

Abstract
Treatment of cancer is a major challenge even though the pathophysiology is becoming clearer with time. A number of new chemical entities are developed to target cancer growth inhibition, but the targeted delivery of these products still needs novel research. This is of utmost importance not only for higher efficacy but also for a reduction in systemic toxicity and cost of treatment. Although multiple novel targets and molecules are being researched, most of them could not pass the regulatory approval process, due to low benefit–risk ratio and lack of target specificity. Failure of a majority of these drugs was in part due to their superiority claimed via surrogate markers. Despite these, currently, more than 100 chemotherapeutic agents are in practice. This review paper discusses in detail the molecular basis, drug discovery, and pros and cons over conventional treatment approaches of three novel approaches in cancer therapy, i.e., (i) antibody–drug conjugates, (ii) cancer immunotherapy, and (iii) metronomic chemotherapy. All the drugs developed using these three novel approaches were compared against the established treatment regimens in clinical trials with clinical end points, such as overall survival, progression-free survival, and quality of life.
Keywords: Antibody–drug conjugates, cancer immunotherapy, metronomic chemotherapy, tisagenlecleucel, trastuzumab emtansine
Introduction
Worldwide, cancer contributes to the second foremost cause of death after cardiovascular diseases.[1] As per a recent study, out of 10 million deaths that occurred worldwide due to cancer, nearly 700,000 cases have been reported in India.[2] It is expected that more than 20 million new cases of cancer will be diagnosed worldwide by 2025, which will lead to 80% of the burden on low- and lower-middle–income countries.[3] Cancer is a multifactorial disease with uncontrolled cell division and a potential to spread to other cells known as metastasis, which is the reason for 90% of deaths from cancer.[4]
The most common cancers detected in the Indian population belong to carcinoma of lungs, colon, breast, rectum, liver, and stomach.[5] Some conventional treatment approaches, such as surgery, chemotherapy, and radiation therapy, are most commonly employed worldwide for the management of cancer, and the choice of treatment depends on the cancer type, site or location, and severity of disease progression. Chemotherapy is a systemic approach that imposes the apoptosis of cancerous cells and ceases their ability to divide. Nowadays, more than 100 drugs are being used as chemotherapeutic agents either as monotherapy or in combination therapy, and they are classified into different categories such as alkylating agents, antimetabolites, mitotic inhibitors, and anthracyclines. However, these chemotherapeutic agents have limitations such as (1) failure of therapy, (2) effect on healthy tissue and lack of targeted delivery, and (3) adverse drug reactions (ADR) including systemic toxicity involving multiple organ systems.[6]
The chemotherapeutic approach may result in a high rate of failure due to factors, such as multidrug resistance and poor response.[7] According to the American Cancer Society, the 5-year survival rate in different cancers are as follows: small-cell lung carcinoma - 7%, nonsmall-cell lung carcinoma - 21%, bladder cancer - 5%–34%, breast cancer - 89%, acute myeloid leukemia - 6%–50%, and chronic myeloid leukemia - 31%–63%.[8]
Along with targeting of the cancerous cells, these chemotherapeutics also affect healthy cells which lead to several adverse effects, such as nausea, vomiting, myelosuppression, alopecia, and mucositis.[9] Consequently, the physical and psychological well-being, as well as the subjects’ quality of life (QOL), gets deteriorated. Recently, a retrospective study has opined that chemotherapy-induced ADRs are more prevalent in patients with multidrug treatment as compared to that of monotherapy (74% vs. 26%). The most common ADRs reported were alopecia - 17.27%, hyperpigmentation of the skin and nail - 14.65%, thrombophlebitis - 9.42%, myelosuppression - 8.37%, vomiting - 8.376%, and diarrhea - 4.71%.[10] These facts highlight the need for novel therapeutic approaches with enhanced efficacy and potentially minimal adverse effects for the management of cancers.
Nonselective conventional chemotherapy has long been the mainstay of treatment for the majority of patients in the developing world. Different regimens that combine multiple cytotoxic agents have been tested for different cancers, and the most effective regimens are in practice. However, this also increases the risk of adverse events such as bone marrow toxicity, weight loss, mucositis, peripheral neuropathy, renal failure, and sterility.[11] These factors, combined with the nonavailability of an effective drug regimen, worsen the prognosis of a person diagnosed with cancer. Adding to this, a recent retrospective cohort study of drug approvals (2009–2013) by the European Medicines Agency (EMA) published in 2017 in the British Medical Journal states that based on the real-world evidence, after 3 years of use, there is lack of evidence that these anticancer agents improve survival in a clinical setting.[12]
This article reviews three novel therapeutic methods: (i) antibody–drug conjugates (ADCs), (ii) cancer immunotherapy, and (iii) metronomic chemotherapy, which were tested in clinical trials with the objectives of not only increasing the efficacy parameters but also improving QOL. There were statistically significant results noticed in the clinical trials, which may translate to clinically meaningful outcomes in the real world.
Methodology
A literature search was performed in PubMed (2000–2019); in addition, a clinical trial registry (ClinicalTrials.gov) was searched. The search strategies included various combinations of text words such as antibody-drug conjugates, oncolytic virus therapy, dendritic cell vaccine, CAR-T cell therapy, metronomic chemotherapy, and cancer immunotherapy. Studies involving participants with advanced metastatic carcinoma, in both genders, and all age groups were considered for this review. This review considered the articles on completed clinical trial/studies, observational studies, and meta-analysis. Two authors have independently searched, and one reviewer has done the final assessment of the studies included. The reference list of all papers selected from the literature search was also inspected. Two authors have independently searched the databases for collection of studies, and one reviewer screened the studies to find out potentially relevant studies satisfying the inclusion criteria and to reduce duplication of studies by checking the titles and abstracts obtained from the search. All authors resolute differences about the inclusion criteria by discussion.
Results
The number of papers found in the PubMed database was 1470, and in ClinicalTrials.gov, it was 253. After screening for potential results, 64 studies (62 from the PubMed database, 2 from ClinicalTrials.gov) were included in this review. The research question was focused to include only three therapeutic mechanisms, i.e., ADCs, cancer immunotherapy, and metronomic chemotherapy. However, several other treatment modalities were not considered in this review. The studies published after the year 2000 are included in this review, so there may be chance of missing some studies published earlier.
Antibody–drug conjugates
The concept of ADC originated from the “magic bullet” idea of Paul Ehrlich about 100 years back. Ehrlich established the concept of targeted drug delivery to the specific disease sites which later led the foundation for the development of ADC.[13] ADC is the novel therapeutic approach that combines the monoclonal antibodies (mAbs) with cytotoxic drugs to ensure specific targeted drug delivery into the cancerous cells through the antibody–antigen interaction, and hence, the normal tissues get spared from chemotherapeutic damage.[14]
The first mAb was licensed almost 30 years ago.[15] However, many drugs that were successful in animal models had poor clinical efficacy. As of 2016, there are around 40 mAbs approved by either the EMA or the United States Food and Drug Administration (USFDA).[16] With 10 of them being approved just in 2016, the total number of mAbs available for clinical use is expected to increase and so will the broader use of each agent with each successful clinical trial. The multiple mechanisms through which a mAb induces tumor cell death are direct effects including downregulation and induction of cell death, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and phagocytosis.[17]
One emerging model of anticancer pharmacotherapy is a subtype of mAb: ADC. These agents combine the specificity of a mAb along with the cytotoxic potency of conventional agents. This implies that these newer agents will have an increased therapeutic window compared to their unconjugated ancestors.[18] The structure of ADC comprises three parts, namely the mAb (giving rise to the drugs’ specificity), the cytotoxic “payload” (for increased potency compared to unconjugated mAb), and a linker molecule that attaches these two units [Figure 1]. Some of the ADCs in common use are discussed briefly in this article [Table 1].
