

FOXA1 is a nonhistone substrate of EZH2, promoting cell growth and sensitizing some prostate cancer to enzymatic EZH2 inhibitors.
Abstract
Forkhead box protein A1 (FOXA1) is essential for androgen-dependent prostate cancer (PCa) growth. However, how FOXA1 levels are regulated remains elusive and its therapeutic targeting proven challenging. Here, we report FOXA1 as a nonhistone substrate of enhancer of zeste homolog 2 (EZH2), which methylates FOXA1 at lysine-295. This methylation is recognized by WD40 repeat protein BUB3, which subsequently recruits ubiquitin-specific protease 7 (USP7) to remove ubiquitination and enhance FOXA1 protein stability. They functionally converge in regulating cell cycle genes and promoting PCa growth. FOXA1 is a major therapeutic target of the inhibitors of EZH2 methyltransferase activities in PCa. FOXA1-driven PCa growth can be effectively mitigated by EZH2 enzymatic inhibitors, either alone or in combination with USP7 inhibitors. Together, our study reports EZH2-catalyzed methylation as a key mechanism to FOXA1 protein stability, which may be leveraged to enhance therapeutic targeting of PCa using enzymatic EZH2 inhibitors.
INTRODUCTION
Forkhead box protein A1 (FOXA1), a forkhead (FKHD) family transcription factor, is a pioneer factor of the androgen receptor (AR) and is essential for the development and maturation of prostate luminal epithelial cells (1). As an epithelial differentiation regulator, FOXA1 plays at least two major roles in the adult prostate: (i) induction of androgen-dependent cell growth and (ii) maintenance of a differentiated cellular state, at least in part, by suppression of dedifferentiation pathways (2, 3). FOXA1 gene is frequently mutated and its expression strongly up-regulated in prostate cancer (PCa) as compared to benign prostates (3–6). We and others have shown that FOXA1 induces androgen-dependent cell growth by transcriptional regulation of genes related to cell cycle process, DNA replication, cell division, and apoptosis as well as other death pathways (2, 7). FOXA1 not only drives G1-S progression via regulation of CCNE2 and E2F1 but also promotes G2-M cell cycle progression via regulation of UBE2C and CDK1 in PCa cells (7). Despite these important roles, how FOXA1 levels are regulated in PCa remains largely unknown.
Polycomb group (PcG) protein EZH2 (enhancer of zeste homolog 2) harbors a SET (Su[var]3-9, enhancer of zeste, Trithorax) domain on its C terminus and is the enzymatic subunit of the polycomb repressive complex 2 (PRC2) (8). EZH2 cooperates with other core subunits of the PRC2 complex, embryonic ectoderm development (EED) and suppressor of zeste 12 protein homolog (SUZ12), to trimethylate histone H3 at lysine-27 (H3K27me3) for epigenetic silencing of target genes (9, 10). Extensive studies have shown primary roles of PRC2 in maintaining the undifferentiated state of embryonic stem cells via suppression of developmental regulators through bivalent chromatin modifications (11–13). In recent years, EZH2 has been reported to be overexpressed in many cancer types including PCa (14). EZH2 was shown to promote PCa tumorigenesis by epigenetically silencing a wide range of tumor suppressor genes, including DAB2IP, ADRB2, CDH1, RKIP, SLIT2, and TIMP2/3 (15–20). However, how EZH2 activates the expression of many cell cycle genes remains unclear, although it has been shown as a target of the pRB-E2F pathway that plays important roles in mediating cell cycle progression (21).
Several recent studies have reported nonhistone substrates of EZH2. For example, EZH2 has been shown to methylate its cofactor jumonji and AT-rich interaction domain containing 2 (JARID2) to further enhance PRC2 activities through an autoregulatory feedback loop (22). Likewise, EZH2 methylates the RNA polymerase II transcription elongation factor, Elongin A, contributing to the repression of a subset of PRC2 target genes (23). During cardiac morphogenesis, EZH2 interacts with cardiac transcription factor GATA binding protein 4 (GATA4) to catalyze GATA4 methylation, which reduces GATA4 interaction with and acetylation by p300 (24). In addition, EZH2-mediated methylation of some substrates, such as retinoic acid-related orphan receptors alpha (RORα) and promyelocytic leukemia zinc finger (PLZF), generates methyl-degrons, resulting in protein degradation (25, 26). EZH2 has also been shown to activate some pathways. In glioblastoma stem-like cells, phosphorylation of EZH2 methylates signal transducers and activators of transcription 3 (STAT3) to activate STAT3 signaling and promote tumorigenicity (27). In metastatic castration-resistant PCa (CRPC), EZH2 can be phosphorylated, and it interacts with AR to activate its transcriptional program through PRC2-independent mechanisms (28). However, nonhistone substrates of EZH2 in PCa are yet to be characterized.
Ubiquitin (Ub)–specific protease 7 (USP7 or HAUSP) is a member of nearly 100 deubiquitinases that remove ubiquitination on selected substrate proteins, sparing them from degradation (29). Previously reported substrates of USP7 include p53 regulator mouse double minute 2 homolog (MDM2) (30), transcription factors FOXO4 and RE1-silencing transcription factor (REST) (31, 32), and mitotic regulator Aurora A kinase (33). In PCa, USP7 is overexpressed and has been shown to mediate phosphatase and TENsin homolog deleted on chromosome 10 (PTEN) deubiquitination and subsequent trafficking involving the adaptor protein death-associated protein 6 (DAXX) (34). Many USP proteins have been shown to interact with proteins containing the WD40 repeat, which is a short structural motif of approximately 40 amino acids, often terminating in a tryptophan-aspartic acid (WD) dipeptide. Large-scale mass spectrometry analyses have reported USP7 interaction with multiple WD40 repeat proteins, including BUB3, budding uninhibited by benzimidazoles 3 homolog (35). BUB3 is a mitotic checkpoint protein that regulates the spindle assembly checkpoint and plays essential roles in the formation of correct microtubule-kinetochore attachments for proper cell divisions (36). However, the role of BUB3 acting as a scaffold for protein interaction in multiprotein complexes has not been investigated.
Here, we report FOXA1 as a nonhistone substrate of EZH2. We found that EZH2 interacts with FOXA1 protein to catalyze FOXA1 methylation at lysine-295 (K295). The methylated FOXA1 is recognized by BUB3, which subsequently recruits USP7 to remove ubiquitination of FOXA1 and increase protein stability. Similar to H3K27me3, EZH2-mediated methylation of FOXA1 is dependent on PRC2 and the methyltransferase (MTase) activity of EZH2, which is targeted by clinically available enzymatic EZH2 inhibitors.
RESULTS
PcG protein EZH2 interacts with FOXA1 and increases protein stability
Although substantial up-regulation of FOXA1 has been reported in PCa, where it was shown to promote cell cycle and tumor growth, little is known regarding how FOXA1 level is regulated. Gene set enrichment analysis (GSEA) of FOXA1-induced genes (3) showed a significant (false discovery rate < 0.001) enrichment for down-regulation upon EZH2 knockdown (KD) (37), while FOXA1-repressed genes enriched for up-regulation, suggesting a positive molecular cross-talk between these two proteins (fig. S1, A and B). To examine whether EZH2 increases FOXA1 levels, we performed EZH2 KD in LNCaP cells using two independent short hairpin RNAs (shRNAs): one targeting EZH2 coding region (shEZH2-C) and the other the 3′untranslated region (shEZH2-3′). EZH2 KD led to a marked decrease in FOXA1 protein levels (Fig. 1A). This was confirmed in five additional PCa cell lines, including androgen-dependent Vertebral-Cancer of the Prostate (VCaP) and Los Angeles Prostate Cancer-4 (LAPC4), androgen-independent C4-2B and 22Rv1, and AR-negative PC3 cells (Fig. 1B), and in cells treated with small interfering RNAs (siRNAs) for EZH2 KD (fig. S1C). On the other hand, ectopic overexpression of EZH2 wild type (WT), but not its SET domain deletion mutant (ΔSET), increased FOXA1 proteins (Fig. 1C and fig. S1D). In contrast, FOXA1 mRNA levels were not altered upon EZH2 deregulation, suggesting a mechanism involving posttranscriptional regulation (fig. S1E). A time-course cycloheximide (CHX) experiment revealed that FOXA1 protein half-lives were markedly decreased, from approximately 9 hours in control LNCaP cells to less than 2 hours in EZH2-KD cells (Fig. 1D and fig. S1F), suggesting that EZH2 increases FOXA1 protein stability.
Fig. 1. EZH2 increases FOXA1 protein stability through physical interaction.
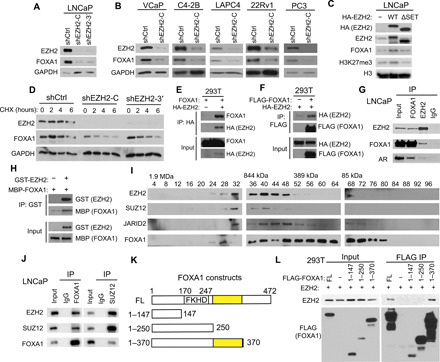
(A and B) PCa cells were infected with control (shCtrl) or two independent shEZH2 lentiviruses (shEZH2-C and shEZH2-3′) for 72 hours and subjected to WB. (C) LNCaP cells were infected with the indicated EZH2 lentivirus for 96 hours and analyzed by WB. (D) LNCaP cells were treated with protein synthesis inhibitor CHX (150 μg/ml) for 0, 2, 4, and 6 hours before they were collected for WB. (E and F) 293T cells were transfected with indicated plasmids for 48 hours. Coimmunoprecipitation (co-IP) was performed using a hemagglutinin (HA) (E) or FLAG antibody (F), followed by WB. (G) Co-IP with FOXA1, EZH2, or immunoglobulin G (IgG) antibody in LNCaP nuclear lysates followed by WB. (H) Purified recombinant GST–green fluorescent protein (GFP) or GST-EZH2 coupled to GST beads were used to pull-down purified MBP-FOXA1, which was analyzed by WB using an MBP antibody. (I) Nuclear extracts from LNCaP cells were fractionated and subjected to WB. (J) Co-IP using LNCaP nuclear lysates was performed with FOXA1, SUZ12, or IgG antibody. (K and L) EZH2 was cotransfected into 293T cells along with various FLAG-tagged FOXA1 domain constructs (K). Co-IP assay was performed with anti-FLAG M2 beads followed by WB using anti-EZH2 (L). EZH2-binding domains are highlighted in yellow.
