

The engineered nanoparticles mitigate osteoarthritis progression.
Abstract
Treating osteoarthritis (OA) remains a major clinical challenge. Despite recent advances in drug discovery and development, no disease-modifying drug for knee OA has emerged with any notable clinical success, in part, due to the lack of valid and responsive therapeutic targets and poor drug delivery within knee joints. In this work, we show that the amount of secretory phospholipase A2 (sPLA2) enzyme increases in the articular cartilage in human and mouse OA cartilage tissues. We hypothesize that the inhibition of sPLA2 activity may be an effective treatment strategy for OA. To develop an sPLA2-responsive and nanoparticle (NP)–based interventional platform for OA management, we incorporated an sPLA2 inhibitor (sPLA2i) into the phospholipid membrane of micelles. The engineered sPLA2i-loaded micellar NPs (sPLA2i-NPs) were able to penetrate deep into the cartilage matrix, prolong retention in the joint space, and mitigate OA progression. These findings suggest that sPLA2i-NPs can be promising therapeutic agents for OA treatment.
INTRODUCTION
Osteoarthritis (OA) is a painful and debilitating disease of the articular cartilage, leading to joint pain, loss of joint function, and deleterious effects on the quality of daily life. It occurs in approximately 20 to 30 million adults in the United States alone, with staggering societal and economic costs (60 billion per year) (1). Current treatments for OA include nonpharmacological treatment (e.g., diet or exercise), pharmacological approaches, and surgical intervention (2). Although many pharmacologic approaches have been extensively pursued and some drugs have shown promise in preclinical studies, none has emerged with any notable clinical success, and there are no disease-modifying therapies available to delay and/or limit OA development and progression (3). Accordingly, there is a great unmet medical need to develop new interventional platforms for greater effectiveness in OA treatment.
The etiology of OA is broad and includes various mechanical, biochemical, and genetic factors (4, 5). Recent work has indicated that chronic unresolved inflammation has a critical role in OA development and progression (6, 7). Analyses of tissues from both human patients and animal models of OA have revealed that different inflammatory mediators have been implicated in OA pathogenesis (4). However, the most widely used nonsteroidal anti-inflammatory drugs only provide the short-term management of the pain symptoms in OA. Moreover, they have substantial renal and cardiovascular toxicities and limited efficacy without repeated administration (8). Thus, the identification of new anti-inflammatory targets may provide benefits in the treatment of OA disease.
Among the potent inflammatory mediators involved in the development of OA is the family of secreted phospholipase A2 (sPLA2) enzymes. sPLA2 is a heterogeneous group of enzymes that specifically recognize and catalytically hydrolyze the sn-2 ester bond of glycerophospholipids, releasing free fatty acids such as arachidonic acid (AA) and lysophospholipids (9), which are well-known mediators of inflammation and tissue damage (10–16). sPLA2 is normally present at low levels in healthy knee joint tissues. However, under pathological conditions, sPLA2 can be induced by multiple cascades and effector molecules including inflammatory cytokines (17, 18) and free radicals (19). A high expression level and activity of sPLA2 are observed in the synovial membrane, synovial fluid, and articular cartilage of human OA patients (20–23). Hence, we postulated that inhibition of sPLA2 enzyme activity by sPLA2 inhibitor (sPLA2i) can be exploited as a promising therapeutic strategy for OA treatment. Note that several sPLA2i compounds have already been developed and used in clinical trials for treating other inflammatory diseases (24). However, few studies have examined the role of sPLA2 in OA, and no study has sought to harness the sPLA2 activity for the treatment of OA (18).
Clinical management of OA is further complicated by lack of effective drug delivery systems (25, 26). Rapid clearance of the drugs by the joint (i.e., a short half-life) and therapeutic target sites deep within the cartilage not accessible to drugs pose significant delivery challenges for many promising drugs (27, 28). Recently, nanoparticle (NP)–based targeted drug delivery has been exploited in the treatment of OA (1, 27, 29). The main benefits of these delivery systems are that they can increase the retention of drugs in the joints, thus lowering the therapeutic dose, reducing the frequency of administration, increasing therapeutic efficacy, and reducing off-target toxicity. Here, we examined whether inhibition of sPLA2 enzyme activity by sPLA2i-loaded NPs could be an effective OA therapy. Specifically, a lipid-based sPLA2i, thioetheramide-PC [molecular weight (MW), 735.1 Da], was incorporated into nanometer-sized phospholipid micelles. The joint retention, cartilage penetration, biodistribution, and toxicity of the sPLA2i-loaded micellar NPs (sPLA2i-NPs) were subsequently characterized. Using an in vitro cartilage explant model and two animal models of OA, we investigated the effectiveness of sPLA2i-NPs in inhibiting the inflammatory signals and attenuating the OA progression following direct delivery into knee joints.
