | tlrl-eblps |
| R848 | Invivogen | tlrl-r848 |
| IL-1β | R&D Systems | 201-LB-005 |
| IL-2 | R&D Systems | 202-IL-010 |
| IL-7 | R&D Systems | 207-IL-005 |
| PMA | Sigma-Aldrich | P1585 |
| Ionomycin | Sigma-Aldrich | I3909 |
| Deposited Data | ||
| The Cancer Genome Atlas, TCGA | Gemonic Data Commons at National Cancer, TCGA version by January 2018 | NA |
| Cancer Cell Line Encyclopedia, CCLE | Broad Institute, https://portals.broadinstitute.org/ccle doi: 10.1038/nature11003 | NA |
| Clinical Proteomic Tumor Analysis Consortium (CPTAC) | National Cancer Institute, https://proteomics.cancer.gov/data-portal Two analysed studies are published: doi: 10.1016/j.cell.2019.03.030 doi: 10.1016/j.cell.2020.01.026 |
NA |
| Proteomics Identifications Database (PRIDE) | EMBL-EBI, https://www.ebi.ac.uk/pride/ Results used are published: doi: 10.1016/j.cell.2020.01.026 | NA |
| Immune Epitope Database (IEDB) | National Institute of Allergy and Infectious Diseases, https://www.iedb.org | NA |
| MSI-H immunotherapy cohort | T.Chan lab Results published: doi: 10.1126/science.aau0447 doi: 10.1056/NEJMoa1500596 |
NA |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247 |
| HeclB | ATCC | HTB-113 |
| LOVO | ATCC | CCL-229 |
| Hec59 | AddexBio Technologies | C0026001 |
| Oligonucleotides | ||
| oligonucleotide primers for target genomic loci amplification | Integrated DNA Technologies, https://www.idtdna.com/pages. full list is provided in the Data S1 | NA |
| Software and Algorithms | ||
| PRISM 8 | Graphpad, https://www.graphpad.com/scientific-software/prism/ | NA |
| FlowJo v. 10.6.2 | BD, FlowJo, https://www.flowjo.com/ | NA |
| ImmunoSpot | ImmunoSpot, http://www.immunospot.com/ImmunoSpot-analyzers- | NA |
| software | ||
| R v.3.6.0 | The R Project for Statistical Computing, https://www.r-project.org | NA |
| samtools v.1.7 | http://www.htslib.org | NA |
| blast v.2.6.0 | https://www.ncbi.nlm.nih.gov/books/NBK279671/ | NA |
| last-align, last-1061 version |
http://last.cbrc.jp doi: 10.1101/gr.113985.110 |
NA |
| NetMHC v.4.0 | DTU Health Tech, http://www.cbs.dtu.dk/services/NetMHC/ doi: 10.1093/bioinformatics/btv639 | NA |
| Optitype | https://github.com/FRED-2/OptiType | NA |
| doi: 10.1093/bioinformatics/btu548 | ||
| bam-readcount, v.0.8.0 | https://github.com/genome/bam-readcount | NA |
| pepquery, v.1.4.1 | http://www.pepquery.org doi: 10.1101/gr.235028.118 | NA |
| msconvert, proteinwizard package, v.3.0 | http://proteowizard.sourceforge.net/tools.shtml doi: 10.1093/bioinformatics/btn323 | NA |
| frameshift neoantigen caller | https://github.com/VladimirRoudko/shared frameshift neoantigen | NA |
Highlights.
MSI-H tumors are enriched in recurrent shared immunogenic frameshifts
Shared frameshifts are expressed on RNA and protein levels
Shared frameshifts produce exceptional T cell responses
Tumors that have high levels of mutations within microsatellites (MSI-H) demonstrate specific frameshifts that are then expressed at the RNA and protein levels across endometrial, colorectal and stomach cancers. Epitopes from these frameshifts yield neoantigens that are distinct from self and viral antigens and elicit T cell responses.