Mass spectrometry analyses revealed that FOXA1 was enriched in the immunoprecipitates of WT EZH2, but not of its ΔSET mutant, that were expressed ectopically in LNCaP cells (fig. S1G and tables S1 and S2). Reciprocal coimmunoprecipitation (co-IP) confirmed a strong interaction between ectopic EZH2 and FOXA1 proteins in 293T cells (Fig. 1, E and F). Similar interaction was observed between endogenous proteins in PCa cells, and their interaction was independent of RNA or DNA (Fig. 1G and fig. S1H). To investigate whether this is a direct interaction, we performed in vitro glutathione S-transferase (GST) pull-down assay with recombinant GST-EZH2 and maltose-binding protein (MBP)–tagged FOXA1 proteins and found that FOXA1 proteins directly interacted with immobilized EZH2 proteins in vitro (Fig. 1H and fig. S1I). Although most EZH2-interacting proteins are associated with the PRC2 complex, some have been reported to interact with EZH2 only (38, 39). To determine how FOXA1 interacts with EZH2, we analyzed LNCaP nuclear extracts using size exclusion chromatography. Western blot (WB) analysis revealed that some FOXA1 proteins copurified with the PRC2 complex proteins including EZH2, SUZ12, and JARID2 at about 844 kDa (Fig. 1I). Reciprocal co-IP confirmed that FOXA1 interacts with both EZH2 and SUZ12 in LNCaP cells (Fig. 1J). To map the FOXA1 domains critical for its interaction with EZH2, we generated a series of FOXA1 truncation mutants with a FLAG tag (Fig. 1K), which were cotransfected along with EZH2 into 293T or LNCaP cells. Co-IP experiment showed that full-length (FL) FOXA1 and the FOXA1 amino acids 1 to 370 fragment, but not the amino acids 1 to 147 or amino acids 1 to 250 fragments, copurified with EZH2 (Fig. 1L and fig. S1J). These data suggest that the region between amino acids 250 and 370 (amino acids 250 to 370), which is immediately next to the FKHD DNA binding domain (amino acids 170 to 247) (40), is crucial for FOXA1 interaction with EZH2.
EZH2 reduces FOXA1 protein ubiquitination requiring its MTase activities
To test whether PRC2 interaction with FOXA1 and regulation of its protein stability is dependent on EZH2 MTase activities, we treated LNCaP cells with GSK-126 and EPZ-6438, two selective, S-adenosyl-l-methionine–competitive inhibitors of EZH2 (41, 42). Both catalytic inhibitors of EZH2 decreased H3K27me3 levels as expected, and they also markedly reduced FOXA1 protein levels in a dosage-dependent manner (Fig. 2A). In contrast, FOXA1 mRNA levels were not decreased (fig. S2A). Further, time-course CHX treatment revealed that, similar to EZH2 KD (Fig. 1D), inhibition of EZH2 MTase activities substantially shortened FOXA1 protein half-lives by more than sixfold (Fig. 2, B and C). We hypothesized that this is due to accelerated FOXA1 protein degradation upon EZH2 inhibition, as the 26S proteasome system is a primary pathway for intracellular protein degradation. To test this, we treated control and EZH2-KD LNCaP cells with 20 μM MG132, a proteasome inhibitor, for 6 hours. WB analysis confirmed FOXA1 degradation upon EZH2 KD, which, was rescued by MG132 (Fig. 2D). Similarly, MG132 also rescued FOXA1 levels in cells treated with EPZ-6438 (fig. S2B). The 26S proteasome system degrades target proteins by recognizing ubiquitination. To examine FOXA1 ubiquitination, we treated LNCaP cells with either vehicle control or 20 μM MG132 for 16 hours and performed co-IP using an FOXA1 antibody, followed by WB with Ub antibody, which revealed an accumulation of ubiquitinated FOXA1 proteins in MG132-treated cells (Fig. 2E). Next, we attempted to investigate whether EZH2 regulates FOXA1 ubiquitination. Control or EZH2-KD LNCaP cells were treated with 20 μM MG132 for 16 hours and then subjected to co-IP using an anti-FOXA1 antibody, followed by WB analysis of Ub. Our data showed marked increase in ubiquitinated FOXA1 in EZH2-KD cells, suggesting that EZH2 reduces FOXA1 ubiquitination (Fig. 2F). Ectopic EZH2 overexpression abolished FOXA1 ubiquitination (Fig. 2G). In contrast, H689A, a catalytically dead mutant of EZH2 (43), failed to reduce FOXA1 ubiquitination, suggesting a dependence on methylation. In agreement with this, EZH2 MTase inhibitors greatly restored FOXA1 ubiquitination in LNCaP cells (Fig. 2H).
Fig. 2. EZH2 attenuates FOXA1 ubiquitination and degradation requiring its MTase activities.
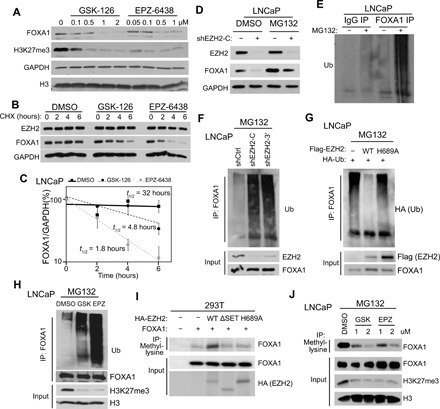
(A) LNCaP cells were treated with EZH2 inhibitors at indicated doses for 7 days before WB analysis. (B and C) LNCaP cells were treated with 2 μM EZH2 inhibitors for 96 hours and CHX (150 μg/ml) over indicated time course before WB (B). FOXA1 protein levels were plotted in (C) and half-lives estimated. Error bars, means ± SEM, n = 3. (D) LNCaP with stable control or EZH2 KD was treated with 20 μM MG132 for 6 hours before WB. (E) LNCaP cells were treated with either DMSO or 20 μM MG132. Co-IP was performed with IgG or FOXA1 antibody, followed by WB using anti-Ub. (F) LNCaP with stable control or EZH2 KD were treated with 20 μM MG132. Co-IP was performed with FOXA1 antibody and WB by anti-Ub. (G) LNCaP cells expressing indicated plasmids were treated with 20 μM MG132 and then subjected to co-IP by anti-FOXA1 and WB by anti-HA (Ub). (H) LNCaP cells were treated with 2 μM EZH2 inhibitors for 6 days before Co-IP. (I) 293T cells were transfected with indicated plasmids. Co-IP was performed with a pan-methyl lysine antibody and WB by anti-FOXA1. (J) LNCaP cells were treated with EZH2 inhibitors at the indicated doses for 72 hours, followed by 20 μM MG132 treatment before co-IP.
Because EZH2 is a lysine-specific MTase, we first tested whether lysine residues of FOXA1 could undergo methylation in PCa cells. Co-IP with a pan-methyl lysine antibody followed by subsequent WB with a FOXA1 antibody detected FOXA1 proteins with lysine methylation (fig. S2C). To assess whether EZH2 methylates FOXA1, we cotransfected 293T cells with FOXA1, along with control, WT, ΔSET, or H689A EZH2. Co-IP using a pan-methyl lysine antibody followed by WB using anti-FOXA1 indicated that overexpression of WT EZH2, but not ΔSET or H689A, increased FOXA1 methylation (Fig. 2I). Similar results were observed in LNCaP cells by reciprocal co-IP using a FLAG (FOXA1) antibody, followed by WB using anti–pan-methyl lysine (fig. S2D). Moreover, we treated LNCaP cells with dimethyl sulfoxide (DMSO), GSK-126, or EPZ-6438 for 72 hours and assessed FOXA1 protein methylation levels. Co-IP performed in the presence of the proteasome inhibitor MG132 to prevent unmethylated FOXA1 from degradation demonstrated that both GSK-126 and EPZ-6438 markedly reduced FOXA1 protein methylation in a dose-dependent manner (Fig. 2J). Together, these data suggest that EZH2 regulates FOXA1 protein ubiquitination and stability through methylating FOXA1.
EZH2 methylates FOXA1 protein at K295
To identify the lysine sites on the FOXA1 protein that is methylated by EZH2, we immunoprecipitated FOXA1 from 293T cells with ectopic FOXA1 and EZH2 expression and analyzed posttranslational modifications by mass spectrometry. Our data showed a total of eight peptides bearing methylation on K295 of FOXA1 (fig. S3A and table S3). K295 is located within the EZH2-interacting domain (amino acids 250 to 370) of FOXA1 as identified in Fig. 1L. Further, K295 and adjacent amino acid sequences of FOXA1 are highly conserved among vertebrates (Fig. 3A). Notably, an arginine residue at position −1 (R294) of FOXA1 resembles the arginine residue at position −1 of H3K27, the main target of EZH2 MTase activities. Previous studies have shown that arginine at −1 forms a series of hydrogen bonds with complementary side chains on the SET domain of EZH2 and is critical for EZH2 methylation of its substrates (23, 44). To assess the essentiality of K295 for FOXA1 methylation, we used site-directed mutagenesis to generate two FLAG-tagged FOXA1 mutants where alanine (K295A) or arginine (K295R) replaced K295. We transfected 293T or PCa cells with control, FOXA1 WT, K295A, or K295R and assessed total methylation levels of immunoprecipitated FOXA1 proteins. Compared with WT, both K295A and K295R mutations markedly reduced overall FOXA1 methylation levels, suggesting that K295 is a major lysine site for FOXA1 methylation (Fig. 3B and fig. S3B). Considering that EZH2 MTase activity is required to maintain FOXA1 protein stability, we next investigated whether the methyl-defective FOXA1 mutants are less stable than WT FOXA1. Time-course CHX treatment demonstrated that K295A proteins exhibited substantially shorter half-lives than WT FOXA1 (Fig. 3C). In agreement with these, K295A FOXA1 was also found to have a higher level of ubiquitination as compared to WT FOXA1 (Fig. 3D). Chromatin IP sequencing (ChIP-seq) of hemagglutinin (HA)–FOXA1 in isogenic FOXA1-KD cell lines with reexpression of HA-FOXA1 WT or K295A revealed that over 97% WT FOXA1–binding sites were also occupied by K295A, with the latter showing slightly reduced ChIP-seq intensity than the WT, likely due to the impaired protein stability of K295A (fig. S3, C to E). As the difference between WT FOXA1– and K295A FOXA1–binding sites is comparable to that observed between replicate FOXA1 ChIP-seq (fig. S3, F and G), we concluded that K295A did not reprogram FOXA1 cistrome. Accordingly, RNA sequencing (RNA-seq) analysis demonstrated that, K295A, when reexpressed at equivalent protein levels, fully rescued FOXA1-regulated gene expression in FOXA1-KD cells, resembling WT FOXA1 (Fig. 3E). Together, these findings suggest that methylation at K295 plays principal role in regulating FOXA1 ubiquitination and protein stability without clearly altering its target gene preference.
Fig. 3. EZH2 methylates FOXA1 protein at K295.