RESULTS
sPLA2 amount in OA cartilage
To confirm the elevated levels of sPLA2 enzyme in OA-related tissues, we performed immunohistochemistry (IHC) to investigate a commonly studied isoform sPLA2-IIA in knees. In healthy young and adult human cartilage, the level of sPLA2-IIA was very low at the top and middle region of the articular cartilage (Fig. 1, A and B). However, its amount was markedly elevated in the OA cartilage at early and middle stages. Abundant staining was found in the cartilage matrix and chondrocytes. Mouse cartilage showed similar results (Fig. 1, C and D). While normal mouse articular cartilage displayed only a weak staining of sPLA2-IIA, the cartilage in mouse knees receiving destabilization of the medial meniscus (DMM) surgery a month earlier showed greatly enhanced staining throughout the cartilage. The marked increase in sPLA2 in human and mouse OA cartilage suggested its potential role in OA development.
Fig. 1. sPLA2 expression in human and mouse OA cartilage.
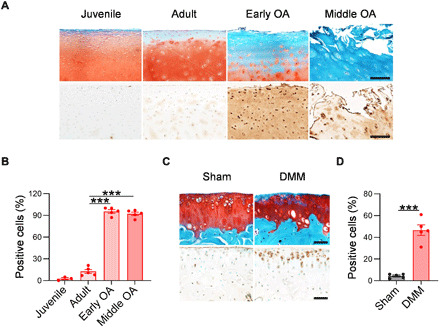
(A) Representative images of Safranin O/Fast Green staining (top) and IHC of sPLA2 (bottom) in healthy juvenile, healthy adult, early-stage, and middle-stage human cartilage tissues. Scale bars, 200 μm. (B) Quantification of sPLA2-positive chondrocytes as a proportion of total chondrocytes in healthy and OA human articular cartilage tissues (n = 5). (C) Representative images of Safranin O/Fast Green staining (top) and IHC of sPLA2 (bottom) in the tibial articular cartilage of WT mice at 1 month after sham or after DMM surgery. Scale bars, 50 μm. (D) Quantification of sPLA2-positive chondrocytes in the tibial articular cartilage of sham- or DMM-operated joints (n = 5). Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s post hoc test for (B) and paired two-tailed t test for (D). Data presented as means ± SEM. ***P < 0.001.
sPLA2i-NP synthesis and characterization
sPLA2i-NPs were prepared by incorporating 25 mole percent (mol %) thioetheramide-PC into phospholipid micelles with lipid composition 10 mol % 1,2-dioleoyl-3-trimethylammonium propane (DOTAP)/65 mol % 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) (Fig. 2A). Because of its amphiphilic nature, lipid-based sPLA2i was easily doped into the phospholipid film during NP preparation, and the sPLA2i loading efficiency was almost 100%. Dynamic light scattering (DLS) measurements revealed that sPLA2i-NPs had a mean hydrodynamic diameter of 10 nm and a relatively narrow size distribution (Fig. 2B). sPLA2i-NPs observed by transmission electron microscopy (TEM) were approximately spherical in shape together with some worm-like structures (fig. S1). As shown in Fig. 2C, the presence of cationic DOTAP within sPLA2i-loaded NPs transitioned NPs from a negative surface charge (−8 mV) to a positive surface charge (+2 mV) in phosphate-buffered saline (PBS) solution, which could increase the retention and penetration ability of sPLA2i-NPs within the cartilage via electrostatic interactions between the NPs and the anionic glycosaminoglycans (GAGs) in the cartilage. It should also be noted that the NP surface charge is highly dependent on its surrounding medium. For example, zeta potential of DOTAP-doped sPLA2i-NPs in 10 mM Hepes buffer (pH 7.4) was −14.5 mV. The surface charge of DOTAP-doped sPLA2i-NPs in synovial fluid may be different from the one in PBS or Hepes. The stability of the sPLA2i-NPs was evaluated in PBS and bovine synovial fluid. There was no observable change in the hydrodynamic diameter of sPLA2i-NPs in PBS for at least 1 week (Fig. 2D) or in bovine synovial fluid for 5 days (fig. S2).
Fig. 2. Preparation and characterization of sPLA2i-NPs.
