

Abstract
The 7-azaindenoisoquinolines are cytotoxic topoisomerase I (Top1) inhibitors. Previously reported representatives bear a 3-nitro group. The present report documents the replacement of the potentially genotoxic 3-nitro group by 3-chloro and 3-fluoro substituents, resulting in compounds with high Top1 inhibitory activities and potent cytotoxicities in human cancer cell cultures and reduced lethality in an animal model. Some of the new Top1 inhibitors also possess moderate inhibitory activities against tyrosyl-DNA phosphodiesterase 1 (TDP1) and tyrosyl-DNA phosphodiesterase 2 (TDP2), two enzymes that are involved in DNA damage repair resulting from Top1 inhibitors, and they produce significantly more DNA damage in cancer cells than in normal cells. Eighteen of the new compounds had cytotoxicity mean-graph midpoint (MGM) GI50 values in the submicromolar (0.033–0.630 μM) range. Compounds 16b and 17b are the most potent in human cancer cell cultures with MGM GI50 values of 0.063 and 0.033 μM, respectively. Possible binding modes to Top1 and TDP1were investigated by molecular modeling.
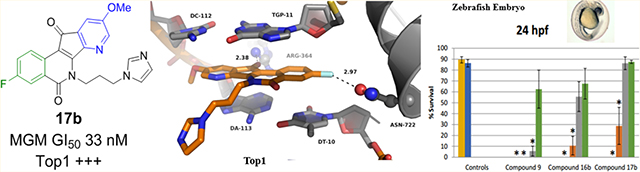
Graphical Abstract
INTRODUCTION
The camptothecin (1) analogues topotecan (2) and irinotecan (3) are topoisomerase I (Top1) inhibitors that are in clinical use as anticancer agents (Figure 1).1 Both of them inhibit the DNA religation reaction in the ternary covalent Top1–DNA–drug complex and are therefore classified as Top1 “poisons” as opposed to Top1 “suppressors”, which inhibit the ability of Top1 to cleave DNA.2 Topotecan (2) is currently used in the treatment of ovarian and lung cancers, while irinotecan (3) is used in colon cancer in combination with other chemotherapies.3,4 Unfortunately, the safety profiles of these compounds are compromised by inherent toxicity. Their side effects such as diarrhea and neutropenia are dose-limiting and may lead to interruption of treatment.3–5 Moreover, these drugs suffer from formulation and administration drawbacks such as limited solubility and the need for prolonged IV infusion to achieve maximal therapeutic effect.5–9 In addition, the emergence of resistance against these camptothecin derivatives is a major problem.6,10 Consequently, significant investments have been expended and are currently being made to develop new Top 1 inhibitors without these disadvantages.11,12
Figure 1.

Top1 inhibitors.
Top1 poisons stabilize the Top1–DNA cleavage complex (Top1cc) by DNA intercalation at the DNA break site and highly specific hydrogen bonding to Top1 amino acid residues in the enzyme active site.2,13,14 For example, in the case of the indenoisoquinolines and their analogues, a key hydrogen bond is formed between the C-11 carbonyl oxygen and the guanidinium group of the Arg364 side chain (Figure 4).2 Collision of the replication fork with the single-strand DNA breaks leads to DNA double-strand breaks and cancer cell death.1 To counteract this, cancer cells possess ubiquitous DNA repair mechanisms that work to reduce the number of Top1-DNA cleavage complexes and religate the broken DNA strands.15–18 DNA ligases cannot work directly on the Top1 peptide–DNA adducts derived from proteolysis of the Top1cc, and the peptide adduct must therefore be removed in order to make DNA a substrate for the ligase.19,20 Tyrosyl-DNA phosphodiesterase 1 (TDP1) performs the first step of the repair mechanism by cleaving the phosphodiester bond between the Top1-derived peptide and DNA.21,22 This limits the cytotoxicity of Top1 poisons.12,20,23–25
Figure 4.

Hypothetical binding mode of compound 16d in the Top1–DNA–drug ternary complex (derived from PDB ID 1K4T). The dashed lines indicate hydrogen or halogen bonding interactions between the ligand and the protein with distances indicated in Å units.
Indenoisoquinolines (e.g., 4–6) have been developed as Top1 inhibitors based on their improved chemical and biological properties compared to the camptothecins.26,27 They have greater chemical stability than the camptothecins, which contain a lactone ring that undergoes facile hydrolysis within minutes at physiological pH. The camptothecin ring-open lactone hydrolysis product is reported to be inactive,28–30 although both the lactone and its hydrolysis product appear in the crystal structure of the ternary topotecan–DNA–Top1 complex.31 In addition, the ternary drug–Top1–DNA–drug complexes induced by indenoisoquinolines have greater stabilities than those induced by camptothecin.9,32 Moreover, the DNA cleavage site specificities of indenoisoquinolines differ from those of camptothecins,9,28 suggesting that different genes and thus tumors could be targeted with indenoisoquinolines.1 Finally, some of the indenoisoquinolines retain activity against Top1 enzymes bearing mutations that confer resistance to camptothecin, and some of them are also active in cancer cells that are resistant to camptothecins because of upregulated ABCG2 efflux transporters.9,33 Compounds 4 (LMP400, aka indotecan) and 5 (LMP776, aka indimitecan)27 have recently completed phase 1 clinical trials at the National Cancer Institute (NCI),34,35 and cancer patients are currently being recruited for a new phase I clinical trial at the NCI involving LMP744 (6).26,36
A recent study involved the systematic incorporation of nitrogen into the eight possible locations within the two aromatic rings of the indenoisoquinoline heterocyclic ring system.37–39 The investigation was driven by the hypothesis that the incorporation of nitrogen would stabilize drug–Top1–DNA ternary complexes through enhanced charge transfer complex formation involving the donation of electron density from the flanking DNA base pairs to the drug. The most potent compounds in this class regarding cytotoxicity and Top1 inhibitory activity bear a nitro group at position 3 and methoxyl group at position 9 as shown in compounds 9 and 10.39 Although nitro groups are present in some clinically used medications, compounds with nitro groups on aromatic systems are not generally included in drug discovery pipelines due to their potential toxicity.40 Aryl nitro groups can be metabolized to toxic aryl nitroso compounds and hydroxylamine sulfates that can covalently modify the proteins and DNA of healthy cells.41 Accordingly, the NCI-60 cytotoxicity screening service recently modified its policy to discourage submission of molecules containing functionalities with known toxicophores, including nitro groups.42 The 3-nitro group in the indenoisoquinoline class of Top1 inhibitors can be replaced with chlorine or fluorine atoms without sacrificing the potent anticancer activity profile (e.g., compounds 7 vs 8).43,44 These results led to the hypothesis that replacement of the nitro group in the 3-nitro-7-azaindenoisoquinolines with chlorine or fluorine atoms would lead to positive results.
In the present work, the two strategies involving nitrogen incorporation and replacement of the 3-nitro group with halogens were combined to produce a new series of compounds in the 7-azaindenoisoquinoline class that bear a chlorine or a fluorine atom in the 3-position instead of a nitro group. In addition, the lactam ω-aminopropyl group was varied by employing ten different amines that had previously displayed good activity in the indenoisoquinoline class (Figure 2).
Figure 2.

Design of 7-azaindenoisoquinolines.
CHEMISTRY
The anhydrides 11a and 11b (Scheme 1) were prepared by published literature procedures.45,46 Bromination of 5-methoxy-3-methylpicolinonitrile in the presence of the radical initiator AIBN produced intermediate bromide 12,38 which was used directly in the next step without additional purification. The condensation of 12 and 11a,b in acetonitrile promoted by Et3N afforded compounds 13a,b.37 Oxidation of 13a,b with selenium dioxide provided azaindenoisoquinoline intermediates 14a,b. Treating compounds 14a,b with NaH in DMF at 0 °C, followed by reaction with 1-chloro-3-bromopropane, yielded the common intermediates 15a,b. The common intermediates 15a,b were used for the synthesis of the final compounds 16a–j and 17a–i by alkylation of the corresponding amines in DMF as shown in Scheme 2.
Scheme 1a.

aReagents and conditions: (a) Et3N, CH3CN, reflux; (b) SeO2, 1,4-dioxane, reflux, 24 h; (c) (i) NaH, DMF, 0 °C to RT, 3 h; (ii) 1-bromo-3-chloropropane, −10 to 0 °C, 24 h.
Scheme 2.

BIOLOGICAL RESULTS AND DISCUSSION
All of the new compounds (16a–j and 17a–i) were tested in a Top1-mediated DNA cleavage assay to assess Top1 poisoning activity.11,47 In addition, they were tested for antiproliferative activity in the NCI-60 human tumor cell line screen.48 The Top1-mediated DNA cleavage assay scores the activity of Top1 poisons with a rubric based on the activity of 1 μM camptothecin. Test agents are incubated at 0.1, 1, 10, and 100 μM concentrations with a 3′-[32P]-labeled double-stranded DNA fragment and Top1 enzyme. Top1 poisons bind to and trap Top1–DNA cleavage complexes. The DNA cleavage pattern is then documented by gel electrophoresis.9,49 Visual comparison of the lanes produced with 1 μM CPT indicates the activity of the new compounds. Finally, a semiquantitative score that ranges from 0 (no activity) to ++++ (activity equal to that of 1 μM CPT) is used to describe the activity of the new compounds (see Table 1 caption for a complete description of the scoring rubric). A representative gel is shown in Figure 3, and the biological activities of the compounds are summarized in Table 1. The new series of compounds showed good to moderate Top1 inhibitory activity. Of the 19 compounds synthesized, compound 16d displayed the best activity with a score of ++++, while five other compounds exhibited good activity with a score of +++. Five compounds displayed weak activity with only a “+” score. Some of the compounds act as Top1 suppressors at high concentration, which likely results from the binding of the drug to the DNA at high drug concentration, making the DNA a poorer substrate for the cleavage reaction (e.g., see the 100 μM lanes for compounds 16b–d in Figure 3).
Table 1.