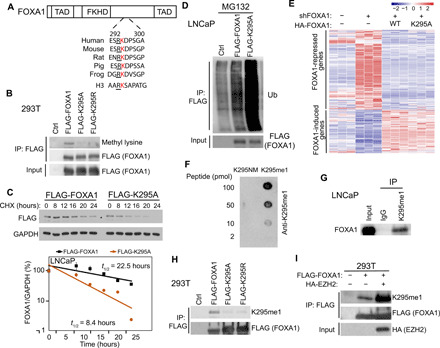
(A) Amino acid sequences surrounding FOXA1 K295 from different species comparing to human H3K27 site. TAD, transactivation domain. (B) 293T cells were transfected with control, FLAG-FOXA1, FLAG-K295A, or FLAG-K295R. Co-IP was performed with anti-FLAG M2 beads. (C) LNCaP cells stably expressing FLAG-FOXA1 or FLAG-K295A were treated with CHX (150 μg/ml) for indicated time course and collected for WB. FLAG (FOXA1) protein levels were measured and half-lives estimated. Error bars, means ± SEM, n = 3. (D) LNCaP cells were transfected with control, FLAG-FOXA1, or FLAG-K295A and treated with 20 μM MG132 for 16 hours before co-IP. (E) FOXA1-KD LNCaP cells were infected with control, HA-FOXA1, or HA-K295A lentiviruses and subjected to RNA-seq. Heatmaps show genes that were up- or down-regulated by WT FOXA1 reexpression. (F) Nitrocellulose membrane blotted with nonmethylated (K295NM) or K295-methylated peptides at different picomoles were subjected to WB with the K295me1 antibody. (G) Co-IP using LNCaP nuclear lysates was performed with either IgG or K295me1 antibody. (H) 293T cells were transfected with control, FLAG-FOXA1, FLAG-K295A, or FLAG-K295R before co-IP by anti-FLAG. (I) 293T cells were transfected with the indicated plasmids. Co-IP was performed with anti-FLAG M2 beads.
To directly test FOXA1 methylation at K295, we proceeded with the generation of polyclonal antibodies that specifically recognize methylated K295. As only monomethylation by EZH2 has been detected on almost all of its nonhistone substrates (23–25), we chose to generate antibodies specific for FOXA1 protein with K295 monomethylation. Dot blot assays confirmed that our FOXA1 monomethyl K295 (K295me1) antibody was specific for the methylated K295 peptide and did not cross-react with the nonmethylated K295 peptide (K295NM) (Fig. 3F). In addition, co-IP using this antibody successfully pulled down FOXA1 protein, confirming that K295me1 antibody recognized methylated FOXA1 proteins (Fig. 3G). Moreover, we performed co-IP analysis of 293T cells with overexpression of FLAG-tagged WT, K295A, or K295R FOXA1 using an anti-FLAG antibody, followed by WB using the K295me1 antibody. Consistent with the pan-methyl lysine antibody experiment shown in Fig. 3B, the K295me1 antibody detected FOXA1 methylation in WT, but not K295A- or K295R-expressing cells (Fig. 3H). In agreement with this, immunofluorescence staining with the K295me1 antibody detected methylated FOXA1 proteins in the majority of cells expressing WT FOXA1, but not in cells expressing K295 mutant (fig. S3H). Last, we used the K295me1 antibody to confirm if its level is increased by EZH2. The 293T cells were transfected with FLAG-FOXA1, along with either control or HA-EZH2. Co-IP using anti-FLAG (FOXA1) followed by WB using the K295me1 antibody showed markedly increased levels of FOXA1 methylation following EZH2 overexpression (Fig. 3I). We therefore conclude that EZH2 methylates FOXA1 protein at K295 to prevent its ubiquitination and increase protein stability.
Methylated FOXA1 interacts with USP7 for protein deubiquitination
We next sought to understand the molecular mechanisms by which K295 methylation on FOXA1 protects itself from ubiquitination. To identify potential enzymes that could deubiquitinate FOXA1, we performed mass spectrometry analysis of FOXA1-associated proteins in LNCaP cells, which revealed multiple deubiquitinases, including USP7, USP39, USP10, and USP9X, that interact with FOXA1 (fig. S4A and table S4). A small shRNA-based screening further pinpointed USP7 as the candidate FOXA1 deubiquitinase (fig. S4, B and C). In particular, USP7 KD decreased FOXA1 protein levels, but not FOXA1 mRNA levels (Fig. 4A and fig. S4D). Accordingly, FOXA1 protein half-life in USP7-KD cells, as compared to the control cells, was greatly reduced, suggesting that USP7 maintains FOXA1 protein stability (fig. S4E). Further, co-IP demonstrated interaction between ectopic FOXA1 and USP7 proteins expressed in 293T cells (Fig. 4B). In concordance with the observed regulation of FOXA1 protein stability, USP7 expression decreased the levels of FOXA1 ubiquitination (Fig. 4C). Together, these data suggest that USP7 interacts with FOXA1 to reduce its ubiquitination and increase its protein stability.
Fig. 4. Deubiquitinase USP7 interacts with methylated FOXA1 to remove FOXA1 protein ubiquitination.
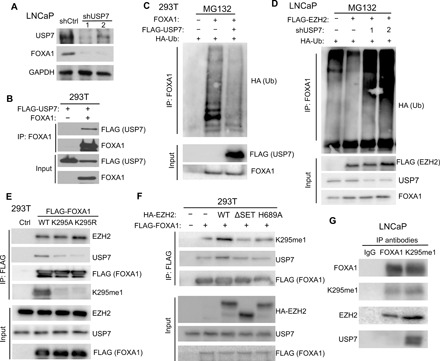
(A) LNCaP cells were infected with either shCtrl or two independent shUSP7 lentiviruses (shUSP7-1 and shUSP7-2) and subjected to WB. (B) 293T cells were transfected with FLAG-USP7, either alone or together with FOXA1. Co-IP was performed with a FOXA1 antibody. (C) 293T cells were transfected with the indicated plasmids and treated with 20 μM MG132 for 16 hours before co-IP by anti-FOXA1 and WB by anti-HA (Ub). (D) LNCaP cells stably expressing the indicated proteins were transfected with HA-Ub for 72 hours, treated with 20 μM MG132 for 16 hours, and then subjected to co-IP. (E) 293T cells were transfected with control, FLAG-FOXA1, FLAG-K295A, or FLAG-K295R. Co-IP was performed with anti-FLAG M2 beads. (F) 293T cells were transfected with FLAG-FOXA1 along with different HA-EZH2 constructs. Co-IP was performed using anti-FLAG M2 beads. (G) Co-IP using LNCaP nuclear lysates was performed with IgG, FOXA1, or K295me1 antibody.
Next, we asked whether USP7 is involved in EZH2-mediated reduction of FOXA1 ubiquitination. As expected, EZH2 expression in LNCaP cells reduced endogenous FOXA1 ubiquitination. Concurrent KD of USP7 in these cells unleashed FOXA1 ubiquitination, indicating that USP7 is required for EZH2 to reduce FOXA1 ubiquitination (Fig. 4D). Given that EZH2 requires its MTase activity to stabilize FOXA1 protein, we asked whether USP7 specifically interacts with methylated FOXA1. Co-IP analysis revealed markedly reduced interactions between USP7 and FOXA1 K295 mutants that are methylation resistant (Fig. 4E and fig. S4F). EZH2, on the other hand, showed similar interactions with FOXA1 WT and K295 mutants. Accordingly, USP7 interaction with FOXA1 was induced by overexpression of WT EZH2, but not ΔSET or H689A EZH2 that have impaired ability to catalyze FOXA1 methylation (Fig. 4F and fig. S4G). Further IP of FOXA1-K295me1 remarkably pulled down more USP7, as compared to IP of total FOXA1 (Fig. 4G). In summary, EZH2-catalyzed FOXA1 methylation recruits USP7 to mediate FOXA1 protein deubiquitination and increase its protein stability.
BUB3 recruits USP7 to methylated FOXA1 protein, and they coregulate cell cycle genes
Methyl-lysine–binding proteins, such as WD40 repeat proteins, recognize (“read”) methyl-lysines in their partner proteins and link them to downstream events (45). These reader proteins are expected to have stronger interaction with FOXA1 in the presence of MTase, e.g., EZH2. We thus performed mass spectrometry analysis of FOXA1-expressing 293T cells with control or EZH2 overexpression (table S5). Our data demonstrated increased FOXA1 association with a number of proteins that contain the WD40 methyl-lysine–binding motif, including WDR5, BUB3, WDR61, and WDR18, in cells with EZH2 overexpression (fig. S5A). To determine which of these proteins have preferential binding to methylated FOXA1, we performed co-IP analysis of 293T cells with ectopic expression of FOXA1 WT or mutants and found that the interaction between WT FOXA1 with BUB3, but not with WDR5 or WDR61, was abolished by K295A and K295R mutations (Fig. 5A). Similar K295-dependent interaction between FOXA1 and BUB3 was also observed in PCa cells (fig. S5B). Moreover, BUB3 was coenriched along with USP7 by FOXA1 proteins in mass spectrometry analysis (table S4). Reciprocal co-IP experiments of LNCaP cells confirmed FOXA1 interacting with both BUB3 and USP7 and USP7 enriching both BUB3 and FOXA1 (Fig. 5B). By contrast, while EZH2 interacts with FOXA1, it did not show interaction with BUB3, suggesting that BUB3 is recruited by methylated FOXA1 independently of EZH2 (Fig. 5C).
Fig. 5. BUB3 recruits USP7 to methylated FOXA1 for cell cycle gene regulation.
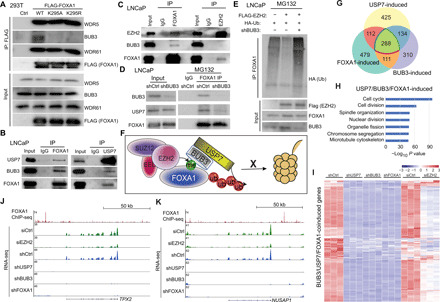
(A) Co-IP in 293T cells transfected with indicated plasmids was performed using anti-FLAG M2 beads as shown in Fig. 4E, followed by WB. (B and C) Co-IP was performed in LNCaP cells using FOXA1, USP7, or IgG antibody (B) or FOXA1, EZH2, or IgG antibody (C). (D) LNCaP cells were infected with indicated lentivirus and treated with 20 μM MG132 for 16 hours. Co-IP was performed with IgG or FOXA1 antibody. (E) LNCaP cells expressing indicated plasmids were treated with 20 μM MG132 before co-IP with a FOXA1 antibody. (F) A working model showing that PRC2 methylates (me) FOXA1, which recruits BUB3 and USP7 to remove ubiquitination (Ub), thus preventing FOXA1 degradation by the proteasome system (barrel shape structure). (G) Venn diagram showing overlap between USP7-, BUB3-, and FOXA1-induced gene sets derived from RNA-seq (adjusted P < 0.05 and fold change > 3). (H) Gene ontology (GO) analysis of USP7/BUB3/FOXA1-coinduced genes. (I) Heatmap of USP7/BUB3/FOXA1-coinduced genes in LNCaP cells with indicated gene KD. (J and K) Genome browser view of two cell cycle genes showing FOXA1 ChIP-seq (top tracks), RNA-seq in siCtrl or EZH2-KD (green tracks), and RNA-seq in shCtrl, USP7-KD, BUB3-KD, and FOXA1-KD (bottom 4) LNCaP cells.