Figure 1.
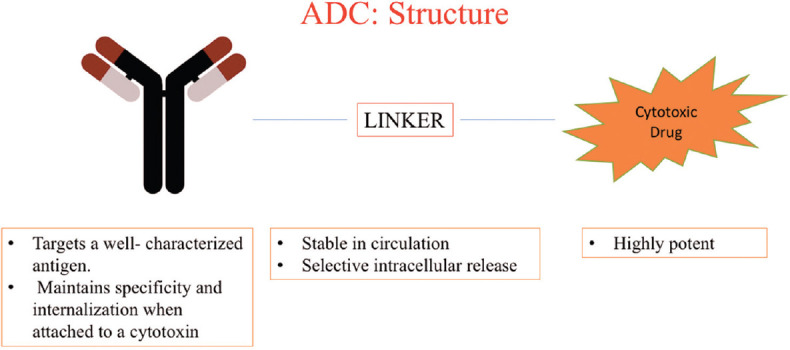
The structure of antibody–drug conjugate comprises three parts; namely the monoclonal antibody (giving rise to the drugs specificity), the cytotoxic “payload” (for increased potency compared to unconjugated monoclonal antibody), and a linker molecule that attaches these two units
Table 1.
List of approved antibody- drug conjugates
| ADC | mAb | Linker | Cytotoxic drug | Indication | Current Status | Reference |
|---|---|---|---|---|---|---|
| Gemtuzumab ozagamicin | Gemtuzumab: humanized anti-CD33 monoclonal antibody (IgG4 k antibody) | 4-(4-acetylphenoxy)butanoic acid (AcBut) | Ozagamicin (calcheamicin): produces site-specific double-strand breaks at minor groove of DNA by forming p-benzene diradical | Newly-diagnosed CD33-positive AML in adults and relapsed or refractory CD33-positive AML in adults and in pediatric patients 2 years and older | Approved by the USFDA in September 2017, EMA in February 2018 | [19,20,21,22] |
| Trastuzumab emtansine | Trastuzumab: humanized anti-HER2 IgG1 | SMCC | Maytansinoid (emtansine or DM1): A tubulin polymerization inhibitor which interferes with mitosis and promotes apoptosis. | HER-2–positive MBC patients who have previously received trastuzumab and taxane, separately or in combination. Patients should have either: Received prior therapy for metastatic disease, or developed disease recurrence during or within 6 months of completing adjuvant therapy |
Approved by the USFDA in 2013, EMA in 2013. | [23,24,25,26,27,28,29,30] |
| Systemic anaplastic large cell lymphoma after failure of at least one prior multi-agent chemotherapy regimen | ||||||
| Brentuximab vedotin | Brentuximab: chimeric anti-CD30 antibody. | Dipeptide valine–citrulline linker. | MMAE: disrupts the microtubule network within the cell, which, in turn, induces cell cycle arrest and results in apoptotic death. | Hodgkins lymphoma after failure of ASCT or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not ASCT candidates | Approved by USFDA in March 2018, EMA in November 2017. | [31,32,33,34] |
| Inotuzumab ozagamicin | Humanized anti-CD22 antibody | Acid-labile 4-(4¢-acetylphenoxy) butanoic acid (acetyl butyrate) linker | See Gemtuzumab ozagamicin | Adults with relapsed of refractory Philadelphia chromosome positive B-cell ALL | Approved by the USFDA in August 2017, EMA in June 2017 | [35,36] |
| Polatuzumab vedotin-piiq | Humanized anti-CD79b-antibody | maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl (mc-vc-PAB) | MMAE | Adults with relapsed or refractory diffuse large B-cell lymphoma, not otherwise specified, after at least two prior therapies | Approved by the USFDA in June 2019 | [37] |
ADC: Antibody drug conjugate, mAb: Monoclonal antibody, CD: Cluster differentiation, AML: Acute myeloid leukemia, USFDA: United States Food and Drug Administration, EMA: European Medicines Agency, HER: Human epidermal growth factor receptor – 2, MBC: Metastatic breast cancer, MMAE: Monomethyl auristatin E, ASCT: Autologous stem cell transplant, ALL: Acute lymphoid leukemia, SMCC: N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate
Gemtuzumab ozogamicin
The target antigen of gemtuzumab (humanized IgG mAb) is CD33, a marker of malignant myeloid cells. It is conjugated to a cytotoxic agent: ozogamicin, a calicheamicin derivative that binds to DNA on its minor groove and proceeds to destroy the tumor cell.[19] It received accelerated approval in 2000 for the use in older (>60 years) patients with relapsed acute myeloid leukemia after a Phase 2 trial showed a 26% overall response rate (ORR).[20] It was marketed as Mylotarg by Pfizer. Once the drug is guided to its target antigen via the mAb component, it is internalized. The acid-sensitive linker (activated by low pH) releases ozogamicin, leading to cell death.[21] Mylotarg was voluntarily withdrawn by Pfizer in 2010 after postapproval studies showed a lack of clinical efficacy and increased adverse events which were the results of early release (extracellular) of the cytotoxic payload from the mAb due to an unstable linker.[22]
Following results from a Phase 3 open-label trial, gemtuzumab ozogamicin was reapproved in September 2017 for the management of newly diagnosed acute myeloid leukemia (including relapsed or refractory cases) in adults and children (>2 years) in a decreased dose compared to the dose approved in 2000. The guidelines also allow the drug to be used in a combination with other cytotoxic drugs such as cytarabine and daunorubicin.
Trastuzumab emtansine
Trastuzumab (humanized mAb) is a successful mAb used to treat HER-2–positive metastatic breast cancer and was approved by the USFDA in 2006 based on trials that showed significant evidence of increased disease-free survival. Despite trastuzumab therapeutic use and its duration of administration, still a subset of women subjects did not come across change in the overall survival (OS) pattern or progression-free survival (PFS).[23] Trastuzumab was therefore conjugated to a cytotoxic payload: a maytansinoid (emtansine or DM1) which is a microtubule disrupting agent, the active derivative of which upon internalization by HER-2–positive cells is released via proteolytic degradation within the lysosome [Figure 2].[24]
Figure 2.
