

Abstract
Factor VII-activating protease (FSAP) is a novel plasma-derived serine protease structurally homologous to tissue-type and urokinase-type plasminogen activators. We demonstrate that plasminogen activator inhibitor-1 (PAI-1), the predominant inhibitor of tissue-type and urokinase-type plasminogen activators in plasma and tissues, is an inhibitor of FSAP as well. We detected PAI-1·FSAP complexes in addition to high levels of extracellular RNA, an important FSAP cofactor, in bronchoalveolar lavage fluids from patients with acute respiratory distress syndrome. Hydrolytic activity of FSAP was inhibited by PAI-1 with a second-order inhibition rate constant (Ka) of 3.38 ± 1.12 × 105m–1·s–1. Residue Arg346 was a critical recognition element on PAI-1 for interaction with FSAP. RNA, but not DNA, fragments (>400 nucleotides in length) dramatically enhanced the reactivity of PAI-1 with FSAP, and 4 μg·ml–1 RNA increased the Ka to 1.61 ± 0.94 × 106m–1·s–1. RNA also stabilized the active conformation of PAI-1, increasing the half-life for spontaneous conversion of active to latent PAI-1 from 48.4 ± 8 min to 114.6 ± 5 min. In contrast, little effect of DNA on PAI-1 stability was apparent. Residues Arg76 and Lys80 in PAI-1 were key elements mediating binding of nucleic acids to PAI-1. FSAP-driven inhibition of vascular smooth muscle cell proliferation was antagonized by PAI-1, suggesting functional consequences for the FSAP-PAI-1 interaction. These data indicate that extracellular RNA and PAI-1 can regulate FSAP activity, thereby playing a potentially important role in hemostasis and cell functions under various pathophysiological conditions, such as acute respiratory distress syndrome.
Factor VII-activating protease (FSAP),2 also known as plasma hyaluronan-binding protein or plasma hyaluronan-binding serine protease, is a recently described plasma serine protease (1, 2, 3), expressed primarily in the liver and found in endothelial and epithelial cells of the lung, kidney, placenta, and pancreas (1, 4). The enzyme circulates as a 64-kDa single-chain zymogen (scFSAP) in plasma, which is autoactivated to an enzymatically active two-chain form (tcFSAP). Two-chain FSAP consists of a 46-kDa heavy chain connected by a disulfide bridge to a 29-kDa light chain containing the catalytic domain (5, 6, 7). Autoactivation of scFSAP to tcFSAP is promoted in the presence of negatively charged substances, such as heparin, dextran sulfate, phosphatidylethanolamine, or extracellular RNA (5, 6, 7, 8).
The role of FSAP in different physiological and pathophysiological conditions is not fully understood. A dual role for FSAP in vitro in hemostasis has been suggested (3, 9). FSAP is a potent activator of coagulation factor VII, thus contributing to the initiation of blood coagulation via the extrinsic pathway (3). Furthermore, FSAP also activates prourokinase-type plasminogen activator (uPA) and thus contributes to plasminogen activation as well (9). Additional links between FSAP and the plasminogen activation system exist; FSAP is highly homologous to uPA and tissue-type plasminogen activator (tPA), and uPA is a potential physiological activator of FSAP (5). Additionally, recent in vitro studies have revealed that hemostatic serine protease inhibitors (serpins), such as C1 inhibitor and α2-antiplasmin, are inhibitors of FSAP proteolytic activity (7, 9). These observations prompted us to explore the role of PAI-1, the primary member of this inhibitor family in plasma and tissues (10, 11), as a potential regulator of FSAP. It is known that PAI-1 inhibits uPA and tPA by forming SDS-resistant, catalytically inactive complexes with proteases (12, 13). In this reaction, the reactive center residues P1-P1′ (Arg346-Met347) in PAI-1 function as an exposed “bait” for the protease by mimicking a putative cleavage site (14, 15, 16, 17). In addition to inhibiting serine proteases, PAI-1 also interacts with different components of the extracellular matrix, including heparin (18, 19) and vitronectin (20, 21, 22). In the presence of heparin, the selectivity of PAI-1 for the inhibition of tPA and uPA is compromised, resulting in a 2-fold increased rate of association between PAI-1 and thrombin (18). This leads to neutralization of PAI-1 due to the formation of inactive SDS-stable PAI-1-thrombin complexes and subsequent cleavage of PAI-1 by thrombin (18). Similarly, binding of PAI-1 to vitronectin broadens the specificity of the inhibitor, converting it into a potent inhibitor of activated protein C (23) and thrombin (21). Moreover, vitronectin stabilizes PAI-1 in its active conformational state and mediates the binding of the inhibitor to fibrin clots (24). In complexes with vitronectin, PAI-1 serves as an antiadhesive factor independent of its protease-inhibitory capacity (25).
In the present study, we identified PAI-1·FSAP complexes in bronchoalveolar lavage (BAL) fluids of patients with the acute respiratory distress syndrome (ARDS). Furthermore, formation of a 1:1 stoichiometric complex between PAI-1 and FSAP and dose-dependent binding of PAI-1 to FSAP in vitro was observed. The kinetics of FSAP inhibition by PAI-1 were investigated in the absence and presence of different cofactors (RNA, DNA, vitronectin, and heparin). The presence of extracellular RNA, but not DNA, dramatically enhanced the reactivity of PAI-1 against FSAP. RNA served as a novel stabilizer of the active conformation of PAI-1. These data are particularly relevant, since we also describe elevated levels of extracellular RNA in the BAL fluids of ARDS patients. We also demonstrate that heparin and RNA share the same binding sites on PAI-1. Using site-directed mutagenesis, the critical importance of Arg346 in PAI-1 as a mediator of FSAP inhibition was demonstrated. Additionally, the FSAP-mediated inhibition of vascular smooth muscle cell proliferation was significantly attenuated in the presence of PAI-1. These results suggest that PAI-1-mediated inhibition of FSAP might play an important role in the regulation of hemostasis and vascular cell functions under a variety of physiological and pathological conditions, such as ARDS.
EXPERIMENTAL PROCEDURES
Materials—The purification of FSAP from human plasma by affinity chromatography, together with the conversion of scFSAP to tcFSAP, was performed as described previously (5, 26). Urokinase-type plasminogen activator and α1-proteinase inhibitor were from ZLB-Behring (Marburg, Germany). Aprotinin was from Bayer (Leverkusen, Germany). Recombinant human wild-type (WT) PAI-1 and PAI-1 variant R346A were expressed in Escherichia coli as described previously (27). The PAI-1 WT as well as PAI-1 variants K65A, K69A, R76A, K80A, and K88A, expressed in E. coli, were a gift from Dr. H. Pannekoek (19) (University of Amsterdam, Academic Medical Center, Amsterdam, The Netherlands). Tissue-type plasminogen activator, C1 inhibitor, antithrombin, and α2-antiplasmin were obtained from American Diagnostica (Pfungstadt, Germany). Vitronectin was prepared from human plasma as described previously (20). Heparin was purchased from Ratiopharm (Ulm, Germany). The extraction of RNA from HepG2 cells was performed using QIAzol™ lysis reagent (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Genomic DNA was isolated from HepG2 cells using Genomic DNA Kit (Qiagen) following the manufacturer's instructions. For visual detection, isolated nucleic acids were subjected to electrophoresis on a 1% agarose gel followed by ethidium bromide staining. The concentrations of nucleic acids were determined using a Gene Quant photometer (Amersham Biosciences, Freiburg, Germany).
Bronchoalveolar Lavage—BAL fluids were obtained by flexible fiberoptic bronchoscopy from spontaneously breathing healthy volunteers without any history of cardiac or lung disease and with normal pulmonary function (n = 8) and from mechanically ventilated patients with early ARDS (<120 h after onset of the disease; n = 8) as recently described (28). Diagnosis of ARDS was made on the basis of the ARDS American-European consensus criteria (29). Written informed consent was obtained from either the patients or their next-of-kin.
Determination of RNA and DNA Concentration in BAL Fluid—RNA and DNA were extracted from BAL fluid using QIAzol™ lysis reagent and QIAamp® DNA minikit, respectively (both from Qiagen). The RNA concentration was measured using one-step real time quantitative reverse transcription-PCR for GAPDH. The amplification primers were GAPDH-F (5′-CCA CAT CGC TCA GAC ACC AT-3′) and GAPDH-R (5′-GGC AAC AAT ATC CAC TTT ACC AGA G-3′) and a dual-labeled fluorescent probe was GAPDH-P (5′-AAG GTC GGA GTC AAC GGA TTT GGT CG-3′). The reverse transcription-PCRs were set up with the GeneAmp EZ rTth RNA PCR kit (PE Applied Biosystems, Foster City, CA) according to the manufacturer's protocol using 5 μl of extracted RNA. The DNA concentration was assessed using real time quantitative PCR for β-globin. The amplification primers were β-globin-F (5′-GTG CAC CTG ACT CCT GAG GAG-3′) and β-globin-R (5′-CCT TGA TAC CAA CCT GCC CAG-3′), and a dual-labeled fluorescent probe was β-globin-P (5′-AAG GTG AAC GTG GAT GAA GTT GGT GG-3′). The PCRs were set up with a Quan-i Tect probe PCR kit (Qiagen) according to the manufacturer's protocol using 5 μl of extracted DNA. Amplification data were collected and analyzed with an ABI Prism 7700 Sequence Detector (PE Applied Biosystems). Each sample was analyzed in triplicate, and multiple negative water blanks were included in each analysis. A calibration curve was prepared using serial dilutions of a commercially available human control RNA or human control DNA (both from PE Applied Biosystems).