As BUB3 and USP7 have previously been shown to form a WD40 repeat protein–containing deubiquitin complex (29), it is likely that BUB3 recognizes and binds to methylated FOXA1 and subsequently recruits USP7. To test whether BUB3 bridges the interaction between USP7 and FOXA1, we performed BUB3 KD in LNCaP cells (Fig. 5D). Co-IP using anti-FOXA1 demonstrated reduced coprecipitation of not only BUB3 protein but also USP7 in BUB3-KD cells, suggesting that BUB3 is required for FOXA1 interaction with USP7. Further, BUB3 depletion led to markedly increased FOXA1 ubiquitination even in cells with EZH2 overexpression, suggesting that EZH2 reduces FOXA1 ubiquitination requiring BUB3 (Fig. 5E). Together, these results suggest a working model wherein BUB3 recognizes and binds to methylated FOXA1 and subsequently recruits USP7 to catalyze FOXA1 deubiquitination, a process that is required for EZH2 to reduce FOXA1 ubiquitination and increase FOXA1 protein stability (Fig. 5F).
To examine downstream genes/pathways of USP7, BUB3, and FOXA1, we performed RNA-seq analysis of LNCaP cells with USP7 or BUB3 KD, which was integrated with FOXA1-KD RNA-seq data. Comparative analysis revealed remarkable overlap of the genes that were induced by USP7, FOXA1, or BUB3 (Fig. 5G). Of 959 USP7-induced and 843 BUB3-induced genes, 422 genes were coinduced by both. Further, a significant subset of this, namely, 288 genes representing 68%, was also induced by FOXA1, supporting FOXA1 as a key downstream effector of the USP7/BUB3 axis despite its many other potential substrates. Gene ontology (GO) analysis of the 288 USP7/BUB3/FOXA1-coinduced gene set showed enrichment in cell cycle, cell division, spindle organization, and other growth-promoting cellular processes (Fig. 5H). Integrative analysis with RNA-seq data of EZH2-KD cells revealed that almost all of these genes were also induced by EZH2, strongly supporting the functional relevance of the EZH2-BUB3/USP7-FOXA1 axis (Fig. 5I and table S6). For instance, similar to FOXA1 KD, loss of either USP7 or BUB3 leads to down-regulation of key cell cycle genes such as spindle assembly factors TPX2 (targeting protein for Xklp2) and NUSAP1 (nucleolar and spindle-associated protein 1) (Fig. 5, J and K). ChIP-seq showed FOXA1 binding near both genes, suggesting that they are likely direct targets of FOXA1. Further, quantitative real-time polymerase chain reaction (qRT-PCR) analysis of a subset of genes confirmed their decreased expression in LNCaP cells with USP7, BUB3, or FOXA1 KD (fig. S5C). Together, these data indicate FOXA1 as a key mediator of EZH2 and USP7/BUB3 transcriptome in PCa cells.
FOXA1 mediates EZH2’s role in promoting cell cycle progression and PCa growth
To examine the clinical relevance of EZH2-mediated stabilization of FOXA1 protein, we first confirmed reduced FOXA1 protein levels in two independent transgenic mouse models with EZH2 knockout in (i) bladder urothelial cells during development (46) and (ii) PCa cells of adult mouse (Fig. 6A and fig. S6A) (47). As immunostaining of EZH2 in human PCa tissues has provided variable results (14, 28), we determined to use H3K27me3 as a readout for EZH2 catalytic activity. Further, we examined total FOXA1 protein since the K295me1 antibody failed to provide specific signal during immunohistochemistry (IHC) staining of human tissue sections. Tissue microarray analyses (TMAs) of adjacent sections revealed strong nuclear staining of FOXA1 and H3K27me3, and their staining intensities were significantly correlated (Pearson correlation coefficient = 0.49, P < 0.001) (Fig. 6B and table S7). Next, to identify essential target genes downstream of EZH2-mediated FOXA1 methylation, we first identified the genes that are induced by EZH2 MTase activity, likely through FOXA1 methylation and stabilization, by selecting an overlapping set that was decreased in EZH2 inhibition not only by KD but also by EPZ-6438 treatment in LNCaP cells. This set of genes was further selected for up-regulation by FOXA1, resulting in 49 genes that were coinduced by both EZH2 MTase activity and FOXA1, defined as EZH2/FOXA1-induced genes (Fig. 6C). To gain insights into the biological functions of the EZH2-FOXA1 axis, we conducted GO analysis, which revealed significant enrichment of cell cycle processes including “mitotic cell cycle,” “nuclear division,” and “mitotic sister chromatid segregation” (Fig. 6D). Being consistent with the EZH2-FOXA1 axis entailing BUB3 and USP7, this EZH2/FOXA-induced gene set was significantly enriched for up-regulation by BUB3 and USP7 (fig. S6, B and C). Moreover, analysis of multiple publically available datasets (48–50) revealed that the expression levels of most EZH2/FOXA1-induced genes were indeed increased in PCa with high EZH2 expression, supporting patient relevance (Fig. 6E and fig. S6D). Further, Kaplan-Meier analysis revealed that PCa with high EZH2/FOXA1-induced gene signature was significantly associated with shorter biochemical recurrence–free survivals than those with lower signature scores (Fig. 6F and fig. S6E). In summary, these data show the pathological relevance of the EZH2-FOXA1 axis in human PCa, suggesting that EZH2 inhibitors may be useful in therapeutic targeting for FOXA1-driven tumors.
Fig. 6. EZH2 and FOXA1 induce cell cycle progression and PCa growth.
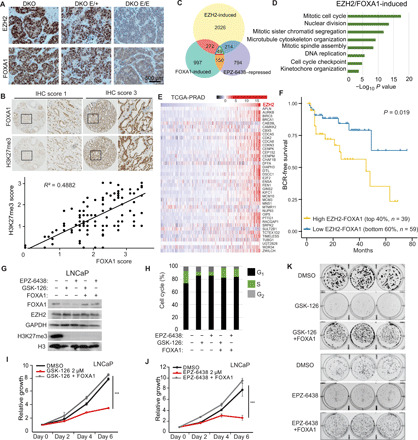
(A) PCa tumor sections from 14-week-old double knockout (DKO) (PBCre4:Ptenf/f:Rb1f/f), DKO E/+ (PBCre4:Ptenf/f:Rb1f/f:Ezh2+/f), and DKO E/E mice (PBCre4:Ptenf/f:Rb1f/f:Ezh2f/f) were stained with Ezh2 or Foxa1 antibodies. (B) IHC was performed in TMA of primary prostate tumors (n = 123). Shown are representative images (top) of FOXA1 and H3K27me3 staining with IHC scores of 1 or 3 and overall correlation (bottom) between FOXA1 and H3K27me3 immunostaining intensities. (C) Venn diagram showing overlap between EZH2-induced, EPZ-6438–repressed, and FOXA1-induced gene sets derived from previous studies (3, 37). (D) GO analysis of EZH2/FOXA1-induced genes. (E) Heatmap showing the levels of EZH2/FOXA1-induced genes in the The Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) samples (66) sorted by EZH2. (F) Kaplan-Meier curves showing biochemical recurrence (BCR)–free survival of patients in the TCGA-PRAD dataset with high versus low expression of EZH2/FOXA1-induced genes. Log-rank test, P < 0.05. (G to K) LNCaP cells stably overexpressing control or FOXA1 were treated with 2 μM GSK-126 or EPZ-6438 for 5 days before WB (G) and cell cycle analysis (H). Cell growths were measured with WST-1 reagent every 2 days (I and J). Data were normalized to day 0. Error bars, means ± SEM, n = 3. Cells were analyzed for colony formation assay for 2 weeks (K). n = 3.
We have previously shown that EPZ-6438 treatment of PCa cells leads to cell cycle arrest and a marked reduction of cells in the S phase (37). Considering our current findings of FOXA1 as a new substrate of EZH2 MTase activity, in addition to H3K27, we sought to understand the degree to which FOXA1 targeting contributes to the efficacy of these inhibitors in PCa. We treated LNCaP cells with 2 μM EPZ-6438 or GSK-126, followed by reintroduction of ectopic FOXA1 (Fig. 6G). Flow cytometry analysis revealed that FOXA1 reexpression substantially rescued a diminished S phase caused by GSK-126 or EPZ-6438 treatment (Fig. 6H) and the growth of inhibitor-suppressed LNCaP cells (Fig. 6, I and J). Similarly, colony formation assays showed that long-term treatment of GSK-126 or EPZ-6438 abolished LNCaP cell growth, which was rescued by concurrent expression of ectopic FOXA1 (Fig. 6K and fig. S6F). Together, these findings suggest that FOXA1 is an important therapeutic target of EZH2 enzymatic inhibitors in PCa.
Therapeutic targeting of FOXA1 using combinatorial EZH2 and USP7 inhibitors
Being consistent with the essential role of USP7 and BUB3 in EZH2-mediated FOXA1 stabilization and cell cycle promotion, we found significant up-regulation of USP7 and BUB3 mRNA in PCa as compared to benign prostate in multiple publically available datasets (Fig. 7A and fig. S7A). Further, PCa with high expression of USP7 or BUB3 showed significantly worse prognoses in multiple large patient cohorts (Fig. 7B and fig. S7, B and C). The EZH2/FOXA1-induced gene signature was also markedly up-regulated in human prostate tumors with high USP7 or BUB3 expression (fig. S7D). To examine USP7 and BUB3 at protein levels in PCa, we performed TMA analysis and observed abundant nuclear staining of BUB3 and USP7 in PCa cells, which showed positive correlation with FOXA1 staining intensities (Fig. 7C, fig. S7E, and table S7). These data suggest that EZH2-FOXA1 axis may be therapeutically targeted by pharmaceutical inhibitors of USP7 or BUB3.
Fig. 7. Therapeutic targeting of FOXA1-driven PCa using EZH2 and/or USP7 inhibitors.