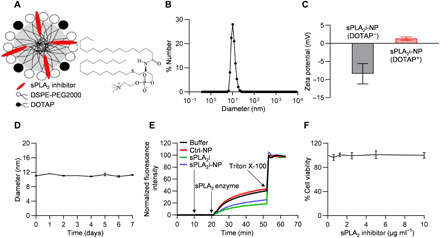
(A) Schematic diagram of sPLA2i-NPs in which a lipid-based sPLA2i was incorporated into nanometer-sized phospholipid NPs (DSPE-PEG2000) with cationic lipid DOTAP doped. (B) DLS measurement of the hydrodynamic diameter of sPLA2i-NPs. (C) Zeta potential of sPLA2i-NPs in the presence or absence of cationic lipid DOTAP in 0.1 M PBS (pH 7.4). (D) The stability of sPLA2i-NPs in 0.1 M PBS (pH 7.4) was accessed by monitoring the hydrodynamic diameter for up to 1 week. (E) In vitro response of sPLA2i-NPs (final sPLA2i concentration, 500 nM) to sPLA2 enzyme experiment showed a significant inhibition effect. (F) The cytotoxicity of sPLA2i-NPs was determined by measuring the cell viability of primary chondrocytes after coincubation with sPLA2i-NPs at various concentrations. In all datasets, n = 3 biologically independent experiments. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc test. Data presented as means ± SEM.
To examine whether sPLA2i-NPs can inhibit sPLA2 enzyme, a fluorescence dequenching assay with liposomes that contain a self-quenching concentration of the fluorescent lipid was used (30). Phospholipid hydrogenated soy phosphatidylcholine (HSPC) liposomes doped with 20 mol % fluorescent lipid 1-palmitoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine (NBD-PC) were prepared in which the NBD fluorescence was self-quenched (fig. S3). NBD-PC is a fluorescent phospholipid with the fluorophore NBD attached to the lipid backbone using an sPLA2-responsive sn-2 acyl bond. When sPLA2 enzyme was added to the NBD-PC–doped liposome sample in the absence of sPLA2i or in the presence of Ctrl-NPs (i.e., NPs without sPLA2i), sPLA2 catalytically hydrolyzed the sn-2 acyl bond of NBD-PC, resulting in the release of NBD into the surrounding bulk solution. The released NBD led to fluorescence dequenching, resulting in a higher fluorescence intensity (Fig. 2E). When sPLA2 enzyme was added to the liposome sample in the presence of sPLA2i, including free sPLA2i and sPLA2i-NPs, the reduced sPLA2 activity led to the low release of NBD into the surrounding bulk solution, resulting in a reduction in the extent of fluorescence increase (Fig. 2E). Further studies showed that free sPLA2i or sPLA2i-NPs inhibited the sPLA2 activity in a concentration-dependent manner with half-maximal inhibitory concentration (IC50) values of 2.5 and 0.59 μM, respectively (fig. S4), indicating that sPLA2 inhibition by sPLA2i-NPs was more potent than the free sPLA2i.
The cytotoxic effects of sPLA2i-NP were examined in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay. Specifically, various concentrations of sPLA2i-NPs were incubated with primary mouse chondrocytes for 24 hours. The cell viability for each group was normalized to a control group with no sPLA2i-NPs. In general, sPLA2i-NPs had little effect on the viability of cells up to a sPLA2i concentration of 10 μg ml−1 (Fig. 2F), suggesting their physiological biocompatibility.
sPLA2i-NP penetration
Because mouse knee cartilage is very thin and not an appropriate model for studying cartilage penetration, bovine cartilage explants, mimicking human knee cartilage (~2 to 4 mm), were used to examine whether the sPLA2i-NPs could penetrate into deep layer of cartilage tissue. sPLA2i-NPs were incubated with bovine cartilage tissue ex vivo for different times including 2, 4, 6, and 8 days. Confocal fluorescence images of cartilage section were acquired preincubation and at various time points after incubation with sPLA2i-NPs (Fig. 3A). In the preincubation images, there was little intrinsic tissue fluorescence. At 2 days following incubation with the sPLA2i-NPs, a strong fluorescence signal was observed within the superficial zone of the cartilage. The fluorescence signal was distributed in the entire cartilage tissue (1 to 2 mm in thickness) as early as day 4 and became stronger over time, indicating that the sPLA2i-NPs indeed moved into the deep zone of the cartilage. Control experiments were performed by incubation of bovine cartilage explants with sPLA2i-NPs that were not doped with the positively charged DOTAP. In these tissues, a strong fluorescence signal was only observed within the superficial zone of the cartilage, even up to 8 days. Similarly, when bovine cartilage explants were incubated with free fluorescent dye rhodamine, very little fluorescence signal was observed in the deep zone of the cartilage. Analysis of fluorescence images revealed that DOTAP-doped sPLA2i-NPs exhibited a great improvement in cartilage penetration at all time points compared with non-DOTAP–doped sPLA2i-NPs or free rhodamine (Fig. 3B and fig. S5). In addition, the area under the curve (AUC) in the cartilage achieved by sPLA2i-NPs (DOTAP+) was much larger than that of non-DOTAP–doped sPLA2i-NPs or free rhodamine (Fig. 3C and fig. S5). These results indicate that the DOTAP-doped sPLA2i-NPs are able to penetrate into the deep zone of the articular cartilage and exhibit high cartilage accumulation.
