

Abstract
Porcine circovirus type 3 (PCV3) has been reported in many countries such as USA, China, Korea and many European countries during 2015–2018. The six PCV3 strains named IH, SJ, N5, N10, N13 and N62 were detected out of 220 samples by PCR methods while the prevalence our study was conducted in 2017 to 2018. The six detected strains were hard to genotype with reference viruses due to their diverse phylogenetic relationship. PCV3 capsid, ORF3 and replicase protein coding genes were reassembled at the nucleotide sequence level, then 16 new reassembled PCV3 sequences were generated. Based on the maximum likelihood mapping analysis of 303 PCV3 sequences a model with a combination of replicase, ORF3 and capsid protein coding genes was selected as the most appropriate target for genotyping, which provided the best support for the clade classification into three genotypes and several subtypes (genotype 1, genotype 2; subtype: a and b, genotype 3; subtype a, b, c, d, e, f, g, h). This study, the IH_Korea_2017 and N62_Korea_2018 strains belong to genogroup 3 (subtype a) the SJ_Korea_2017 strain genogroup 3 (subtype g) and the N5, N10, N13 Korea_ 2018 strains genogroup 3 (subtype f), respectively. In conclusion, this study may provide insights to classification of PCV3 genotypes around the world.
Keywords: genetic analysis, likelihood mapping, porcine circovirus type 3, South Korea
1. INTRODUCTION
The porcine circovirus type 3 (PCV3) genome is an ambisense, single‐stranded, closed‐ circular DNA containing 2,000 nucleotides (nt) (Ku et al., 2017; Palinski et al., 2017; Zheng et al., 2017). The 2,000 nt ambisense genome contains three open reading frames (ORFs) encoding less than 200‐amino acids (aa) and synthesizing the following: capsid proteins (CAP), replicase proteins (REP) and a protein of unknown function (ORF3) (Palinski et al., 2017). PCV3 was firstly reported from swine farms in the United States in 2016 (Palinski et al., 2017). In the same year, other cases showed similar PCV2 clinical symptoms with porcine dermatitis and severe failure of reproductive (Palinski et al., 2017). Another case showed no clinical symptoms with a high positive rate in China (Zheng et al., 2017). Despite PCV3 being identified in the United States, China, Italy, Poland, etc. (Ku et al., 2017; Stadejek et al., 2017), the debate regarding the pathogenicity of PCV3s is still ongoing.
PCV3 KU strains showed a high prevalence from piglets to sow from nine provinces in South Korea, 2016 ~ 2017. The strains were 98.9% ~ 99.8% identical to the above United States strains (Kwon et al., 2017).
Until now, many researchers have studied the genotyping and topology of PCV3s and molecular genetics analyses (Fu et al., 2018; Ku et al., 2017; Kwon et al., 2017; Li et al., 2018; Liu et al., 2020; Vargas‐Bermudez et al., 2019). To date, there are two PCV3 clades (3a, 3b), and a new clade (3c) has been recently suggested (Fu et al., 2018; Ku et al., 2017). However, to the best of our knowledge, there are no published results supporting the most suitable tree models and clarifying the differences of clades within PCV3.
Therefore, in this study, we used reassembled PCV3 viral gene sequences for the best phylogenetic tree model analysis, including maximum likelihood mapping and p‐distance analysis to compare the statistical significance of PCV3 genotyping.
2. MATERIAL AND METHODS
2.1. Sample information and complete genome sequencing
We randomly collected pooled organs (lymph nodes, heart, lung, spleen and kidney, n = 220) causing respiratory problems from January 2017 to February 2018 from 68 commercial farms in nine provinces in Korea. Age groups ranged from suckling to sow pigs. DNA extraction was individually done from each tissue, and all samples were conducted for the extraction as previously described (Kim et al., 2014).
We proceeded to screen the PCV3 specific primers, designated as PCV3‐1‐F [5′‐ TAGTATTACCCGGCACCTCGGAACC −3′], and PCV3‐1‐R [5′‐ ACAGGTAAACGCCCTCGCATGTGGG −3′] which amplified 649‐bp (Ku et al., 2017).
The PCV3 positive samples of the six strains (IH, SJ, N5, N10, N13, N62), were completely sequenced by a primer walking method using three pairs of overlapping primers (Table S1). The specific PCR products were purified by using the gel extraction method and further processed for TA cloning and transformation (Kim et al., 2014). The full‐length genome of the six of PCV3 strains (in this study) were registered in GenBank (accession numbers; shown Table 1).
