

Abstract
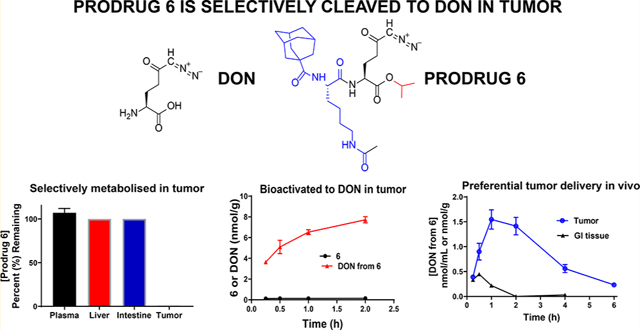
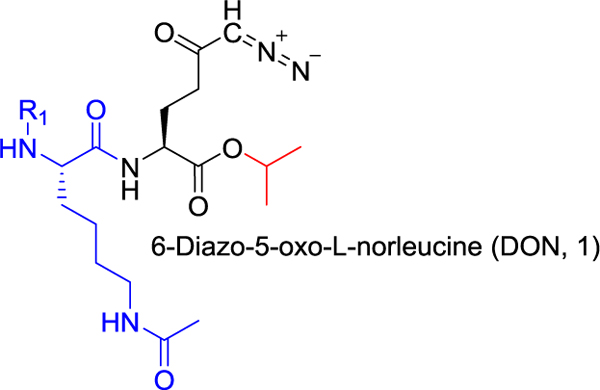
6-Diazo-5-oxo-l-norleucine (DON) is a glutamine antagonist with robust anticancer efficacy; however, its therapeutic potential was hampered by its biodistribution and toxicity to normal tissues, specifically gastrointestinal (GI) tissues. To circumvent DON’s toxicity, we synthesized a series of tumor-targeted DON prodrugs designed to circulate inert in plasma and preferentially activate over DON in tumor. Our best prodrug 6 (isopropyl 2-(6-acetamido-2-(adamantane-1-carboxamido)hexanamido)-6-diazo-5-oxohexanoate) showed stability in plasma, liver, and intestinal homogenates yet was readily cleaved to DON in P493B lymphoma cells, exhibiting a 55-fold enhanced tumor cell-to-plasma ratio versus that of DON and resulting in a dose-dependent inhibition of cell proliferation. Using carboxylesterase 1 knockout mice that were shown to mimic human prodrug metabolism, systemic administration of 6 delivered 11-fold higher DON exposure to tumor (target tissue; AUC0–t = 5.1 nmol h/g) versus GI tissues (toxicity tissue; AUC0–t = 0.45 nmol h/g). In summary, these studies describe the discovery of a glutamine antagonist prodrug that provides selective tumor exposure.
Graphical Abstract
INTRODUCTION
The transcription factor proto-oncogene c-Myc drives certain cancers to change their energy metabolism requirements and become “glutamine addicted” for their growth and survival.1–3 The glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON) broadly blocks glutamine-utilizing reactions critical for the synthesis of nucleic acids,4 proteins,5 and the generation of α-ketoglutarate for energy metabolism6 and has shown robust efficacy in both preclinical animal models and exploratory clinical studies.7–18 Although promising, clinical development of DON was halted due to its dose-limiting toxicities, many of which were gastrointestinal (GI)-related (e.g., nausea, vomiting, diarrhea)7,9,10 as the GI system is highly dependent on glutamine utilization.19 Given DON’s promising efficacy in treating glutamine-addicted tumors, it would be of clinical importance to identify a strategy to deliver it preferentially to the tumor while minimizing its peripheral exposure and thus toxicity to nontumor tissues.
We recently described DON prodrugs with enhanced brainto-plasma ratios as a treatment strategy for brain malignancies20 and neurological disorders.21 The brain-targeted prodrugs with substituted N-(pivaloyloxy)alkoxy-carbonyl esters were designed to circulate inert in plasma and be taken up and activated to DON in the brain. When evaluated in swine and primates, these changes led to a 10–15-fold increased CSF-to-plasma ratio20 and a 9-fold enhanced brain-to-plasma ratio,21 respectively, when directly compared with equivalent doses of DON. These data support the feasibility of a prodrug strategy for targeted delivery of DON.
On the basis of a similar yet distinct principle, we hypothesized that a DON prodrug designed for tumor-targeted delivery could enhance its therapeutic index for the treatment of peripheral cancers. Our initial strategy was based on a recently reported prodrug of puromycin utilizing a ε-acetylated lysine promoiety.22,23 The ε-acetylated lysine was designed to be sequentially activated by histone deacetylase (HDAC) and cathepsin L (CTSL), which are known to be enriched in tumor cells.22,23 The authors also showed that addition of lipophilic moieties, such as tert-butyloxycarbonyl (Boc) and fluorenylmethyloxycarbonyl (Fmoc), to the amine of the lysine provided enhanced cell penetration and efficacy;22 however systematic optimization of this secondary lipophilic moiety was not described.
To emulate the puromycin prodrug strategy with DON, we initially synthesized a prototypical prodrug with ε-acetylated lysine on DON’s amine and isopropyl ester on the carboxylate; the latter promoiety was previously shown to have excellent chemical and plasma stability.21 Using this scaffold, we then substituted the lysine α-amine with lipophilic groups employing carbamate and amide linkages to enhance prodrug cell penetration and efficacy. Twelve analogues were synthesized and evaluated in a comprehensive in vitro screening paradigm, which included multispecies plasma stability, intestinal and liver homogenate stability, as well as cancer cell partitioning. We identified several stable compounds, the best of which exhibited ~100% stability in swine and human plasma and intestinal homogenate yet was readily taken up and cleaved to DON in human lymphoma cells. In a cytotoxicity assay, the prodrug was found to be cytotoxic to tumor cells with an improved potency compared to that of DON. Using a mouse model that was shown to exhibit prodrug metabolism similar to that of human, we showed that the prodrug delivered approximately 5-fold higher tumor exposure of DON compared with plasma and 11-fold higher tumor exposure versus that of GI tissues. These studies describe discovery of a novel tumor-targeted glutamine antagonist, paving a path for DON’s clinical translation. In addition, we introduced a murine model that mimics human metabolism of ester-containing prodrugs and can be broadly used for development of other similar prodrugs.
CHEMISTRY
Analogous to puromycin prodrugs reported previously,22 our initial prodrugs 2 and 3 were synthesized using the standard tert-butyloxycarbonyl (Boc) and fluorenylmethyloxycarbonyl (Fmoc) protecting groups on the α-amino group of Lys(Ac) residue. As presented in Scheme 1, prodrugs 2 and 3 were synthesized by a coupling reaction of DON-isopropyl ester (1a)21 and Boc-l-Lys(Ac)-OH or Fmoc-l-Lys(Ac)-OH, respectively, in presence of N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIEA). Compound 2 was isolated in 79% yield and 3 was obtained in 67% yield.
Scheme 1. Synthesis of Prodrugs 2 and 3a.

aReagents and conditions: (i) Boc-l-Lys(Ac)-OH, HBTU, DIEA, dichloromethane (DCM), 0 °C to room temperature (rt), 1.5 h, 79%; (ii) Fmoc-l-Lys(Ac)-OH, HBTU, DIEA, DCM, 0 °C to rt, 1.5 h, 67%.
The common intermediate for the synthesis of compounds 4–13 was 3a, which was prepared by the deprotection of Fmoc moiety in 3 and was isolated in 92% yield. Compounds 4–7 were designed to enhance lipophilicity and cell permeability by attaching either linear chain fatty acids (4 and 5) or more compact hydrophobic adamantyl moieties (6 and 7) to the α-amino Lys(Ac) residue. Inclusion of fatty acids has been successfully employed to enhance the delivery of biologically active peptides,24,25 whereas adamantyl moieties have been used in Food and Drug Administration-approved antiviral and neurological agents, including amantadine, memantine, and saxagliptin.26,27 These latter groups are hypothesized to both enhance lipophilicity and impede the access of hydrolytic enzymes, thereby increasing prodrug stability.28 Compounds 4 and 5 were prepared by coupling of 3a with palmitic or caprylic acid and were isolated in 63 and 65% yield, respectively (Scheme 2). Compounds 6 and 7 with adamantyl moieties were synthesized by reaction with 1-adamantanecarboxylic acid or adamantan-1-yl carbonochloridate, yielding both amide 6 and carbamate 7 in 82 and 55% yields, respectively.
Scheme 2. Deprotection of Intermediate 3 and Synthesis of Prodrugs 4–8a.

aReagents and conditions: (i) diethylamine, DCM, rt, 20 h, 92%; (ii) for 4: palmitic acid, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), DIEA, DCM, 0 °C to rt, 1 h, 63%; for 5: octanoic acid, HATU, DIEA, DCM, 0 °C to rt, 1 h, 65%; for 6: 1-adamantanecarboxylic acid, HATU, DIEA, DCM, 0 °C to rt, 1 h, 82%; for 7: adamantan-1-yl carbonochloridate, triethylamine, DCM, 0 °C to rt, 1 h, 55%; for 8: indolylacetic acid, HATU, DIEA, DCM, 0 °C to rt, 4 h, 53%.
Compounds 8–13 were designed to target intracellular enzymes that are overexpressed in cancer cells such as cathepsins,29,30 and which often prefer cleavage sites in post-aromatic amino acids and postproline peptidases.31 1,2,3,4-Tetrahydroisoquinoline carboxylate is a secondary amino acid resembling both proline and phenylalanine. It has been used frequently as a template in the design of drugs.32–34 Using these, we designed compounds containing both free α amino groups (9 and 12) as well as protecting the amine by acetyl group (10 and 13) or removing it entirely (8). Further, we attempted to slow down prodrug hydrolysis by substituting the acetyl of 10 with a bulky trimethyl acetyl group (11). Indolylacetic acid was used in the synthesis of prodrug 8, which was isolated in 53% yield (Scheme 2). Compounds 9–13 containing tryptophan (Trp) or 1,2,3,4-tetrahydroisoqui-nolinecarboxylic acid moiety (Tic) were prepared by coupling with Fmoc-protected amino acids in presence of HATU and DIEA in dimethylformamide (DMF) and following deprotection of Fmoc group using diethylamine (Scheme 3). Compound 9 was isolated with 52% yield. Compound 9 was transformed to 10 by a reaction with acetanhydride in presence of pyridine, with yield of 80%. Compound 11 was prepared starting with 9 by coupling with trimethylacetyl chloride in 48% yield. Compound 12, synthesized by coupling of Fmoc-l-Tic-OH and deprotection with diethylamine, was isolated in 57% yield. Compound 13 was synthesized by acetylation of the amino group in 12 with 77% yield.
Scheme 3. Synthesis of Prodrugs 9–13a.

aReagents and conditions: (i) Fmoc-l-Trp-OH, HATU, DIEA, DMF, 0 °C to rt, 4 h, then diethylamine, rt, overnight, 52%; (ii) for 10: Ac2O, pyridine, DMF, rt, 2 h, 80%; for 11: trimethylacetyl chloride, DIEA, DMF, rt, 5 h, 48%; (iii) Fmoc-l-Tic-OH, HATU, DIEA, DMF, 0 °C to rt, 4 h, then diethylamine, rt, overnight, 57%; (iv) Ac2O, pyridine, DMF, rt, 3 h, 83%.
RESULTS AND DISCUSSION
Metabolic Stability.