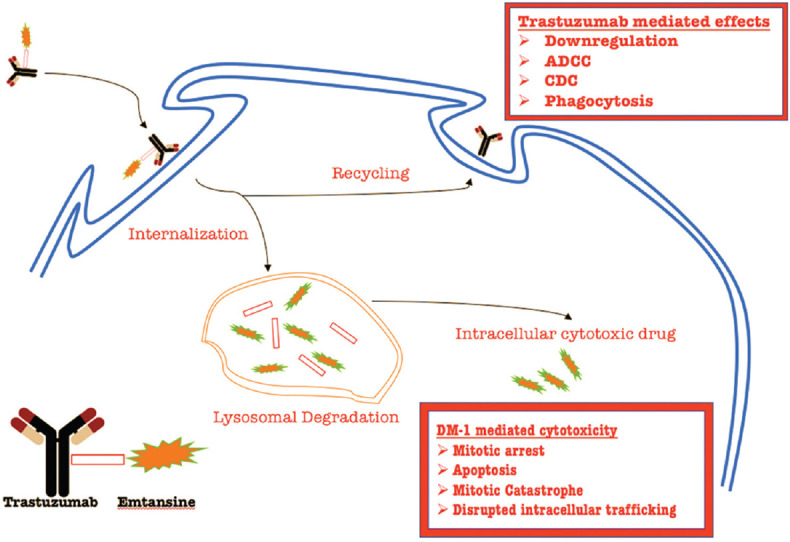
Trastuzumab emtansine (T-DM1) in action. The monoclonal antibody component helps T-DM1 locate the HER-2-neu positive cells. After internalization, the antibody component is recycled via the endosome. The antibody component is capable of causing cell death via downregulation, antibody-dependent cell cytotoxicity, complement-dependent cytotoxicity, and phagocytosis. The remaining portion of the conjugate undergoes lysosomal degradation. The cytotoxic component (emtansine) is capable of causing cell death via mitotic arrest, apoptosis, mitotic catastrophe, and disrupted intracellular trafficking
The superiority of trastuzumab emtansine (T-DM1) was shown by EMILIA TRIAL conducted between 2009 and 2011.[25] T-DM1 was compared with capecitabine and lapatinib combination. The findings suggested that patients who were administered T-DM1 had an increased OS (median, 29.9 vs. 25.9 months) and a PFS (median, 9.6 vs. 6.4 months). Further, in a research performed later, the TH3R3SA trial,[26] it was found that the difference in median OS was statistically significant with T-DM1 compared to the physician’s choice of treatment (22.7 vs. 15.8 months). T-DM1 (trade name: Kayla) was approved in 2013 by the USFDA for use as a monotherapy in patients with metastatic breast cancer which is HER-2–positive with a relapse after previous trastuzumab or taxane therapy or disease recurrence within 6 months of prior adjuvant chemotherapy.[27]
The primary goal of the development of any ADC is to provide stability to the linker to prevent the dissociation of the drug in the circulation. However, it should be susceptible to degradation mechanisms once it enters its targeted cells, the journey to that target being aided by the mAb to which it is conjugated. In comparison to the first-generation ADC (gemtuzumab ozogamicin), the second-generation ADC (trastuzumab emtansine) has a wider therapeutic window, a more effective cytotoxic payload, a more stable linker, and better chemistry, manufacturing, and control characteristics.[28] The linker in T-DM1 is a thioether molecule (nonreducible), N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC).[29] Compared to the disulfide linker, a thioether linker is more potent as it can hold 3–3.6 moieties of DM1. A recent trial (2017) compared its efficacy against taxanes in HER-2–positive, unresectable, or metastatic gastric cancer (GATSBY trial).[30] Results stated the nonsuperiority of T-DM1 over taxanes.
Brentuximab vedotin
The target antigen of brentuximab (chimeric mAb) is CD30-positive cells (reed Sternberg cells) which are expressed in high quantity in Hodgkin’s disease (HD). A tubulin inhibitor monomethyl auristatin E is connected to this mAb via a cleavable peptide.[31] On binding, the ADC will internalize and succumb to lysosomal mechanisms that proteolytically cleave the linker, leading to the release of the cytotoxic component.
With regard to the treatment of HD, a Phase 2 study showed 75% ORR (complete response [CR] in 34%) with PFS of 5.6 months (median) for all patients. In this study, out of the 102 subjects who participated, 31 subjects became free of progressive disease at the end of 1.5 years.[32] Accelerated approval was granted in 2011 when this Phase 2 study was compared against a meta-analytic study of existing therapeutic options.[33] Results showed an overall CR rate of 11.1% for historical data versus 33.3% for brentuximab vedotin (P < 0.0001). Its superiority in primary cutaneous anaplastic large cell lymphoma (ALCL) was confirmed with the ALCANZA trial. Improvement in ORR (56% vs. 12%) and median PFS (17 vs. 4 months) were noticed in the brentuximab vedotin arm versus methotrexate or bexarotene.[34]
The drug was granted accelerated approval by the USFDA (marketed as Adcetris) for the treatment in HD who had relapsed after an autologous stem cell transplant (ASCT) or trial of two chemotherapeutic treatments (non-ASCT candidates) and as a second-line agent in ALCL.
Inotuzumab ozogamicin
Inotuzumab targets CD22-positive cells which are present in B-cell malignancies.[35] Its cytotoxic payload is the same as gemtuzumab ozogamicin, i.e., a calicheamicin derivative. In a Phase 3 trial (INO-VATE ALL), when compared with standard therapy, inotuzumab ozogamicin was shown to have a higher rate of CR (80.7% vs. 29.4%), increased remission period (4.6 vs. 3.1 months), increased PFS (5 vs. 1.8 months), and increased OS (7.7 vs. 6.7 months).[36] Following the success of this trial, the USFDA granted the drug approval in August 2017 (trade name: Besponsa) for the indication of relapsed or refractory Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (ALL) in adults.
Polatuzumab vedotin-piiq
Polatuzumab vedotin-piiq (POLIVY) was given accelerated approval in June 2019 by the USFDA and in November 2019 by the EMA. It is a CD79b-directed ADC used in conjunction with bendamustine and a product of rituximab (a combination known as BR) for the management of adult subjects with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). The clinical trial assessing the efficacy of POLIVY comprised 40 patients with DLBCL, and they were randomized into two groups as POLIVY plus BR and BR-alone groups based on the intervention given. In this study, 63% of POLIVY plus BR group showed partial remission of tumors and 40% showed complete remission at the end of treatment. On the other hand, 25% of the BR-alone group had partial remission and 18% had complete remission.[37]
Trastuzumab deruxtecan
In 2017, a Phase 1 study adopting dose-escalation method investigated the safety and tolerability of an ADC trastuzumab deruxtecan (DS-8201) in subjects with advanced gastric or gastroesophageal and breast tumors and found the antitumor activity of DS-8201 even in low HER-2–expressing tumors. There were no dose-limiting toxic effects or death following DS-8201 therapy and disease control was achieved in 91% of patients.[38]
Another part of this study was done in 2019 (dose-expansion study) where the dose was expanded based on the previous study results (dose-escalation study). This study result has shown tumor shrinkage in 80% of subjects and also reported manageable safety profile of DS-8201.[39]
Rovalpituzumab tesirine
Rovalpituzumab tesirine is an ADC which acts against delta-like protein 3 (DLL3), and this protein is detected in tumor-initiating cells. About 80% of patients with small-cell lung cancer express DLL3. As per a multicenter ópen-label Phase 1 clinical trial, conducted to assess the safety and activity of rovalpituzumab tesirine, this drug was observed to possess tolerable safety concerns and antitumor activity in subjects with small-cell lung carcinoma.[40]
Cancer immunotherapy
There is a broad history behind the development of cancer immunotherapy which started with “Coley’s toxin.” In 1891, William Coley, an American surgeon observed that Erysipelas infection following injection of heat-inactivated bacteria resulted in long-term regression of sarcoma.[41] Morales et al. have reported the usefulness of Bacillus Calmette-Guérin (BCG) bacterium in the management of superficial or early bladder cancer.[42] In 1975, the first mAb was manufactured by Köhler and Milstein.[43] The first personalized (autologous) immunotherapy-based cancer vaccine was approved by the USFDA in 2010 for the treatment of metastatic castration-resistant prostate cancer. This was based on accumulated evidence of promising results from several immunotherapy based research including sipuleucel-T. The most efficient method of genomic editing, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system was discovered in 2012, and it was first used in eukaryotic cells in 2013. CRISPR/Cas9 system consists of two components: CRISPR which acts as a guide RNA and protein Cas9 which is a nonspecific CRISPR-associated endonuclease. Cas9 is a defense mechanism of the immune system in bacteria and acts to resist interference from the external environment and viruses. It has a wide variety of applications such as genome editing, developing disease models for genetic disorders, and correcting disease-related mutations.[44] Recently, in 2016, the CRISPR gene editing technique has been used in humans for the first time for chimeric antigen receptor T (CAR T)-cell therapy.[45]
Cancer cells can escape the body’s immune response owing to many factors such as chemicals (IL-10) released by the tumor cell in its microenvironment that dampens T-cell activity,[46] conversion of effector T-cells into tumor-infiltrating regulatory T-cells (Tregs),[47] overexpression of tumor self-antigens,[48] and camouflaging antigens to escape an immune response.[49]
Cancer immunotherapy aims to boost the natural defense system, i.e., support and assist the patient’s immune cells in fighting the cancer cells. Some of the mechanisms that have had initial success in the field of cancer immunotherapy are high-dose interleukin-2 injections for advanced renal cell carcinoma and intravesical BCG vaccine irrigation for superficial bladder cell carcinoma. The recent advancements in the arena of vaccination strategy are enumerated in Table 2.