The presence of nucleic acids in BAL fluids from ARDS patients and healthy controls was also assessed by a filter-binding assay, where a nylon membrane was soaked with buffer A (10 mm Tris-Cl (pH 7.5), 50 mm NaCl, 1 mm EDTA, and 1× Denhardt's solution (0.02% (m/v) Ficoll, 0.02% (m/v) polyvinylpyrrolidone, and 0.02% (m/v) BSA)) and fixed into a slot-blot apparatus. The RNA and DNA extracted from the BAL fluid from eight ARDS patients and from eight healthy volunteers was biotinylated and then applied to the filter in 100 μl of TBS for 10 min, aspirated through the filter, and cross-linked for 10 min by exposure to ultraviolet light (254 nm). After washing with buffer A, the membrane was incubated with peroxidase-labeled streptavidin (DAKO, Glostrup, Denmark), and detection of RNA and DNA was performed using enhanced chemiluminescence (Pierce). Biotinylation of RNA and DNA was performed with EZ-Link™ Psoralen-PEO-biotin from Pierce according to the manufacturer's instructions.
Western Blotting Analysis of BAL Fluids from ARDS Patients—Bronchoalveolar lavage fluids (25 μl) were combined with nonreducing sample buffer containing 6 m guanidine hydrochloride, and after SDS-PAGE on 10% gels, proteins were transferred to a polyvinylidene difluoride membrane. After blocking with 5% (m/v) nonfat milk in Tris-buffered saline (25 mm Tris-Cl, 150 mm NaCl, pH 7.5) containing 0.1% (v/v) Tween 20 (TBS-T), the membrane was incubated at 4 °C overnight with a rabbit antibody raised against FSAP (Aventis Behring, Marburg, Germany), followed by incubation with horseradish peroxidase-labeled secondary antibody (DAKO). Immune complexes were detected using enhanced chemiluminescence (Pierce). For detection of PAI-1, the membrane was stripped using stripping buffer (2% (m/v) SDS, 100 mm β-mercaptoethanol in TBS) and reprobed with a rabbit anti-PAI-1 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Co-immunoprecipitation Experiments—Prior to co-immunoprecipitation, albumin was removed from BAL fluid samples using a ProteoSeek™ antibody-based albumin/IgG removal kit from Pierce according to the manufacturer's instructions. Bronchoalveolar lavage fluid (100 μl) was incubated at 4 °C overnight with 5 μg of murine monoclonal anti-FSAP antibody or IgG as an isotype control. Samples were transferred to tubes containing 50 μl of protein A-Sepharose™ CL-4B beads (Amersham Biosciences). After a 2-h incubation at room temperature, the immunoprecipitates were washed five times (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% (v/v) Triton X-100), boiled in 40 μl of SDS sample buffer, separated by SDS-PAGE (10% gels) under reducing conditions, and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% (m/v) nonfat dry milk in TBS-T and then incubated at 4 °C overnight with rabbit antibodies directed against FSAP, PAI-1 (Santa Cruz Biotechnology), α1-proteinase inhibitor (Aventis Behring), C1 inhibitor, α2-antiplasmin, and antithrombin, respectively (all obtained from DAKO). After incubation with peroxidase-labeled secondary antibody, proteins were detected by enhanced chemiluminescence.
Inhibition of FSAP by C1 Inhibitor, α1-Proteinase Inhibitor, Antithrombin III, α2-Antiplasmin, and PAI-1—Fourteen nm (by mass) tcFSAP was added to C1 inhibitor (50 nm), α1-proteinase inhibitor (50 μm), antithrombin (50 μm), α2-antiplasmin (100 nm), and PAI-1 (50 nm) in TBS, pH 7.4, in a total volume of 100 μl. After a 5-min incubation at 37 °C, 10 μl of 3 mm chromogenic substrate S-2288 (H-d-isoleucyl-l-prolyl-l-arginyl-para-nitroanilide dihydrochloride; Chromogenix, Mölndal, Sweden) was added, and the hydrolysis of S-2288 by FSAP was measured spectrophotometrically at 405 nm in an EL 808 microtiter plate reader (BioTek Instruments, Highland Park, VT).
Complex Formation between FSAP and C1 Inhibitor, α1-Proteinase Inhibitor, Antithrombin III, α2-Antiplasmin, and PAI-1—Two-chain FSAP (1 μg) was combined with C1 inhibitor (2 μg), α1-proteinase inhibitor (3 μg), antithrombin (2 μg), α2-antiplasmin (2 μg), or PAI-1 (4 μg) in TBS, pH 7.4, in a total volume of 100 μl and incubated at room temperature for 30 min. Subsequently, nonreducing sample buffer was added, and the samples were subjected to 10% SDS-PAGE. The proteins were stained with Coomassie Brilliant Blue R-250.
Titration of Wild-type PAI-1 and PAI-1 R346A—Increasing amounts of wild-type PAI-1 and PAI-1 R346A were incubated at 37 °C for 30 min in a total volume of 100 μl with 3 nm tPA (active concentration) in TBS buffer. Chromogenic substrate (10 μl of a 3 mm S-2288 solution) was added, and residual tPA activity was determined from a linear plot of the increase of absorbance at 405 nm over time.
Analysis of PAI-1 Binding to FSAP—A microtiter plate was coated with 50 μl of 140 nm tcFSAP or scFSAP in 50 mm NaHCO3, pH 9.6, at 4 °C overnight. The plate was washed three times with TBS-T buffer, and nonspecific binding sites were blocked with 3% (m/v) BSA in TBS-T at room temperature for 1 h. Increasing concentrations (0–20 nm) of PAI-1 in TBS-T containing 0.3% (m/v) BSA alone or in the presence of 2 μg·ml–1 rabbit antibody against FSAP were added to the wells, and PAI-1 was allowed to bind to FSAP at room temperature for 2 h. After extensive washing with TBS-T, bound PAI-1 was detected using a rabbit polyclonal antibody against PAI-1 followed by peroxidase-labeled secondary antibody. Final detection was performed using 3,3′,5,5′-tetramethylbenzidine (TMB), with a TMB substrate kit (Pierce) according to the manufacturer's instructions. Binding of PAI-1 to BSA-coated wells was employed as a background control, against which values were normalized. The uPA-PAI-1 complexes were generated by incubation of equimolar quantities of two-chain uPA and active PAI-1 at room temperature for 30 min and subsequently purified by Superdex 200 gel filtration chromatography (Amersham Biosciences).
Complex Formation between FSAP and PAI-1—Twenty ng of tcFSAP or scFSAP were incubated with increasing amounts of PAI-1 (0–80 ng) at room temperature for 30 min. Samples were mixed with either nonreducing or reducing sample buffer, and after SDS-polyacrylamide gel electrophoresis on 10% polyacrylamide gels, proteins were transferred to a polyvinylidene difluoride membrane. Detection of proteins was performed with a rabbit polyclonal antibody directed against FSAP or with murine monoclonal antibodies directed against heavy and light chain FSAP, respectively. Subsequently, the membranes were stripped and reprobed with a rabbit anti-PAI-1 antibody.
NH2-terminal Sequencing—The amino-terminal sequence of peptides contained in the PAI-1·FSAP complex was determined by automated Edman degradation using an Applied Biosystems 492 pulsed liquid phase sequencer equipped with an on-line 785A phenylthiohydantoin derivative analyzer. Ten cycles of Edman degradation were performed, and the amino acids detected at each cycle were aligned with the FSAP sequence (GenBank® accession number NP_004123).
Enzymatic Analysis of FSAP Inhibition by PAI-1—Reaction samples contained 14 nm (by mass) tcFSAP, increasing concentrations of PAI-1 (0–24 nm; by active site titration against tPA) and 0.3 mm of the chromogenic substrate S-2288 in 100 μlof TBS, pH 7.4. The hydrolysis of S-2288 by FSAP was measured spectrophotometrically at 405 nm every 30 s at 37 °C for 30 min in an EL 808 microtiter plate reader.
To investigate the influence of vitronectin, heparin, RNA, or DNA on the PAI-1-mediated inhibition of FSAP amidolytic activity, 14 nm (by mass) tcFSAP or scFSAP was mixed with 10 nm PAI-1 (by active site titration against tPA) and increasing concentrations of vitronectin (0–40 nm), heparin (0–24 nm), RNA (0–4 μg·ml–1), or DNA (0–4 μg·ml–1). Hydrolysis of S-2288 (0.3 mm) was measured spectrophotometrically as described above.
The second-order inhibition rate constant Ka was determined using a continuous progress curve method essentially as described by Futamura and Gettins (30). The change in absorbance resulting from hydrolysis of 1.5 mm S-2288 by FSAP (8 nm, by mass) in the presence of PAI-1 (0–200 nm, by active site titration against tPA) was monitored at 405 nm. A pseudo-first order inhibition rate constant, k obs, was determined by fitting data to a monoexponential equation [P]t = [P]∞(1 – ek obs· t), where [P]t and [P]∞ are the product concentrations at time t and infinite time, respectively, proportional to the recorded A 405 values. The Ka was then calculated from the equation, correcting for the presence of competing substrate, by k = k obs(1 + [S-2288]0/Km)/[PAI-1]0, where [S-2288]0 is the concentration of S-2288 at time 0, and [PAI-1]0 is the PAI-1 concentration at time 0. Under the conditions used, the Km was determined to be 70.4 ± 4.4 μm (n = 5).
