

RXNmapper constructs coherent atom-mapping rules from raw chemical reactions using unsupervised training of neural networks.
Abstract
Humans use different domain languages to represent, explore, and communicate scientific concepts. During the last few hundred years, chemists compiled the language of chemical synthesis inferring a series of “reaction rules” from knowing how atoms rearrange during a chemical transformation, a process called atom-mapping. Atom-mapping is a laborious experimental task and, when tackled with computational methods, requires continuous annotation of chemical reactions and the extension of logically consistent directives. Here, we demonstrate that Transformer Neural Networks learn atom-mapping information between products and reactants without supervision or human labeling. Using the Transformer attention weights, we build a chemically agnostic, attention-guided reaction mapper and extract coherent chemical grammar from unannotated sets of reactions. Our method shows remarkable performance in terms of accuracy and speed, even for strongly imbalanced and chemically complex reactions with nontrivial atom-mapping. It provides the missing link between data-driven and rule-based approaches for numerous chemical reaction tasks.
INTRODUCTION
Humans leverage domain-specific languages to communicate and record a variety of concepts. Every language contains structural patterns that can be formalized as a grammar, i.e., a set of rules that describe how words can be combined to form sentences. Through the use of these rules, it is possible to create an infinite number of comprehensible clauses (knowledge) using a set of domain characteristic elements (words) obeying domain-specific rules (grammar and syntax). When applied to scientific and technical domains, a language is often more a method of computation than a method of communication.
Organic chemistry rules, for instance, have been developed over two centuries, in which experimental observations were translated into a specific language where molecular structures are words and reaction templates the grammar. These grammar rules illustrate the outcome of chemical reactions and are routinely taught using specific diagrammatic representation (Markush representations). More convenient representations like reaction SMILES (1) also exist for information technologies applied to synthesis planning and reaction prediction. In both Markush and SMILES representations, the grammar rules are present as latent knowledge in the historical corpus of raw reaction data.
The digitization of these rules proved to be a successful approach to design modern computer programs (2) aiding chemists in synthetic laboratory tasks. Compiling reaction rules from domain data is tedious, requiring decades of labor hours and challenging to scale. The availability of an automatic and reliable method for annotating how atoms rearrange in chemical reactions, a process known as atom-mapping, could change profoundly the way organic chemistry is currently digitized. However, the process of atom-mapping is an NP-hard problem, dealt with computational technologies since 1970s (3, 4). Most atom-mapping solutions are either structure based (5–10) or optimization based (11–15). The current state of the art is a combination of heuristics, a set of expert-curated rules that precompute candidates for complex reactions, and a graph-theoretical algorithm to generate the final mapping as developed by Jaworski et al. (16). Nonetheless, brittle preprocessing steps, closed-source code, computationally intensive strategies (more than 100 s for some reactions), and the need for expert-curated rules hinder its wider adoption. Most public reaction data come with rule-based Indigo atom-maps (17), which are taken as ground truth for subsequent work (18–23), irrespective of the explicit warnings about atom-maps quality issues (24).
Natural language processing (NLP) models (25) are among the few neural network architectures showing a substantial impact on synthetic chemistry (26) and not relying on atom-mapping algorithms. Their ability to encode latent knowledge from a training set of molecules and reactions represented as text [SMILES (1)] avoids the need to codify the chemical reaction grammar. Molecular Transformer models, a recent addition to the NLP family, are the state of the art for forward reaction prediction tasks, achieving an accuracy higher than 90% (27–30). Understanding the reasons for this performance requires the analysis of the neural network’s hidden weights, which introduces the inherent complexity of interpreting neural networks.
Here, we report the evidence that Transformer encoder models (31, 32) learn atom-mapping as a key signal when trained on unmapped reactions on the self-supervised task of predicting the randomly masked parts in a reaction sequence, a process depicted in Fig. 1A. Transformer architectures can learn the underlying atom-mapping of chemical reactions, without any human labeling or supervision, solely from a large training set of reaction SMILES tokenized by atoms (28, 33). After establishing an attention-guided atom-mapper and introducing a neighbor attention multiplier, we were able to achieve 99.4% correct full atom-mappings on a test set of 49k strongly unbalanced patent reactions (34) with high-quality atom-maps (35).
Fig. 1. Overview.
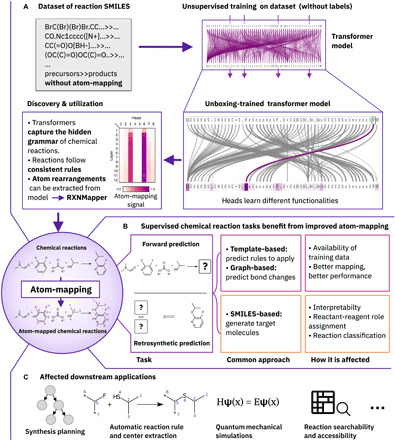
(A) Process that led to the discovery of the atom-mapping signal and ultimately to the development of RXNMapper. (B) Directly affected chemical reaction prediction tasks. (C) Importance of atom-mapping in affected downstream applications.