Given that species-specific metabolic differences are well-described for prodrugs,20,21,35 we developed an in vitro multispecies screening platform to identify prodrugs that were stable (and thus inert) in plasma and liver ( highly metabolic sites) and gastrointestinal tract (toxicity site)7,9,10 yet readily taken up and cleaved to DON in tumor cells (efficacy target site). All compounds were tested for in vitro plasma stability in mouse, swine, and human plasma (Table 1). For tissue stability studies, we utilized swine liver and intestines due to their accessibility and the fact that swine is a model organism that we21 and others36 have shown as closely recapitulating human drug metabolism and pharmacokinetics.
Table 1.
 |
| Compound # | R1 | Plasma Stability (%Remaining at 1 h) | ||
|---|---|---|---|---|
| Mouse | Swine | Human | ||
| 2 |  |
1 ± 0 | 85 ± 1 | 95 ± 3 |
| 3 |  |
2 ± 1 | 78 ± 1 | 93 ± 3 |
| 4 | 30 ± 4 | 88 ± 3 | 84 ± 4 | |
| 5 | 0 ± 0 | 91 ± 6 | 95 ± 3 | |
| 6 | 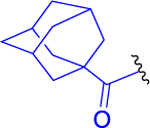 |
0 ± 0 | 108 ± 5 | 107 ± 4 |
| 7 | 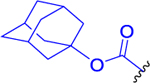 |
4 ± 1 | 77 ± 3 | 100 ± 5 |
| 8 | 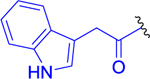 |
0 ± 0 | 98 ± 5 | 94 ± 3 |
| 9 | 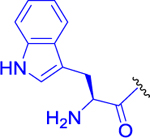 |
0 ± 0 | 19 ± 0 | 7 ± 1 |
| 10 | 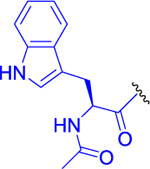 |
0 ± 0 | 75 ± 2 | 102 ± 3 |
| 11 | 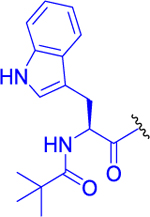 |
0 ± 0 | 95 ± 3 | 88 ± 6 |
| 12 | 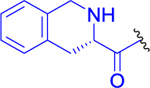 |
0 ± 0 | 62 ± 4 | 95 ± 2 |
| 13 | 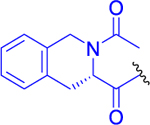 |
0 ± 0 | 98 ± 2 | 110 ± 4 |
All compounds were stable in swine and human plasma (>50% remaining at 1 h; Table 1) with the exception of 9 with a pivaloyl-l-tryptophan that was unstable in both human and swine plasma. By contrast, all prodrugs were unstable in mouse plasma (<50% remaining at 1 h; Table 1). Differences in mouse and human metabolism are not uncommon in prodrug discovery where increased metabolism in rodents is well documented.37,38
In swine liver homogenates (Figure 1A), several prodrugs showed instability (<50% remaining at 1 h), including 3 with an Fmoc substitution, 5 with caprylic acid, 9 and 11 with Nα-free or Nα-pivaloyl tryptophan, and 12 with 1,2,3,4-tetrahydroisoquinoline-3-carboxylate (Tic) substitution. Notably, 3 with substitution similar to that described on puromycin22 was highly unstable in swine liver (19% remaining at 1 h). Compound 8 with 3-indolyl acetate and 10 with Nα-acetyl tryptophan imparted enhanced stability compared with unsubstituted tryptophan analogue 9 (40 and 67% remaining (8, 10) vs 0% remaining (9) at 1 h). Similarly, 13 with N-acetyl Tic residue imparted stability to liver metabolism versus 12 (72% remaining vs 0% remaining at 1 h, respectively). Prodrug 6 imparted higher liver stability versus 7 (100% remaining vs 28% remaining at 1 h, respectively), suggesting adamantyl carboxamide was preferable over carbamate. The stability of 6 was further analyzed in both human liver microsomes and S9 fractions. High stability of 6 (~100% remaining at 1 h; Figure S2) in human liver fractions was observed.
Figure 1.

In vitro metabolic stability studies in swine (A) liver and (B) intestinal homogenates. In liver homogenate, 2, 4, 6, 10, and 13 were found to be stable (>50% remaining at 1 h). In intestinal homogenate, 2, 5, 6, 7, 8, 10, and 13 were stable (>50% remaining at 1 h). The most desirable prodrugs were those stable in plasma, liver, and intestines. On the basis of the plasma (Table 1) and liver and intestinal stability results, 2, 6, 10, and 13 were prioritized into tumor cell partitioning studies.
In swine intestinal homogenates (Figure 1B), several prodrugs also showed instability, including 3, 9, 11, and 12. Interestingly, compound 4 with a palmitic acid substituent was stable in plasma and liver yet was unstable in intestinal homogenates. The most desirable prodrugs were those stable in all three matrices, including plasma, liver, and intestinal homogenates. These prodrugs included 2 with Boc substitution, 6 with adamantyl-1-carboxamide, 10 with Nα-acetyl tryptophan, and 13 with N-acetyl Tic.
The results from these multiple in vitro metabolic stability assays were critical in the selection of prodrugs that had the least liability for presystemic metabolism and delivery of DON to sites of known toxicity such as GI tissues. Additionally, stability in plasma and liver tissues was crucial in selecting prodrugs that could survive the abundant plasma and liver hydrolytic enzymes and enable prodrugs (e.g., 2, 6, 10, and 13) to circulate intact so that they could be delivered to the tumor.
Human Tumor Cell-to-Plasma Partitioning.
Prodrugs 2, 6, 10, and 13 that passed the stability criteria were next evaluated for their ability to permeate and be cleaved to DON in tumor cells (efficacy target site) using human P493B lymphoma cells suspended in human plasma as a model system. Figure 2 shows the results of DON release in tumor cells and plasma following a 1 h incubation with 2, 6, 10, and 13 versus equimolar DON. With the exception of 13, all compounds showed partitioning into and biotransformation to DON inside the lymphoma cells, achieving a concentration similar to that obtained following DON incubation (Figure 2A). Notably, all four prodrugs were stable in human plasma, causing minimal DON release (Figure 2B). When directly compared to DON, which displayed a tumor cell-to-plasma partitioning of 0.4, the DON tumor cell-to-plasma partitioning ratio from 2, 6, 10, and 13 was improved by 32-, 55-, 30-, and 115-fold, respectively (Figure 2C). Despite compound 13 affording the best tumor cell-to-plasma partitioning ratio, compound 6 delivered enhanced DON tumor levels compared with 13 (8.81 ± 0.02 vs 0.12 ± 0.04 μm).
Figure 2.

Human tumor cell-to-plasma partitioning. DON (1), 2, 6, 10, and 13 were incubated for 1 h in 10 million P493B lymphoma cells suspended in 1 mL of human plasma. (A) 2, 6, and 10 showed DON release in tumor cells similar to equimolar DON, whereas 13 showed minimal DON release. (B) All prodrugs showed minimal release of DON in human plasma. (C) DON exhibited a human tumor cell-to-plasma ratio of 0.4, whereas the DON tumor cell-to-plasma partitioning ratio from 2, 6, 10, and 13 was 13, 22, 11, and 46, respectively. Although 13 afforded the best tumor cell-to-plasma partitioning ratio, 6 delivered enhanced DON tumor levels compared with 13 (8.81 ± 0.02 vs 0.12 ± 0.04 μM) and thus was selected for further evaluation in a time course experiment.
Although P493B cells were employed as a model system for in vitro evalution, DON has been shown to be efficacious in multiple cancer types.9–11 To confirm that the tumor cell activation was not specific to P493B cells, the best prodrug 6 was also tested in additional human cancer cell lines, including H69 (lung cancer) and DU4475 (breast cancer) cells. Employing experimental conditions similar to those for P493B cells, it was confirmed that DON release was similar in all cell lines with 9.95 ± 0.4 μM (H69), 11.8 ± 0.9 μM (DU4475), and 8.81 ± 0.02 μM (P493B) (Figure S3A). Plasma levels of DON from 6 were also similar (0.25–0.5 μM) upon 1 h incubation (Figure S3B). Again, these results established that prodrug uptake and activation was not specific to lymphoma cells and was consistent against multiple cell lines.
Finally, to evaluate if the tumor-to-plasma ratio was sustained, we next measured intact 6 and released DON from 6 in plasma and P493B cells over a 2 h incubation period. 6 remained intact in plasma with minimal DON release (Figure 3A). In contrast, 6 showed partitioning into and released DON in the tumor cells with minimal measurable intact prodrug (Figure 3B).
Figure 3.

Time-dependent human tumor cell-to-plasma partitioning of 6 and DON (1) release. 6 was incubated in P493B lymphoma cells suspended in human plasma, and at 0.25, 0.5, 1, and 2 h, aliquots were removed and evaluated for 6 and DON released from 6 using liquid chromatography (LC)–mass spectrometry (MS). (A) In human plasma, 6 was stable over the 2 h incubation period with minimal DON released. (B) In contrast, 6 partitioned into P493B tumor cells and was biotransformed to DON. These results confirm selective uptake and bioactivation of 6 to DON in tumor cells over 2 h.
Metabolite Identification (MET ID) for 6 in Tumor Cells.
We conducted MET ID studies using high-resolution mass spectrometry (HRMS) to determine the mechanism of biotransformation of 6 to DON in tumor cells (Figure 4A–F). Bioactivation of 6 was found to occur by two distinct pathways. First, in the minor pathway, 6 underwent deacetylation on the lysine to liberate 6a followed by removal of the adamantane-1carbonyl lysine 6b resulting in DON-isopropyl ester (1a). In the second pathway, hydrolysis of the entire adamantane-1carbonyl-ε-acetyl-lysine promoiety 6c occurred to form 1a directly. In both cases, 1a was subsequently hydrolyzed to DON inside the tumor cells. These data suggest that unlike the sequential bioactivation hypothesized for the Fmoc-Lys(Ac)-puromycin-prodrug22 where deacetylation by HDAC was proposed prior to proteolysis of lysine by CTSL, 6 may also undergo one-step cleavage of the entire promoiety.
Figure 4.

Metabolic identification (MET ID) of 6 in tumor cells. 6 was incubated for 1 h in 10 million P493B lymphoma cells suspended in 1 mL of human plasma. Cell pellets were subsequently obtained, and HRMS was performed to determine the pathway of prodrug metabolism. Bioactivation of 6 was found to occur by two distinct pathways (A). First (minor pathway), 6 (B) underwent deacetylation on the lysine to liberate 6a (C) followed by removal of the adamantane-1-carbonyl lysine 6b (D), resulting in DON-isopropyl ester 1a (F). In the second pathway (major pathway), hydrolysis of the entire adamantane-1-carbonyl-ε-acetyl-lysine promoiety 6c (E) occurred to directly form 1a. In both cases, 1a was subsequently hydrolyzed to DON (1).
In an attempt to characterize the specific mechanism of bioactivation of 6, human recombinant cathepsin B and L and histone deacetylase (HDAC) enzymes were used. Cathepsin L and HDAC were selected due to our prodrug design similarity to the previously reported Boc-Lys(Ac)-puromycin and Fmoc-Lys(Ac)-puromycin-prodrugs22,23 that were hypothesized to be sequentially hydrolyzed by these enzymes. Of the multiple HDACs, HDAC2 was selected due to its increased expression in tumor cells.39 We also studied cathepsin B as it is overexpressed in various cancers39,40 and is associated with several prodrug bioactivation processes.41,42 Using these enzymes it was observed that 6 was metabolized faster in the presence of recombinant cathepsin L (36% remaining) than cathepsin B (64% remaining) following a 3 h incubation (Figure 5); moreover, when incubated in presence of both cathepsin B and L, metabolism was nearly complete (9% remaining at 3 h). Interestingly, 6 was relatively stable in presence of HDAC2 (>95% remaining at 3h), suggesting deacetylation prior to deamidation was not rate limiting, as previously described for puromycin prodrug22,23 where the ε-acetylated lysine was designed to be sequentially activated by histone deacetylase (HDAC) and cathepsin L (CTSL). These results corroborate the tumor cell activation data and MET ID observed in P493B cells, suggesting that tumor cell-enriched proteases, such as cathepsin B and L, were involved in prodrug bioactivation.