TABLE 1.
Detection of porcine circoviruses type 3 from Korean domestic pigs in 2017 to 2018
|
Name of sample/ Specimens |
Farm | Clinical symptoms | Region | Pig group* | Collection date |
Sow Scale |
Virus a | Bacteria b | GenBank | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
PCV3 |
PCV2 | PRRSV | SIV | EMCV | PPV | CSFV | JEV | Strep | HPS | (Accession number) | |||||||
| IH/ Lung | IH | Wasting, Respiratory disorders, Death | Kyunggido | Suckling | 7.April.2017 | 140 | + | − | − | − | − | − | − | − | + | − | MF448445 |
| SJ/ Lung | SJ | Respiratory disorders, Death | Jeju | Grower | 17.April.2017 | 270 | + | − | − | − | − | − | − | − | + | − | MF448446 |
| N5/ Lung | DB | Respiratory disorders, Death | Chungbuk | Grower | 19.Jan. 2018 | 200 | + | + | + | − | − | − | − | − | − | − | MH231552 |
| N10/ Lung | NY | Respiratory disorders, Death | Kyunggido | Grower | 30. Jan. 2018 | 300 | + | − | − | − | − | − | − | − | − | − | MH231553 |
| N13/ Lung | Respiratory disorders, Death | Grower | + | − | − | − | − | − | − | − | − | − | MH231554 | ||||
| N62/ Lung | DC | Respiratory disorders, Death | Kyoungbuk | Suckling | 21. Feb. 2018 | 260 | + | − | − | − | − | − | − | − | − | − | MH231555 |
Porcine circovirus type 3 (PCV3), Porcine circovirus type 2 (PCV2), Porcine reproductive and respiratory syndrome virus (PRRSV), Swine influenza virus (SIV), Encephalomyelitis virus (EMCV), Porcine Parvovirus (PPV), Classical swine fever virus (CSFV), Japanese encephalitis virus (JEV).
Streptococcus suis (Strep), Hemophilus parasuis (HPS)
Samples were classified into six groups of sow, suckling pigs (<30 days), weaner (30–60 days), grower (60–90 days) and finisher (≥90 days)
2.2. Screening of other pathogens in the PCV3‐positive samples
For the PCV3 positive samples of the six strains (IH, SJ, N5, N10, N13, N62), extensive testing was performed (on the basic of pathological changes) in order to figure out a possible cause(s) of the death. Viruses and bacteria which are known to induce neurological disorder in pigs were examined, including: Classical swine fever virus (CSFV), Encephalomyelitis virus (EMCV), Japanese encephalitis virus (JEV), Hemophilus parasuis (HPS), Streptococcus suis and verotoxin‐2e producing Escherichia coli (VTEC). Pathogens that associated with the porcine respiratory disease complex were investigated, such as: Porcine reproductive and respiratory syndrome virus (PRRSV), Swine influenza virus (SIV), Porcine circovirus type 2 (PCV2). Methods for detection of EMCV, JEV, PRRSV and SIV were based on commercial single or multiplex PCR (RT‐PCR) kits (catalogue 3,033, 3,031, 3,024 and 3,011, Median Diagnostics, Korea). The other's pathogen‐specific primers were mapped following previous publications (Okwumabua et al., 2003; Oliveira et al., 2001).
2.3. Likelihood mapping
To find the best phylogenetic tree model, three protein coding genes (ORF3; 663nt, capsid; 642nt, replicase; 888nt) and complete genome (1997nt) of PCV3s were selected for likelihood mapping, respectively (Figure 3a). Each protein coding gene was reassembled by connecting the nucleotide sequences with different combinations, such as capsid and ORF3, capsid, ORF3 and replicase and so on. (Figure 1). When reassembling, all nucleotide stop codon sequences were removed. Reference sequences from GenBank with information on collection date and country of origin were included in the analysis. Overall, the final dataset contained 303 PCV3 sequences originating from Asia (China, Korea, Thailand, Taiwan, Inia, Malaysia), America (USA, Brazil, Columbia), and Europe (Italy, Germany, Denmark, Spain, Chile, Sweden, Hungary, Russia) covering a sampling period from 1998 to 2018 each of the three coding genes were aligned by BioEdit version 7.2.5. Collectively, a total of 16 reassembled sequences were obtained.
