

Abstract
The separation of xylenes is one of the most important processes in the petrochemical industry. In this article, the competitive adsorption from a fluid-phase mixture of xylenes in zeolites is studied. Adsorption from both vapor and liquid phases is considered. Computations of adsorption of pure xylenes and a mixture of xylenes at chemical equilibrium in several zeolite types at 250 °C are performed by Monte Carlo simulations. It is observed that shape and size selectivity entropic effects are predominant for small one-dimensional systems. Entropic effects due to the efficient arrangement of xylenes become relevant for large one-dimensional systems. For zeolites with two intersecting channels, the selectivity is determined by a competition between enthalpic and entropic effects. Such effects are related to the orientation of the methyl groups of the xylenes. m-Xylene is preferentially adsorbed if xylenes fit tightly in the intersection of the channels. If the intersection is much larger than the adsorbed molecules, p-xylene is preferentially adsorbed. This study provides insight into how the zeolite topology can influence the competitive adsorption and selectivity of xylenes at reaction conditions. Different selectivities are observed when a vapor phase is adsorbed compared to the adsorption from a liquid phase. These insight have a direct impact on the design criteria for future applications of zeolites in the industry. MRE-type and AFI-type zeolites exclusively adsorb p-xylene and o-xylene from the mixture of xylenes in the liquid phase, respectively. These zeolite types show potential to be used as high-performing molecular sieves for xylene separation and catalysis.
1. Introduction
Xylenes are hydrocarbons that are mainly produced by the catalytic reforming of crude oil.1 From this process, xylenes are usually mixed with benzene and toluene (BTX mixture).2 Further extraction and distillation processes yield mixtures of xylenes that typically contain 53% m-xylene, 24% o-xylene, and 23% p-xylene.3 Of the xylene isomers, p-xylene has the highest economic value.4,5 It is a core raw material for manufacturing poly(ethylene terephthalate),6,7 commonly known as PET. o-Xylene is mostly used in the production of phthalic anhydride.8m-Xylene—the main component of the mixture—has a limited end use and is preferably isomerized into p-xylene.9,10 Xylenes are components of solvents, cleaning agents, paint thinners, varnish, and corrosion preventives.11−13
The different practical applications and economic value of xylene isomers make the separation of the isomers from the mixture a relevant industrial process.14,15 Separation of xylenes is a major challenge.16−18 Commonly, the separation of xylenes is performed by distillation processes2 or by fractional crystallization.19,20 The similar boiling points of xylene isomers make separation using distillation a very difficult21 and energy-intensive process.22 Sholl and Lively23 have underlined that improvements to the separation processes of benzene and C8 aromatics would greatly benefit the world.
Separation using adsorption in porous materials is an attractive alternative to the distillation for the separation of chemicals.24 The use of solid adsorbents for separation yields higher separation efficiencies and lower energy consumption than traditional separation processes.25,26 The separation of xylenes in porous materials can be achieved by adsorption zeolites such as FAU-type27−36 and metal–organic frameworks.37,38 Many industrial applications strongly rely on the selective hosting capabilities of zeolites.39−44
Zeolites have been widely used in many processes for the transformation of hydrocarbons.45−47 These materials are considered the most important heterogeneous catalysts in the petrochemical industry.48,49 Zeolites have a porous structure with well-defined pores and channel systems.50 These systems offer large void volumes that are able to host a wide variety of molecules while maintaining mechanical stability.51,52
Several mechanisms influence the selective adsorption of molecules in zeolites.53 These mechanisms are based on enthalpy, shape, and entropy differences between the competing guest molecules. The enthalpy difference between competing molecules is relevant when the change in enthalpy of the molecules transferred from the bulk phase into the zeolite is even slightly different. In this mechanism, the adsorption is driven by the attractions between the molecules and the zeolite atoms. At low loadings, when interactions among adsorbates can be neglected, there is selectivity toward molecules with stronger attractive interactions with the zeolite.54 In the shape selectivity mechanism, the adsorption of molecules that do not fit in the pores of the zeolite is hindered.55 Instead, only molecules that are smaller than the zeolite pores—and that can diffuse through the pores—are adsorbed. This size exclusion mechanism is related to entropic effects.56 In the entropic selectivity mechanism, packing effects drive the selectivity. The efficiency with which the adsorbed molecules are arranged within the channels of the zeolite determines which molecules are preferentially adsorbed.56,57 This mechanism is particularly important at high loadings.58
Molecular simulations are an extensively used tool to predict the thermodynamic properties of a wide diversity of systems.59 Molecular simulations have been extensively used to predict the adsorption properties of hydrocarbons in zeolites60−68 and other nanoporous materials.69−72 Commonly, Monte Carlo simulations (MCs) in the grand-canonical ensemble (GCMC) can be used to compute sorbate loadings in a zeolite for different temperatures and pressures.73−75 Several studies where MCs are used to study adsorption of aromatics in zeolites can be found in the literature.44,76−86 Most molecular simulation studies of adsorption of hydrocarbons in zeolites have focused on adsorption from a vapor phase.78 This is due to the difficult insertion and deletion of molecules at conditions close to saturation, leading to inefficient simulations.87 However, advanced biasing techniques—such as continuous fractional component (CFCMC) or configurational-bias (CBMC)88—can be used nowadays in Monte Carlo simulations to overcome this issue. In practice, most C8 processing technologies have classically considered adsorption from a vapor phase. More recently, there has been interest in adsorption from a liquid phase.89 This leads to an efficient use of all of the pore volume of the zeolite. Liquid-phase adsorption is preferred due to its operational, maintenance, and environmental advantages over other processing technologies.90
The adsorption of xylene isomers in zeolites and the mechanisms that drive adsorption are studied using Monte Carlo simulations. This article aims to study the competitive adsorption of xylene isomers in selected zeolite frameworks at conditions relevant for industrial purposes. The prediction of the adsorption equilibrium of multiple components is one of the most challenging problems in adsorption.91−93 Typical processing of xylenes uses a mixture of xylenes at chemical equilibrium as feedstock. For this reason, the composition of a mixture of xylene isomers at chemical equilibrium is computed by MC simulations and used as an input for GCMC simulations of adsorption in zeolites. The calculations of the compositions of the mixtures at chemical equilibrium also yield the chemical potentials and fugacity coefficients of xylene isomers.94 For the grand-canonical ensemble (μVT), there is thermodynamic equilibrium between the adsorbed molecules and the reservoir.95 The fugacity coefficients and the composition of the mixture at chemical equilibrium are used to describe the reservoir for GCMC simulations. As there is chemical equilibrium in the reservoir, there must be chemical equilibrium for the molecules adsorbed in the zeolite framework. The adsorption of xylenes in zeolites from a vapor and a liquid phase is computed at 523 K as a function of the total pressure. This temperature is chosen as it is high enough to promote isomerization of xylene isomers catalyzed by acid sites in zeolites.96−99 Pressures of interest for industrial processing of xylenes in the liquid phase using zeolites at temperatures close to 523 K are in the range of 15–40 bar.100−102 In this work, the pressures considered for the simulations are in the range of 0.03–300 bar. To investigate the dependence of xylene adsorption on the zeolite structure, nine different zeolite types are chosen: FAU, MWW, AFI, MEL, MOR, BEA, MRE, MFI, and MTW. These zeolite types are considered as part of the most commonly used in the industry, including four out of the so-called “big five” zeolite types.103,104 Also, a characterization of the selected zeolite frameworks is presented.
This article is organized as follows. The methods are explained in Section 2. The characterization of the selected zeolite frameworks, computed heats of adsorption, and adsorption isotherms of xylenes in the zeolite frameworks are reported and discussed in Section 3. It is shown that the type and topology of the zeolite framework influence the mechanisms for selective adsorption of xylene isomers in different zeolites. The concluding remarks regarding the competitive adsorption of xylenes in zeolites are discussed in Section 4.
2. Methods
The compositions of the mixture at chemical equilibrium and densities of the mixture of xylene isomers are obtained using force-field-based Monte Carlo simulations in the isothermal–isobaric ensemble (NPT), combined with the reaction ensemble.105−108 In the reaction ensemble, trial moves are attempted to transform reactant molecules of a chemical reaction into reaction product molecules. This method considers chemical reactions as Monte Carlo trial moves, without considering kinetics or the reaction mechanism. Brick–CFCMC software94 is used for these simulations. The continuous fractional component Monte Carlo (CFCMC)109,110 algorithm is used to achieve efficient insertions and deletions of molecules in the system.88,110,111 Fugacity coefficients of xylene isomers in the mixture are obtained from the chemical potential computed with the CFCMC algorithm.108,111,112 Details about the conversion of chemical potentials to fugacity coefficients can be found in the Supporting Information. A MC cycle is defined as the maximum between 20 and N-move-attempts, with N being the number of molecules in the system.94,113 In the CFCMC algorithm, the interactions of a fractional molecule are scaled by the so-called λ parameter. The λ parameter scales the interactions of the fractional molecule in the range 0–1 (0 for no interactions with surrounding molecules, and 1 for full interaction with surrounding molecules). The λ-trial moves are biased to obtain a flat probability distribution of λ. The simulations are initialized with 105 MC cycles, and only translation and rotation trial moves are attempted. After initialization, an equilibration run of 5 × 105 MC cycles is performed. All of the types of trial moves considered are allowed, and the biasing factors for the λ-trial moves of the CFCMC algorithm are calculated. After equilibration, ensemble averages are obtained in a 5 × 105 MC cycle production stage. The reported errors account for the 95% confidence interval calculated by dividing the production run into five parts and computing the standard deviation. The initial system contains a total of 300 molecules (200 m-xylene, 50 o-xylene, and 50 p-xylene). The simulations are performed at pressures ranging between 0.03 and 300 bar. This pressure range considers both a vapor and a liquid phase. The initial volume of the simulation box for the simulations in the vapor phase is estimated assuming ideal gas behavior. The initial volume of the simulation box for the simulations of the liquid phase is 403 Å3. Two isomerization reactions are considered: m-xylene ↔ o-xylene and m-xylene ↔ p-xylene. The simulations using the reaction ensemble require the partition functions of the isolated molecules of all of the species involved in the reaction as input. The partition functions of isolated molecules can be obtained using standard thermodynamic tables (e.g., JANAF tables114) or by quantum mechanics calculations.115 Mullen and Maginn116 computed the composition of xylene mixtures at chemical equilibrium as a function of temperature using the reaction ensemble Monte Carlo simulations. These authors found that the use of tabulated free energies of reaction for the calculation of partition functions yields the best agreement with experimental composition of the bulk fluid phase of xylene mixtures. In this work, the isolated molecule partition functions are obtained computing the change in the ideal gas free energy ΔGA↔Bideal of a reaction A ↔ B using thermodynamic tables.117−120 The changes in the ideal gas free energy ΔGA↔B of a reaction A ↔ B computed with tabulated enthalpies and entropies of formation for the reactions considered in this work are listed in the Supporting Information.
The composition of the mixture at chemical equilibrium and the fugacity coefficients of xylene isomers computed in the NPT ensemble combined with the reaction ensemble are used as inputs for the computations of adsorption in zeolites. The computations of adsorption are performed using the CFCMC109,110 algorithm in the grand-canonical ensemble. The RASPA software113,121 is used for all of the simulations of adsorption. The all-silica zeolite structures are obtained from the IZA-SC Database of Zeolite Structures.122 The volume of the simulation box is kept fixed. Periodic boundary conditions are applied. The computations of adsorption do not consider reaction trial moves.
The interactions between the zeolite and guest hydrocarbons are modeled using the TraPPE-zeo model.123 In this force field, all oxygen and silicon atoms are modeled with Lennard-Jones (LJ) interactions and partial charges. It is known that framework flexibility plays a significant role in the adsorption of aromatics in zeolites.80 For very flexible zeolite frameworks, loadings up to two times larger than in a rigid zeolite framework are obtained at a given pressure.124 However, models for framework flexibility should not be blindly applied to zeolites.80 The intra-framework interactions in flexible framework models induce small but important changes in the atom positions of the zeolite and hence in the adsorption isotherms.80 The effects of using models for framework flexibility in different zeolite types are unknown. For these reasons, the zeolite frameworks are considered rigid. Force fields for the interactions between aromatic molecules are typically fitted to model the vapor–liquid equilibrium with LJ potentials or a combination of LJ and electrostatic interactions.125,126 Guest–guest force fields that use electrostatic interactions (such as OPLS127,128) have been used to a great extent for the simulation of adsorption of aromatics in zeolites.76−78,129 The electrostatic interactions of guest–guest force fields of aromatics are fitted for VLE and not for interaction with a host framework.80 Also, the electrostatic interactions included in the TraPPE-zeo model123 are fitted and tested for the adsorption of CO2, and not for aromatic/zeolite systems. It is convenient to use a guest–guest force field that does not include electrostatic interactions. In this work, the guest–guest interactions are modeled using the TraPPE-UA130,131 force field. This force field considers a single uncharged interaction site representing each CHx group in the aromatic molecules. Electrostatic interactions are not considered in this work. Although the presence of nonframework cations may change the adsorption of xylenes,132 this is not considered in this work as it would interfere with understanding the effect of zeolite type on adsorption. The interactions between different atom types are obtained using Lorentz–Berthelot mixing rules.133 A cutoff radius of 14 Å is applied for all LJ interactions, and analytic tail corrections are used.134 All force field parameters are listed in the Supporting Information.