Acknowledgements
N.B. receives research funds from the National Institutes of Health grants R01CA201189, R01CA180913 and R01AI08184, the Tisch Cancer Institute Cancer Center Support Grant, the Department of Defense, the Parker Institute for Cancer Immunotherapy, the Melanoma Research Alliance, the Leukemia and Lymphoma Society, the Pershing Square Sohn Foundation and NYSTEM; B.G. receives research funds from the National Institutes of Health grants R01AI081848, U01CA224175, R01CA240924 and U01CA228963; the Memorial Sloan Kettering Cancer Center Grant; a collaboration by Stand Up To Cancer, a program of the Entertainment Industry Foundation, the Society for Immunotherapy of Cancer, and the Lustgarten Foundation; the V Foundation for Cancer Research; and the Pershing Square Sohn Foundation. B.G. and V.R. were supported by The Pershing Square Sohn Prize—Mark Foundation Fellowship supported by funding from The Mark Foundation for Cancer Research. C.C.B. receives research funds from the Parker Institute for Cancer Immunotherapy. The authors would like to thank the members of the Bhardwaj and Greenbaum laboratories for many discussions.
Footnotes
Declaration of Interests
N.B. is an extramural member of the Parker Institute for Cancer Immunotherapy (PICI), receives research funds from Genentech, Oncovir, Regeneron and Dragonfly Therapeutics, and is on the advisory boards of Novartis, Roche, Avidea, Boehringer Ingelheim, Rome Therapeutics, BreakBio, Carisma Therapeutics, Roswell Park, and the Cancer Research Institute. C.B. is a PICI Bridge scholar. B.G. has received honoraria for speaking engagements from Merck, Bristol–Meyers Squibb, and Chugai Pharmaceuticals; has received research funding from Bristol-Meyers Squibb; and has been a compensated consultant for PMV Pharma and Rome Therapeutics of which he is a cofounder.
V.R., C.C.B., N.B., B.G., S.B., and T.O have a pending provisional patent application (No. 62/813,829, filled on March 5, 2019). The other authors have not declared any competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed D et al. (2013) ‘Epigenetic and genetic features of 24 colon cancer cell lines.’, Oncogenesis, 2(9), pp. 1–9. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM et al. (2015) ‘Genomic correlates of response to CTLA-4 blockade in metastatic melanoma’, Science, 350(6257), pp. 207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M and Nielsen M (2016) ‘Gapped sequence alignment using artificial neural networks: application to the MHC class I system’, Bioinformatics, 32(4), pp. 511–517. Available at: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apcher S et al. (2011) ‘Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation.’, Proceedings of the National Academy of Sciences of the United States of America, 108(28), pp. 11572–7. doi: 10.1073/pnas.1104104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran VP et al. (2017) ‘Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer’, Nature. Nature Publishing Group, 551(7681), pp. S12–S16. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J et al. (2012) ‘The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity’, Nature, 483, pp. 4–11. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M et al. (2015) ‘Mass spectrometry of human leukocyte antigen class i peptidomes reveals strong effects of protein abundance and turnover on antigen presentation’, Molecular and Cellular Proteomics, 14(3), pp. 658–673. doi: 10.1074/mcp.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald G et al. (2010) ‘Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex.’, Proceedings of the National Academy of Sciences of the United States of America, 107(22), pp. 10050–10055. doi: 10.1073/pnas.1000993107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M, Blattmann P and Aebersold R (2018) ‘Systematic characterization of pan-cancer mutation clusters’, Molecular systems biology, 14(e7974), pp. 1–19. doi: 10.15252/msb.20177974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carethers JM et al. (2015) ‘Advances in Colorectal Cancer Lynch syndrome and Lynch syndrome mimics : The growing complex landscape of hereditary colon cancer’, World Journal of Gastroenterology, 21(31), pp. 9253–9261. doi: 10.3748/wjg.v21.i31.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoentong P et al. (2017) ‘Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade: Cell Reports’, Cell Reports. ElsevierCompany., 18(1), pp. 248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Chung DC and Rustgi AK (2019) ‘The Hereditary Nonpolyposis Colorectal Cancer Syndrome: Genetics and Clinical Implications’, Annals of Internal Medicine, 138(7). [DOI] [PubMed] [Google Scholar]