Figure 5.

Stability of 6 in the presense of human recombinant enzymes. 6 was spiked in incubations containing no enzyme, cathepsin B, cathepsin L, a mixture of cathepsin B and L, or HDAC2, and prodrug disappearance was measured over a period of 3 h via LC–MS. 6 showed the highest rate of disappearance in the presense of combined cathepsin B and L. HDAC2 did not cause prodrug metabolism. Negative control without enzymes showed no metabolism, confirming 6 to be chemically stable.
P493B Cell Viability.
To ensure that 6 was able to maintain the antiproliferative effect of DON, 6 and equimolar DON were evaluated in a cell viability assay using P493B lymphoma cells. A dose-dependent decrease in cell proliferation was observed following a 72 h incubation with both DON and 6 (Figure 6). Consistent with the minor increase in DON tumor cell exposure following 6 versus that in equimolar DON (Figure 2a), 6 caused a slight leftward shift in the viability curve; nonlinear regression analysis of the log-transformed data gave EC50 values for DON and 6 at 10.0 ± 0.11 and 5.0 ± 0.12 μM, respectively.
Figure 6.

Cell viability assay using P493B lymphoma cells. DON (1) and 6 were incubated with P493B lymphoma cells for 72 h, and cell viability was measured. Nonlinear regression analysis of the log-transformed data gave EC50 values for DON and 6 of 10.0 ± 0.11 and 5.0 ± 0.12 μM, respectively. EC50 value for 6 was 2-fold lower than that for DON, suggesting that the prodrug delivers higher DON tumor cell levels compared with DON itself, thus leading to better potency.
Pharmacokinetics of 6 in Carboxylesterase 1 Knock-out (CES1)−/− Mice.
Given the excellent stability, tumor partitioning, and cytotoxicity data from 6, we next desired to evaluate its pharmacokinetic profile in tumor-bearing animals. One challenging aspect of prodrug development is the selection of an appropriate animal model, as it can be confounded by interspecies variation in metabolism.43 This is exemplified in the plasma metabolism studies where we observed complete stability of 6 in human and swine plasma but instability in mouse plasma (Table 1). We hypothesized that the rapid metabolism in mice was due to the de-esterification of the isopropyl ester via the carboxylesterase enzyme, carboxylesterase 1 (CES1), which is highly abundant in rodent plasma but not present in human plasma.44 This was confirmed by directly comparing metabolism of 6 in plasma from C57BL/6 CES1−/− mouse, wild-type (WT) mouse, and human. 6 exhibited similar stability in CES1−/− mouse and human plasma but not in wild-type mouse plasma (Figure 7A). Employing MET ID studies, we confirmed that the instability in wild-type mouse plasma was due to de-esterification of the carboxylate ester (Figure S4). Given the similarity to human metabolism, pharmacokinetic evaluation of 6 was performed in CES1−/− mice bearing flank murine EL4 (mouse lymphoma cell line) lymphoma tumors. We first confirmed that prodrug 6 showed similar differential partitioning in the murine EL4 and determined that the tumor-cell-partitioning ratio was 16. Next, 6 was dosed via a subcutaneous (SC) route at 3.2 mg/kg (1 mg/kg DON equivalent dose) and plasma, tumor, and GI tissues were collected at predetermined time points. Tissues were analyzed for both intact 6 and DON levels. Following subcutaneous dosing, 6 exhibited an excellent pharmacokinetic profile, delivering DON at the highest exposure to tumor (Figure 7B). Notably, 6 delivered approximately 5-fold higher tumor exposure of DON (area under curve (AUC) = 5.1 nmol h/g) versus that of plasma (1.1 nmol h/mL) and 11-fold higher tumor exposure versus that of jejunum (toxicity site; AUC = 0.45 nmol h/g) (Figure 7B,C). Moreover, whereas the intact prodrug was measurable in plasma (AUC = 0.4 nmol h/mL), minimal intact 6 was measured in tumor and jejunum levels were below the limit of quantification (0.001 nmol/mL). These in vivo results were in congruence with the in vitro stability analysis of 6 and confirmed preferential tumor activation of 6.
Figure 7.

(A) In vitro stability of 6 in wild-type mouse, CES1−/− mouse, and human plasma. (B) Pharmacokinetic profile of 6 and DON (1) release from 6 in EL4 tumor-bearing CES1−/− mice following SC dosing at 3.2 mg/kg (1 mg/kg of DON equivalent). (C) Pharmacokinetic parameters of 6 and released DON (1) calculated using noncompartmental analysis in WinNonlin, with data expressed as mean or mean ± standard error of the mean (n = 3 mice/time point). BLQ: below the limit of quantification.
CONCLUSIONS
DON is a glutamine antagonist with robust anticancer efficacy, yet its therapeutic potential has been hampered by its biodistribution and toxicity to normal tissues. To increase DON’s therapeutic index, we synthesized a series of DON prodrugs with ε-acetylated lysine on DON’s amine to target tumor-enriched proteases such as HDAC and CTSL. Using this scaffold, we added lipophilic groups on the α-amino group of the ε-acetyl-lysine to enhance prodrug cell penetration and efficacy. Twelve prodrugs were synthesized and evaluated in a comprehensive in vitro screening paradigm. Our best prodrug, 6, showed stability in plasma, liver, and intestinal homogenates but was readily cleaved to DON in multiple tumor cells, providing a 55-fold enhanced tumor cell-to-plasma ratio when directly compared with DON. The mechanism of 6 bioactivation was shown to involve both cathepsin B and L. Further, using CES1−/− mice that recapitulated human metabolism, we showed that 6 was preferentially bioactivated in tumor, affording 5- and 11-fold higher tumor exposures versus those in plasma and intestinal tissues, respectively. These studies provide a path for the development of a better tolerated tumor-targeted glutamine antagonist for cancer therapy. In addition, the studies highlight the utility of using CES1−/− mice to mimic human metabolism of ester-containing prodrugs.
EXPERIMENTAL SECTION
Commercially available high-performance liquid chromatography (HPLC) grade acetonitrile, catalysts, and reagent grade materials were used for synthesis of compounds described. Thin-layer chromatography was performed on Silica gel 60 F254-coated aluminum sheets (Merck). Flash chromatography was performed on silica gel 60 (0.040–0.063 mm, Fluka). All chemicals were purchased from Sigma-Aldrich, TCI, Combi-Blocks, or Iris Biotech GmbH and were used without further purification. The 1H NMR spectra were measured at 400.1 MHz and 13C NMR spectra at 100.8 MHz (see Sections S3–S16, Supporting Information). For standardization of 1H NMR spectra, the internal signals of tetramethylsilane (δ 0.0, CDCl3) or residual signals of CDCl3 (δ 7.26), dimethyl sulfoxide (DMSO)-d6 (δ 2.50, 3.33), or MeOD-d4 (δ 3.31, 4.87) were used. In the case of 13C NMR spectra (attached proton test experiments), the residual signal of CDCl3 (δ 77.16), DMSO-d6 (δ 39.52), or MeOD-d4 (δ 49.00) was used. The chemical shifts are given in δ scale; the coupling constant J is given in hertz. The electrospray ionization (ESI) mass spectra were recorded using a ZQ micromass mass spectrometer (Waters) equipped with an ESCi multimode ion source and controlled by MassLynx software. The low-resolution ESI mass spectra were recorded using a quadrupole orthogonal acceleration time-of-flight tandem mass spectrometer (Q-TOF Micro, Waters), and high-resolution ESI mass spectra, using a hybrid Fourier-transform (FT) mass spectrometer combining a linear ion trap MS and the Orbitrap mass analyzer (LTQ Orbitrap XL, Thermo Fisher Scientific). The conditions were optimized for suitable ionization in the ESI Orbitrap source (sheath gas flow rate 35 au, aux gas flow rate 10 au of nitrogen, source voltage 4.3 kV, capillary voltage 40 V, capillary temperature 275 °C, tube lens voltage 155 V). The samples were dissolved in methanol and applied by direct injection. The purity of all compounds subjected to biological testing was established using a reversed-phase HPLC (Jasco Inc.), with a Reprosil 100 C18, 5 μm, 250 × 4 mm2 column (see Sections S17–S22, Supporting information) and UV detection, with λ = 210 nm. The purity of all compounds subjected to biological testing was over 95%, unless otherwise noted. Optical rotations were measured in CHCl3, CH3OH, or DMF using an Autopol IV instrument (Rudolph Research Analytical). IR spectra were measured in CHCl3 or KBr (pellets) on an Fourier-transform infrared spectrometer.
Isopropyl 2-(6-Acetamido-2-((tert-butoxycarbonyl)amino)-hexanamido)-6-diazo-5-oxohexanoate (2).
Boc-l-Lys(Ac)-OH (446 mg, 1.55 mmol, 1.1 equiv) and HBTU (641 mg, 1.69 mmol, 1.2 equiv) were suspended in anhydrous DCM (8 mL), and the suspension was cooled to 0 °C. DIEA (546 mg, 735 μL, 4.22 mmol, 3 equiv) was added. The reaction mixture was stirred for 5 min, and then the solution of DON-OiPr21 (1a) (300 mg, 1.41 mmol, 1 equiv) in anhydrous DCM (2 mL) was added by a syringe over 5 min. The reaction mixture was stirred for 0.5 h at 0 °C and for 1 h at room temperature under an inert atmosphere. DCM (30 mL) was added and the solution was washed with H2O (2 × 50 mL) and brine (50 mL) and dried over MgSO4. The organic solvent was evaporated in vacuo. The residue was chromatographed on silica gel (DCM/MeOH, 25:1) to afford the desired product 2 (535 mg, 79%) as a light yellow amorphous solid. 1H NMR (400 MHz, CDCl3): 1.22 (3H, d, J = 6.3), 1.24 (3H, d, J = 6.3), 1.36–1.44 (2H, m), 1.42 (9H, s), 1.46–1.56 (2H, m), 1.59–1.71 (1H, m), 1.73–1.87 (1H, m), 1.95 (3H, s), 1.97–2.04 (1H, m), 2.12–2.24 (1H, m), 2.34–2.47 (2H, m), 3.22 (2H, dq, J = 6.5, 3.4), 4.09 (1H, q, J = 7.0), 4.36–4.55 (1H, m), 5.00 (1H, hept, J = 6.3), 5.26 (1H, d, J = 7.3), 5.34 (1H, s), 6.05 (1H, bs), 7.12 (1H, d, J = 7.0). 13C NMR (101 MHz, CDCl3): 21.82, 21.82, 22.41, 23.32, 26.90, 28.43 (3C), 28.87, 32.22, 36.56, 38.94, 52.18, 54.24, 55.11, 69.53, 80.00, 155.79, 170.42, 171.26, 172.29, 194.08. IR (CHCl3): 3431, 3325, 3116, 2984, 2937, 2867, 2110, 1728, 1708, 1668, 1635, 1518, 1499, 1468, 1387, 1394, 1376, 1369, 1241, 1147, 1166, 1105 cm−1. Optical rotation: [α]D22 −22.6° (c 0.212, CH3OH). ESI MS: 506 ([M + Na]+). High-resolution (HR) ESI MS: calcd for C22H37O7N5Na 506.25852; found 506.25866.