Fig. 3. Cartilage penetration and joint retention of sPLA2i-NPs.
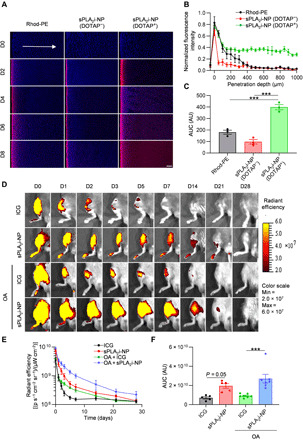
(A) Representative confocal microscope images of cross sections of bovine cartilage explants incubated with free rhodamine dye or rhodamine-labeled sPLA2i-NPs in the presence or absence of cationic lipid DOTAP for 0, 2, 4, 6, and 8 days. Scale bar, 200 μm. (B) Semiquantitative analysis of the fluorescence intensity of the above different formulations over the entire explant sections after 8 days of incubation (n = 3). (C) Semiquantitative analysis of the AUC based on the fluorescence intensity profiles in (B) (n = 3). (D) Representative IVIS images of healthy and OA (OA was induced surgically 8 weeks before) mouse knee joints over 28 days after single intra-articular injection of free ICG or Cy7-labeled sPLA2i-NPs. (E) Semiquantitative analysis of time-course fluorescent radiant efficiency within healthy and OA mouse knee joints over 28 days (n = 5). (F) Semiquantitative analysis of the AUC based on the fluorescence intensity profiles in (E) (n = 5). Rhod-PE, free rhodamine dye; sPLA2i-NPs (DOTAP−), sPLA2i-NPs in the absence of DOTAP; sPLA2i-NPs (DOTAP+), sPLA2i-NPs in the presence of DOTAP. AU, arbitrary units. Statistical analysis was performed using one-way ANOVA with Turkey’s post hoc test. Data presented as means ± SEM. ***P < 0.001. Photo credit: Yulong Wei, University of Pennsylvania.
To further assess the total accumulation of sPLA2i-NPs within the cartilage, we acquired fluorescence images of the bovine cartilage 24 hours following the incubation of DOTAP-doped sPLA2i-NPs, non-DOTAP–doped sPLA2i-NPs, or free rhodamine. As expected, the fluorescent intensity of DOTAP-doped sPLA2i-NPs in the cartilage tissue was significantly higher than that of non-DOTAP–doped sPLA2i-NPs or free rhodamine (fig. S6, A and B). This convincingly demonstrates the benefit of including DOTAP moieties in the sPLA2i-NPs. Because of the ability to penetrate the cartilage, the DOTAP-doped sPLA2i-NPs were chosen for subsequent studies.
sPLA2i-NP retention
In vivo fluorescence imaging was used to study the retention of Cyanine 7 (Cy7)–labeled sPLA2i-NPs in the knee joints after intra-articular injection. Although this technique has limitations due to the limited depth of penetration in tissues and the sensitivity of fluorescent probes to their surrounding environment, it is still a very practical method for providing semiquantitative information for NP joint retention and in vivo biodistribution. To show the difference in retention between NPs and small-molecule agents, we used a low-MW fluorescent dye indocyanine green (ICG; MW, 775 Da) for comparison. Their retention within the joints was evaluated in the knee under healthy and early OA conditions. Fluorescence images of mouse knee joints were acquired at various time points after sample injection (Fig. 3D). With a single intra-articular injection, the fluorescence intensity of sPLA2i-NPs in joints was significantly higher than that of free ICG at all time points, indicating that the retention time of sPLA2i-NPs in joints is much longer than that of free ICG. Analysis of fluorescence images showed that the retention of sPLA2i-NPs in joints with OA condition was even greater than that of healthy joints (Fig. 3, E and F). This finding is consistent with previous reports that NPs are retained in the OA joint much longer than in the healthy joint (31, 32), possibly due to synovial thickening (31) and altered lymphatic vessels in OA knees (33). A longer retention time of sPLA2i-NPs over small molecules within knee joints was also confirmed when rats were used (fig. S7), suggesting that long retention of sPLA2i-NPs in the joints could be achieved for different animals.