The advantage of this approach is its unsupervised nature. In contrast to supervised approaches, here, the atom-mapping signal is learned during training as a consistent pattern hidden in the reaction datasets, without ever seeing any example of atom-mapped reactions. As a consequence, the quality of this approach is not limited by the quality of labeled data generated by an existing annotation tool. Moreover, the unsupervised nature allows scaling the extraction of chemical reaction grammar without the need of increasing human resources.
Numerous deep learning methods developed for organic chemistry, like forward and backward reaction prediction, will benefit from better atom-mapping (Fig. 1B). Examples range from template-based approaches that use atom-mapping to automatically extract the templates from chemical reaction datasets (18, 36–38), to graph-based approaches, predicting bond changes or graph edits, that require atom-mapped reactions to extract the labels used for training the models (19, 21). Even the predictions of atom-mapping–independent and template-free SMILES-2-SMILES approaches (28, 33) may benefit from better atom-mapping, thus becoming more transparent and interpretable. In SMILES-2-SMILES approaches, the models generate the product structures sequentially atom-by-atom given the precursors or vice versa, generate the precursors given the product, without any support from atom-mapping information. After adding the atom-mapping in a postprocessing step, predictions can be linked back to training reactions with the same reaction template. The atom-maps also enable the use of quantum mechanical simulations to compute reaction energies and the mechanism without human intervention by providing the corresponding atom pairs between precursors and products.
Moreover, our contributions will lead to improvements in the downstream applications that depend on better atom-mapping and chemical reaction rules (Fig. 1C): retrosynthesis planning methods (36, 38, 39), chemical reactivity predictions using graph neural network algorithms (21), reactant-reagent role assignments (34), interpretation of predictions (28), and knowledge extraction from reaction databases (40).
The attention-guided reaction mapper (henceforth referred to as RXNMapper) can handle stereochemistry and unbalanced reactions and is, in terms of speed and accuracy, the state-of-the-art open-source tool for atom-mapping, providing an effective alternative to the time-intensive human extraction of chemical reaction rules. We release RXNMapper together with the atom-mapped public reaction dataset of Lowe (24) and a set of retrosynthetic rules (18, 36–38) extracted from it. The observed atom-mapping performance indicates that a consistent set of atom-mapping grammar rules exists as latent information in large datasets of chemical reactions, providing the link between data-driven/template-free and rule-based systems.
RESULTS
Attention-guided chemical reaction mapping
Self-attention is the major component of algorithms called Transformers that are setting records on NLP benchmarks, e.g., BERT (31) and ALBERT (32), and even creating breakthroughs in the chemical domain (28, 33, 41). Transformers use several self-attention modules, called heads, across multiple layers to learn how to represent each token in an input—e.g., each atom and bond in a reaction SMILES—given the tokens around it. Each head learns to attend to the inputs independently. When applied to chemical reactions, Transformers use attention to focus on atoms relevant to understand important molecular structures, describe the chemical transformation, and detect useful latent information. Fortunately, the internal attention mechanisms are intuitive to visualize and interpret using interactive tools (42–44). Through visual analysis, we observed that some Transformer heads learn distinct chemical features. Specific heads learned how to connect product atoms to reactant atoms, the process defined above as atom-mapping. We call these Transformer heads atom-mapping heads.
Throughout this work, our Transformer architecture of choice is ALBERT (32). ALBERT’s primary advantage over its predecessor BERT (31) is that it shares network weights across layers during training. This both makes the model smaller and keeps the functionality learned by a head the same across layers and consistent across inputs. Learned functions such as forward and backward scanning of the sequence, focusing on nonatomic tokens (ring openings/closures), and atom-mapping all perform similarly, irrespective of the input.
From raw attention to atom-mapping
To quantify our observations, we developed an attention-guided algorithm that converts the bidirectional attention signal of an atom-mapping head into a products-to-reactants atom-mapping. This specific mapping order ensures that each atom in the products corresponds to an atom in the reactants, which is important given that the most sizable open-source reaction datasets (24, 45) report only major products and show reactions that have fewer product atoms than reactant atoms.
The product atoms are mapped to reactant atoms one at a time, starting with product atoms that have the largest attention to an identical atom in the reactants. At each step, we introduce a neighbor attention multiplier that increases the attention connection from adjacent atoms of the newly mapped product atom to adjacent atoms of the newly mapped reactant atom, boosting the likelihood of an atom having the same adjacent atoms in reactants and products. This process continues until all product atoms are mapped to corresponding reactant atoms. The constraint of mapping only to equivalent atoms led to negligible improvements in terms of atom-mapping correctness, indicating that the model had already learned this rule in its atom-mapping function.