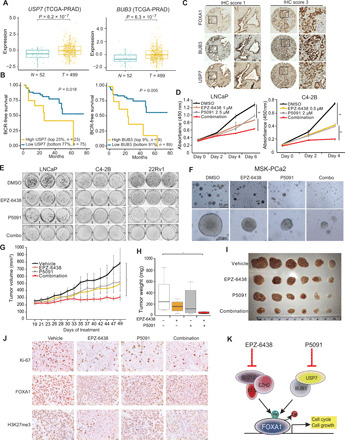
(A) USP7 and BUB3 levels in benign adjacent (N) and localized PCa (T). (B) Kaplan-Meier analyses of patients in the TCGA-PRAD dataset stratified by USP7 and BUB3 expressions. (C) IHC of FOXA1, BUB3, and USP7 was performed in adjacent sections of TMA of primary prostate tumors (n = 110). Shown are representative images with IHC scores of 1 or 3. (D) WST-1 growth assay of PCa cells treated with EZH2 or USP7 inhibitors. Error bars, means ± SEM, n = 3. (E) Colony formation assay of PCa cells treated with DMSO, EPZ-6438 (1 μM in LNCaP and 0.5 μM in C4-2B and 22Rv1), P5091 (3 μM in LNCaP and 2 μM in C4-2B and 22Rv1), or their combination for 2 weeks. (F) Representative images of mitogen- and stress-activated protein kinase (MSK)–PCa2 organoids after 10 days of treatment by DMSO, 10 μM EPZ-6438, 5 μM P5091, or combination. (G to J) C4-2B xenograft growth in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice received vehicle, EPZ-6438 (200 mg/kg) (twice a day; oral gavage), P5091 (10 mg/kg) (twice a week; intravenous), or both for 30 days (G). Tumor weights were measured at end point (H and I), and sections were stained with indicated antibodies (J). Error bars, means ± SEM, n = 6, and *P < 0.05 by analysis of variance (ANOVA). (K) A model depicting the therapeutic strategies to target FOXA1-driven PCa.
A potent and selective inhibitor (P5091) targeting deubiquitylating activity of USP7 has been tested in multiple preclinical studies (51, 52). P5091 has exhibited antitumor activities in different cancers including myeloma cells and T cell leukemia (51, 52). To determine whether targeting EZH2 and/or USP7 inhibits FOXA1 activities, we treated LNCaP cells with P5091 and EPZ-6438, either alone or in combination and confirmed suppression of FOXA1 protein levels (fig. S7F). Accordingly, ChIP-seq revealed decreased FOXA1 occupancy at the chromatin in cells treated with either inhibitors alone or their combination (fig. S7, G and H). Moreover, qRT-PCR analysis of representative genes demonstrated that the expression of FOXA1 target genes was suppressed by both inhibitors (fig. S7I).
Next, we examined functional consequences of pharmacological inhibitions of EZH2 and/or USP7. We found that C4-2B, an androgen-independent derivative of LNCaP that expresses five times more FOXA1 than LNCaP (53), was much more sensitive to EZH2 inhibition, requiring only half of the doses needed for LNCaP to achieve comparable growth inhibition (Fig. 7D). Moreover, PC3 and DU145 cells, which respectively express low or no FOXA1, showed minimal or no response to EPZ-6438 even at 10 μM dosage, suggesting that FOXA1 high cells are more sensitive to EZH2 inhibitors (fig. S7J). The cell lines showed a similar trend of varying sensitivity to USP7 inhibitor P5091, albeit in a much lesser degree, in correlation with FOXA1 levels. The combinatorial effects of P5091 and EPZ-6438 on cell proliferation were the most apparent in the FOXA1-positive LNCaP and C4-2B cells. In agreement with this, colony formation assay demonstrated similar combinatorial effects of EPZ-6438 and P5091 in reducing LNCaP, C4-2B, and 22Rv1 cell growth over long-term drug treatment (Fig. 7E and fig. S7K). Again, 22Rv1 cells, which express much lower FOXA1 than C4-2B (53), also exhibited less sensitivity to EZH2 inhibition. In summary, these data support that FOXA1 is an important therapeutic target of pharmacological inhibitors of EZH2 MTase activity and USP7.
To investigate EPZ-6438 and P5091 combination in more clinically relevant models, we performed growth assays using a patient-derived organoid culture. We treated MSK-PCa2, a prostate organoid from the acetabulum with metastatic PCa (54), with EZH2 inhibitors, either alone or in combination with USP7 inhibitors and observed a robust reduction in the growth of the organoids in response to drug combinations, similar to PCa cell lines (Fig. 7F and fig. S7L). Last, to determine the efficacy of EPZ-6438 and P5091 combination in vivo, we inoculated C4-2B cells subcutaneously into the dorsal flanks of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice, which were randomized to receive vehicle control, EPZ-6438 at 200 mg/kg daily, P5091 at 10 mg/kg twice weekly, or their combination for 30 days (Fig. 7G). We found that EPZ-6438 or P5091 alone showed a trend in reducing tumor growth as compared to vehicle treatment, albeit not statistically significant, whereas their combination significantly reduced tumor growth (P < 0.05) (Fig. 7, G to I). Ki-67 IHC staining confirmed on-target drug effects in reducing FOXA1 and H3K27me3 levels and demonstrated a substantial decrease in proliferating tumor cells by drug treatment (Fig. 7J and fig. S7M). In summary, these data demonstrated that inhibition of EZH2 and USP7 activities synergistically targets FOXA1 and suppresses PCa tumor growth.
DISCUSSION
Although EZH2 is best known for its role in epigenetically silencing tumor suppressive genes via catalyzing H3K27me3 (8, 9), growing evidence supports the notion that EZH2 can also methylate nonhistone substrates, such as transcription factors STAT3 and GATA4 (24, 27). Here, we present evidences to support FOXA1, an essential prostatic developmental regulator, as a previously unknown nonhistone substrate of EZH2 (Fig. 7K). Further, EZH2-mediated methylation of FOXA1 is polycomb dependent, given that other core PRC2 subunit SUZ12 also interacts with FOXA1 and that this methylation can be abolished by enzymatic EZH2 inhibitors. This mechanism is in contrast to the polycomb-independent role of EZH2 in acting as a coactivator of the AR protein to induce a CRPC cell–specific AR transcriptional program (28). Moreover, our findings suggest that EZH2 does not induce FOXA1 transcription, thus mechanistically different from our previously reported role of EZH2 in transcriptional activation of the AR gene (37). Further, this newly identified role of EZH2 in regulating FOXA1 posttranslational modification and protein stability is most important in primary PCa, wherein both FOXA1 and EZH2 transcription levels are high.
There are a few examples where WD40 repeat proteins have been shown to bind to methyl-lysines, including EED that recognizes H3K27me3 to enhance PRC2 function (55). Our mass spectrometry and subsequent analysis indicate that EZH2 methylation of FOXA1 recruits a different member of WD40 repeat proteins, BUB3. Subsequently, BUB3 recruits deubiquitinase USP7 to methylated FOXA1 to remove protein ubiquitination, thereby preventing its degradation by the 26S proteasome system. In line with the function of FOXA1 in cellular proliferation, BUB3 is a mitotic checkpoint protein that plays essential roles in regulating cell division and cell cycle progression (36). In addition, a previous study has reported that USP7 regulates PTEN nuclear exclusion and thus inhibits PTEN tumor suppressor function, contributing to PCa growth (34). Although there are limited studies on USP7 and BUB3 in PCa, emerging evidence link them to aggressive PCa (34, 56). It will be interesting to further understand the roles of BUB3 and USP7 as PCa progresses to CRPC.
FOXA1 is an essential regulator for androgen-dependent PCa cell growth (2, 57), and yet therapeutic targeting of FOXA1 has been challenging. Similar to FOXA1, EZH2 has been shown to induce the expression of many cell cycle genes (21). As the primary role of EZH2 and PRC2 is to maintain embryonic stem cell identity or cancer cell stemness (11, 12, 18, 58), while FOXA1 belongs to developmental regulators that are often repressed by PRC2, a cross-talk between these two proteins has not been previously envisaged. Our data show PRC2-mediated methylation and stabilization of a prostate-specific transcription factor FOXA1 and that their functions converge at the activation of cell cycle genes. This supports an interesting model wherein a stem cell maintenance factor hijacks an epithelial developmental regulator to promote cell cycle progression and PCa growth and that EZH2 inhibitors are effective in abolishing FOXA1-driven tumor growth (Fig. 7K).
Because of its overexpression and established oncogenic functions in PCa, EZH2 has been known as an important therapeutic target for decades. Several EZH2 enzymatic inhibitors are under active preclinical or clinical development (41, 59). Previous studies have suggested that the main antitumor effect of these inhibitors is mediated by blocking H3K27me3 and epigenetic silencing of tumor suppressor genes (60). However, these inhibitors have shown limited efficacy in suppressing PCa, and recent studies have suggested that the activities of these EZH2 enzymatic inhibitors may also rely on the AR status of a PCa and can be sensitized by AR inhibitors (28, 37, 61, 62). Here, we uncover FOXA1 as a novel and important target of EZH2, further contributing to the antitumor activities of EZH2 enzymatic inhibitors. We show that the growth inhibitory effects of EZH2 enzymatic inhibitors in AR-positive PCa cells, most of which are also FOXA1-positive, are dependent largely on FOXA1 degradation, as they are fully rescued by the reexpression of FOXA1. Moreover, we found that PCa cells with high FOXA1 expression levels show higher sensitivity to enzymatic EZH2 inhibitors, likely due to targeted therapy against the EZH2-FOXA1 axis in these cells. In summary, our study identifies FOXA1 as a novel substrate of EZH2 MTase activities, demonstrating the convergence of a stem cell regulator and a development regulator in regulating cell cycle and shedding light on potentially important roles of BUB3 and USP7 in PCa. Moreover, our study provides important insights into the use of enzymatic EZH2 inhibitors in the treatment of selected patient populations with FOXA1-driven PCa.
MATERIALS AND METHODS
Antibodies
The following antibodies were used in this study: rabbit anti-EZH2 [5246S, Cell Signaling Technology (CST)], mouse anti-EZH2 (05-1319, Millipore), rabbit anti-FOXA1 (ab23738, Abcam), mouse anti-FOXA1 (sc-101058, Santa Cruz Biotechnology), mouse anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab9484, Abcam), rabbit anti-HA (ab9110, Abcam), rabbit anti-H3K27me3 (9733S, CST), rabbit anti-H3 (ab1791, Abcam), mouse anti-FLAG M2 Affinity Gel (A2220, Sigma-Aldrich), rabbit anti-FLAG (F7425, Sigma-Aldrich), rabbit anti-AR (06-680, Millipore), rabbit anti-SUZ12 (3737S, CST), rabbit anti-JARID2 (13594S, CST), rabbit anti-Ub (3933S, CST), rabbit antimethylated lysine (ab76118, Abcam), rabbit antimethylated lysine (ab23366, Abcam), rabbit anti-USP7 (A300-033A-M, Bethyl), mouse anti-WDR5 (sc-393080, Santa Cruz Biotechnology), mouse anti-WDR61 (sc-100897, Santa Cruz Biotechnology), mouse anti-BUB3 (sc-376506, Santa Cruz Biotechnology), rabbit anti-MBP (homemade), rabbit anti-BUB3 (ab133699, Abcam), and rabbit anti-Ki67 (9027T, CST).