We also examined the biodistribution of sPLA2i-NPs in joint components, internal organs, and blood. At 24 hours after injection, fluorescence signals were detected on the cartilage surfaces of patellar, femoral condyles, and tibial plateau as well as on the meniscus (fig. S8, A and B). By 24 hours after injection, the accumulation of the sPLA2i-NPs was mainly observed in the liver and kidney, but no signal was detected in blood at that time, indicating that sPLA2i-NPs were nearly cleared from circulation (fig. S9, A and B). One month later, no fluorescence was observed in the liver and kidney.
sPLA2i-NPs block cartilage degeneration in OA cartilage explants
The articular cartilage explants represent an in vitro model to study OA progression in a three-dimensional (3D) environment (34). We harvested femoral head cartilage explants from 2-month-old wild-type (WT) mice and then stimulated them with OA-associated proinflammatory cytokine interleukin-1β (IL-1β) (35). The penetration of sPLA2i-NPs was studied by acquiring fluorescence images of IL-1β–stimulated cartilage explants that were incubated with rhodamine-labeled sPLA2i-NPs for 24 or 48 hours. Fluorescent signal was mainly present in the cartilage surface at 24 hours (Fig. 4A), while a strong fluorescence signal was observed in the deep calcified layer at 48 hours (Fig. 4, B and C), indicating that sPLA2i-NPs are able to penetrate into femoral head cartilage treated with IL-1β. These results were consistent with those presented above for sPLA2i-NPs penetration within bovine cartilage explants.
Fig. 4. Penetration and chondroprotective effects of sPLA2i-NPs in mouse femoral heads.
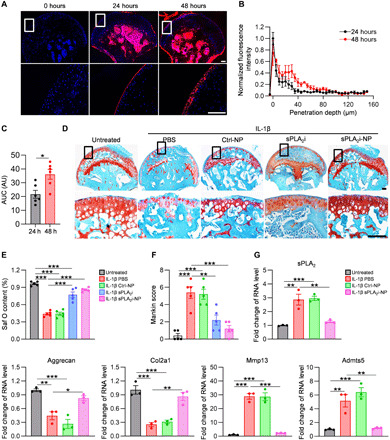
(A) Representative fluorescence images of cross sections of mouse femoral heads stimulated with recombinant mouse IL-1β and incubated with rhodamine-labeled sPLA2i-NPs for 0, 24, or 48 hours. Bottom: Magnified images of the white boxed areas. Scale bars, 100 μm. (B) Semiquantitative analysis of rhodamine-labeled sPLA2i-NPs penetration depth into IL-1β–stimulated mouse femoral heads (n = 6). (C) Quantification of the AUC based on the fluorescence intensity profiles in (B) (n = 6). (D) Representative images of Safranin O/Fast Green staining on the sections of untreated and PBS-, Ctrl-NP–, sPLA2i-, and sPLA2i-NP–treated IL-1β–stimulated mouse femoral heads. Bottom: Magnified images of the black boxed areas. Scale bars, 100 μm. (E) Safranin O (Saf O)–positive area among the above groups was quantified (n = 5). (F) The OA severity was accessed by Mankin score (n = 5). (G) The relative gene expression of sPLA2-IIA, aggrecan, Col2a1, Mmp13, and Adamts5 was examined by quantitative reverse transcription polymerase chain reaction (PCR) in the untreated and PBS-, Ctrl-NP–, and sPLA2i-NP–treated IL-1β–stimulated mouse femoral heads (n = 3). Statistical analysis was performed using paired two-tailed t test for (C) and one-way ANOVA with Turkey’s post hoc test for (E), (F), and (G). Data presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
To test the therapeutic effects of sPLA2i-NPs in vitro, we divided femoral head cartilage explants into five groups that received PBS, IL-1β and PBS, IL-1β and Ctrl-NPs, IL-1β and sPLA2i, or IL-1β and sPLA2i-NP treatments, respectively, for 8 days. As shown in Fig. 4 (D to F), IL-1β treatment led to an OA-like phenotype in the cartilage, featured by surface fibrillation and proteoglycan loss. Notably, sPLA2i-NPs and free sPLA2i, but not Ctrl-NPs, blocked cartilage degeneration induced by IL-1β, leading to comparable Safranin O content and Mankin score as the untreated group. sPLA2i-NPs, but not PBS or Ctrl-NPs, attenuated IL-1β–induced sPLA2-IIA expression in the cartilage (Fig. 4G). Furthermore, the catabolic effects of IL-1β, such as decreasing the expression of matrix proteins, Col2a1 and aggrecan, and increasing the expression of proteinases, matrix metalloproteinase 13 (Mmp13), and Adamts5, were effectively reversed by sPLA2i-NPs (Fig. 4G). Together, these data provide ex vivo evidence that sPLA2i-NPs have protective action on chondrocytes against OA-inducing insults.