- Cimen Bozkus C et al. (2019) ‘Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms’, Cancer Discovery, 9(9), pp. 1192–1207. doi: 10.1158/2159-8290.cd-18-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SA, Pritchard CC and Jarvik GP (2019) ‘Lynch Syndrome : From Screening to Diagnosis to Treatment in the Era of Modern Molecular Oncology’, Annual Revew of Genomics and Human Genetics, 20(4), pp. 1–15. [DOI] [PubMed] [Google Scholar]
- Conti E and Izaurralde E (2005) ‘Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species’, Current Opinion in Cell Biology, 17(3), pp. 316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Cortes-Ciriano I et al. (2017) ‘A molecular portrait of microsatellite instability across multiple cancers’, Nature Communications. Nature Publishing Group, 8, pp. 1–12. doi: 10.1038/ncomms15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JM et al. (1998) ‘Hypermethylation of the hMLHl Promoter in Colon Cancer with Microsatellite Instability’, (507). [PubMed] [Google Scholar]
- Diaz-Padilla I et al. (2013) ‘Mismatch repair status and clinical outcome in endometrial cancer: A systematic review and meta-analysis’, Critical Reviews in Oncology/Hematology. Elsevier Ireland Ltd, 88(1), pp. 154–167. doi: 10.1016/j.critrevonc.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Dou Y et al. (2020) ‘Proteogenomic Characterization of Endometrial Carcinoma.’, Cell, pp. 729–748. doi: 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley JC et al. (2016) ‘Microsatellite instability as a biomarker for PD-1 blockade’, Clinical Cancer Research, 22(4), pp. 813–820. doi: 10.1158/1078-0432.CCR-15-1678. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA et al. (2002) ‘An mRNA surveillance mechanism that eliminates transcripts lacking termination codons.’, Science (New York, N.Y.), 295(5563), pp. 2258–61. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Gao J et al. (2016) ‘Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy’, Cell. Elsevier, 167(2), pp. 397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe Y, Maletzki C and Linnebacher M (2011) ‘An MSI Tumor Specific Frameshift Mutation in a Coding Microsatellite of MSH3 Encodes for HLA-A0201-Restricted CD8+ Cytotoxic T Cell Epitopes’, PLoS ONE, 6(11), pp. 2–9. doi: 10.1371/journal.pone.0026517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatalica Z et al. (2016) ‘High microsatellite instability ( MSI-H ) colorectal carcinoma : a brief review of predictive biomarkers in the era of personalized medicine’, Familial Cancer. Springer Netherlands, 15(3), pp. 405–412. doi: 10.1007/s10689-016-9884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M et al. (2017) ‘Universal Patterns of Selection in Cancer and Somatic Tissues’, Cell, 171, pp. 1029–1041. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M et al. (2019) ‘Next-generation characterization of the Cancer Cell Line Encyclopedia’, Nature. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M et al. (2014) ‘RNF43 is frequently mutated in colorectal and endometrial cancers’, Nature Genetics. Nature Publishing Group, 46(12), pp. 1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo J, Martincorena I and Koonin EV (2018) ‘Cancer-mutation network and the number and specificity of driver mutations’, PNAS, 115(26), pp. 6010–6019. doi: 10.1073/pnas.1803155115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O and Maquat LE (2008) ‘The multiple lives of NMD factors: balancing roles in gene and genome regulation.’, Nature reviews. Genetics, 9(9), pp. 699–712. doi: 10.1038/nrg2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin DB et al. (2018) ‘Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial’, Nature. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Laird PW and Park PJ (2013) ‘The landscape of microsatellite instability in colorectal and endometrial cancer genomes’, Cell. Elsevier Inc., 155(4), pp. 858–868. doi: 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J and Plasterk RHA (2019) ‘A library of Neo Open Reading Frame peptides ( NOPs ) as a sustainable resource of common neoantigens in up to 50 % of cancer patients’, Scientific Reports. Springer US, (October 2018), pp. 1–8. doi: 10.1038/s41598-019-42729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Popp MW and Maquat LE (2019) ‘Quality and quantity control of gene expression by nonsense-mediated mRNA decay’, Nature Reviews Molecular Cell Biology. Springer US. doi: 10.1038/s41580-019-0126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT et al. (2015) ‘PD-1 Blockade in Tumors with Mismatch-Repair Deficiency’, New England Journal of Medicine, 372(26), pp. 2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT et al. (2017) ‘Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade’, Science, 413(July), pp. 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksza M et al. (2017) ‘A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy’, Nature. Nature Publishing Group, 551(7681), pp. 517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletzki C et al. (2013) ‘Frameshift-derived neoantigens constitute immunotherapeutic targets for patients with microsatellite-instable haematological malignancies: Frameshift peptides for treating MSI+ blood cancers’, European Journal of Cancer. Elsevier Ltd, 49(11), pp. 2587–2595. doi: 10.1016/j.ejca.2013.02.035. [DOI] [PubMed] [Google Scholar]