Table 2.
Various cancer immunotherapies in clinical practice
| Drug | Mechanism | Indication | Current status | References |
|---|---|---|---|---|
| Sipuleucel-T | Vaccine: dendritic cells approach | Metastatic castrate-resistant (hormone refractory) prostate cancer | Approved by the USFDA in 2010, by the EMA in 2013 but withdrawn in 2015 due to commercial reasons. | [51,52,53,54] |
| T-VEC | Vaccine: Oncolytic virus approach | Unresectable cutaneous, subcutaneous, and nodal lesions in patients with melanoma recurrent after initial surgery | Approved by the USFDA and the EMA in 2015. | [58,59,60] |
| Tisagenlecleucel | CAR T-cell therapy | Treatment of patients up to 25 years of age with B-cell precursor ALL that is refractory or in the second or later relapse | Approved by the USFDA in August 2017, by the EMA in November 2017 | [64,65,66,67] |
| Adult patients with relapsed or refractory (r/r) large B-cell lymphoma | Approved by the USFDA on May 1, 2018, by the EMA on August 22, 2018 | [68] | ||
| After two or more lines of systemic therapy including | ||||
| DLBCL not otherwise specified, high grade B-cell | ||||
| Lymphoma and DLBCL arising from follicular lymphoma |
DC: Dendritic cell, USFDA: United States Food and Drug Administration, EMA: European Medicines Agency, CAR: Chimeric antigen receptor, ALL: Acute lymphoid leukemia, DLBCL: Diffuse large B-cell lymphoma, T-VEC: Talimogene laherparepvec
Dendritic cell approach: Sipuleucel-T
The most potent antigen-presenting cells in human body, i.e., the dendritic cells, are instrumental in initiating MHC-restricted T-cell responses.[50] Unlike the administration of interleukins and BCG, vaccines are supposed to be specific to a particular antigen (tumor antigen in this case). For the production of a dendritic cells vaccine, a patient’s dendritic cells are collected following leukapheresis. These cells are then cultured for the next couple of days where they are made tumor ready and instilled back into the patient [Figure 3].
Figure 3.
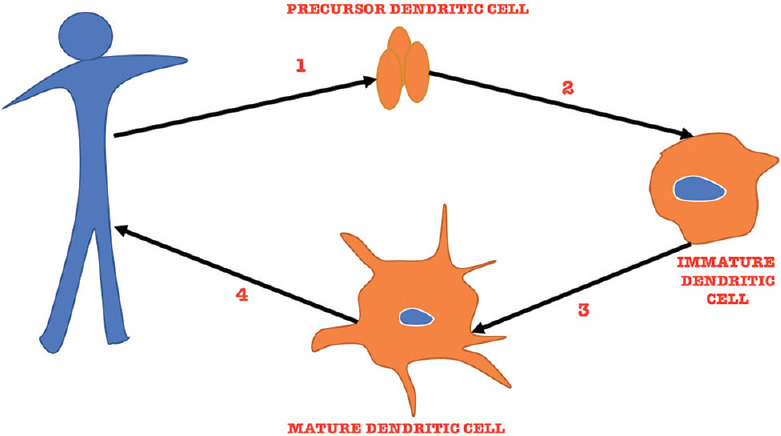
Description of the steps involved in production of therapeutic DCs. Step 1 involves leukapheresis followed by selection of precursor DC. In Step 2, precursor DC is cultured in vitro under the influence of cytokines where they transform into immature DC. In Step 3, they are exposed to the tumor Ag, which helps them transform into mature DC. In Step 4, these mature DC (capable of fighting cancer cells) are transfused back into the patient. DC: Dendritic cell
In the case of sipuleucel-T, the target antigen is prostatic acid phosphatase (PAP), which is an enzyme secreted by the epithelial cells of the prostate gland and converts orthophosphoric monoester to alcohol and orthophosphate. Unlike prostate-specific antigen (PSA), the physiological role of PAP is not well understood and it is used only as a marker in high-risk prostate cancer, wherein the PAP levels serve as important predictor of recurrence following radical prostatectomy.[51] The adjuvant used for the production of this therapeutic vaccine is granulocyte macrophage-colony stimulating factor (GM-CSF).[52] After introduction of this vaccine into the patient, mature dendritic cells prompt a cytotoxic T-lymphocyte-mediated action against PAP-positive prostate carcinoma cells.
This vaccine got the USFDA approval in 2010 for the indication of metastatic castration-resistant prostate cancer treatment, following a Phase 3 trial where it showed superiority with OS of 4.1 months (median value).[53] Since these subjects were castration resistant (hormone refractory), there is no other viable treatment strategy left to offer. Thus, therapeutically boosting their immune system via a vaccine will be a novel therapeutic approach.[54]
Oncolytic virus approach: Talimogene laherparepvec (T-VEC)
Specificity to an antigen is of prime concern for the production of a vaccine. Like the dendritic cells approach mentioned above, there is another approach that harnesses the power of an oncolytic virus to destroy cancer cells. Viruses possess cell-specific tropisms such as influenza for respiratory epithelium,[55] rabies virus for acetylcholine receptor in the nervous system,[56] and herpes simplex virus for skin.[57] This characteristic is not seen with dendritic cells. Second, viruses kill tumor cells via other mechanisms such as cell lysis due to viral replication and capturing the cellular death pathways.