sPLA2i-NPs attenuate joint destruction in a surgery-induced mouse OA model
To study the in vivo therapeutic effects of sPLA2i-NPs, we used two mouse OA models. The first is a DMM surgery–induced OA model with high reliability and reproducibility (Fig. 5A). After DMM, WT knees started to show cartilage damage, including surface fibrillation and loss of proteoglycan, at 2 months, and exhibited severe cartilage erosion up to the entire uncalcified zone at 4 months, resulting in Mankin scores of 5.5 and 8.9, at these two time points, respectively (Fig. 5, B and C, and figs. S10 and S11). Intra-articular injection of sPLA2i-NPs into DMM knees, once every two weeks, starting immediately after the surgery, greatly improved the morphology and structure of the articular cartilage, leading to an almost intact cartilage surface with no proteoglycan loss even at 4 months after surgery (Fig. 5B and figs. S10 and S11B). These therapeutic effects were not observed with free sPLA2i or Ctrl-NP treatments.
Fig. 5. Therapeutic effects of sPLA2i-NPs for attenuation of DMM-induced chronic OA.
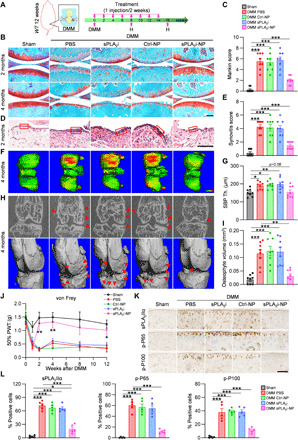
(A) The study design of sPLA2i-NP treatment for DMM-induced OA mice. (B) Safranin O/Fast Green staining of sham- or DMM-operated knee joints with 2 or 4 months of treatment. Scale bar, 200 μm. (C) The OA severity was accessed by Mankin score at 2 months after surgery (n = 8). (D) Hematoxylin and eosin (H&E) staining of synovium tissue at 2 months after surgery. Scale bar, 100 μm. Red boxed areas indicate the synovial lining layer. (E) Synovial inflammation was evaluated (n = 8). (F) 3D color maps of femoral SBP thickness at 4 months after surgery. Color ranges from 0 μm (blue) to 320 μm (red). (G) Quantification of SBP thickness (Th.) at the medial posterior site (n = 8). (H) Osteophytes (red arrows) were revealed by 2D (top) and 3D (bottom) micro–computed tomography (micro-CT) images. (I) Osteophyte volume was quantified (n = 8). (J) von Frey assay was performed at 1, 2, 4, 8, and 12 weeks after surgery (n = 8). The data of day 0 were acquired before surgery. PWT, paw withdrawal threshold. (K) IHC staining of the tibial articular cartilage at 2 months after surgery. Scale bar, 100 μm. (L) Quantification of sPLA2-IIa–, p-P65–, and p-P100–positive chondrocytes (n = 5). Statistical analysis was performed using one-way ANOVA with Turkey’s post hoc test. Data presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 in (C), (E), (G), (I), and (L). *P < 0.05 and **P < 0.01 for sPLA2i-NPs versus PBS in (J).
The thickness of synovium is an indicator of inflammation in the knee joint (36, 37). PBS-, sPLA2i-, and Ctrl-NP–treated DMM knees exhibited a significantly thickened synovial lining with more than 10-fold increase in the synovitis score compared with sham knees. In contrast, sPLA2i-NP–treated DMM knees had only a fourfold increase (Fig. 5, D and E), suggesting that sPLA2i-NPs generate an anti-inflammatory effect in the injured knees.
We further characterized some of the late OA symptoms including subchondral bone plate (SBP) sclerosis and osteophyte formation. At 4 months after DMM surgery, the thickness of the SBP at the femoral medial site in the knee joint was significantly increased in PBS-treated (1.25-fold), sPLA2i-treated (1.31-fold), and Ctrl-NP–treated (1.29-fold) groups compared to that in sham joints (Fig. 5, F and G). However, this SBP sclerosis was not observed in the sPLA2i-NP–treated group. Furthermore, osteophytes were also frequently found in DMM knees in control-treated groups but not in the sPLA2i-NP–treated group (Fig. 5, H and I). These data clearly indicate that sPLA2i-NPs prevent OA progression into a late stage.
