

SUMMARY
MicroRNAs are important regulators of immune responses. Here, we showed miR-221 and miR-222 modulate the intestinal Th17 cell response. Expression of miR-221 and miR-222 was induced by proinflammatory cytokines and repressed by the cytokine TGF-β. Molecular targets of miR-221 and miR-222 included Maf and Il23r, and loss of miR-221 and miR-222 expression shifted the transcriptomic spectrum of intestinal Th17 cells to a proinflammatory signature. Although the loss of miR-221 and miR-222 was tolerated for maintaining intestinal Th17 cell homeostasis in healthy mice, Th17 cells lacking miR-221 and miR-222 expanded more efficiently in response to IL-23. Both global and T cell specific deletion of miR-221 and miR-222 rendered mice prone to mucosal barrier damage. Collectively, these findings demonstrate that miR-221 and miR-222 are an integral part of intestinal Th17 cell response that are induced after IL-23 stimulation to constrain the magnitude of proinflammatory response.
Keywords: miRNA, miR-221, miR-222, Helper T cells, Th17, Maf, IL23r, intestine, mucosal barrier damage, negative feedback
Graphical Abstract
eTOC Blurb:
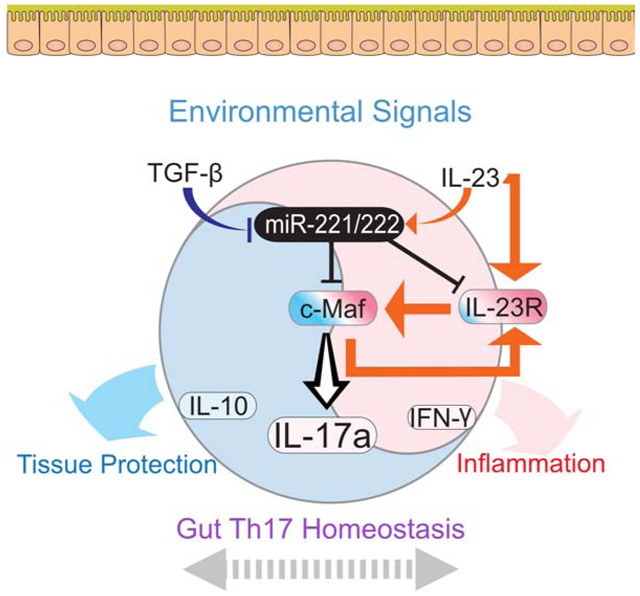
Mikami et al examine the role of miR-221/222 in helper T cells in the gut. MiR-221/222 are induced by IL-23 and suppressed by TGFb, targeting Maf and IL23r for degradation. During inflammation, these miRNAs serve as a negative feedback rheostat to constrain IL23-Th17 cell responses.
Introduction
Helper T (Th) cells play a major role in immunoregulation, host defense, and autoimmune pathogenesis by differentiating into specialized subsets with distinct functionalities (Zhu et al., 2010). Major Th subsets include T effector cells that produce specialized cytokines (Th1, Th2, Th17), regulatory T cells (Treg) that suppress T effectors, and follicular B helper T cells (Tfh) that interact with and facilitate B cell differentiation (O’Shea and Paul, 2010). At intestinal mucosal barriers at which commensal bacteria reside, resident cells and commensal bacteria constantly interact and communicate not only physically but also through soluble factors such as cytokines (Ivanov et al., 2009; Omenetti et al., 2019). In particular, interleukin-17 (IL-17) producing Th17 cells provide a unique and important role in preserving immune homeostasis in the gut (Korn et al., 2009; Littman and Rudensky, 2010).
Multiple cytokines and transcription factors take part in the generation of Th17 cells. Th17 cell-instructive cytokines include IL-6, IL-21, and IL-23, which mainly act via the transcription factor STAT3 alongside other STATs (Langrish et al., 2005; Yang et al., 2007). Transforming growth factor-β (TGF-β) coupled with IL-6 supports in vitro differentiation of IL-17 producing cells, whereas other multi-cytokine combinations, such as IL-23, IL-6 and IL-1β in the absence of TGF-β, promote Th17 cells with prominent inflammation signature (Bettelli et al., 2006; Esplugues et al., 2011; Gaublomme et al., 2015; Ghoreschi et al., 2010; Lee et al., 2012). Thus, the diverse combinatorial cytokine milieu in vivo is likely to instruct a spectrum of Th17 cells in tissues (Stockinger and Omenetti, 2017).
Cytokine signaling subsequently initiates intracellular transcription factor networks that further drive the Th17 cell program, including the lineage-determining transcription factor RORγt (Ivanov et al., 2006), which coordinates with IRF4 (Schraml et al., 2009), BATF (Schraml et al., 2009), AHR (Quintana et al., 2008; Veldhoen et al., 2008), c-Maf (Rutz et al., 2011; Tanaka et al., 2014) and other factors (Ciofani et al., 2012; Yosef et al., 2013). The phenotypic outcome of Th17 cell differentiation driven by combination of multiple transcription factors represents an additional aspect of heterogeneity in gene expression profiles. In particular, the degree to which inflammatory signature manifests is relevant to understanding the physiology of intestinal mucosal immunity at healthy steady state as well as pathological autoimmune conditions (Gaffen et al., 2014; Patel and Kuchroo, 2015; Stockinger and Omenetti, 2017). Furthermore, post-transcriptional regulators are likely to impact the heterogeneity of Th17 cells. We hypothesize that microRNAs (miRNAs) with selective expression in different Th17 cell conditions might play such regulatory roles.
Named after their small size of around 21-25 nucleotides, miRNAs are non-coding RNAs that negatively regulate gene expression in a target sequence-specific manner at the post-transcriptional level (Bartel, 2018). Depending on the degree of complementarity between a miRNA and its target, a miRNA can target multiple mRNAs to influence diverse arrays of developmental and physiological processes, including helper T cell differentiation (Baumjohann and Ansel, 2013). By conducting unbiased expression analysis of miRNAs across immune cells, we have reported distinct miRNA signatures of various immune cells (Kuchen et al., 2010), in which expression of miR-221 and miR-222, two miRNAs generated from a single pri-microRNA transcript, was detected in subsets of innate and adaptive lymphocytes. Recently, miR-221 and miR-222 were reported to play a role in macrophages to induce lipopolysaccharide tolerization by silencing inflammatory genes (Seeley et al., 2018). In contrast, the cell intrinsic function of miR-221 and miR-222 in adaptive cells such as T helper cells has not been fully investigated.
In this study, we examined the functional roles of miR-221 and miR-222 in T cells in mice in which these miRNAs were deleted globally and in a T cell specific manner. We found that a notable phenotype in these mice was related to response of Th17 cell populations in the gut in inflammatory conditions. MiR-221 and miR-222 targeted Maf and Il23r to control Th17 cell populations and limited the expansion of IL-17+ RORγt+ cell population in response to IL-23. Upon intestinal barrier perturbation with dextran sodium sulfate (DSS), T cell specific loss of miR-221 and miR-222 was associated with exacerbated damage. Beyond the impact on innate immune cells, our observations collectively support an important role of miR-221 and miR-222 in regulating intestinal Th17 cells and indicate that miR-221 and miR-222 act as negative feedback regulators downstream of IL-23 to modulate the magnitude of intestinal Th17 cell response in inflammatory milieu.
Results
Transcriptional regulation of miR-221 and miR-222 in CD4+ T cells
Using previously reported global miRNA expression profiling (Kuchen et al., 2010), miR-221 and miR-222 were found to be expressed dynamically among various blood cells (Fig.S1A). Focusing on CD4+ T helper cell subsets, miR-221 and miR-222 expression was noted to be high in Th1, Th17, and Tfh cells, and low in Th2 and Treg cells, suggesting that miR-221 and miR-222 expression is differentially regulated by diverse cytokine signals. To test this, epigenetic histone marks at the Mir221 and Mir222 genomic loci and miR-221 and miR-222 expression were examined in in vitro differentiated Th1 and Th17 cells; Th1 cells were differentiated with IL-12 and Th17 cells were differentiated under TGF-β or IL-23 conditions (see Experimental Procedures for details). Examination of the chromatin landscape in Th1 and Th17 cells revealed multiple enhancer activities upstream of the Mir221 and Mir222 loci marked by p300, H3K4me1, and H3K4me3 (Fig.1A). These regions were highly conserved across mammalian species (Fig. S1B) and overlapped with regions identified as superenhancer loci in human T cells (Fig. S1C). Under Th1 conditions, p300-prominent enhancers were also positive for STAT4 binding (Fig1A), the predominant STAT protein preferentially activated by IL-12 (Kaplan et al., 1996). In the absence of STAT4, active chromatin marks and miR-221 and miR-222 expression were reduced (Fig.1A-B), indicating a positive regulatory role for STAT4 driving miR-221 and miR-222 expression in Th1 cells. Similarly, STAT3 binding overlapped with active enhancer marks in Th17 cells from both cytokine conditions (Fig.1A) (Ghoreschi et al., 2010), and both enhancer activity and miR-221 and miR-222 expression were decreased when STAT3 was absent (Fig.S1D, Fig.1C). Therefore, miR-221 and miR-222 expression is apparently controlled by STAT proteins at an extended regulatory region in Th1 and Th17 lymphocytes.
Figure 1. Regulation of miR-221 and miR-222 expression in Th1 and Th17 cells.

(A) Chromatin landscape of the extended Mir221 and Mir222 locus (boxed in red) in Th1 cells (above Mir221 and Mir222 gene track) and Th17 cells (below Mir221 and Mir222 track). For Th1 condition, wild type (WT) and STAT4 deficient (S4KO) cells were evaluated. For Th17 condition, TGF-β (TGF-β+IL-6) and IL-23 (IL-6+IL-23) conditions were used with WT cells. ChIP-seq for binding of p300, STAT4, STAT3, H3K4me1,H3 K4me3, H3K27me3 are shown (Table S1; data are derived from GSE40463 (Vahedi et al., 2012), GSE22104 (Wei et al., 2010), GSE23681 (Ghoreschi et al., 2010), and GSE65621 (Hirahara et al., 2015). Putative regulatory regions with enhancer activity are marked at the bottom.