FIGURE 3.
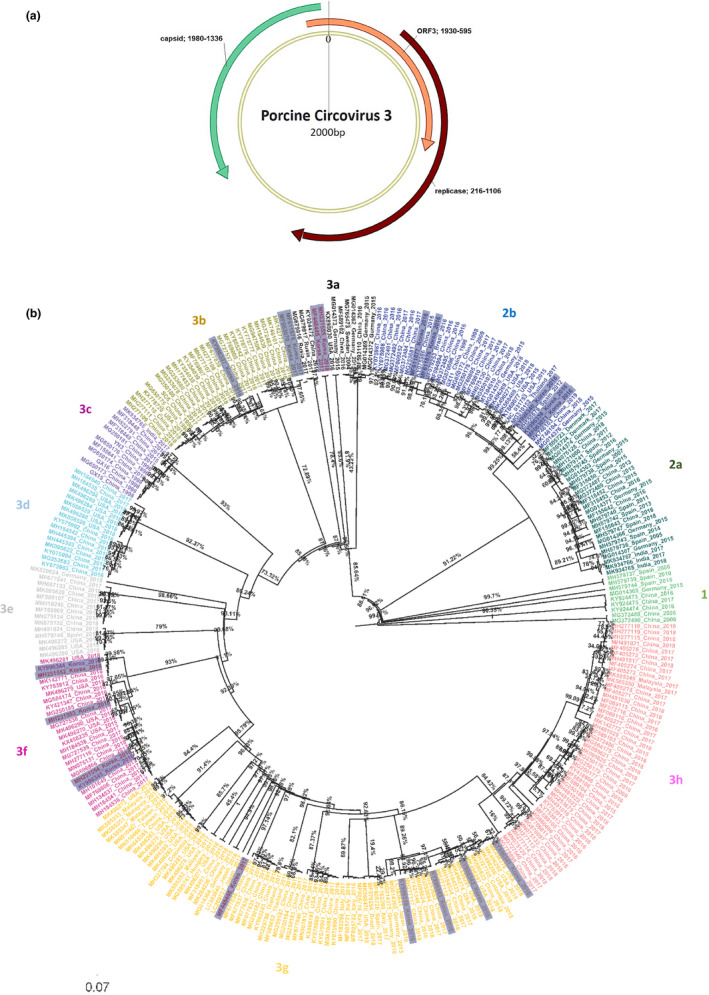
The genomic organization and phylogeny of PCV3. (a) The 2,000‐nt ambisense genome contains three ORFs, encoding capsid (1930–592 nt), replicase (1980–1336 nt) proteins and a protein of unknown function ORF3 (1930–2000(0)‐592 nt). (b) Maximum clade credibility phylogenetic tree of PCV3 based on replicase, ORF3 and capsid protein coding genes. The Maximum likelihood trees of genomes with bootstrap 1,000, automatically best fitting model selected by IQ‐TREE. South Korea strains were highlighted with gray colour, the IH, SJ, N5, N10, N13 and N62 strains (in this study) in red colour and the posterior supported values represented in the node bar
FIGURE 1.
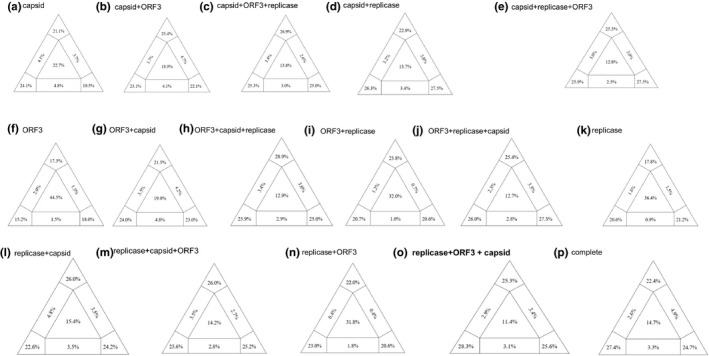
Likelihood mapping of the PCV3 15 recombinations coding (a ~ o) and complete genome (p) datasets of nucleotide sequences. Shown as a whose corner values indicate percentage of well‐resolved phylogenies for all possible quartets, whereas central and lateral values are percentages of unresolved phylogenies (noisy signal). Selected as the best model (0) replicase, ORF3 and capsid visualized a greater emphasis from any mapping recombination model
The phylogenetic signal of the reassembled sequences was tested by performing a likelihood mapping analysis using IQ‐TREE version 1.3.8 (Nguyen et al., 2014). To select the best nucleotide substitution likelihood mapping model was selected based on option mapping bootstrap 1,000 (‐lmap 1,000). For each alignment, it was determined automatically by specifying the ‘‐m TESTONLY’ option. Among 16 reassembled models, each of the analysis visualizes the phylogenetic content of aligned sequences by plotting probability vectors of any quartets of taxa in an equilateral triangle. Results are also shown with a partitioned triangular graph. Three tips of the triangle represent the resolved percentages of quartets. Three rectangles on the sides represent quartets with network evolution (conflicting signal). The central region of the triangle represents star‐like evolution (noisy signal).