Inhibition of FSAP by PAI-1 R346A—Two-chain FSAP (14 nm) was incubated with wild-type PAI-1 or PAI-1 R346A (24 nm active PAI-1, titrated against tPA) in a total volume of 100 μlof TBS buffer. After 30 min at 37 °C, 10 μl of 0.3 mm S-2288 was added, and FSAP activity was determined from the linear increase of absorbance at 405 nm. The increase of absorbance measured for the sample containing FSAP alone was taken as 100%.
Binding of RNA and DNA to PAI-1—For fragmentation of nucleic acids, 1 mg of biotinylated RNA or DNA was dissolved in 1 ml of RNase-, DNase-free water and sonicated for 2, 4, 6, 8, 10, or 12 s. At the indicated time points, 100-μl samples were withdrawn, and nucleic acids were precipitated with ethanol. The RNA and DNA were dissolved in RNase-, DNase-free water and tested for PAI-1-binding capacity (see below). Parallel samples were subjected to agarose gel electrophoresis and visualized by ethidium bromide staining.
The wells of a microtiter plate were coated at 4 °C overnight with wild-type PAI-1 at 200 nm (by mass) in 50 mm NaHCO3, pH 9.6. Nonspecific binding sites were blocked by incubation with 3% (m/v) BSA in TBS at room temperature for 1 h. Binding assays were performed by adding increasing concentrations of biotinylated RNA (0–25 μg·ml–1 in TBS) or DNA (0–5 μg·ml–1 in TBS) to immobilized wild-type PAI-1. After a 2-h incubation at room temperature, the plate was extensively washed with TBS, and bound nucleic acids were detected using peroxidase-labeled streptavidin. Final detection was performed with a TMB substrate kit from Pierce. The binding of nucleic acids to BSA-coated wells was employed as a blank in all experiments and was subtracted from experimental values to obtain specific binding. Binding of biotinylated RNA or DNA to PAI-1 in the presence of 1 mm heparin, a 100-fold excess of unlabeled RNA or DNA, or 10 mm tranexamic acid (Sigma) served as controls. To determine the RNA and DNA binding sites on PAI-1, the wells of a microtiter plate were coated at 4 °C overnight with PAI-1 variants K65A, K69A, R76A, K80A, and K88A all at 200 nm (by mass) in 50 mm NaHCO3, pH 9.6. After blocking with 3% (m/v) BSA in TBS, 10 μg·ml–1 of biotinylated RNA or 3 μg·ml–1 of DNA were added, and the plate was incubated at room temperature for 2 h. Final detection of PAI-1-bound RNA or DNA was performed as described above. Since PAI-1 is partially denatured by adsorption to plastic at 4 °C overnight, data obtained in the microplate assay reflect nucleic acid binding to denatured PAI-1. To assess nucleic acid binding to active PAI-1, a filter-binding assay was performed, where nitrocellulose membranes were soaked with buffer A (see above) and fixed into a slot-blot apparatus. Increasing concentrations (0–6 μg·ml–1) of either wild-type PAI-1 or PAI-1 variants (R76A and K80A) were mixed with 10 μg·ml–1 of RNA or 3 μg·ml–1 of DNA, incubated on the filters in 100 μl of TBS for 15 min, and then aspirated through the filters. After extensive washing with buffer A, the membrane was incubated with peroxidase-labeled streptavidin, and PAI-1-nucleic acid complexes were detected by enhanced chemiluminescence (Pierce).
Assessment of PAI-1 Activity—Samples of PAI-1 (24 nm, by active site titration against tPA) dissolved in TBS, pH 7.4, were incubated at 37 °C for 1–5 h in the absence or presence of either vitronectin (70 nm), RNA (4 μg·ml–1), or DNA (4 μg·ml–1). Inhibition of uPA by PAI-1 was measured with the peptide-derived chromogenic substrate S-2444 (l-pyroglutamyl-l-glycyl-l-argininyl-para-nitroanilide; Chromogenix) at various time points. A ratio was determined for the residual PAI-1 activity at time t (vt) versus the initial, uninhibited activity (v 0). Linear regression of a semilog plot of ln(vt/v 0) versus time yielded a graph with a slope = k, where k is the rate of disappearance of active PAI-1. The half-life for inactivation of PAI-1, t ½(inact), is given by t ½(inact) = ℓn2/k.
Cell Proliferation Assay—Proliferation of mouse vascular smooth muscle cells (VSMC) in the presence of different test reagents (FSAP (12 μg·ml–1), PDGF-BB (20 ng·ml–1), PAI-1 (60 μg·ml–1)) or combinations thereof was determined by DNA synthesis assays based on the uptake of bromodeoxyuridine as previously described (26).
Statistics—Data are presented as mean ± S.D. The statistical analyses were performed with SPSS version 9.0.1 (SPSS Inc., Chicago, IL). Differences between two groups were tested with Student's t test.
RESULTS
Complex Formation between FSAP and PAI-1 and RNA Levels in ARDS BAL Fluids—Dysregulated proteolysis is an emerging area of interest in the pathogenesis and management of acute lung injury (31, 32, 33). During the course of a broad screen of proteolytic enzymes in patients with ARDS, we detected significantly increased levels of FSAP protein and FSAP activity in BAL fluids of these patients. Remarkably, almost no FSAP was detectable in BAL fluids from healthy patients (data not shown). Upon electrophoretic analysis and Western blotting, BAL fluids from ARDS patients exhibited a high molecular mass band of 120 kDa, together with an expected band of 64 kDa, when probed with anti-FSAP antibodies (Fig. 1A ). These data suggested the presence of high molecular mass complexes of FSAP with other proteins in the BAL fluids of ARDS patients. Most likely, these complexes consisted of FSAP bound to a cognate inhibitor also present in the lung. Therefore, to identify potential interaction partners of FSAP, co-immunoprecipitation studies of the BAL fluids with a murine monoclonal antibody against FSAP, as well as antibodies directed against a spectrum of serine protease inhibitors, were performed. As is evident from Fig. 2A , PAI-1 co-immunoprecipitated with FSAP, whereas C1 inhibitor, α1-proteinase inhibitor, antithrombin, and α2-antiplasmin did not. Interestingly, PAI-1·FSAP complexes precipitated as a 70 kDa band. To examine whether 70 kDa bands represent PAI-1·FSAP complexes, the blots were stripped and reprobed with FSAP-specific and PAI-1-specific antibodies, respectively. The 70 kDa band was detected by both of these antibodies, indicating PAI-1·FSAP complex formation in the BAL fluids from ARDS patients (data not shown). We were not able to co-immunoprecipitate C1 inhibitor with FSAP from BAL fluids from ARDS patients, although C1 inhibitor is known to be a strong inhibitor of FSAP in plasma (7, 9). It may well be that such complexes do exist and simply could not be detected because the complex formation masks an essential epitope and prevents recognition by the antibodies employed. Alternatively, C1 inhibitor may not be a relevant FSAP inhibitor in the BAL fluids (contrasting with the important role of C1 in the plasma with respect to FSAP inhibition). Indeed, we did observe formation of SDS-stable complexes between FSAP and PAI-1, and also between FSAP and C1 inhibitor and between FASP and α2-antiplasmin, in a purified system, utilizing nonreducing SDS-PAGE followed by Coomassie staining (Fig. 2B). In contrast, no complex formation between FSAP and α1-proteinase inhibitor or between FSAP and antithrombin was noted (Fig. 2B), despite the fact that α1-proteinase inhibitor levels are dramatically elevated in lavage fluids from ARDS patients (34). To further validate these complex formation data, we assessed the inhibition of FSAP catalytic activity by these five protease inhibitors. The amidolytic activity of FSAP was potently inhibited by PAI-1, C1 inhibitor, and α2-antiplasmin, although α1-proteinase inhibitor and antithrombin were largely without effect (Fig. 2C). These trends parallel the trends observed in the in vitro FSAP-serpin complex formation assays (Fig. 2B). Together, our data suggest that FSAP and PAI-1 form a 1:1 stoichiometric complex in the lavage fluids of ARDS patients. This was the only FSAP-serpin complex that we were able to detect in the lavage fluid of ARDS patients, although other serpins were able to inhibit and form complexes with FSAP when assessed in purified systems in vitro. For this reason, we elected to further explore the interaction of FSAP with PAI-1 in the context of ARDS.
FIGURE 1.

FSAP·PAI-1 complexes and extracellular RNA are present in the BAL fluids of ARDS patients.A, BAL fluids from seven ARDS patients were subjected to SDS-PAGE under nonreducing conditions after treatment with 6 m guanidine hydrochloride, and FSAP or PAI-1 was detected on Western blots using an anti-FSAP antibody (left) or anti-PAI-1 antibody (right). Seven representative patients of 12 are illustrated. B, the nucleic acid levels in BAL fluids of ARDS patients and healthy controls were determined by filter-binding assay, where nucleic acids were extracted from BAL fluids from ARDS patients and healthy controls (n = 8 per group), biotinylated, and incubated on nitrocellulose filters. Bound nucleic acids were detected using peroxidase-labeled streptavidin and enhanced chemiluminescence. C, the nucleic acid levels in BAL fluids were also assessed by real time quantitative reverse transcription-PCR for GAPDH (RNA) and for β-globin (DNA). *, p = 0.005 (ARDS versus healthy controls).