Isopropyl 2-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)-amino)-6-acetamidohexanamido)-6-diazo-5-oxohexanoate (3).
Fmoc-l-Lys(Ac)-OH (6.35 g, 15.5 mmol, 1.1 equiv) and HBTU (6.41 g, 16.9 mmol, 1.2 equiv) were suspended in anhydrous DCM (80 mL), and the suspension was cooled to 0 °C. DIEA (5.46 g, 7.35 mL, 42.2 mmol, 3 equiv) was added. The reaction mixture was stirred for 5 min, and then the solution of DON-OiPr21 (1a) (3.00 g, 14.1 mmol, 1 equiv) in anhydrous DCM (20 mL) was added by a syringe over 10 min. The reaction mixture was stirred for 0.5 h at 0 °C and for 1 h at room temperature under an inert atmosphere. DCM (30 mL) was added, and the solution was washed with H2O (2 × 50 mL) and brine (50 mL) and dried over MgSO4. The organic solvent was evaporated in vacuo. The residue was chromatographed on silica gel (CHCl3/MeOH, 30:1) to afford the desired product 3 (5.72 g, 67%) as a light yellow amorphous solid. 1H NMR (400 MHz, DMSO-d6): 1.15 (3H, d, J = 4.2), 1.17 (3H, d, J = 4.2), 1.21–1.44 (4H, m), 1.47–1.67 (2H, m), 1.77 (3H, s), 1.77–1.85 (1H, m), 1.89–2.03 (1H, m), 2.31–2.43 (2H, m), 2.94–3.06 (2H, m), 3.90–4.09 (1H, m), 4.10–4.35 (4H, m), 4.87 (1H, hept, J = 6.3), 6.01 (1H, bs), 7.32 (2H, t, J = 7.4), 7.42 (2H, t, J = 7.1), 7.47 (1H, d, J = 8.1), 7.73 (2H, dd, J = 8.8, 1.6), 7.79 (1H, t, J = 5.4), 7.89 (2H, d, J = 7.5), 8.26 (1H, d, J = 7.5). 13C NMR (101 MHz, DMSO-d6): 21.46, 21.50, 22.64, 22.99, 25.87, 28.93, 31.60, 36.20, 38.41, 46.67, 51.48, 54.32, 54.92, 65.62, 67.98, 120.12 (2C), 125.34 (2C), 127.06 (2C), 127.65 (2C), 140.71, 140.72, 143.80, 143.91, 155.95, 168.93, 171.07, 172.34, 194.06. IR (CHCl3): 3450, 3420, 3318, 3270, 3117, 3069, 2986, 2939, 2865, 2110, 1727, 1668, 1638, 1579, 1504, 1479, 1465, 1451, 1386, 1377, 1233, 1184, 1146, 1105, 1033, 622, 426 cm−1. Optical rotation: [α]D22 −18.9° (c 0.111, CH3OH). ESI MS: 628 ([M + Na]+). HR ESI MS: calcd for C32H39O7N5Na 628.27417; found 628.27430.
Isopropyl 2-(6-Acetamido-2-aminohexanamido)-6-diazo-5-oxohexanoate (3a).
Compound 3 (5.00 g, 8.26 mmol, 1 equiv) was dissolved in anhydrous DCM (150 mL), and diethylamine (3.51 g, 4.97 mL, 41.3 mmol, 5 equiv) was added. The reaction mixture was stirred overnight at room temperature (20 h). The solvent was evaporated, and the crude product was purified on silica gel to afford 2.91 g (92%) of yellow solid compound 3a. 1H NMR (400 MHz, CDCl3): 1.20 (3H, d, J = 6.0), 1.21 (3H, d, J = 6.0), 1.33–1.53 (4H, m), 1.56–1.68 (1H, m), 1.72–1.84 (1H, m), 1.92 (3H, s), 1.91–2.02 (1H, m), 2.09–2.21 (1H, m), 2.28–2.48 (2H, m), 3.10–3.25 (2H, m), 3.43–3.73 (3H, m), 4.43 (1H, td, J = 8.1, 4.7), 4.97 (1H, hept, J = 6.0), 5.46 (1H, bs), 6.51 (1H, bs), 8.05 (1H, d, J = 7.7). 13C NMR (101 MHz, CDCl3): 21.76 (2C), 22.57, 23.22, 27.29, 29.06, 33.92, 36.70, 39.06, 51.84, 54.53, 55.13, 69.40, 170.66, 171.24, 174.15, 194.15. IR (KBr): 3432, 2110, 1731, 1635, 1615, 1547, 1453, 1377, 1217, 1106 cm−1. Optical rotation: [α]D22 −8.8° (c 0.283, DMF). ESI MS: 406 ([M + Na]+). HR ESI MS: calcd for C17H29O5N5Na 406.20609; found 406.20612.
Isopropyl 2-(6-Acetamido-2-palmitamidohexanamido)-6-diazo-5-oxohexanoate (4).
Palmitic acid (184 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were suspended in anhydrous DCM (10 mL), and the suspension was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added. The reaction mixture was stirred for 5 min, and then the solution of 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DCM (5 mL) was added by a syringe over 2 min. The reaction mixture was stirred for 15 min at 0 °C and 45 min at room temperature under an inert atmosphere. DCM (30 mL) was added, and the solution was washed with H2O (2 × 50 mL) and brine (50 mL) and dried over MgSO4. The organic solvent was evaporated in vacuo. The residue was chromatographed on silica gel (CHCl3/MeOH, 20:1) to afford the desired product 4 (255 mg, 63%) as a light yellow solid. 1H NMR (400 MHz, CDCl3): 0.86 (3H, t, J = 6.8), 1.18–1.32 (30H, m), 1.34–1.44 (2H, m), 1.47–1.64 (4H, m), 1.65–1.74 (1H, m), 1.77–1.88 (1H, m), 1.96 (3H, s), 1.97–2.05 (1H, m), 2.13–2.24 (3H, m), 2.33–2.48 (2H, m), 3.23 (2H, q, J = 6.3), 4.48 (2H, m), 5.00 (1H, hept, J = 6.3), 5.35 (1H, bs), 6.14 (1H, t, J = 5.8), 6.45 (1H, d, J = 7.5), 7.35 (1H, d, J = 6.4). 13C NMR (101 MHz, CDCl3): 14.24, 21.83, 21.85, 22.11, 22.80, 23.31, 25.86, 26.59, 28.76, 29.47 (2C), 29.49, 29.64, 29.77 (2C), 29.78, 29.81 (3C), 32.03, 32.07, 36.53, 36.71, 38.75, 52.35, 52.70, 55.18, 69.52, 170.58, 171.23, 172.08, 173.58, 194.13. IR (CHCl3): 3447, 3417, 3314, 2928, 2871, 2855, 2110, 1731, 1659, 1525, 1507, 1467, 1456, 1385, 1377, 1367, 1236, 1105 cm−1. Optical rotation: [α]D20 −6.0° (c 0.380, CHCl3). ESI MS: 644 ([M + Na]+). HR ESI MS: calcd for C33H59O6N5Na 644.43576; found 644.43572.
Isopropyl 2-(6-Acetamido-2-octanamidohexanamido)-6-diazo-5-oxohexanoate (5).
Octanoic acid (103 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were suspended in anhydrous DCM (10 mL), and the suspension was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added. The reaction mixture was stirred for 5 min, and then the solution of 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DCM (5 mL) was added by a syringe over 2 min. The reaction mixture was stirred for 15 min at 0 °C and 45 min at room temperature under an inert atmosphere. DCM (30 mL) was added, and the solution was washed with H2O (2 × 50 mL) and brine (50 mL) and dried over MgSO4. DCM was evaporated, and the residue was chromatographed on silica gel (CHCl3/MeOH, 20:1) to afford the desired product 5 (216 mg, 65%) as a light yellow solid. 1H NMR (400 MHz, CDCl3): 0.87 (3H, t, J = 7.0), 1.24 (3H, d, J = 6.3), 1.27 (3H, d, J = 6.3), 1.26−1.32 (6H, m), 1.35–1.45 (2H, m), 1.49–1.60 (2H, m), 1.60–1.75 (5H, m), 1.80–1.91 (1H, m), 1.97 (3H, s), 1.99–2.07 (1H, m), 2.15–2.21 (1H, m), 2.22 (2H, t, J = 6.9), 2.37–2.49 (2H, m), 3.25 (2H, q, J = 6.2), 4.44 (2H, m), 5.03 (1H, hept, J = 6.3), 5.34 (1H, bs), 5.97 (1H, t, J = 6.3), 6.31 (1H, d, J = 7.7), 7.17 (1H, d, J = 5.5). 13C NMR (101 MHz, CDCl3): 14.12, 21.78 (2C), 22.13, 22.65, 23.21, 25.81, 26.58, 28.74, 29.07, 29.33, 29.74, 31.74, 32.08, 36.60, 38.75, 52.27, 52.67, 55.08, 69.40, 170.56, 171.19, 172.15, 173.56, 194.04. IR (CHCl3): 3449, 3417, 3313, 2931, 2858, 2827, 2110, 1731, 1659, 1524, 1508, 1387, 1377, 1370, 1235, 1106 cm−1. Optical rotation: [α]D20 −4.8° (c 0.227, CHCl3). ESI MS: 532 ([M + Na]+). HR ESI MS: calcd for C25H43O6N5Na 532.31056, found 532.31063.
Isopropyl 2-(6-Acetamido-2-adamantane-1-carboxamido)-hexanamido)-6-diazo-5-oxohexanoate (6).
1-Adamantanecarboxylic acid (129 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were suspended in anhydrous DCM (10 mL), and the suspension was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added. The reaction mixture was stirred for 5 min, and then the solution of 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DCM (10 mL) was added by a syringe over 2 min. The reaction mixture was stirred for 15 min at 0 °C and for 45 min at room temperature under an inert atmosphere. DCM (30 mL) was added, and the solution was washed with H2O (2 × 50 mL) amd brine (50 mL) and dried over MgSO4. DCM was evaporated in vacuo. The residue was chromatographed on silica gel (CHCl3/MeOH, 20:1) to afford the desired product 6 (292 mg, 82%) as a light yellow solid. 1H NMR (400 MHz, CDCl3): 1.24 (3H, d, J = 6.3), 1.27 (3H, d, J = 6.3), 1.33–1.44 (2H, m), 1.49–1.61 (2H, m), 1.64–1.79 (7H, m), 1.82–1.89 (7H, m), 1.97 (3H, s), 1.99–2.07 (4H, m), 2.13–2.25 (1H, m), 2.33–2.48 (2H, m), 3.15–3.34 (2H, m), 4.37–4.49 (2H, m), 5.02 (1H, hept, J = 6.3), 5.33 (1H, bs), 6.04 (1H, bs), 6.36 (1H, d, J = 6.8), 7.18 (1H, bs). 13C NMR (101 MHz, CDCl3): 21.72, 21.74, 22.05, 23.13, 26.47, 28.04 (3C), 28.57, 32.34, 36.42 (3C), 36.58, 38.82, 39.16 (3C), 40.64, 52.15, 52.24, 54.99, 69.30, 170.44, 171.15, 172.21, 178.14, 193.94. IR (CHCl3): 3448, 3419, 3316, 2931, 2912, 2853, 2110, 1731, 1658, 1640, 1541, 1518, 1504, 1453, 1387, 1377, 1370, 1347, 1319, 1236, 1105, 976 cm−1. Optical rotation: [α]D20 −4.1° (c 0.220, CHCl3). ESI MS: 568 ([M + Na]+). HR ESI MS: calcd for C28H43O6N5Na 568.31056; found 568.31055.