Cell lines, drugs, and plasmids
PCa cell lines LNCaP, VCaP, C4-2B, 22Rv1, PC3, and human embryonic kidney cell line HEK293T cells were obtained from American Type Culture Collection. LNCaP, C4-2B, 22Rv1, and PC3 cells were cultured in RPMI 1640 with 10% fetal bovine serum (FBS). VCaP cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS. LAPC4 cells were provided by S. Thaxton (Northwestern University) and cultured in Improved Minimum Essential Medium (IMEM) with 10% FBS and 1 nM fresh R1881. All cell lines were authenticated (Genetica DNA Laboratories) and were free of mycoplasma (Mycoalert detection kit, NC9719283, Thermo Fisher Scientific). GSK-126 (S7061) and EPZ-6438 (S7128) were purchased from Selleck Chemicals. P5091 (T6925) was purchased from TargetMol. Overexpression constructs for FOXA1 were generated as previously described (53). FOXA1 domain constructs and K295A and K295R mutants were generated using the Q5 Site-Directed Mutagenesis Kit (E0552S, New England Biolabs). Oligonucleotide sequences shEZH2-C and shEZH2-3′ were ligated with the pLKO.1 vector (#10878, Addgene) and subjected to transformation using DH5a cells. All other shRNA and siRNA were purchased from Sigma-Aldrich. The siRNAs were transfected into cells using Lipofectamine RNAiMAX. The shRNA lentiviruses were generated from 293T cells transfected with lentiviral shRNA plasmid, psPAX2, and pMD2.G by polyethylenimine transfection reagents. Viruses were collected 48 hours after transfection and filtered with 0.45-μm filters. All plasmids/constructs were validated by sequencing. All primers used in the study for mutagenesis, shRNA, and qRT-PCR are listed in table S8.
Generation of FOXA1 K295me1 antibody
New England Peptide performed peptide synthesis, rabbit immunization, serum collections, enzyme-linked immunosorbent assay (ELISA) analysis, and affinity purification for the FOXA1 K295me1 antibody. Briefly, New Zealand White rabbits were immunized three times with FOXA1 K295me1 peptide Acetyl(Ac)-CPESR[methyllysine(KMe)]DPSG-amide. After three immunizations, antiserum was subjected to an ELISA analysis to measure relative amount of antigen-specific antibody present. Antiserum from a rabbit with a higher methyllysine (KMe):methyl (Me) titer ratio was used for double affinity purification to yield polyclonal FOXA1 K295me1-specific antibody.
Ubiquitination assay
Transfected, infected, or drug-treated cells were treated with 20 μM MG132 for 12 hours before cell lysis in radioimmunoprecipitation assay buffer [25 mM tris HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, and 0.1% SDS]. Lysates were incubated overnight with appropriate antibodies (or Sigma-Aldrich M2 preconjugated beads for FLAG IP). Dynabeads Protein A (Life Technologies) were added and incubated for 1 hour at 4°C. Bound proteins were eluted from the beads with 1.5× sample buffer for 10 min at 37°C, and the supernatant was transferred to a new tube to boil at 95°C for another 2 min before SDS–polyacrylamide gel electrophoresis (PAGE) analysis.
Co-IP and gel filtration chromatography assays
Endogenous Co-IP was performed using nuclear lysates. Cytoplasmic fractions were removed with lysis buffer (10 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, and 1 mM EDTA), and the remaining nuclear pellets were lysed with nuclear protein extraction buffer [20 mM tris-HCl (pH 7.5), 420 mM NaCl, 1.5 mM MgCl2, 1% Triton X-100, 10% glycerol, and 1 mM EDTA] by incubating for 1 hour at 4°C. Nuclear lysates were subjected to centrifugation, and the supernatant was diluted with dilution buffer [20 mM tris-HCl (pH 7.5), 1.5 mM MgCl2, 1% Triton X-100, 10% glycerol, and 1 mM EDTA]. Ectopic co-IP was performed with whole-cell lysates using co-IP buffer 50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100. Either nuclear or whole-cell lysates were incubated overnight with antibodies (or M2 FLAG preconjugated beads for FLAG IP) and 1 hour with appropriate Dynabeads the following day. Bound proteins were eluted with 1.5× sample buffer for 10 min at 95°C before SDS-PAGE analysis. For gel filtration chromatography, nuclear extracts from LNCaP cells were fractionated by HiPrep Sephacryl S-400 HR column.
GST pulldown
To generate a bacterially expressed and purified FOXA1 protein, a construct encoding MBP-FOXA1 was transformed into Escherichia coli BL21. Bacterial cells were collected after 6 hours induction with 0.6 mM isopropyl-b-d-1-thiogalactopyranoside at 30°C and resuspended in binding buffer containing 20 mM tris (pH 7.4), 0.2 M NaCl, 1 mM EDTA, and protease inhibitor. After lysis by pressure homogenizer, MBP fusion proteins were affinity purified by Amylose Resin (New England Biolabs) and eluted with 10 mM maltose. For a GST pulldown assay, 0.5 μg of GST-EZH2 was conjugated to 30 μl of glutathion sepharose beads by incubating in buffer A [20 mM tris-HCl (pH7.9), 20% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.1% NP-40,1 mM dithiothreitol (DTT), and 0.1 M NaCl] for 2 hours at 4°C. EZH2-bound GST beads were washed twice with buffer B [20 mM tris-HCl (pH 7.9), 20% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.1% NP-40, 1 mM DTT, and 1 M NaCl] and another two times with buffer C [20 mM tris-HCl (pH 7.9), 20% glycerol, 5 mM MgCl2, 5 mM CaCl2, 0.1% NP-40, 1 mM DTT, and 0.1 M NaCl]. Then, 10 μg of MBP–green fluorescent protein (GFP) or MBP-FOXA1 proteins were incubated with EZH2-bound GST beads in 30 μl of buffer C for 2 hours at 4°C. After four washes with buffer C, proteins were eluted with 1.5× SDS buffer for 10 min at 95°C.
Immunohistochemistry
IHC was performed on paraffin-embedded mouse tissue sections. After deparaffinization, rehydration, and antigen retrieval with citrate buffer, tissues were permeabilized with 0.5% Triton X-100 and sequentially blocked with 0.3% H2O2, avidin/biotin, and 2% bovine serum albumin. Then, slides were incubated with anti-EZH2 (1:800; 5246, Cell Signaling Technology) or anti-FOXA1 (1:200; EPR10881, Abcam) overnight at 4°C. After extensive washing with 1× tris-buffered saline, 0.1% Tween 20 (TBST), slides were incubated with biotin-conjugated secondary antibody (1:200) for 1 hour at room temperature. Slides were washed again with TBST and incubated with streptavidin–horseradish peroxidase (1:500) for 15 min at room temperature. After another round of extensive wash with TBST, slides were incubated with 3,3′-Diaminobenzidine (DAB) substrate for 5 to 10 min at room temperature. Slides were then counterstained with hematoxylin for 1 s, washed with water, dehydrated in ethanol, cleared with xylene, and mounted with permount.
Immunofluorescence
Cells growing on poly-d-lysine–coated coverslips were transfected with appropriate plasmids for 48 hours. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature, followed by permeabilization with 0.1% Triton X-100 for 15 min at room temperature. After three washes with phosphate-buffered saline (PBS), cells were incubated with blocking buffer (5% normal goat serum in PBS) for 30 min. Then, the cells were incubated with primary antibody in blocking buffer overnight at 4°C. This was followed by three washes and incubation with secondary antibodies conjugated with Alexa Fluor 488 or Alex Fluor 594 (A11034 or A11037, Invitrogen) for 1 hour. After three washes with PBS, coverslips were mounted on glass slides, and the cells were imaged by Nikon A1 Confocal Laser Microscope System.
Tissue acquisition and TMA analysis
TMAs of primary PCa (n = 51 patients; n = 143 sites) were generated at the Northwestern University Pathology Core through the prostate SPORE program and approved by the Northwestern University Institutional Review Board. Studies were performed in compliance with the institutional guidelines of Northwestern University. Human TMA IHC staining was conducted using the Dako Autostainer Link 48 with enzyme-labeled biotin streptavidin system and the SIGMAFAST DAB Map Kit (MilliporeSigma). Antibodies used in IHC include anti-FOXA1 (1:400; ab23738, Abcam), anti-H3K27me3 (1:200; 9733, CST), anti-BUB3 (1:100; ab133699, Abcam), and anti-USP7 (1:100; A300-033A, Bethyl). Images were captured with TissueFAXS PLUS from TissueGnostics, exported to TissueFAXS viewer, and analyzed using Photoshop CS4 (Adobe). For each site, percentage of stained cells (with total of 100% for each site) and their staining intensity score ranging from 0 to 3 were determined. The overall score for each site was calculated on the basis of the product of the percentage of cells and their staining intensity and restratified to a range of 0 to 3.
Cell cycle analysis
A total of 1 × 106 cells were washed twice with 1 ml of PBS. The washed cells were fixed with 70% ethanol in double-distilled H2O at −20°C overnight. Fixed cells were washed twice with 1 ml of PBS and stained with propidium iodide staining solution (20 mg/ml; 2 mg of ribonuclease A, 0.1% Triton X-100, and 2 mM EDTA) for 20 min at 37°C. Cells were subjected to flow cytometry analysis using LSRFortessa cell analyzer (BD Biosciences) and analyzed by ModFit LT (Verity Software).
WST-1 cell proliferation and colony formation assay
Cell proliferation was measured with tetrazolium salt (WST-1) (Promega), as described by the manufacturer. Briefly, cells seeded in 96-well plates were incubated with tetrazolium salt (WST-1) for 2 hours at 37°C and 5% CO2. The absorbance was measured at 440 nm using the KC4 microplate reader (BioTek Instruments) and normalized to the media control. For colony formation assay, cells were infected with appropriate lentivirus for 72 hours before 8000 LNCaP cells per well were seeded into a six-well plate. Seeded colonies were treated with the indicated concentrations of DMSO, GSK-126/EPZ-6438, or P5091 for 14 days. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature, followed by staining with 0.025% crystal violet for 2 hours. The ColonyArea ImageJ plugin was used to calculate colony area percentages, and the comparisons were performed by analysis of variance (ANOVA) with Bonferroni’s multiple comparison test.
RNA isolation and qRT-PCR
RNA was isolated from cells using NucleoSpin RNA isolation kit (MACHEREY-NAGEL). The complementary DNA (cDNA) was synthesized from 500 ng of RNA using qScript cDNA Synthesis SuperMix (Quanta BioSciences). The Applied Biosystems StepOnePlus was used to perform a real-time PCR.
Quantification and statistical analysis
For protein half-life measurement, FOXA1 protein levels were estimated by ImageJ, normalized to GAPDH loading control, converted to a percentage relative to corresponding values at t = 0, and plotted on a log scale. FOXA1 half-lives were calculated using linear regression analyses. For qRT-PCR analysis of KD or overexpression experiments, Student’s t test (two-tailed unpaired) was used to determine statistical significance. Error bars are presented as the means ± SD from triplicate samples. For statistical analysis between multiple groups, we performed one-way ANOVA with Tukey post hoc test at the significance level of α equals 0.05.