Since talimogene laherparepvec (T-VEC) is a vaccine of pathogenic origin (HSV-1), genetic modifications had to be done to reduce its pathogenicity while preventing it from losing its pathogenicity. Specifically, the ICP34.5 gene was deleted to abolish the affinity that HSV-1 holds for trigeminal ganglion, thereby preventing latent infections. ICP47 gene was also deleted to assist antigen presentation, thereby promoting antiviral and antitumor activities.[58] In addition, the viral genome was inserted with two copies of GM-CSF genes which help the virus in releasing GM-CSF in a paracrine fashion, thereby engaging local dendritic cells into antitumor activity.[59] Once inside the target cell, T-VEC replicates and soon leads to lysis of the cell. Another virulent factor it possesses is the release of various pathogen-associated molecular patterns postcell lysis that sensitizes the patient’s immune system to fight the cancer cells. T-VEC was approved by the USFDA in 2015 for local management of unresectable lesions in subjects with recurrent melanoma (postinitial surgery).[60] Several other drugs are in pipeline such as pexastimogene devacirepvec (Pexa-Vec; JX-594) indicated for hepatocellular carcinoma, Toca511 for recurrent high-grade glioma, DNX-2401 for recurrent malignant glioma, PVS-RIPO for Grade IV malignant glioma, and CAVATAK for Stage IIIC–IVM1c melanoma.[61]
Adoptive cell transfer: Chimeric antigen receptor T-cell therapy
CAR T-cell therapy embodies the personification of personalized cancer care, where a patient’s T-cells are reformed to fight the cancer cells. The treatment starts with the collection of the patient’s blood. T-cells are separated via leukapheresis and engineered to express receptors (chimerically-CAR) on its surface [Figure 4]. These are expanded in culture and transfused back into the patient, making it a one-time treatment (similar to bone marrow transplant).[62] Viral/nonviral inclusions into the T-cells make them express antigens that are specific to the type of cancer. The chimeric receptor holds a greater affinity for the cancer cells than its unmodified ancestor. Further potency of this treatment relies on the action of T-cells. These modified T-cells identify and eradicate normal and malignant cells with CD19 expression.[63]
Figure 4.
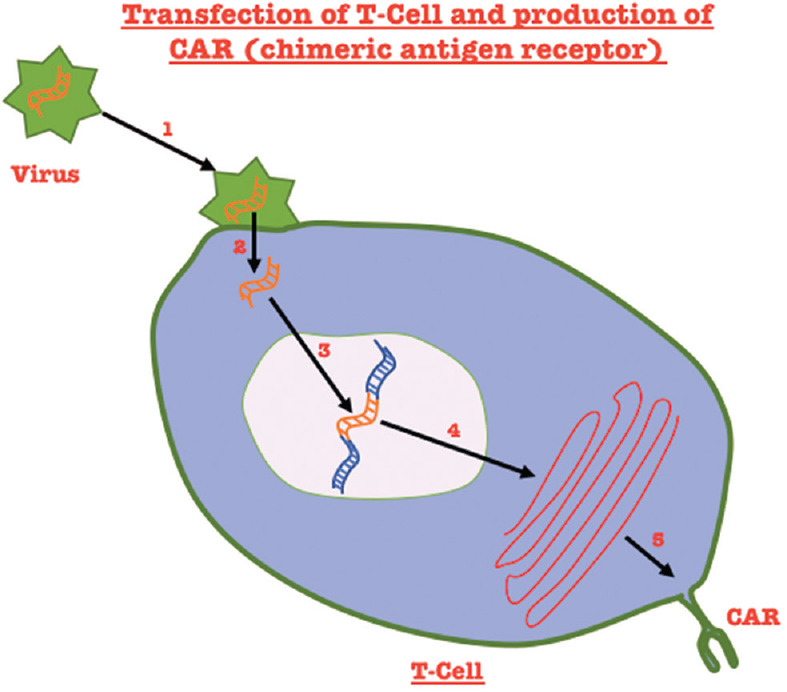
Steps involved in the production of a chimeric antigen receptor: Step 1: Binding of the virus containing the gene for the chimeric antigen to the T-cell. Step 2: Entry of the virus into the T-cell and uncoating of the chimeric antigen gene. Step 3: Integration of the chimeric gene into the T-cell gene. Step 4: Transcription of the gene. Step 5: Protein expression
Tisagenlecleucel (Kymriah), a genetically modified autologous T-cell immunotherapy/CAR T-cell therapy, got the USFDA approval in August 2017 based on the results of a single-arm trial for relapsed or refractory pediatric precursor B-cell ALL. Post bone marrow depleting therapy, patients received tisagenlecleucel intravenously, which contains autologous T-cells transfected with CD19 CAR genes (CD19 is a pan B-cell marker). The ORR in patients was 82% including CR in 63%, and 19% of CR was with incomplete hematological recovery.[64,65] ALL shows up to 85% OS in pediatric cases. It is only approved in cases of refractory/relapsed pediatric ALL and adult ALL,[66] and there have been no changes in OS in the last 20 years.[67] On May 1, 2018, tisagenlecleucel got the USFDA approval based on the result of a Phase 2 trial, i.e., JULIET study for the treatment of relapsed or refractory large B-cell lymphoma in adults after trial of two or more lines of systemic therapy. The indication includes DLBCL in adult patients. In JULIET trial, the ORR was 50% and the CR rate (relapse-free survival) was 32%.[68] The most common adverse reactions after administration of tisagenlecleucel was cytokine release syndrome (incidence >20%) and tocilizumab has been approved for its treatment.
It is important to note that even though the first CAR-T cell therapy was approved only recently, these molecules have undergone a series of evolution with subsequent generations, showing increasing degrees of co-stimulatory action and therefore possessing increased potency.[69] The latest addition to this family is axicabtagene ciloleucel (trade name: Yescarta), which is approved for use since November 2017 in adults with relapsed or refractory large B-cell lymphoma only after failure of two regimens of systemic therapy. The benefit of such a treatment apart from improved OS is that using a patient’s cells would eliminate the chances of graft versus host disease.
Metronomic chemotherapy
In 2000, Fidler and Ellis proposed the idea that “cancer is a chronic disease and should be treated like other chronic diseases.”[70] Metronomic is a therapeutic philosophy where unlike usual cytotoxic chemotherapy is given intravenously at the maximum tolerated doses followed by prolonged periods of rest; chemotherapeutic agents are administered in chronic, usually oral, equally spaced, low doses without the extended rest periods[71] [Figure 5]. Metronomic chemotherapy has shown superiority over conventional chemotherapy in some instances and does hold significance in developing countries such as India where a majority of patients are unable to bear standard treatment costs. Studies have shown that when cytotoxic agents are given in a metronomic fashion, they target endothelial cells which are persistently proliferating in tumor vasculature thus showing anti-angiogenic effects.[72] Many pharmacological strategies employ anti-vascular endothelial growth factor (anti-VEGF) therapies (e.g., bevacizumab) which prolong OS in patients with terminal cancer by only few months until relapse occurs. Apart from its anti-angiogenic properties, metronomic chemotherapy has also been noted to possess “immunomodulatory” as well as “continuous cytotoxic” properties.[73]
Figure 5.