Top1, TDP1, and TDP2 Inhibitory Activities and Cytotoxicities of 7-Azaindenoisoquinolines
| cytotoxicitya (GI50, μM) |
enzyme inhibitory activity |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | lung HOP-62 | colon HCT-116 | CNS SF-539 | melanoma UACC-62 | ovarian OVCAR-3 | renal SN12C | prostate DU-145 | breast MCF-7 | MGM | Top1b | TDP1c | TDP2c |
| 1 | 0.010 | 0.030 | 0.010 | 0.010 | 0.22 | 0.020 | 0.010 | 0.013 | 0.040 | ++++ | 0 | 0 |
| 7 | 0.19 | 0.274 | 0.016 | 0.012 | 0.864 | 0.015 | 0.017 | 2.17 | 0.370 | ++++ | d | d |
| 8 | 0.186 | 0.107 | 0.204 | 0.074 | 0.407 | 0.110 | 0.309 | 0.031 | 0.229 | +++ | d | d |
| 9 | <0.01 | <0.01 | <0.01 | <0.01 | 0.025 | <0.01 | <0.01 | <0.01 | 0.0208 | ++++(+) | d | d |
| 10 | 0.051 | 0.050 | 0.035 | 0.040 | 0.11 | 0.043 | 0.040 | 0.020 | 0.085 | ++++ | d | d |
| 16a | 0.27 | 0.16 | 0.23 | 0.18 | 0.78 | d | 0.41 | 0.09 | 0.10 | +++ | >111 | >111 |
| 17a | 0.187 | 0.115 | 0.228 | 0.235 | 0.250 | 0.320 | 0.341 | 0.044 | 0.269 | +++ | >111 | >111 |
| 16b | 0.02 | 0.014 | 0.038 | 0.028 | 0.12 | d | 0.043 | <0.01 | 0.063 | +++ | >111 | >111 |
| 17b | <0.01 | <0.01 | 0.024 | 0.016 | 0.043 | <0.01 | <0.01 | <0.01 | 0.033 | +++ | >111 | >111 |
| 16c | 0.042 | 0.35 | 0.47 | 0.095 | 1.3 | d | 0.092 | 0.036 | 0.39 | ++ | 11.9 ± 3.9 | 32.9 ± 5.7 |
| 17c | 0.059 | 0.082 | 0.305 | 0.344 | 0.567 | 0.238 | 0.144 | 0.033 | 0.269 | ++ | >111 | 61.6 ± 14 |
| 16d | 0.094 | 0.098 | 0.43 | 0.19 | 1.42 | d | 0.27 | 0.042 | 0.66 | ++++ | 14 ± 1 | >111 |
| 17d | 0.142 | 0.089 | 0.295 | 0.240 | 0.254 | 0.189 | 0.174 | 0.031 | 0.199 | + | >111 | >111 |
| 16e | 0.20 | 0.298 | 1.33 | 1.43 | 1.43 | 0.489 | 0.242 | 0.089 | 0.58 | + | 20.2 ± 3.7 | >111 |
| 17e | 0.066 | 0.091 | 0.532 | 1.26 | 0.335 | 0.080 | 0.062 | 0.034 | 0.218 | + | >111 | 107 ± 5.2 |
| 16f | 0.070 | 0.138 | 0.554 | 0.303 | 1.39 | 0.417 | 0.173 | 0.037 | 0.380 | ++ | 33.3 ± 4.2 | >111 |
| 17f | 0.107 | 0.093 | 0.248 | 0.271 | 0.190 | 0.164 | 0.210 | 0.031 | 0.165 | ++ | >111 | >111 |
| 16g | 0.220 | 0.550 | 0.730 | 0.574 | 1.41 | 1.18 | 0.331 | 0.046 | 0.60 | ++/+++ | 16 ± 3.2 | >111 |
| 17g | 0.142 | 0.143 | 0.368 | 0.608 | 0.263 | 0.211 | 0.202 | 0.034 | 0.245 | + | >111 | 108 ± 3 |
| 16h | 0.122 | 0.203 | 0.506 | 0.532 | 2.13 | 0.675 | 0.208 | <0.01 | 0.426 | ++/+++ | 22.2 ± 6.4 | >111 |
| 17h | 0.149 | 0.136 | 0.320 | 0.341 | 0.282 | 0.151 | 0.215 | 0.042 | 0.229 | ++ | >111 | 104 |
| 16i | 0.046 | 0.104 | 1.98 | 2.09 | 5.97 | 2.05 | 0.186 | 0.023 | 0.630 | ++ | 27.5 ± 0.6 | >111 |
| 17i | 0.212 | 0.153 | 0.360 | 0.471 | 0.307 | 0.241 | 0.211 | 0.048 | 0.288 | +++ | >111 | >111 |
| 16j | 0.950 | 0.980 | 2.15 | 1.81 | 15.1 | 10.2 | 3.18 | 0.487 | 2.95 | + | >111 | >111 |
The cytotoxicity GI50 values listed are the concentrations corresponding to 50% growth inhibition, and for compounds 8 and 10–16j, the values are the result of single determinations.
Compound-induced DNA cleavage resulting from Top1 inhibition is graded by the following semiquantitative scale relative to 1 μM MJ-III-65 (6) and 1 μM camptothecin (1): 0, no detectable activity; +, weak activity; ++, activity less than that of MJ-III-65 (6); +++, activity equal to that of 6; ++++, activity equipotent to 1.
TDP1 and TDP2 IC50 values (μM) were determined in duplicate.
The data are not available.
Figure 3.

(A) Representative Top1 cleavage assay gel. Top1-mediated DNA cleavage induced by indenoisoquinolines 16a–d. From left to right: lane 1, DNA alone; lane 2, DNA + Top1; lane 3, CPT, 1 μM; lane 4, MJ-III-65 (6, LMP744), 1 μM; Lanes 5–20, compounds 16a–d (each at 0.1, 1.0, 10, and 100 μM). Numbers and arrows on the left indicate cleavage site positions (see Experimental Section). MJ-III-65 is the positive indenoisoquinoline control. (B) Sequence of the 3′-[32P]-labeled 117-bp DNA (labeled guanine with the asterisk) with the indicated Top1 cleavage site positions.9,49
Molecular modeling was used to investigate the possible binding modes of the active compounds in the DNA break site at the interface between Top1 and DNA in the Top1cc. Figure 4 shows the hypothetical binding mode of the most potent halogenated Top1 inhibitor 16d between the DNA base pairs at the DNA cleavage site in the Top1cc. The model was derived by docking and energy minimization of 16d in the topotecan binding site of the topotecan–Top1–DNA ternary complex (PDB ID 1K4T). The 7-azaindenoisoquinoline scaffold is stacked between the base pairs of DNA with a hydrogen bond between the ketone carbonyl and a side chain of Arg364. The chlorine atom is very close to the Asn722 residue suggesting a possible halogen bond between the chlorine atom and the amide oxygen.50 The hydroxyl group of the 4-hydroxypiperidine side chain is involved in a hydrogen bond with the backbone carbonyl oxygen of the Tyr426 residue, which might explain the superior activity of this compound against Top1 (++++). In addition, the hydrophilic nature of this side chain facilitates the positioning and stabilization of the 7-azaindenoisoquinoline system between the DNA base pairs. This model is consistent with the crystal structure of a related indenoisoquinoline–Top1–DNA complex.2
In addition to the Top1 cleavage assay, the compounds were tested against the two tyrosyl–DNA phosphodiesterases (TDP1 and TDP2). As mentioned before, TDP1 is a DNA-repair enzyme that is specifically involved in the recognition and repair of DNA lesions induced by Top1 poisons. It has been argued that adding a TDP1 inhibitor to Top1 poisons would enhance their cytotoxicity and reduce resistance frequency.20 A representative TDP1 inhibition assay electrophoresis gel is shown in Figure 5. In addition, TDP2 is also involved in the DNA repair of damaged DNA caused by stalled topoisomerase II (Top2) cleavage complexes.51 Recent reports have documented that TDP2 can repair DNA damage caused by Top1 in the absence of TDP1.52 This supports the idea that TDP2 inhibitors could also enhance the cytotoxic activity of Top1 poisons. TDP1 and TDP2 inhibitors may also be useful in radiation and antiviral therapies, where DNA breakage with blocking 3′-ends are induced to create a therapeutic effect.53,54 Some of the compounds have moderate activities against both phosphodiesterases while the others have no activity at all. Compound 16c has the best activity against both enzymes with IC50 values of 11.9 μM against TDP1 and 32.9 μM against TDP2. The activity profiles of the new compounds also reveal that compounds with a 3-fluoro substituent have no activity against both phosphodiesterases, with the exception of 17c and 17e, which exhibit very low inhibitory activities vs TDP2 (61.6 and 107 μM, respectively). The data in Table 1 do not indicate a cytotoxicity enhancement due to TDP1 and TDP2 inhibition, which may reflect the moderate potencies of the inhibitors of these two DNA repair enzymes.
Figure 5.

TDP1 assay electrophoresis gel showing conversion of N14Y substrate (single-strand 3′-phosphotyrosine DNA) to tyrosine and N14P product (free 3′-phosphate N14P) in the presence of inhibitors 16g and 16c. Bands (from left to right): N14Y DNA only, N14Y+TDP1, N14Y +TDP1+16g (eight concentrations 0.05, 0.15, 0.46, 1.4, 4.1, 12.3, 37, 111 μM, increasing from left to right as depicted by black wedge), N14Y +TDP1+16c (eight concentrations 0.05, 0.15, 0.46, 1.4, 4.1, 12.3, 37, 111 μM, increasing from left to right as depicted by black wedge).
The active hits were docked in the active site of TDP1 (PDB 1NOP) in order to investigate their binding modes. The hypothetical structure of the most active TDP1 inhibitor 16c bound to TDP1 is shown in Figure 6. The 7-azaindenoisoquinoline scaffold π-stacks with the Phe259 benzene ring. The lactam carbonyl oxygen and the side chain amino group are involved in hydrogen bonding with the Ser518 hydroxyl, an important residue for the binding of the DNA–peptide substrate to the enzyme.55 Finally, the 2-amino-2-thiazoline side chain of the compound is directed toward the catalytic core of the enzyme.
Figure 6.

Hypothetical binding mode of compound 16c to TDP1 (derived from PDB ID 1NOP). The dashed lines indicate hydrogen bonding interactions between the ligand and the protein with distances indicated in Å units.