(B-D) RT-qPCR analysis of miR-221- and miR-222-3p expression in Th1 conditions comparing WT and S4KO (B), Th17-full conditions (TGF-β, IL-23 and others; see Materials and Methods) comparing WT and S3KO (C), and Th1 conditions +/− TGF-β (D). (*p<0.05, **p<0.01, ***p<0.005, ****p<0.001; Student’s t-test) Data are representative of at least 2 independent experiments. Data are shown as means with SD. See also Figure S1.
Additionally, gene expression and histone epigenetic marks suggest that TGF-β negatively regulates miR-221 and miR-222 expression. The presence of TGF-β in the Th17 polarization cocktail substantially enhanced deposition of repressive H3K27me3 marks (Fig.1A). This is consistent with expression data demonstrating that miR-221 and miR-222 are induced in Th17 cells generated by IL-23 but not TGF-β conditions (Fig.S2B); this differential response of miR-221 and miR-222 to IL-23 vs TGF-β will be further addressed below. Furthermore, miR-221 and miR-222 expression was downregulated when TGF-β was added to Th1 cultures (Fig.1D). Collectively, our data demonstrates that miR-221 and miR-222 expression is induced by proinflammatory cytokines and repressed by TGF-β.
Effect of Mir221 and Mir222 deletion on T cell development and in vitro T helper differentiation
We first examined the function of miR-221 and miR-222 in lymphocytes using germline Mir221 and Mir222 gene deletion mice. We aimed for simultaneous deletions of juxtaposing Mir221 and Mir222 loci via a pair of flox sites and generated Mir221Mir222 fl/fl mice (Flox allele in Fig.S2A). Subsequently Mir221Mir222fl/fll mice (denoted as wild-type (WT) hereafter) were crossed to Pgk1-cre mice to create germline deletion of both Mir221 and Mir222 (designated miR-221/222-KO mice) (KO allele in Fig.S2A). MiR-221/222-KO mice were fertile and developed normally. MicroRNA-seq of various in vitro generated T helper lineages confirmed that miR-221 and miR-222 were successfully deleted (Fig.S2B-C). Specifically, the expression of miR-221/222-3p (from the 3’-arms of the precursor miRNA) in WT cells was high in Th1 and IL-23-treated Th17 cells and low in TGF-β-treated Th17 and iTreg cells (Fig.S2B). Because miRNAs may regulate other miRNAs, we sought to determine whether deletion of miR-221 and miR-222 would impact miRNAs relevant to T cells. Importantly, deleting the Mir221 and Mir222 loci did not alter expression of other miRNAs when evaluated globally (Fig. S2C) or specifically (iTreg specific miR-10a; Fig. S2D) (Takahashi et al., 2012).
To determine potential developmental effects of miR-221 and miR-222 deletion, we examined T populations in the thymus and spleen in miR-221/222-KO mice. While the proportions of specific thymocyte subsets were slightly altered in KO mice, the populations of lymphocytes in the spleen remained unchanged (Fig. 2A-D), indicating that miR-221 and miR-222 deletion does not dramatically alter T cell development or the composition of splenic lymphocytes. Next, in vitro T helper differentiation of naïve cells derived from secondary lymphoid tissues was evaluated comparing WT and miR-221/222 KO cells. When cell proliferation was measured by CFSE dilution, a slight impairment was detected for miR-221/222-KO cells in T helper null condition (Fig. S2E, F). Under Th1 conditions, the production of IFN-γ as well as the expression of T-bet was decreased in miR-221/222-KO CD4+ T cells (Fig. 2E). In contrast, the proportion of Th17 cells, generated under full cocktail conditions (see Methods), was significantly increased in miR-221/222-KO CD4+ T cells, as assessed by IL-17 production and RORγt expression (Fig. 2F). Interestingly, the observed difference in IL-17 production was not due to differences in gut microbiome among mice, as microbiome profiles were variable but comparable between WT and miR-221/222 KO mice (Fig.S2G). Collectively, our data indicate that deletion of miR-221 and miR-222 allows for normal development of lymphocytes but differentially impacts in vitro CD4+ T helper differentiation of Th1 and Th17 subtypes.
Figure 2. Effect of miR-221 and miR-222 deletion on T cell development and in vitro T helper differentiation.

(A-B) Flow cytometric analyses comparing WT and miR-221/222 KO thymocyte subsets. Plots show the frequency of CD4SP and CD8SP cells, γδT cells (from CD4−CD8− gate), and DN subsets (from CD4−CD8− TCRγδ- gate). CD25 and CD44 distinguish DN subsets (DN1: CD25−CD44+; DN2: CD25+CD44+; DN3: CD25+CD44−; DN4 CD25−CD44−). (CD) Flow cytometric analyses comparing WT and miR-221/222 KO splenocytes: B cells (NKp46− CD19+), NK cells (NKp46+ CD19−), αβT cells (TCRβ+ NKp46− CD19−), CD4+ and CD8+ T cells. (E) Naïve CD4+ T cells of indicated genotypes were cultured under Th1 condition for 3 days. Flow cytometric analysis of cells expressing IFN-γ and T-bet are shown. The left panel shows a representative result from biological triplicates with cumulative data shown on the right (**p<0.01; Student’s t-test). Data are represented as mean with SEM. Data are representative of at least 3 independent experiments. (F) Naïve CD4+ T cells of indicated genotypes were cultured under Th17-full conditions for 3 days. Flow cytometric analysis of cells expressing IL-17A and RORγt are shown. Panels shows a representative result from biological triplicates with cumulative data shown in plots (*p<0.05; Student’s t-test). Data are represented as mean with SEM. See also Figure S2.
Loss of miR-221 and miR-222 enhances activation-dependent IL-17 production in intestinal CD4+ T cells
To determine the impact of miR-221 and miR-222 on CD4+ T helper subsets in vivo, small intestine lamina propria CD4+ T lymphocyte subsets were examined in co-housed miR-221/222-KO and WT mice. Using flow cytometry to identify subsets based on transcription factor expression (Fig. 3A), there was no significant difference in the frequency of CD4+ T cells subsets (Fig. 3B), but the number of Treg and Th1 lymphocytes were reduced in miR-221/222 mice compared to WT mice (Fig. 3C). Since CD4+ T helper subsets are also identified by cytokine production, intestinal lymphocytes were stimulated with PMA and ionomycin ex vivo to measure IFN-γ and IL-17a production. While there was no difference in the frequency of IFN-γ+ lymphocytes between mice, miR-221/222-deficient CD4+ T cells exhibited an increase in the proportion of IL-17a+ cells compared to WT mice (Fig.3D). Furthermore, the frequency of RORγt+ IL-17+ CD4+ T cells was increased in miR-221/222-KO mice with no difference in the frequency of RORγt+ cells (Fig. 3E), suggesting that miR-221 and miR-222 are required to constrain IL-17a production upon activation. Of note, IL-17+ cells from miR-221/222-KO mice also had higher levels of IL-17 protein on a per cell basis compared to WT cells, as measured by MFI (Fig. 3E). Collectively, our data indicate that deletion of miR-221 and miR-222 impacts activation-dependent cytokine production of peripheral Th17 cells derived from the gut.
Figure 3. Loss of miR-221 and miR-222 enhances IL-17 production in activated intestinal CD4+ T cells.

(A-E) Lymphocytes were isolated from the small intestine of healthy WT and miR-221/222-KO mice and analysed by FACS. (A) Representative FACS plots show T helper subsets defined as naïve, Treg, Th1 and Th17. (B-C) Pooled data comparing the frequencies (B) and cell number (C) of T helper subsets between WT (n=8) and miR-221/222-KO (n=7) according to A. (D) Proportion of CD4+ T cells expressing IL-17a and IFN-γ following PMA-I stimulation. (E) Proportion of CD4+ T cells expressing RORγt and IL-17a after PMA-I stimulation. (B-E) Data are represented as mean with SEM. Plots are pooled from 6-8 mice/group from at least 3 independent experiments. (**p<0.01; Student’s t-test) (F-I) Il17-gfp+ cells from the small intestine of healthy WT and miR-221/222-KO mice were used for scRNA-seq (see Fig. S3A for gating strategy). (F) UMAP plot depicts 8 clusters separated in an unbiased manner. The clusters 6 and 7 contained less than 10 cells of unknown origin and removed from further analysis. (G) Dot plot shows the expression of representative marker genes to define cluster identities. (H) Stacked bar plots depict the frequency of each cluster between WT and miR-221/222-KO mice.
To obtain a clearer picture of the impact of miR-221 and miR-222 deletion on unperturbed tissue-resident IL-17-producing CD4+ T cells, healthy IL-17-GFP reporter mice (IL17-GFP-WT and IL17-GFP-miR-221/222-KO) were used to isolate intestinal IL-17-GFP+ CD4+ cells (Fig.S3A) for single cell RNA-seq analysis. Comparable numbers of Il17-gfp+ CD4+ cells were captured from WT and miR-221/222-KO mice (WT: 688 cells; KO: 526 cells). Differential gene expression resolved 8 clusters, two of which represent minor cell contamination of unknown origin and thus, were excluded from further analysis (clusters 6-7; Fig. 3F). While all cells were captured based on the presence of “proxy” GFP protein that was driven by endogenous Il17a gene promoter, there were considerable differences in endogenous Il17a transcript expression amongst individual cells, as well as other key phenotype-defining transcripts (Fig. 3G-H). We identified cells expressing predominantly Il17a and Il22 (17 single positive (SP) effector), both Il17a and Ifng (double positive (DP) effector), quiescent cells with minimal cytokine expression (double negative (DN) Quiescent), actively cycling cells expressing Mki67, Il17a and Ifng (DP cycling), cells expressing Ifng, Gzma, Hspa1a and Ccl5 (gSP cytotoxic), and cells expressing Il17a along with Il10 and Foxp3 (17SP Treg) (Fig. 3G). The frequencies of each Th17 cell subset (clusters 0 to 5) were comparable between WT and miR-221/222-KO samples (Fig. 3H), indicating that the overall balance of heterogeneity among intestinal Th17 cells is maintained in the absence of miR-221 and miR-222 in healthy mice.