At each MC cycle, trial moves attempt to rotate, displace, randomly reinsert, and insert/remove adsorbates. Also, λ-trial moves scale the interactions of the fractional molecule (via the CFCMC algorithm109,110). The simulations use 105 MC cycles to initialize the system. The initialization run only allows translation, rotation, and insertion/deletion and reinsertion trial moves. After initialization, a stage of 5 × 105 MC cycles is used to equilibrate the CFCMC biasing factors. Ensemble averages are obtained in a 5 × 105 MC cycle production stage. The reported errors account for the 95% confidence interval calculated by dividing the production run into five parts and computing the standard deviation.
The pore size distribution (PSD) of each zeolite structure considered in this work is calculated geometrically with the method of Gelb and Gubbins.135,136 The heat of adsorption of xylenes at infinite dilution in the zeolite structures is calculated via Widom’s test-particle insertion method.137 The heat of adsorption is computed in a 105 MC cycle production run. Enthalpies of adsorption at 523 K from the mixture at chemical equilibrium to the zeolite frameworks at 0.3 and 30 bar are computed by grand-canonical Monte Carlo simulations in a production stage of 5 × 106 MC cycles. The helium void fraction (HVF) and the gravimetric surface area of the zeolite frameworks used in this work are determined using iRASPA visualization software.138 The HVF is determined by probing the framework with a nonadsorbing helium molecule using Widom’s test-particle insertion method.137 The surface area is determined by probing the zeolite framework with a nitrogen molecule. The HVF, surface area, and the amount of unit cells considered for each zeolite framework used in this work are listed in the Supporting Information. The discussion of the zeolite types is ordered based on the maximum diameter of a sphere that can be included in the zeolite framework.122 This diameter is listed in Table S3 of the Supporting Information.
For comparison with the adsorption of the mixture of xylene isomers, the ideal adsorbed solution theory (IAST)139,140 is used to predict the loadings of the xylene mixture using single-component adsorption isotherms. The single-component adsorption isotherms are computed using the fugacity coefficients for each xylene isomer as inputs. The IAST predictions are computed using pyIAST software.141
Multiple linear regression142,143 is used to compute changes in enthalpy and entropy due to the transfer of one xylene molecule from the bulk phase to the zeolite framework. Details about these calculations can be found in the Supporting Information.
3. Results and Discussion
3.1. Properties of Mixture of Xylenes
The properties of the mixture of xylene isomers at chemical equilibrium are calculated by Monte Carlo simulations in the NPT ensemble, combined with the reaction ensemble. Figure 1 shows the computed equilibrium composition, density of the mixture, and fugacity coefficients of xylenes as a function of pressure at 523 K. The simulations suggest that the composition of xylene isomers in the mixture at chemical equilibrium does not significantly vary with changes in pressure at 523 K. Taylor et al.144 reported that at 500 K the C8 aromatic mixture has an equilibrium composition of 3.7% ethylbenzene, 52.8% m-xylene, 20.4% o-xylene, and 23.2% p-xylene. In this work, the equilibrium composition of the mixture of xylenes at 0.94 bar and 523 K is 54.6% m-xylene, 21.5% o-xylene, and 23.9% p-xylene. The computed composition of the mixture at chemical equilibrium is in agreement with the experimental data. The computed composition of the mixture at chemical equilibrium is in excellent agreement with the equilibrium composition estimated by Mullen and Maginn116 at 1 bar by MC simulations in the reaction ensemble.
Figure 1.

Properties of the mixture of xylene isomers at 523 K as computed in this work by Monte Carlo simulations in the NPT ensemble combined with the reaction ensemble. (a) Composition of the mixture of xylene isomers at chemical equilibrium as a function of pressure. Crossed symbols denote the mole fractions of xylenes and ethylbenzene (purple) in the mixture at chemical equilibrium determined from experiments by Taylor et al.144 at 500 K. The mole fraction of ethylbenzene in the experiments is very low, and the computed mole fractions of xylenes match the experimental mole fractions well. (b) Density of the mixture of xylene isomers as a function of pressure. (c) Fugacity coefficients of xylene isomers as a function of pressure. Computed data are listed in the Supporting Information.
Given the similarity of the thermodynamic properties of xylene isomers,125,126 it is expected that the vapor pressure of the mixture of xylenes is close to the vapor pressure of the xylene isomers. From experiments, it is known that the vapor pressure of m-xylene (at 523 K) is 9.85 bar,145 for o-xylene (at 523 K) it is 8.83 bar,146 and for p-xylene (at 520 K) it is 9.419 bar.147 It is expected that the vapor pressure of the mixture of xylenes at chemical equilibrium is close to these pressures. From the computed densities of the mixture of xylene isomers, it is observed that the mixture is in the vapor phase when the pressure is lower than 5.3 bar and that the mixture is in the liquid state when the pressure is higher than 16.8 bar. The density of the mixture of xylenes as a function of pressure suggests that the phase change from vapor to liquid occurs at some point between 5.3 and 16.8 bar. From the bulk phase simulations, only an estimation of the composition of the mixture at chemical equilibrium and the fugacity coefficients are required. As such, the properties for pressures close to the phase change at 523 K are not computed. The computed fugacity coefficients significantly decrease from pressures higher than 5.3 bar. The fugacity coefficients of the three xylene isomers are nearly identical at 523 K and pressures between 0.03 and 300 bar.
3.2. Zeolite Structures
The pore size distribution (PSD) of the zeolite frameworks considered in this work is shown in Figure 2. The PSD is calculated geometrically with the method of Gelb and Gubbins.135,136 In this method, the diameter of the largest sphere that does not intercept any framework atoms is computed for each point in the void space of the framework. In a PSD of zeolite frameworks, a peak in the distribution denotes the diameter of a cavity or a channel. For FAU-type zeolites, the cages are denoted at a diameter of ca. 9.5 Å. For MWW-type zeolites, the two independent pore systems are represented by two ranges of diameters in the pore size distribution. The 10-ring pore system is denoted between 3 and 5 Å approximately. The 12-ring pore system is denoted between 6 and 8.5 Å. For AFI-type zeolites, the channel is denoted at a diameter of approx. 7 Å. For MEL-type zeolites, a peak at a diameter of approx. 4.5 Å denotes the channels, and the distribution at diameters larger than 5.5 Å denotes the intersections of the channels. For MOR-type zeolites, the peak at a diameter of approx. 5.5 Å denotes the 12-ring channels. The small peak at a diameter of approx. 4 Å denotes the 8-ring side pockets that link the 12-ring channels. For BEA-type zeolites, the channel and the intersection of the channels are denoted at a diameter of approx. 5.5 Å. For MFI-type zeolites, two peaks are observed: one at a diameter of approx. 3.5 Å and one at approx. 5 Å. These peaks correspond to the channels (zigzag and straight) and intersection of the channels, respectively. For MRE-type and MTW-type zeolites, the peaks at diameters of approx. 5 and 4.5 Å denote the channels, respectively.
Figure 2.
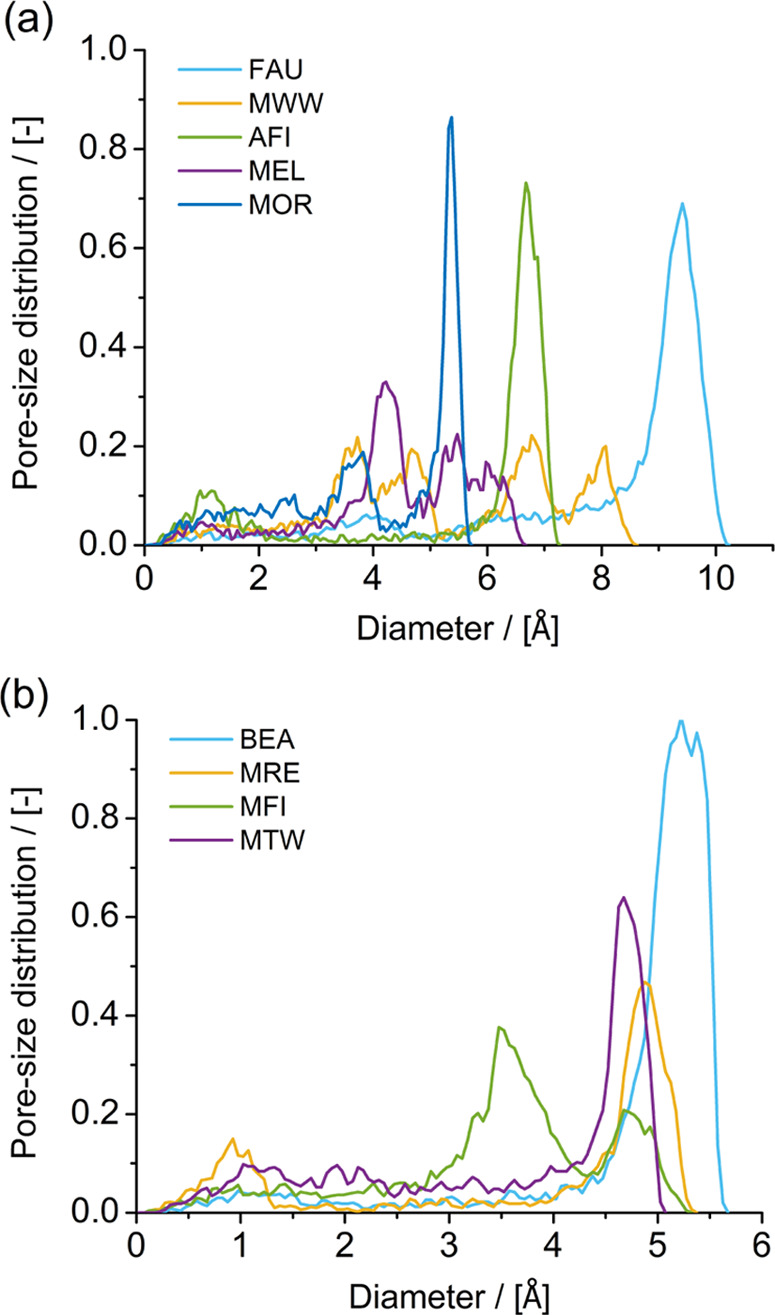
Pore size distribution (PSD) of the zeolite frameworks considered in this work computed with the method of Gelb and Gubbins.135 In this method, the diameter of the largest sphere that does not intercept any framework atoms is computed for each point in the void space of the zeolite framework. (a) PSDs for FAU-type, MWW-type, AFI-type, MEL-type, and MOR-type zeolites. (b) PSDs for BEA-type, MRE-type, MFI-type, and MTW-type zeolites.
3.3. Adsorption of Xylenes in Zeolites
The heats of adsorption at infinite dilution of xylene isomers at 523 K in the zeolite frameworks considered in this work are shown in Figure 3. In FAU-type zeolites, there is enthalpic preference for m-xylene and o-xylene over p-xylene. In AFI-type, MWW-type, BEA-type, MRE-type, and MTW-type zeolites, there is an enthalpic preference for p-xylene over o-xylene and m-xylene. For MOR-type zeolites, p-xylene and o-xylene are preferentially adsorbed based on enthalpy. For MFI-type and MEL-type zeolites, there is an enthalpic preference for m-xylene. In the following section, the location of the xylene molecules in each zeolite framework is discussed and linked to the heat of adsorption and changes in enthalpy and entropy due to the transfer of xylenes from the bulk mixture to the zeolite. Table 1 lists the preferential adsorption of xylene isomers based on the heat of adsorption at infinite dilution. The computed heat of adsorption values of xylenes adsorbed from the mixture at chemical equilibrium at 30 bar and 523 K in the zeolite types considered in this work are shown in Figure S2 of the Supporting Information.
Figure 3.