The NCI-60 human tumor cell line screen provides cytotoxicity data across 60 cancer cell lines originating from human breast, colon, central nervous system (CNS), melanoma, and other tissues.48,56 Compounds that cause significant growth inhibition in the preliminary screening at 10 μM concentration are selected for accurate GI50 determination using five concentrations of the test compound. The five testing concentrations range from 0.01 to 100 μM, and the resulting dose–response curves are used to calculate the concentration required for 50% growth inhibition relative to control, or GI50, for each cell line. Compounds whose GI50 activities fall outside the testing concentration range are recorded as having potencies of 0.01 or 100 μM. The mean GI50 calculated this way for all cell lines tested is called the mean graph midpoint (MGM) GI50.
The activities are listed in Table 1. All of the compounds have submicromolar MGM GI50 values with the exception of compound 16j. Compounds with an imidazole side chain have the best activity in the series with MGM GI50 values of 63 nM for the chloro compound 16b and 33 nM for the fluoro compound 17b. These two compounds have Top1 inhibitory activities of +++. The most active compound against Top1, compound 16d, has an MGM GI50 value of 660 nM. As a general trend, compounds in the 17 (fluoro) series have higher cytotoxicities as represented by their lower MGM GI50 values. The fluorinated compound 17b (MGM 33 nM) has cytotoxicity that is comparable to 9 (MGM 21 nM), documenting that the genotoxic 3-nitro group can be replaced by a 3-fluoro substituent with retention of anticancer potency. The cytotoxicity of the chloro compound 16a (MGM 100 nM) is close to that of the corresponding 3-nitro compound 10 (MGM 85 nM).
This study was based on the prediction that the replacement of the potentially genotoxic 3-nitro group in the 7-azaindenoisoquinoline system with metabolically stable halogens would result in decreased cytotoxicity to normal, noncancer cells that would translate into decreased mammalian toxicity in vivo. In order to access the validity of this prediction, toxicology studies were performed in a zebrafish animal model.57 The zebrafish toxicology studies involved exposure of the fish to four different concentrations of the 3-nitro compound 9, the corresponding 3-chloro compound 16b, and the 3-fluoro analogue 17b in the media, and data were recorded at five different time points (Table 2). The two analogues 16b and 17b were chosen because they are the most cytotoxic of the analogues of 9 in human cancer cell cultures (Table 1). Each determination was carried out with 40 fish with 4 replicates, for a total of 160 animals contributing to each one of the 70 percentages listed in Table 2. As expected, the 3-nitro compound 9 was significantly more lethal than either the 3-chloro compound 16b or the 3-fluoro analogue 17b. Although the numbers listed in Table 2 for compounds 16b and 17b at 0.01 μM concentration suggest that the 3-fluoro compound 17b is less toxic than the 3-chloro compound 16b, a detailed statistical analysis has revealed that the lethality difference between them is not significant. Lastly, general morphology assessment revealed increases in bent tail and yolk sac edema phenotypes for compound 16b at a concentration of 0.1 μM. There were no significant differences in hatching rate among surviving embryos at any time point (data not shown).
Table 2.
Zebrafish Survival Percentages as a Function of Compound Concentration in the Medium and Time after Fertilizationa
| hours after fertilization |
|||||
|---|---|---|---|---|---|
| medium | 24 h | 48 h | 72 h | 96 h | 120 h |
| fish water | 89.4 ± 7.2 | 88.1 ± 6.9 | 87.5 ± 7.9 | 87.5 ± 7.9 | 87.5 ± 7.9 |
| 0.1% DMSO | 86.2 ± 8.5 | 84.4 ± 8.5 | 82.5 ± 8.4 | 82.5 ± 8.4 | 82.5 ± 8.4 |
| 9 (10, μM) | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 9 (1 μM) | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 9 (0.1, μM) | 5.6 ± 11.3* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 9 (0.01, μM) | 62.5 ± 43.7 | 55.6 ± 43.4 | 45.0 ± 52.0* | 45.0 ± 52.0* | 45.0 ± 52.0* |
| 16b (10 μM) | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 16b (1 μM) | 10.6 ± 21.3* | 1.2 ± 2.5* | 1.2 ± 2.5* | 1.2 ± 2.5* | 1.2 ± 2.5* |
| 16b (0.1 μM) | 55.6 ± 34.2 | 54.4 ± 33.3 | 51.9 ± 32.3 | 50.6 ± 31.5 | 50.6 ± 31.5 |
| 16b (0.01, μM) | 67.5 ± 35.2 | 65.6 ± 38.9 | 63.1 ± 37.3 | 61.8 ± 36.5 | 61.9 ± 36.5 |
| 17b (10, μM) | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 17b (1, μM) | 28.8 ± 42.5* | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0* |
| 17b (0.1, μM) | 86.2 ± 14.5 | 62.5 ± 44.3 | 45.6 ± 52.9* | 45.6 ± 52.9* | 45.6 ± 52.9* |
| 17b (0.01, μM) | 87.5 ± 3.5 | 87.5 ± 3.5 | 87.5 ± 3.5 | 87.5 ± 3.5 | 87.5 ± 3.5 |
Each survival percentage is the average based on 40 zebrafish embryos per determination with 4 replicates, so that a total of 160 embryos were assessed to determine each average survival percentage. Chemical exposures were started 1 h after fertilization. The “±” denotes standard deviation, and the asterisks (*) specify p < 0.05 from solvent control.
The biological activities of the compounds in cancer cells versus normal cells were contrasted by comparing human lymphoblastic leukemia CCRF-CEM cells to normal lymphocytes (PBMCs) with respect to their sensitivity to DNA damage induced by camptothecin (1) and three 7-azaindenoisoquinolines 6, 16d, and 17h at two different concentrations (100 and 1 μM). Detection of DNA damage was performed by monitoring the ability of the drugs to induce phosphorylation of histone H2AX on serine 139 (γ-H2AX), which is an established consequence of DNA damage.58 As shown in Figure 7, camptothecin (1) and the azaindenoisoquinolines 6, 16d, and 17h produced significantly more DNA damage in cancer cells as opposed to normal cells after the cells were exposed to the drugs for 2 h. The DNA damage was apparent at both low (100 μM) and high (1 μM) drug concentrations. As is evident in Figure 7, camptothecin (1) and the azaindenoisoquinolines cause significantly more DNA damage in cancer cells vs normal cells, which may reflect upregulation of Top1 in cancer cells vs normal cells.59
Figure 7.

γ-H2AX foci (green) formation in PBMCs (lymphocytes) and acute lymphoblastic leukemia CCRF-CEM cells treated with indenoisoquinolines 1, 6, 16d, and 17b at 100 nM and 1 μM for 2 h. Representative confocal microscopy images are shown. DNA was stained blue with DAPI.
CONCLUSION
Previously optimized 7-azaindenoisoquinoline Top1 inhibitors were very active in cancer cell cultures, but they contained a 3-nitro group that could possibly be reduced metabolically to genotoxic aryl nitroso and hydroxylamine sulfate species that might result in greater toxicity to animals. The present study documents an attempt to replace it while retaining the anticancer activity and reducing in vivo toxicity. The structure–activity relationships within the resulting series of Top1 inhibitors were explored within an array of 19 compounds. Chlorine and fluorine atoms can replace the nitro group with retention of Top1 inhibitory activity and antiproliferative activity in human cancer cell cultures with reduced general toxicity as monitored by lethality in a zebrafish animal model. The contribution of the amino group in the lactam side chain to the biological activity was investigated with ten different terminal amines. Imidazole analogues 16b and 17b possess the highest anticancer activity in the tested series, while the hydroxypiperidine combined with chlorine in position 3 (compound 16d) had the most potent Top1 inhibitory activity at the ++++ level. The molecular modeling study showed the importance of a hydroxypiperidine for optimal Top1 inhibitory activity within this class. Moreover, docking studies provided insight into the importance of the aminothiazole side chain for inhibitory activity against TDP1. The hypothesis that the 3-chloro and 3-fluoro 7-azaindenoisoquilines would be less toxic in vivo than the 3-nitro lead compounds was proven to be correct through zebrafish lethality studies. In addition, γ-H2AX monitoring of DNA damage revealed that camptothecin (1) and the 7-azaindenoisoquinolines produce significantly more DNA damage in an acute lymphoblastic leukemia CCRF-CEM cancer cell line than in a normal PBMC lymphocyte cell line. An important aspect of this study is that it provides new halogenated 7-azaindenoisoquinolines that are selectively cytotoxic to cancer cells without the presence of a nitro group that is likely to confer undesirable toxicity in humans.
EXPERIMENTAL SECTION
General.
Melting points were determined with a Mel-Temp apparatus using capillary tubes and are uncorrected. Proton nuclear magnetic resonance spectra (1H NMR) were recorded using an ARX300 300 MHz Bruker NMR spectrometer. IR spectra were obtained with a PerkinElmer 1600 series FTIR spectrometer. The purities of all of the biologically tested compounds were estimated by HPLC, and in each case, the major peak accounted for ≥95% of the combined total peak area when monitored by a UV detector at 254 nm. HPLC analyses were performed on a Waters 1525 binary HPLC pump/Waters 2487 dual λ absorbance detector system. HPLC analyses were performed on a Sunrise C-18 column with dimensions of 16 × 4.6 cm and 5 μm particle size. Analytical thin-layer chromatography was conducted on Baker-flex silica gel IB2-F plates, and compounds were visualized with UV light at 254 nm. Silica gel flash chromatography was performed using 230–400 mesh silica gel. The anhydrides 11a and 11b and compound 12 were prepared according to the literature and showed similar spectroscopic data.38,45
7-Aza-3-chloro-5,6-dihydro-9-methoxy-5-oxo-11H-indeno[1,2-c]isoquinoline (13a).