We selected the best performing model/layer/head combination after evaluation on a curated set of 1k patent reactions by Schneider et al. (34) originally mapped with the rule-based NameRXN tool (35). We used the remaining 49k reactions as a test set. We consider the atom-maps in NameRXN (35) to be of high quality because they are a side product of successfully matched reaction rules humanly designed. We used our best ALBERT model (12 layers and 8 heads) configuration (at layer 11, head 6, and multiplier 90) for RXNMapper.
Atom-mapping evaluation
The predominant use case for atom-mapping algorithms is to map heavily imbalanced reactions, such as those in patent reaction datasets (24, 45) or those predicted by data-driven reaction prediction models (28). After training RXNMapper on unmapped reactions (24), we investigated the chemical knowledge our model had extracted by comparing our predicted atom-maps to a set of 49k test reactions (34). The majority (96.8%) of the atom-mappings matched the reference, including methylene transfers, epoxidations, and Diels-Alder reactions (Fig. 2). We manually annotated the remaining discrepancies to find edge cases where RXNMapper seemingly failed. A more careful analysis showed that of the 1551 nonmatching reactions, only 284 predictions were incorrect. In 415 reactions, RXNmapper gave atom-maps equivalent to the original (e.g., tautomers), and in 436, the atom-maps were better than the reference. In 369 cases, the original reaction was questionable and likely wrongly extracted from patents. For 47 reactions, the key reagents to determine the reaction mechanisms were missing. After removing questionable reactions from the statistics and counting the equivalent mappings as correct, the overall correctness increased to 99.4%.
Fig. 2. Reaction map and examples.
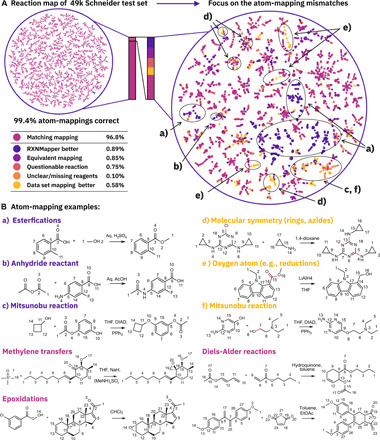
(A) Visualizing the results on the whole 49k Schneider test set with a focus on the mismatched atom-mappings (together with 1.5 k matches for context) using reaction tree maps (TMAPs) (41, 58). (B) Examples of atom-mappings generated by RXNMapper. Reactants and reagents were not separated in the inputs.
Among the most frequent failures of RXNMapper, we find examples of wrong atom ordering in rings and azide compounds (Fig. 2B, d). In others, the model assigns wrong mappings to a single oxygen atom, like in reductions (Fig. 2B, e) or in Mitsunobu reactions (Fig. 2B, f), where the phenolic oxygen should become part of the product, but the model maps the primary or secondary alcohol instead.
We also observed counterexamples of Mitsunobu reactions (Fig. 2B, c) for which our model correctly mapped the reacting oxygen, while the rule-based reference contained the wrong mapping as a result of the reaction not matching the Mitsunobu reaction rule. Although the overall quality of the reference atom-maps in the 49k test set (46) is high, we were able to identify few important advantages of using RXNMapper instead of the rule-based mapped dataset. RXNMapper correctly assigns the oxygen of the primary alcohols to be part of the major product for esterification reactions (Fig. 2B, a) like Fischer-Speier and Steglich esterifications as opposed to the annotated ground truth. It also correctly recognizes anhydrides (Fig. 2B, b) and peroxides as reactants in acylation and oxidation reactions where the ground truth favored formic acid and water.
RXNMapper not only excels on patent reactions but also performs remarkably well on reactions involving rearrangements of the carbon skeleton where humans require an understanding of the reaction mechanism to correctly atom-map. Notable examples include an intramolecular Claisen rearrangement used to construct fused seven- to eight-membered ring in the synthesis of the natural product micrandilactone A (Fig. 3A) (47, 48) and the tandem Palladium-catalyzed semipinacol rearrangement/direct arylation used for a stereoselective synthesis of benzodiquinanes from cyclobutanols (Fig. 3B) (49). In both cases, RXNMapper completes the correct atom-mapping despite the entirely rearranged carbon skeletons resulting in different ring sizes and connections. ReactionMap, Marvin, ChemDraw, and Indigo failed at this atom-mapping task. RXNMapper also succeeds in atom-mapping the ring rearrangement metathesis of a norbornene to form a bicyclic enone under catalysis by Grubbs-(I) catalyst (Fig. 3C) (50). In this case, ChemDraw successfully completes the mapping, while the other tools failed. Furthermore, RXNMapper performs well with multicomponent reactions such as the Ugi four-component condensation of isonitriles, aldehydes, amines, and carboxylic acids to form acylated aminoacid amides (Fig. 3D) (51). Here, RXNmapper maps all atoms correctly except for the carbonyl oxygen atom of the isonitrile-derived carboxamide. RXNMapper assigns this oxygen atom to the oxygen atom of the carbonyl group of the aldehyde reagent, although this atom actually comes from the hydroxyl group of the carboxylic acid reagent. All other tools failed this atom-mapping task except for Mappet.