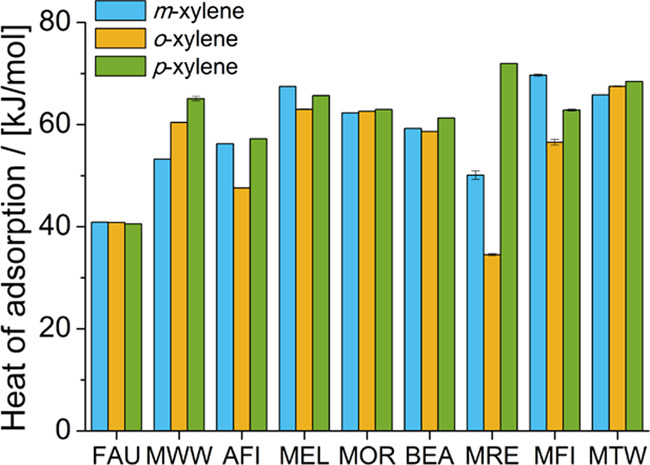
Heat of adsorption at infinite dilution of xylene isomers at 523 K in the zeolite framework types considered in this work as computed by Monte Carlo simulations.
Table 1. Summary of the Observed Preferential Adsorption of Xylene Isomers in Zeolites for Pure Components and for the Adsorption of the Mixture of Xylenes at Chemical Equilibrium at 523 Ka.
| zeolite framework type | observed
preferential adsorption of xylenes as single components |
observed
preferential adsorption of xylenes for the mixture at chemical equilibrium |
|||
|---|---|---|---|---|---|
| 0.03 ≤ P ≤ 5.3 bar | 16.8 ≤ P ≤ 300 bar | 0.03 ≤ P ≤ 5.3 bar | 16.8 ≤ P ≤ 300 bar | preference based on the heat of adsorption at infinite dilution | |
| FAU | m = o = p | m = o = p | m > o ∼ p | m ≫ o > p | m ∼ o > p |
| AFI | o > m ∼ p | o ≫ m ∼ p | o ∼ m ∼ p | o ≫ m > p | p > m ∼ o |
| MWW | o > p > m | m > p > o | o > m ∼ p | o > m ∼ p | p > o > m |
| MEL | m > p > o | m ∼ p∼ o | m ≫ p > o | m ≫ p > o | m > p > o |
| MFI | m > p > o | m > p > o | m ≫ p ∼ o | m ≫ p ∼ o | m > p > o |
| BEA | p > m ∼ o | o > p > m | p ∼ m > o | p > m > o | p > m > o |
| MRE | p ≫ m ∼ o | p ≫ m ∼ o | p ≫ m ∼ o | p ≫ m ∼ o | p > m > o |
| MTW | p > o > m | m = o > p | p > m > o | p ∼ m > o | p > o > m |
| MOR | m ∼ o ∼ p | o > m ∼ p | m ∼ p > o | m > o ∼ p | p > o > m |
The preferences are observed for two pressure ranges: 0.03 ≤ P ≤ 5.3 bar (i.e., adsorption from a vapor phase) and 16.8 ≤ P ≤ 300 bar (i.e., adsorption from a liquid phase). The preferential adsorption based on the heat of adsorption at infinite dilution is also listed. The xylene isomers are represented as m for m-xylene, o for o-xylene, and p for p-xylene.
The adsorption isotherms of single-component xylene isomers and the mixture of isomers at chemical equilibrium in FAU-type zeolite at 523 K are shown in Figure 4. The single-component isotherms of the three isomers suggest that there is no preferential adsorption for any isomer. Zheng et al.82 reported the adsorption of aromatics in FAU-type zeolites. The same packing efficiency of C8 aromatics in the cages of FAU-type zeolites is observed. In Figure 4a, it is observed that nearly identical loadings of the three xylene isomers in FAU-type zeolites as a function of pressure are computed. This is expected due to the large pore size of FAU-type zeolites.
Figure 4.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in FAU-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of total pressure for the mixture at chemical equilibrium adsorbed in the FAU-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the FAU-type zeolite at 0.3 and 30 bar. (e) Typical snapshot of the simulation of adsorption of the mixture of xylene isomers in FAU-type zeolites at 523 K and 300 bar. m-Xylene is shown in blue, p-xylene in gray, and o-xylene in orange. The snapshot shows how several xylenes are hosted in the cages of FAU-type zeolites.
Figure 4b shows the adsorption isotherm of the mixture of xylenes at chemical equilibrium in FAU-type zeolites. It is observed that the loadings of m-xylene are higher than for the other isomers. This is related to the high mole fraction of m-xylene in the bulk phase, compared to the mole fractions of the other isomers. The loadings of o-xylene in FAU-type zeolites are higher than the loadings of p-xylene.
Figure 4c shows the composition of the mixture of xylenes adsorbed in FAU-type zeolites compared to the composition of mixture in the bulk phase (Figure 1a). It is observed that the composition of the adsorbed fluid does not change with pressure. The mole fractions of p-xylene in the adsorbed phase are lower than the mole fractions of p-xylene in the bulk phase. Figure 4d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the FAU-type zeolite for 0.3 and 30 bar and 523 K. At 0.3 bar, adsorption is driven by enthalpic changes. At 30 bar, the changes in entropy (multiplied by the absolute temperature) are larger than the changes in enthalpy. This suggests that for adsorption from the liquid phase, changes in entropy significantly influence adsorption and the arrangement of xylenes in the cages of FAU-type zeolites. The changes in enthalpy and entropy due to the transfer of xylene molecules from the bulk phase to the zeolite (either at 0.3 or at 30 bar) do not show any selectivity for a xylene isomer.
A typical snapshot of the simulation of adsorption of the mixture of xylene isomers at 523 K and 300 bar is shown in Figure 4e. It is observed that xylene molecules are almost fluidlike in the cages of FAU-type zeolites. At high pressure, or when molecules are adsorbed from the liquid phase, each cage hosts two or more xylene molecules. The loadings predicted with IAST are in agreement with the simulations of adsorption of the mixture of xylenes. This suggests that xylene molecules compete for the same adsorption sites in the FAU-type zeolite.
The adsorption isotherms of xylene isomers as single components and the mixture of xylenes at chemical equilibrium in MWW-type zeolites at 523 K are shown in Figure 5. The single-component isotherms show that for pressures lower than 5.3 bar (adsorption from the vapor phase), the loadings of o-xylene are higher than for p-xylene and m-xylene. For pressures higher than 16.8 bar (i.e., adsorption from the liquid phase), the loadings of m-xylene are higher than for p-xylene and o-xylene.
Figure 5.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MWW-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of total pressure for the mixture at chemical equilibrium adsorbed in the MWW-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MWW-type zeolite at 0.3 and 30 bar. (e) Typical snapshot of the simulation of the mixture of xylene isomers at chemical equilibrium at 523 K and 300 bar. m-Xylene is shown in blue, p-xylene in gray, and o-xylene in orange. o-Xylene is located in the 12-ring cages of the MWW-type zeolite. m-Xylene and p-xylene are located in the 10-ring channels.
For the adsorption isotherm of the mixture of xylenes at chemical equilibrium, there is a preferential adsorption of o-xylene over m-xylene and p-xylene. Figure 5c shows the composition of the mixture of xylenes adsorbed in MWW-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that for the adsorbed molecules, the mole fractions of m-xylene and p-xylene are nearly identical. The heat of adsorption of xylenes in MWW-type zeolites at infinite dilution shows an energetic preference for p-xylene over the other isomers. Figure 5d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the MWW-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption is driven by enthalpy and there is an enthalpic preference for p-xylene over o-xylene and m-xylene. At 30 bar, the changes in entropy (multiplied by the absolute temperature) are larger than the changes in enthalpy for each xylene isomer. The adsorption of o-xylene is almost equally driven by enthalpic and entropic changes. For m-xylene and p-xylene, adsorption is hindered by entropic effects. For pressures higher than 16.8 bar, the loadings predicted with IAST are not in agreement with the simulations of adsorption of the mixture of xylenes. IAST does not provide a suitable prediction of the component loadings if there is a segregation of the preferential adsorption sites.148−150 For the adsorption of xylene isomers in MWW-type zeolites, the isomers do not compete for the same adsorption sites. o-Xylene is preferentially adsorbed in the 12-ring cages. m-Xylene and p-xylene are preferentially adsorbed in the 10-ring channels. Figure 5e shows a typical snapshot of the simulation of adsorption of the mixture of xylenes at chemical equilibrium at 523 K and 300 bar. It is observed that for the mixture of xylenes at chemical equilibrium, the 12-ring cages preferentially host o-xylene. m-Xylene and p-xylene are hosted in the 10-ring channels. Typical snapshots of the adsorption of o-xylene in MWW-type zeolites at 0.94 and 300 bar can be found in Figure S3 of the Supporting Information.
The adsorption isotherms of xylene isomers as single components and the mixture of isomers at chemical equilibrium in AFI-type zeolites at 523 K are shown in Figure 6. The single-component isotherms show a significant adsorption selectivity of o-xylene over m-xylene and p-xylene. This is in agreement with the observations of Chiang et al.,151 who studied the adsorption of xylenes in AlPO4-5 (an AFI-type zeolite) via experiments.
Figure 6.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in AFI-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of total pressure for the mixture at chemical equilibrium adsorbed in the AFI-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the AFI-type zeolite at 0.3 and 30 bar. Typical snapshots of the simulation of adsorption from the mixture of xylenes at chemical equilibrium at 523 K and 300 bar showing (e) m-xylene + p-xylene and (f) o-xylene. m-Xylene is shown in blue, p-xylene in gray, and o-xylene in orange. The snapshots show the face-to-face stacking of o-xylene molecules, in comparison to how m-xylene and p-xylene molecules are arranged in the channels of AFI-type zeolites.
The adsorption isotherm of the mixture of xylenes at chemical equilibrium shows a significant selectivity of o-xylene over m-xylene and p-xylene for pressures higher than 16.8 bar, i.e., adsorption from the liquid phase. The total loading of the mixture is lower than the single-component isotherm for o-xylene (Figure 6a). This suggests that the preferential adsorption of o-xylene from the mixture is affected by guest–guest interactions between o-xylene and the other isomers. The loadings predicted with IAST agree with the simulations of adsorption of the mixture of xylenes. Figure 6c shows the composition of the mixture of xylenes adsorbed in AFI-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that for pressures higher than 0.53 bar, the composition of the adsorbed molecules significantly changes as a function of the pressure. At low pressures, the adsorption of m-xylene is higher than for the other isomers. At pressures higher than 0.53 bar, the mole fraction of o-xylene is significantly higher than for the other isomers. At 300 bar, the mole fraction of o-xylene in the adsorbed phase is 0.92. The heats of adsorption of xylene isomers at infinite dilution in AFI-type zeolites show an energetic preference for p-xylene. Figure 6d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the AFI-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption is driven by enthalpic changes due to the transfer of xylenes to the zeolite. At 30 bar, adsorption of m-xylene and p-xylene is significantly affected by entropic changes. For o-xylene, adsorption is almost equally driven by entropic and enthalpic changes. Figure 6e shows a typical snapshot of the adsorption of m-xylene and p-xylene in AFI-type zeolites. At least one methyl group of these molecules is aligned with the channel direction. Figure 6f shows a typical snapshot of the adsorption of o-xylene in AFI-type zeolites. Face-to-face stacking of o-xylene molecules is observed. The aromatic ring of o-xylene is perpendicular to the direction of the channel. This arrangement of o-xylene molecules has been previously observed by Torres-Knoop et al.152 for the adsorption of an equimolar mixture of xylene isomers and ethylbenzene in AFI-type zeolites. In the literature, the face-to-face stacking has been related to entropic effects,57,152 i.e., how efficiently xylene molecules are arranged. The simulations from this work suggest that the preferential adsorption of o-xylene over the other isomers is related to enthalpic changes affecting adsorption of the other isomers.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in MEL-type zeolites at 523 K are shown in Figure 7. The single-component isotherms show that the loadings of m-xylene and p-xylene are higher than the loadings of o-xylene for pressures lower than 5.3 bar. For pressures higher than 16.8 bar (adsorption from the liquid phase), almost identical loadings are obtained for the three isomers. This suggests that the three xylene isomers are adsorbed on the same sites of MEL-type zeolites.
Figure 7.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MEL-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of total pressure for the mixture at chemical equilibrium adsorbed in the MEL-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MEL-type zeolite at 0.3 and 30 bar. (e) Typical snapshot of the simulation of adsorption of the mixture of xylenes in MEL-type zeolites at 300 bar and 523 K. m-Xylene is shown in blue, p-xylene in gray, and o-xylene in orange. The snapshot shows how xylenes are located in the intersection of the channels of MEL-type zeolites.
For the mixture of xylene isomers at chemical equilibrium, there is a strong selectivity of m-xylene adsorption over o-xylene and p-xylene. Figure 7c shows the composition of the mixture of xylenes adsorbed in MEL-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that the composition of the adsorbed phase does not depend on the pressure. The mole fractions of p-xylene in the adsorbed phase are the same as in the bulk phase.