5-Methoxy-3-methylpicolinonitrile (1.06 g, 7.2 mmol), NBS (1.41 g, 7.92 mmol), and AIBN (118 mg, 0.72 mmol) were diluted with 1,2-dichloroethane (40 mL), and the mixture was heated at reflux for 24 h. The reaction mixture was filtered, and the filtrate was evaporated to dryness under reduced pressure to give crude 12. The residue was redissolved in acetonitrile (70 mL). 7-Chloroisochroman-1,3-dione (11a, 2.12 g, 10.8 mmol) was added, followed by triethylamine (1.2 mL, 9.36 mmol), and the solution was heated at reflux for 24 h. The hot solution was filtered, and the precipitate was washed with boiling acetonitrile (3 × 40 mL) to provide a gray solid, 13a (698 mg, 32%): mp 258–262 °C. The product was introduced into the next step without additional purification. APCIMS m/z (rel intensity): 299 (MH+, 100).
7-Aza-3-fluoro-5,6-dihydro-9-methoxy-5-oxo-11H-indeno[1,2-c]isoquinoline (13b).
5-Methoxy-3-methylpicolinonitrile (2.09 g, 14.12 mmol), NBS (2.6 g, 14.60 mmol), and AIBN (214 mg, 1.31 mmol) were diluted with CCl4 (100 mL), and the mixture was heated at reflux for 24 h. The reaction mixture was filtered, and the filtrate was evaporated to dryness under reduced pressure to give crude 12. The residue was redissolved in acetonitrile (140 mL). 7-Fluoroisochroman1,3-dione (11b, 2.8 g, 14.26 mmol) was added, followed by triethylamine (1.6 mL, 12.48 mL), and the solution was heated at reflux for 24 h. The hot solution was filtered, and the precipitate was washed with boiling acetonitrile (3 × 60 mL) to provide a gray solid, 13b (1.26 g, 31.6%): mp 304–310 °C. The product was introduced into the next step without additional purification. 1H NMR (300 MHz, DMSO-d6) δ 12.31 (s, 1 H), 8.43–8.17 (m, 2 H), 7.91 (ddd, J = 14.1, 9.2, 4.0 Hz, 3 H), 7.80–7.55 (m, 2 H), 3.93 (s, 3 H). APCIMS m/z (rel intensity) 283.0 (MH+, 100).
7-Aza-3-chloro-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (14a).
7-Aza-3-chloro-5,6-dihydro-9-methoxy-5-oxo-11H-indeno[1,2-c]isoquinoline (13a, 698 mg, 2.34 mmol) and SeO2 (525 mg, 4.68 mmol) were diluted with 1,4-dioxane (40 mL), and the mixture was heated at reflux for 24 h. The reaction mixture was filtered while hot, and the precipitate was washed with hot dioxane (4 × 30 mL). The combined filtrates were evaporated to dryness under reduced pressure to yield 14a (628 mg, 86%) as an orange solid: mp >300 °C. IR (thin film) 3434, 1650, 1481, 1307, 1017 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.38 (d, J = 9.1 Hz, 1 H), 8.25 (d, J = 2.4 Hz, 1 H), 8.10 (d, J = 2.3 Hz, 1 H), 7.87 (dd, J = 9.3, 2.6 Hz, 1 H), 7.54 (d, J = 2.7 Hz, 1 H), 3.92 (s, 3 H). APCIMS m/z (rel intensity): 311.3 (MH+, 100).
7-Aza-3-fluoro-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (14b).
7-Aza-3-fluoro-5,6-dihydro-9-methoxy-5-oxo-11H-indeno-[1,2-c]isoquinoline (13b, 1.26 g, 4.47 mmol) and SeO2 (0.94 g, 8.38 mmol) were diluted with 1,4-dioxane (170 mL), and the mixture was heated at reflux for 24 h. The reaction mixture was filtered while hot, and the precipitate was washed with hot dioxane (3 × 50 mL). The combined filtrate was evaporated to dryness under reduced pressure to yield 14b as an orange solid (0.81 g, 40.5%): mp 331–334 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.41 (dd, J = 8.8, 5.4 Hz, 1 H), 8.24 (d, J = 2.6 Hz, 1 H), 7.85 (dd, J = 9.5, 2.8 Hz, 1 H), 7.72 (dd, J = 10.2, 7.4 Hz, 1 H), 7.52 (d, J = 2.7 Hz, 1 H), 3.93 (s, 3 H). IR (thin film) 2994, 1682, 1612, 1574, 1560, 1537 cm−1. ESIMS m/z (rel intensity) 295.0 (M – H, 100).
3-Chloro-6-(3-chloropropyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (15a).
Sodium hydride (62 mg, 2.5 mmol) and sodium iodide (24 mg, 0.162 mmol) were added to a suspension of 7-aza-3-chloro-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]-isoquinoline (14a, 270 mg, 0.86 mmol) in dry DMF (30 mL) at 0 °C. After the reaction mixture had been warmed to room temperature and stirred for 1.5 h, a dark-red solution formed. The solution was cooled to 0 °C again, and 1-bromo-3-chloropropane (0.4 g, 2.9 mmol) was added. The solution was stirred for 24 h, and the reaction was quenched with water (100 mL), followed by extraction with chloroform (3 × 50 mL). The combined extracts were washed with water (3 × 50 mL) and brine (50 mL), dried with sodium sulfate, and evaporated to dryness under reduced pressure. The residue was triturated with diethyl ether to yield a red solid product (0.09 g, 26%): mp 195–197 °C. IR (thin film) 3062, 1659, 1607, 1483 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 8.6 Hz, 1 H), 8.01 (d, J = 2.1 Hz, 1 H), 7.84 (d, J = 2.7 Hz, 1 H), 7.36 (dd, J = 8.7, 2.2 Hz, 1 H), 7.09 (d, J = 2.8 Hz, 1 H), 4.82–4.67 (m, 2 H), 3.65 (s, 3 H), 3.24 (t, J = 7.0 Hz, 2 H), 2.18–2.03 (m, 2 H). MALDIMS m/z (rel intensity) 389/391 (MH+, 100); HRESIMS calcd for C19H15Cl2N2O3 (MH+) 389.0460, found 389.0452.
6-(3-Chloropropyl)-3-fluoro-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (15b).
Sodium hydride (90 mg, 3.56 mmol) and sodium iodide (26 mg, 0.18 mmol) were added to a suspension of 7-aza-3-fluoro-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c] isoquinoline (14b, 0.536 g, 1.81 mmol) in dry DMF (45 mL) at 0 °C. After the reaction mixture had been warmed to room temperature and stirred for 2 h, a dark-red solution was formed. The solution was cooled to 0 °C again, and 1-bromo-3-chloropropane (726 mg, 2.32 mmol) was added. The solution was stirred for 24 h and quenched with water (150 mL), followed by extraction with ethyl acetate (3 × 70 mL) and brine (1 × 70 mL). The combined extracts were dried with sodium sulfate and evaporated to dryness under reduced pressure. The residue was subjected to column chromatography (silica gel), eluting with hexaneethyl acetate (2:1), to yield the red solid product (0.31 g, 46%): mp 244–248 °C. IR (thin film) 3053, 1701, 1653, 1589, 1549, 1507, 1482, 1435, 1285 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.50 (dd, J = 8.9, 5.4 Hz, 1 H), 8.26 (d, J = 2.7 Hz, 1 H), 7.87 (dd, J = 9.5, 2.7 Hz, 1 H), 7.73 (td, J = 8.8, 2.9 Hz, 1 H), 7.52 (d, J = 2.8 Hz, 1 H), 4.91 (t, J = 7.1 Hz, 2 H), 3.94 (s, 3 H), 3.76 (t, J = 6.7 Hz, 2 H), 2.28–2.15 (m, 2 H). MALDIMS m/z (rel intensity) 373.0 (MH+, 100). HRESIMS calcd for C19H15ClFN2O3 (MH+) 373.0755, found 373.0748.
General Procedures for the Preparation of Compounds 16a–j.
Compound 15a (0.1 mmol), K2CO3 (0.05 g, 1 mmol), NaI (40 mg), and the appropriate amine (10 equiv) were dissolved in DMF (5 mL). The mixture was stirred for 12 h at 90 °C and then cooled to room temperature. Water (10 mL) was added to the reaction mixture, and then the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (3 × 15 mL) and brine (15 mL), dried over Na2SO4, and evaporated under vacuum. The residue was purified using silica gel column chromatography (MeOH–CHCl3, 5:95) to yield compounds 16a–j.
3-Chloro-9-methoxy-6-(3-morpholinopropyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16a).
This compound was isolated as a red powder (0.03 g, 66%): mp 253–256 °C. IR (thin film) 3065, 2941, 2808, 1698, 1661, 1608, 1590, 1562, 1537, 1500 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.47 (d, J = 8.7 Hz, 1 H), 8.26 (d, J = 2.8 Hz, 1 H), 8.13 (d, J = 2.3 Hz, 1 H), 7.88 (dd, J = 8.7, 2.3 Hz, 1 H), 7.59 (d, J = 2.8 Hz, 1 H), 4.83 (m, 2 H), 3.94 (s, 3 H), 3.89 (d, J = 6.2 Hz, 2 H), 3.73–3.56 (m, 3 H), 3.20 (m, 2 H), 3.03 (m, 3 H), 2.20 (m, 2 H). MALDIMS m/z (rel intensity) 440 (MH+, 100). HRESIMS calcd for C23H22ClN3O4 (MH+) 440.1322, found 440.1372. HPLC purity, 96.2% (MeOH–H2O, 85:15).
3-Chloro-6-(3-(1H-Imidazol-1-yl)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16b).
This compound was isolated as an orange-red solid (0.018 g, 41%): mp 284–286 °C. IR (thin film) 2918, 1696, 1673, 1607, 1566, 1535, 1501 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.37 (d, J = 8.7 Hz, 1 H), 8.08 (s, 1 H), 8.04 (d, J = 2.2 Hz, 1 H), 7.78 (d, J = 6.3 Hz, 1 H), 7.62–7.48 (m, 1 H), 7.46 (d, J = 2.7 Hz, 1 H), 7.17–7.01 (m, 1 H), 6.85–6.70 (m, 1 H), 4.71 (m, 2 H), 4.02 (m, 2 H), 2.10 (m, 2 H). MALDIMS m/z (rel intensity) 421 (MH+, 100). HRESIMS calcd for C22H17ClN4O3 (MH+) 421.1068, found 421.1059. HPLC purity, 97.17% (MeOH–H2O, 85:15).