Fig. 3. Atom-mapping on complex reactions.
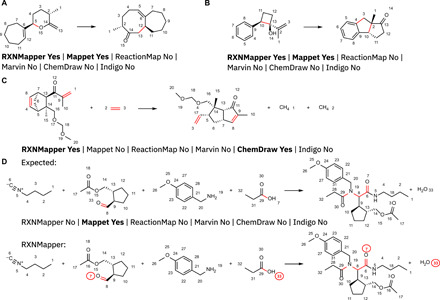
Examples and results for commercially available tools from the complex reactions dataset by Jaworski et al. (16). (A) Bu3Al-promoted Claisen rearrangement (47, 48). (B) Palladium-catalyzed semipinacol rearrangement and direct arylation (49). (C) Grubbs-catalyzed ring rearrangement metathesis reaction (50). (D) Ugi reaction (51).
Similar to Jaworski et al. (16), we analyzed the atom-mapping in United States Patent and Trademark Office (USPTO) patent reactions according to the number of bond changes (Fig. 4A ). RXNMapper performs better than Mappet (16) on all reactions except for those involving only one bond change. With an average time to solution of 7.7 ms per reaction on graphics processing unit (GPU) accelerators and 36.4 ms per reaction on central processing unit (CPU), RXNMapper’s speed is similar to the Indigo toolkit (17) on balanced reactions and far exceeds Indigo on unbalanced ones (Fig. 4B). As a comparison, Mappet (16) takes more than 10 s per reaction for 3.2% of their balanced test set reactions and for few of the reactions even more than 100 s per reaction. In addition, RXNMapper outputs a confidence score for the generated atom-maps. An analysis of the confidence scores and more detailed comparisons are available in the Supplementary Materials.
Fig. 4. Comparison with other tools.
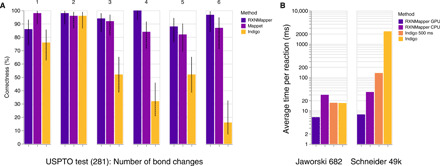
(A) Comparison of RXNMapper, Mappet (16), and the original Indigo mapping from the USPTO dataset (281 reactions). The error bars show the Wilson confidence interval (59). (B) Mapping speed comparison between RXNMapper and Indigo (17), which is orders of magnitude faster than Mappet (16). For Indigo of 500 ms, we set a timeout of 500 ms, after which the tool would return an incomplete mapping. We averaged the timing on the imbalanced reactions for Indigo without timeout on 20k reactions.
The advantages of RXNMapper compared to the open-source Indigo (17) and the closed-source Mappet (16) are summarized in Table 1. RXNMapper is noticeably faster than other tools, handles strongly unbalanced reactions, performs well even on complex reactions, and is open-source. It can also be used for compiling retrosynthetic rules, which are of crucial importance for several reaction and retrosynthesis prediction schemes. For instance, in the Chematica project (2), numerous Ph.D. students and Postdocs across 15 years continuously worked to extract reactions from literature and convert them into retrosynthetic rules. With unsupervised schemes such as RXNMapper, the extraction of retrosynthetic rules can be completed in a matter of weeks, with little human intervention. We demonstrate such an extraction by atom-mapping the entire USPTO datasets and by extracting the retrosynthetic rules using the approach described by Thakkar et al. (38). We make available the corresponding atom-mappings of the USPTO dataset and the 21k most frequently extracted retrosynthetic rules along with the most commonly used reagents, the corresponding patent numbers, and the first year of appearance. The application of unsupervised schemes demonstrates the feasibility of running a completely unassisted construction of retrosynthetic rules in just a few days—three orders of magnitude faster than previous human curation protocols. The use of unsupervised schemes will facilitate the compilation of previously unidentified retrosynthetic rules in existing rule-based systems.
Table 1. Comparison of different atom-mapping tools.
DISCUSSION
We have shown that the application of unsupervised, attention-based language models to a corpus of organic chemistry reactions provides a way to extract the organic chemistry grammar without human intervention. We unboxed the neural network architecture to extract the rules governing atom rearrangements between products and reactants/reagents. Using this information, we developed an attention-guided reaction mapper that exhibits remarkable performance in both speed and accuracy across many different reaction classes. We showed how to create a state-of-the-art atom-mapping tool within 2 days of training without the need for tedious and potentially biased human encoding or curation. Because the entire approach is completely unsupervised, the use of specific reaction datasets can improve the atom-mapping performance on corner cases. The resulting atom-mapping tool is significantly faster and more effective than existing tools, especially for strongly imbalanced reactions. Last, our work provides evidence that unannotated collections of chemical reactions contain all the relevant information necessary to construct a coherent set of atom-mapping rules. Numerous applications built on atom-mapping will immediately benefit from our findings (21, 36, 38), and others will become more interpretable exploiting the potential of unsupervised atom-mappings (28, 33).