Collectively, our findings indicate an interesting dichotomy regarding the phenotype of intestinal Th17 cells from miR-221/222 KO mice. Whereas loss of miR-221 and miR-222 was tolerated in healthy, unperturbed mice and intestinal Th17 cell homeostasis was maintained, intestinal Th17 cells devoid of miR-221 and miR-222 produced more IL-17a upon activation. Therefore, miR-221 and miR-222 may regulate the Th17 cell response under certain cellular environments that occur upon pathological provocation.
miR-221/222-deficient Th17 cells exhibit a cell intrinsic proinflammatory Th17 transcriptomic signature
As a variety of intrinsic and extrinsic factors may influence Th17 cells in the gut, mixed bone marrow (BM) chimera mice were first used to test the cell intrinsic roles of miR-221 and miR-222. Irradiated Rag2−/− mice were reconstituted with bone marrow obtained from miR-221/222-KO and WT congenic mice at a 1:1 ratio (Fig.4A). After 6 to 7 weeks following BM reconstitution, CD4+ T cells were isolated from the small and large intestine lamina propria and congenic markers were used to evaluate WT and KO cells separately. The absence of miR-221 and miR-222 had no significant impact on lymphopoiesis, as co-transferred WT and miR-221/222-KO T cells populated gut mucosal tissues similarly (Fig. 4B). While stem cell transplantation in the setting of lymphopenia provokes inflammatory responses, cells are exposed to the same local microenvironment. In this setting, we noted the presence of more IL-17-producing Th17 cells from miR-221/222-KO fractions compared to WT fractions in the small intestine and large intestine (Fig. 4C-D). Therefore, loss of miR-221 and miR-222 resulted in more gut IL-17-producing cells in a cell intrinsic manner.
Figure 4. miR-221/222-deficient Th17 cells exhibit a cell intrinsic proinflammatory Th17 transcriptomic signature.

(A) Diagram of mixed bone marrow (BM) chimera experiment using congenic WT and miR-221/222-KO BM cells. Irradiated Rag2-KO hosts were reconstituted with 1:1 ratio of WT (CD45.1) and miR-221/222-KO (CD45.2) BM. Th cells populating the gut were analyzed at 7-8 weeks post BM transfer and identified by congenic markers. (B-F) Chimerism (B) and proportions of Th cells expressing IL-17 and RORγt from the small intestine (C) or large intestine (D) are depicted. Plots represent mean with SEM of pooled samples isolated from 27 mice (small intestine) and 29 mice (large intestine) from 2 independent experiments. (E-F) CD4+ T cells isolated from the small intestine of BM chimera mice were separated into WT and miR-221/222-KO fractions and mRNA-seq was performed. (E) Summary of 5 GSEA (Broad Institute) analyses for signature genes of Th cell subsets. Signature gene sets were extracted from published data. Also see Table S2. (F) GSEA plots for signature genes of Th17 and Th2 subsets. Replicates were compared for significance, ns (not significant), *p<0.05, **p<0.01, and ****p<0.001. See also Figure S4.
Next, mRNA-seq was used to determine the transcriptomic signature of WT and miR-221/222-deficient CD4+ T cells from bone marrow-reconstituted mice (n = 3; Fig. 4A, S4). With a cutoff of 1.5-fold change and a p value of <0.05, 28 genes were found to be differentially regulated between WT and KO cells (Fig. S4). In order to further assess differential gene expression in “bulk” CD4+ T cell samples, gene lists were generated from multiple publicly available datasets to perform gene set enrichment analysis (GSEA). These datasets utilized include: (1) Th17 genes (Ciofani et al., 2012), (2) IL-10-positive Th17 genes, (3) IL-10-negative Th17 genes (Aschenbrenner et al., 2018), (4) c-Maf-induced genes (Ciofani et al., 2012), and (5) Th2 genes (Ranzani et al., 2015) (Fig.4E and Table S1). While the Th2 signature was not enriched in miR-221/222-deficient T lymphocytes, Th17 signatures were significantly enriched compared to WT CD4+ T lymphocytes (Fig. 4E-F). Notably, the IL-10 negative Th17 signature was more prominent than IL-10 positive Th17 signature, indicating a more pro-inflammatory nature of miR-221/222-deficient cells. In addition, the c-Maf-induced gene signature was enriched in miR-221/222-KO cells (Fig. 4E), suggesting that c-Maf may be a key target downregulated by miR-221 and miR-222.
miR-221 and miR-222 target 3’UTR of Maf and Il23r for degradation to constrain Th17 response to IL-23
To examine the basis of the proclivity of miR-221/222-deficient Th17 cells to generate a pro-inflammatory signature, potential targets for miR-221 and miR-222 that are relevant to Th17 function were explored. A total of 2827 putative targets were identified from an in silico prediction algorithm (microRNA.org: (Betel et al., 2008)), which were then cross-referenced to a list of core Th17 cell signature genes (Ciofani et al., 2012). Among the nine putative targets identified (Fig. 5A), Maf and Il23r had potential target sequences in their 3’ untranslated region (3’ UTR) for miR-221 and miR-222 (Fig.S5A-B)(Betel et al., 2008). Maf was of interest due to its essential role in regulating Th17 function and enrichment of its target genes in miR-221/222 KO cells (Fig.4E). Il23r drew our attention for its proinflammatory nature in Th17 cells and its upregulated expression in miR-221/222 KO cells (Fig.S4) (Ciofani et al., 2012; Cua et al., 2003; Ghoreschi et al., 2010; Pfeifle et al., 2017; Tanaka et al., 2014) To validate Maf and Il23r as direct targets of miR-221 and miR-222, reporter constructs were generated with the 3’ UTR of Maf or Il23r fused to luciferase. Overexpression of miR-221 and miR-222 reduced luciferase activity compared to control miRNA (Fig. 5B), indicating that both the 3’ UTR of Maf and Il23r mediated miR-221/222-dependent mRNA degradation of heterologous reporters.
Figure 5. miR-221 and miR-222 target Maf and Il23r to regulated Th17 response.

(A) Overlap of curated Th17-signature genes (Ciofani et al., 2012) and predicted target genes for miR-221 and miR-222 (microRNA.org: (Betel et al., 2008)). (B) HEK293 cells were transfected with luciferase reporter fused to 3’ UTR of Il23r or Maf together with miR-221 plus miR-222 or control miRNA (random sequences). Luciferase activity was evaluated at 24 hr post-transfection. Data are representative of 2 independent experiments. (C) Flow cytometric analysis of c-Maf in WT and miR-221/222-KO Th cells, gated according to Figure 3A. Data show representative flow cytometry plots and the pooled data with statistical evaluation. (D-E) Ex vivo intestinal lymphocytes were stimulated in vitro with IL-23 or IL-23 and IL-1β for 6 hrs and subjected to flow cytometric analysis to measure the induction of c-Maf expression in Th17 cells (D; 3-4 mice/group) and the frequency of IL-17-producing RORγt+ CD4+ T cells in WT and miR-221/222-KO mice (E; 6-7 mice/group). See also Figure S5.
Next, flow cytometry was used to test whether deletion of miR-221 and miR-222 resulted in elevated c-Maf protein expression in intestinal Th cells from WT and miR-221/222-KO mice. Consistent with previous studies, c-Maf was expressed higher in Treg and Th17 lymphocytes compared to naïve CD4+ T cells and Th1 cells from both WT and miR-221/222-KO mice (Fig. 5C). Deletion of miR-221 and miR-222 resulted in increased c-Maf protein expression only in Th17 cells (Fig.5C). Of note, the expression of Maf and Il23r transcripts were largely comparable between WT and KO Th17 cells in steady-state condition, confirming the expected action of miRs at post-transcriptional and translational levels (Fig.S5C). Next, to determine whether proinflammatory cytokine conditions further promote c-Maf expression in miR-221/222-deficient Th17 cells, small intestine lamina propria lymphocytes were stimulated ex vivo with IL-23 or IL-23 plus IL-1β. While these cytokines did not induce a change in c-Maf expression in Th17 cells from WT mice, c-Maf protein was significantly induced in Th17 from miR-221/222-KO mice (Fig.5D). In the same experiment, stimulation also led to an increase in the frequency of IL-17-producing Th17 cells from miR-221/222-KO mice compared to WT mice (Fig.5E). Collectively, these results suggest that miR-221 and miR-222 function as negative feedback regulators in a proinflammatory Th17 signaling circuit by directly downregulating a signal entry step (IL-23R) as well as reducing a downstream transcription factor (c-Maf).
Deficiency of miR-221 and miR-222 increases the susceptibility of DSS-induced colitis in mice
In the gut, Th17 cells can be protective and promote homeostasis or mediate pathology depending upon the circumstance. The pathogenic role of IL-23R on T lymphocytes is well established and IL23R has been identified as an important susceptibility gene in inflammatory bowel diseases (Duerr et al., 2006). In healthy miR-221/222 deficient mice, we found no broad disruption of T cell homeostasis in the gut (Fig. 3A-C, 3F-I). Nonetheless, over time, miR-221/222 deficient Th17 lymphocytes may still contribute to spontaneous intestinal inflammation due to their enhanced response to IL-23 in mice. Accordingly, when small intestine histology was evaluated in WT and miR-221/222-KO mice living in a standard pathogen free facility (Fig. S6A), the average pathology score was higher in miR-221/222-KO mice compared to WT mice. However, the differences did not reach a statistical significance. In contrast, we consistently found that miR-221/222-deficient T cells had exaggerated Th17 responses once activated by TCR mimic, inflammatory cytokines or lymphopenic expansion environment, (Fig. 2E-F, 3D-E, 4, 5E). Thus, we hypothesized that miR-221 and miR-222, while not being critical in the steady state, may be more important for constraining inflammation during pathological perturbation. Using the DSS colitis model, miR-221/222-KO mice showed a significant reduction in body weight and a greater severity of colitis compared to the WT mice (Fig.6A-B). Correspondingly, deficiency of miR-221 and miR-222 also resulted in a significant increase in the total number of lamina propria infiltrating cells and number of Th17 lymphocytes compared to WT mice (Fig.6C-F), demonstrating that miR-221/222 KO are more susceptible to DSS-induced colitis.
Figure 6. miR-221/222-KO and CD4-cre miR-221/222-conditional KO mice exhibit increased susceptibility to DSS-induced colitis.