3-Chloro-6-(3-((4,5-dihydrothiazol-2-yl)amino)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16c).
This compound was isolated as a red solid (0.012 g, 25%): mp 295–298 °C. IR (thin film) 2924, 1666, 1605, 1536, 1501 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.50 (d, J = 8.5 Hz, 1 H), 8.26 (d, J = 2.8 Hz, 1 H), 8.15 (d, J = 2.0 Hz, 1 H), 7.90 (d, J = 8.7 Hz, 1 H), 7.62 (d, J = 2.8 Hz, 1 H), 4.85 (m, 2 H), 4.06 (m, 2 H), 3.96 (s, 3 H), 3.88–3.78 (m, 1 H), 3.70–3.62 (m, 2 H), 3.55–3.38 (m, 2 H), 2.09 (m, 2 H). MALDIMS m/z (rel intensity) 457 (MH+, 100), 455. HRESIMS calcd for C22H19ClN4O3S (MH+) 455.0945, found 455.0928. HPLC purity, 100% (MeOH–H2O, 85:15).
3-Chloro-6-(3-(4-hydroxypiperidin-1-yl)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16d).
This compound was isolated as deep red solid (0.018 g, 38%): mp 279–281 °C. IR (thin film) 3320, 2940, 1698, 1666, 1610, 1589, 1563, 1538, 1500 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.44 (d, J = 8.7 Hz, 1 H), 8.24 (d, J = 2.8 Hz, 1 H), 8.11 (d, J = 2.3 Hz, 1 H), 7.84 (dd, J = 8.7, 2.3 Hz, 1 H), 7.53 (d, J = 2.7 Hz, 1 H), 4.87 (m, 2 H), 4.58–4.49 (m, 1 H), 3.92 (s, 3 H), 2.60–2.58 (m, 2 H), 1.90–1.85 (m, 2 H), 1.49–1.42 (m, 2 H), 1.16–1.00 (m, 2 H). MALDIMS m/z (rel intensity) 454 (MH+, 100). HRESIMS calcd for C24H24ClN3O4 (MH+) 454.1534, found 454.1524. HPLC purity, 96.77% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(piperazin-1-yl)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16e).
This compound was isolated as a red powder (0.044 g, 78%): mp 264–268 °C. IR (thin film) 2937, 2808, 2469, 1697, 1669, 1606, 1565, 1536, 1502 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.46 (d, J = 8.7 Hz, 1 H), 8.25 (d, J = 2.7 Hz, 1 H), 8.12 (d, J = 2.3 Hz, 1 H), 7.86 (dd, J = 8.7, 2.3 Hz, 1 H), 7.56 (d, J = 2.8 Hz, 1 H), 4.87 (m, 2 H), 4.10 (m, 2 H), 3.94 (s, 3 H), 2.76 (m, 4 H), 2.35 (m, 4 H), 1.89 (m, 2 H). MALDIMS m/z (rel intensity) 439 (MH+, 100). HRESIMS calcd for C23H23ClN4O3 (MH+) 439.1537, found 439.1525. HPLC purity, 98.56% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(isopropylamino)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16f).
The reaction to prepare this compound was performed in a 45 mL pressure vessel, and the product was isolated as red powder (0.032 g, 60%): mp 250–251 °C. IR (thin film) 3065, 2942, 2782, 1700, 1664, 1609, 1590, 1563, 1539, 1499 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.46 (d, J = 8.6 Hz, 1 H), 8.28 (s, 1 H), 8.13 (s, 1 H), 7.89 (d, J = 8.5 Hz, 1 H), 7.58 (s, 1 H), 4.85 (m, 2 H), 3.95 (s, 3 H), 3.06 (s, 1 H), 2.88 (m, 2 H), 2.03 (m, 2 H), 1.10 (d, J = 5.6 Hz, 6 H). MALDIMS m/z (rel intensity) 412 (MH+, 100). HRESIMS calcd for C22H22ClN3O3 (MH+) 412.1428, found 412.1420. HPLC purity, 100.00% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(methylamino)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16g).
The reaction to prepare this compound was performed in a 45 mL pressure vessel, and the product was isolated as red powder (0.038 g, 77%): mp 274–275 °C. IR (thin film) 2941, 2777, 1699, 1669, 1608, 1565, 1535, 1500 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.48 (d, J = 8.7 Hz, 1 H), 8.29 (d, J = 2.7 Hz, 1 H), 8.15 (d, J = 2.4 Hz, 1 H), 7.90 (d, J = 8.6 Hz, 1 H), 7.60 (d, J = 2.7 Hz, 1 H), 4.85 (m, 2 H), 3.95 (s, 3 H), 3.33 (s, 3 H), 2.91 (m, 2 H), 2.05 (m, 2 H). MALDIMS m/z (rel intensity) 384 (MH+, 100). HRESIMS calcd for C20H18ClN3O3 (MH+) 384.1115, found 384.1108. HPLC purity, 95.81% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(ethylamino)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16h).
The reaction to prepare this compound was performed in a 45 mL pressure vessel, and the product was isolated as red powder (0.04 g, 80%): mp 264–266 °C. IR (thin film) 2945, 2756, 2497, 1710, 1661, 1608, 1566, 1537, 1499 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.47 (d, J = 8.7 Hz, 1 H), 8.28 (d, J = 2.7 Hz, 1 H), 8.14 (d, J = 2.3 Hz, 1 H), 7.88 (dd, J = 8.7, 2.3 Hz, 1 H), 7.57 (d, J = 2.7 Hz, 1 H), 4.84 (d, J = 7.0 Hz, 2 H), 3.96 (s, 3 H), 2.83 (t, J = 7.4 Hz, 2 H), 2.73 (q, J = 7.3 Hz, 2 H), 2.00 (m, 2 H), 1.06 (t, J = 7.2 Hz, 3 H). MALDIMS m/z (rel intensity) 398 (MH+, 100). HRESIMS calcd for C21H20ClN3O3 (MH+) 398.1272, found 398.1258. HPLC purity, 97.07% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(pyrrolidin-1-yl)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16i).
This compound was isolated as red powder (0.045 g, 83%): mp 242–244 °C. IR (thin fim) 3063, 2934, 2787, 1699, 1664, 1608, 1565, 1537, 1501 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.45 (d, J = 8.7 Hz, 1 H), 8.27 (d, J = 2.7 Hz, 1 H), 8.12 (d, J = 2.2 Hz, 1 H), 7.86 (dd, J = 8.7, 2.3 Hz, 1 H), 7.55 (d, J = 2.8 Hz, 1 H), 4.90 (m, 2 H), 4.11 (m, 2 H), 3.95 (s, 3 H), 3.16 (d, J = 5.1 Hz, 4 H), 1.96 (m, 2 H), 1.57 (m, 4 H). MALDIMS m/z (rel intensity) 424 (MH+, 100). HRESIMS calcd for C23H22ClN3O3 (MH+) 424.1428, found 424.1419. HPLC purity, 96.93% (MeOH–H2O, 85:15).
3-Chloro-9-methoxy-6-(3-(4-methylpiperazin-1-yl)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (16j).
This compound was isolated as red powder (0.042 g, 72%): mp 253–255 °C. IR (thin film) 2931, 2789, 1699, 1666, 1607, 1565, 1537, 1501 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 1 H), 8.23 (d, J = 2.8 Hz, 1 H), 8.08 (d, J = 2.2 Hz, 1 H), 7.83 (dd, J = 8.7, 2.3 Hz, 1 H), 7.51 (d, J = 2.7 Hz, 1 H), 4.89 (m, 2 H), 3.93 (s, 3 H), 2.37 (m, 2 H), 2.12 (m, 8 H), 1.96 (s, 3 H), 1.87 (m, 2 H). MALDIMS m/z (rel intensity) 453 (MH+, 100). HRESIMS calcd for C24H25ClN4O3 (MH+) 453.1694, found 453.1686. HPLC purity, 95.88% (MeOH–H2O, 85:15).
General Procedures for Preparation of Compounds 17a–i.
Compound 15b (50 mg, 0.134 mmol), K2CO3 (0.05 g, 1 mmol), NaI (40 mg), and the appropriate amine (10 equiv) were mixed in DMF (5 mL). The mixture was stirred overnight at 90 °C and then cooled to room temperature. Water (30 mL) was added to the reaction flask, and then the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (3 × 15 mL) and brine (15 mL), dried over Na2SO4, and evaporated to give a red residue. The residue was purified by silica gel column chromatography (MeOH–CHCl3, 5:95) to yield compounds 17a–i. Compounds 17f–h were prepared using the same procedures, but the reaction was performed in a 15 mL pressure vessel.
3-Fluoro-9-methoxy-6-(3-morpholinopropyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2- c]isoquinoline-5,11(6H)-dione (17a).
This compound was isolated as a dark red solid (46 mg, 84%): mp 175–178 °C. IR (thin film) 3070, 2856, 2806, 1698, 1658, 1593, 1548, 1511 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.50 (dd, J = 9.0, 5.4 Hz, 1 H), 8.22 (d, J = 2.8 Hz, 1 H), 7.86 (dd, J = 9.5, 2.7 Hz, 1 H), 7.72 (td, J = 8.8, 2.8 Hz, 1 H), 7.52 (d, J = 2.7 Hz, 1 H), 4.88 (t, J = 7.4 Hz, 2 H), 3.94 (s, 3 H), 3.34 (m, 4 H), 2.40 (t, J = 6.4 Hz, 2 H), 2.20 (m, 4 H), 1.91 (d, J = 6.7 Hz, 2 H). MALDI m/z (rel intensity) 424 (MH+, 100). HRMS-ESI m/z MH+ calcd for C23H23FN3O4 424.1673, found 424.1665. HPLC purity: 96.06% (MeOH–H2O, 85:15).
3-Fluoro-6-(3-(1H-imidazol-1-yl)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]-cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17b).