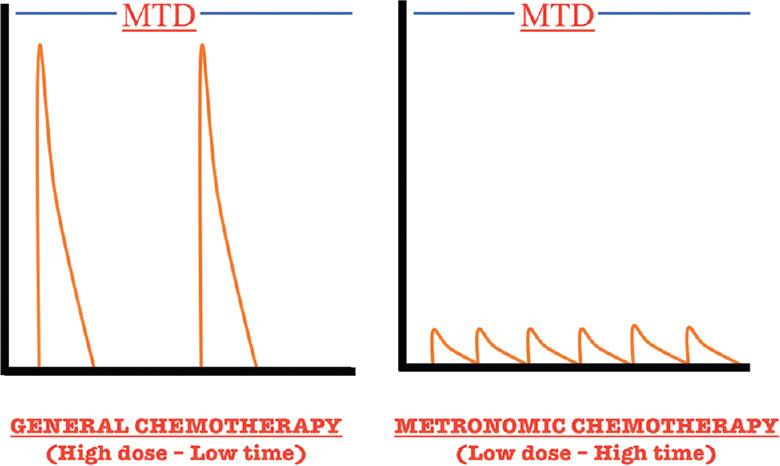
Dose–response curve showing the difference between general chemotherapy and metronomic chemotherapy. MTD: Maximum tolerated dose
In a Phase 1/2 trial, metastatic breast cancer patients who were administered metronomic chemotherapy comprising dalteparin, cyclophosphamide, and prednisone (once daily), along with methotrexate (twice-weekly), showed a nonsignificant decrease in angiogenesis marker VEGF levels. Simultaneously, soluble VEGF receptor-1 and 2 levels augmented significantly after 2 weeks of this therapy.[74] In another randomized Phase 2 trial in elderly breast malignancy patients, letrozole was compared to a combination of letrozole and oral metronomic cyclophosphamide. Results showed higher ORR in the metronomic group (87.7% vs. 71.9%).[75]
Metronomic chemotherapy has also shown success in metastatic colorectal carcinoma,[76] advanced non-small cell lung cancer,[77] advanced gastric carcinoma,[78] advanced epithelial ovarian cancer,[79] and recurrent malignant gliomas in adults[80] as well as in children with recurrent embryonal brain tumors.[81] It is to be noted that, to check the effectiveness of metronomic agents, or to show their superiority, the patient population must be homogenous to a disease subset. In a randomized clinical trial,[82] to check the efficacy of metronomic chemotherapy (thalidomide, celecoxib, etoposide, and cyclophosphamide) in progressive pediatric solid malignant tumors versus best supportive care, researchers found no benefit of metronomic treatment in terms of 6-month PFS (primary end point). However, in subgroup analysis (post hoc), it was found that patients with nonbone sarcomas and those able to withstand more than three cycles of treatment benefited from metronomic chemotherapy [Table 3].
Table 3.
Examples of drug combinations used in metronomic chemotherapy regimens
| MCTh regimen | Clinical phase | Indication | Significant results | Reference |
|---|---|---|---|---|
| Oral vinorelbine 50 mg three times weekly | Phase 2 | Stage IIIB–IV NSCLC | Treatment was well tolerated with rare serious toxicity | [77] |
| Capecitabine 625 mg/m2/d BD for 3 weeks plus bevacizumab 7.5 mg/kg i.v. every 3 weeks | Phase 2 | mCRC | PFS duration was longer in treatment group. However, benefits not seen once bevacizumab is withdrawn | [76] |
| Thalidomide 3 mg/kg OD, Celecoxib 100 mg BD for patients <20 kg 200 mg BD for patients 20–50 kg 400 mg BD for patients >50 kg |
Phase 3 | Refractory/progressive non-hematopoietic extracranial solid tumors following treatment with at least 2 lines of chemotherapy | Longer PFS seen only in patients without bone sarcomas (post hoc analysis) and those able to tolerate more than 3 cycles (9 weeks) benefit | [82] |
OD: Once daily, BD: Twice daily, PFS: Progression-free survival, NSCLC: Nonsmall-cell lung carcinoma, mCRC: Metastatic colorectal carcinoma, TNBC: Triplenegative b breast cancer, DFS: Disease-free survival, OS: Overall survival, MCTh: Metronomic chemotherapy
A recent study compared standard chemotherapy with metronomic cyclophosphamide plus low dose of corticosteroids in subjects with metastatic castration-resistant prostate cancer. This study resulted in ≥50% decline in the PSA level in 51% of patients. With the result of PFS 11 months (median) and OS 28 months, metronomic-based therapy is projected as the safe alternative to standard treatment.[83]
Another Phase 2 study compared capecitabine metronomic chemotherapy plus bevacizumab versus bevacizumab alone as maintenance therapy in subjects of metastatic colorectal cancer who have completed FOLFOXIRI plus bevacizumab for eight cycles. This study however did not find any significant difference in PFS and OS among these two.[84]
Conclusion
Metastatic cancer is a burden on the society. Epidemiologically, cancer morbidity is a major contributor to the DALY causing diseases.[85,86] Since the inception of cancer chemotherapy (nitrogen mustards) in the 1940s,[87] medical science has come a long way. Advancements in cancer care are currently in progress, some of which have been discussed in this article. These methods not only increase the PFS and OS but also have been shown to decrease adverse events in some instances. These new strategies encompass highly expensive treatment regimens such as tisagenlecleucel[88] for refractory B-cell ALL to cheap oral medication administered in the form of metronomic chemotherapy. We hope that future research into alternatives to the current cytotoxic chemotherapy will continue and the cost of these treatments will reduce over time. However, further trials are needed to establish the efficacy of these newer agents, along with the efforts to propel pharmacovigilance of these drugs to elucidate their safety.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Feigin VL, Roth GA, Naghavi M, Parmar P, Krishnamurthi R, Chugh S, et al. Global burden of stroke and risk factors in 188 countries, during 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016;15:913–24. doi: 10.1016/S1474-4422(16)30073-4. [DOI] [PubMed] [Google Scholar]
- 2.Behera P, Patro BK. Population based cancer registry of India The challenges and opportunities. Asian Pac J Cancer Prev. 2018;19:2885–9. doi: 10.22034/APJCP.2018.19.10.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tangka FK, Subramanian S, Edwards P, Cole-Beebe M, Parkin DM, Bray F, et al. Resource requirements for cancer registration in areas with limited resources: Analysis of cost data from four low- and middle-income countries. Cancer Epidemiol. 2016;45(Suppl 1):S50–8. doi: 10.1016/j.canep.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbas Z, Rehman S. An overview of cancer treatment modalities. In: Shahzad H, editor. Neoplasm. 1st ed. London: IntechOpen; 2018. pp. 139–57. [Google Scholar]
- 5.Chatterjee S, Chattopadhyay A, Senapati SN, Samanta DR, Elliott L, Loomis D, et al. Cancer registration in India Current scenario and future perspectives. Asian Pac J Cancer Prev. 2016;17:3687–96. [PubMed] [Google Scholar]
- 6.Saraswat N, Verma R, Neema S, Kumar S. A case of capecitabine-induced dermatomyositis. Indian J Pharmacol. 2018;50(6):350–53. doi: 10.4103/ijp.IJP_356_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda H, Khatami M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin Transl Med. 2018;7:11. doi: 10.1186/s40169-018-0185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.American Cancer Society. Cancer Treatment & Survivorship Facts & Figures 2016-2017. Atlanta: American Cancer Society; 2016. [[Last acessed on 2019 Jul 11]]. Available from: https://wwwcancerorg/research/cancer-facts-statistics/survivor-facts-figureshtml . [Google Scholar]
- 9.Alam A, Farooq U, Singh R, Dubey VP, Kumar S, Kumari R, et al. Chemotherapy treatment and strategy schemes: A review. Open Access J Toxicol. 2018;2:555600. [Google Scholar]
- 10.Sharma PK, Misra AK, Gupta A, Singh S, Dhamija P, Pareek P. A retrospective analysis of reporting of adverse drug reactions to oncology drugs: An experience from a national center of clinical excellence. Indian J Pharmacol. 2018;50:273–8. doi: 10.4103/ijp.IJP_544_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataria PS, Kendre PP, Patel AA, Tahiliani N, Bhargav V, Parekh H. Rare occurrence of hand-foot syndrome due to paclitaxel: A rare case report. Indian J Pharmacol. 2018;50(5):284–86. doi: 10.4103/ijp.IJP_547_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: Retrospective cohort study of drug approvals 2009-13. BMJ. 2017;359:j4530. doi: 10.1136/bmj.j4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8:473–80. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 14.Abdollahpour-Alitappeh M, Lotfinia M, Gharibi T, Mardaneh J, Farhadihosseinabadi B, Larki P, et al. Antibody-drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J Cell Physiol. 2019;234:5628–42. doi: 10.1002/jcp.27419. [DOI] [PubMed] [Google Scholar]