(A-F) WT (n=4) and miR-221/222-KO (n=5) mice were challenged with 2% DSS in drinking water to provoke intestinal inflammation. Data are representative of at least 3 independent experiments. (A) Body weight change is expressed as a percentage of initial weight in WT and miR-221/222-KO mice. Statistically significant differences between WT and miR-221/222-KO mice are designated by asterisks. (B) Representative H&E staining of colon specimens at day 10. A scale bar at the bottom left corner of each image is 100 μm. (C) Absolute cell numbers of colonic LPLs isolated from WT and miR-221/222-KO mice. (D-F) Frequency (D, E) and absolute number (F) of RORγt+ and IL-17+ CD4+ lamina propria cells from colons of WT and miR-221/222-KO mice. Colonic lamina propria cells were stimulated with PMA-I for 4 hr before staining. Data are represented as mean with SEM. Replicates were compared for significance, *p<0.05, **P<0.01, Student’s t-test. (G) WT (n=6) and CD4-cre miR-221/222-conditional KO (n=8) mice were challenged with 2% DSS in drinking water to provoke intestinal inflammation. Data provided represent mean ± SEM of the % body weight of pooled data from 2 independent experiments (*p < 0.05, **p < 0.01). (H) Data provided represent mean ± SEM of the % body weight of Rag2-KO (n=5) and Rag2-Mir221Mir222 double KO (n=6) mice pooled from 2 independent experiments. See also Figure S6.
To assess whether the abnormalities seen in the DSS model with global miR-221/222-KO mice could be attributed mainly to T cell fractions or other fractions (innate cells and epithelial lining cells), we generated two more genetic models of miR-221/222 deletion in T cells (CD4-cre Mir221Mir222 conditional KO (cKO)) and non-T cell fractions (Rag2-Mir221Mir222 dual KO (DKO)). While no major defect was noted in major lymphocyte populations in cKO mice (Fig.S6B), cKO mice were also more susceptible to DSS colitis compared to WT mice (Fig. 6G). By contrast, DKO mice did not exhibit enhanced weight loss compared to Rag2-KO control (Fig. 6H), indicating that deletion of miR-221 and miR-222 in innate cell compartments or epithelial cells did not compromise mice to DSS challenge to the same degree as deletion in T cells. Collectively, our findings indicate that miR-221 and miR-222 regulate response to DSS-mediated barrier damage mostly, if not exclusively, in a T cell intrinsic manner via modulating the magnitude of proinflammatory Th17 response induced by injury.
Discussion
A growing body of evidence supports important regulatory roles of miRNAs in a wide range of biological and pathological processes by modulating multiple target mRNAs at a post-transcriptional stage (Bartel, 2018). MiR-221 and miR-222, which reside in close proximity on chromosome X, have previously been dubbed as “onco-miRNAs” reflecting their expression in malignant cells and potential roles in cell proliferation, initiation, and progression of cancer (Garofalo et al., 2012). MiR-221 and miR-222 were reported to play an important regulatory role in macrophage tolerance to LPS, and its high expression is relevant to predict immunoparalysis and poor prognosis of sepsis patients (Seeley et al., 2018). In this study, we reveal a previously unappreciated role of miR-221 and miR-222 in controlling Th17 cell response in the intestinal mucosa under proinflammatory conditions. MiR-221 and miR-222 are critical negative feedback components that constrain IL-23-induced Th17 response by downregulating IL-23R and c-Maf. Accordingly, loss of miR-221 and miR-222 compromised the ability of miR-221/222-defficient mice to protect against mucosal barrier damage.
To date, several miRNAs have been reported to contribute to Th17 cell differentiation (Baumjohann and Ansel, 2013). Loss of mature miRNAs in Dicer-deficient CD4+ T cells shows reduced IL-17 production under Th17(β) condition (Cobb et al., 2006). Similarly, knocking out or knocking down miR-155, miR-326, miR-301a, miR-183c, and miR-132 and miR-212 results in marked reduction of Th17 cells in vitro and in vivo (Du et al., 2009; Escobar et al., 2014; Ichiyama et al., 2016; Mycko et al., 2012; O'Connell et al., 2010). In contrast, some miRNAs have been shown to repress the development of Th17 cells, including miR-15b, miR-17~92 and miR-18a that target Ogt, Rora, and Smad4 respectively (Liu et al., 2017). Our data add miR-221 and miR-222 to the list of Th17 regulating miRNAs with a very specialized window of action. miR-221 and miR-222 operate in a proinflammatory signal selective (IL-23) condition but their action is muted in TGF-β dominant condition. This unique feature of miR-221 and miR-222 has an illuminating implication in understanding the regulatory mechanisms balancing divergent proinflammatory vs regulatory actions of Th17 cells.
Early on, phenotypic description of Th17 subpopulations was defined in vitro by different cytokines (Bettelli et al., 2006; Ghoreschi et al., 2010). TGF-β is thought to represent a regulatory feature of Th17 cells, whereas IL-23 represents a proinflammatory feature (Ghoreschi et al., 2010). From human Th17 clones, IL-10 was identified as a key marker representing an immunoregulatory and tissue resident program, whereas IL-10-negative Th17 cells were proinflammatory (Aschenbrenner et al., 2018). In vivo Th17 studies provided further insight into protective versus inflammatory features of tissue-resident Th17 cells, which underpinned differences in the microbiome. Homeostatic Th17 cells induced by commensal bacteria maintained muted memory-cell-like metabolism and did not participate in inflammatory reaction, whereas pathogen induced Th17 cells were glycolytic, inflammatory, and produced IFN-γ (Omenetti et al., 2019). In this study, the observation that deficiency of miR-221 and miR-222 is tolerated and Th17 homeostasis is maintained in healthy hosts suggests that commensal-induced non-pathological Th17 cells are similar to TGF-β induced regulatory Th17 cell; expression of miR-221 and miR-222 is muted under homeostatic conditions and functionally less critical. In contrast, miR-221 and miR-222 are more relevant during proinflammatory conditions, as they are upregulated by IL-23 and target Il23r for downregulation to constrain an otherwise feedforward circuit of proinflammatory Th17 response.
Another key target of miR-221 and miR-222 is Maf (protein c-Maf). Th17 cells express this transcription factor highly, and it is critical for several important aspects of Th17 differentiation and function (reviewed in (Imbratta et al., 2020)). In miR-221/222-KO mice, c-Maf expression was enhanced in Th17 cells and not naïve, Th1, or Treg fractions isolated from the gut. In addition, ex vivo stimulation of miR-221/222-deficient gut lymphocytes with IL-23 resulted in more c-Maf and more IL-17-producing Th17 cells. C-Maf is a major regulator of cytokine loci, especially Il17a in Th17 cells and γδT cells (Ciofani et al., 2012; Tanaka et al., 2014; Zuberbuehler et al., 2019), suggesting that elevated c-Maf results in more IL-17 production in miR-221/222-deficient Th17 cells. Elevated c-Maf may also induce more IL-23R expression in miR-221/222-deficient Th17 cells, perpetuating a proinflammatory positive feedback loop. The Il23r gene contains a MARE-like sequence (Sato et al., 2011), but whether c-Maf induces or represses IL-23R expression remains unclear. Immunoregulatory or proinflammatory conditions may explain this discrepancy, as c-Maf represses Il23r expression in IL-10-positive Th17 cells (Aschenbrenner et al., 2018) and induces Il23r under IL-23 culture conditions (Bauquet et al., 2009). Considering the latter, our data suggests that c-Maf induces IL-23R in miR-221/222-deficient Th17 cells stimulated with IL-23, and normally miR-221 and miR-222 function to prevent this feedforward loop. As c-Maf shows broad, robust expression across an array of Th17 cells and has its own network of target genes, miR-221 and miR-222-mediated regulation may act via c-Maf independent of IL-23 signalling (Ciofani et al., 2012; Imbratta et al., 2020). Nonetheless, our data demonstrates that a relevant action of miR-221 and miR-222 is in the proinflammatory arm of Th17 response and they act as inflammation-induced negative feedback regulators directly downregulating a signal entry step (IL-23R), as well as constraining a downstream transcription factor (c-Maf).
In a broader context, the current study, which places miR-221 and miR-222 as negative regulators of intestinal inflammatory Th17 response, aligns well with reported action of these miRs in innate cells. In macrophages, miR-221 and miR-222 induce transcriptional silencing of inflammatory genes downstream of LPS signalling, albeit through a different target molecule (Brg1) (Seeley et al., 2018). In this prior report, evaluation of sepsis patients revealed an association between higher expression of these miRs and poor clinical outcome (Seeley et al., 2018). Altogether, miR-221 and miR-222 may serve as a generalizable biomarker of immune response to proinflammatory environment adapted by both innate and adaptive cells. Further study is warranted to pursue action of these miRs in broad array of immune cells as well as their potential as a biomarker of inflammation in various type of clinical settings.
Limitations of the study
This study is limited by its focus on evaluating the action of miR-221 and miR-222 in selected loss-of-function experimental settings and concentrating on cells isolated from the gut in mice. As inflammatory Th17 responses play a role beyond intestine, investigation into other forms of pathological inflammation in the skin or brain will be important to understand the broader impact of miR-221 and miR-222. Moreover, microRNAs typically target numerous gene transcripts, so our analysis undoubtedly misses many other relevant targets in a wide variety of cells in diverse environments. Furthermore, the relevance of miR-221 and miR-222 in human inflammatory bowel diseases has not been addressed in this study.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
John O’Shea (john.oshea@nih.gov).
Material Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John O’Shea, at NIH.