This compound was isolated as a dark red solid (37.5 mg, 69%): mp 176–180 °C. IR (thin film) 3543, 2969, 1707, 1657, 1592, 1572, 1508 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.53 (dd, J = 9.0, 5.4 Hz, 1 H), 8.15 (d, J = 2.8 Hz, 1 H), 7.95–7.69 (m, 2 H), 7.55 (d, J = 2.8 Hz, 1 H), 7.28 (s, 1 H), 6.98 (s, 1 H), 4.81 (t, J = 7.4 Hz, 2 H), 4.14 (t, J = 7.0 Hz, 2 H), 3.95 (s, 3 H), 2.27–2.16 (m, 2 H). MALDI m/z (rel intensity) 405 (MH+, 100). HRMS-ESI m/z MH+ calcd for C22H18FN4O3 405.1363, found 405.1357. HPLC purity: 96.60% (MeOH–H2O, 85:15).
3-Fluoro-6-(3-((4,5-dihydrothiazol-2-yl)amino)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17c).
This compound was isolated as a dark red solid (25 mg, 43%): mp 226–250 °C. IR (thin film) 2937, 1699, 1657, 1593, 1549, 1510 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.56 (d, J = 5.5 Hz, 1 H), 8.25 (d, J = 2.7 Hz, 1 H), 7.90 (d, J = 9.5 Hz, 1 H), 7.79 (d, J = 8.8 Hz, 1 H), 7.59 (d, J = 2.7 Hz, 1 H), 4.85 (m, 2 H), 4.03 (d, J = 7.3 Hz, 2 H), 3.96 (s, 3 H), 3.68 (m, 2 H), 3.51 (d, J = 7.4 Hz, 2 H), 2.09 (m, 2 H). ESIMS m/z (rel intensity) 439 (MH+, 100). HRMS-ESI m/z MH+ calcd for C22H20FN4O3S 439.1240, found 439.1233. HPLC purity: 95.01% (MeOH–H2O, 85:15).
3-Fluoro-6-(3-(4-hydroxypiperidin-1-yl)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17d).
This compound was isolated as a dark red solid (30 mg, 50%): mp 205–209 °C. IR (thin film) 3263, 2942, 1668, 1591, 1551, 1510 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.54–8.44 (m, 1 H), 8.24 (d, J = 2.1 Hz, 1 H), 7.87 (d, J = 6.9 Hz, 1 H), 7.73 (s, 1 H), 7.53 (s, 1 H), 4.88 (m, 2 H), 4.47 (m, 1 H), 3.94 (s, 3 H), 2.57 (m, 1 H), 2.40 (m, 2 H), 1.89 (m, 4 H), 1.50 (m, 2 H), 1.07 (m, 2 H). ESIMS m/z (rel intensity) 437 (MH+, 100). HRMS-ESI m/z MH+ calcd for C24H24FN3O4 437.1989, found 437.1981. HPLC purity: 95.24% (MeOH–H2O, 85:15).
3-Fluoro-9-methoxy-6-(3-(piperazin-1-yl)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17e).
This compound was isolated as a dark red solid (44 mg, 78%): mp 237–239 °C. IR (thin film) 2936, 2808, 1699, 1658, 1593, 1550, 1510 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.51 (dd, J = 9.0, 5.4 Hz, 1 H), 8.23 (d, J = 2.8 Hz, 1 H), 7.86 (dd, J = 9.5, 2.8 Hz, 1 H), 7.73 (td, J = 8.8, 2.8 Hz, 1 H), 7.53 (d, J = 2.8 Hz, 1 H), 4.87 (t, J = 7.3 Hz, 2 H), 3.94 (s, 3 H), 2.69 (m, 4 H), 2.43 (t, J = 6.5 Hz, 2 H), 2.30 (m, 4 H), 1.90 (t, J = 6.9 Hz, 2 H). MALDI m/z (rel intensity) 423 (MH+, 100). HRMS-ESI m/z MH+ calcd for C23H23N4O3F 423.1833, found 423.1824. HPLC purity: 95.28% (MeOH–H2O, 85:15).
3-Fluoro-6-(3-(isopropylamino)propyl)-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17f).
This compound was isolated as a dark red solid (41 mg, 75%): mp 285–292 °C. IR (thin film) 2714, 1705, 1659, 1588, 1552, 1511 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.52 (dd, J = 8.9, 5.5 Hz, 1 H), 8.36–8.22 (m, 2 H), 7.89 (dd, J = 9.4, 2.8 Hz, 1 H), 7.75 (td, J = 8.7, 2.7 Hz, 1 H), 7.57 (t, J = 2.5 Hz, 1 H), 4.85 (t, J = 7.1 Hz, 2 H), 3.95 (s, 3 H), 3.05 (m, 1 H), 2.86 (d, J = 7.8 Hz, 2 H), 2.04 (m, 2 H), 1.10 (d, J = 6.5 Hz, 6 H). MALDI m/z (rel intensity) 396 (MH+, 100). HRMS-ESI m/z MH+ calcd for C22H23FN3O3 396.1724, found 396.1719. HPLC purity: 96.70% (MeOH–H2O, 85:15).
3-Fluoro-9-methoxy-6-(3-(methylamino)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17g).
This compound was isolated as a dark red solid (31 mg, 63%): mp 259–265 °C. IR (thin film) 1667, 1554, 1513, 1484, 1454 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.56 (dd, J = 8.9, 5.3 Hz, 1 H), 8.29 (d, J = 2.7 Hz, 1 H), 7.92 (dd, J = 9.4, 2.7 Hz, 1 H), 7.86–7.75 (m, 1 H), 7.61 (d, J = 2.7 Hz, 1 H), 4.87 (m, 2 H), 3.96 (s, 3 H), 3.00 (t, J = 7.7 Hz, 2 H), 2.54 (s, 3 H), 2.11 (m, 2 H). ESIMS m/z (rel intensity) 368 (MH+, 100). HRMS-ESI m/z MH+ calcd for C20H19FN3O3 368.1411, found 368.1402. HPLC purity: 95.05% (MeOH–H2O, 85:15).
6-(3-(Ethylamino)propyl)-3-fluoro-9-methoxy-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17h).
This compound was isolated as a dark red solid (18.8 mg, 37.1%): mp 286–288 °C. IR (thin film) 3451, 2924, 2851, 2783, 1700, 1671, 1616, 1592, 1573, 1552, 1511 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.55 (dd, J = 9.0, 5.5 Hz, 1 H), 8.27 (d, J = 2.8 Hz, 1 H), 7.91 (dd, J = 9.4, 2.8 Hz, 1 H), 7.84–7.72 (m, 1 H), 7.60 (d, J = 2.8 Hz, 1 H), 4.86 (t, J = 6.9 Hz, 2 H), 3.96 (s, 3 H), 3.04–2.82 (m, 4 H), 2.10 (d, J = 8.2 Hz, 2 H), 1.26–1.08 (m, 3 H). MALDI m/z (rel intensity) 382 (MH+, 100). HRMS-ESI m/z MH+ calcd for C21H21FN3O3 382.1567, found 382.1558. HPLC purity: 95.58% (MeOH–H2O, 85:15).
3-Fluoro-9-methoxy-6-(3-(pyrrolidin-1-yl)propyl)-5H-pyrido[3′,2′:4,5]cyclopenta[1,2-c]isoquinoline-5,11(6H)-dione (17i).
This compound was isolated as a dark red solid (20 mg, 37%): mp 157–162 °C. IR (thin film) 1702, 1663, 1614, 1572, 1550, 1510, 1480, 1446, 1434 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.51 (dd, J = 8.9, 5.4 Hz, 1 H), 8.26 (t, J = 2.6 Hz, 1 H), 7.88 (dd, J = 9.5, 2.8 Hz, 1 H), 7.74 (td, J = 8.7, 2.8 Hz, 1 H), 7.54 (t, J = 2.5 Hz, 1 H), 4.90 (t, J = 6.9 Hz, 2 H), 3.95 (s, 3 H), 2.73 (m, 4 H), 1.98 (m, 3 H), 1.60 (m, 5 H). ESIMS m/z (rel intensity) 408 (MH+, 100). HRMS-ESI m/z MH+ calcd for C27H23FN3O3 408.1724, found 408.1717. HPLC purity: 95.08%. (MeOH–H2O, 85:15).
Topoisomerase I-Mediated DNA Cleavage Reactions.
A 3′-[32P]-labeled 117-bp DNA oligonucleotide was prepared as previously described.49 The oligonucleotide contains previously identified Top1 cleavage sites in 161-bp pBluescript SK(−) phagemid DNA. Approximately 2 nM radiolabeled DNA substrate was incubated with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25 °C for 20 min in the presence of various concentrations of test compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a phosphoimager and ImageQuant software (Molecular Dynamics). Cleavage sites are numbered to reflect actual sites on the 117 bp oligonucleotide.60
Recombinant TDP1 Assay.61,62
A 5′-[32P]-labeled single-stranded DNA oligonucleotide containing a 3′-phosphotyrosine (N14Y)61 was incubated at 1 nM with 10 pM recombinant TDP1 in the absence or presence of inhibitor for 15 min at room temperature in the LMP1 assay buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20.62 Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE with multiple loadings at 12 min intervals. Gels were dried and exposed to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometry analyses were performed using the ImageQuant software (GE Healthcare).
Recombinant TDP2 Assay.63
TDP2 reactions were carried out as described previously with the following modifications. The 18-mer single-stranded oligonucleotide DNA substrate (TY18, α−32P-cordycepin, 3′-labeled) was incubated at 1 nM with 25 pM recombinant human TDP2 in the absence or presence of inhibitor for 15 min at room temperature in the LMP2 assay buffer containing 50 mM Tris-HCl, pH 7.5, 80 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween 20. Reactions were terminated and treated similarly to recombinant TDP1 reactions (see above).
Molecular Modeling.
The PDB files for the X-ray crystal structures of Top1, TDP1, and TDP2 were obtained using the protein data bank codes 1K4T, 1NOP, and 5J3S, respectively. The protein structures were cleaned and inspected for errors and missing residues, hydrogens were added, and the water molecules were deleted. The 7-azaindenoisoquinolines were constructed using ChemBioDraw Ultra 13, saved in SDF file format, and corrected and optimized using Accelry’s Discovery Studio 2.5 software. GOLD 4.1 was used for docking with the default parameters except that the iterations were increased to 300000 and the early termination option was disabled. The centroids of the binding sites were defined by the ligands in the cocrystal structures. The top 10 docking poses per ligand were inspected visually following the docking runs. Energy minimizations were performed for selected ligand poses. The CHARMM force field was utilized within the Accelrys Discovery Studio 2.5 for energy minimization.