In WT mice, mechanical allodynia was observed in the operated hind paw after DMM surgery but not following sham surgery (38). Administration of PBS, sPLA2i, or Ctrl-NPs into DMM knees did not alter the OA pain (Fig. 5J). DMM knees receiving sPLA2i-NPs initially exhibited pain response at 1 week, probably due to the persistent postsurgical pain. At 2 weeks after surgery, sPLA2i-NP treatment greatly increased the paw withdrawal threshold (PWT) to a level close to that in sham knees and maintained that level throughout the experimental period, implying that sPLA2i-NP treatment relieves OA pain.
We further studied the mechanism of sPLA2 inhibition for treating OA. IHC showed that sPLA2i-NPs blocked the up-regulation of sPLA2-IIa after OA induction (Fig. 5, K and L). Consequently, while DMM elevated the inflammatory signaling pathway, such as phospho (p)–p65 and p-p100, sPLA2i-NP treatment greatly decreased the amounts of these inflammatory indicators in the cartilage at 2 and 4 months after DMM, thus protecting cartilage from OA insults (Fig. 5, K and L, and fig. S12). Collectively, these results suggest that the sPLA2 enzyme mediates the pathogenesis of OA, while sPLA2i-NPs inhibit OA inflammation in DMM knee joints.
To assess the toxicity of sPLA2i-NPs in mice, we evaluated the histopathology of tissues from the liver, spleen, kidney, lung, heart, and knee joint of mice 2 months after serial intra-articular injections of PBS, sPLA2i, Ctrl-NPs, or sPLA2i-NPs. We did not observe any histopathological abnormalities in any of the mouse organs and knee joints that received these injections directly (figs. S13 and S14). Furthermore, we evaluated toxic effects on blood parameters of mice following treatment and did not observe any clinical signs of toxicity (fig. S15). These results suggest that sPLA2i-NPs at the adopted dosages show no toxicity in mice.
sPLA2i-NPs block joint damage in a single load-induced mouse posttraumatic OA model
The second mouse OA model we tested is a noninvasive mechanical loading model, which is clinically relevant to posttraumatic OA (PTOA) (Fig. 6A). This injury model allows for the study of early events after impact, at a time when inflammation is thought to be particularly important (29). We applied a single loading episode, composed of 60 cycles of 6- or 9-N peak load, on the mouse tibia to induce joint translation and anterior cruciate ligament (ACL) rupture. Two weeks later, a lesion was seen alongside with proteoglycan loss in the lateral femoral articular cartilage surface (Fig. 6B). Quantification of the length of cartilage injury revealed significant damage areas in PBS-, sPLA2i-, and Ctrl-NP–treated knees (Fig. 6C and fig. S16). As expected, 9 N generated more severe damage than 6 N in all groups. In this model, chondrocyte apoptosis at the loading site contributes significantly to OA development. TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining revealed that sPLA2i-NP treatment greatly reduced the number of apoptotic chondrocytes in the cartilage following loading (Fig. 6, D and E, and fig. S17). Examining the inflammatory pathway again revealed a reduction of p-P65 and p-P100 in sPLA2i-NP–treated knee joints, but not in sPLA2i or Ctrl-NP groups (Fig. 6, F and G, and fig. S18). Similar to the above DMM model, we found that sPLA2i-NPs suppressed the amount of sPLA2-IIA enzyme in the cartilage. Synovitis scores suggested significant synovitis in PBS-, sPLA2i-, and Ctrl-NP–treated knees (Fig. 6, H and I, and fig. S19). von Frey assay clearly showed that mice in these groups experienced OA pain (Fig. 6J and fig. S20). In contrast, injections of sPLA2i-NPs into knee joints immediately and 48 hours after loading reversed these PTOA symptoms regardless of loading force. Specifically, compared to PBS-treated knees, the length of cartilage injury in sPLA2i-NP–treated knees was decreased by 67% (6 N) and 59% (9 N), synovitis score was reduced by 71% (6 N) and 56% (9 N), and pain threshold was increased to a level similar to sham knees. Hence, our data demonstrate a joint protective role of sPLA2i-NPs in load-induced OA.
Fig. 6. Therapeutic effects of sPLA2i-NPs for attenuation of load-induced PTOA.