FIGURE 2.

FSAP co-immunoprecipitates with PAI-1 in the BAL fluids of ARDS patients.A, immunoprecipitation (IP) was performed from BAL fluids (100 μl) using a mouse anti-FSAP or mouse anti-PAI-1 antibody, followed by Western blot (WB) analysis of the immunoprecipitate with antibodies directed against different serine protease inhibitors or FSAP, as indicated. Each reference protein is indicated in lane 1 of each blot. B, two-chain FSAP (1 μg) was incubated with C1 inhibitor (C1 INH), α1-proteinase inhibitor (α1PI), antithrombin (AT), α2-antiplasmin (α2AP), and PAI-1 (2–4 μg each) for 30 min. The samples were run on a 10% polyacrylamide gel under nonreducing conditions and stained with Coomassie Blue R-250. Arrowheads, FSAP-inhibitor complexes. C, two-chain FSAP (14 nm) was incubated with C1 INH (50 nm), α1PI (50 μm), AT (50 μm), α2AP (100 nm), and PAI-1 (50 nm) for 5 min. Remaining FSAP activity was measured with the conversion of the chromogenic substrate S-2288 at 405 nm. FSAP activity is depicted as percentage of control (100% – activity of FSAP in the absence of an inhibitor). Data represent the mean ± S.D. (n = 5). ***, p = 0.0004; **, p = 0.002 versus FSAP in the absence of an inhibitor.
We have previously demonstrated that extracellular RNA but not DNA is an important cofactor that mediates the autoactivation of scFSAP to tcFSAP (8). For this reason, we also screened the BAL fluids from ARDS patients for extracellular nucleic acids. Significantly elevated levels of RNA were found in BAL fluids from ARDS patients, as assessed by a filter-binding assay (Fig. 1B). Real time quantitative reverse transcription-PCR analysis also demonstrated a significant increase in GAPDH RNA levels in these ARDS BAL fluids in comparison with those from healthy volunteers (Fig. 1C). While the levels of DNA in the BAL fluids from ARDS patients exhibited trends toward an increase, the increase did not achieve statistical significance (Fig. 1C).
Complex Formation and Binding between Purified PAI-1 and FSAP—Complex formation between FSAP and PAI-1 was assessed by incubation of both two-chain (Fig. 3 ) and single-chain (Fig. 4 ) forms of FSAP with increasing amounts of PAI-1 for 30 min, followed by Western blot analysis with anti-FSAP and anti-PAI-1 antibodies. No complex formation between PAI-1 and scFSAP was observed (Fig. 4A). However, PAI-1 formed complexes with tcFSAP, detectable as a 120 kDa band after separation by SDS-PAGE under nonreducing conditions (Fig. 3A). Interestingly, under reducing conditions, formation of a 70-kDa complex between PAI-1 and tcFSAP occurred (Fig. 3B). To identify the components of the 70 kDa band, Western blots with FSAP heavy chain- and light chain-specific antibodies were performed. The 70-kDa band was detected by the FSAP light chain-specific antibody and by PAI-1-specific antibody but not by the antibody directed against heavy chain of FSAP (Fig. 3C). In line with this observation, the 70 kDa band was analyzed by amino-terminal sequencing, and this led to the identification of two separate NH2 termini in the band: IYGGFKSTAG (the NH2 terminus of light chain FSAP (Fig. 3D)) and the other from PAI-1. In contrast, no complex formation between PAI-1 and the FSAP heavy chain was observed (Fig. 3C).
FIGURE 3.

PAI-1 forms SDS-stable complexes with two-chain FSAP. Complexes between PAI-1 (applied at different concentrations) and tcFSAP, were formed for 30 min, and samples were analyzed by SDS-PAGE under nonreducing (A) and reducing (B) conditions, followed by Western blot analysis with a rabbit antibody directed against FSAP (top) and a rabbit antibody directed against PAI-1 (bottom). C, the tcFSAP (14 nm) was incubated with 50 nm PAI-1 for 30 min. After gel electrophoresis under reducing conditions, proteins were transferred to a polyvinylidene difluoride membrane, and complexes were immunodetected with anti-heavy- and light-chain FSAP-specific monoclonal antibodies (top panel) and a PAI-1-specific polyclonal antibody (bottom). D, schematic structure of human FSAP. The 10 NH2-terminal residues of the light chain of FSAP are indicated. E, binding of PAI-1 in the absence (▪) or presence (•) of a rabbit anti-FSAP antibody or of preformed PAI-1-uPA complexes (▴) to immobilized tcFSAP was performed in microtiter plates, and bound PAI-1 was quantified with a rabbit polyclonal antibody directed against PAI-1. Data represent the mean ± S.D. (n = 5) of a single representative experiment of three separate experiments. hcFSAP, heavy chain FSAP; lcFSAP, light chain FSAP, E1, E2, and E3, EGF-like domain 1, 2, and 3, respectively.
FIGURE 4.

PAI-1 does not form complexes with single-chain FSAP.A, complexes between PAI-1 (applied at different concentrations) and scFSAP were formed for 30 min, and samples were analyzed by SDS-PAGE under nonreducing conditions, followed by Western blot analysis with a FSAP-specific antibody (top) and a PAI-1-specific antibody (bottom). B, binding of PAI-1 in the absence (▪) or presence (•) of a rabbit anti-FSAP antibody or of preformed PAI-1-uPA complexes (▴) to immobilized scFSAP was performed in microtiter plates, and bound PAI-1 was quantified with a rabbit polyclonal antibody directed against PAI-1. Inset, autoconversion of scFSAP to tcFSAP (indicated by the appearance of the FSAP heavy chain (hcFSAP)) after a 16-h incubation at 4 °C. Data represent the mean ± S.D. (n = 5) of a single representative experiment of three independent experiments.
Binding experiments conducted in microtiter plates employing isolated PAI-1 and FSAP revealed a dose-dependent and saturable interaction between PAI-1 and tcFSAP (Fig. 3E). Binding of PAI-1 to tcFSAP was almost completely abrogated by a polyclonal anti-FSAP antibody or when PAI-1 was complexed to uPA prior to combination with FSAP (Fig. 3E). Likewise, PAI-1 binding to scFSAP was noted, yet to a lesser degree (Fig. 4B), which would be expected, since scFSAP is largely proteolytically inactive and therefore effectively unable to interact with protease inhibitors. Indeed, the apparent binding of PAI-1 to scFSAP that was observed probably reflects the binding of PAI-1 to tcFSAP, since small amounts of tcFSAP were generated by autoactivation of scFSAP over the course of the binding reaction at 4 °C (Fig. 4B, inset).
Inhibition of FSAP Amidolytic Activity by Wild-type PAI-1 and a PAI-1 R346A Variant—To characterize the inhibitory capacity of PAI-1 against FSAP amidolytic activity, tcFSAP was incubated with increasing amounts of active and inactivated wild-type PAI-1. Active PAI-1 inhibited the serine protease activity of tcFSAP in a dose-dependent manner (Fig. 5A ). The second-order inhibition rate constant (Ka) for the FSAP inhibition by PAI-1 was calculated as 3.38 ± 1.12 × 105 m –1·s–1 (Table 1 ).
FIGURE 5.