Data and Code Availability
All sequencing data generated during this study are available at Gene Expression Omnibus under the accession number GSE160250. Other source data used in the paper are summarized in supplemental table S1 and analysis pipelines used are listed in Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-K4m1 antibody | Abcam | ab8895; RRID: AB_306847 |
| Anti-K4m3 antibody | Abcam | ab8580; RRID:AB_306649 |
| Anti-K27m3 antibody | Millipore | 07-449; RRID:AB_310624 |
| Anti-p300 antibody | Santa Cruz Biotechnology | sc-585; RRID:AB_2231120 |
| Anti-STAT3 antibody | Thermo Fisher Scientific | 14-6727-81; AB_468247 |
| anti-CD3 antibody | Thermo Fisher Scientific | 16-0031-86; RRID:AB_468849 |
| anti-CD3 antibody | BioxCell | BE0001-1; RRID:AB_1107634 |
| anti-CD28 antibody | Thermo Fisher Scientific | 16-0281-86; RRID:AB_468923 |
| anti-CD28 antibody | BioxCell | BE0015-1; RRID:AB_1107624 |
| anti-IFN-γ antibody | BioXCell | BE0055; RRID:AB_1107694 |
| anti-IL-4 antibody | BioXCell | BE0045; RRID:AB_1107707 |
| anti-CD4 antibody | BD Biosciences | 550954; RRID:AB_393977 |
| anti-CD8 antibody | BD Biosciences | 551162; RRID:AB_394081 |
| anti-CD25 antibody | Thermo Fisher Scientific | 17-0251-81; RRID:AB_469365 |
| anti-CD44 antibody | Thermo Fisher Scientific | 12-0441-81; RRID:AB_465663 |
| anti-CD45.1 antibody | BD Biosciences | 17-0453-81; RRID:AB_469397 |
| anti-CD45.2 antibody | BioLegend | 48-0454-80; RRID:AB_11039533 |
| anti-CD62L antibody | Thermo Fisher Scientific | 25-0621-81; RRID:AB_469632 |
| anti-c-Kit antibody | BD Biosciences | 553354; RRID:AB_394805 |
| anti-Sca-1 antibody | BioLegend | 108112; RRID:AB_313349 |
| anti-Nkp46antibody | Thermo Fisher Scientific | 46-3351-82; RRID:AB_1834441 |
| anti-RORgt antibody | BD Biosciences | 562607; RRID:AB_11153137 |
| anti-T-bet antibody | Thermo Fisher Scientific | 25-5825-82; RRID:AB_11042699 |
| anti-Foxp3 antibody | Thermo Fisher Scientific | 48-5773-82; RRID:AB_1518812 |
| anti-IL-17A antibody | BioLegend | 506922; RRID:AB_2125010 |
| anti-IFN-γ antibody | Thermo Fisher Scientific | 17-7311-81; RRID:AB_469503 |
| anti-IL-4 antibody | BioLegend | 504120; RRID:AB_2562102 |
| anti-IL-13 antibody | Thermo Fisher Scientific | 50-7133-82; RRID:AB_2574279 |
| anti-TCR γ/δ antibody | BioLegend | 118118; RRID:AB_10612756 |
| anti-IL-2 antibody | BioxCell | BE0043-1; RRID:AB_1107705 |
| anti-c-MAF Monoclonal Antibody (sym0F1), PE | Thermo Fisher Scientific | Cat # 12-9855-42; RRID:AB_2572747 |
| Prime Flow c-Maf probe (Assay ID: VB6-14037-PF), Alexa Fluor 750 | Thermo Fisher Scientific | Cat # PF-204 |
| Prime Flow Il23r probe (Assay ID: VB1-17154-PF), Alexa Fluor 647 | Thermo Fisher Scientific | Cat # PF-204 |
| PrimeFlow™ RNA Assay Kit, 40 tests | Thermo Fisher Scientific | Cat # 88-18005-204 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human TGF-β1 | R&D Systems | 240-B-010 |
| Recombinant Mouse IL-1 beta | R&D Systems | 401-ML-005 |
| Recombinant Mouse IL-12 | R&D Systems | 419-ML-010 |
| Recombinant Mouse IL-23 | R&D Systems | 1887-ML-010 |
| Recombinant Mouse IL-6 | R&D Systems | 406-ML-005 |
| Human IL-2 | National Cancer Institute | N/A |
| Dextran sulfate sodium salt | MP Biomedicals | 160110 |
| Critical Commercial Assays | ||
| TruSeq RNA Sample Prep Kit v2 | Illumina | RS-122-2001 |
| TruSeq Small RNA Sample Prep Kit | illumina | RS-200-0012 |
| Luc-Pair Duo-Luciferase Assay Kits 2.0 | GeneCopoeia | LF-001 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher Scientific | A12621 |
| TaqMan Reverse Transcription Kit | Thermo Fisher Scientific | 4366597 |
| TaqMan® Universal PCR Master Mix, No AmpErase® UNG | Thermo Fisher Scientific | 4364341 |
| TRIzol | Thermo Fisher Scientific | 15596018 |
| mirVana miRNA Isolation Kit | Thermo Fisher Scientific | AM1560 |
| Chromium Single Cell 3’ Reagent kits v2 | 10x Genomics | PN-120237 |
| Zombie NIR Fixable Viability kit | Biolegend | Cat #: 423105 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE160250 |
| ChIP-seq samples for p300 and K4me1 (Th1) | Vahedi G, et al., 2012 | GEO: GSE40463 |
| ChIP-seq samples for STAT4 (Th1) | Wei L, et al., 2010 | GEO: GSE22104 |
| ChIP-seq samples for STAT3 and K27me3 (Th17) | Ghoreschi K, et al., 2010 | GEO: GSE23681 |
| ChIP-seq samples for calling super enhancers (Th cells) | Hnisz D, et al., 2013 | GEO: GSE17312 |
| Consensus coding sequence | Pruitt KD, et al., 2009 | Genome Res. 2009 Jul; 19(7): 1316–1323. |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J (WT) | The Jackson Laboratory | #000664 |
| B6.SJL-Ptprca/BoyAiTac (CD45.1+) | Taconic | #4007 |
| C57BL/6-Il17atm1Bcgen/J (Il17a-GFP) | The Jackson Laboratory | #018472 |
| C.B6 (Cg)-Rag2tm1.1Cgn/J (Rag2−/−) | The Jackson Laboratory | #008448 |
| B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | # 017336 |
| CD4-Cre-STAT3fl/fl | Dr. David E. Levy (NYU) | N/A |
| STAT4 KO | Dr. Mark Kaplan (IU) | N/A |
| C57BL/6-Mir221/222 fl/fl | This paper | N/A |
| Mir221/222 KO | This paper | N/A |
| CD4-Cre-mir221/222 fl/fl (conditional KO: cKO) | This paper | N/A |
| Rag2-miR-221/222 (double KO: DKO) | This paper | N/A |
| IL17-GFP miR-221/222 KO | This paper | N/A |
| Oligonucleotides | ||
| TaqMan® miRNA Assay (snoRNA202) | Thermo Fisher Scientific | 4427975-001232 |
| TaqMan® miRNA Assay (hsa-miR-221) | Thermo Fisher Scientific | 4427975-000524 |
| TaqMan® miRNA Assay (hsa-miR-222) | Thermo Fisher Scientific | 4427975-002276 |
| miRIDIAN microRNA Mimic (mmu-miR-221-3p) | Dharmacon | C-310583-07-0005 |
| miRIDIAN microRNA Mimic (mmu-miR-222-3p) | Dharmacon | C-310584-07-0005 |
| miRIDIAN microRNA Mimic Negative Control | Dharmacon | CN-001000-01-05 |
| Recombinant DNA | ||
| miRNA 3' UTR target clones for Maf | GeneCopoeia | MmiT024436-MT06 |
| miRNA 3' UTR target clones for Il23r | GeneCopoeia | MmiT038074-MT06 |
| miRNA Target clone control vector for pEZX-MT06 | GeneCopoeia | CmiT000001-MT06 |
| Software and Algorithms | ||
| BEDTOOLS | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/index.html |
| bedGraphToBigWig | Kent et al., 2010 | https://genome.ucsc.edu/goldenpath/help/bigWig.html |
| wigToBigWig | Kent et al., 2010 | https://genome.ucsc.edu/goldenpath/help/bigWig.html |
| The Integrative Genomics Viewer (IGV) | Thorvaldsdottir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| Bowtie v1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cufflinks v 2.2.1 | Trapnell et al., 2012 | https://github.com/cole-trapnell-lab/cufflinks |
| MACS v 1.4.3 | Y. Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/index.html |
| Seurat R package 3.1.5 | Satija et al., 2015 | http://satijalab.org/seurat/ |
| R studio | RStudio | https://www.rstudio.com/ |
| Partek Genome Suite v6.6 | Partek Incorporated. | http://www.partek.com/ |
| Prism software | GraphPad | RRID: SCR_002798 |
| Cytoscape 3.6.0 | Nepusz et al., 2012 | RRID: SCR_003032 |
| ClueGO 2.5.0 | Bindea et al., 2009 | RRID: SCR_005748 |
| FlowJo v 9, v 10 | BD Biosciences | RRID: SCR_008520 |
| Other | ||
| NGS data generated in this study | GEO | GSE160250 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were performed in the AAALAC-accredited animal housing facilities at NIH. All animal studies were performed according to the NIH guidelines for the use and care of live animals and were approved by the Institutional Animal Care and Use Committee of NIAMS. Mice of 6 -12 weeks old were used in all experiments. For sample size, see corresponding figure legends.
METHOD DETAILS
Mice
C57BL/6J, B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ, C.B6 (Cg)-Rag2tm1.1Cgn/J (Rag2-KO), and C57BL/6-Il17atm1Bcgen/J (Il17a-GFP) were purchased from Jackson Laboratory. B6.SJLPtprca/BoyAiTac (CD45.1+) were purchased from Taconic. Stat3fl/fl mice were from Dr. David Levy (Lee et al., 2002) and bred with CD4-Cre Tg mice. Stat4 KO mice were from Dr. Mark Kaplan (Indiana University). C57BL/6-Mir221Mir222 fl/fl mice and miR-221/222-KO (Mir221Mir222 KO) mice were generated as described in Fig.2A and Fig.S2A. C57BL/6-Mir221Mir222 fl/fl mice were bred with CD4-Cre to generate miR-221/222 conditional KO (cKO). Germline Mir221Mir222 KO mice were bred with Rag2-KO to generate Rag2-miR-221/222 double KO mice. Il17a-GFP mice were bred with Mir221Mir222 KO to generate IL17-GFP-miR-221/222-KO.
For some experiments, WT (Mir221Mir222 fl/fl) and miR-221/222 KO mice were cohoused for at least 12 weeks starting at 3-4 weeks old so that microbiota of the gut is shared among them. Cohoused mice were used for the following experiments (Fig.3A-C, Fig.5C-E).
For generation of bone marrow chimera mice Rag2-KO recipient mice were conditioned with 450 Rads prior to injection of 3 million donor BM cells (CD45.1+ and miR-221/222-KO). Mice were then fed Trimethoprim/Sulfamethoxazole antibiotics via drinking water for 5 weeks.
Preparation of cell suspensions from tissues
All cells were cultured in RPMI medium with 10% (vol/vol) FCS, 2 mM glutamine, 100 IU/mL of penicillin, 0.1 mg/mL, of streptomycin and 20 mM HEPES buffer, pH 7.2-7.5, 1 mM sodium pyruvate, nonessential amino acids (all from Thermo Fisher Scientific), and 2 μM β–mercaptoethanol (Sigma-Aldrich).