The loadings predicted with IAST are in agreement with the simulations of adsorption of the mixture. Based on the heat of adsorption at infinite dilution, there is a preferential adsorption of m-xylene > p-xylene > o-xylene. Figure 7d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the MEL-type zeolite for 0.3 and 30 bar at 523 K. At both 0.3 and 30 bar, adsorption is driven by enthalpic changes due to the transfer of xylenes to the zeolite. The changes in enthalpy due to the transfer of xylenes between the fluid phase and the MEL-type zeolite (as computed using eq S-4) become positive when xylenes are adsorbed from the liquid phase. Figure S2 of the Supporting Information shows the computed heat of adsorption of xylenes in MEL-type zeolites at 30 bar and 523 K. It can be observed that the heat of adsorption of xylenes in MEL-type zeolites at 30 bar is significantly lower than the heat of adsorption at infinite dilution (see Figure 3). This is in agreement with the observations from experiments of m-xylene adsorption in ZSM-11 (an MEL-type zeolite) at 315 K by Guil et al.,153 where this decrease in the heat of adsorption at high loadings compared to low loadings is related to the complete filling of the pores of MEL-type zeolites. At high pressures, the heat of adsorption of xylenes in the bulk phase is higher than the heat of adsorption of xylenes in MEL-type zeolites (due to the complete pore filling), yielding a positive change in enthalpy. Figure 7e shows a typical snapshot of the simulation of adsorption of the mixture of xylenes at chemical equilibrium at 523 K and 300 bar. The snapshot shows that the three xylene isomers are located in the intersections of the channels of MEL-type zeolites.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in MOR-type zeolites at 523 K are shown in Figure 8. The single-component isotherms (Figure 8a) show that for pressures lower than 0.94 bar, the loadings of the three xylene isomers are nearly identical. For pressures higher than 0.94 bar, the loadings of o-xylene are higher than for the other isomers. The preferential adsorption of xylene isomers in MOR-type zeolites based on the heat of adsorption is p-xylene > o-xylene > m-xylene. This suggests that entropic effects are important for the arrangement of o-xylene molecules in the 12-ring channels of MOR-type zeolites for adsorption from the liquid phase.
Figure 8.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MOR-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of the total pressure for the mixture at chemical equilibrium adsorbed in the MOR-type zeolites and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MOR-type zeolite at 0.3 bar and 30 bar. Typical snapshots of the simulations of adsorption of (e) o-xylene and (f) p-xylene in MOR-type zeolites at 300 bar and 523 K. p-Xylene is shown in gray, and o-xylene is shown in orange. The snapshots show the typical configuration of xylene isomers in the channels of MOR-type zeolites.
For the adsorption isotherm of the mixture of xylenes at chemical equilibrium in MOR-type zeolites (Figure 8b), there is a preferential adsorption of m-xylene and p-xylene for pressures lower than 5.3 bar (i.e., adsorption from the vapor phase). For adsorption from the liquid phase, there is preferential adsorption of m-xylene and nearly identical loadings of o-xylene and p-xylene. Figure 8c shows the composition of the mixture of xylenes adsorbed in MOR-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that the composition of the adsorbed fluid changes as a function of pressure. For pressures higher than 0.94 bar, the mole fractions of p-xylene in MOR-type zeolites decrease with increasing total pressure, while the mole fractions of o-xylene increase with increasing total pressure. The mole fractions of p-xylene for the mixture adsorbed in MOR-type zeolites do not change as a function of the total pressure. Figure 8d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the MOR-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption is driven by changes in enthalpy due to the transfer of xylenes to the zeolite. At 30 bar, adsorption of xylene isomers is affected by entropic changes. However, there is a small enthalpic preference for o-xylene over the other xylene isomers. This suggests that at higher pressures there is a competition between enthalpic and entropic effects for accommodating p-xylene and o-xylene molecules in MOR-type zeolites.
Figure 8e,f shows typical snapshots of the adsorption at 523 K and 300 bar of o-xylene and p-xylene, respectively. At high pressures, adsorption of o-xylenes is favored by the alignment of its methyl groups. These can be aligned to the direction of the channel (c-crystallographic axis) or perpendicular to the channel (a-crystallographic axis). This favors an efficient pore volume occupancy of o-xylenes in MOR-type zeolites. The total loading of the mixture is lower than the single-component isotherm for o-xylene (Figure 8a). The preferential adsorption of o-xylene from the mixture is affected by guest–guest interactions between o-xylene and the other isomers. This leads to a disagreement between the computed loadings of o-xylenes with the predictions of IAST. For p-xylene, the methyl groups are typically aligned with the channel direction. The grooves of the surface of the 12-ring channel also typically allow the methyl groups of p-xylene to align with an inclination of ca. 45° from the channel direction.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in BEA-type zeolites at 523 K are shown in Figure 9. The single-component isotherms show that for pressures lower than 1 bar, there is p-xylene selectivity over m-xylene and o-xylene. For the adsorption from a liquid phase (pressures higher than 16.8 bar), loadings of o-xylene higher than p-xylene and m-xylene are obtained.
Figure 9.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in BEA-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of total pressure for the mixture at chemical equilibrium adsorbed in the BEA-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the BEA-type zeolite at 0.3 and 30 bar. Typical snapshot of the simulation of adsorption of the mixture of xylenes in BEA-type zeolites at 523 K and 30 bar showing (e) m-xylene and (f) p-xylene. m-Xylene is shown in blue and p-xylene in gray. The snapshot shows how m-xylene and p-xylene are arranged in the intersection of the channels.
The adsorption isotherm of the mixture at chemical equilibrium suggests that there is a preferential adsorption of p-xylene and m-xylene over o-xylene. The loadings predicted with IAST are in agreement with the simulations of adsorption of the mixture of xylenes. Figure 9c shows the composition of the mixture of xylenes adsorbed in BEA-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that the composition of the adsorbed phase does not significantly vary with pressure. Based on the heat of adsorption at infinite dilution, there is a preferential adsorption of p-xylene > m-xylene > o-xylene. Figure 9d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the BEA-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption is driven by changes in enthalpy due to the transfer of xylenes to the zeolite. At 30 bar, adsorption of xylene isomers is driven by changes in entropy. However, there is a small enthalpic preference for the adsorption of p-xylene over the other isomers. Figure 9e,f shows typical snapshots of the simulation of adsorption of m-xylene and p-xylene at 30 bar and 523 K. It is observed that m-xylene molecules are located in the intersection of the channels and that the methyl groups align with the two intersecting channels. For p-xylene molecules, the methyl groups can align either with the channel direction or with an intersecting channel. This suggests that the differences between the loadings of the xylene isomers in the adsorbed mixture are related to the shape of the molecules.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in MRE-type zeolite at 523 K are shown in Figure 10. The single-component isotherms show a strong preference for the adsorption of p-xylene over the adsorption of m-xylene and o-xylene. The loadings of m-xylene and o-xylene are below 0.05 mol/kg for the considered pressure range. This can be related to the size of the channel of MRE-type zeolites. Figure 10c shows the composition of the mixture of xylenes adsorbed in MRE-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. The composition of the adsorbed phase does not vary as a function of pressure. The mole fraction of p-xylene in the adsorbed phase is 0.999 for the considered pressure range. The loadings predicted with IAST are in agreement with the simulations of adsorption of the mixture of xylenes. The PSD (Figure 2) shows a peak in the distribution for MRE-type zeolites at a diameter of ca. 5 Å, which suggests that there are size restriction and shape selectivity for molecules larger than p-xylene. Figure 10d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the MRE-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption of p-xylene is driven by changes in enthalpy due to the transfer to the zeolite. For m-xylene and o-xylene, the shape exclusion is shown as changes in entropy. This is also observed for adsorption at 30 bar, as only p-xylene is adsorbed from the mixture of xylenes at chemical equilibrium. Figure 10e shows a snapshot of the simulation of adsorption of the mixture of xylenes. It is observed that the methyl groups of p-xylene are aligned with the channel.
Figure 10.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MRE-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of the total pressure for the mixture at chemical equilibrium adsorbed in the MRE-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MRE-type zeolite at 0.3 and 30 bar. (e) Typical snapshot of the simulation of adsorption of the mixture of xylenes at chemical equilibrium in MRE-type zeolites at 300 bar and 523 K. p-Xylene is shown in gray. The snapshot shows how p-xylene molecules are aligned with the channel of MRE-type zeolites.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in MFI-type zeolites at 523 K are shown in Figure 11. The single-component isotherms show that the loadings of m-xylene are higher than the loadings of p-xylene and o-xylene in the considered pressure range. In this pressure range, the adsorption site for all isomers is nearly identical. It is known that p-xylene molecules are hosted in the channels of MFI-type zeolites when the loadings are higher than 4 molecules/unit cell (ca. 0.693 mol/kg).76,80 The simulations show that all of the molecules are located in the intersections of the zigzag and straight channels. The preference for m-xylene is due to an alignment of the methyl groups with the zigzag and straight channels. Figure 11e shows how as one methyl group of m-xylene is aligned in the direction of the straight channel (b-crystallographic axis), the other methyl group is aligned in the direction of the zigzag channel (c-crystallographic axis). It can also be observed how this arrangement of the methyl groups is difficult for o-xylene molecules. p-Xylenes are aligned with the straight channel (b-crystallographic axis).
Figure 11.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MFI-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of the total pressure for the mixture at chemical equilibrium adsorbed in the MFI-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MFI-type zeolite at 0.3 and 30 bar. (e) Typical snapshot of the simulation of adsorption of the mixture of xylenes in the MFI-type zeolite at 300 bar and 523 K. m-Xylene is shown in blue, p-xylene in gray, and o-xylene in orange. The snapshot shows how xylenes are located in the intersection of the zigzag and straight channels of MFI-type zeolites.
For the mixture of xylene isomers at chemical equilibrium, there is a strong selectivity of m-xylene adsorption over o-xylene and p-xylene. Figure 11c shows the composition of the mixture of xylenes adsorbed in MFI-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that the composition of the adsorbed phase does not depend on the pressure. The loadings predicted with IAST are in agreement with the simulations of adsorption of the mixture. Based on the heat of adsorption at infinite dilution, there is a preferential adsorption of m-xylene > p-xylene > o-xylene. Figure 11d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and the MFI-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption of m-xylene and o-xylene is almost equally driven by changes in enthalpy and entropy due to the molecule’s transfer to the zeolite. For p-xylene, adsorption is driven by changes in enthalpy. For adsorption of m-xylene at 30 bar, the changes in enthalpy and entropy (multiplied by the absolute temperature) due to the transfer to the zeolite are almost equal.
The adsorption isotherms of xylene isomers as single components and the mixture at chemical equilibrium in MTW-type zeolite at 523 K are shown in Figure 12. The single-component isotherms show that for pressures lower than 5.3 bar, the loadings of p-xylene are higher than for o-xylene and m-xylene. For pressures higher than 16.8 bar, the loadings of m-xylene are higher than for o-xylene and p-xylene.
Figure 12.

Adsorption isotherms of xylene isomers as (a) single components (total loading mixture is the sum of the loadings of xylenes from the mixture at chemical equilibrium) and (b) a mixture at chemical equilibrium in MTW-type zeolites at 523 K. (c) Mole fractions of xylene isomers as a function of the total pressure for the mixture at chemical equilibrium adsorbed in the MTW-type zeolite and for the bulk phase. The composition in the bulk phase follows from Figure 1a. (d) Changes in enthalpy ΔHtr,i and entropy TΔStr,i at 523 K due to the transfer of xylene i from the fluid-phase mixture at chemical equilibrium to the MTW-type zeolite at 0.3 and 30 bar. (e) and (f) are typical snapshots of the simulation of adsorption of the mixture of xylene isomers in MTW-type zeolites at 300 bar and 523 K. m-Xylene is shown in blue, p-xylene is shown in gray, and o-xylene is shown in orange. The snapshots show the typical configuration of xylene isomers in the channels of MTW-type zeolites.
For the adsorption of the mixture of xylenes at chemical equilibrium, there is a preferential adsorption of p-xylene over m-xylene and o-xylene. The isotherm predicted with IAST is in agreement with the simulations of adsorption of the mixture of xylenes at chemical equilibrium. This implies that the adsorbed molecules compete for the same adsorption sites. Figure 12c shows the composition of the mixture of xylenes adsorbed in MTW-type zeolites compared to the composition of the mixture in the bulk phase as a function of pressure. It is observed that the phase composition of the adsorbed fluid changes as a function of the pressure. The mole fractions of p-xylene for the mixture adsorbed in MTW-type zeolites decrease from 0.6 at 0.03 bar to 0.4 at 300 bar. For m-xylene, the mole fractions increase from 0.27 at 0.03 bar to 0.4 at 300 bar. Based on the heat of adsorption at infinite dilution, there is a preferential adsorption of p-xylene > o-xylene > m-xylene. Figure 12d shows the changes in enthalpy and entropy due to the transfer of a xylene molecule between the bulk mixture and MTW-type zeolite for 0.3 and 30 bar at 523 K. At 0.3 bar, adsorption of xylenes is driven by changes in enthalpy due to the transfer of molecules between the bulk mixture and the MTW-type zeolite. For 30 bar, the adsorption of xylenes is influenced by changes in entropy. The changes in entropy and enthalpy due to the transfer of xylenes to MTW-type zeolites suggest that the preferential adsorption of p-xylene is due to entropic effects and that the preferential adsorption of m-xylene over o-xylene is due to its large mole fraction in the bulk phase. Figure 12e,f shows typical snapshots of the location of xylene molecules in the channel of an MTW-type zeolite. It is observed that m-xylene and o-xylene molecules fit tightly in the channel due to the direction of the methyl groups.