- 15.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 16.Cai HH. Therapeutic monoclonal antibodies approved by FDA in 2016. MOJ Immunol. 2017;5:1–2. [Google Scholar]
- 17.Golay J, Introna M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys. 2012;526:146–53. doi: 10.1016/j.abb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Kim EG, Kim KM. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol Ther (Seoul) 2015;23:493–509. doi: 10.4062/biomolther.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, Bitto E, Goff RD, Singh S, Bingman CA, Griffith BR, et al. Biochemical and structural insights of the early glycosylation steps in calicheamicin biosynthesis. Chem Biol. 2008;15:842–53. doi: 10.1016/j.chembiol.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–54. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 21.Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13:47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 22.ten Cate B, Bremer E, de Bruyn M, Bijma T, Samplonius D, Schwemmlein M, et al. A novel AML-selective TRAIL fusion protein that is superior to Gemtuzumab Ozogamicin in terms of in vitro selectivity, activity and stability. Leukemia. 2009;23:1389–97. doi: 10.1038/leu.2009.34. [DOI] [PubMed] [Google Scholar]
- 23.Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert JM, Chari RV. Ado-trastuzumab Emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem. 2014;57:6949–64. doi: 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- 25.Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:732–42. doi: 10.1016/S1470-2045(17)30312-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krop IE, Kim SB, Martin AG, LoRusso PM, Ferrero JM, Badovinac-Crnjevic T, et al. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017;18:743–54. doi: 10.1016/S1470-2045(17)30313-3. [DOI] [PubMed] [Google Scholar]
- 27.Amiri-Kordestani L, Blumenthal GM, Xu QC, Zhang L, Tang SW, Ha L, et al. FDA approval: Ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res. 2014;20:4436–41. doi: 10.1158/1078-0432.CCR-14-0012. [DOI] [PubMed] [Google Scholar]
- 28.Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 29.Barok M, Joensuu H, Isola J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014;16:209. doi: 10.1186/bcr3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thuss-Patience PC, Shah MA, Ohtsu A, Van Cutsem E, Ajani JA, Castro H, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): An international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol. 2017;18:640–53. doi: 10.1016/S1470-2045(17)30111-0. [DOI] [PubMed] [Google Scholar]
- 31.Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30:631–7. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 32.Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30:2183–9. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonthapally V, Wu E, Macalalad A, Yang H, Shonukan O, Liu Y, et al. Brentuximab vedotin in relapsed/refractory Hodgkin lymphoma post-autologous transplant: Meta-analysis versus historical data. Curr Med Res Opin. 2015;31:993–1001. doi: 10.1185/03007995.2015.1030378. [DOI] [PubMed] [Google Scholar]
- 34.Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): An international, open-label, randomised, phase 3, multicentre trial. Lancet. 2017;390:555–66. doi: 10.1016/S0140-6736(17)31266-7. [DOI] [PubMed] [Google Scholar]
- 35.Lamb YN. Inotuzumab ozogamicin: First global approval. Drugs. 2017;77:1603–10. doi: 10.1007/s40265-017-0802-5. [DOI] [PubMed] [Google Scholar]
- 36.Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375:740–53. doi: 10.1056/NEJMoa1509277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.FDA Approves Polatuzumabvedotin-Piiq for Diffuse Large B-Cell Lymphoma. [[Last accessed on 2019 Nov 06]]. Available from: https://wwwfdagov/drugs/resources-information-approved-drugs/fda-approves-polatuzumab-vedotin-piiq-diffuse -large-b-cell-lymphoma .
- 38.Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017;18:1512–22. doi: 10.1016/S1470-2045(17)30604-6. [DOI] [PubMed] [Google Scholar]
- 39.Shitara K, Iwata H, Takahashi S, Tamura K, Park H, Modi S, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive gastric cancer: A dose-expansion, phase 1 study. Lancet Oncol. 2019;20:827–36. doi: 10.1016/S1470-2045(19)30088-9. [DOI] [PubMed] [Google Scholar]
- 40.Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18:42–51. doi: 10.1016/S1470-2045(16)30565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;262:3–11. [PubMed] [Google Scholar]
- 42.Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116:180–3. doi: 10.1016/s0022-5347(17)58737-6. [DOI] [PubMed] [Google Scholar]
- 43.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity.1975. J Immunol. 2005;174:2453–5. [PubMed] [Google Scholar]
- 44.Zhang J, Zhou W, Wanga X, Wang L. The CRISPR-Cas9 system: A promising tool for discovering potential approaches to overcome drug resistance in cancer. RSC Adv. 2018;58:33464–72. doi: 10.1039/c8ra04509g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oiseth SJ, Aziz MS. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. 2017;3:250–61. [Google Scholar]
- 46.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:e1792. doi: 10.1038/cddis.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward-Hartstonge KA, Kemp RA. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunology. 2017;6:e154. doi: 10.1038/cti.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuvaraj N, Arul V. Sulfated polysaccharides of seagrass Halophila ovalis suppresses tumor necrosis factor-α-induced chemokine interleukin-8 secretion in HT-29 cell line. Indian J Pharmacol. 2018;50(6):336–43. doi: 10.4103/ijp.IJP_202_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: Tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–71. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossi M, Young JW. Human dendritic cells: Potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J Immunol. 2005;175:1373–81. doi: 10.4049/jimmunol.175.3.1373. [DOI] [PubMed] [Google Scholar]
- 51.Muniyan S, Chaturvedi NK, Dwyer JG, Lagrange CA, Chaney WG, Lin MF. Human prostatic acid phosphatase: Structure, function and regulation. Int J Mol Sci. 2013;14:10438–64. doi: 10.3390/ijms140510438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 53.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 54.Hammerstrom AE, Cauley DH, Atkinson BJ, Sharma P. Cancer immunotherapy: Sipuleucel-T and beyond. Pharmacotherapy. 2011;31:813–28. doi: 10.1592/phco.31.8.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.García-Sastre A. Influenza virus receptor specificity. Am J Pathol. 2010;176:1584–5. doi: 10.2353/ajpath.2010.100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Albertini AA, Baquero E, Ferlin A, Gaudin Y. Molecular and cellular aspects of rhabdovirus entry. Viruses. 2012;4:117–39. doi: 10.3390/v4010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh A, Preiksaitis J, Ferenczy A, Romanowski B. The laboratory diagnosis of herpes simplex virus infections. Can J Infect Dis Med Microbiol. 2005;16:92–8. doi: 10.1155/2005/318294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. 2017;18:1–5. doi: 10.1007/s40257-016-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toda M, Martuza RL, Rabkin SD. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol Ther. 2000;2:324–9. doi: 10.1006/mthe.2000.0130. [DOI] [PubMed] [Google Scholar]
- 60.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 61.Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18:689–706. doi: 10.1038/s41573-019-0029-0. [DOI] [PubMed] [Google Scholar]
- 62.Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkins O, Keeler AM, Flotte TR. CAR T-cell therapy: Progress and prospects. Hum Gene Ther Methods. 2017;28:61–6. doi: 10.1089/hgtb.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Study of Efficacy and Safety of CTL019 in Pediatric ALL Patients. ClinicalTrialsgov. 2017. [[Last accessed on 2020 Oct 12]]. Available from: https://clinicaltrialsgov/ct2/show/NCT02228096 .