Isopropyl 2-(6-Acetamido-2-((((adamantan-1-yl)oxy)-carbonyl)amino)hexanamido)-6-diazo-5-oxohexanoate (7).
Compound 3a (250 mg, 0.652 mmol, 1 equiv) was dissolved in anhydrous DCM (10 mL), and reaction mixture was cooled to 0 °C. Triethylamine (79 mg, 109 μL, 0.782 mmol, 1.2 equiv) followed by a solution of adamantanyl carbonochloridate (168 mg, 0.782 mmol, 1.2 equiv) in anhydrous DCM (3 mL) was added. The resulting mixture was stirred for 15 min at 0 °C and for 45 min at room temperature. DCM was evaporated, and the crude product was purified by LC (CHCl3/MeOH, 20:1). The desired compound 7 was obtained in 55% yield (201 mg) as a light yellow solid. 1H NMR (400 MHz, CDCl3): 1.24 (3H, d, J = 6.3), 1.26 (3H, d, J = 6.3), 1.37–1.46 (2H, m), 1.49–1.58 (2H, m), 1.59–1.69 (7H, m), 1.76–1.89 (1H, m), 1.97 (3H, s), 2.06–2.11 (6H, m), 2.11–2.18 (4H, m), 2.18–2.26 (1H, m), 2.30–2.52 (2H, m), 3.16–3.32 (2H, m), 4.08 (1H, q, J = 6.9), 4.47 (1H, ddd, J = 8.5, 7.5, 4.4), 5.03 (1H, hept, J = 6.3), 5.16 (1H, d, J = 7.2), 5.32 (1H, bs), 5.86 (1H, bs), 6.96 (1H, d, J = 6.5). 13C NMR (101 MHz, CDCl3): 21.79 (2C), 22.42, 23.28, 26.88, 28.81, 30.89 (3C), 36.22 (3C), 36.53, 38.93, 41.69 (3C), 45.40, 52.13, 54.15, 55.06, 69.48, 79.84, 155.41, 170.42, 171.25, 172.31, 194.05. IR (CHCl3): 3598, 3445, 3420, 2933, 2918, 2854, 2110, 1730, 1706, 1669, 1637, 1518, 1494, 1456, 1386, 1377, 1367, 1354, 1318, 1272, 1236, 1111, 1104, 1086, 1057, 968, 551 cm−1. Optical rotation: [α]D20 −2.0° (c 0.151, CHCl3). ESI MS: 584 ([M + Na]+). HR ESI MS: calcd for C28H43O7N5Na 584.30547; found 584.30551.
Isopropyl 2-(2-(2-(1H-Indol-3-yl)acetamido)-6-acetamidohexanamido)-6-diazo-5-oxohexanoate (8).
3-Indolylacetic acid (126 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were dissolved in anhydrous DCM (8 mL), and the reaction mixture was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added, and the reaction mixture was stirred for 15 min at the same temperature. Finally, the solution of compound 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DCM (4 mL) was added dropwise. The mixture was stirred at 0 °C for a further 30 min and at room temperature for 3 h. DCM (30 mL) was added, and the organic phase was washed with 10% NaHCO3 (50 mL), H2O (50 mL), and brine and dried over MgSO4, and the solvent was evaporated. The crude product was purified by column chromatography (CHCl3/MeOH, 20:1). The desired product 8 was obtained as a light yellow solid (189 mg, 53% yield). 1H NMR (400 MHz, CDCl3): 1.22 (3H, d, J = 6.3), 1.24 (3H, d, J = 6.3), 1.20–1.26 (2H, m), 1.35–1.47 (2H, m), 1.48–1.61 (1H, m), 1.70–1.81 (1H, m), 1.86–1.95 (1H, m), 1.93 (3H, s), 2.08–2.19 (1H, m), 2.21–2.37 (2H, m), 3.13 (2H, q, J = 6.5), 3.75 (2H, bs), 4.33–4.45 (2H, m), 5.00 (1H, hept, J = 6.3), 5.14 (1H, bs), 5.82 (1H, bs), 6.37 (1H, d, J = 7.6), 7.06 (1H, d, J = 6.4), 7.13 (1H, ddd, J = 8.0, 7.1, 1.1), 7.17 (1H, d, J = 2.1), 7.21 (1H, ddd, J = 8.0, 7.1, 1.1), 7.38 (1H, dd, J = 8.1, 0.9), 7.58 (1H, dd, J = 7.8, 1.0), 8.43 (1H, bs). 13C NMR (101 MHz, CDCl3): 21.83 (2C), 22.24, 23.29, 26.64, 28.73, 31.77, 33.50, 36.39, 38.99, 52.24, 52.95, 55.07, 69.53, 108.73, 111.69, 118.66, 119.89, 122.48, 123.94, 127.14, 136.53, 170.61, 171.20, 171.87, 172.10, 194.23. IR (KBr): 3304, 3081, 3062, 2106, 1732, 1647, 1542, 1458, 1437, 1375, 1215, 1106 cm−1. Optical rotation: [α]D20 −1.9° (c 0.260, CHCl3). ESI MS: 563 ([M + Na]+). HR ESI MS: calcd for C27H36O6N6Na 563.25885; found 563.25890.
Isopropyl 2-(6-Acetamido-2-(2-amino-3-(1H-indol-3-yl)-propanamido)hexanamido)-6-diazo-5-oxohexanoate (9).
Fmoc-l-Trp-OH (306 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were dissolved in anhydrous DMF (8 mL), and the reaction mixture was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added, and the reaction mixture was stirred for 15 min at the same temperature. Finally, the solution of 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DMF (4 mL) was added dropwise. The mixture was stirred at 0 °C for a further 30 min and at room temperature for 3 h. Diethylamine (477 mg, 674 μL, 6.52 mmol, 10 equiv) was added to the mixture to remove the Fmoc protecting group, and the solution was stirred overnight at room temperature under an inert atmosphere. DMF was evaporated, and the crude product was purified by LC (CHCl3/MeOH, 7:1). The desired product 9 (194 mg, 52% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, CDCl3): 1.24 (3H, d, J = 6.3), 1.26 (3H, d, J = 6.3), 1.35–1.51 (3H, m), 1.53–1.60 (2H, m), 1.63 (2H, bs), 1.73–1.85 (1H, m), 1.94–2.05 (1H, m), 2.00 (3H, s), 2.12–2.23 (1H, m), 2.30–2.48 (2H, m), 3.11 (1H, dq, J = 13.0, 6.1), 3.24 (2H, d, J = 5.4), 3.29 (1H, dq, J = 13.7, 7.0), 3.79 (1H, t, J = 5.5), 4.40 (2H, dt, J = 12.9, 6.3), 5.02 (1H, hept, J = 6.3), 5.31 (1H, bs), 5.83 (1H, t, J = 5.1), 7.05 (1H, d, J = 2.2), 7.07–7.14 (2H, m), 7.18 (1H, dt, J = 7.1, 1.3), 7.40 (1H, d, J = 8.1), 7.65 (1H, d, J = 7.8), 7.77 (1H, d, J = 8.1), 9.10 (1H, bs). 13C NMR (101 MHz, DMSO-d6): 21.45, 21.50, 22.34, 22.62, 25.76, 28.98, 30.37, 32.36, 38.43, 41.56, 51.52, 51.68, 54.80, 56.03, 68.00, 110.05, 111.33, 118.23, 118.45, 120.90, 123.99, 127.39, 136.24, 168.95, 170.97, 171.71, 173.59, 194.03. IR (KBr): 3277, 3084, 2105, 1731, 1666, 1643, 1545, 1457, 1439, 1375, 1212, 1106 cm−1. Optical rotation: [α]D20 −37.6° (c 0.226, CHCl3). ESI MS: 570 ([M + H]+). HR ESI MS: calcd for C28H40O6N7 570.30346; found 570.30349.
Isopropyl 2-(6-Acetamido-2-(2-acetamido-3-(1H-indol-3-yl)propanamido)hexanamido)-6-diazo-5-oxohexanoate (10).
Compound 9 (150 mg, 0.263 mmol, 1 equiv) was dissolved in anhydrous DMF (5 mL) and pyridine (42 mg, 42 μL, 0.527 mmol, 2 equiv) followed by adding acetanhydride (32 mg, 30 μL, 0.316 mmol, 1.2 equiv). The resulting mixture was stirred for 2 h at room temperature under an inert atmosphere. DMF was evaporated, and the residue was purified by LC (CHCl3/MeOH, 10:1) to obtain compound 10 (129 mg, 80%) as a light yellow solid. 1H NMR (400 MHz, MeOD-d4): 1.25 (3H, d, J = 6.3), 1.26 (3H, d, J = 6.3), 1.30–1.37 (2H, m), 1.42–1.54 (2H, m), 1.55–1.68 (1H, m), 1.72–1.84 (1H, m), 1.85–1.93 (1H, m), 1.93 (3H, s), 1.94 (3H, s), 2.10–2.22 (1H, m), 2.36–2.47 (2H, m), 3.08–3.18 (3H, m), 3.28 (1H, dd, J = 9.2, 6.0), 4.27 (1H, dd, J = 8.4, 5.7), 4.33 (1H, dd, J = 9.5, 4.9), 4.69 (1H, dd, J = 7.6, 5.9), 5.01 (1H, hept, J = 6.3), 5.72 (1H, bs), 7.02 (1H, t, J = 7.4), 7.10 (1H, t, J = 7.4), 7.14 (1H, s), 7.34 (1H, d, J = 8.0), 7.61 (1H, d, J = 7.8). 13C NMR (101 MHz, MeOD-d4): 21.99 (2C), 22.57, 22.60, 23.96, 27.59, 28.73, 29.93, 32.50, 37.25, 40.19, 53.19, 54.62, 55.66, 56.22, 70.34, 110.83, 112.30, 119.34, 119.81, 122.44, 124.53, 128.86, 137.99, 172.42, 173.20, 173.26, 174.07 (2C), 196.98. IR (KBr): 3295, 3090, 3063, 2107, 1729, 1660, 1644, 1542, 1459, 1437, 1375, 1231, 1106 cm−1. Optical rotation: [α]D20 −18.0° (c 0.260, DMF). ESI MS: 634 ([M + Na]+). HR ESI MS: calcd for C30H41O7N7Na 634.29597; found 634.29598.
Isopropyl 2-(2-(3-(1H-Indol-3-yl)-2-pivalamidopropanamido)-6-acetamidohexanamido)-6-diazo-5-oxohexanoate (11).