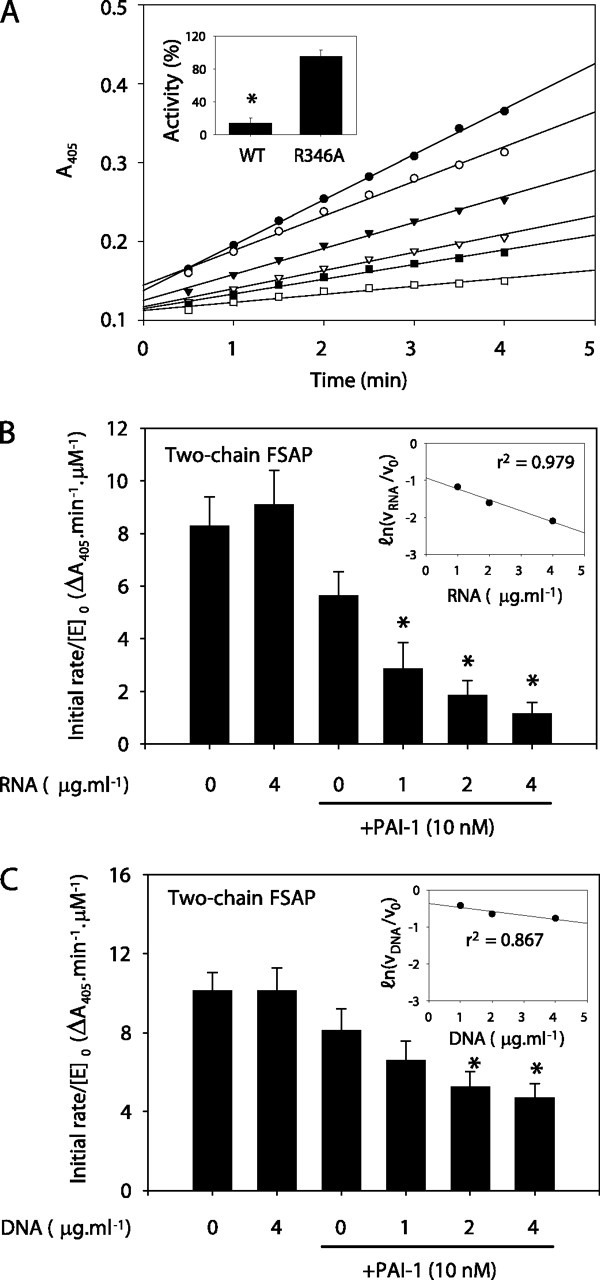
Inhibition of FSAP hydrolytic activity by PAI-1.A, two-chain FSAP (14 nm, by mass) was incubated in the absence (•) or in the presence of active wild-type PAI-1 at a concentration of 10 nm (▾), 14 nm (▿), 19 nm (▪), or 24 nm (□) or in the presence of 19 nm heat-inactivated PAI-1 (○) in 100 μl of TBS buffer. Amidolytic activity was followed as the conversion of the chromogenic substrate S-2288 at 405 nm. A single representative experiment of seven is illustrated. Amidolytic activity of tcFSAP (14 nm, by mass) against S-2288 was evaluated after a 30-min preincubation with WT PAI-1, or PAI-1 R346A (both at 24 nm, by mass) as described above (inset). Data represent mean ± S.D. (n = 7). *, p < 0.001 between PAI-1 WT- and R346A-treated tcFSAP. The effect of RNA (B) or DNA (C) on the inhibition of tcFSAP by PAI-1 was also assessed. In both instances, the linear dependence of PAI-1 inhibitory capacity on RNA and DNA concentration was assessed by a semilog plot of residual enzyme activity versus RNA or DNA concentration (insets). The correlation coefficient, r2, is indicated in the insets. Data represent mean ± S.D. (n = 7). B, *, p < 0.001 versus PAI-1 in the absence of nucleic acids.
TABLE 1.
Inhibition of tcFSAP activity by PAI-1 in the presence of modulators
| Modulator | Ka |
|---|---|
| m-1·s-1 | |
| PAI-1 alone | 3.38 ± 1.12 × 105 |
| PAI-1 + RNAa (4 μg·m-1) | 1.61 ± 0.94 × 106b |
| PAI-1 + DNAa (4 μg·m-1) | 6.23 ± 1.17 × 105b |
| PAI-1 + heparina (24 nm)c | 4.43 ± 1.29 × 105 |
| PAI-1 + vitronectina (40 nm)c |
5.58 ± 1.02 × 105b |
No significant effect on base-line FSAP activity was observed for any modulator when applied in the absence of PAI-1.
p < 0.01, compared with PAI-1 alone (n = 4).
Concentrations reflect saturating concentration of these modulators. No additive effect was observed at higher concentrations.
FSAP preferentially attacks substrates with an Arg or Lys residue at P1 (35). Analysis of PAI-1 mutants has previously demonstrated that a basic residue (Arg or Lys) at P1 is required for the inhibitory activity of PAI-1 toward uPA (36). A PAI-1 R346A variant exhibited a 20-fold reduced inhibitory capacity against FSAP, compared with wild-type PAI-1 (Fig. 5A, inset), indicating that a basic residue at P1 in the reactive center of PAI-1 was required for FSAP inhibition as well.
Influence of Cofactors on FSAP Inhibition by PAI-1—We demonstrate in this study not only that FSAP·PAI-1 complexes are the principal FSAP-serpin complexes formed in the BAL fluids of ARDS patients but also that large amounts of extracellular RNA, but not DNA, are present in these BAL fluids. Other reports have demonstrated that double-stranded nucleic acids prevent the inhibition of serine proteinases (such as cathepsin G) by their cognate serpins, including α1-proteinase inhibitor and α1-antichymotrypsin (37, 38). Therefore, we felt it relevant to explore whether the presence of large amounts of extracellular RNA impacted the FSAP-PAI-1 interaction.
The presence of RNA significantly increased the ability of PAI-1 to inhibit tcFSAP (Fig. 5B). A similar trend was observed for DNA; however, DNA was significantly less potent than was RNA (Fig. 5C). The inhibition of tcFSAP by PAI-1 exhibited a linear dose dependence on RNA and DNA concentration over the nucleic acid concentration range 1–4 μg·ml–1 when assessed under second-order conditions (Fig. 5, B (inset) and C (inset)).
The second-order inhibition rate constant (Ka) for the inhibition of tcFSAP by PAI-1 was increased from 3.38 ± 1.12 × 105 to 1.61 ± 0.94 × 106 m –1·s–1 in the presence of RNA (4 μg·ml–1) and to 6.23 ± 1.17 × 105 m –1·s–1 in the presence of DNA (4 μg·ml–1) (Table 1). The PAI-1-mediated inhibition of FSAP amidolytic activity was also enhanced by vitronectin (where the Ka was increased to 5.58 ± 1.02 × 105 m –1·s–1) and thus to a lesser extent than that observed for RNA (Table 1). Heparin also has a cofactor function for the autoactivation of scFSAP to tcFSAP but, in contrast to RNA and vitronectin, did not significantly influence PAI-1-mediated inhibition of FSAP proteolytic activity (Table 1).
Interaction of PAI-1 with RNA and DNA—Solid-phase binding assays revealed a dose-dependent and saturable interaction between PAI-1 and RNA (Fig. 6A ) and between PAI-1 and DNA (Fig. 6B). Binding of biotinylated RNA or DNA to PAI-1 was almost completely abrogated by the lysine analog tranexamic acid or when unlabeled RNA or DNA was added in a 100-fold molar excess. Additionally, RNA, as well as DNA, did not bind to PAI-1 in the presence of 1 mm heparin, suggesting that the binding sites on PAI-1 for nucleic acids and heparin may be the same or are located in close proximity to each other. The major determinants for interaction of PAI-1 with heparin are the amino acid residues Lys65, Lys69, Arg76, Lys80, and Lys88 located in and around helix D of PAI-1 (19). Therefore, PAI-1 variants (K65A, K69A, R76A, K80A, and K88A) were subsequently evaluated for nucleic acid-binding capacity in a solid-phase (microtiter plate-based) binding assay. A significant reduction in the RNA-binding capacity of PAI-1 variants K69A, R76A, and K80A was observed when compared with wild-type PAI-1 (Fig. 6C). The most dramatic reduction in binding capacity was observed for variants R76A and K80A, suggesting a particularly critical role in RNA binding for the amino acid residues Arg76 and Lys80 (Fig. 6C). Since PAI-1 is partially denatured by adsorption to plastic at 4 °C overnight, data obtained in the microplate assay reflect nucleic acid binding to denatured PAI-1. To assess nucleic acid binding to active PAI-1, a filter-binding assay was performed, where essentially the same trends were observed. In a slot-blot filter-binding assay, although the R76A PAI-1 variant retained weak RNA binding activity, no RNA binding was observed for the K80A variant (Fig. 6D). Similarly, DNA interaction with PAI-1 was dependent upon amino acid residue Arg76 and was almost completely abrogated when residue Lys80 was replaced by alanine (Fig. 6C). In a slot-blot filter-binding assay, almost negligible binding of DNA to the K80A PAI-1 variant was noted (Fig. 6E). The ability of PAI-1 to bind nucleic acids was also dependent upon the size of the RNA and DNA fragments. Shearing of RNA and DNA fragments by sonication (Fig. 7A ) led to a gradual loss of RNA and DNA binding by PAI-1 (Fig. 7B). This was particularly evident when RNA or DNA fragments were sheared to less than 400 ribonucleotide or deoxyribonucleotide bases in length (Fig. 7B). These data suggest that binding of nucleic acids to PAI-1 is achieved only when RNA or DNA fragments are sufficiently long (>400 bases for RNA and >500 bases for DNA). These data also indicate that RNA and DNA can bind to both active and latent forms of PAI-1.
FIGURE 6.

PAI-1 binds nucleic acids. Increasing amounts of biotinylated RNA (0–12.5 μg·ml–1)(A) or DNA (0–5 μg·ml–1)(B) were added to wells of a microtiter plate coated with wild type PAI-1 (200 nm) in the absence (▪) or presence of 10 mm tranexamic acid (♦), 1 mm heparin (▴), or a 100-fold excess of unlabeled RNA (•) or DNA (•), respectively. Bound nucleic acids were detected using peroxidase-labeled streptavidin and TMB as the substrate. Data represent mean ± S.D. (n = 7) of a typical experiment of three. C, biotinylated RNA (10 μg·ml–1; black bars) or DNA (5 μg·ml–1; open bars) was added to wells of a microtiter plate coated with either WT PAI-1 or PAI-1 variant K65A, K69A, R76A, K80A, or K88A (200 nm) and incubated for 2 h at room temperature. Bound nucleic acids were detected using peroxidase-labeled streptavidin and TMB as the substrate and are depicted as percentage of control. Controls represent amounts of RNA or DNA bound to wild type PAI-1, set at 100%. Data represent mean ± S.D. (n = 7) of a typical experiment of three. *, p < 0.001; **, p < 0.0001; ***, p < 0.00001 versus wild type PAI-1. D, filter-binding assays with increasing amounts (0–6 μg·ml–1) of either WT PAI-1 or PAI-1 variant R76A or K80A and 10 μg·ml–1 of biotinylated RNA were performed. E, filter-binding assay with increasing amounts (0–6 μg·ml–1) of either WT PAI-1 or PAI-1 variant K80A and 3 μg·ml–1 of biotinylated DNA was performed.