Table 1 lists a summary of the observed preferential adsorption of the single components and for the mixture at chemical equilibrium for each zeolite type considered in this work. It is observed that m-xylene is preferentially adsorbed in FAU-, MEL-, and MFI-type zeolites. The preferential adsorption of m-xylene in the FAU-type zeolites is related to the large mole fraction of m-xylene in the bulk phase, compared to the mole fraction of the other isomers. In MEL- and MFI-type zeolites, the preferential adsorption of m-xylene is related to the shape of the adsorption site in which xylene molecules are located. Xylene molecules are located in the intersections of two channel systems. The methyl groups of m-xylene are aligned with these two channel systems. For p-xylene, the methyl groups are aligned with one of the channels. One of the methyl groups of o-xylene is in alignment with one of the channels, while the other methyl group is not.
The BEA-type zeolite shows a preferential adsorption of p-xylene. Also, significant adsorption of m-xylene is observed. This is related to m-xylene molecules located in the intersection of the channels and that the methyl groups are aligned with the two intersecting channels as well as to the large mole fraction of m-xylene in the bulk mixture of xylenes. For p-xylene molecules, the methyl groups can align either with the channel direction or with an intersecting channel.
The PSD of BEA-type zeolites shows a peak of the distribution at a diameter of approx. 5.5 Å. This diameter is larger than for MFI- and MEL-type zeolites (intersections with diameters of approx. 5 Å). This suggests that the preferential adsorption of m-xylene in zeolites with an intersecting channel system depends on the size of such channel systems. If the channel intersection is large, molecules such as p-xylene can be accommodated in configurations different than in alignment with the channel direction. This influences the preferential adsorption of p-xylene over the other xylene isomers in zeolites with intersecting channel systems. The preferential adsorption of m-xylene in zeolites with intersecting channel systems occurs when the molecules fit tightly in the adsorption site.
For MRE-type zeolites, only p-xylene molecules are adsorbed in the 10-ring channel. m-Xylene and o-xylene are not adsorbed. In this zeolite type, the methyl groups of p-xylene are in alignment with the 10-ring channel. Steric effects hinder the adsorption of m-xylene and o-xylene in MRE-type zeolites. For MTW-type zeolites, p-xylene is preferentially adsorbed. At high pressures, significant loadings of m-xylene are observed. MTW-type zeolites have small 12-ring one-dimensional channels.154 In these channels, the methyl groups of p-xylene are in alignment with the channel. Also, these 12-ring channels can host m-xylene and o-xylene when one of its methyl groups is accommodated in the cavity (Figure 12f). In this case, there is an enthalpic preference for p-xylene.
MOR-type zeolites have a 12-ring channel wider than the one-dimensional channel of MTW-type zeolites (see Figure 2). In these channels, entropic effects favor the adsorption of o-xylenes. This is caused by the alignment of its methyl groups. These can be aligned with the direction of the channel or perpendicular to the channel. For the adsorption of the mixture at chemical equilibrium, this entropic effect is hindered by the other isomers in the neighborhood. The adsorption is driven by a competition between entropic and enthalpic effects.
The AFI-type zeolite has a 12-ring one-dimensional channel even wider than the channel of the MOR-type zeolite (see Figure 2). The size of such a channel can host o-xylene molecules that align perpendicular to the channel direction. In such a channel, m-xylene and p-xylene molecules accommodate in alignment with the direction of the channel. In this case, the o-xylene preference is related to entropic effects.
In zeolites with one-dimensional channel systems, the size of such channels determines the selective mechanism for a particular xylene isomer. p-Xylene is preferentially adsorbed in the 10-ring channel of MRE-type zeolites due to size exclusion of the other isomers. Entropic and enthalpic factors compete for the preferential adsorption of xylenes in the 12-ring channels of MTW-type and MOR-type zeolites. Entropic effects yield a preferential adsorption of o-xylene in the 12-ring channels of AFI-type zeolites.
In the case of the MWW-type zeolite, there is a preferential adsorption of o-xylene and significant loadings of m-xylene and p-xylene. It is observed that for the adsorption of the mixture of xylenes at chemical equilibrium, o-xylene molecules are located in the 12-ring cages. The 10-ring channels host m-xylene and p-xylene. MWW-type zeolites can accommodate xylene isomers in different channel systems depending on the size of the channel.
The insight from this work can be considered for the design and/or improvement of applications of zeolites for the processing of xylenes. However, it is known that the predictions of adsorption of aromatics in zeolites are sensitive to the flexibility of the framework,124,155 to the flexibility of aromatic molecules,156 and to the partial charges used for the models.62 These factors are not considered in this work. Models that capture the physics behind the flexibility of the zeolite and aromatics in confinements are needed.80 The insights shown here are based on the thermodynamic stability of the isomers inside the zeolite pores. Thus, diffusion limitations of xylenes in the channels of the zeolite types considered should be thoroughly assessed. As such, future work on experimental testing of these findings is encouraged.
4. Conclusions
The adsorption of xylenes as single components and as a mixture at chemical equilibrium in several zeolites has been computed by Monte Carlo simulations. The simulations show the role of the different mechanisms that drive adsorption in several zeolite types. It is observed that for zeolites with one-dimensional channels, the selectivity for a xylene isomer is determined by a competition between entropic and enthalpic effects. Each of these effects is related to the diameter of the zeolite channel. Shape and size selectivity entropic effects are predominant for small one-dimensional systems. Entropic effects due to the efficient arrangement of xylenes become relevant for large one-dimensional systems. For zeolites with two intersecting channels, the selectivity is determined by a competition between enthalpic and entropic effects. Such effects are related to the orientation of the methyl groups of the competing xylenes. m-Xylene is preferentially adsorbed if xylenes fit tightly in the intersection of the channels. If the intersection is much larger than the adsorbed molecules, p-xylene molecules are adsorbed and can be efficiently arranged in the void volumes of the zeolite. The simulations show that different selectivity mechanisms are predominant when the vapor phase mixture of xylenes is adsorbed in a zeolite compared to the adsorption from the liquid phase. This suggests that screening studies that consider adsorption only from a vapor phase may have overlooked well-performing candidates for C8 aromatics processing. MRE-type and AFI-type zeolites exclusively adsorb p-xylene and o-xylene from the mixture of xylenes in the liquid phase, respectively. These zeolite types show potential to be used as high-performing molecular sieves for xylene separation and catalysis.
Acknowledgments
The authors gratefully acknowledge the financial support from Shell Global Solutions International B.V. This work was sponsored by NWO Exacte Wetenschappen (Physical Sciences) for the use of supercomputer facilities, with financial support from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research, NWO). T.J.H.V. acknowledges NWO-CW for a VICI grant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcc.0c09411.
All force field parameters used in this work; change in the ideal gas free energy ΔGA↔Bideal of a reaction A ↔ B computed using tabulated data for the reactions m-xylene ↔ o-xylene and m-xylene ↔ p-xylene; computed compositions of mixtures of xylene isomers at chemical equilibrium as a function of pressure at 523 K; computed fugacity coefficients of xylene isomers as a function of pressure at 532 K; computed densities of the mixture of xylene isomers at chemical equilibrium as a function of pressure at 523 K; computed heats of adsorption at infinite dilution of xylene isomers at 523 K in the zeolite types considered in this work; selectivities for adsorption of xylenes from the mixture at chemical equilibrium at 523 K in the zeolite frameworks considered in this work; computed changes in free energy ΔGtr,i, changes in enthalpy ΔHtr,i, and changes in entropy ΔStr,i, due to transferring one xylene i between the bulk phase at chemical equilibrium at 523 K to the zeolite types considered in this work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Niziolek A. M.; Onel O.; Floudas C. A. Production of benzene, toluene, and xylenes from natural gas via methanol: Process synthesis and global optimization. AIChE J. 2016, 62, 1531–1556. 10.1002/aic.15144. [DOI] [Google Scholar]
- Yang Y.; Bai P.; Guo X. Separation of xylene isomers: A review of recent advances in materials. Ind. Eng. Chem. Res. 2017, 56, 14725–14753. 10.1021/acs.iecr.7b03127. [DOI] [Google Scholar]
- Das J.; Bhat Y.; Halgeri A.. Aromatization of C4-C6 Hydrocarbons to Benzene, Toluene and Para Xylene Over Pore Size Controlled ZnO-HZSM-5 Zeolite. In Recentadvances in Basic and Applied Aspects of Industrial Catalysis; Rao T. S. R. P.; Dhar G. M., Eds.; Elsevier: Amsterdam, 1998; Vol. 113, pp 447–453. [Google Scholar]
- Egan C. J.; Luthy R. V. Separation of xylenes. Ind. Eng. Chem. 1955, 47, 250–253. 10.1021/ie50542a028. [DOI] [Google Scholar]
- Caro J.; Kärger J. From computer design to gas separation. Nat. Mater. 2020, 19, 374–375. 10.1038/s41563-020-0630-y. [DOI] [PubMed] [Google Scholar]
- Tomás R. A. F.; Bordado J. C. M.; Gomes J. F. P. p-Xylene oxidation to terephthalic acid: A literature review oriented toward process optimization and development. Chem. Rev. 2013, 113, 7421–7469. 10.1021/cr300298j. [DOI] [PubMed] [Google Scholar]
- Maneffa A.; Priecel P.; Lopez-Sanchez J. A. Biomass-derived renewable aromatics: Selective routes and outlook for p-xylene commercialisation. ChemSusChem 2016, 9, 2736–2748. 10.1002/cssc.201600605. [DOI] [PubMed] [Google Scholar]