Compound 9 (150 mg, 0.263 mmol, 1 equiv) was dissolved in anhydrous DMF (6 mL) and DIEA (68 mg, 92 μL, 0.527 mmol, 2 equiv), followed by adding trimethylacetyl chloride (41 mg, 42 μL, 0.342 mmol, 1.3 equiv). The resulting mixture was stirred at room temperature under an inert atmosphere for 5 h. DMF was evaporated, and the crude product was purified by LC (CHCl3/MeOH, 10:1) to afford 83 mg (48%) of a yellowish solid compound 11. 1H NMR (400 MHz, CDCl3/MeOD-d4): 1.13 (9H, s), 1.24 (3H, d, J = 6.3), 1.26 (3H, d, J = 6.3), 1.21–1.30 (2H, m), 1.35–1.50 (2H, m), 1.51–1.62 (1H, m), 1.74–1.85 (1H, m), 1.93–2.04 (1H, m), 2.00 (3H, s), 2.11–2.21 (1H, m), 2.27–2.44 (2H, m), 3.08 (1H, dt, J = 13.7, 6.8), 3.21 (1H, dd, J = 14.6, 4.8), 3.29 (1H, dt, J = 13.7, 6.8), 3.42 (1H, dd, J = 14.6, 4.8), 4.25–4.44 (2H, m), 4.73 (1H, td, J = 7.1, 4.8), 5.02 (1H, hept, J = 6.3), 5.35 (1H, bs), 6.01 (1H, t, J = 5.9), 6.41 (1H, d, J = 7.3), 6.44 (1H, d, J = 7.3), 6.90 (1H, d, J = 7.6), 7.09 (1H, d, J = 2.4), 7.13 (1H, ddd, J = 8.0, 7.1, 1.1), 7.19 (1H, ddd, J = 8.2, 7.0, 1.2), 7.41 (1H, d, J = 8.0), 7.69 (1H, d, J = 7.8), 9.44 (1H, bs). 13C NMR (101 MHz, CDCl3): 21.82 (2C), 21.95, 23.33, 26.98, 27.48 (3C), 29.20, 29.79, 31.64, 36.53, 38.82, 39.17, 52.14, 53.07, 54.42, 55.32, 69.48, 109.50, 111.84, 118.39, 119.57, 122.09, 123.73, 127.57, 136.49, 170.91, 171.15, 171.34, 171.72, 179.05, 194.43. IR (CHCl3): 3477, 3447, 3418, 3326, 3117, 3084, 3062, 2967, 2110, 1731, 1660, 1620, 1603, 1548, 1524, 1504, 1495, 1458, 1419, 1398, 1385, 1376, 1368, 1321, 1262, 1236, 1105, 1069, 1011 cm−1. Optical rotation: [α]D20 −18.3° (c 0.104, CHCl3). ESI MS: 676 ([M + Na]+). HR ESI MS: calcd for C33H47O7N7Na 676.34292; found 676.34300.
Isopropyl 2-(6-Acetamido-2-(1,2,3,4-tetrahydroisoquinoline-3-carboxamido)hexanamido)-6-diazo-5-oxohexanoate (12).
Fmoc-l-Tic-OH (286 mg, 0.717 mmol, 1.1 equiv) and HATU (298 mg, 0.782 mmol, 1.2 equiv) were dissolved in anhydrous DMF (8 mL), and the reaction mixture was cooled to 0 °C. DIEA (253 mg, 341 μL, 1.96 mmol, 3 equiv) was added, and the reaction mixture was stirred for 15 min at the same temperature. Finally, the solution of 3a (250 mg, 0.652 mmol, 1 equiv) in anhydrous DMF (4 mL) was added dropwise. The mixture was stirred at 0 °C for a further 30 min and at room temperature for 3 h. Diethylamine (477 mg, 674 μL, 6.52 mmol, 10 equiv) was added to the mixture to remove the Fmoc protecting group, and the solution was stirred overnight at room temperature under an inert atmosphere. DMF was evaporated, and the crude product was purified by LC (CHCl3/MeOH, 10:1). The desired product 12 (202 mg, 57% yield) was obtained as a light yellow solid. 1H NMR (400 MHz, DMSO-d6): 1.17 (3H, d, J = 6.3), 1.19 (3H, d, J = 6.3), 1.20–1.28 (2H, m), 1.29–1.41 (2H, m), 1.46–1.58 (1H, m), 1.59–1.71 (1H, m), 1.77 (3H, s), 1.77–1.86 (1H, m), 1.92–2.03 (1H, m), 2.35–2.47 (2H, m), 2.72 (1H, dd, J = 16.3, 10.0), 2.92 (1H, dd, J = 16.3, 4.9), 2.98 (2H, q, J = 6.9), 3.46 (1H, dd, J = 9.7, 4.8), 3.82–3.95 (2H, m), 4.17 (1H, ddd, J = 9.3, 7.3, 5.1), 4.33 (1H, dt, J = 8.2, 5.1), 4.89 (1H, hept, J = 6.3), 6.07 (1H, bs), 7.01–7.08 (1H, m), 7.08–7.16 (4H, m), 7.77 (1H, t, J = 5.6), 7.93 (1H, d, J = 8.3), 8.38 (1H, d, J = 7.4). 13C NMR (101 MHz, DMSO-d6): 21.46, 21.50, 22.43, 22.60, 25.76, 28.92, 30.84, 32.32, 36.20, 38.37, 46.51, 51.49, 51.67, 55.63, 55.71, 68.01, 125.62, 125.70, 125.87, 128.81, 134.44, 136.21, 168.87, 170.97, 171.75, 172.28, 194.04. IR (KBr): 3302, 3089, 2106, 1735, 1663, 1630, 1543, 1512, 1451, 1434, 1386, 1375, 1214, 1107, 703 cm−1. Optical rotation: [α]D20 −43.2° (c 0.118, CHCl3). ESI MS: 543 ([M + H]+). HR ESI MS: calcd for C27H39O6N6 543.29256; found 543.29255.
Isopropyl 2-(6-Acetamido-2-(2-acetyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamido)hexanamido)-6-diazo-5-oxohexanoate (13).
Compound 12 (150 mg, 0.276 mmol, 1 equiv) was dissolved in anhydrous DMF (5 mL) and pyridine (44 mg, 45 μL, 0.553 mmol, 2 equiv), followed by adding acetanhydride (34 mg, 31 μL, 0.332 mmol, 1.2 equiv). The resulting mixture was stirred for 3 h at room temperature under an inert atmosphere. DMF was evaporated, and the residue was purified by LC (CHCl3/MeOH, 15:1) to obtain compound 13 (135 mg, 83%) as a light yellow solid (mixture of isomers ~5:1). 1H NMR (400 MHz, CDCl3, major isomer): 1.23 (3H, d, J = 6.3), 1.24 (3H, d, J = 6.3), 1.28–1.54 (3H, m), 1.64–1.75 (1H, m), 1.95 (3H, s), 1.95–2.03 (2H, m), 2.09–2.18 (2H, m), 2.28 (3H, s), 2.32–2.44 (2H, m), 3.07 (1H, dd, J = 9.3, 6.2), 3.12 (2H, q, J = 6.2), 3.30 (1H, dd, J = 15.4, 4.9), 4.30 (1H, td, J = 8.0, 4.6), 4.39 (1H, ddd, J = 9.1, 7.7, 4.3), 4.53–4.70 (2H, m), 4.96 (1H, dd, J = 5.2, 4.0), 5.00 (1H, hept, J = 6.3), 5.38 (1H, bs), 6.23 (1H, t, J = 5.7), 6.67 (1H, d, J = 7.8), 7.09 (1H, d, J = 7.7), 7.13–7.25 (4H, m). 13C NMR (101 MHz, CDCl3, major isomer): 21.75, 21.83, 21.84, 22.51, 23.34, 26.70, 28.60, 31.43, 32.95, 36.71, 38.79, 47.42, 52.33, 52.60, 54.89, 58.00, 69.48, 125.97, 127.17, 128.17, 128.19, 133.10, 134.06, 170.52, 171.03, 171.25, 171.44, 171.67, 194.44. IR (CHCl3): 3449, 3413, 3337, 3116, 3073, 2109, 1731, 1661, 1636, 1514, 1460, 1437, 1385, 1377, 1365, 1321, 1262, 1235, 1118, 1105, 968, 823 cm−1. Optical rotation: [α]D20 −5.8° (c 0.188, CHCl3). ESI MS: 607 ([M + Na]+). HR ESI MS: calcd for C29H40O7N6Na 607.28507; found 607.28511.
Metabolic Stability Studies.
The in vitro plasma (mouse, swine, human), tissue homogenate (swine jejunum/liver), and liver fraction (human microsomes/S9 fractions) stability assays were performed following our previously reported methods.45 In brief, for the swine jejunum and liver homogenate studies, washed tissues were diluted 10-fold in 0.1 M potassium phosphate buffer and homogenized using a probe sonicator. To evaluate the stability of the intact prodrug over time, 1 mL aliquots were made of each matrix and the prodrug was spiked to a final assay concentration of 20 μm. Stability in liver fractions was assessed for 6 at a final prodrug concentration of 1 μm. Protein concentrations used in the incubations were 0.2 and 0.5 mg/mL for microsomes and S9 fractions, respectively. Spiked samples were incubated in an orbital shaker at 37 °C for 1 h, following which reactions were quenched in triplicate with three volumes of acetonitrile containing the internal standard (IS; losartan: 0.5 μm). The samples were vortex-mixed for 30 s and centrifuged at 10 000g for 10 min at 4 °C. Fifty microliters of the supernatant was diluted with 50 μL of water and transferred to a 250 μL polypropylene vial sealed with a Teflon cap. Prodrug disappearance was monitored over time using liquid chromatography tandem mass spectrometry (LC–MS/MS), as described below in the bioanalysis section.
Human Tumor Cell-to-Plasma Partitioning Assays.
The tumor cell-to-plasma partitioning assays were conducted using P493B lymphoma cells (obtained from Dr. Chi Dang, Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA).46 In brief, cells were grown in 150 cm2 cell T-flasks (Falcon, Cat. #08-772-48) with Roswell Park Memorial Institute Medium 1640 1× with L-glutamine (Corning, Cat. #10-040-CV) supplemented with 10% (v/v) fetal bovine serum (Gibco, Cat. # 26140079) and 1% antimycotic/antibiotic (Gibco, Cat. #15240062). All cells were incubated at 37 °C in a humidified atmosphere with 5% CO2. Cell confluency was monitored visually using an Axiovert 25 optical microscope. Upon confluency, cell suspensions were collected and centrifuged at 200g for 5 min at 25 °C to collect the cell pellet. The supernatant media was decanted, and cell pellets were resuspended in 20 mL of Dulbecco’s phosphate-buffered saline (Gibco, Cat. #14-190-144) maintained at 37 °C.
Cell counts were determined using an automated cell counter (BioRad) following 1:1 dilution of an aliquot with 0.4% trypan blue solution (Bio-Rad). The cell suspension in Dulbecco’s phosphate-buffered saline was centrifuged at 200g for 5 min at 25 °C to collect the cell pellet, following which cell pellets were resuspended in human plasma (Innovative Research) to obtain a cell density of 10 million cells/mL of plasma. For analysis of cell partitioning, 1 mL of the cellplasma suspension was preincubated at 37 °C for 5 min, following which the prodrug was spiked for a final concentration of 20 μm and reincubated at 37 °C for 1 h. Following incubation, the cell suspension was centrifuged at 10 000g for 10 min at 4 °C and supernatant plasma was collected and stored at −80 °C until bioanalysis. The cell pellet was washed once with ice-cold Dulbecco’s phosphate-buffered saline, followed by centrifugation and stored at −80 °C for prodrug and/or DON bioanalysis (described in the Bioanalysis section).
Similarly, tumor-to-plasma partitioning was assessed in additional human tumor cell lines, including H69 (small cell lung cancer cells, ATCC HTB-119) and DU4475 cells (breast cancer cells obtained from metastatic site: skin, ATCC HTB-123). Cells were cultured and treated similar to P493B cells.
For the time-dependent tumor cell partitioning study, a similar protocol was used except that the samples were incubated for predetermined times, including 0.25, 0.5, 1, and 2 h, following which samples were collected for bioanalysis and analyzed for intact 6 and DON, as described below in the Bioanalysis section.