FIGURE 7.
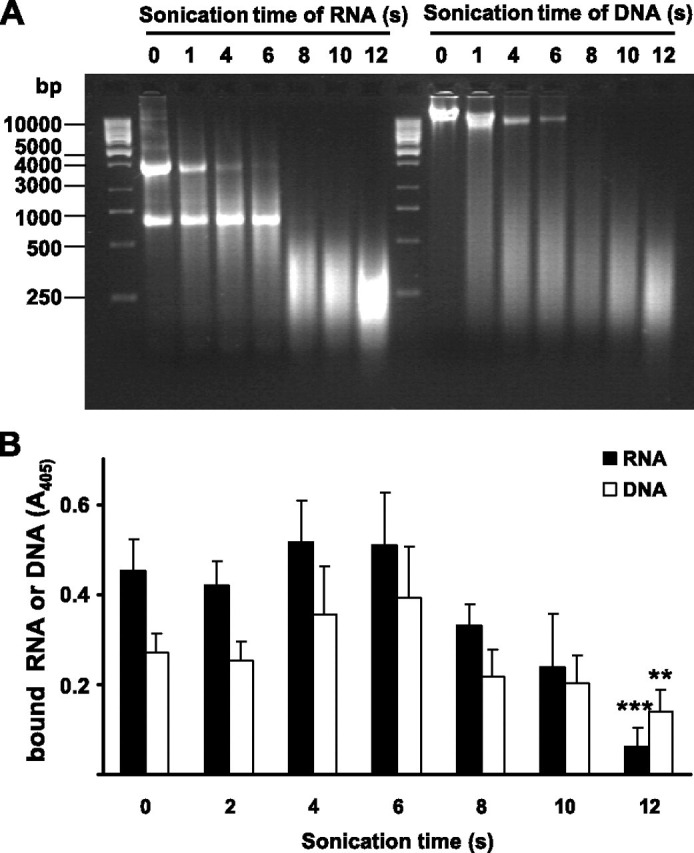
Binding of RNA and DNA fragments to PAI-1.A, biotinylated RNA or DNA was sonicated for the indicated times, subjected to agarose gel electrophoresis, and visualized by ethidium bromide staining. B, after sonication, the same RNA (black bars) or DNA (open bars) samples were added to wells of a microtiter plate coated with wild type PAI-1 and incubated for 2 h at room temperature. Bound nucleic acids were detected using peroxidase-labeled streptavidin and TMB as substrate. Data represent mean ± S.D. (n = 8) of a typical experiment of three. **, p < 0.0001; ***, p < 0.00001 versus nonsonicated RNA or DNA.
We next investigated the effects of RNA binding on PAI-1 stability. Incubation of PAI-1 alone for 5 h at 37 °C resulted in a complete loss of inhibitory activity toward uPA (Fig. 8A ), with a half-life for the spontaneous inactivation (t ½(inact)) of PAI-1 of 48.4 ± 8 min. This phenomenon was prevented by vitronectin (Fig. 8B) (41, 42), where the t ½(inact) for PAI-1 was increased to 138.6 ± 9 min in the presence of 70 nm vitronectin (Fig. 8B). We demonstrate here that RNA was also a potent stabilizer of PAI-1, since the t ½(inact) for PAI-1 in the presence of RNA (4 μg·ml–1) was increased to 114.6 ± 5 min (Fig. 8C). In contrast to RNA, DNA was a markedly weaker stabilizer of the active conformation of PAI-1, since the t ½(inact) for PAI-1 in the presence of DNA (4 μg·ml–1) was increased to 60.8 ± 7 min (Fig. 8D). These data indicate that RNA, as well as vitronectin, is an effective stabilizer of the active conformation of PAI-1.
FIGURE 8.

Stabilization of the active conformational state of PAI-1 by nucleic acids. PAI-1 was incubated alone (A) or together with vitronectin (B), RNA (C), or DNA (D) for 1–5 h at 37 °C prior to evaluation of PAI-1 inhibitory activity against uPA. Data points indicate the mean ± S.D. (n = 5 for each time point). The dashed lines indicate the 95% confidence intervals of the regression analysis. The calculated half-life for spontaneous inactivation (t½(inact)) of PAI-1 is indicated within the plots.
PAI-1-mediated Inhibition of Cellular Activities of FSAP—Cellular activities of tcFSAP have been recently described, particularly with respect to the inhibition of PDGF-BB-induced proliferation of VSMC (26). In the absence of PAI-1, FSAP inhibited PDGF-BB-induced VSMC proliferation (Fig. 9 ). This effect was significantly attenuated in the presence of 10 nm PAI-1 (Fig. 9). In the absence of FSAP, PAI-1 did not influence VSMC proliferation (not shown). Together, these data clearly indicate that the inhibition of FSAP by PAI-1 alters the cellular effects of FSAP.
FIGURE 9.

Influence of PAI-1 on the FSAP-mediated inhibition of vascular smooth muscle cell proliferation. Cell proliferation was determined by a DNA synthesis assay based on the uptake of bromodeoxyuridine in the presence of different test reagents in the cell culture medium, as indicated. DNA synthesis is depicted as absorbance at 405 nm. Data represent the mean ± S.D. (n = 5) of a typical experiment of three. Levels of significance are indicated by p < 0.001 (***) and p = 0.004 (**).
DISCUSSION
Plasminogen activator inhibitor-1 in a stabilized conformation is a rapid and specific inhibitor of tPA and uPA (10, 11, 12, 13). Thus, PAI-1 represents the major plasminogen activator inhibitor in plasma and tissues and regulates fibrinolysis and other processes mediated by plasmin (10, 11, 12, 13). In the present study, PAI-1 was identified as a novel inhibitor of the recently described plasma-derived serine protease FSAP. In particular, the presence of large amounts of FSAP, in free as well as in complexed high molecular mass forms in the BAL fluids of patients with ARDS, led us to search for serpins that can form complexes with FSAP. Co-immunoprecipitation studies revealed that PAI-1, levels of which are elevated in the circulation and in the alveolar compartment of ARDS patients (39), binds to FSAP in BAL fluids from ARDS patients and may, therefore, function as an important regulator of FSAP activity in the alveolar space under inflammatory conditions. These findings were underscored by in vitro studies, demonstrating the formation of a 1:1 stoichiometric complex between PAI-1 and FSAP and dose-dependent binding of purified PAI-1 to purified FSAP. Interestingly, we were not able to co-immunoprecipitate FSAP from BAL fluids from ARDS patients with other known inhibitors of this protease, including C1 inhibitor and α2-antiplasmin (7, 9). However, formation of SDS-stable complexes between FSAP and C1 inhibitor and FSAP and α2-antiplasmin, as well as between FSAP and PAI-1, were observed in a purified in vitro system. Additionally, C1 inhibitor was a potent inhibitor of FSAP activity, exhibiting an inhibitory capacity comparable with that of PAI-1, thus supporting previous findings demonstrating C1 inhibitor as a major inhibitor of FSAP in the plasma (7). Since C1 inhibitor is known to be elevated in BAL fluids from ARDS patients (40), we cannot exclude the presence of FSAP-C1 inhibitor complexes in the alveolar space under inflammatory conditions, although they were not detected in our study.
We demonstrate in the current study the requirement for a basic amino acid residue at P1 in the reactive center of PAI-1 for FSAP inhibition. Substitution of Arg346 with Ala at P1 in the reactive center of PAI-1 abolished the inhibitory activity of PAI-1 toward FSAP, indicating that Arg346 at P1 is a crucial recognition site on the serpin facilitating interaction with FSAP. The requirement for a basic residue at P1 of PAI-1 has also been demonstrated for the inhibition of uPA; however, it was not obligatory for the inhibition of tPA (36, 43).
PAI-1 inhibited FSAP activity with a second-order inhibition rate constant (Ka) of 3.38 ± 1.12 × 105 m –1·s–1. The presence of RNA enhanced the reactivity of PAI-1 with FSAP 6-fold, thereby yielding a second-order inhibition rate constant comparable with that observed for the reaction between PAI-1 and uPA. In contrast, DNA weakly affected (causing a 1.8-fold elevation) the rate of inhibition of FSAP by PAI-1. These data are particularly relevant, since the levels of extracellular RNA in the BAL fluids from ARDS patients were significantly elevated compared with healthy controls. In contrast, levels of DNA were not significantly increased. We further demonstrated that RNA, but not DNA, stabilized the active conformation of PAI-1 and that PAI-1 residues Arg76 and Lys80 were major determinants of nucleic acids binding to the PAI-1 molecule. We have also documented that the interactions between FSAP and PAI-1 have functional relevance, since PAI-1 can antagonize cellular activities mediated by FSAP, particularly those related to cell proliferation.