- Mandal R et al. (2019) ‘Genetic diversity of tumors with mismatch repair deficiency influences anti - PD-1 immunotherapy response’, Science, 491(May), pp. 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty R et al. (2017) ‘MHC-I Genotype Restricts the Oncogenic Mutational Landscape’, Cell. Elsevier Inc., 0(0), pp. 1–12. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcgranahan N et al. (2016) ‘Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade’, Science, 351(6280), pp. 1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittica G et al. (2017) ‘Checkpoint inhibitors in endometrial cancer : preclinical rationale and clinical activity’, Oncotarget, 8(52), pp. 90532–90544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B et al. (2016) ‘Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability’, Immunity, 44(3), pp. 698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- Nangalia J et al. (2013) ‘Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2’, New England Journal of Medicine, 369(25), pp. 2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M and Andreatta M (2016) ‘NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets’, Genome Medicine. Genome Medicine, 8(1), pp. 1–9. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA et al. (2017) ‘An immunogenic personal neoantigen vaccine for patients with melanoma’, Nature. Nature Publishing Group. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman LP, Vonderheide RH and Rech AJ (2019) ‘Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade’, Cell Systems. Elsevier Inc., 9(4), pp. 375–382.e4. doi: 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA et al. (2015) ‘Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer’, Science, 348(6230), pp. 124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh W et al. (2017) ‘Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance’, Science Translational Medicine, 9(379). doi: 10.1126/scitranslmed.aah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM et al. (2019) ‘Tumor mutational load predicts survival after immunotherapy across multiple cancer types’, Nature Genetics. Springer US, 51(2), pp. 202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert B et al. (2014) ‘OptiType : precision HLA typing from next-generation sequencing data’, Bioinformatics, 30(23), pp. 3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN and Schreiber RD (2015) ‘Neoantigens in cancer immunotherapy’, Science, 348(6230), pp. 69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Schweingruber C et al. (2013) ‘Nonsense-mediated mRNA decay - mechanisms of substrate mRNA recognition and degradation in mammalian cells.’, Biochimica et biophysica acta, 1829(6–7), pp. 612–23. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Schwitalle Y et al. (2008) ‘Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers’, Gastroenterology, 134(4), pp. 988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Sharma P et al. (2017) ‘Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy’, Cell. Elsevier Inc., 168(4), pp. 707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A et al. (2014) ‘Genetic basis for clinical response to CTLA-4 blockade in melanoma’, New England Journal of Medicine, 371(23), pp. 2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J et al. (2019) ‘The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis’, Scientific Reports, 9(1), pp. 1–12. doi: 10.1038/s41598-019-54931-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S et al. (2017) ‘Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis’, The Lancet Oncology. The Author(s). Published by Elsevier Ltd. This is an Open Access article under the CC BY 4.0 license, 18(8), pp. 1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- Vasaikar S et al. (2019) ‘Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities’, Cell, 177(4), pp. 1035–1049.e19. doi: 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen H et al. (1996) ‘Cancer Risk in Families With Hereditary Nonpolyposis Colorectal Cancer Diagnosed by Mutation Analysis’, Gastroenterology, 110, pp. 1020–1027. [DOI] [PubMed] [Google Scholar]