The use of symbolic representations and the means to learn autonomously from rich chemical data led to the design of valuable assistants in chemical synthesis (26). A strengthened trust between human and interpretable data-driven assistants will spark the next revolutions in chemistry, where domain patterns and knowledge can be easily extracted and explained from the inner architectures of trained models.
MATERIALS AND METHODS
Transformers
Transformers are a class of deep neural network architectures that relies on multiple and sequential applications of self-attention layers (27). These layers are composed of one or more heads, each of which learns a square attention matrix A∈RN × N of weights that connect each token’s embedding Yi in an input sequence Y of length N to every other token’s embedding Yj. Thus, each element Aij is the attention weight connecting Yi to Yj. This formulation makes the attention weights in the Transformer architecture amenable to visualizations as the curves connecting an input sequence to itself, where a thicker, darker line indicates a higher attention value.
The calculation of the attention matrix of each head can be easily interpreted as a probabilistic hashmap or lookup table over all other elements Yj. Each head in a self-attention layer will first convert the vector representation of every token Yi into a key, query, and value vector using the following operations
| (1) |
where Wk ∈Rdk × de, Wq ∈Rdk × de, and Wv ∈Rdv × de are learnable parameters. Ai, or the vector of attention out of token Yi, is then a discrete probability distribution over the other input tokens, and it is calculated by taking a dot product over that token’s query vector and every other token’s key vector followed by a softmax to convert the information into probabilities
| (2) |
Note that one can define input sequence Y as an N × de matrix and matrix Wk as a dk × de matrix, where de is the embedding dimension of each token and dk is the embedding dimension shared by the query and the key.
Each head must learn a unique function to accomplish the masked language modeling task, and some of these functions are inherently interpretable to the domain of the data. For example, in NLP, it has been shown that certain heads learn dependency and part of speech relationships between words (52, 53). Using visual tools can make exploring these learned functions easier (42).
Model details
For our experiments, we used PyTorch (v1.3.1) (54) and huggingface transformers (v2.5.0) (55). The ALBERT model was trained for 48 hours on a single Nvidia P100 GPU with the hyperparameters stated in the Supplementary Materials. Schwaller et al. (28) developed the tokenization regex used to tokenize the SMILES. We expect further performance improvements when using more extensive datasets (e.g., commercially available ones). The RXNMapper model uses 12 layers, 8 heads, a hidden size of 256, an embedding size of 128, and an intermediate size of 512. In contrast to ALBERT base (32) with 12M parameters, our model is small and contains only 770k trainable parameters.
Data
The work by Lowe (24) provides the datasets used for training, composed of chemical reactions extracted from both grants and patent applications. We removed the original atom-mapping from this dataset, canonicalized the reactions with RDKit (56), and removed any duplicate reactions. The dataset includes reactions with fragment information twice, once with and once without fragment bonds, as defined in the work of Schwaller et al. (33). The final training set for the masked language modeling task contained a total of 2.8M reactions. For the evaluation and the model selection, we sampled 996 random reactions from the dataset of Schneider et al. (34).
To test our models, we first used the remaining 49k reactions from the Schneider 50k patents dataset (34). We do not distinguish between reactants and reagents in the inputs of our models. We also used the human-curated test sets that were introduced by Jaworski et al. (16) to compare our approach to previous methods. Table 2 shows an overview of the test sets. Note that patent reactions differ from the reactions in Jaworski et al. (16) because the latter removes most reactants and reagents in an attempt to balance the reactions.
Table 2. Test datasets.
Datasets used for the comparison with other tools.
Attention-guided atom-mapping algorithm
The attention-guided algorithm relies on the construction of the attention matrix for a selected layer and head, where we sum the product-to-reactant and the corresponding reactant-to-product atom attentions. Algorithm 1 provides the exact atom-mapping algorithm. By default, after matching a product-reactant pair, the attentions to those atoms are zeroed. Optionally, atoms in product and reactants can have multiple corresponding atoms. We always mask out attention to atoms of different types.
Atom-mapping curation
Chemically equivalent atoms exist in many chemical reactions. Most of the chemically equivalent atoms could be matched after canonicalizing the atom-mapped reaction using RDKit (56, 57). Exceptions were atoms of the same type connected to another atom with different bond types, which would form a resonance structure with delocalized electrons. We manually curated these exceptions and added them as alternative maps in the USPTO bond changes test set (16).