As a consequence of the cellular disruption sustained during acute lung injury, such as that which occurs in ARDS, we speculated that nucleic acids may be released into the extracellular milieu. In line with this idea, the release of extracellular DNA by activated neutrophils has already been reported in a murine model of pneumococcal pneumonia (44). Additionally, high concentrations of DNA have been found in the airway secretions of cystic fibrosis patients (45). In this report, we show, for the first time, that levels of nucleic acids, particularly RNA, are elevated in BAL fluids from ARDS patients. Our data indicate two separate, but related, phenomena that may impact PAI-1 activity as a consequence of this RNA release. First, we have demonstrated that, in the long term (30 min to 5 h), RNA can stabilize the active conformation of PAI-1 and thus dramatically decrease the rate of spontaneous inactivation of PAI-1. Thus, in the presence of RNA, PAI-1 inhibitory activity would be prolonged in vivo, and this would have the overall effect of increasing the active concentration of PAI-1 in the lung. Second, in the short term (0–4 min), RNA also promoted a dramatic and significant increase in the rate at which PAI-1 associates with FSAP, which would have the overall effect of increasing the inhibitory potency of PAI-1 against FSAP and other proteases. Since the effect of RNA on the Ka is evident within seconds of exposure of PAI-1 to RNA, during which time little effect of RNA on PAI-1 stability is observed, the increased Ka cannot be attributed to increased PAI-1 stability but may well be explained by a template mechanism, such as that previously proposed for RNA-assisted autoactivation of FSAP (8). A similar mechanism has been also described for other negatively charged molecules and seems to be involved, for instance, in the heparin-induced acceleration of the inhibition of thrombin by the serpin antithrombin (18, 19). The RNA-binding domain in FSAP has been recently identified; the amino acid residues Arg170, Arg171, Ser172, and Lys173 within the epidermal growth factor-like domain 3 were found to be essential for RNA binding (46). In contrast, the possible RNA-binding domains in PAI-1 have been investigated for the first time in the present study. We have demonstrated that the basic amino acid residues Arg76 and Lys80, located in the helix D subdomain of PAI-1, that have been previously identified to be critical for the interaction of PAI-1 and heparin (18, 19) are also of critical importance for the interaction of PAI-1 and RNA. Additional PAI-1 residues clearly also play a role in RNA binding, since PAI-1 variants containing nonconservative substitutions in Lys65, Lys69, and Lys88 exhibited moderately reduced RNA-binding capacity.
To a much lesser extent than that observed for RNA, the inhibition of FSAP by PAI-1 was also enhanced in the presence of the extracellular matrix protein vitronectin (1.7-fold) and DNA (1.8-fold), whereas heparin had no effect on this interaction. These data are in line with other reports indicating that heparin did not enhance the inhibitory potency of other FSAP inhibitors, such as aprotinin, C1 inhibitor, and α2-antiplasmin (9). In contrast, antithrombin inhibits FSAP only in the presence of heparin (9). Our data contrast sharply with observations made concerning the effect of DNA on the interactions between cathepsin G and α1-antichymotrypsin and between cathepsin G and α1-proteinase inhibitor (38), where cathepsin G-DNA complexes were resistant to the inhibition by α1-antichymotrypsin and α1-proteinase inhibitor. It remains unclear why RNA is a much better enhancer of the FSAP-PAI-1 interaction than is DNA, although it might be speculated that single-stranded RNA is less sterically constrained than is double-stranded DNA. This flexibility might facilitate the FSAP-PAI-1 interaction directly, or it may alter the conformation of either FSAP or PAI-1 or both molecules, facilitating enhanced interaction. We do not currently have any data to support either suggestion.
The role of FSAP inhibition by PAI-1 under different physiological and pathophysiological conditions is presently unclear. A dual role for FSAP in hemostasis was recently reported; FSAP activates components of both the coagulation and the fibrinolytic systems in vitro (3, 9). Which of these two hemostatic pathways predominates in vivo may depend on local conditions (e.g. the presence or absence of different components of the coagulation and fibrinolytic system, including protease inhibitors, such as PAI-1). Therefore, FSAP inhibition by PAI-1, as demonstrated in the present study, might be an important regulatory mechanism at play in hemostasis under physiological and pathophysiological conditions. In acute inflammatory lung diseases, such as ARDS, alterations to the alveolar hemostatic balance toward procoagulant activity and overwhelming and persistent pulmonary fibrin deposition occur and are thought to contribute to the impairment of gas exchange and to the induction of fibroproliferative processes (47, 48, 49, 50, 51). Whether increased FSAP levels3 and interactions between FSAP and PAI-1 (as demonstrated in the present study) in the lungs of patients with ARDS contribute to these phenomena is presently unresolved and warrants further investigation.
Apart from a potential role for FSAP in hemostasis, cellular activities of FSAP have been described. It was recently shown that FSAP reduces cell proliferation of human umbilical vein endothelial cells (52) as well as VSMC (26). Together with the observed presence of FSAP in atherosclerotic lesions and the identification of a single nucleotide polymorphism (Marburg I) as a risk predictor of carotid stenosis (53), FSAP was suggested to be an important inhibitor of the proatherogenic phenotype of VSMC (26). In the present study, we demonstrated that PAI-1 can antagonize the FSAP-mediated inhibition of VSMC proliferation, underscoring the functional relevance of FSAP inhibition by PAI-1 and its possible involvement in cellular activities associated with cell proliferation. Whether inhibition of FSAP by PAI-1 by interfering with FSAP-mediated inhibition of cell proliferation contributes to the development of atherothrombotic events needs to be investigated in future studies.
To summarize, this study documents for the first time that PAI-1 is an inhibitor of FSAP, potentially serving as an important regulatory mechanism in hemostasis and vascular cell functions, particularly in processes such as inflammation and atherosclerosis. Moreover, under a variety of experimental conditions, we have demonstrated that extracellular RNA but not DNA enhances the reactivity of PAI-1 with FSAP and stabilizes the active conformational state of PAI-1. Therefore, in addition to vitronectin, extracellular RNA may provide a reservoir of active PAI-1, particularly during inflammation, when cell damage occurs and RNA is released.
Acknowledgments
We thank Gisela Müller and Horst Thiele for excellent technical assistance.
Footnotes
The abbreviations used are: FSAP, factor VII-activating protease; ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage fluid; PAI-1, plasminogen activator inhibitor-1; PDGF, platelet-derived growth factor; scFSAP, single-chain FSAP; TBS, Tris-buffered saline; tcFSAP, two-chain FSAP; TMB, 3,3′,5,5′-tetramethylbenzidine; tPA, tissue-type plasminogen activator; VSMC, vascular smooth muscle cell(s); uPA, urokinase-type plasminogen activator; WT, wild-type; BSA, bovine serum albumin; TBS, Tris-buffered saline; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