2.4. Calculating genetic distance
Genetic distance was calculated on selected reassembled PCV3s with a combination of replicase, ORF3 and capsid (n = 303 aligned strains). The p‐distance was estimated by the MEGA 7 program (Kumar et al., 2016). The genetic distance was represented as a frequency histogram.
2.5. Combination of replicase, ORF3 and capsid dataset‐ phylogenetic analysis
The best model was chosen based on the central value (low noise) and the value of the three triangular peak sums of bootstrap supporting values (Strimmer & Von Haeseler, 1997). To find the best nucleotide substitution model, the reassembled sequence (replicase, ORF3, capsid) was selected automatically by specifying the ‘‐m TEST’ option in IQ‐TREE version 1.3.8 (Nguyen et al., 2014). In this study, the Bayesian skyline plot model (HKY + G4) was determined as the best for phylogenetic analysis.
The Bayesian coalescent‐based Markov chain Monte Carlo (MCMC) was performed using Beast package v1.8.2 under the following assumptions: (i) HKY + G4 nucleotide substitution model in codon based SRD06 (Firth et al., 2009), (ii) a constant population size for the coalescent prior, and (iii) the molecular clock model of uncorrelated lognormal distribution.
For Bayesian phylo‐geographical analysis a framework (Lemey et al., 2009) was applied to reconstruct the spatial‐temporal diffusion history of PCV3 strains. Basically, the spatial diffusion of the time‐scaled genealogy is modelled as a standard continuous time Markov chain (CTMC) process over discrete sampling locations. A Bayesian stochastic search variable selection (BSSVS) approach, which allows the exchange rates in the CTMC to be zero with some prior probability, was used to find a parsimonious set of rates explaining the diffusions in the phylogeny. The analysis was performed using BEAST package v1.8.2 under the following assumptions (i) a codon based SRD06 nucleotide substitution model, (ii) a constant population size for the coalescent prior, and (iii) the molecular clock model of uncorrelated lognormal distribution.
These analyses were run for 100 million generations, sampling every 10,000 iterations. The phylogenic trees were summarized with TreeAnnotator v1.7.2 to find the maximum clade credibility (MCC) tree, which was depicted using FigTree v1. 4.3 program.
3. RESULTS
3.1. PCR‐based detection of PCV3 and other pathogens
The screening analysis by PCR showed that six samples from five swine farms were PCV3 positive among 220 samples in 2017 to 2018, and the six strains (IH, SJ, N5, N10, N13, N62) were completely sequenced for genetic characterization. The strains complete genome size is 2000nt.
IH_Korea_2017 and SJ_Korea_2017 strains were streptococcus positive in addition to PCV3 from other respiratory pathogen examinations. (Table 1). The N5_Korea_2018 strain also detected PRRSV and PCV2.
3.2. Likelihood mapping analysis
PCV3 protein coding genes were reassembled in diverse combination, which were then analysed for genotype classification (16 reassembled sequences). To find the optimal phylogenetic classification model of PCV3, the phylogenetic noise of the dataset was investigated by means of likelihood mapping using the reassembled PCV3 sequences. As shown in Figure 1, the central areas of the map represent a star like noisy signal (the region in which the star tree is optimal). The most proper fitting model conditions should show tree triangles vertex sum values greater than 70% (bootstrap value), and central nosy value less than 20% (Salemi et al., 2009; Zhaxybayeva & Gogarten, 2002). Based on this strategy, among the 16 reassembled sequences, the model showed less than 20% tree noisy values in Figure 1 (b, c, d, e, g, h, j, l, m, o, p), but Figure 1 (o); the replicase, ORF3 and capsid reassembled viral coding genes model had a bootstrap supporting value (79.2%) of the three vertex points and in the central area of the triangles the lowest 11.4% value indicated a fully resolved phylogenetic signal in both case conditions. The model, selected (Figure 1 (o)) in this study can be used as an important basis for future topology PCV3 analysis.