Cells from bone marrow, liver, lymph node and spleen were obtained by mechanical disruption. Cells from intestinal lamina propria were isolated after incubating fine-cut intestine in HBSS solution with 0.5 mg/ml DNase I (10104159001, Sigma-Aldrich) and 0.25 mg/ml Liberase TL (05401020001, Sigma-Aldrich) followed by filtering with 100 μm cell strainer and purification with 40% Percoll (6505, GE) (Sciumè et al., 2012). Isolated cells were subjected to sorting CD4 T cells and ILCs by using FACS Aria III, FACSAria or Fusion (BD).
Cell culture
CD4+ T cells from spleens and/or lymph nodes of 6- to 12-week-old mice were purified by negative selection and magnetic separation (Miltenyi Biotec) followed by sorting of naïve CD4+CD62L+CD44−CD25− population using FACSAria III or FACSAria Fusion (BD). Naïve CD4+ T cells were activated by plate-bound anti-CD3 (10 μg/mL, Clone: 145-2C11) and anti-CD28 (10μg/mL, 37.51) in media for 3 days with 5 different conditions. (1) Th17(β) cell-polarization with IL-6 (20 ng ml−1, R&D Systems), human TGF-β1 (2.5 ng ml−1, R&D Systems), anti-IFN-γ neutralizing antibodies (10 μg ml−1, BioXCell), and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell); (2) Th17(23) cell-polarization with IL-6 (20 ng ml−1, R&D Systems), IL-23 (50 ng ml−1, R&D Systems), anti-IFN-γ neutralizing antibodies (10 μg ml−1, BioXCell), and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell); (3) Th17 cell-full polarization with IL-6 (20 ng ml−1, R&D Systems), IL-23 (50 ng ml−1, R&D Systems), IL-1β (20 ng ml−1, R&D Systems), human TGF-β1 (2.5 ng ml−1, R&D Systems), anti-IL-2 neutralizing antibodies (10 μg ml−1, BioXCell), anti-IFN-γ neutralizing antibodies (10 μg ml−1, BioXCell), and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell); (4) Th1 cell-polarization with IL-12 (20 ng ml−1, R&D Systems) and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell).; (5) iTreg cell-polarization with human TGF-β1 (2.5 ng ml−1, R&D Systems), human IL-2 (100 IU ml−1, National Cancer Institute), anti-IFN-γ neutralizing antibodies (10 μg ml−1, BioXCell), and anti-IL-4 neutralizing antibodies (10 μg ml−1, BioXCell).
Flow cytometry
Flow cytometry analysis and sorting was performed on a FACSVerse, FACSAria III, FACSAria Fusion (BD) or Cytek Aurora (Cytek Biosciences). Acquired data were analyzed with FlowJo software (TreeStar). For cell surface staining, the following anti–mouse antibodies were used: anti-CD4 (GK1.5 or RM4-5), anti-CD8 (53-6.7), anti-CD25 (PC61.5), anti-CD44 (IM7), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD45 (30-F11), anti-CD62L (MEL-14), anti-NKp46 (29A1.4), and anti-TCR-β (H57-597). For intracellular cytokine and transcription factor staining, cells were fixed and permeabilized with Foxp3 / Transcription Factor Staining Buffer Set (Thermo Fisher Scientific), and were stained with anti-RORγt (Q31-378), anti-T-bet (eBio4B10), anti-Foxp3 (FJK-16s), anti-IL-17A (TC11-18H10.1 or eBio17B7), anti-IFN-γ (XMG1.2), and anti-c-Maf (sym0F1). Zombie NIR Fixable Viability kit (Biolegend) was used according to the manufacturer’s protocol to exclude dead cells from analysis.
Library preparation for RNA Sequencing (small RNA-seq and mRNA-seq)
RNA-seq and small RNA-seq was performed and analyzed as described previously (Kuchen et al., 2010; Shih et al., 2016). Total RNA was prepared from approximately 1 million cells by using TRIzol or mirVana miRNA Isolation Kit (Thermo Fisher Scientific Inc.) and 200 ng or 1000 ng of total RNA were used to prepare libraries for RNA-seq (with TruSeq SR RNA sample prep kit (FC-122-1001, Illumina)) or small RNA-seq (with TruSeq Small RNA Sample Prep Kit (RS-200-0012, Illumina)) respectively by following manufacturer’s protocol. The libraries were sequenced for 50 cycles (single read) with a HiSeq 2000 or HiSeq 2500 (Illumina).
Library preparation for Single cell RNA-sequencing (scRNA-seq)
For scRNA-seq analysis, Th cells were isolated from small intestinal lamina propria of Il17agfp/− x Mir221Mir222wt/Y (IL17-GFP/WT) and Il17agfp/− x Mir221Mir222 KO/Y (IL17-GFP/miR-221/222-KO) male mice. Th17 cells (Il17-gfp+ CD4+ TCRβ+ CD44+ CD62L−) were purified by FACS sorting by FACSAria III or FACSAria Fusion (BD) and scRNA-seq libraries were prepared by using Chromium Single Cell 3’ Reagent Kits v2 (10x Genomics) by following manufacturer’s protocol. The libraries were sequenced on an Illumina HiSeq 3000 (Illumina, San Diego, CA).
Chromatin immunoprecipitation sequencing (ChIP-seq)
Cells cultured under indicated conditions were cross-linked for 10 minutes with 1% formaldehyde and harvested. Cells were lysed by sonication and immunoprecipitated with anti-H3K4me1 (ab8895, AbCam), anti-H3K4me3 (ab8580, AbCam), anti-H3K27me3 (07-449, Millipore), anti-STAT3 (14-6727, eBiosciences), and anti-p300 (sc-585, Santa Cruz Biotechnology) antibodies as previously described (Shih et al., 2016). Recovered DNA fragments were blunt-end ligated to the Illumina adaptors, amplified, and sequenced by using Genome Analyzer (Illumina, San Diego, CA).
RT-qPCR
Quantification of miRNA expression was performed as previously described (Takahashi et al., 2012). Total RNA was prepared by using TRIzol or mirVana miRNA Isolation Kit (Thermo Fisher Scientific). For reverse transcription and quantification of miRNA, TaqMan Reverse Transcription Kit was used in combination with TaqMan miRNA assays for snoRNA202 and hsa-miR-221 and -222 (Thermo Fisher Scientific Inc.). Results were properly normalized to snoRNA202 levels.
Luciferase assay
The vectors carrying 3' UTR of Maf or Il23r cloned into a firefly/Renilla Duo-Luciferase reporter vector (pEZX-MT06) (GeneCopoeia) were transfected in HEK293T cells with miR-221/222 mimics or a control mimic (Dharmacon) by Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific). Firefly luciferase and Renilla luciferase activity were measured with Luc-Pair Duo-Luciferase Assay Kits 2.0 (GeneCopoeia).
DSS-induced colitis
Mice were received 2.0 % dextran sulfate sodium salt (MW 36-50kD, 160110, MP Biomedicals) dissolved in sterile distilled water ad libitum for 7 days followed by regular drinking water for 3 days. Three sets of WT and miR-221/222 KO mice were tested; (1) Mir221Mir222 fl/fl mice (n=4) and germline Mir221Mir222 KO (n=5); (2) Mir221Mir222 fl/fl mice (n=6) and CD4-Cre Mir221Mir222 fl/fl(n=8); (3) Rag2-KO (n=5) and Rag2 Mir221Mir222 DKO (n=6)
Histology
Isolated small intestines from Mir221Mir222fl/fl or Mir22Mir/222 KO were flushed with ice cold PBS and divided into sections corresponding to duodenum, jejunum and ileum. Each section was opened longitudinally, then rolled into a ‘Swiss roll’, fixed with 10% neutral formalin solution. Samples were processed for paraffin sections and stained with H&E or periodic acid-Schiff (PAS) at Histoserv (Germantown, MD). Intestines were evaluated by an investigator with experimental conditions masked, using the following criteria; 0: no visible infiltrate; 0.5: infiltrate in <10% of sections; 1: infiltrate in <25% of sections; 1.5: infiltrate in <35% of sections; 2: infiltrate in <50% of sections.
Microbiome analysis
Microbiota composition of feces was determined by 16S rRNA analysis (ZymoBIOMICS Services) as briefly described in the followings. DNA was extracted from fecal samples stabilized in DNA/RNA Shield (Zymo Research, CA, US) with ZR Fecal DNA Miniprep (Zymo Research) according to the manufacturer’s protocol. Bacterial 16S ribosomal RNA gene targeted sequencing was performed as previously described with slight modification (Kozich et al., 2013). The general Bacterial 16S primers, 341f (CCTACGGGNGGCWGCAG) and 805r (GACTACHVGGGTATCTAATCC), were used to amplify the v3-4 region of the 16S rRNA gene. PCR amplicons were purified using Select-a-Size DNA Clean & Concentrator (Zymo Research). The 16S rRNA was sequenced using Illumina MiSeq with v2 reagent kit (500 cycles, with 10% PhiX mix, and in paired-end mode). Reference sequences were obtained from the workflow of pick_open_reference_otus.py using SILVA (v. 123) (Quast et al., 2013). Taxa that have an abundance significantly different among groups were identified by LEfSe with default settings (p>0.05 and LDA effect size >2) (Segata et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
mRNA-seq analysis
Raw sequencing data were processed with CASAVA 1.8.2 to generate FastQ files. Sequence reads for RNA-seq were mapped onto the mouse genome build mm9 using TopHat 2.1.0. Gene expression values (FPKM, fragments per kilobase exon per million mapped reads) were calculated with Cufflinks 2.2.1. (“Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks.,” 2012)).
microRNA-seq analysis
For small RNA-seq, 5’ 19 base sequence reads were mapped onto mm9 with Bowtie (0.12.8) (Langmead et al., 2009), allowing no mismatch. Gene expression values (RPKM, reads per kilobase exon per million mapped reads) were calculated by Partek Genomics Suite (6.6/6.14.0514). BigWig tracks were generated from Bam files and converted into Bedgraph format using BEDTOOL. These were further reformatted with the UCSC tool bedGraphToBigWig. We used genes that are included in consensus coding sequences set (released: 8/27/2017)(The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes) and are expressed over 10 FPKM in at least one condition. The differential gene expression was calculated by Partek Genomics Suite (6.6/6.14.0514).
scRNA-seq analysis
10x Genomics CellRanger and Illumina Bcl2fastq software were used to demultiplex and generate FastQ files. CellRanger and STAR were used to align to mouse mm10 genome. CellRanger was used to generate counts and output matrix files were loaded into R for analysis using the Seurat 3.1.5 package. Low-quality cells were filtered out based on >5% mitochondrial gene expression and number of genes per cell < 200 or > 2500. Data were integrated, normalized, and transformed for downstream analysis. Cells were clustered using Seurat’s graph-based clustering and visualized using UMAP. Gene expression was subsequently determined in each cluster. Data were visualized using ggplot2 3.3.0.