Metabolic Stability Using Recombinant Human Enzymes.
Human recombinant cathepsins and histone deacetylase2 (HDAC2) were purchased from R&D Systems (Minneapolis, MN). Compound 6 was incubated at 1 μm concentration in a reaction mixture (50 μL) consisting of enzymes (HDAC2, cathepsin B, or cathepsin L) and 50 mM 2-(N-morpholino)ethanesulfonic acid (pH 6) containing 5 mM dithiothreitol. The reaction was performed at 37 °C with constant shaking. The final concentration of enzymes in the reaction mixture was 10 μg/mL (HDAC2), 1 μg/mL (cathepsin B), or 0.1 μg/mL (cathepsin L). Following incubation, the reaction was quenched at each time point (0.25, 0.5, 1, and 3 h), by aliquoting 10 μL of the reaction mixture to a new tube containing 30 μL of methanol containing 0.5 μm losartan (internal standard). Samples were vortex-mixed thoroughly and centrifuged at 16 000g for 5 min at 4 °C. Supernatants were transferred to LC vials and analyzed using LC–MS.
P493B Cell Viability Assay.
Cell proliferation assays were performed using CellTiter 96 AQueous One Solution Cell Proliferation reagents following the manufacturers’ instruction (Promega). Briefly, P493B lymphoma cells were plated in 96-well plates at a density of 20 000 cells/well in a final volume of 100 μL of growth media. Test compound stocks were made in DMSO and were added to cells in a 1:10 serial dilution with a final concentration of 0.2% DMSO. Cells were allowed to proliferate for 72 h in the presence of test compounds. Thereafter, 20 μL of CellTiter 96 AQueous (Promega #3580) was added per well and incubated for 2 h. Absorbance was measured at 490 nm.
Pharmacokinetics of 6 in CES1−/− Mice.
The C57BL/6 CES1−/− mice were obtained as a generous gift from the United States Army Medical Research Institute of Chemical Defense, Maryland. Breeding was conducted at the Johns Hopkins Animal Facility. The pharmacokinetic study in C57BL/6 CES1−/− mice was conducted according to protocols reviewed and approved by the Johns Hopkins Institutional Animal Care and Use Committee in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care International and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals (PHS Policy). Briefly, naïve male and female C57BL/6 CES1−/− mice (weighing between 25 and 30 g) 6–8 weeks of age were used. The animals were maintained on a 12 h light–dark cycle with ad libitum access to food (certified laboratory food: Teklad 18% protein extruded rodent diet) and water. EL4 mouse lymphoma cells were obtained as a gift from the laboratory of Dr. Jonathan Powell (Johns Hopkins University, Baltimore, MD) and maintained in Roswell Park Memorial Institute 1640 medium 1× (Corning, Cat. #10–040-CV) with 10% (v/v) fetal bovine serum (Corning, Cat. #35–011-CV), 1% (v/v) antimycotic/antibiotic (Corning, Cat. #30–004-CI), 2 mM L-glutamine (Corning, Cat. #25–005-CI), and 10 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (Corning, Cat. #25–060-CI) in a 5% (v/v) CO2 and 95% (v/v) air incubator prior to subcutaneous (SC) injection (1 × 106 cells in 0.2 mL of phosphate-buffered saline) in one location on the flank of each mouse. When tumors grew to a mean volume of around 400 mm3, mice were used for pharmacokinetic study (n = 3 mice per time point, 2 males and 1 female). Prior to dosing, the interscapular region was wiped with alcohol gauze. 6 was dissolved immediately prior to dosing in ethanol/Tween 80/saline (5:10:85 v/v/v) and was administered to mice as a single SC dose of 3.2 mg/kg (1 mg/kg DON equivalent dose). The mice were euthanized with carbon dioxide at specified time points post drug administration, blood samples (~0.8 mL) were collected in heparinized microtubes by cardiac puncture, and jejunum as well as xenograft tumors were removed and flash-frozen on dry ice. Blood samples were centrifuged at a temperature of 4 °C at 3000g for 10 min. All samples were kept chilled throughout processing. Plasma samples (~300 μL) were collected in polypropylene tubes and stored at −80 °C until bioanalysis. Flash-frozen jejunum and tumor samples were also stored at −80 °C until bioanalysis.
Bioanalysis. Metabolic Stability Studies.
Samples were analyzed on a Thermo Scientific Accela ultra-HPLC (UHPLC) system coupled to an Accela open autosampler with an Agilent Eclipse Plus column (100 × 2.1 mm i.d.; maintained at ambient temperature) packed with a 1.8 μm C18 stationary phase. The autosampler was temperature-controlled and operated at 10 °C. The mobile phase used for chromatographic separation was composed of acetonitrile and water, each containing 0.1% formic acid. Pumps were operated at a flow rate of 0.4 mL/min for 5 min using gradient elution. Peak area counts of analyte and internal standard were measured using a TSQ Vantage triple-quadrupole mass-spectrometric detector, equipped with an electrospray probe set in positive ionization mode. Disappearance of prodrugs was measured from the ratio of peak areas of analyte to IS. Mass transitions of prodrugs including 6 can be found in Table S1 (see Supporting Information page S23).
Tumor Cell-to-Plasma Partitioning Bioanalysis.
Cell pellets and plasma samples were quantified for intact prodrug(s) and DON. The standard curves for quantification of analytes were prepared using naive P493B cells or human plasma. For extraction of analytes, pellets̈ were weighed and suspended in a known quantity of water. To 50 μL of cell suspension or plasma, ice-cold methanol containing internal standards (losartan: 0.5 μm and glutamate-d5: 10 μm) was added in a 5:1 ratio. Samples were vortex-mixed for 30 s and centrifuged at 10 000g for 10 min at 4 °C. The supernatant was collected and analyzed for both DON and intact prodrug.
For DON analysis, supernatants were dried at 45 °C under vacuum for 1 h. To each tube, 50 μL of 0.2 M sodium bicarbonate buffer (pH 9.0) and 100 μL of 10 mM dabsyl chloride was added. After vortex-mixing, samples were incubated at 60 °C for 15 min to derivatize, followed by centrifugation at 16 000g for 5 min at 4 °C. One hundred microliters of the supernatant was transferred to a 96-well plate, diluted with 400 μL of water and injected onto LC–MS/MS.21 DON was analyzed on a Dionex ultra high-performance LC system coupled with a Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation was achieved at 35 °C using an Agilent Eclipse Plus column (100 × 2.1 mm2, i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Pumps were operated at a flow rate of 0.3 mL/min for 3.5 min using gradient elution. The mass spectrometer controlled by Xcalibur software 4.0.27.13 (Thermo Scientific) was operated with a heated ESI ion source in positive ionization mode. Quantification was performed in parallel-reaction monitoring mode.
For intact prodrug analysis, 50 μL of the supernatant obtained after centrifugation was diluted with 50 μL of water and transferred to a 250 μL polypropylene vial sealed with a Teflon cap and analyzed using LC–MS/MS, as described above (see the Metabolic Stability section).
Calibration curves were constructed over the range 0.03–100 nmol/mL for DON and 0.001–50 nmol/mL for prodrugs. Linear regression with 1/(nominal concentration) weighting factor was used for fitting the calibration curve. A correlation coefficient greater than 0.99 was considered to be acceptable in the analytical runs for all analyses.
Metabolic Stability Using Recombinant Human Enzymes.
Samples were injected on an Agilent UHPLC coupled to a 6250 Q-TOF mass spectrometer (Agilent). Chromatographic separation was achieved using gradient conditions on a Zorbax Eclipse Plus C18 column. The total run time was 7.5 min. Samples were analyzed in positive mode with electrospray ionization. The mobile phase consists of 0.1% formic acid in water and 0.1% formic acid in acetonitrile as aqueous and organic modifiers, respectively. Peaks and spectra were analyzed using Agilent MassHunter Qualitative Analysis software.
Metabolite Identification (MET ID) for 6 in Tumor Cells.
The cell samples used for the intact prodrug analysis were also analyzed for prodrug intermediates qualitatively. Briefly, the analysis of prodrug intermediates was performed on an Agilent 1290 LC system coupled to an Agilent 6520B Q-TOF mass spectrometer. Separation was achieved on an Agilent Eclipse plus C18 RRHD 2.1 × 100 mm2, 1.8 μm column maintained at 35 °C. The mobile phase consisted of a mixture of A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) with a flow rate of 0.3 mL/min. The exact mass measurement was based on external calibration performed on the same day. Initial full scans were performed between m/z 50 and 1600. Metabolites were identified in the full-scan mode (from m/z 50 to 1600) by comparing t = 0 samples with t = 60 min samples, and structural information was generated from collision-induced dissociation spectra of molecular ions (M + H)+.
Pharmacokinetic Analysis.
For quantifying intact 6 in the pharmacokinetic samples, plasma samples (25 μL) were processed using a single liquid extraction method by addition of 150 μL of acetonitrile containing internal standard (losartan: 0.5 μm), followed by vortex-mixing for 30 s and then centrifugation at 10 000g for 10 min at 4 °C. Jejunum and tumor tissues were diluted 1:2 w/v with acetonitrile containing losartan (0.5 μm) and homogenized, followed by vortex-mixing and centrifugation at 10 000g for 10 min at 4 °C. A 25 μL aliquot of the supernatant was diluted with 25 μL of water and transferred to 250 μL polypropylene autosampler vials sealed with Teflon caps. Then, 3 μL of the sample was injected into the LC–MS/MS system.
For bioanalysis of DON in pharmacokinetic samples, DON was extracted from plasma samples by protein precipitation using methanol. Briefly, standards, QCs, and samples (50 μL) were mixed with 250 μL of methanol containing 10 μm glutamate-d5 (internal standard) in low-retention microcentrifuge tubes. Jejunum and tumor samples were weighed in TaKaRa BioMasher tubes. Five microliters of methanol containing 10 μm glutamate-d5 was added per milligram of the tissue sample and homogenized by a pestle. For plasma, jejunum, and tumor, the mixture was vortex-mixed and centrifuged at 16 000g for 5 min at 4 °C. The supernatant (100 μL) was transferred to a new tube and dried under vacuum at 45 °C for 1 h. The samples were subjected to dabsyl chloride derivatization and analyzed by LC–MS/MS, as described above.
Calibration curves were constructed over the range 0.03–100 nmol/mL for DON and 0.001–50 nmol/mL for 6 in plasma, jejunum, and tumor tissues. Linear regression with a 1/(nominal concentration) weighting factor was used for fitting the calibration curve. A correlation coefficient of greater than 0.99 was considered to be acceptable in the analytical runs for all analyses.
Mean plasma and tissue concentrations of 6 and DON were analyzed using the noncompartmental method, as implemented in the computer software program Phoenix WinNonlin version 7.0 (Certara USA, Inc., Princeton, NJ). The maximum plasma and tissue concentration (Cmax) and time to Cmax (Tmax) were the observed values. The area under the plasma and tissue concentration time curve (AUC) value was calculated to the last quantifiable sample (AUClast) by use of the log-linear trapezoidal rule.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH Grant R01CA193895 (to B.S.S. and R.R.), R01CA229451 (to B.S.S. and R.R.), R01NS103927 (to B.S.S. and R.R.), Bloomberg Kimmel Institute for Cancer Immunotherapy (to B.S.S. and R.R.), LTAUSA18166 by the Ministry of Education, Youth and Sports of the Czech Republic, program INTER-EXCEL-LENCE (to P.M.), and by the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic, v.v.i. (RVO 61388963).