- Veigl M et al. (1998) ‘Biallelic inactivation of hMLH 1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers’, PNAS, 95(July), pp. 8698–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Mullins S C and Linnebacher M (2018) ‘Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens’, World Journal of Gastroenterology, 24(48), pp. 5418–5432. doi: 10.1007/s12519-013-0433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A et al. (2009) ‘Genetic prognostic and predictive markers in colorectal cancer’, Nature Reviews Cancer, 9(7), pp. 489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Wen B et al. (2020) ‘Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis’, Nature Communications. Springer US, 11(1), pp. 1–14. doi: 10.1038/s41467-020-15456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wang X and Zhang B (2019) ‘PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations’, Genome Research, 29(3), pp. 485–493. doi: 10.1101/gr.235028.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis JA et al. (2019) ‘Immune Activation in Mismatch Repair-Deficient Carcinogenesis: More Than Just Mutational Rate’, Clinical Cancer Research, (24), pp. 11–18. doi: 10.1158/1078-0432.ccr-18-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner SM et al. (2003) ‘Pathogenesis of DNA repair-deficient cancers: A statistical meta-analysis of putative Real Common Target genes’, Oncogene, 22(15), pp. 2226–2235. doi: 10.1038/sj.onc.1206421. [DOI] [PubMed] [Google Scholar]
- Zhang B et al. (2014) ‘Proteogenomic characterization of human colon and rectal cancer’, Nature, 513(7518), pp. 382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J et al. (2019) ‘The combination of neoantigen quality and T lymphocyte infiltrates identifies glioblastomas with the longest survival’, Communications Biology, pp. 1–10. doi: 10.1038/s42003-019-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zighelboim I et al. (2007) ‘Microsatellite instability and epigenetic inactivation of MLH1 and outcome of patients with endometrial carcinomas of the endometrioid type’, Journal of Clinical Oncology, 25(15), pp. 2042–2048. doi: 10.1200/JC0.2006.08.2107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mutational analysis of TCGA cancers and cancer cell lines from the CCLE, Related to Figure 1. A. Mutational load (insertion, deletion, somatic variant count or missense mutation) per patient across different tumor types in TCGA as quantified from WES. COAD, STAD and UCEC tumor types are shown in red. B. Comparison of frequencies of shared somatic indels and missense point mutations in human cancer cell lines from CCLE. LEFT – number of cancer cell lines sharing the same fs-mutation or missense point mutation from all CCLE (fs-all and ms-all, respectively) or from stomach, colon and endometrium cell lines (fs-S/C/E and ms-S/C/E respectively). Statistical significance determined by non-parametric Mann-Whitney, one-tailed test. RIGHT – absolute number of shared mutations from all CCLE (fs-all and ms-all respectively) or stomach, colon and endometrium cell lines (fs-S/C/E and ms-S/C/E respectively).
Figure S2. Properties of shared frameshift peptides, related to Figure 2. A. Distribution of fs-peptide frequency per 10% somatic score bins. Somatic score is defined as a ratio of the number of times each frameshift mutation has passed the filters of somatic mutation callers to the total number that each frameshift mutation has been called. Black lines indicate the mean frequency. B. Distribution of fs-peptide length across all predicted fs-peptides in MSI-H tumors. C. Combined criteria applied to select shared fs-mutation events in MSI-H UCEC, COAD and STAD tumors. Selection function is a multiplication of somatic score, number of pMHC interactions (pMHC) and average fs-epitope frequency (AEF). Fs-mutations shared by at least 20% of patients are ranked according to the selection function. Cutoff value is shown by the red line. D. Estimation of “translatability” and correctness of 9 shared fs-peptides in MSI-H UCEC. In the reduced approximation, any upstream mutation (upstream fs-mutation uFM or upstream point mutation uPM) may alter the fs-peptide if uFM appears somewhere between a start codon and a apredicted shared fs-mutation or if uPM leads to an abortive stop codon; alternatively, any downstream mutation (downstream fs-mutation dFM or downstream point mutation dPM) may alter the shared fs-peptide if dFM occurs between the shared mutation and a novel stop codon, defined by the frame of fs-peptide or if dPM occurs within the predicted T cell epitope or leads to an abortive stop codon. E. Quantification of detrimental events around shared fs-mutations. F. Probabilistic function to estimate conditional probability of fs-peptide being correctly translated given that this fs-mutation has occurred; table showing estimated posterior probabilities for each nine shared fs-peptide. G. Allele frequencies of 46 shared deletion events in MSI-H and MSS TCGA samples determined by bam-read count analysis workflow. Mutations are classified as one-, two-, three- or four-nucleotide deletion events. Majority of shared frameshifts are one-nucleotide deletions, with statistically significant higher allele frequency in tumor samples. H. Shared tumor allele frequency in MS locus of RNF43 gene. T – Tumor; TN – Tissue Normal; BN – Blood Normal. MSIH and MSS stands for MS unstable and stable tumors respectively.