ChIP-seq analysis
We aligned ChIP-seq reads to the mouse genome (build mm9) with Bowtie (v0.12.8) (Langmead et al., 2009), allowing two mismatches. We then identified peaks using MACS (v 1.4.2; default p-value threshold of 1E-5) (Zhang et al., 2008). We visualized each ChIP-seq dataset by counting positional coverage across the genome (BEDTOOLS v2.24) (Quinlan and Hall, 2010), reformatting to bigWig (bedGraphToBigWig), and viewing in IGV (Thorvaldsdottir et al., 2013).
Gene Set Enrichment Analysis (GSEA)
GSEA from the Massachusetts Institute of Technology (www.broad.mit.edu/gsea) was used. For analyzing in vivo Th cells derived from bone marrow chimera experiment, we first extracted gene signatures from public data for the followings; (1) Th17 (Ciofani, 2012), (2) human IL-10+ Th17 (Aschenbrenner, 2019), (3) human IL-10− Th17 (Aschenbrenner, 2019), (4) c-Maf-induced (Ciofani, 2012), (5) Th2 (Ranzani, 2015) (also see Table S2), then run GSEA analyses for enrichment against gene expression profiles of WT and miR-221/222 KO gut CD4+ T cells (Fig.5F and S5A).
Statistics
For calculation of comparison between groups, unpaired t test was used. Values about WT and miR-221/222 KO CD4+ T cells within each mouse were compared using the paired t test (Fig.4C, D, Fig.S4). P-values and Log2 fold change (Log2 ((mean WT FPKM)+1)/((mean KO FPKM)+1) ) was projected to a volcano plot (Fig.S4). In all bar graphs, error bars represent SEM. Statistical analysis was performed using the Prism software (GraphPad). P values less than 0.05 were considered significantly different.
DATA AND SOFTWARE AVAILABILITY
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number 160250.
Supplementary Material
Table S1. Deep sequencing data list used in the study. Related to Figures 1, S1, S2, 3, S3, 4 and S4.
Table S2. Gene list of modules used for GSEA analysis. Related to Figure 4.
Highlights:
miR-221/222 are induced by proinflammatory cytokines and repressed by TGF-β
Intestinal Th17 homeostasis is maintained without miR-221/222 in healthy hosts
miR-221/222 target Maf and Il23r to constrain IL-23-induced Th17 cell response
T cell-dependent miR-221/222 are protective against DSS-induced mucosal damage
Acknowledgements
We thank J. Simone, J. Lay, K. Tinsley (NIAMS) for flow cytometry, G. Gutierrez-Cruz, S. Dell’Orso, F. Naz (NIAMS) for deep sequencing, K. Jiang and Dimitrios Anastasakis (NIAMS) for bioinformatics support, Ivan Fuss (NIAID) for histopathologic scoring and the NIAMS LACU staff and Y. Morimoto (NIAMS) for their technical support to handle mice. We thank Vanja Lazarevic (NCI) and Mark Ansel (UCSF) for critical reading, the members of the O’Shea laboratory for helpful discussions and Tomoya Kanno for graphics. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH. This work was supported by the Intramural Research Programs of NIAMS and NIAID. Y.M. was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid (B) 20H03666 and Japanese Researcher Fellowships at NIH. R.L.P. was supported by a Postdoctoral Research Associate (PRAT) fellowship from the National Institute of General Medical Sciences (NIGMS), award number 1Fi2GM137942-01.
Footnotes
Declaration of Interests The authors declare no competing financial interests. John O’Shea is a member of the advisory board of Immunity.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers. All sequencing data have been deposited to Gene Expression Omnibus under the accession number GSE160250.
References
- Aschenbrenner D, Foglierini M, Jarrossay D, Hu D, Weiner HL, Kuchroo VK, Lanzavecchia A, Notarbartolo S, Sallusto F, 2018. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat Immunol 19, 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, 2018. Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumjohann D, Ansel KM, 2013. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol 13, 666–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho I-C, Sharpe AH, Kuchroo VK, 2009. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol 10, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, Marks DS, Sander C, 2008. The microRNA.org resource: targets and expression. Nucleic Acids Res 36, D149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Bonneau R, Littman DR, 2012. A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BS, Hertweck A, Smith J, O'Connor E, Graf D, Cook T, Smale ST, Sakaguchi S, Livesey FJ, Fisher AG, Merkenschlager M, 2006. A role for Dicer in immune regulation. J Exp Med 203, 2519–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD, 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748. [DOI] [PubMed] [Google Scholar]
- Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, Li Z, Wu Z, Pei G, 2009. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 10, 1252–1259. [DOI] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH, 2006. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, Bhairavabhotla RK, Northrup D, Zahr R, Burr P, Liu X, Zhao K, Sher A, Zhu J, Muljo SA, 2014. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity 40, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O'Connor W, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, 2011. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, Cua DJ, 2014. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G, 2012. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr. Mol. Med 12, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H, Regev A, 2015. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 163, 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun H-W, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ, 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciumè G, Laurence A, Sun H-W, Brooks SR, Vahedi G, Shih H-Y, Gutierrez-Cruz G, Iwata S, Suzuki R, MIKAMI Y, Okamoto Y, Nakayama T, Holland SM, Hunter CA, Kanno Y, O’Shea JJ, 2015. Asymmetric Action of STAT Transcription Factors Drives Transcriptional Outputs and Cytokine Specificity. Immunity 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Gonzalez-Martin A, Kim B-S, Jin HY, Jin W, Xu W, Sabouri-Ghomi M, Xu S, Zheng P, Xiao C, Dong C, 2016. The MicroRNA-183-96-182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity 44, 1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbratta C, Hussein H, Andris F, Verdeil G, 2020. c-MAF, a Swiss Army Knife for Tolerance in Lymphocytes. Front. Immunol 11, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR, 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ, 1996. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382, 174–177. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK, 2009. IL-17 and Th17 Cells. Annu. Rev. Immunol 27, 485–517. [DOI] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD, 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, Wei L, Laurence A, Yasuda T, Peng S, Hu-Li J, Lu K, Dubois W, Kitamura Y, Charles N, Sun H-W, Muljo S, Schwartzberg PL, Paul WE, O'Shea J, Rajewsky K, Casellas R, 2010a. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity 32, 828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL, 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ, 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK, 2012. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR, Rudensky AY, 2010. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140, 845–858. [DOI] [PubMed] [Google Scholar]
- Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao J, Jiang W, Zhang F, Li D, Han B, Han R, Qiu R, Huang W, Wang Y, Hao J, 2017. MicroRNA-15b Suppresses Th17 Differentiation and Is Associated with Pathogenesis of Multiple Sclerosis by Targeting O-GlcNAc Transferase. J. Immunol 198, 2626–2639. [DOI] [PubMed] [Google Scholar]
- Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW, 2012. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A 109, E1248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Kahn D, Gibson WSJ, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D, 2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, Li Y, Kelly G, Chakravarty P, Shoaie S, Gutierrez MG, Stockinger B, 2019. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 51, 77–89.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, Paul WE, 2010. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DD, Kuchroo VK, 2015. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 43, 1040–1051. [DOI] [PubMed] [Google Scholar]
- Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, Ackermann JA, Seefried M, Kleyer A, Uderhardt S, Haugg B, Hueber AJ, Daum P, Heidkamp GF, Ge C, Böhm S, Lux A, Schuh W, Magorivska I, Nandakumar KS, Lönnblom E, Becker C, Dudziak D, Wuhrer M, Rombouts Y, Koeleman CA, Toes R, Winkler TH, Holmdahl R, Herrmann M, Blüml S, Nimmerjahn F, Schett G, Kronke G, 2017. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol 18, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO, 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL, 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. [DOI] [PubMed] [Google Scholar]
- Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, Gruarin P, Provasi E, Sugliano E, Marconi M, De Francesco R, Geginat J, Bodega B, Abrignani S, Pagani M, 2015. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol 16, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, Hackney J, Ding J, Singh H, Ouyang W, 2011. Transcription factor c-Maf mediates the TGF-β-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol 12, 1238–1245. [DOI] [PubMed] [Google Scholar]
- Sato K, Miyoshi F, Yokota K, Araki Y, Asanuma Y, Akiyama Y, Yoh K, Takahashi S, Aburatani H, Mimura T, 2011. Marked induction of c-Maf protein during Th17 cell differentiation and its implication in memory Th cell development. J. Biol. Chem 286, 14963–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml BU, Hildner K, Ise W, Lee W-L, Smith WA-E, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, Hatton RD, Stormo GD, Weaver CT, Russell JH, Murphy TL, Murphy KM, 2009. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciumè G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, O’Shea JJ, 2012. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, Freedberg DE, Ghosh S, 2018. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature 13, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C, 2011. Metagenomic biomarker discovery and explanation. Genome biology 12, R60–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H-Y, Sciumè G, Mikami Y, Guo L, Sun H-W, Brooks SR, Urban JF, Davis FP, Kanno Y, O’Shea JJ, 2016. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, Omenetti S, 2017. The dichotomous nature of T helper 17 cells. Nat Rev Immunol 17, 535–544. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciumè G, Muljo SA, Kuchen S, Casellas R, Wei L, Kanno Y, O’Shea JJ, 2012. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol 13, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Suto A, Iwamoto T, Kashiwakuma D, Kagami S-I, Suzuki K, Takatori H, Tamachi T, Hirose K, Suzuki J, Ohara O, Yamashita M, Nakajima H, 2014. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J Exp Med 211, 1857–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP, 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun H-W, Sartorelli V, Kanno Y, O’Shea JJ, 2012. STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld J-C, Stockinger B, 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109. [DOI] [PubMed] [Google Scholar]
- Wei L, Vahedi G, Sun H-W, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, Peng W, O’Shea JJ, Kanno Y, 2010. Discrete Roles of STAT4 and STAT6 Transcription Factors in Tuning Epigenetic Modifications and Transcription during T Helper Cell Differentiation. Immunity 32, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C, 2007. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem 282, 9358–9363. [DOI] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK, Regev A, 2013. Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based analysis of ChIP-Seq (MACS). 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Paul WE, 2010. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol 28, 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberbuehler MK, Parker ME, Wheaton JD, Espinosa JR, Salzler HR, Park E, Ciofani M, 2019. The transcription factor c-Maf is essential for the commitment of IL-17-producing γδ T cells. Nat Immunol 20, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Deep sequencing data list used in the study. Related to Figures 1, S1, S2, 3, S3, 4 and S4.