Acknowledgments
We thank the RXN for Chemistry team, and the Reymond group for insightful discussions and comments. H. Strobelt is a visiting research scientist at MIT. Funding: This work was supported by IBM Research. Author contributions: The project was conceived and planned by P.S. and B.H. and supervised by J.-L.R., H.S., and T.L. P.S. implemented and trained the models. B.H. and H.S. developed the visualization tools. P.S. and B.H. built RXNMapper. P.S., T.L., and J.L.R. analyzed and compared the atom-mapping. All the authors were involved in discussions on the project and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All our generated atom-mappings, including those for the largest open-source patent dataset (24), the unmapped training, validation, and test set reactions, can be found in the following repository https://github.com/rxn4chemistry/rxnmapper. The code is available at https://github.com/rxn4chemistry/rxnmapper and a demo at http://rxnmapper.ai.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe4166/DC1
REFERENCES AND NOTES
- 1.Weininger D., SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988). [Google Scholar]
- 2.Klucznik T., Mikulak-Klucznik B., McCormack M. P., Lima H., Szymkuc S., Bhowmick M., Molga K., Zhou Y., Rickershauser L., Gajewska E. P., Toutchkine A., Dittwald P., Startek M. P., Kirkovits G. J., Roszak R., Adamski A., Sieredzinska B., Mrksich M., Trice S. L., Grzybowski B. A., Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 4, 522–532 (2018). [Google Scholar]
- 3.Chen W. L., Chen D. Z., Taylor K. T., Automatic reaction mapping and reaction center detection. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 560–593 (2013). [Google Scholar]
- 4.Gonzalez G. A. P., El Assal L. R., Noronha A., Thiele I., Haraldsdóttir H. S., Fleming R. M., Comparative evaluation of atom mapping algorithms for balanced metabolic reactions: Application to Recon 3D. J. Cheminform. 9, 39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch M. F., Willett P., The automatic detection of chemical reaction sites. J. Chem. Inf. Comput. Sci. 18, 154–159 (1978). [Google Scholar]
- 6.McGregor J. J., Willett P., Use of a maximum common subgraph algorithm in the automatic identification of ostensible bond changes occurring in chemical reactions. J. Chem. Inf. Comput. Sci. 21, 137–140 (1981). [Google Scholar]
- 7.T. E. Moock, J. G. Nourse, D. Grier, W. D. Hounshell, The implementation of Fapping and related features in the reaction access system (REACCS), in Chemical Structures, W. A. Warr, Ed. (Springer, Berlin, Heidelberg, 1988), pp. 303–313. [Google Scholar]
- 8.Funatsu K., Endo T., Kotera N., Sasaki S.-I., Automatic recognition of reaction site in organic chemical reactions. Tetrahedron Comput. Method. 1, 53–69 (1988). [Google Scholar]
- 9.Korner R., Apostolakis J., Automatic determination of reaction mappings and reaction center information. 1. The imaginary transition state energy approach. J. Chem. Inf. Model. 48, 1181–1189 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Apostolakis J., Sacher O., Korner R., Gasteiger J., Automatic determination of reaction mappings and reaction center information. 2. Validation on a biochemical reaction database. J. Chem. Inf. Model. 48, 1190–1198 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Jochum C., Gasteiger J., Ugi I., The principle of minimum chemical distance (PMCD). Angew. Chem. Int. Ed. 19, 495–505 (1980). [Google Scholar]
- 12.Akutsu T., Efficient extraction of mapping rules of atoms from enzymatic reaction data. J. Comput. Biol. 11, 449–462 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Crabtree J. D., Mehta D. P., Automated reaction mapping. ACM J. Exp. Algor. 13, 1.15 (2009). [Google Scholar]
- 14.First E. L., Gounaris C. E., Floudas C. A., Stereochemically consistent reaction mapping and identification of multiple reaction mechanisms through integer linear optimization. J. Chem. Inf. Model. 52, 84–92 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Latendresse M., Malerich J. P., Travers M., Karp P. D., Accurate atom-mapping computation for biochemical reactions. J. Chem. Inf. Model. 52, 2970–2982 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Jaworski W., Szymkuć S., Mikulak-Klucznik B., Piecuch K., Klucznik T., Kaźmierowski M., Rydzewski J., Gambin A., Grzybowski B. A., Automatic mapping of atoms across both simple and complex chemical reactions. Nat. Commun. 10, 1434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Indigo Toolkit (2020); https://lifescience.opensource.epam.com/indigo/ [accessed 02 Apr 2020].
- 18.Coley C. W., Barzilay R., Jaakkola T. S., Green W. H., Jensen K. F., Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 3, 434–443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.W. Jin, C. Coley, R. Barzilay, T. Jaakkola, Predicting organic reaction outcomes with weisfeiler-lehman network, in Advances in Neural Informations Processing Systems (NIPS, 2017), pp. 2607–2616. [Google Scholar]
- 20.J. Bradshaw, M. Kusner, B. Paige, M. Segler, J. Hernandez-Lobato, A generative model for electron paths, in Proceedings of ICLR (2019).