3.3. Pairwise genetic distances of PCV3s
The p‐distance among 303 ‘replicase, ORF3 and capsid’ reassembled sequences of the six strains (IH, SJ, N5, N10, N13, N62) and reference PCV3 strains were 0.001 to 0.089. The Chinese PCV3 strains showed the widest genetic variations (0.001–0.089). The genetic distance between PCV3 strains circulating in each country Germany – Germany (0.003 – 0.021), Korea – Korea (0.001 – 0,011), USA – USA (0.001 – 0.015), Italy – Italy (0.001 – 0.004), Japan – Japan (0.005 – 0.006) and Thailand – Thailand (0.004 – 0.01) were almost within the rage of those in China (Figure 2).
FIGURE 2.
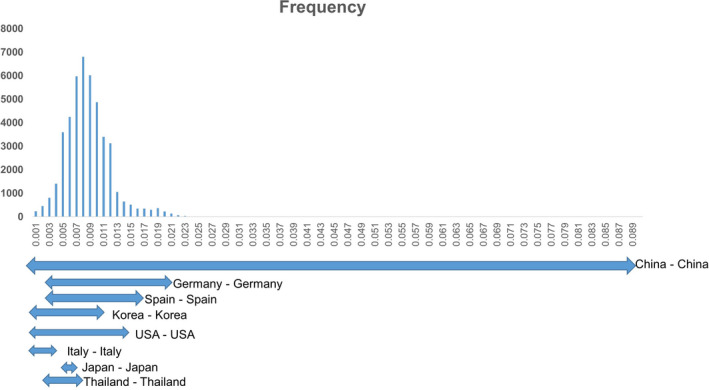
The combination of replicase, ORF3 and capsid protein coding genes, based on genetic distance including several other county PCV3 strains. Frequency of each p‐distance is shown as a column. The genetic distance of the virus circulating in each country is indicated by arrows
3.4. Phylogenetic classification of PCV3s and geographical bayesian analysis
Of the phylogenetic tree topology (Figure 3), the replicase, ORF3 and capsid reassembled dataset consistently divided PCV3s into three genotypes (1, 2, 3) and several subtypes (2a and 2b, 3a to 3h) with a posterior probability of above 70%. Based on the above genotyping method, the six strains (in this study) belonged to genotype 3. More specifically, IH_Korea_2017 strain and N62_Korea_2018 belonged to subtype a (3a) closely related to the FuJian512 _ China _2016 strain (accession number. ky924472). The SJ_Korea_2017 strain was located in the g subtype (3g), and the N5, N10, and N13 korea_2018 stains belonged to the f (3f) subtypes.
Of the virus circulating in South Korea (collected in 2016–2018, Figure 3), the phylogenetic trees reconstructed from the replicase, ORF3 and capsid reassembled dataset showed that they were grouped within genotype 2 in subtype b (2b) and genotype 3 in subtypes 3a, 3b, 3f, 3g (highlighted gray color Figure 3).
Based on selecting the replicase, ORF3 and capsid reassembled dataset of PCV3s, the evolutionary rate was estimated at 1.562 × 10–4 substitutions per site per year (95% highest posterior density (HPD) intervals: 7.590 × 10–5 to 2.369 × 10–4.
The current data supports China being the most probable originate of PCV3 in the world. The most recent common ancestor of PCV3 was predicted to emerge around the year 1945. Based on the described pattern of geographical distribution, this study predicted that Korean PCV3s do not tend to cluster, rather than they originated from China strains (Figure 4).
FIGURE 4.