- 65.Determine Efficacy and Safety of CTL019 in Pediatric Patients with Relapsed and Refractory B-cell ALL and High risk B-cell ALL at First Relapse. Determine Feasibility and Safety of CTL019 Therapy in Pediatric Patients with High Risk B-cell ALL that Relapsed<6 Months Post All-HSCT. ClinicalTrials.gov. 2017. [[Last accessed on 2020 Oct 12]]. Available from: https://clinicaltrials.gov/ct2/show/NCT02435849 .
- 66.Pulte D, Jansen L, Gondos A, Katalinic A, Barnes B, Ressing M, et al. Survival of adults with acute lymphoblastic leukemia in Germany and the United States. PLoS One. 2014;9:e85554. doi: 10.1371/journal.pone.0085554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia. 2008;22:2142–50. doi: 10.1038/leu.2008.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 69.Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22. doi: 10.1186/s40364-017-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fidler IJ, Ellis LM. Chemotherapeutic drugs More really is not better. Nat Med. 2000;6:500–02. doi: 10.1038/74969. [DOI] [PubMed] [Google Scholar]
- 71.Hanahan D, Bergers G, Bergsland E. Less is more, regularly: Metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045–7. doi: 10.1172/JCI9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: Changing the paradigm that more is better. Curr Oncol. 2009;16:7–15. doi: 10.3747/co.v16i2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banys-Paluchowski M, Schütz F, Ruckhäberle E, Krawczyk N, Fehm T. Metronomic chemotherapy for metastatic breast cancer A systematic review of the literature. Geburtshilfe Frauenheilkd. 2016;76:525–34. doi: 10.1055/s-0042-105871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong NS, Buckman RA, Clemons M, Verma S, Dent S, Trudeau ME, et al. Phase I/II trial of metronomic chemotherapy with daily dalteparin and cyclophosphamide, twice-weekly methotrexate, and daily prednisone as therapy for metastatic breast cancer using vascular endothelial growth factor and soluble vascular endothelial growth factor receptor levels as markers of response. J Clin Oncol. 2010;28:723–30. doi: 10.1200/JCO.2009.24.0143. [DOI] [PubMed] [Google Scholar]
- 75.Bottini A, Generali D, Brizzi MP, Fox SB, Bersiga A, Bonardi S, et al. Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J Clin Oncol. 2006;24:3623–8. doi: 10.1200/JCO.2005.04.5773. [DOI] [PubMed] [Google Scholar]
- 76.Jung YH, Lee WJ, Byeon JH, Lee IK, Han CW, Woo IS. Metronomic chemotherapy with capecitabine for metastatic colorectal cancer in very elderly patients. Korean J Intern Med. 2017;32:926–9. doi: 10.3904/kjim.2015.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Camerini A, Puccetti C, Donati S, Valsuani C, Petrella MC, Tartarelli G, et al. Metronomic oral vinorelbine as first-line treatment in elderly patients with advanced non-small cell lung cancer: Results of a phase II trial (MOVE trial) BMC Cancer. 2015;15:359. doi: 10.1186/s12885-015-1354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He S, Shen J, Hong L, Niu L, Niu D. Capecitabine “metronomic” chemotherapy for palliative treatment of elderly patients with advanced gastric cancer after fluoropyrimidine-based chemotherapy. Med Oncol. 2012;29:100–6. doi: 10.1007/s12032-010-9791-x. [DOI] [PubMed] [Google Scholar]
- 79.Samaritani R, Corrado G, Vizza E, Sbiroli C. Cyclophosphamide “metronomic” chemotherapy for palliative treatment of a young patient with advanced epithelial ovarian cancer. BMC Cancer. 2007;7:65. doi: 10.1186/1471-2407-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kesari S, Schiff D, Doherty L, Gigas DC, Batchelor TT, Muzikansky A, et al. Phase II study of metronomic chemotherapy for recurrent malignant gliomas in adults. Neuro Oncol. 2007;9:354–63. doi: 10.1215/15228517-2007-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peyrl A, Chocholous M, Kieran MW, Azizi AA, Prucker C, Czech T, et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer. 2012;59:511–7. doi: 10.1002/pbc.24006. [DOI] [PubMed] [Google Scholar]
- 82.Pramanik R, Agarwala S, Gupta YK, Thulkar S, Vishnubhatla S, Batra A, et al. Metronomic chemotherapy vs best supportive care in progressive pediatric solid malignant tumors: A randomized clinical trial. JAMA Oncol. 2017;3:1222–7. doi: 10.1001/jamaoncol.2017.0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Calvani N, Morelli F, Naglieri E, Gnoni A, Chiuri VE, Orlando L, et al. Metronomic chemotherapy with cyclophosphamide plus low dose of corticosteroids in advanced castration-resistant prostate cancer across the era of taxanes and new hormonal drugs. Med Oncol. 2019;36:80. doi: 10.1007/s12032-019-1304-y. [DOI] [PubMed] [Google Scholar]
- 84.Cremolini C, Marmorino F, Bergamo F, Aprile G, Salvatore L, Masi G, et al. Phase II randomised study of maintenance treatment with bevacizumab or bevacizumab plus metronomic chemotherapy after first-line induction with FOLFOXIRI plus Bevacizumab for metastatic colorectal cancer patients: The MOMA trial. Eur J Cancer. 2019;109:175–82. doi: 10.1016/j.ejca.2018.12.028. [DOI] [PubMed] [Google Scholar]
- 85.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–48. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Munshi R, Panchal F, Kulkarni V, Chaurasia A. Methylenetetrahydrofolate reductase polymorphism in healthy volunteers and its correlation with homocysteine levels in patients with thrombosis. Indian J Pharmacol. 2019;51(4):248–54. doi: 10.4103/ijp.IJP_215_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munshi R, Panchal F, Kulkarni V, Chaurasia A. Methylenetetrahydrofolate reductase polymorphism in healthy volunteers and its correlation with homocysteine levels in patients with thrombosis. Indian J Pharmacol. 2019;51(4):248–54. doi: 10.4103/ijp.IJP_215_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bach PB, Giralt SA, Saltz LB. FDA approval of tisagenlecleucel: Promise and complexities of a $475,000 cancer drug. JAMA. 2017;318:1861–2. doi: 10.1001/jama.2017.15218. [DOI] [PubMed] [Google Scholar]