- Minceva M.; Rodrigues A. E. Adsorption of xylenes on faujasite-type zeolite: Equilibrium and kinetics in batch adsorber. Chem. Eng. Res. Des. 2004, 82, 667–681. 10.1205/026387604323142739. [DOI] [Google Scholar]
- Cannella W. J.Xylenes and Ethylbenzene; Kirk-Othmer Encyclopedia of Chemical Technology; Wiley-Interscience: New York, NY, 2007. [Google Scholar]
- Young L. B.; Butter S. A.; Kaeding W. W. Shape selective reactions with zeolite catalysts: III. Selectivity in xylene isomerization, toluene-methanol alkylation, and toluene disproportionation over ZSM-5 zeolite catalysts. J. Catal. 1982, 76, 418–432. 10.1016/0021-9517(82)90271-8. [DOI] [Google Scholar]
- Stoye D.; Freitag W.. Paints, Coatings and Solvents, 2nd ed.; Wiley-Blackwell: Weinheim, 2007. [Google Scholar]
- Fishbein L. An overview of environmental and toxicological aspects of aromatic hydrocarbons III. Xylene. Sci. Total Environ. 1985, 43, 165–183. 10.1016/0048-9697(85)90039-7. [DOI] [PubMed] [Google Scholar]
- Kandyala R.; Raghavendra S. P.; Rajasekharan S. Xylene: An overview of its health hazards and preventive measures. J. Oral Maxillofac. Pathol. 2010, 14, 1–5. 10.4103/0973-029X.64299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denayer J. F. M.; De Vos D.; Leflaive P.. Separation of Xylene Isomers. In Metal-Organic Frameworks: Applications from Catalysis to Gas Storage; Farrusseng D., Ed.; John Wiley & Sons: Weinheim, 2011; Chapter 8, pp 171–190. [Google Scholar]
- Silva M. S. P.; Mota J. P. B.; Rodrigues A. E. Fixed-bed adsorption of aromatic C8 isomers: Breakthrough experiments, modeling and simulation. Sep. Purif. Technol. 2012, 90, 246–256. 10.1016/j.seppur.2012.02.034. [DOI] [Google Scholar]
- Gu Z.-Y.; Yan X.-P. Metal-organic framework MIL-101 for high-resolution gas-chromatographic separation of xylene isomers and ethylbenzene. Angew. Chem., Int. Ed. 2010, 49, 1477–1480. 10.1002/anie.200906560. [DOI] [PubMed] [Google Scholar]
- Ashraf M. T.; Chebbi R.; Darwish N. A. Process of p-xylene production by highly selective methylation of toluene. Ind. Eng. Chem. Res. 2013, 52, 13730–13737. 10.1021/ie401156x. [DOI] [Google Scholar]
- Ma Y.; Zhang F.; Yang S.; Lively R. P. Evidence for entropic diffusion selection of xylene isomers in carbon molecular sieve membranes. J. Membr. Sci. 2018, 564, 404–414. 10.1016/j.memsci.2018.07.040. [DOI] [Google Scholar]
- Dye S. R.; Ng K. M. Fractional crystallization: Design alternatives and tradeoffs. AIChE J. 1995, 41, 2427–2438. 10.1002/aic.690411109. [DOI] [Google Scholar]
- Mohameed H.; Jdayil B. A.; Takrouri K. Separation of para-xylene from xylene mixture via crystallization. Chem. Eng. Process. 2007, 46, 25–36. 10.1016/j.cep.2006.04.002. [DOI] [Google Scholar]
- Zhou Y.; Wu J.; Lemmon E. W. Thermodynamic properties of o-xylene, m-xylene, p-xylene, and ethylbenzene. J. Phys. Chem. Ref. Data 2012, 41, 023103 10.1063/1.3703506. [DOI] [Google Scholar]
- Humphrey J. L.; Keller G.. Separation Process Technology; McGraw-Hill: New York, 1997. [Google Scholar]
- Sholl D. S.; Lively R. P. Seven chemical separations to change the world. Nature 2016, 532, 435–437. 10.1038/532435a. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Taylor M. K.; Jiang L.; Ren H.; Zhu G. Light hydrocarbon separations using porous organic framework materials. Chem. - Eur. J. 2020, 26, 3205–3221. 10.1002/chem.201904455. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Peh S. B.; Zhao D. Alternatives to cryogenic distillation: Advanced porous materials in adsorptive light olefin/paraffin separations. Small 2019, 15, 1900058 10.1002/smll.201900058. [DOI] [PubMed] [Google Scholar]
- Cui W.-G.; Hu T.-L.; Bu X.-H. Metal-organic framework materials for the separation and purification of light hydrocarbons. Adv. Mater. 2020, 32, 1806445 10.1002/adma.201806445. [DOI] [PubMed] [Google Scholar]
- Rodrigues A. E.; Pereira C.; Minceva M.; Pais L. S.; Ribeiro A. M.; Ribeiro A.; Silva M.; Graça N.; Santos J. C.. The Parex Process for the Separation of p-Xylene. In Simulated Moving Bed Technology; Rodrigues A. E.; Pereira C.; Minceva M.; Pais L. S.; Ribeiro A. M.; Ribeiro A.; Silva M.; Graça N.; Santos J. C., Eds.; Butterworth-Heinemann: Oxford, 2015; pp 117–144. [Google Scholar]
- Broughton D. B.; Neuzil R. W.; Pharis J. M.; Brearley C. S. The Parex process for recovering paraxylene. Chem. Eng. Prog. 1970, 66, 70–75. [Google Scholar]
- Ash G.; Barth K.; Hotier G.; Mank L.; Renard P. Eluxyl: a new paraxylene separation process. Rev. Inst. Fr. Pet. 1994, 49, 541–549. 10.2516/ogst:1994035. [DOI] [Google Scholar]
- Jin W.; Wankat P. C. Hybrid simulated moving bed processes for the purification of p-xylene. Sep. Sci. Technol. 2007, 42, 669–700. 10.1080/01496390601173978. [DOI] [Google Scholar]
- Minceva M.; Rodrigues A. E. Understanding and revamping of industrial scale SMB units for p-xylene separation. AIChE J. 2007, 53, 138–149. 10.1002/aic.11062. [DOI] [Google Scholar]
- Cottier V.; Bellat J.-P.; Simonot-Grange M.-H.; Méthivier A. Adsorption of p-xylene/m-xylene gas mixtures on BaY and NaY zeolites. Coadsorption equilibria and selectivities. J. Phys. Chem. B 1997, 101, 4798–4802. 10.1021/jp9640033. [DOI] [Google Scholar]
- Bellat J.-P.; Simonot-Grange M.-H. Adsorption of gaseous p-xylene and m-xylene on NaY, KY, and BaY zeolites. Part 2: Modeling. Enthalpies and entropies of adsorption. Zeolites 1995, 15, 219–227. 10.1016/0144-2449(94)00048-W. [DOI] [Google Scholar]
- Cottier V.; Pilverdier E.; Simonot-Grange M.-H.; Bellat J.-P. Derivative enthalpies of adsorption of p-xylene and m-xylene onto NaY and BaY Zeolites at 150 °C: Contribution to the prediction of adsorption selectivity. J. Therm. Anal. Calorim. 1999, 58, 121–128. 10.1023/A:1010191820215. [DOI] [Google Scholar]
- Daems I.; Leflaive P.; Méthivier A.; Denayer J.; Baron G.. Size and Packing Related Adsorption Effects in the Liquid Phase Adsorption of Aromatics and Alkenes on FAU Type Zeolites. In MolecularSieves: From Basic Research to Industrial Applications; čejka J.; Žilková N.; Nachtigall P., Eds.; Elsevier, 2005; Vol. 158, pp 1177–1184. [Google Scholar]
- Daems I.; Leflaive P.; Méthivier A.; Baron G. V.; Denayer J. F. Influence of Si:Al-ratio of faujasites on the adsorption of alkanes, alkenes and aromatics. Microporous Mesoporous Mater. 2006, 96, 149–156. 10.1016/j.micromeso.2006.06.029. [DOI] [Google Scholar]
- Gu Z.-Y.; Jiang D.-Q.; Wang H.-F.; Cui X.-Y.; Yan X.-P. Adsorption and separation of xylene isomers and ethylbenzene on two Zn-terephthalate metal-organic frameworks. J. Phys. Chem. C 2010, 114, 311–316. 10.1021/jp9063017. [DOI] [Google Scholar]
- Torres-Knoop A.; Krishna R.; Dubbeldam D. Separating xylene isomers by commensurate stacking of p-xylene within channels of MAF-X8. Angew. Chem. 2014, 126, 7908–7912. 10.1002/ange.201402894. [DOI] [PubMed] [Google Scholar]
- Zeolites in Industrial Separation and Catalysis, 1st ed.; Kulprathipanja S., Ed.; John Wiley & Sons: Weinheim, 2010. [Google Scholar]
- Vermeiren W.; Gilson J. P. Impact of zeolites on the petroleum and petrochemical industry. Top. Catal. 2009, 52, 1131–1161. 10.1007/s11244-009-9271-8. [DOI] [Google Scholar]
- Ackley M. W.; Rege S. U.; Saxena H. Application of natural zeolites in the purification and separation of gases. Microporous Mesoporous Mater. 2003, 61, 25–42. 10.1016/S1387-1811(03)00353-6. [DOI] [Google Scholar]
- Ramirez A.; Dutta Chowdhury A.; Dokania A.; Cnudde P.; Caglayan M.; Yarulina I.; Abou-Hamad E.; Gevers L.; Ould-Chikh S.; De Wispelaere K.; et al. Effect of zeolite topology and reactor configuration on the direct conversion of CO2 to light olefins and aromatics. ACS Catal. 2019, 9, 6320–6334. 10.1021/acscatal.9b01466. [DOI] [Google Scholar]
- Shi J.; Wang Y.; Yang W.; Tang Y.; Xie Z. Recent advances of pore system construction in zeolite-catalyzed chemical industry processes. Chem. Soc. Rev. 2015, 44, 8877–8903. 10.1039/C5CS00626K. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gil V.; Noya E. G.; Sanz A.; Khatib S. J.; Guil J. M.; Lomba E.; Marguta R.; Valencia S. Experimental and simulation studies of the stepped adsorption of toluene on pure-silica MEL zeolite. J. Phys. Chem. C 2016, 120, 8640–8652. 10.1021/acs.jpcc.6b00402. [DOI] [Google Scholar]
- Martínez A.; Arribas M. A.; Moussa S.. Application of Zeolites in the Production of LightOlefins and BTX Petrochemical Intermediates. In Properties and Applications; čejka J.; Morris R. E.; Nachtigall P., Eds.; The Royal Society of Chemistry: London, 2017; pp 351–408. [Google Scholar]
- Uslamin E. A.; Saito H.; Kosinov N.; Pidko E.; Sekine Y.; Hensen E. J. M. Aromatization of ethylene over zeolite-based catalysts. Catal. Sci. Technol. 2020, 10, 2774–2785. 10.1039/C9CY02108F. [DOI] [Google Scholar]
- Rigutto M.Cracking and Hydrocracking. In Zeolites and Catalysis; čejka J.; Corma A.; Zones S., Eds.; John Wiley & Sons: Weinheim, 2010; pp 547–584. [Google Scholar]
- Vogt E. T. C.; Weckhuysen B. M. Fluid catalytic cracking: recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. 10.1039/C5CS00376H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Li L.; Yu J. Applications of zeolites in sustainable chemistry. Chem. 2017, 3, 928–949. 10.1016/j.chempr.2017.10.009. [DOI] [Google Scholar]
- Davis M. E. Zeolites and molecular sieves: not just ordinary catalysts. Ind. Eng. Chem. Res. 1991, 30, 1675–1683. 10.1021/ie00056a001. [DOI] [Google Scholar]
- Coasne B.; Haines J.; Levelut C.; Cambon O.; Santoro M.; Gorelli F.; Garbarino G. Enhanced mechanical strength of zeolites by adsorption of guest molecules. Phys. Chem. Chem. Phys. 2011, 13, 20096–20099. 10.1039/c1cp22477h. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cao H.; Yu J. Toward a new era of designed synthesis of nanoporous zeolitic materials. ACS Nano 2018, 12, 4096–4104. 10.1021/acsnano.8b02625. [DOI] [PubMed] [Google Scholar]
- Smit B.; Maesen T. L. M. Molecular simulations of zeolites: Adsorption, diffusion, and shape selectivity. Chem. Rev. 2008, 108, 4125–4184. 10.1021/cr8002642. [DOI] [PubMed] [Google Scholar]
- Denayer J. F. M.; Ocakoglu R. A.; De Meyer K.; Baron G. V. Exploiting pore or cavity size and shape in separating linear and branched hydrocarbons by inverse selectivity: Enthalpy, entropy and packing effects. Adsorption 2005, 11, 49–53. 10.1007/s10450-005-5897-4. [DOI] [Google Scholar]
- Smit B.; Maesen T. L. M. Towards a molecular understanding of shape selectivity. Nature 2008, 451, 671–678. 10.1038/nature06552. [DOI] [PubMed] [Google Scholar]