- 21.Coley C. W., Jin W., Rogers L., Jamison T. F., Jaakkola T. S., Green W. H., Barzilay R., Jensen K. F., A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 10, 370–377 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.W. W. Qian, N. T. Russell, C. L. Simons, Y. Luo, M. D. Burke, J. Peng, Integrating Deep Neural Networks and Symbolic Inference for Organic Reactivity Prediction (2020); https://arxiv.org/abs/2006.07038.
- 23.V. R. Somnath, C. Bunne, C. W. Coley, A. Krause, R. Barzilay, Learning Graph Models for Template-Free Retrosynthesis (2020); 10.26434/chemrxiv.11659563. [DOI]
- 24.D. Lowe, Chemical reactions from US patents (1976-Sep2016) (2017); https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873.
- 25.Ozturk H., Ozgur A., Schwaller P., Laino T., Ozkirimli E., Exploring chemical space using natural language processing methodologies for drug discovery. Drug Discov. Today 25, 689–705 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Almeida A., Moreira R., Rodrigues T., Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 3, 589–604 (2019). [Google Scholar]
- 27.A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser, I. Polosukhin, Attention is all you need, in Advances in Neural Information Processing Systems (NIPS, 2017), pp. 5998–6008. [Google Scholar]
- 28.Schwaller P., Laino T., Gaudin T., Bolgar P., Hunter C. A., Bekas C., Lee A. A., Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5, 1572–1583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.P. Schwaller, T. Laino, Data-Driven Learning Systems for Chemical Reaction Prediction: An Analysis of Recent Approaches, in Machine Learning in Chemistry: Data-Driven Algorithms, Learning Systems, and Predictions (ACS Publications, Washington, 2019), pp. 61–79. [Google Scholar]
- 30.Tetko I. V., Karpov P., Van Deursen R., Godin G., State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 11, 5575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.J. Devlin, M.-W. Chang, K. Lee, K. Toutanova, BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding, in Proceeding of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (Assoiciation for Computational Linguistics, Minneapolis, Minnesota, 2019), vol. 1, pp. 4171–4186. [Google Scholar]
- 32.Z. Lan, M. Chen, S. Goodman, K. Gimpel, P. Sharma, R. Soricut, ALBERT: A lite BERT for self-supervised learning of language representations, in Proceedings of 8th International Conference on Learning Representations ( ICLR, Ethiopia, 2020). [Google Scholar]
- 33.Schwaller P., Petraglia R., Zullo V., Nair V. H., Haeuselmann R. A., Pisoni R., Bekas C., Iuliano A., Laino T., Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 11, 3316–3325 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider N., Stiefl N., Landrum G. A., What’s what: The (nearly) definitive guide to reaction role assignment. J. Chem. Inf. Model. 56, 2336–2346 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Nextmove Software NameRXN (2020); www.nextmovesoftware.com/namerxn.html [accessed 02 April 2020].
- 36.Segler M. H., Preuss M., Waller M. P., Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Coley C. W., Green W. H., Jensen K. F., RDChiral: An RDKit wrapper for handling stereochemistry in retrosynthetic template extraction and application. J. Chem. Inf. Model. 59, 2529–2537 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Thakkar A., Kogej T., Reymond J.-L., Engkvist O., Bjerrum E. J., Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain. Chem. Sci. 11, 154–168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortunato M., Coley C. W., Barnes B. C., Jensen K. F., Data augmentation and pretraining for template-based retrosynthetic prediction in computer-aided synthesis planning. J. Chem. Inf. Model. 60, 3398–3407 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Chen L., Nourse J. G., Christie B. D., Leland B. A., Grier D. L., Over 20 years of reaction access systems from MDL: A novel reaction substructure search algorithm. J. Chem. Inf. Comput. Sci. 42, 1296–1310 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Schwaller P., Probst D., Vaucher A. C., Nair V. H., Kreutter D., Laino T., Reymond J.-L., Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach. Intell. 3, 144–152 (2021). [Google Scholar]
- 42.B. Hoover, H. Strobelt, S. Gehrmann, exBERT: A visual analysis tool to explore learned representations in transformer models, in Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics: System Demonstration (ACL, 2020), pp. 187–196. [Google Scholar]
- 43.S. Wiegreffe, Y. Pinter, Attention is not not explanation, in Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (Association for Computational Linguistics, 2019), pp. 11–20. [Google Scholar]