Figure S3. Recall of 46 shared fs-mutations by bamreadcount analysis pipeline, related to Figures 2 and 3. A. Heatmap of 46 shared fs-mutation allele frequencies in MSI-H tumor (LEFT) and normal (RIGHT) WES samples from TCGA. B. Box-plot representation of allele frequencies per each fs-mutation in MSI-H UCEC and COAD/STAD tumor and normal samples. C. Classification of shared fs-mutations by allele frequency bins, as defined by conventional thresholds. Majority of shared fs-mutations are subclonal in MSI-H tumors (VAF < 0.25), however nearly every patient has at least one shared fs-mutation with intermediate-clonal range allele frequency.
Figure S4. Genomic and proteomic detection of shared fs-mutations in CPTAC colon and endometrial cancers, related to Figure 3. A. Heatmap of shared fs-mutations identified in the CPTAC MSI-H UCEC cohort. B. Distribution of population frequencies of all and shared fs-mutations in COAD and UCEC CPTAC cohorts. C. Representative MS/MS spectra of tryptic peptides derived from predicted shared fs-peptides identified by Pepquery analysis in whole cell MS proteomic datasets in CPTAC.
Figure S5. Comparison of missense epitopes and fs-epitopes in MSI-H patients with the human proteome and annotated viral epitopes (IEDB), related to Figure 3. A. Quantification of epitopes per missense or frameshift mutation. On average, frameshift mutations generate 4 epitopes, while missense about 2. B. Mapping missense and fs-epitopes back to the human proteome (ensemble, 2016). “Epitope load” indicates the total amount of predicted neoantigens from missense and frameshift mutations. “Similar match” shows the number of epitopes 8 amino acids in length successfully aligned to the human proteome, allowing 1 mismatch. “Perfect match” indicates the number of 9 amino acid long epitopes perfectly matched to the human proteome with no mismatches. C. BLAST comparison of missense epitopes and fs-epitopes with viral epitopes from IEDB, allowing different number of mismatches (marked as “miss” on the x axis). Missense-derived epitopes are more similar to viral epitopes then frameshift-derived on average.
Figure S6. Survival analysis and other confounding parameter distributions of MSI-H patients with high and low shared frameshift load, related to Figure 5. A. Kaplan-Meier plot of survival analysis and Cox’s proportional hazards model of MSI-H UCEC, COAD and STAD patients, classified by shared fs-epitope load (HIGH vs LOW). B. LEFT - distribution of patients’ age in all, shared fs-epitope high and shared fs-epitope low MSI-H UCEC, COAD and STAD cohorts. RIGHT – patients’ frequency (y axis) across tumor stages (x axis) in the same cohorts.