The authors declare the following competing financial interest(s): Under a license agreement between Dracen Pharmaceuticals, Inc. and the Johns Hopkins University, B.S.S., R.R., and J.A. are entitled to royalty distributions related to technology used in the research described in this publication. B.S.S., P.M. and R.R. are also co-founders of and hold equity in Dracen Pharmaceuticals, Inc. which is clinically developing glutamine antagonist prodrugs. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
ABBREVIATIONS
- BLQ
below the limit of quantification
- Boc
tert-butyloxycarbonyl
- CES1
carboxylesterase 1
- CES1−/−
carboxylesterase 1 knockout
- CTSL
cathepsin L
- DIEA
N,N-diisopropylethylamine
- DON
6-diazo-5-oxo-l-norleucine
- Fmoc
fluorenylmethyloxycarbonyl
- GI
gastrointestinal
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HDAC
histone deacetylase
- HRMS
high-resolution mass spectrometry
- MET ID
metabolite identification
- SC
subcutaneous
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylate
- Trp
tryptophan
- WT
wild-type
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b02009.
Details of 1H and 13C NMR and HPLC analysis, 1H and 13C NMR spectra, HPLC chromatograms; table containing mass transitions for LC–MS analysis; figures showing molecular structures of internal standards, stability of 6 in human liver fractions, tumor cell/plasma partitioning in three different cell lines, HRMS chromatogram and spectra of 6 and its de-esterified product (PDF)
Molecular formula strings and associated biological data (CSV)
REFERENCES
- (1).Reitzer LJ; Wice BM; Kennell D Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [PubMed] [Google Scholar]
- (2).Wise DR; DeBerardinis RJ; Mancuso A; Sayed N; Zhang XY; Pfeiffer HK; Nissim I; Daikhin E; Yudkoff M; McMahon SB; Thompson CB Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu W; Le A; Hancock C; Lane AN; Dang CV; Fan TW; Phang JM Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8983–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ahluwalia GS; Grem JL; Hao Z; Cooney DA Metabolism and action of amino acid analogue anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [DOI] [PubMed] [Google Scholar]
- (5).Thangavelu K; Chong QY; Low BC; Sivaraman J Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci. Rep. 2014, 4, No. 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tran TQ; Ishak Gabra MB; Lowman XH; Yang Y; Reid MA; Pan M; O’Connor TR; Kong M Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, No. e2002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Magill GB; Myers WP; Reilly HC; Putnam RC; Magill JW; Sykes MP; Escher GC; Karnofsky DA; Burchenal JH Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957, 10, 1138–1150. [DOI] [PubMed] [Google Scholar]
- (8).Sullivan MP; Nelson JA; Feldman S; Van Nguyen B Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-l-norleucine) in children. Cancer Chemother. Pharmacol. 1988, 21, 78–84. [DOI] [PubMed] [Google Scholar]
- (9).Earhart RH; Amato DJ; Chang AY; Borden EC; Shiraki M; Dowd ME; Comis RL; Davis TE; Smith TJ Phase II trial of 6-diazo-5-oxo-l-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest. New Drugs 1990, 8, 113–119. [DOI] [PubMed] [Google Scholar]
- (10).Rahman A; Smith FP; Luc PT; Woolley PV Phase I study and clinical pharmacology of 6-diazo-5-oxo-l-norleucine (DON). Invest. New Drugs 1985, 3, 369–374. [DOI] [PubMed] [Google Scholar]
- (11).Eagan RT; Frytak S; Nichols WC; Creagan ET; Ingle JN Phase II study on DON in patients with previously treated advanced lung cancer. Cancer Treat. Rep. 1982, 66, 1665–1666. [PubMed] [Google Scholar]
- (12).Earhart RH; Koeller JM; Davis HL Phase I trial of 6-diazo-5-oxo-l-norleucine (DON) administered by 5-day courses. Cancer Treat. Rep. 1982, 66, 1215–1217. [PubMed] [Google Scholar]
- (13).Kovach JS; Eagan RT; Powis G; Rubin J; Creagan ET; Moertel CG Phase I and pharmacokinetic studies of DON. Cancer Treat. Rep. 1981, 65, 1031–1036. [PubMed] [Google Scholar]
- (14).Lynch G; Kemeny N; Casper E Phase II evaluation of DON (6-diazo-5-oxo-l-norleucine) in patients with advanced colorectal carcinoma. Am. J. Clin. Oncol. 1982, 5, 541–543. [PubMed] [Google Scholar]
- (15).Ovejera AA; Houchens DP; Catane R; Sheridan MA; Muggia FM Efficacy of 6-diazo-5-oxo-l-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res. 1979, 39, 3220–3224. [PubMed] [Google Scholar]
- (16).Rubin J; Sorensen S; Schutt AJ; van Hazel GA; O’Connell MJ; Moertel CG A phase II study of 6-diazo-5-oxo-l-norleucine (DON, NSC-7365) in advanced large bowel carcinoma. Am. J. Clin. Oncol. 1983, 6, 325–326. [DOI] [PubMed] [Google Scholar]
- (17).Shelton LM; Huysentruyt LC; Seyfried TN Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int. J. Cancer 2010, 127, 2478–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tarnowski GS; Stock CC Effects of combinations of azaserine and of 6-diazo-5-oxo-l-norleucine with purine analogues and other antimetabolites on the growth of two mouse mammary carcinomas. Cancer Res. 1957, 17, 1033–1039. [PubMed] [Google Scholar]
- (19).Hensley CT; Wasti AT; DeBerardinis RJ Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013, 123, 3678–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rais R; Jancarik A; Tenora L; Nedelcovych M; Alt J; Englert J; Rojas C; Le A; Elgogary A; Tan J; Monincova L; Pate K; Adams R; Ferraris D; Powell J; Majer P; Slusher BS Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J. Med. Chem. 2016, 59, 8621–8633. [DOI] [PubMed] [Google Scholar]
- (21).Nedelcovych MT; Tenora L; Kim BH; Kelschenbach J; Chao W; Hadas E; Jancarik A; Prchalova E; Zimmermann SC; Dash RP; Gadiano AJ; Garrett C; Furtmuller G; Oh B; Brandacher G; Alt J; Majer P; Volsky DJ; Rais R; Slusher BS N-(Pivaloyloxy)alkoxy-carbonyl prodrugs of the glutamine antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a potential treatment for HIV associated neurocognitive disorders. J. Med. Chem. 2017, 60, 7186–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ueki N; Wang W; Swenson C; McNaughton C; Sampson NS; Hayman MJ Synthesis and preclinical evaluation of a highly improved anticancer prodrug activated by histone deacetylases and cathepsin L. Theranostics 2016, 6, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ueki N; Lee S; Sampson NS; Hayman MJ Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat. Commun. 2013, 4, No. 2735. [DOI] [PubMed] [Google Scholar]
- (24).Gras-Masse H Lipid vector for the delivery of peptides towards intracellular pharmacological targets. J. Mol. Recognit. 2003, 16, 234–239. [DOI] [PubMed] [Google Scholar]
- (25).Hutchinson J; Burholt S; Hamley I Peptide hormones and lipopeptides: from self-assembly to therapeutic applications. J. Pept. Sci. 2017, 23, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rogawski MA; Wenk GL The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003, 9, 275–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hubsher G; Haider M; Okun M Amantadine The journey from fighting flu to treating Parkinson disease. Neurology 2012, 78, 1096–1099. [DOI] [PubMed] [Google Scholar]
- (28).Liu J; Obando D; Liao V; Lifa T; Codd R The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011, 46, 1949–1963. [DOI] [PubMed] [Google Scholar]
- (29).Zhang W; Wang S; Wang Q; Yang Z; Pan Z; Li L Overexpression of cysteine cathepsin L is a marker of invasion and metastasis in ovarian cancer. Oncol. Rep. 2014, 31, 1334–1342. [DOI] [PubMed] [Google Scholar]
- (30).Cui F; Wang W; Wu D; He X; Wu J; Wang M Overexpression of Cathepsin L is associated with gefitinib resistance in non-small cell lung cancer. Clin. Transl. Oncol. 2016, 18, 722–727. [DOI] [PubMed] [Google Scholar]
- (31).Henderson JM; Zhang HE; Polak N; Gorrell MD Hepatocellular carcinoma: Mouse models and the potential roles of proteases. Cancer Lett. 2017, 387, 106–113. [DOI] [PubMed] [Google Scholar]
- (32).Zhang Y; Fang H; Xu W Applications and modifications of 1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic acid (Tic) in peptides and peptidomimetics design and discovery. Curr. Protein Pept. Sci. 2010, 11, 752–758. [DOI] [PubMed] [Google Scholar]
- (33).Majer P; Slaninová J; Lebl M Synthesis of methylated phenylalanines via hydrogenolysis of corresponding 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids: Synthesis and biological activity of oxytocin analogues with methylated phenylalanines in position 2. Int. J. Pept. Protein Res. 1994, 43, 62–68. [DOI] [PubMed] [Google Scholar]
- (34).Kaplan HR; Taylor D; Olson S Quinapril: overview of preclinical data. Clin. Cardiol. 1990, 13, VII-6–VII-12. [DOI] [PubMed] [Google Scholar]
- (35).Testa B; Mayer JM Concepts in Prodrug Design To Overcome Pharmacokinetic Problems. In Pharmacokinetic Optimization in Drug Research; Testa, B. et al. , Ed.; Wiley-VCH, 2001; pp 85–95. [Google Scholar]
- (36).Swindle M; Makin A; Herron A; Clubb F Jr.; Frazier K Swine as models in biomedical research and toxicology testing. Vet. Pathol. 2012, 49, 344–356. [DOI] [PubMed] [Google Scholar]
- (37).Fu J; Pacyniak E; Leed MGD; Sadgrove MP; Marson L; Jay M Interspecies differences in the metabolism of a multi-ester prodrug by carboxylesterases. J. Pharm. Sci. 2016, 105, 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Van Gelder J; Shafiee M; De Clercq E; Penninckx F; Van den Mooter G; Kinget R; Augustijns P Species-dependent and site-specific intestinal metabolism of ester prodrugs. Int. J. Pharm. 2000, 205, 93–100. [DOI] [PubMed] [Google Scholar]
- (39).Krämer OH HDAC2: a critical factor in health and disease. Trends Pharmacol Sci. 2009, 30, 647–655. [DOI] [PubMed] [Google Scholar]
- (40).Gondi CS; Rao JS Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhong Y-J; Shao L-H; Li Y Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Seymour LW; Ulbrich K; Steyger PS; Brereton M; Subr V; Strohalm J; Duncan R Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. Br. J. Cancer 1994, 70, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rautio J; Kumpulainen H; Heimbach T; Oliyai R; Oh D; Jarvinen T; Savolainen J Prodrugs: design and clinical applications.¨ Nat. Rev. Drug. Discovery 2008, 7, 255. [DOI] [PubMed] [Google Scholar]
- (44).Li B; Sedlacek M; Manoharan I; Boopathy R; Duysen EG; Masson P; Lockridge O Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 2005, 70, 1673–1684. [DOI] [PubMed] [Google Scholar]
- (45).Zimmermann SC; Tichy T; Vávra J; Dash RP; Slusher CE; Gadiano AJ; Wu Y; Jančařík A; Tenora L; Monincova L N-substituted prodrugs of mebendazole provide improved aqueous solubility and oral bioavailability in mice and dogs. J. Med. Chem. 2018, 61, 3918–3929. [DOI] [PubMed] [Google Scholar]
- (46).Gao P; Tchernyshyov I; Chang TC; Lee YS; Kita K; Ochi T; Zeller KI; De Marzo AM; Van Eyk JE; Mendell JT; Dang CV c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.