- 44.J. Vig, A multiscale visualization of attention in the transformer model, in Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics: system Demonstrations (Association for Computational Linguistics, Florence, Italy, 2019), pp. 37–42. [Google Scholar]
- 45.D. M. Lowe, Extraction of chemical structures and reactions from the literature, Ph.D. thesis, University of Cambridge (2012). [Google Scholar]
- 46.Schneider N., Lowe D. M., Sayle R. A., Landrum G. A., Development of a novel fin-gerprint for chemical reactions and its application to large-scale reaction classification and similarity. J. Chem. Inf. Model. 55, 39–53 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y.-D., Ren W.-W., Lan Y., Xiao Q., Wang K., Xu J., Chen J.-H., Yang Z., Stereoselective construction of an unprecedented 7- 8 fused ring system in micrandilactone a by [3, 3]-sigmatropic rearrangement. Org. Lett. 10, 665–668 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Sun T.-W., Ren W.-W., Xiao Q., Tang Y.-F., Zhang Y.-D., Li Y., Meng F.-K., Liu Y.-F., Zhao M.-Z., Xu L.-M., Chen J.-H., Yang Z., Diastereoselective total synthesis of (±)-Schindilactone A, Part 1: Construction of the ABC and FGH ring systems and initial attempts to construct the CDEF ring system. Chem. Asian J. 7, 2321–2333 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Schweinitz A., Chtchemelinine A., Orellana A., Synthesis of benzodiquinanes via tandem palladium-catalyzed semipinacol rearrangement and direct arylation. Org. Lett. 13, 232–235 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Acharyya R. K., Rej R. K., Nanda S., Exploration of ring rearrangement metathesis reaction: A general and flexible approach for the rapid construction [5, n]-fused bicyclic systems en route to linear triquinanes. J. Org. Chem. 83, 2087–2103 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Moni L., Banfi L., Basso A., Carcone L., Rasparini M., Riva R., Ugi and Passerini reactions of biocatalytically derived chiral aldehydes: Application to the synthesis of bicyclic pyrrolidines and of antiviral agent telaprevir. J. Org. Chem. 80, 3411–3428 (2015). [DOI] [PubMed] [Google Scholar]
- 52.J. Vig, Y. Belinkov, Analyzing the structure of attention in a transformer language model, in Proceedings of the 2019 ACL Workshop BlackboxNLP: Analyzing and Interpreting Neural Networks for NLP (Association for Computational Linguistics, Florence, Italy, 2019), pp. 63–76. [Google Scholar]
- 53.K. Clark, U. Khandelwal, O. Levy, C. D. Manning, What does BERT look at? An analysis of BERT’s attention, in Proceedings of the 2019 ACL Workshop BlackboxNLP: Analyzing and Interpreting Neural Networks for NLP (Florence, Italy, 2019), pp. 276–286. [Google Scholar]
- 54.A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. De Vito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai, S. Chintala, PyTorch: An imperative style, high-performance deep learning library, in Advances. in Neural Information Processing Systems (2019), pp. 8024–8035.
- 55.T. Wolf, J. Chaumond, L. Debut, V. Sanh, C. Delangue, A. Moi, P. Cistac, M. Funtowicz, J. Davison, S. Shleifer, P. von Platen, C. Ma, Y. Jernite, J. Plu, C. Xu, T. L. Scao, S. Gugger, M. Drame, Q. Lhoest, A. Rush, Transformers: State-of-the-art natural language processing, in Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations (Association for Computational Linguistics, 2020), pp. 38–45. [Google Scholar]
- 56.G. Landrum, P. Tosco, B. Kelley, S. Riniker, P. Gedeck, NadineSchneider, R. Vianello, A. Dalke, R. R. Schmidt, B. Cole, A. Savelyev, S. Turk, M. Swain, A. Vaucher, D. Nealschneider, M. Wojcikowski, A. Pahl, J.-P. Ebejer, F. Berenger, A. Stretton, N. O’Boyle, D. Cosgrove, P. Fuller, J. H. Jensen, G. Sforna, K. Leswing, S. Leung, J. vanSanten, rdkit/rdkit: 2019 03 4 (Q1 2019) Release (2019); 10.5281/zenodo.3366468. [DOI]
- 57.Schneider N., Sayle R. A., Landrum G. A., Get your atoms in order - An open-source implementation of a novel and robust molecular canonicalization algorithm. J. Chem. Inf. Model. 55, 2111–2120 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Probst D., Reymond J.-L., Visualization of very large high-dimensional data sets as minimum spanning trees. J. Chem. 12, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallis S., Binomial confidence intervals and contingency tests: Mathematical fundamentals and the evaluation of alternative methods. J. Quant. Ling. 20, 178–208 (2013). [Google Scholar]
- 60.J. S. Marvin, ChemAxon (2020); https://chemaxon.com [accessed 02 April 2020].
- 61.Fooshee D., Andronico A., Baldi P., ReactionMap: An efficient atom-mapping algorithm for chemical reactions. J. Chem. Inf. Model. 53 (11), 2812–2819 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe4166/DC1