RNA-seq and ChIP-seq
Total RNA was isolated from cells using NucleoSpin RNA isolation kit (MACHEREY-NAGEL). RNA-seq and ChIP-seq were performed using previously described protocols (37).
Gene expression and signature analysis
Reference datasets were prepared from our previous studies of EZH2 and FOXA1 and retrieved from the National Center for Biotechnology Information gene expression omnibus (GEO) with accession numbers GSE107778 and GSE128882, as previously described (3). GSEA was performed as previously described (64, 65). To identify the EZH2/FOXA1-coinduced gene set, a Venn diagram (Venny 2.1) was used to overlap identified gene sets derived from our previous studies (3, 37). Kaplan-Meier test was used for survival analysis using gene set compound scores. First, we calculated Pearson correlation coefficients between the expression level of EZH2 and each of the EZH2/FOXA1-induced genes for every dataset. A weighted score for each sample was generated as the summation of products of the expression of each gene multiplied by its corresponding correlation coefficient (the weight) with EZH2. On the basis of their weighted scores, samples were then rank ordered and stratified using the criteria indicated in the figures for survival analyses and Kaplan-Meier plots using survival and survminer R packages.
Human prostate organoid
Human prostate organoid MSK-PCa2 was derived from a patient with PCa with metastatic adenocarcinoma and was a gift from Y. Chen (54). After treatment with TrypLE (Sigma-Aldrich) for 5 min at 37°C, organoids were seeded at 1:3 ratio in the Matrigel and cultured in Advanced DMEM/F12 containing epithelial growth factor (50 ng/ml), 5% (v/v) R-spondin 1, 10% (v/v) Noggin, fibroblast growth factor 10 (FGF10) (10 ng/ml), FGF2 (1 ng/ml), 1 nM dihydrotestosterone, 10 mM nicotninamide, 0.5 μM A83-01, 10 μM Y-27632, 1× B27 additive, 1.25 mM N-acetyl-l-cysteine, 2 mM GlutaMAX, 10 mM Hepes, and 1:100 (v/v) primocin. Organoids were treated with indicated inhibitors 48 hours after seeding and replaced with fresh medium with inhibitors every 2 days.
Xenograft experiments
All procedures involving mice were approved by the Institutional Animal Care and Use Committee at Northwestern University in compliance with all relevant ethical regulations. NOD/SCID (Charles River Laboratory, Wilmington, MA, USA) mice of 5 to 6 weeks of age were used for Xenograft. A total of 2 × 106 of C4-2B cells were suspended in 200 μl of PBS with 50% Matrigel (BD Biosciences) and injected subcutaneously into the dorsal flank of the mice. Mice were randomly divided into four different groups and treated with 200 μl of vehicle control, EPZ-6438 (200 mg/kg; oral gavage), P5091 (10 mg/kg; intravenous), or in combination. EPZ-6438 were administered twice a day, and P5091 were given twice a week. Tumor volumes were measured with digital caliper once every 2 days in a blinded fashion and calculated with the formula, V = p/6 (length × width2). After 30 days of treatments, mice were euthanized, tumors were excised and weighed. The effects of drug treatment in suppressing target pathways were examined by IHC analysis. Tumor sections were fixed with formalin and embedded in paraffin. Formalin-fixed and paraffin-embedded tumor section were then stained with Ki-67, FOXA1, and H3K27me3 antibodies.
Acknowledgments
We thank E. Mong for constructive criticism of the manuscript. We thank C. Marinaccio for assistance with the flow cytometry. We thank X. Yang (University of Texas Southwestern Medical Center) for help with some mass spectrometry experiments. Size exclusion chromatography was conducted by the Recombinant Protein Production Core (rPPC), Northwestern University. TMA staining and mouse tumor sectioning and embedding were conducted by the Pathology Core of Northwestern University. Funding: Mass spectrometry services were provided by the Taplin Mass Spectrometry Facility, Harvard Medical School, and Northwestern Proteomics Core Facility, supported by NCI CCSG P30CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S100D025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics (P41 GM108569). This work was supported in part by the NIH/NCI training grant T32CA09560 (to S.H.P.), the American Cancer Society IRG-18-163-24 (to K.-w.F.), R01CA172384 and R01CA227918 (to J.Y.), PCa SPORE P50CA180995 (to J.Y.), R50CA211271 (to J.C.Z.), R01CA234162 (to D.W.G.), R01CA207757 (to D.W.G.), R01DK110477(to X. Li), GM121662 (to X. Liu), GM136308 (to X. Liu), Welch Foundation research grant I-1790 (to X. Liu), Department of Defense grants W81XWH-17-1-0405 and W81XWH-17-1-0578 (to J.Y.), and Prostate Cancer Foundation 2017CHAL2008 (to J.Y. and J.C.Z.). Author contributions: J.Y. and S.H.P. conceived the project and designed the experiments. S.H.P., K.-w.F., J.K., F.W., X. Lu, K.W., and C.G. performed the experiments. J.C.Z., S.H.P., and J.Y. conducted the bioinformatics and statistics analysis. X. Li and D.W.G. assisted with mouse tissue acquisition. X. Liu, Y.L., S.A.A., J.D.C., D.F., P.N., and Y.W. provided critical discussions of the project. J.Y., S.H.P., and L.T.B. generated the figures and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-seq and ChIP-seq data that were generated in this study have been uploaded to GEO database under GSE161519. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe2261/DC1
REFERENCES AND NOTES
- 1.Gao N., Ishii K., Mirosevich J., Kuwajima S., Oppenheimer S. R., Roberts R. L., Jiang M., Yu X., Shappell S. B., Caprioli R. M., Stoffel M., Hayward S. W., Matusik R. J., Forkhead box A1 regulates prostate ductal morphogenesis and promotes epithelial cell maturation. Development 132, 3431–3443 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Jin H.-J., Zhao J. C., Ogden I., Bergan R. C., Yu J., Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 73, 3725–3736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu B., Song B., Lu X., Kim J., Hu M., Zhao J. C., Yu J., Altered chromatin recruitment by FOXA1 mutations promotes androgen independence and prostate cancer progression. Cell Res. 29, 773–775 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin H.-J., Zhao J. C., Wu L., Kim J., Yu J., Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 5, 3972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams E. J., Karthaus W. R., Hoover E., Liu D., Gruet A., Zhang Z., Cho H., DiLoreto R., Chhangawala S., Liu Y., Watson P. A., Davicioni E., Sboner A., Barbieri C. E., Bose R., Leslie C. S., Sawyers C. L., FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature 571, 408–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parolia A., Cieslik M., Chu S.-C., Xiao L., Ouchi T., Zhang Y., Wang X., Vats P., Cao X., Pitchiaya S., Su F., Wang R., Feng F. Y., Wu Y.-M., Lonigro R. J., Robinson D. R., Chinnaiyan A. M., Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C., Wang L., Wu D., Chen H., Chen Z., Thomas-Ahner J. M., Zynger D. L., Eeckhoute J., Yu J., Luo J., Brown M., Clinton S. K., Nephew K. P., Huang T. H.-M., Li W., Wang Q., Definition of a FoxA1 cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 71, 6738–6748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y., Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Cao R., Zhang Y., SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Margueron R., Reinberg D., The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee T. I., Jenner R. G., Boyer L. A., Guenther M. G., Levine S. S., Kumar R. M., Chevalier B., Johnstone S. E., Cole M. F., Isono K., Koseki H., Fuchikami T., Abe K., Murray H. L., Zucker J. P., Yuan B., Bell G. W., Herbolsheimer E., Hannett N. M., Sun K., Odom D. T., Otte A. P., Volkert T. L., Bartel D. P., Melton D. A., Gifford D. K., Jaenisch R., Young R. A., Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyer L. A., Plath K., Zeitlinger J., Brambrink T., Medeiros L. A., Lee T. I., Levine S. S., Wernig M., Tajonar A., Ray M. K., Bell G. W., Otte A. P., Vidal M., Gifford D. K., Young R. A., Jaenisch R., Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Bernstein E., Duncan E. M., Masui O., Gil J., Heard E., Allis C. D., Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 26, 2560–2569 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varambally S., Dhanasekaran S. M., Zhou M., Barrette T. R., Kumar-Sinha C., Sanda M. G., Ghosh D., Pienta K. J., Sewalt R. G. A. B., Otte A. P., Rubin M. A., Chinnaiyan A. M., The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Chen H., Tu S.-w., Hsieh J.-T., Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J. Biol. Chem. 280, 22437–22444 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Cao Q., Yu J., Dhanasekaran S. M., Kim J. H., Mani R. S., Tomlins S. A., Mehra R., Laxman B., Cao X., Yu J., Kleer C. G., Varambally S., Chinnaiyan A. M., Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27, 7274–7284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren G., Baritaki S., Marathe H., Feng J., Park S., Beach S., Bazeley P. S., Beshir A. B., Fenteany G., Mehra R., Daignault S., Al-Mulla F., Keller E., Bonavida B., de la Serna I., Yeung K. C., Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 72, 3091–3104 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Yu J., Cao Q., Yu J., Wu L., Dallol A., Li J., Chen G., Grasso C., Cao X., Lonigro R. J., Varambally S., Mehra R., Palanisamy N., Wu J. Y., Latif F., Chinnaiyan A. M., The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene 29, 5370–5380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin Y. J., Kim J.-H., The role of EZH2 in the regulation of the activity of matrix metalloproteinases in prostate cancer cells. PLOS ONE 7, e30393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu J., Cao Q., Mehra R., Laxman B., Yu J., Tomlins S. A., Creighton C. J., Dhanasekaran S. M., Shen R., Chen G., Morris D. S., Marquez V. E., Shah R. B., Ghosh D., Varambally S., Chinnaiyan A. M., Integrative genomics analysis reveals silencing of β-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 12, 419–431 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Bracken A. P., Pasini D., Capra M., Prosperini E., Colli E., Helin K., EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22, 5323–5335 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanulli S., Justin N., Teissandier A., Ancelin K., Portoso M., Caron M., Michaud A., Lombard B., da Rocha S. T., Offer J., Loew D., Servant N., Wassef M., Burlina F., Gamblin S. J., Heard E., Margueron R., Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol. Cell 57, 769–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ardehali M. B., Anselmo A., Cochrane J. C., Kundu S., Sadreyev R. I., Kingston R. E., Polycomb repressive complex 2 methylates elongin A to regulate transcription. Mol. Cell 68, 872–884.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He A., Shen X., Ma Q., Cao J., von Gise A., Zhou P., Wang G., Marquez V. E., Orkin S. H., Pu W. T., PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 26, 37–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J. M., Lee J. S., Kim H., Kim K., Park H., Kim J.