Figure S7. Fs-peptide-specific T cells T cell immunity, related to Figure 6. PBMCs from healthy donors (HD) were expanded in vitro following stimulation with fs-peptide OLPs. DMSO was used as vehicle control. A. Representative flow cytometry overlay plots for IFN-γ (y axis) and TNF-α (x axis) production by CD8+ T cells. Cytokine production upon OLP or DMSO stimulation is shown in red or blue, respectively. B. Frequencies of CD8+ T cells producing IFN-γ or TNF-α in response to each fs-peptide OLP pool were plotted for each subject. Green denotes HD producing only TNF-α, blue denotes HD producing only IFN-γ and yellow denotes HD producing both IFN-γ and TNF-α. % IFN-γ values were calculated by subtracting the values obtained after DMSO stimulation from the values after OLP pool stimulation and negative values were set to zero. C. Comparison of predicted total epitope load and experimental response rate. Summary of log2 transformed data for IFN-γ /TNF-α response by CD8+ T cells against fs-peptides and the quantification (log2 transformed) of pMHC interactions for each subject per fs-peptide. Interactions were counted when IC50<500 nM or percentile rank<2. Note, T cell response rates do not correlate with pMHC counts (Pearson correlation coefficient r ~ 0.181). D. Demographics and clinical characteristics of MSI-H patients. Patient data depicted above corresponds to measurements at the time samples were obtained and experiments were run. F: female, NED: no evidence of cancer. Reference values for the measurements indicated in the chart are as follows: HGB: 12-16, HCT: 34-47, WBC: 4.5-11, neutrophil: 40-78, lymphocyte: 15-50, monocyte 2-11. E. Spontaneous T cell immunity against fs-peptides in MSI-H cancer patients. PBMCs from MSI-H patients (Pt 2 with COAD and Pt 3 with UCEC) were seeded at 2.5×105 cells per well and stimulated ex vivo with fs-peptide OLP pools, CEFT pool or the control peptide pool MOG. Stimulation with PMA and ionomycin was performed as positive control. IFN-γ formation was measured by ELISPOT after 48 hours of culture. The spot numbers were calculated by subtracting the number of spots formed after MOG stimulation from the number of spots formed after OLP pool stimulation and negative values were set to zero. Statistical significance was calculated by comparing MOG vs the corresponding test group via t test.
Table S1. Aggregation of indel calling in TCGA COAD, STAD and UCEC samples by TCGA calling pipelines (Mutect, Varscan, Muse, Somatic Sniper) and orthogonal validation with bam-readcount approach. Related to Figures 1 and 2.
Table S2. Frameshift neoantigen prediction using NetMHC.v.4.0. Patient frequencies of fs-mutations, fs-peptides and neoantigens in TCGA cohorts. Selection of 46 shared fs-peptides. Identification of shared fs-peptides in whole tumor MS/MS datasets by Pepquery. Analysis of MHC-I elutions of MSI-H cancer cell lines using Pepquery approach. Overlapping peptide design to test immunogenicity of fs-peptides in vitro. Related to Figures 2, 3 and 6.
Table S3. Analysis of shared fs-mutations in Cancer Cell Line Encyclopedia (CCLE) database. Related to Figure 4.
Table S4. Analysis of shared events in MSI-H immunotherapy cohort generously provided by T. Chan from published studies (Le et al., 2015; Mandal et al., 2019). Related to Figure 5.
Data Availability Statement
Source data for TCGA part (Figures 1–3, S1, S2) is available at GDC data commons and generated by the TCGA Research Network (TCGA, January 2018, https://www.cancer.gov/tcga). Data for CPTAC analysis part (Figures 3, S4, Table S2) is available at the Clinical Proteomic Tumor Analysis Consortium, (https://proteomics.cancer.gov/data-portal). Both datasets used by this study (prospective colon and endometrial cancer samples) are published elsewhere (Vasaikar et al., 2019; Dou et al., 2020). Source data for CCLE analysis part (Figure 4) is available at the Cancer Cell Line Encyclopedia, Broad Institute (https://portals.broadinstitute.org/ccle ) and published elsewhere (Barretina et al., 2012). Source data for MHC-I peptide elution analysis from HCT116 cell line (Figure 4) is available at Proteomics Identification Database (PRIDE) and published elsewhere (Bassani-Sternberg et al., 2015). Source data for viral versus tumor epitope comparisons (Figure S6) is available at Immune Epitope Database (IEDB) at NIAID. Source data for MSI-H immunotherapy cohort (NCT01876511) is available upon request from Timothy Chan and published elsewhere (Le et al., 2015; Mandal et al., 2019).
Custom computer code and pipelines are either described in the Method Detail section or available at GitHub: https://github.com/VladimirRoudko/shared_frameshift_neoantigen/. Alternatively, the code is available upon request by the first author (V.R).