Zebrafish Husbandry.
Embryos were obtained from a breeding colony of wild-type AB strain laboratory zebrafish (Danio rerio). Adult zebrafish are maintained in a Z-Mod System (Aquatic Habitats, Apopka, FL) on a 14:10 light–dark cycle. Water is maintained at 28 °C, the pH at 7.0–7.3, and salinity at 470–550 μS conductivity. Fish and aquaria are monitored twice daily. Adult zebrafish are bred in spawning tanks according to established protocols, and embryos are collected immediately after the breeding interval, approximately at the 4–8 cell stage of embryonic development.64,65 The embryos are rinsed and randomly sorted into groups for experimentation. All embryos used in experiments are incubated at 28 °C through 120 h postfertilization (hpf). All protocols were approved by the Purdue University Animal Care and Use Committee, and all fish were treated humanely with regard to prevention and alleviation of suffering.
Zebrafish Acute Developmental Toxicity Assay.
Stock solutions (10 mM) of compounds 9, 16b, and 17b were made by dissolving the chemical powder in DMSO [compound 9 (2.21 mg) was dissolved in DMSO (552.5 μL), compound 16b (2.6 mg) was dissolved in DMSO (604.65 μL), and compound 17b (2.07 mg) was dissolved in DMSO (492.7 μL)]. Serial dilutions where made to create 10, 1, 0.1, and 0.01 μM treatments of each compound in filtered fish aquaria water (FFW) containing 0.1% DMSO (v/v). Controls included a FFW control and a 0.1% DMSO (v/v) solution. The acute toxicity assay was performed similarly to previous experiments.64,66 In short, four biological replicates (n = 4; defined as being started from a different clutch) of 40 embryos (considered as subsamples) per treatment were sorted into Petri dishes and dosed with 20 mL of 10, 1, 0.1, and 0.01 μM for compound 9, compound 16b, and compound 17b, or a FFW or 0.1% DMSO (v/v) control. Mortality, hatching rates, and gross malformations were observed every 24 h through 120 hpf. An analysis of variance (ANOVA) was performed with SAS 94 software (SAS Institute Inc., Cary, NC) for each observational time point to compare the treatment groups. When the outcome was statistically significant, a least significant difference (LSD) test at α = 0.05 was performed to determine differences between treatment groups.
γ-H2AX Detection.
The experiments resulting in Figure 7 were performed as previously described.67
Supplementary Material
ACKNOWLEDGMENTS
This work was facilitated by the National Institutes of Health (NIH) through support with Research Grant P30CA023168. This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. In vitro cytotoxicity testing was performed by the Developmental Therapeutics Program at the National Cancer Institute under Contract NO1-CO-56000.
ABBREVIATIONS USED
- AIBN
azobisisobutyronitrile
- APCIMS
atmospheric pressure chemical ionization mass spectrometry
- CPT
camptothecin
- DMSO-d6
dimethyl sulfoxide-d6
- ESIMS
electrospray ionization mass spectrometry
- hpf
hours post-fertilization
- HRESIMS
high-resolution electrospray ionization mass spectrometry
- HRMS
high resolution mass spectrometry
- MALDIMS
matrix-assisted laser desorption/ionization mass spectrometry
- NBS
N-bromosuccinimide
- Top1
topoisomerase type I
- TDP1
tyrosyl-DNA phosphodiesterase 1
- TDP2
tyrosyl-DNA phosphodiesterase 2
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01870.
Molecular model of compound 16d in the Top1–DNA–drug ternary complex (PDB)
Molecular model for binding mode of compound 16c to TDP1 (PDB)
SMILES molecular strings (CSV)
REFERENCES
- (1).Pommier Y Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- (2).Staker BL; Feese MD; Cushman M; Pommier Y; Zembower D; Stewart L; Burgin AB Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [DOI] [PubMed] [Google Scholar]
- (3).Hu GH; Zekria D; Cai X; Ni XL Current Status of CPT and Its Analogues in the Treatment of Malignancies. Phytochem. Rev. 2015, 14, 429–441. [Google Scholar]
- (4).Armstrong DK Topotecan Dosing Guidelines in Ovarian Cancer: Reduction and Management of Hematologic Toxicity. Oncologist 2004, 9, 33–42. [DOI] [PubMed] [Google Scholar]
- (5).Ewesuedo RB; Ratain MJ Topoisomerase I Inhibitors. Oncologist 1997, 2, 359–364. [PubMed] [Google Scholar]
- (6).Pommier Y DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem. Rev. 2009, 109, 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Masi G; Falcone A; Di Paolo A; Allegrini G; Danesi R; Barbara C; Cupini S; Del Tacca M A Phase I and Pharmacokinetic Study of Irinotecan Given as a 7-Day Continuous Infusion in Metastatic Colorectal Cancer Patients Pretreated with 5-Fluorouracil or Raltitrexed. Clin. Cancer Res. 2004, 10, 1657–1663. [DOI] [PubMed] [Google Scholar]
- (8).Stein SM; Tiersten A; Hochster HS; Blank SV; Pothuri B; Curtin J; Shapira I; Levinson B; Ivy P; Joseph B; Guddati AK; Muggia F A Phase 2 Study of Oxaliplatin Combined With Continuous Infusion Topotecan for Patients With Previously Treated Ovarian Cancer. Int. J. Gyecol. Cancer 2013, 23, 1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Antony S; Jayaraman M; Laco G; Kohlhagen G; Kohn KW; Cushman M; Pommier Y Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003, 63, 7428–7435. [PubMed] [Google Scholar]
- (10).Rasheed ZA; Rubin EH Mechanisms of Resistance to Topoisomerase I-Targeting Drugs. Oncogene 2003, 22, 7296–7304. [DOI] [PubMed] [Google Scholar]
- (11).Pommier Y Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xu Y; Her CT Inhibition of Topoisomerase (DNA) I (TOP1): DNA Damage Repair and Anticancer Therapy. Biomolecules 2015, 5, 1652–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ioanoviciu A; Antony S; Pommier Y; Staker BL; Stewart L; Cushman M Synthesis and Mechanism of Action Studies of a Series of Norindenoisoquinoline Topoisomerase I Poisons Reveal an Inhibitor with a Flipped Orientation in the Ternary DNA-Enzyme-Inhibitor Complex as Determined by X-ray Crystallographic Analysis. J. Med. Chem. 2005, 48, 4803–4814. [DOI] [PubMed] [Google Scholar]
- (14).Marchand C; Antony S; Kohn KW; Cushman M; Ioanoviciu A; Staker BL; Burgin A; Stewart L; Pommier Y A Novel Norindenoisoquinoline Structure Reveals a Common Interfacial Inhibitor Paradigm for Ternary Trapping of the Topoisomerase I-DNA Complex. Mol. Cancer Ther. 2006, 5, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pourquier P; Pilon AA; Kohlhagen G; Mazumder A; Sharma A; Pommier Y Trapping of Mammalian Topoisomerase I and Recombinations Induced by Damaged DNA Containing Nicks or Gaps - Importance of DNA End Phosphorylation and Camptothecin Effects. J. Biol. Chem. 1997, 272, 26441–26447. [DOI] [PubMed] [Google Scholar]
- (16).Pourquier P; Ueng LM; Kohlhagen G; Mazumder A; Gupta M; Kohn KW; Pommier Y Effects of Uracil Incorporation, DNA Mismatches, and Abasic Sites on Cleavage and Religation Activities of Mammalian Topoisomerase I. J. Biol. Chem. 1997, 272, 7792–7796. [DOI] [PubMed] [Google Scholar]
- (17).van Waardenburg RCAM Tyrosyl-DNA Phosphodiesterase I a Critical Survival Factor for Neuronal Development and Homeostasis. J. Neurol. Neuromed. 2016, 1, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Caldecott KW DNA Single-Strand Break Repair and Spinocerebellar Ataxia. Cell 2003, 112, 7–10. [DOI] [PubMed] [Google Scholar]
- (19).Murai J; Huang SYN; Das BB; Dexheimer TS; Takeda S; Pommier Y Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells. J. Biol. Chem. 2012, 287, 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dexheimer TS; Antony S; Marchand C; Pommier Y Tyrosyl-DNA Phosphodiesterase As a Target for Anticancer Therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang SW; Burgin AB; Huizenga BN; Robertson CA; Yao KC; Nash HA A Eukaryotic Enzyme that Can Disjoin Dead-end Covalent Complexes between DNA and Type I Topoisomerases. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Davies DR; Interthal H; Champoux JJ; Hol WGJ The Crystal Structure of Human Tyrosyl-DNA Phosphodiesterase, Tdp1. Structure 2002, 10, 237–248. [DOI] [PubMed] [Google Scholar]
- (23).Nivens MC; Felder T; Galloway AH; Pena MMO; Pouliot JJ; Spencer HT Engineered Resistance to Camptothecin and Antifolates by Retroviral Coexpression of Tyrosyl DNA Phosphodiesterase-I and Thymidylate Synthase. Cancer Chemother. Pharmacol. 2004, 53, 107–115. [DOI] [PubMed] [Google Scholar]
- (24).Fam HK; Walton C; Mitra SA; Chowdhury M; Osborne N; Choi K; Sun G; Wong PCW; O’Sullivan MJ; Turashvili G; Aparicio S; Triche TJ; Bond M; Pallen CJ; Boerkoel CF TDP1 and PARP1 Deficiency Are Cytotoxic to Rhabdomyosarcoma Cells. Mol. Cancer Res. 2013, 11, 1179–1192. [DOI] [PubMed] [Google Scholar]
- (25).Gao R; Das BB; Chatterjee R; Abaan OD; Agama K; Matuo R; Vinson C; Meltzer PS; Pommier Y Epigenetic and Genetic Inactivation of Tyrosyl-DNA-Phosphodiesterase 1 (TDP1) in Human Lung Cancer Cells from the NCI-60 Panel. DNA Repair 2014, 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cushman M; Jayaraman M; Vroman JA; Fukunaga AK; Fox BM; Kohlhagen G; Strumberg D; Pommier Y Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J. Med. Chem. 2000, 43, 3688–3698. [DOI] [PubMed] [Google Scholar]