-Y., Lee S. H., Kim I. S., Kim J., Lee M., Chung C. H., Seo S.-B., Yoon J.-B., Ko E., Noh D.-Y., Kim K. I., Kim K. K., Baek S. H., EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 48, 572–586 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Vasanthakumar A., Xu D., Lun A. T., Kueh A. J., van Gisbergen K. P., Iannarella N., Li X., Yu L., Wang D., Williams B. R., Lee S. C., Majewski I. J., Godfrey D. I., Smyth G. K., Alexander W. S., Herold M. J., Kallies A., Nutt S. L., Allan R. S., A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. 18, 619–631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim E., Kim M., Woo D.-H., Shin Y., Shin J., Chang N., Oh Y. T., Kim H., Rheey J., Nakano I., Lee C., Joo K. M., Rich J. N., Nam D.-H., Lee J., Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu K., Wu Z. J., Groner A. C., He H. H., Cai C., Lis R. T., Wu X., Stack E. C., Loda M., Liu T., Xu H., Cato L., Thornton J. E., Gregory R. I., Morrissey C., Vessella R. L., Montironi R., Magi-Galluzzi C., Kantoff P. W., Balk S. P., Liu X. S., Brown M., EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villamil M. A., Liang Q., Zhuang Z., The WD40-repeat protein-containing deubiquitinase complex: Catalysis, regulation, and potential for therapeutic intervention. Cell Biochem. Biophys. 67, 111–126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M., Brooks C. L., Kon N., Gu W., A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Huang Z., Wu Q., Guryanova O. A., Cheng L., Shou W., Rich J. N., Bao S., Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat. Cell Biol. 13, 142–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Horst A., de Vries-Smits A. M. M., Brenkman A. B., van Triest M. H., van den Broek N., Colland F., Maurice M. M., Burgering B. M. T., FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 8, 1064–1073 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Giovinazzi S., Morozov V. M., Summers M. K., Reinhold W. C., Ishov A. M., USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death Differ. 20, 721–731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song M. S., Salmena L., Carracedo A., Egia A., Lo-Coco F., Teruya-Feldstein J., Pandolfi P. P., The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W., Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Logarinho E., Resende T., Torres C., Bousbaa H., The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore-microtubule attachments. Mol. Biol. Cell 19, 1798–1813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J., Lee Y., Lu X., Song B., Fong K. W., Cao Q., Licht J. D., Zhao J. C., Yu J., Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S. T., Li Z., Wu Z., Aau M., Guan P., Murthy Karuturi R. K., Liou Y. C., Yu Q., Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 43, 798–810 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Shi B., Liang J., Yang X., Wang Y., Zhao Y., Wu H., Sun L., Zhang Y., Chen Y., Li R., Zhang Y., Hong M., Shang Y., Integration of estrogen and wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 27, 5105–5119 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cirillo L. A., Lin F. R., Cuesta I., Friedman D., Jarnik M., Zaret K. S., Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289 (2002). [DOI] [PubMed] [Google Scholar]
- 41.McCabe M. T., Ott H. M., Ganji G., Korenchuk S., Thompson C., Van Aller G. S., Liu Y., Graves A. P., Iii A. D. P., Diaz E., LaFrance L. V., Mellinger M., Duquenne C., Tian X., Kruger R. G., McHugh C. F., Brandt M., Miller W. H., Dhanak D., Verma S. K., Tummino P. J., Creasy C. L., EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Knutson S. K., Warholic N. M., Wigle T. J., Klaus C. R., Allain C. J., Raimondi A., Porter Scott M., Chesworth R., Moyer M. P., Copeland R. A., Richon V. M., Pollock R. M., Kuntz K. W., Keilhack H., Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 110, 7922–7927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D., Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes Dev. 16, 2893–2905 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Justin N., Zhang Y., Tarricone C., Martin S. R., Chen S., Underwood E., De Marco V., Haire L. F., Walker P. A., Reinberg D., Wilson J. R., Gamblin S. J., Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7, 11316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hyun K., Jeon J., Park K., Kim J., Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49, e324–e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo C., Balsara Z. R., Hill W. G., Li X., Stage- and subunit-specific functions of polycomb repressive complex 2 in bladder urothelial formation and regeneration. Development 144, 400–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wadosky K. M., Wang Y., Ellis L., Goodrich D. W., Abstract 3016: Ezh2 is a dose-dependent mediator of prostate cancer aggressiveness and lineage transformation. Cancer Res. 78, 3016 (2018). [Google Scholar]
- 48.Ross-Adams H., Lamb A. D., Dunning M. J., Halim S., Lindberg J., Massie C. M., Egevad L. A., Russell R., Ramos-Montoya A., Vowler S. L., Sharma N. L., Kay J., Whitaker H., Clark J., Hurst R., Gnanapragasam V. J., Shah N. C., Warren A. Y., Cooper C. S., Lynch A. G., Stark R., Mills I. G., Grönberg H., Neal D. E.; CamCaP Study Group , Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2, 1133–1144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sboner A., Demichelis F., Calza S., Pawitan Y., Setlur S. R., Hoshida Y., Perner S., Adami H.-O., Fall K., Mucci L. A., Kantoff P. W., Stampfer M., Andersson S.-O., Varenhorst E., Johansson J.-E., Gerstein M. B., Golub T. R., Rubin M. A., Andrén O., Molecular sampling of prostate cancer: A dilemma for predicting disease progression. BMC Med. Genomics 3, 8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor B. S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B. S., Arora V. K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J. E., Wilson M., Socci N. D., Lash A. E., Heguy A., Eastham J. A., Scher H. I., Reuter V. E., Scardino P. T., Sander C., Sawyers C. L., Gerald W. L., Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chauhan D., Tian Z., Nicholson B., Kumar K. G. S., Zhou B., Carrasco R., McDermott J. L., Leach C. A., Fulcinniti M., Kodrasov M. P., Weinstock J., Kingsbury W. D., Hideshima T., Shah P. K., Minvielle S., Altun M., Kessler B. M., Orlowski R., Richardson P., Munshi N., Anderson K. C., A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin Q., Martinez C. A., Arcipowski K. M., Zhu Y., Gutierrez-Diaz B. T., Wang K. K., Johnson M. R., Volk A. G., Wang F., Wu J., Grove C., Wang H., Sokirniy I., Thomas P. M., Goo Y. A., Abshiru N. A., Hijiya N., Peirs S., Vandamme N., Berx G., Goosens S., Marshall S. A., Rendleman E. J., Takahashi Y.-H., Wang L., Rawat R., Bartom E. T., Collings C. K., Van Vlierberghe P., Strikoudis A., Kelly S., Ueberheide B., Mantis C., Kandela I., Bourquin J.-P., Bornhauser B., Serafin V., Bresolin S., Paganin M., Accordi B., Basso G., Kelleher N. L., Weinstock J., Kumar S., Crispino J. D., Shilatifard A., Ntziachristos P., USP7 cooperates with NOTCH1 to drive the oncogenic transcriptional program in T-cell leukemia. Clin. Cancer Res. 25, 222–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y. A., Zhao J. C., Fong K.-W., Kim J., Li S., Song C., Song B., Zheng B., He C., Yu J., FOXA1 potentiates lineage-specific enhancer activation through modulating TET1 expression and function. Nucleic Acids Res. 44, 8153–8164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao D., Vela I., Sboner A., Iaquinta P. J., Karthaus W. R., Gopalan A., Dowling C., Wanjala J. N., Undvall E. A., Arora V. K., Wongvipat J., Kossai M., Ramazanoglu S., Barboza L. P., Di W., Cao Z., Zhang Q. F., Sirota I., Ran L., MacDonald T. Y., Beltran H., Mosquera J.-M., Touijer K. A., Scardino P. T., Laudone V. P., Curtis K. R., Rathkopf D. E., Morris M. J., Danila D. C., Slovin S. F., Solomon S. B., Eastham J. A., Chi P., Carver B., Rubin M. A., Scher H. I., Clevers H., Sawyers C. L., Chen Y., Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Margueron R., Justin N., Ohno K., Sharpe M. L., Son J., Drury W. J., Voigt P., Martin S., Taylor W. R., De Marco V., Pirrotta V., Reinberg D., Gamblin S. J., Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ersvær E., Kildal W., Vlatkovic L., Cyll K., Pradhan M., Kleppe A., Hveem T. S., Askautrud H. A., Novelli M., Wæhre H., Liestøl K., Danielsen H. E., Prognostic value of mitotic checkpoint protein BUB3, cyclin B1, and pituitary tumor-transforming 1 expression in prostate cancer. Mod. Pathol. 33, 905–915 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerhardt J., Montani M., Wild P., Beer M., Huber F., Hermanns T., Müntener M., Kristiansen G., FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 180, 848–861 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Yu J., Yu J., Mani R. S., Cao Q., Brenner C. J., Cao X., Wang X., Wu L., Li J., Hu M., Gong Y., Cheng H., Laxman B., Vellaichamy A., Shankar S., Li Y., Dhanasekaran S. M., Morey R., Barrette T., Lonigro R. J., Tomlins S. A., Varambally S., Qin Z. S., Chinnaiyan A. M., An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17, 443–454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knutson S. K., Kawano S., Minoshima Y., Warholic N. M., Huang K. C., Xiao Y., Kadowaki T., Uesugi M., Kuznetsov G., Kumar N., Wigle T. J., Klaus C. R., Allain C. J., Raimondi A., Waters N. J., Smith J. J., Porter-Scott M., Chesworth R., Moyer M. P., Copeland R. A., Richon V. M., Uenaka T., Pollock R. M., Kuntz K. W., Yokoi A., Keilhack H., Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Kim K. H., Roberts C. W. M., Targeting EZH2 in cancer. Nat. Med. 22, 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dardenne E., Beltran H., Benelli M., Gayvert K., Berger A., Puca L., Cyrta J., Sboner A., Noorzad Z., MacDonald T., Cheung C., Yuen K. S., Gao D., Chen Y., Eilers M., Mosquera J. M., Robinson B. D., Elemento O., Rubin M. A., Demichelis F., Rickman D. S., N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30, 563–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ku S. Y., Rosario S., Wang Y., Mu P., Seshadri M., Goodrich Z. W., Goodrich M. M., Labbe D. P., Gomez E. C., Wang J., Long H. W., Xu B., Brown M., Loda M., Sawyers C. L., Ellis L., Goodrich D. W., Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schuur E. R., Henderson G. A., Kmetec L. A., Miller J. D., Lamparski H. G., Henderson D. R., Prostate-specific antigen expression is regulated by an upstream enhancer. J. Biol. Chem. 271, 7043–7051 (1996). [DOI] [PubMed] [Google Scholar]
- 64.Mootha V. K., Lindgren C. M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., Groop L. C., PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer Genome Atlas Research Network , The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe2261/DC1