M. Wygrecka, P. Markart, L. Fink, A. Guenther, and K. T. Preissner, unpublished observations.
References
- 1.Choi-Miura N.H., Tobe T., Sumiya J.I., Nakano Y., Sano Y., Mazda T., Tomita M. J. Biochem. (Tokyo) 1996;119:1157–1165. doi: 10.1093/oxfordjournals.jbchem.a021362. [DOI] [PubMed] [Google Scholar]
- 2.Hunfeld A., Etscheid M., Koenig H., Seitz R., Dodt J. FEBS Lett. 1999;456:290–294. doi: 10.1016/s0014-5793(99)00959-x. [DOI] [PubMed] [Google Scholar]
- 3.Roemisch J., Feussner A., Vermoehlen S., Stoehr H.A. Blood Coagul. Fibrinolysis. 1999;10:471–479. [PubMed] [Google Scholar]
- 4.Knoblauch B., Kellert J., Battmann A., Preissner K.T., Roemisch J. Ann. Haematol. 2002;81:A42. (abstr.) [Google Scholar]
- 5.Kannemeier C., Feussner A., Stoehr H.A., Preissner K.T., Roemisch J. Eur. J. Biochem. 2001;268:3789–3796. doi: 10.1046/j.1432-1327.2001.02285.x. [DOI] [PubMed] [Google Scholar]
- 6.Etscheid M., Hunfeld A., Koenig H., Seitz R., Dodt J. Biol. Chem. 2000;381:1223–1231. doi: 10.1515/BC.2000.150. [DOI] [PubMed] [Google Scholar]
- 7.Choi-Miura N.H., Saito K., Takahashi K., Yoda M., Mazda T., Tomita M. Biol. Pharm. Bull. 2001;24:221–225. doi: 10.1248/bpb.24.221. [DOI] [PubMed] [Google Scholar]
- 8.Nakazawa F., Kannemeier C., Shibamiya A., Song Y., Tzima E., Schubert U., Koyama T., Niepmann M., Trusheim H., Engelmann B., Preissner K.T. Biochem. J. 2005;385:831–838. doi: 10.1042/BJ20041021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roemisch J., Vermoehlen S., Feussner A., Stoehr H.A. Haemostasis. 1999;29:292–299. doi: 10.1159/000022515. [DOI] [PubMed] [Google Scholar]
- 10.Vassalli J.D., Sappino A.P., Belin D. J. Clin. Invest. 1991;88:1067–1072. doi: 10.1172/JCI115405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andreasen P.A., Georg B., Lund L.R., Riccioo A., Stacey S.N. Mol. Cell. Endocrinol. 1990;68:1–19. doi: 10.1016/0303-7207(90)90164-4. [DOI] [PubMed] [Google Scholar]
- 12.Manchanda N., Schwartz B.S. J. Biol. Chem. 1995;270:20032–20035. doi: 10.1074/jbc.270.34.20032. [DOI] [PubMed] [Google Scholar]
- 13.Stratikos E., Gettins P.G. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4808–4813. doi: 10.1073/pnas.96.9.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilczynska M., Fa M., Ohlsson P.I., Ny T. J. Biol. Chem. 1995;270:29652–29655. doi: 10.1074/jbc.270.50.29652. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl T.L., Ohlsson P.I., Wiman B. Biochem. J. 1990;265:109–113. doi: 10.1042/bj2650109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreasen P.A., Riccio A., Welinder K.G., Douglas R., Sartorio R., Nielsen L.S., Oppenheimer C., Blasi F., Dano K. FEBS Lett. 1986;209:213–218. doi: 10.1016/0014-5793(86)81113-9. [DOI] [PubMed] [Google Scholar]
- 17.Lawrence D.A., Ginsburg D., Day D.E., Berkenpas M.B., Verhamme I.M., Kvassman J.O., Shore J.D. J. Biol. Chem. 1995;270:25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 18.Ehrlich H.J., Keijer J., Preissner K.T., Gebbink R.K., Pannekoek H. Biochemistry. 1991;30:1021–1028. doi: 10.1021/bi00218a020. [DOI] [PubMed] [Google Scholar]
- 19.Ehrlich H.J., Gebbink R.K., Keijer J., Pannekoek H. J. Biol. Chem. 1992;267:11606–11611. [PubMed] [Google Scholar]
- 20.Declerck P.J., De Mol M., Alessi M.C., Baudner S., Paques E.P., Preissner K.T., Mueller-Berghaus G., Collen D. J. Biol. Chem. 1988;263:15454–15461. [PubMed] [Google Scholar]
- 21.Ehrlich H.J., Gebbink R.K., Keijer J., Linders M., Preissner K.T., Pannekoek H. J. Biol. Chem. 1990;265:13029–13035. [PubMed] [Google Scholar]
- 22.Lawrence D.A., Palaniappan S., Stefansson S., Olson S.T., Francis-Chmura A.M., Shore J.D., Ginsburg D. J. Biol. Chem. 1997;272:7676–7680. doi: 10.1074/jbc.272.12.7676. [DOI] [PubMed] [Google Scholar]
- 23.Rezaie A.R. J. Biol. Chem. 2001;276:15567–15570. doi: 10.1074/jbc.C100123200. [DOI] [PubMed] [Google Scholar]
- 24.Podor T.J., Peterson C.B., Lawrence D.A., Stefansson S., Shaughnessy S.G., Foulon D.M., Butcher M., Weitz J.I. J. Biol. Chem. 2000;275:19788–19794. doi: 10.1074/jbc.M908079199. [DOI] [PubMed] [Google Scholar]
- 25.Kjoller L., Kanse S.M., Kirkegaard T., Rodenburg K.W., Ronne E., Goodman S.L., Preissner K.T., Ossowski L., Andreasen P.A. Exp. Cell Res. 1997;232:420–429. doi: 10.1006/excr.1997.3540. [DOI] [PubMed] [Google Scholar]
- 26.Kannemeier C., Al-Fakhri N., Preissner K.T., Kanse S.M. FASEB J. 2004;18:728–730. doi: 10.1096/fj.03-0898fje. [DOI] [PubMed] [Google Scholar]
- 27.Wind T., Jensen J.K., Dupont D.M., Kulig P., Andreasen P.A. Eur. J. Biochem. 2003;270:1680–1688. doi: 10.1046/j.1432-1033.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- 28.Gunther A., Siebert C., Schmidt R., Ziegler S., Grimminger F., Yabut M., Temmesfeld B., Walmrath D., Morr H., Seeger W. Am. J. Respir. Crit. Care Med. 1996;153:176–184. doi: 10.1164/ajrccm.153.1.8542113. [DOI] [PubMed] [Google Scholar]
- 29.Bernard G.R., Artigas A., Brigham K.L., Carlet J., Falke K., Hudson L., Lamy M., Legall J.R., Morris A., Spragg R. Am. J. Respir. Crit. Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 30.Futamura A., Gettins P.G.W. J. Biol. Chem. 2000;275:4092–4098. doi: 10.1074/jbc.275.6.4092. [DOI] [PubMed] [Google Scholar]
- 31.Idell S. Crit. Care Med. 2003;31:213–220. doi: 10.1097/01.CCM.0000057846.21303.AB. [DOI] [PubMed] [Google Scholar]
- 32.Moraes T.J., Chow C.W., Downey G.P. Crit. Care Med. 2003;31:189–194. doi: 10.1097/01.CCM.0000057842.90746.1E. [DOI] [PubMed] [Google Scholar]
- 33.Ware L.B. Semin. Respir. Crit. Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 34.Emmett M., Miller J.L., Crowle A.J. Proc. Soc. Exp. Biol. Med. 1987;184:74–82. doi: 10.3181/00379727-184-42448. [DOI] [PubMed] [Google Scholar]
- 35.Kraut J. Annu. Rev. Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 36.Sherman P.M., Lawrence D.A., Yang A.Y., Vandenberg E.T., Paielli D., Olson S.T., Shore J.D., Ginsburg D. J. Biol. Chem. 1992;267:7588–7595. [PubMed] [Google Scholar]
- 37.Belorgey D., Bieth J.G. Biochemistry. 1998;37:16416–16422. doi: 10.1021/bi981536o. [DOI] [PubMed] [Google Scholar]
- 38.Duranton J., Boudier C., Belorgey D., Mellet P., Bieth J.G. J. Biol. Chem. 2000;275:3787–3792. doi: 10.1074/jbc.275.6.3787. [DOI] [PubMed] [Google Scholar]
- 39.Idell S., Koenig K.B., Fair D.S., Martin T.R., McLarty J., Maunder R.J. Am. J. Physiol. 1991;261:L240–L248. doi: 10.1152/ajplung.1991.261.4.L240. [DOI] [PubMed] [Google Scholar]
- 40.Velasco F., Torres A., Guerrero A., Andres P., Guerrero R., Aljama P., Alvarez F. Thromb. Haemost. 1986;55:357–360. [PubMed] [Google Scholar]
- 41.Seiffert D., Loskutoff D.J. J. Biol. Chem. 1991;266:2824–2830. [PubMed] [Google Scholar]
- 42.Mimuro J., Loskutoff D.J. J. Biol. Chem. 1989;264:5058–5063. [PubMed] [Google Scholar]
- 43.Sherman P.M., Lawrence D.A., Verhamme I.M., Paielli D., Shore J.D., Ginsburg D. J. Biol. Chem. 1995;270:9301–9306. doi: 10.1074/jbc.270.16.9301. [DOI] [PubMed] [Google Scholar]
- 44.Beiter K., Wartha F., Albiger B., Normark S., Zychlinsky A., Henriques-Normark B. Curr. Biol. 2006;16:401–407. doi: 10.1016/j.cub.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 45.Potter J.L., Spector S., Matthews L.W., Lemm J. Am. Rev. Respir. Dis. 1969;99:909–916. doi: 10.1164/arrd.1969.99.6.909. [DOI] [PubMed] [Google Scholar]
- 46.Altincicek B., Shibamiya A., Trunsheim H., Tzima E., Niepmann M., Linder D., Preissner K.T., Kanse S.M. Biochem. J. 2006;394:687–692. doi: 10.1042/BJ20051563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertozzi P., Astedt B., Zenzius L. N. Engl. J. Med. 1990;322:890–897. doi: 10.1056/NEJM199003293221304. [DOI] [PubMed] [Google Scholar]
- 48.Idell S., James K.K., Levin E.G., Schwartz B.S., Manchanda N., Maunder R.J., Martin T.R., McLarty J., Fair D.S. J. Clin. Invest. 1989;84:695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guenther A., Mosavi P., Heinemann S., Ruppert C., Muth H., Markart P., Grimminger F., Walmrath D., Temmesfeld-Wollbrueck B., Seeger W. Am. J. Respir. Crit. Care Med. 2000;161:454–462. doi: 10.1164/ajrccm.161.2.9712038. [DOI] [PubMed] [Google Scholar]
- 50.Schermuly R., Guenther A., Ermert M., Ermert L., Ghofrani H.A., Weissmann N., Grimminger F., Seeger W., Walmrath D. Am. J. Physiol. 2001;280:L792–L800. doi: 10.1152/ajplung.2001.280.4.L792. [DOI] [PubMed] [Google Scholar]
- 51.Welty-Wolf K.E., Carraway M.S., Miller D.L., Ortel T.L., Ezban M., Ghio A.J., Idell S., Plantadosi C.A. Am. J. Respir. Crit. Care Med. 2001;164:1988–1996. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- 52.Etscheid M., Beer N., Kress J.A., Seitz R., Dodt J. Eur. J. Cell Biol. 2004;82:597–604. doi: 10.1078/0171-9335-00349. [DOI] [PubMed] [Google Scholar]
- 53.Willeit J., Kiechl S., Weimer T., Mair A., Santer P., Wiedermann C.J., Roemisch J. Circulation. 2003;107:667–670. doi: 10.1161/01.cir.0000055189.18831.b1. [DOI] [PubMed] [Google Scholar]