- Schenk M.; Calero S.; Maesen T. L. M.; Vlugt T. J. H.; van Benthem L. L.; Verbeek M. G.; Schnell B.; Smit B. Shape selectivity through entropy. J. Catal. 2003, 214, 88–99. 10.1016/S0021-9517(03)00023-X. [DOI] [Google Scholar]
- Krishna R. Elucidation and characterization of entropy effects in mixture separations with micro-porous crystalline adsorbents. Sep. Purif. Technol. 2019, 215, 227–241. 10.1016/j.seppur.2019.01.014. [DOI] [Google Scholar]
- Keffer D.; Davis H. T.; McCormick A. V. Effect of loading and nanopore shape on binary adsorption selectivity. J. Phys. Chem. A 1996, 100, 638–645. 10.1021/jp951875g. [DOI] [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulation, 2nd ed.; Academic Press: San Diego, CA, 2002. [Google Scholar]
- Keil F. J.Adsorption and Phase Behaviour in Nanochannels and Nanotubes; Dunne L. J.; Manos G., Eds.; Springer: Dordrecht, 2010. [Google Scholar]
- Babarao R.; Jiang J.. Molecular Computations of Adsorption in Nanoporous Materials. In Adsorptionand Phase Behaviour in Nanochannels and Nanotubes; Dunne L. J.; Manos G., Eds.; Springer: Dordrecht, 2010. [Google Scholar]
- Vlugt T. J. H.; García-Pérez E.; Dubbeldam D.; Ban S.; Calero S. Computing the heat of adsorption using molecular simulations: The effect of strong Coulombic interactions. J. Chem. Theory Comput. 2008, 4, 1107–1118. 10.1021/ct700342k. [DOI] [PubMed] [Google Scholar]
- González-Galán C.; Luna-Triguero A.; Vicent-Luna J.; Zaderenko A.; Sławek A.; Sánchez-de-Armas R.; Calero S. Exploiting the π-bonding for the separation of benzene and cyclohexane in zeolites. Chem. Eng. J. 2020, 398, 125678 10.1016/j.cej.2020.125678. [DOI] [Google Scholar]
- Perez-Carbajo J.; Gómez-Álvarez P.; Bueno-Perez R.; Merkling P. J.; Calero S. Optimisation of the Fischer-Tropsch process using zeolites for tail gas separation. Phys. Chem. Chem. Phys. 2014, 16, 5678–5688. 10.1039/c3cp55255a. [DOI] [PubMed] [Google Scholar]
- Rezlerová E.; Zukal A.; čejka J.; Siperstein F. R.; Brennan J. K.; Lísal M. Adsorption and diffusion of C1 to C4 alkanes in dual-porosity zeolites by molecular simulations. Langmuir 2017, 33, 11126–11137. 10.1021/acs.langmuir.7b01772. [DOI] [PubMed] [Google Scholar]
- Bai P.; Jeon M. Y.; Ren L.; Knight C.; Deem M. W.; Tsapatsis M.; Siepmann J. I. Discovery of optimal zeolites for challenging separations and chemical transformations using predictive materials modeling. Nat. Commun. 2015, 6, 5912 10.1038/ncomms6912. [DOI] [PubMed] [Google Scholar]
- Fuchs A. H.; Cheetham A. K. Adsorption of guest molecules in zeolitic materials: Computational aspects. J. Phys. Chem. B 2001, 105, 7375–7383. 10.1021/jp010702q. [DOI] [Google Scholar]
- Josephson T. R.; Dauenhauer P. J.; Tsapatsis M.; Siepmann J. I. Adsorption of furan, hexanoic acid, and alkanes in a hierarchical zeolite at reaction conditions: Insights from molecular simulations. J. Comput. Sci. 2021, 48, 101267 10.1016/j.jocs.2020.101267. [DOI] [Google Scholar]
- Becker T. M.; Luna-Triguero A.; Vicent-Luna J. M.; Lin L.-C.; Dubbeldam D.; Calero S.; Vlugt T. J. H. Potential of polarizable force fields for predicting the separation performance of small hydrocarbons in M-MOF-74. Phys. Chem. Chem. Phys. 2018, 20, 28848–28859. 10.1039/C8CP05750H. [DOI] [PubMed] [Google Scholar]
- Lamia N.; Jorge M.; Granato M. A.; Almeida Paz F. A.; Chevreau H.; Rodrigues A. E. Adsorption of propane, propylene and isobutane on a metal-organic framework: Molecular simulation and experiment. Chem. Eng. Sci. 2009, 64, 3246–3259. 10.1016/j.ces.2009.04.010. [DOI] [Google Scholar]
- Lennox M. J.; Düren T. Understanding the kinetic and thermodynamic origins of xylene separation in UiO-66(Zr) via molecular simulation. J. Phys. Chem. C 2016, 120, 18651–18658. 10.1021/acs.jpcc.6b06148. [DOI] [Google Scholar]
- Cui W.-G.; Hu T.-L.; Bu X.-H. Metal-organic framework materials for the separation and purification of light hydrocarbons. Adv. Mater. 2020, 32, 1806445 10.1002/adma.201806445. [DOI] [PubMed] [Google Scholar]
- Calero S.Modeling of Transport and Acessibility in Zeolites. In Zeolites and Catalysis; Čejka J.; Corma A.; Zones S., Eds.; John Wiley & Sons: Weinheim, 2010; pp 335–360. [Google Scholar]
- Fang H.; Demir H.; Kamakoti P.; Sholl D. S. Recent developments in first-principles force fields for molecules in nanoporous materials. J. Mater. Chem. A 2014, 2, 274–291. 10.1039/C3TA13073H. [DOI] [Google Scholar]
- Ungerer P.; Tavitian B.; Boutin A.. Applications of Molecular Simulation in the Oil and Gas Industry - Monte-Carlo Methods, 1st ed.; Editions Technip: Paris, 2005. [Google Scholar]
- Snurr R. Q.; Bell A. T.; Theodorou D. N. Prediction of adsorption of aromatic hydrocarbons in silicalite from grand-canonical Monte Carlo simulations with biased insertions. J. Phys. Chem. B 1993, 97, 13742–13752. 10.1021/j100153a051. [DOI] [Google Scholar]
- Torres-Knoop A.; Heinen J.; Krishna R.; Dubbeldam D. Entropic separation of styrene/ethylbenzene mixtures by exploitation of subtle differences in molecular configurations in ordered crystalline nanoporous adsorbents. Langmuir 2015, 31, 3771–3778. 10.1021/acs.langmuir.5b00363. [DOI] [PubMed] [Google Scholar]
- Chempath S.; Snurr R. Q.; Low J. J. Molecular modeling of binary liquid-phase adsorption of aromatics in silicalite. AIChE J. 2004, 50, 463–469. 10.1002/aic.10040. [DOI] [Google Scholar]
- Zeng Y.; Moghadam P. Z.; Snurr R. Q. Pore size dependence of adsorption and separation of thiophene/benzene mixtures in zeolites. J. Phys. Chem. C 2015, 119, 15263–15273. 10.1021/acs.jpcc.5b03156. [DOI] [Google Scholar]
- Caro-Ortiz S.; Zuidema E.; Dekker D.; Rigutto M.; Dubbeldam D.; Vlugt T. J. H. Adsorption of aromatics in MFI-type zeolites: Experiments and framework flexibility in Monte Carlo simulations. J. Phys. Chem. C 2020, 124, 21782. 10.1021/acs.jpcc.0c06096. [DOI] [Google Scholar]
- Lachet V.; Boutin A.; Tavitian B.; Fuchs H. Grand-canonical Monte Carlo simulations of adsorption of mixtures of xylene molecules in faujasite zeolites. Faraday Discuss. 1997, 106, 307–323. 10.1039/a701490b. [DOI] [Google Scholar]
- Zheng H.; Zhao L.; Ji J.; Gao J.; Xu C.; Luck F. Unraveling the adsorption mechanism of mono- and diaromatics in faujasite zeolite. ACS Appl. Mater. Interfaces 2015, 7, 10190–10200. 10.1021/acsami.5b00399. [DOI] [PubMed] [Google Scholar]
- Lucena S. M. P.; Snurr R. Q.; Cavalcante C. L. Monte Carlo and energy minimization studies of binary xylene adsorption in AEL and AFI networks. Adsorption 2007, 13, 477–484. 10.1007/s10450-007-9031-7. [DOI] [Google Scholar]
- Zhao F.; Sun X.; Lu R.; Kang L. Adsorption of methanol, methanal, toluene, ethylbenzene, and styrene in zeolites: A grand-canonical Monte Carlo simulation study. Can. J. Chem. 2017, 95, 1241–1247. 10.1139/cjc-2017-0218. [DOI] [Google Scholar]
- He P.; Liu H.; Li Y.; Zhu J.; Huang S.; Lei Z.; Wang P.; Tian H. Adsorption of benzene and propene in zeolite MCM-22: A grand-canonical Monte Carlo study. Adsorption 2012, 18, 31–42. 10.1007/s10450-011-9378-7. [DOI] [Google Scholar]
- Lachet V.; Boutin A.; Tavitian B.; Fuchs A. H. Molecular simulation of p-xylene and m-xylene adsorption in Y zeolites. Single components and binary mixtures study. Langmuir 1999, 15, 8678–8685. 10.1021/la990305v. [DOI] [Google Scholar]
- Chempath S.; Denayer J. F. M.; De Meyer K. M. A.; Baron G. V.; Snurr R. Q. Adsorption of liquid-phase alkane mixtures in silicalite: Simulations and experiment. Langmuir 2004, 20, 150–156. 10.1021/la035362+. [DOI] [PubMed] [Google Scholar]
- Torres-Knoop A.; Balaji S. P.; Vlugt T. J. H.; Dubbeldam D. A comparison of advanced Monte Carlo methods for open systems: CFCMC vs CBMC. J. Chem. Theory Comput. 2014, 10, 942–952. 10.1021/ct4009766. [DOI] [PubMed] [Google Scholar]
- Perego C.; Ingallina P. Recent advances in the industrial alkylation of aromatics: new catalysts and new processes. Catal. Today 2002, 73, 3–22. 10.1016/S0920-5861(01)00511-9. [DOI] [Google Scholar]
- Rasouli M.; Yaghobi N.; Chitsazan S.; Sayyar M. H. Effect of nanocrystalline zeolite Na-Y on meta-xylene separation. Microporous Mesoporous Mater. 2012, 152, 141–147. 10.1016/j.micromeso.2011.11.045. [DOI] [Google Scholar]
- Talu O. Needs, status, techniques and problems with binary gas adsorption experiments. Adv. Colloid Interface Sci. 1998, 76–77, 227–269. 10.1016/S0001-8686(98)00048-7. [DOI] [Google Scholar]
- Siperstein F. R.; Myers A. L. Mixed-gas adsorption. AIChE J. 2001, 47, 1141–1159. 10.1002/aic.690470520. [DOI] [Google Scholar]
- Siepmann J. I.; Brennecke J. F.; Allen D. T.; Klein M. T.; Savage P. E.; Schatz G. C.; Winnik F. M. ACS virtual issue on multicomponent systems: Absorption, adsorption, and diffusion. J. Chem. Eng. Data 2018, 63, 3651. 10.1021/acs.jced.8b00842. [DOI] [Google Scholar]
- Hens R.; Rahbari A.; Caro-Ortiz S.; Dawass N.; Erdós M.; Poursaeidesfahani A.; Salehi H. S.; Celebi A. T.; Ramdin M.; Moultos O. A.; et al. Brick-CFCMC: Open source software for Monte Carlo simulations of phase and reaction equilibria using the Continuous Fractional Component Method. J. Chem. Inf. Model. 2020, 60, 2678–2682. 10.1021/acs.jcim.0c00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit B. Grand-canonical Monte Carlo simulations of chain molecules: adsorption isotherms of alkanes in zeolites. Mol. Phys. 1995, 85, 153–172. 10.1080/00268979500101011. [DOI] [Google Scholar]
- Collins D. J.; Medina R. J.; Davis B. H. Xylene isomerization by ZSM-5 zeolite catalyst. Can. J. Chem. Eng. 1983, 61, 29–35. 10.1002/cjce.5450610104. [DOI] [Google Scholar]
- Chang X.; Li Y.; Zeng Z. Kinetics study of the isomerization of xylene on HZSM-5 zeolite. 1. Kinetics model and reaction mechanism. Ind. Eng. Chem. Res. 1992, 31, 187–192. 10.1021/ie00001a027. [DOI] [Google Scholar]
- Gonçalves J. C.; Rodrigues A. E. Xylene isomerization in the liquid phase using large-pore zeolites. Chem. Eng. Technol. 2016, 39, 225–232. 10.1002/ceat.201500219. [DOI] [Google Scholar]
- Goła̧bek K.; Tarach K. A.; Góra-Marek K. Xylenes transformation over zeolites ZSM-5 ruled by acidic properties. Spectrochim. Acta A 2018, 192, 361–367. 10.1016/j.saa.2017.11.028. [DOI] [PubMed] [Google Scholar]
- Ou J. D.-Y.; Ross A. D.; Levin D.; Kalyanaraman M.; Lai W. F.. Xylene Isomerization Process and Catalyst Therefor. US Patent US8697929B22014.
- Magne-Drisch J.; Alario F.; Joly J.-F.; Minkkinen A.; Merlen E.. Process for the Production of Paraxylene that Comprises an Adsorption Stage, A Liquid Phase Isomerization Stage and a Gas Phase Isomerization Stage with an EUO-type Zeolite. US Patent US6448459B22002.
- Wolff L.; Leflaive P.; Methivier A.. Process for Co-producing Para-xylene and Styrene. US Patent US7592499B22009.