- (27).Nagarajan M; Morrell A; Ioanoviciu A; Antony S; Kohlhagen G; Agama K; Hollingshead M; Pommier Y; Cushman M Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J. Med. Chem. 2006, 49, 6283–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pommier Y; Cushman M The Indenoisoquinoline Non-camptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol. Cancer Ther. 2009, 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hertzberg RP; Caranfa MJ; Holden KG; Jakas DR; Gallagher G; Mattern MR; Mong S-M; Bartus JO; Johnson RK; Kingsbury WD Modification of the Hydroxy Lactone Ring of Camptothecin: Inhibition of Mammalian Topoisomerase I and Biological Activity. J. Med. Chem. 1989, 32, 715–720. [DOI] [PubMed] [Google Scholar]
- (30).Kingsbury WD; Boehm JC; Jakas DR; Holden KG; Hecht SM; Gallagher G; Caranfa MJ; McCabe FL; Faucette LF; Johnson RK; Hertzberg RP Synthesis of Water-Soluble (Aminoalky1)camptothecin Analogues: Inhibition of Topoisomerase I and Antitumor Activity. J. Med. Chem. 1991, 34, 98–107. [DOI] [PubMed] [Google Scholar]
- (31).Staker BL; Hjerrild K; Feese MD; Behnke CA; Burgin AB; Stewart L The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analog. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kohlhagen G; Paull K; Cushman M; Nagafuji P; Pommier Y Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol. Pharmacol. 1998, 54, 50–58. [DOI] [PubMed] [Google Scholar]
- (33).Antony S; Agama KK; Miao ZH; Takagi K; Wright MH; Robles AI; Varticovski L; Nagarajan M; Morrell A; Cushman M; Pommier Y Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topoisomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007, 67, 10397–10405. [DOI] [PubMed] [Google Scholar]
- (34).Beumer JH; Holleran J; Doroshow J; Chen A; Allen D; Covey J; Tomaszewski J; Pommier Y; Kumar S; Eiseman JL Phase I Pharmacokinetics and Pharmacodynamics of a Novel Indenoisoquinoline Topoisomerase 1 (TOP1) Inhibitor, LMP400, Administered on a Daily x 5 Schedule. Cancer Res. 2014, 74, 4643. [Google Scholar]
- (35).Kummar S; Chen A; Gutierrez M; Pfister TD; Wang LH; Redon C; Bonner WM; Yutzy W; Zhang YP; Kinders RJ; Ji JP; Allen D; Covey JM; Eiseman JL; Holleran JL; Beumer JH; Rubinstein L; Collins J; Tomaszewski J; Parchment R; Pommier Y; Doroshow JH Clinical and Pharmacologic Evaluation of Two Dosing Schedules of Indotecan (LMP400), a Novel Indenoisoquinoline, in Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2016, 78, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Indenoisoquinoline LMP744 in Adults With Relapsed Solid Tumors and Lymphomas. https://clinicaltrials.gov/ct2/show/NCT03030417 (accessed May 15, 2017).
- (37).Kiselev E; Agama K; Pommier Y; Cushman M Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents: A Systematic Study of Structure-Activity Relationships. J. Med. Chem. 2012, 55, 1682–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kiselev E; DeGuire S; Morrell A; Agama K; Dexheimer TS; Pommier Y; Cushman M 7-Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents. J. Med. Chem. 2011, 54, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kiselev E; Sooryakumar D; Agama K; Cushman M; Pommier Y Optimization of the Lactam Side Chain of 7-Azaindenoisoquinoline Topoisomerase I Inhibitors and Mechanism of Action Studies in Cancer Cells. J. Med. Chem. 2014, 57, 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kazius J; McGuire R; Bursi R Derivation and Validation of Toxicophores for Mutagenicity Prediction. J. Med. Chem. 2005, 48, 312–320. [DOI] [PubMed] [Google Scholar]
- (41).Smith GF Designing Drugs to Avoid Toxicity. In Progress in Medicinal Chemistry, Lawton G, Witty DR, Eds.; Elsevier: Amsterdam, 2011; Vol. 50, pp 1–47. [DOI] [PubMed] [Google Scholar]
- (42).https://dtp.cancer.gov/discovery_development/nci-60/guidelines.htm (accessed December 16, 2016).
- (43).Morrell A; Placzek M; Parmley S; Antony S; Dexheimer TS; Pommier Y; Cushman M Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J. Med. Chem. 2007, 50, 4419–4430. [DOI] [PubMed] [Google Scholar]
- (44).Beck DE; Abdelmalak M; Lv W; Reddy PVN; Tender GS; O’Neill E; Agama K; Marchand C; Pommier Y; Cushman M Discovery of Potent Indenoisoquinoline Topoisomerase I Poisons Lacking the 3-Nitro Toxicophore. J. Med. Chem. 2015, 58, 3997–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Randamp D.; Bardiot D.; Blanche E.; Chaltin P.; Koukni M.; Leyssen P.; Neyts J.; Marchand A.; Vliegen I. Novel Inhibitors of Flavivirus Replication. International Patent WO2010/55164 A2, 2010.
- (46).Kang BR; Wang J; Li H; Li Y; Mei QB; Zhang SQ Synthesis and Antitumor Activity Evaluation of 2-Arylisoquinoline1,3(2H,4H)-diones In Vitro and In Vivo. Med. Chem. Res. 2014, 23, 1340–1349. [Google Scholar]
- (47).Nitiss JL; Soans E; Rogojina A; Seth A; Mishina M Topoisomerase Assays. Current Protocols in Pharmacology; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; pp 3.3.1–3.3.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shoemaker RH The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- (49).Dexheimer TS; Pommier Y DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat. Protoc. 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- (51).Pommier Y; Huang SN; Gao R; Das BB; Murai J; Marchand C Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zeng Z; Sharma A; Ju L; Murai J; Umans L; Vermeire L; Pommier Y; Takeda S; Huylebroeck D; Caldecott KW; El-Khamisy SF TDP2 Promotes Repair of Topoisomerase I-Mediated DNA Damage in the Absence of TDP1. Nucleic Acids Res. 2012, 40, 8371–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Huang SYN; Murai J; Dalla Rosa I; Dexheimer TS; Naumova A; Gmeiner WH; Pommier Y TDP1 Repairs Nuclear and Mitochondrial DNA Damage Induced by Chain-terminating Anticancer and Antiviral Nucleoside Analogs. Nucleic Acids Res. 2013, 41, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Menon V; Povirk LF End-processing Nucleases and Phosphodiesterases: An Elite Supporting Cast for the Non-homologous End Joining Pathway of DNA Double-strand Break Repair. DNA Repair 2016, 43, 57–68. [DOI] [PubMed] [Google Scholar]
- (55).Davies DR; Interthal H; Champoux JJ; Hol WGJ Crystal Structure of a Transition State Mimic for Tdp1 Assembled from Vanadate, DNA, and a Topoisomerase I-derived Peptide. Chem. Biol. 2003, 10, 139–147. [DOI] [PubMed] [Google Scholar]
- (56).Reinhold WC; Sunshine M; Varma S; Doroshow JH; Pommier Y Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin. Cancer Res. 2015, 21, 3841–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hill AJ; Teraoka H; Heideman W; Peterson RE Zebrafish As a Model Vertebrate for Investigating Chemical Toxicity. Toxicol. Sci. 2005, 86, 6–19. [DOI] [PubMed] [Google Scholar]
- (58).Bonner WM; Redon CE; Dickey JS; Nakamura AJ; Sedelnikova OA; Solier S; Pommier Y OPINION Gamma H2AX and Cancer. Nat. Rev. Cancer 2008, 8, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Giovanella BC; Stehlin JS; Wall ME; Wani MC; Nicholas AW; Liu LF; Silber R; Potmesil M DNA Topoisomerase-I Targeted Chemotherapy of Human-Colon Cancer in Xenografts. Science 1989, 246, 1046–1048. [DOI] [PubMed] [Google Scholar]
- (60).Antony S; Marchand C; Stephen AG; Thibaut L; Agama KK; Fisher RJ; Pommier Y Novel High-Throughput Electrochemiluminescent Assay for Identification of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors and Characterization of Furamidine (NSC 305831) as an Inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Marchand C; Lea WA; Jadhav A; Dexheimer TS; Austin CP; Inglese J; Pommier Y; Simeonov A Identification of Phosphotyrosine Mimetic Inhibitors of Human Tyrosyl-DNA Phosphodiesterase I by a Novel AlphaScreen High-throughput Assay. Mol. Cancer Ther. 2009, 8, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Nguyen TX; Morrell A; Conda-Sheridan M; Marchand C; Agama K; Bermingam A; Stephen AG; Chergui A; Naumova A; Fisher R; O’Keefe BR; Pommier Y; Cushman M Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J. Med. Chem. 2012, 55, 4457–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Gao R; Huang S.-y. N.; Marchand C; Pommier Y Biochemical Characterization of Human Tyrosyl-DNA Phosphodiesterase 2 (TDP2/TTRAP) A Mg2+/Mn2+-dependent Phosphodiesterase Specific for the Repair of Topoisomerase Cleavage Complexes. J. Biol. Chem. 2012, 287, 30842–30852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Peterson SM; Zhang J; Weber G; Freeman JL Global Gene Expression Analysis Reveals Dynamic and Developmental Stage-Dependent Enrichment of Lead-Induced Neurological Gene Alterations. Environ. Health Perspect. 2011, 119, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th ed.; University of Oregon Press: Eugene, OR, 2007. [Google Scholar]
- (66).Weber GJ; Sepulveda MS; Peterson SM; Lewis SS; Freeman JL Transcriptome Alterations Following Developmental Atrazine Exposure in Zebrafish Are Associated with Disruption of Neuroendocrine and Reproductive System Function, Cell Cycle, and Carcinogenesis. Toxicol. Sci. 2013, 132, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wang P; Elsayed MSA; Plescia CB; Ravji A; Redon CE; Kiselev E; Marchand C; Zeleznik O; Agama K; Pommier Y; Cushman MJ Med. Chem. 2017, 60, 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.