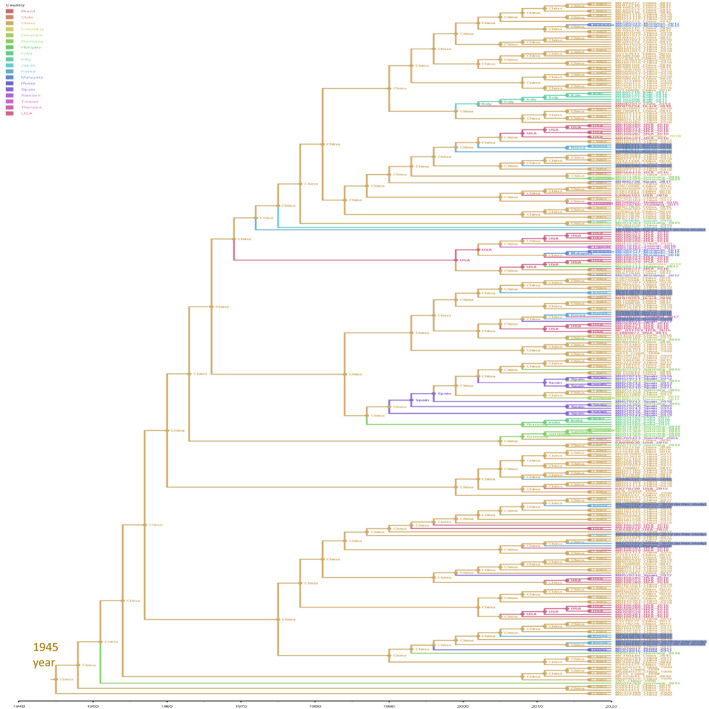
Bayesian phylo‐geographical analysis of PCV3s on the combination of replicase, ORF3 and capsid protein coding genes. Branches were colour‐ coded according to the geographical locations (external branches) and to the most probable geographical origins (internal branches). For clarity, only sequences of Korean PCV3 were highlighted
4. DISCUSSION
The emergence of PCV3s are being reported consecutively worldwide (Li et al., 2018). The present study also shows that PCV3 was continuously detected in Korea in 2017 and 2018.
In Table 1, unlike previously reported (Zheng et al., 2017), six strains (IH, SJ, N5, N10, N13, N62) were collected from the carcass with severe multi‐clinical symptoms. Among strains, the N5_Korea_2018 strain was detected with both PRRSV and PCV2.
The previous studies (Fu et al., 2018; Li et al., 2018; Liu et al., 2020) did not include all PCV3 strains, and since the posterior supporting value was low, it was difficult to distinguish the genotypes. The maximum likelihood mapping analysis in Figure 1 showed that the replicase, ORF3 and capsid protein coding genes model well‐supported and helped in genotyping PCV3s.
As shown in Figure 2, in the case of porcine circovirus type 2, a p‐distance of 0.035 was proposed to be the cut‐off value for genotype delimitation (Grau‐Roma et al., 2008; Trible & Rowland, 2012). Under this point of view, the genetic distances (0.001– 0.089) among PCV3 strains were considered genetic diversity.
Interestingly, a bootstrap value of above 70% (solid value, Figure 3b) supported a separation between distinct genogroups of PCV3‐1, 3‐2a and 2b, 3‐3a to 3h. The China strains are located within all the genogroups, with a fairly high genetic diversity. Especially, the Korean strains showed predominance with geno group 3. Overall, since 2015, the PCV3s has been located in genogroup 3 unlike old strains, which were in genogroup 1.
Based on the replicase, ORF3 and capsid protein coding genes model of PCV3, the evolutionary rate was estimated at 1.562 × 10–4 substitutions per site per year (95% highest posterior density (HPD) intervals: 7.590 × 10–5 to 2.369 × 10–4). Despite similar evolution rate analysis data of PCV3s (Franzo et al., 2019) in a previous study, the low evolutionary rate of the capsid coding gene of PCV3 was comparable with that of PCV2 (1.440 × 10–3) (Collins et al., 2017; Firth et al., 2009), a known pathogen which causes porcine circovirus‐associated disease.
Based on the current dataset (Figure 4), in which all of the PCV3s were sampled from 1998 to 2018, the estimated clock rate gave the TMRCA of PCV3s to be approximately 75 years ago, and is similar to the study results (Saraiva et al., 2018). However, this result should be interpreted with caution as there were no available sequences of PCV3 from archive samples as far back as 1998 (Collins et al., 2017).
The present study supported for China being the most probable origin of PCV3s all over the world. This finding was in line with the previous finding that Korean PCV3 strains were scattered without any geographical association based in the country (Kwon et al., 2017).