Table S2. Gene list of modules used for GSEA analysis. Related to Figure 4.
Data Availability Statement
All sequencing data generated during this study are available at Gene Expression Omnibus under the accession number GSE160250. Other source data used in the paper are summarized in supplemental table S1 and analysis pipelines used are listed in Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-K4m1 antibody | Abcam | ab8895; RRID: AB_306847 |
| Anti-K4m3 antibody | Abcam | ab8580; RRID:AB_306649 |
| Anti-K27m3 antibody | Millipore | 07-449; RRID:AB_310624 |
| Anti-p300 antibody | Santa Cruz Biotechnology | sc-585; RRID:AB_2231120 |
| Anti-STAT3 antibody | Thermo Fisher Scientific | 14-6727-81; AB_468247 |
| anti-CD3 antibody | Thermo Fisher Scientific | 16-0031-86; RRID:AB_468849 |
| anti-CD3 antibody | BioxCell | BE0001-1; RRID:AB_1107634 |
| anti-CD28 antibody | Thermo Fisher Scientific | 16-0281-86; RRID:AB_468923 |
| anti-CD28 antibody | BioxCell | BE0015-1; RRID:AB_1107624 |
| anti-IFN-γ antibody | BioXCell | BE0055; RRID:AB_1107694 |
| anti-IL-4 antibody | BioXCell | BE0045; RRID:AB_1107707 |
| anti-CD4 antibody | BD Biosciences | 550954; RRID:AB_393977 |
| anti-CD8 antibody | BD Biosciences | 551162; RRID:AB_394081 |
| anti-CD25 antibody | Thermo Fisher Scientific | 17-0251-81; RRID:AB_469365 |
| anti-CD44 antibody | Thermo Fisher Scientific | 12-0441-81; RRID:AB_465663 |
| anti-CD45.1 antibody | BD Biosciences | 17-0453-81; RRID:AB_469397 |
| anti-CD45.2 antibody | BioLegend | 48-0454-80; RRID:AB_11039533 |
| anti-CD62L antibody | Thermo Fisher Scientific | 25-0621-81; RRID:AB_469632 |
| anti-c-Kit antibody | BD Biosciences | 553354; RRID:AB_394805 |
| anti-Sca-1 antibody | BioLegend | 108112; RRID:AB_313349 |
| anti-Nkp46antibody | Thermo Fisher Scientific | 46-3351-82; RRID:AB_1834441 |
| anti-RORgt antibody | BD Biosciences | 562607; RRID:AB_11153137 |
| anti-T-bet antibody | Thermo Fisher Scientific | 25-5825-82; RRID:AB_11042699 |
| anti-Foxp3 antibody | Thermo Fisher Scientific | 48-5773-82; RRID:AB_1518812 |
| anti-IL-17A antibody | BioLegend | 506922; RRID:AB_2125010 |
| anti-IFN-γ antibody | Thermo Fisher Scientific | 17-7311-81; RRID:AB_469503 |
| anti-IL-4 antibody | BioLegend | 504120; RRID:AB_2562102 |
| anti-IL-13 antibody | Thermo Fisher Scientific | 50-7133-82; RRID:AB_2574279 |
| anti-TCR γ/δ antibody | BioLegend | 118118; RRID:AB_10612756 |
| anti-IL-2 antibody | BioxCell | BE0043-1; RRID:AB_1107705 |
| anti-c-MAF Monoclonal Antibody (sym0F1), PE | Thermo Fisher Scientific | Cat # 12-9855-42; RRID:AB_2572747 |
| Prime Flow c-Maf probe (Assay ID: VB6-14037-PF), Alexa Fluor 750 | Thermo Fisher Scientific | Cat # PF-204 |
| Prime Flow Il23r probe (Assay ID: VB1-17154-PF), Alexa Fluor 647 | Thermo Fisher Scientific | Cat # PF-204 |
| PrimeFlow™ RNA Assay Kit, 40 tests | Thermo Fisher Scientific | Cat # 88-18005-204 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human TGF-β1 | R&D Systems | 240-B-010 |
| Recombinant Mouse IL-1 beta | R&D Systems | 401-ML-005 |
| Recombinant Mouse IL-12 | R&D Systems | 419-ML-010 |
| Recombinant Mouse IL-23 | R&D Systems | 1887-ML-010 |
| Recombinant Mouse IL-6 | R&D Systems | 406-ML-005 |
| Human IL-2 | National Cancer Institute | N/A |
| Dextran sulfate sodium salt | MP Biomedicals | 160110 |
| Critical Commercial Assays | ||
| TruSeq RNA Sample Prep Kit v2 | Illumina | RS-122-2001 |
| TruSeq Small RNA Sample Prep Kit | illumina | RS-200-0012 |
| Luc-Pair Duo-Luciferase Assay Kits 2.0 | GeneCopoeia | LF-001 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher Scientific | A12621 |
| TaqMan Reverse Transcription Kit | Thermo Fisher Scientific | 4366597 |
| TaqMan® Universal PCR Master Mix, No AmpErase® UNG | Thermo Fisher Scientific | 4364341 |
| TRIzol | Thermo Fisher Scientific | 15596018 |
| mirVana miRNA Isolation Kit | Thermo Fisher Scientific | AM1560 |
| Chromium Single Cell 3’ Reagent kits v2 | 10x Genomics | PN-120237 |
| Zombie NIR Fixable Viability kit | Biolegend | Cat #: 423105 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE160250 |
| ChIP-seq samples for p300 and K4me1 (Th1) | Vahedi G, et al., 2012 | GEO: GSE40463 |
| ChIP-seq samples for STAT4 (Th1) | Wei L, et al., 2010 | GEO: GSE22104 |
| ChIP-seq samples for STAT3 and K27me3 (Th17) | Ghoreschi K, et al., 2010 | GEO: GSE23681 |
| ChIP-seq samples for calling super enhancers (Th cells) | Hnisz D, et al., 2013 | GEO: GSE17312 |
| Consensus coding sequence | Pruitt KD, et al., 2009 | Genome Res. 2009 Jul; 19(7): 1316–1323. |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J (WT) | The Jackson Laboratory | #000664 |
| B6.SJL-Ptprca/BoyAiTac (CD45.1+) | Taconic | #4007 |
| C57BL/6-Il17atm1Bcgen/J (Il17a-GFP) | The Jackson Laboratory | #018472 |
| C.B6 (Cg)-Rag2tm1.1Cgn/J (Rag2−/−) | The Jackson Laboratory | #008448 |
| B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | # 017336 |
| CD4-Cre-STAT3fl/fl | Dr. David E. Levy (NYU) | N/A |
| STAT4 KO | Dr. Mark Kaplan (IU) | N/A |
| C57BL/6-Mir221/222 fl/fl | This paper | N/A |
| Mir221/222 KO | This paper | N/A |
| CD4-Cre-mir221/222 fl/fl (conditional KO: cKO) | This paper | N/A |
| Rag2-miR-221/222 (double KO: DKO) | This paper | N/A |
| IL17-GFP miR-221/222 KO | This paper | N/A |
| Oligonucleotides | ||
| TaqMan® miRNA Assay (snoRNA202) | Thermo Fisher Scientific | 4427975-001232 |
| TaqMan® miRNA Assay (hsa-miR-221) | Thermo Fisher Scientific | 4427975-000524 |
| TaqMan® miRNA Assay (hsa-miR-222) | Thermo Fisher Scientific | 4427975-002276 |
| miRIDIAN microRNA Mimic (mmu-miR-221-3p) | Dharmacon | C-310583-07-0005 |
| miRIDIAN microRNA Mimic (mmu-miR-222-3p) | Dharmacon | C-310584-07-0005 |
| miRIDIAN microRNA Mimic Negative Control | Dharmacon | CN-001000-01-05 |
| Recombinant DNA | ||
| miRNA 3' UTR target clones for Maf | GeneCopoeia | MmiT024436-MT06 |
| miRNA 3' UTR target clones for Il23r | GeneCopoeia | MmiT038074-MT06 |
| miRNA Target clone control vector for pEZX-MT06 | GeneCopoeia | CmiT000001-MT06 |
| Software and Algorithms | ||
| BEDTOOLS | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/index.html |
| bedGraphToBigWig | Kent et al., 2010 | https://genome.ucsc.edu/goldenpath/help/bigWig.html |
| wigToBigWig | Kent et al., 2010 | https://genome.ucsc.edu/goldenpath/help/bigWig.html |
| The Integrative Genomics Viewer (IGV) | Thorvaldsdottir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| Bowtie v1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cufflinks v 2.2.1 | Trapnell et al., 2012 | https://github.com/cole-trapnell-lab/cufflinks |
| MACS v 1.4.3 | Y. Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/index.html |
| Seurat R package 3.1.5 | Satija et al., 2015 | http://satijalab.org/seurat/ |
| R studio | RStudio | https://www.rstudio.com/ |
| Partek Genome Suite v6.6 | Partek Incorporated. | http://www.partek.com/ |
| Prism software | GraphPad | RRID: SCR_002798 |
| Cytoscape 3.6.0 | Nepusz et al., 2012 | RRID: SCR_003032 |
| ClueGO 2.5.0 | Bindea et al., 2009 | RRID: SCR_005748 |
| FlowJo v 9, v 10 | BD Biosciences | RRID: SCR_008520 |
| Other | ||
| NGS data generated in this study | GEO | GSE160250 |
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number 160250.