- Bingre R.; Louis B.; Nguyen P. An overview on zeolite shaping technology and solutions to overcome diffusion limitations. Catalysts 2018, 8, 163 10.3390/catal8040163. [DOI] [Google Scholar]
- Van Speybroeck V.; Hemelsoet K.; Joos L.; Waroquier M.; Bell R. G.; Catlow C. R. A. Advances in theory and their application within the field of zeolite chemistry. Chem. Soc. Rev. 2015, 44, 7044–7111. 10.1039/C5CS00029G. [DOI] [PubMed] [Google Scholar]
- Karl Johnson J.; Panagiotopoulos A. Z.; Gubbins K. E. Reactive canonical Monte Carlo. Mol. Phys. 1994, 81, 717–733. 10.1080/00268979400100481. [DOI] [Google Scholar]
- Smith W. R.; Triska B. The reaction ensemble method for the computer simulation of chemical and phase equilibria. I. Theory and basic examples. J. Chem. Phys. 1994, 100, 3019–3027. 10.1063/1.466443. [DOI] [Google Scholar]
- Heath Turner C.; Brennan J. K.; Lísal M.; Smith W. R.; Karl Johnson J.; Gubbins K. E. Simulation of chemical reaction equilibria by the reaction ensemble Monte Carlo method: A review. Mol. Simul. 2008, 34, 119–146. 10.1080/08927020801986564. [DOI] [Google Scholar]
- Poursaeidesfahani A.; Hens R.; Rahbari A.; Ramdin M.; Dubbeldam D.; Vlugt T. J. H. Efficient application of Continuous Fractional Component Monte Carlo in the reaction ensemble. J. Chem. Theory Comput. 2017, 13, 4452–4466. 10.1021/acs.jctc.7b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursaeidesfahani A.; Torres-Knoop A.; Dubbeldam D.; Vlugt T. J. H. Direct free energy calculation in the Continuous Fractional Component Gibbs ensemble. J. Chem. Theory Comput. 2016, 12, 1481–1490. 10.1021/acs.jctc.5b01230. [DOI] [PubMed] [Google Scholar]
- Shi W.; Maginn E. J. Continuous Fractional Component Monte Carlo: An adaptive biasing method for open system atomistic simulations. J. Chem. Theory Comput. 2007, 3, 1451–1463. 10.1021/ct7000039. [DOI] [PubMed] [Google Scholar]
- Rahbari A.; Hens R.; Ramdin M.; Moultos O. A.; Dubbeldam D.; Vlugt T. J. H. Recent advances in the continuous fractional component Monte Carlo methodology. Mol. Simul. 2020, 29, 20 10.1080/08927022.2020.1828585. [DOI] [Google Scholar]
- Rahbari A.; Hens R.; Nikolaidis I. K.; Poursaeidesfahani A.; Ramdin M.; Economou I. G.; Moultos O. A.; Dubbeldam D.; Vlugt T. J. H. Computation of partial molar properties using continuous fractional component Monte Carlo. Mol. Phys. 2018, 116, 3331–3344. 10.1080/00268976.2018.1451663. [DOI] [Google Scholar]
- Dubbeldam D.; Calero S.; Ellis D. E.; Snurr R. Q. RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 2016, 42, 81–101. 10.1080/08927022.2015.1010082. [DOI] [Google Scholar]
- Chase M. W.NIST-JANAF Thermodynamic Tables 4th ed.; Journal of Physical and Chemical Reference Data Monographs; American Chemical Society, American Institute of Physics, 1998. [Google Scholar]
- McQuarrie D. A.; Simon J. D.; Cox H. A.; Choi J.. Physical Chemistry: A Molecular Approach; University Science Books: Herndon, VA, 1997. [Google Scholar]
- Mullen R. G.; Maginn E. J. Reaction ensemble Monte Carlo simulation of xylene isomerization in bulk phases and under confinement. J. Chem. Theory Comput. 2017, 13, 4054–4062. 10.1021/acs.jctc.7b00498. [DOI] [PubMed] [Google Scholar]
- Prosen E. J.; Johnson W. H.; Rossini F. D. Heats of combustion and formation at 25 degrees C of the alkylbenzenes through C10H14, and of the higher normal monoalkylbenzenes. J. Res. Natl. Bur. Stand. 1946, 36, 455–461. 10.6028/jres.036.025. [DOI] [Google Scholar]
- Pitzer K. S.; Scott D. W. The thermodynamics and molecular structure of benzene and its methyl derivatives. J. Am. Chem. Soc. 1943, 65, 803–829. 10.1021/ja01245a019. [DOI] [Google Scholar]
- Draeger J. A. The methylbenzenes II. Fundamental vibrational shifts, statistical thermodynamic functions, and properties of formation. J. Chem. Thermodyn. 1985, 17, 263–275. 10.1016/0021-9614(85)90020-5. [DOI] [Google Scholar]
- Chao J.; Hall K. R.; Marsh K. N.; Wilhoit R. C. Thermodynamic properties of key organic oxygen compounds in the carbon range C1 to C4. Part 2. Ideal gas properties. J. Phys. Chem. Ref. Data 1986, 15, 1369–1436. 10.1063/1.555769. [DOI] [Google Scholar]
- Dubbeldam D.; Torres-Knoop A.; Walton K. S. On the inner workings of Monte Carlo codes. Mol. Simul. 2013, 39, 1253–1292. 10.1080/08927022.2013.819102. [DOI] [Google Scholar]
- Baerlocher C.; McCusker L. B.. Database of Zeolite Structures. http://www.iza-structure.org/databases (accessed May 4, 2020).
- Bai P.; Tsapatsis M.; Siepmann J. I. TraPPE-zeo: Transferable potentials for phase equilibria force field for all-silica zeolites. J. Phys. Chem. C 2013, 117, 24375–24387. 10.1021/jp4074224. [DOI] [Google Scholar]
- Caro-Ortiz S.; Zuidema E.; Rigutto M.; Dubbeldam D.; Vlugt T. J. H. Effects of framework flexibility on the adsorption and diffusion of aromatics in MFI-type zeolites. J. Phys. Chem. C 2020, 124, 24488–24499. 10.1021/acs.jpcc.0c08054. [DOI] [Google Scholar]
- Caro-Ortiz S.; Hens R.; Zuidema E.; Rigutto M.; Dubbeldam D.; Vlugt T. J. H. Molecular simulation of the vapor-liquid equilibria of xylene mixtures: Force field performance, and Wolf vs. Ewald for electrostatic interactions. Fluid Phase Equilib. 2019, 485, 239–247. 10.1016/j.fluid.2018.12.006. [DOI] [Google Scholar]
- Caro-Ortiz S.; Hens R.; Zuidema E.; Rigutto M.; Dubbeldam D.; Vlugt T. J. H. Corrigendum to “Molecular simulation of the vapor-liquid equilibria of xylene mixtures: Force field performance, and Wolf vs. Ewald for electrostatic Interactions” [Fluid Phase Equilib.] 485 (2019) 239–247. Fluid Phase Equilib. 2020, 506, 112370 10.1016/j.fluid.2019.112370. [DOI] [Google Scholar]
- Jorgensen W. L.; Laird E. R.; Nguyen T. B.; Tirado-Rives J. Monte Carlo simulations of pure liquid substituted benzenes with OPLS potential functions. J. Comput. Chem. 1993, 14, 206–215. 10.1002/jcc.540140208. [DOI] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Snurr R. Q.; Bell A. T.; Theodorou D. N. A hierarchical atomistic/lattice simulation approach for the prediction of adsorption thermodynamics of benzene in silicalite. J. Phys. Chem. C 1994, 98, 5111–5119. 10.1021/j100070a028. [DOI] [Google Scholar]
- Wick C. D.; Martin M. G.; Siepmann J. I. Transferable potentials for phase equilibria. 4. United-atom description of linear and branched alkenes and alkylbenzenes. J. Phys. Chem. B 2000, 104, 8008–8016. 10.1021/jp001044x. [DOI] [Google Scholar]
- Martin M. G.; Siepmann J. I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. 10.1021/jp972543+. [DOI] [Google Scholar]
- Lachet V.; Buttefey S.; Boutin A.; Fuchs A. H. Molecular simulation of adsorption equilibria of xylene isomer mixtures in faujasite zeolites. A study of the cation exchange effect on adsorption selectivity. Phys. Chem. Chem. Phys. 2001, 3, 80–86. 10.1039/b007281h. [DOI] [Google Scholar]
- Allen M. P.; Tildesley D.. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, 2017. [Google Scholar]
- Jablonka K. M.; Ongari D.; Smit B. Applicability of tail corrections in the molecular simulations of porous materials. J. Chem. Theory Comput. 2019, 15, 5635–5641. 10.1021/acs.jctc.9b00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb L. D.; Gubbins K. E. Pore size distributions in porous glasses: A computer simulation study. Langmuir 1999, 15, 305–308. 10.1021/la9808418. [DOI] [Google Scholar]
- Sarkisov L.; Harrison A. Computational structure characterisation tools in application to ordered and disordered porous materials. Mol. Simul. 2011, 37, 1248–1257. 10.1080/08927022.2011.592832. [DOI] [Google Scholar]
- Widom B. Some topics in the theory of fluids. J. Chem. Phys. 1963, 39, 2808–2812. 10.1063/1.1734110. [DOI] [Google Scholar]
- Dubbeldam D.; Calero S.; Vlugt T. J. H. iRASPA: GPU-accelerated visualization software for materials scientists. Mol. Simul. 2018, 44, 653–676. 10.1080/08927022.2018.1426855. [DOI] [Google Scholar]
- Myers A. L.; Prausnitz J. M. Thermodynamics of mixed-gas adsorption. AIChE J. 1965, 11, 121–127. 10.1002/aic.690110125. [DOI] [Google Scholar]
- Tarafder A.; Mazzotti M. A method for deriving explicit binary isotherms obeying the Ideal Adsorbed Solution Theory. Chem. Eng. Technol. 2012, 35, 102–108. 10.1002/ceat.201100274. [DOI] [Google Scholar]
- Simon C. M.; Smit B.; Haranczyk M. pyIAST: Ideal adsorbed solution theory (IAST) python package. Comput. Phys. Commun. 2016, 200, 364–380. 10.1016/j.cpc.2015.11.016. [DOI] [Google Scholar]
- Josephson T. R.; Singh R.; Minkara M. S.; Fetisov E. O.; Siepmann J. I. Partial molar properties from molecular simulation using multiple linear regression. Mol. Phys. 2019, 117, 3589–3602. 10.1080/00268976.2019.1648898. [DOI] [Google Scholar]
- Rahbari A.; Josephson T. R.; Sun Y.; Moultos O. A.; Dubbeldam D.; Siepmann J. I.; Vlugt T. J. H. Multiple linear regression and thermodynamic fluctuations are equivalent for computing thermodynamic derivatives from molecular simulation. Fluid Phase Equilib. 2020, 523, 112785 10.1016/j.fluid.2020.112785. [DOI] [Google Scholar]
- Taylor W. J.; Wagman D. D.; Williams M. G.; Pitzer K. S.; Rossini F. D. Heats, equilibrium constants, and free energies of formation of the alkylbenzenes. J. Res. Natl. Bur. Stand. 1946, 37, 95–122. 10.6028/jres.037.035. [DOI] [Google Scholar]
- Mamedov A. M.; Akhundov T. S.; Asadulaeva N. N. Experimentelle Untersuchung der Dichte und Sättigungsdrukes von m-Xylol. Teploenergetika 1967, 5, 81–82. [Google Scholar]
- Ambrose D. Vapour pressures of some aromatic hydrocarbons. J. Chem. Thermodyn. 1987, 19, 1007–1008. 10.1016/0021-9614(87)90048-6. [DOI] [Google Scholar]
- Ambrose D.; Broderick B. E.; Townsend R. The vapour pressures above the normal boiling point and the critical pressures of some aromatic hydrocarbons. J. Chem. Soc. A 1967, 633–641. 10.1039/j19670000633. [DOI] [Google Scholar]
- Murthi M.; Snurr R. Q. Effects of molecular siting and adsorbent heterogeneity on the ideality of adsorption equilibria. Langmuir 2004, 20, 2489–2497. 10.1021/la035556p. [DOI] [PubMed] [Google Scholar]
- Krishna R.; van Baten J. Segregation effects in adsorption of CO2-containing mixtures and their consequences for separation selectivities in cage-type zeolites. Sep. Purif. Technol. 2008, 61, 414–423. 10.1016/j.seppur.2007.12.003. [DOI] [Google Scholar]
- Swisher J. A.; Lin L.-C.; Kim J.; Smit B. Evaluating mixture adsorption models using molecular simulation. AIChE J. 2013, 59, 3054–3064. 10.1002/aic.14058. [DOI] [Google Scholar]
- Chiang A. S. T.; Lee C.-K.; Chang Z.-H. Adsorption and diffusion of aromatics in AlPO4-5. Zeolites 1991, 11, 380–386. 10.1016/0144-2449(91)80306-K. [DOI] [Google Scholar]
- Torres-Knoop A.; Balestra S. R. G.; Krishna R.; Calero S.; Dubbeldam D. Entropic separations of mixtures of aromatics by selective face-to-face molecular stacking in one-dimensional channels of Metal-Organic Frameworks and Zeolites. ChemPhysChem 2015, 16, 532–535. 10.1002/cphc.201402819. [DOI] [PubMed] [Google Scholar]
- Guil J. M.; Guil-López R.; Perdigón-Melón J. A.; Corma A. Determining the topology of zeolites by adsorption microcalorimetry of organic molecules. Microporous Mesoporous Mater. 1998, 22, 269–279. 10.1016/S1387-1811(98)00104-8. [DOI] [Google Scholar]
- Jones C. W.; Zones S. I.; Davis M. E. m-Xylene reactions over zeolites with unidimensional pore systems. Appl. Catal., A 1999, 181, 289–303. 10.1016/S0926-860X(98)00401-3. [DOI] [Google Scholar]
- Clark L. A.; Snurr R. Q. Adsorption isotherm sensitivity to small changes in zeolite structure. Chem. Phys. Lett. 1999, 308, 155–159. 10.1016/S0009-2614(99)00568-0. [DOI] [Google Scholar]
- Lipkowitz K. B.; Peterson M. A. Benzene is not very rigid. J. Comput. Chem. 1993, 14, 121–125. 10.1002/jcc.540140116. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.