In summary, we statistically found that the reassembled viral coding genes (replicase, ORF3 and capsid) could be used for genotype classification of PCV3. Using those reassembled sequences, we could present the reliable genotyping results on the PCV3s in Korea that the PCV3 in this study belonged to genogroup 3 and subtypes a, f, and g of PCV3s. Based on our analysis approach, the reassembled viral coding genes (replicase and ORF3 and capsid) would be used for the reproducible phylogenetic tree model for future genetic research.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This article does not contain any studies with alive animals performed by any of the authors.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Conceptualization: H.C. Chung, B.K. Park and V.G. Nguyen; Formal analysis: H.C. Chung, V.G. Nguyen and Y.H. Park; Investigation: H.C. Chung and V.G. Nguyen; Writing ‐ original draft: H.C. Chung. Writing ‐ review & editing: H.C. Chung and V.G. Nguyen.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/vms3.374.
Supporting information
Table S1
ACKNOWLEDGEMENTS
The authors thank Jung Ah Kim and Eun OK Kim for their excellent technical assistance.
Chung H‐C, Nguyen VG, Park Y‐H, Park B‐K. Genotyping of PCV3 based on reassembled viral gene sequences. Vet Med Sci.2021;7:474–482. 10.1002/vms3.374
Funding information
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (No. 2020R1I1A1A01054539).
Contributor Information
Van Giap Nguyen, Email: nvgiap@vnua.edu.vn.
Yong‐ho Park, Email: parkx026@snu.ac.kr.
Bong‐Kyun Park, Email: parkx026@snu.ac.kr.
REFERENCES
- Collins, P. , McKillen, J. , & Allan, G. (2017). Porcine circovirus type 3 in the UK. Veterinary Record, 181(22), 599. 10.1136/vr.j5505 [DOI] [PubMed] [Google Scholar]
- Firth, C. , Charleston, M. A. , Duffy, S. , Shapiro, B. , & Holmes, E. C. (2009). Insights into the evolutionary history of an emerging livestock pathogen: Porcine circovirus 2. Journal of Virology, 83(24), 12813–12821. 10.1128/JVI.01719-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo, G. , He, W. , Correa‐Fiz, F. , Li, G. , Legnardi, M. , Su, S. , & Segalés, J. (2019). A shift in Porcine circovirus 3 (PCV‐3) history paradigm: Phylodynamic analyses reveal an ancient origin and prolonged undetected circulation in the worldwide swine population. Advanced Science, 6(22), 1901004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X. , Fang, B. , Ma, J. , Liu, Y. , Bu, D. , Zhou, P. , Wang, H. , Jia, K. , & Zhang, G. (2018). Insights into the epidemic characteristics and evolutionary history of the novel porcine circovirus type 3 in southern China. Transboundary and Emerging Diseases, 65(2). 10.1111/tbed.12752 [DOI] [PubMed] [Google Scholar]
- Grau‐Roma, L. , Crisci, E. , Sibila, M. , López‐Soria, S. , Nofrarias, M. , Cortey, M. , Fraile, L. , Olvera, A. , & Segalés, J. (2008). A proposal on porcine circovirus type 2 (PCV2) genotype definition and their relation with postweaning multisystemic wasting syndrome (PMWS) occurrence. Veterinary Microbiology, 128(1–2), 23–35. 10.1016/j.vetmic.2007.09.007 [DOI] [PubMed] [Google Scholar]
- Kim, A. R. , Chung, H. C. , Kim, H. K. , Kim, E. O. , Nguyen, V. G. , Choi, M. G. , Yang, H. J. , Kim, J. A. , & Park, B. K. (2014). Characterization of a complete genome of a circular single‐stranded DNA virus from porcine stools in Korea. Virus Genes, 48(1), 81–88. 10.1007/s11262-013-1003-2 [DOI] [PubMed] [Google Scholar]
- Ku, X. , Chen, F. , Li, P. , Wang, Y. , Yu, X. , Fan, S. , Qian, P. , Wu, M. , & He, Q. (2017). Identification and genetic characterization of porcine circovirus type 3 in China. Transboundary and Emerging Diseases, 64(3), 703–708. 10.1111/tbed.12638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, T. , Yoo, S. J. , Park, C.‐K. , & Lyoo, Y. S. (2017). Prevalence of novel porcine circovirus 3 in Korean pig populations. Veterinary Microbiology, 207, 178–180. 10.1016/j.vetmic.2017.06.013 [DOI] [PubMed] [Google Scholar]
- Lemey, P. , Rambaut, A. , Drummond, A. J. , & Suchard, M. A. (2009). Bayesian phylogeography finds its roots. PLoS Computational Biology, 5(9), e1000520. 10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , He, W. , Zhu, H. , Bi, Y. , Wang, R. , Xing, G. , Zhang, C. , Zhou, J. , Yuen, K.‐Y. , Gao, G. F. , & Su, S. (2018). Origin, genetic diversity, and evolutionary dynamics of novel porcine circovirus 3. Advanced Science, 5(9), 1800275. 10.1002/advs.201800275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Zhang, X. , Li, X. , Ouyang, T. , Niu, G. , Ouyang, H. , & Ren, L. (2020). Genotyping based on complete coding sequences of porcine circovirus type 3 is stable and reliable. Infection, Genetics and Evolution, 78, 104116. 10.1016/j.meegid.2019.104116 [DOI] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2014). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okwumabua, O. , O'Connor, M. , & Shull, E. (2003). A polymerase chain reaction (PCR) assay specific for Streptococcus suis based on the gene encoding the glutamate dehydrogenase. FEMS Microbiology Letters, 218(1), 79–84. [DOI] [PubMed] [Google Scholar]
- Oliveira, S. , Galina, L. , & Pijoan, C. (2001). Development of a PCR test to diagnose Haemophilus parasuis infections. Journal of Veterinary Diagnostic Investigation, 13(6), 495–501. [DOI] [PubMed] [Google Scholar]
- Palinski, R. , Piñeyro, P. , Shang, P. , Yuan, F. , Guo, R. , Fang, Y. , Byers, E. , & Hause, B. M. (2017). A novel porcine circovirus distantly related to known circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. Journal of Virology, 91(1), e01879–e1816. 10.1128/JVI.01879-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi, M. , Lemey, P. , & Vandamme, A.‐M. (2009). The phylogenetic handbook: A practical approach to phylogenetic analysis and hypothesis testing. Cambridge University Press. [Google Scholar]
- Saraiva, G. L. , Vidigal, P. M. P. , Fietto, J. L. R. , Bressan, G. C. , Júnior, A. S. , & de Almeida, M. R. (2018). Evolutionary analysis of Porcine circovirus 3 (PCV3) indicates an ancient origin for its current strains and a worldwide dispersion. Virus Genes, 54(3), 376–384. 10.1007/s11262-018-1545-4 [DOI] [PubMed] [Google Scholar]
- Stadejek, T. , Woźniak, A. , Miłek, D. , & Biernacka, K. (2017). First detection of porcine circovirus type 3 on commercial pig farms in Poland. Transboundary and Emerging Diseases, 64(5), 1350–1353. 10.1111/tbed.12672 [DOI] [PubMed] [Google Scholar]
- Strimmer, K. , & Von Haeseler, A. (1997). Likelihood‐mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proceedings of the National Academy of Sciences, 94(13), 6815–6819. 10.1073/pnas.94.13.6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trible, B. R. , & Rowland, R. R. (2012). Genetic variation of porcine circovirus type 2 (PCV2) and its relevance to vaccination, pathogenesis and diagnosis. Virus Research, 164(1–2), 68–77. 10.1016/j.virusres.2011.11.018 [DOI] [PubMed] [Google Scholar]
- Vargas‐Bermudez, D. S. , Campos, F. S. , Bonil, L. , Mogollon, D. , & Jaime, J. (2019). First detection of porcine circovirus type 3 in Colombia and the complete genome sequence demonstrates the circulation of PCV 3a1 and PCV 3a2. Veterinary Medicine and Science, 5(2), 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , & Rannala, B. (2005). Branch‐length prior influences Bayesian posterior probability of phylogeny. Systematic Biology, 54(3), 455–470. 10.1080/10635150590945313 [DOI] [PubMed] [Google Scholar]
- Zhaxybayeva, O. , & Gogarten, J. P. (2002). Bootstrap, Bayesian probability and maximum likelihood mapping: Exploring new tools for comparative genome analyses. BMC Genomics, 3(1), 4. 10.1186/1471-2164-3-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, S. , Wu, X. , Zhang, L. , Xin, C. , Liu, Y. , Shi, J. , Peng, Z. , Xu, S. , Fu, F. , Yu, J. , Sun, W. , Xu, S. , Li, J. , & Wang, J. (2017). The occurrence of porcine circovirus 3 without clinical infection signs in Shandong Province. Transboundary and Emerging Diseases, 64(5), 1337–1341. 10.1111/tbed.12667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1