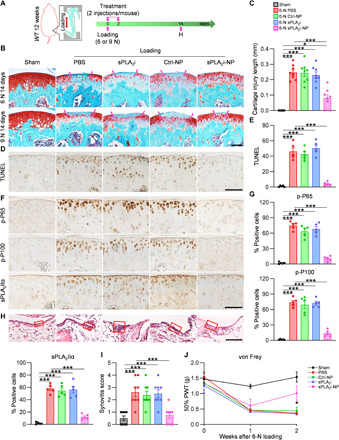
(A) The study design of sPLA2i-NP treatment for loading-induced OA mice. (B) Safranin O/Fast Green staining of the articular cartilage in sham or loaded (6 or 9 N) knee joints with 14-day treatment. Scale bar, 100 μm. Dashed lines indicate the range of loss of staining. (C) The length of cartilage lesion in sham and 6-N loaded knee joints was quantified. (D) TUNEL staining in the tibial articular cartilage from sham and 6-N loaded knee joints. Scale bar, 100 μm. (E) Quantification of TUNEL-positive chondrocytes (n = 5). (F) IHC staining of the tibial articular cartilage from sham and 6-N loaded knee joints. Scale bar, 100 μm. (G) Quantification of sPLA2-IIa–, p-P65–, and p-P100–positive chondrocytes (n = 5). (H) H&E staining of synovium tissue. Scale bar, 100 μm. Red boxed areas indicate the synovial lining layer. (I) Synovial inflammation was evaluated (n = 8). (J) von Frey assay at 1 and 2 weeks after 6-N loading (n = 8). The data of day 0 were acquired before loading. Statistical analysis was performed using one-way ANOVA with Turkey’s post hoc test. Data presented as means ± SEM. *P < 0.05 and ***P < 0.001 in (C), (E), (G), and (I) and *P < 0.05 for sPLA2i-NPs versus PBS in (J).
DISCUSSION
Growing evidence suggests that inflammation has a critical role in the pathogenesis of OA. Classical anti-inflammatory agents have limited utility to slow the progression of OA. In this work, we show that the level of sPLA2 is significantly increased in the articular cartilage in both human and mouse OA knee tissues. Because the release of free AA from membrane phospholipids by sPLA2 is one of the major contributors to producing potent inflammatory mediators such as eicosanoids or platelet-activating factor, we therefore hypothesized that sPLA2 could act as a promising therapeutic target for OA treatment. Our strategy was to develop a sPLA2i-based approach for inhibition sPLA2 activity. Compared to classical anti-inflammatory agents, inhibiting sPLA2 enzyme activity by sPLA2i could offer the potential to block production of a more complete set of inflammatory substances through targeting upstream inflammatory pathways, leading to a safer and more effective alternative to conventional anti-inflammatory agents.
Great efforts from pharmaceutical companies and academic laboratories have recently been devoted to developing potent and selective sPLA2i for the treatment of inflammation-related diseases, with several ongoing clinical trials (39). One clinical trial used the sPLA2i LY315920 to treat patients with rheumatoid arthritis (RA) (40). There was a significant short-term improvement after several days of treatment; however, no significant longer-term effect was observed after weeks of treatment (41). This failure is largely due to the lack of sufficient inhibitor concentration in the joint (41, 42), because human synovial fluid has a rapid turnover in the joint, which could flush the administrated small sPLA2i molecules out of joint through capillaries and draining lymphatic vessels (26, 43). Our results also supported this limitation (i.e., fast clearance of sPLA2i from the joint), showing that the intra-articular injection of free sPLA2i alone did not have a protective effect on cartilage degeneration in OA animals.
Drug delivery systems using NPs are increasingly being used to improve therapeutic delivery due to the favorable pharmacokinetics, biodistribution, and solubility of NPs compared with free drugs. Currently, several NPs have been developed and tested in preclinical OA studies, including liposomes, dendrimers, and poly(lactic-co-glycolic acid) particles. Because of the high density of negatively charged GAG in the cartilage, cationic drug carriers are often explored for enhancing cartilage penetration and retention based on electrostatic interactions (44–46). In this study, we have addressed the sPLA2i delivery challenges by incorporating small sPLA2i into nanometer-sized phospholipid micelles. By tuning the surface charge, the sPLA2i-NPs showed deep cartilage penetration and prolonged residence time in knee joints. This is especially important because the therapeutic target sPLA2 is located within deep cartilage tissue.
Most NPs developed and tested in preclinical OA studies, so far, have some weaknesses. Because of their relatively large size (100 to 200 nm in diameter), liposomes have prolonged residence times in joints but are inaccessible to deep cartilage tissues (47), which could become problematic if therapeutic targets (i.e., sPLA2, the therapeutic target in this study) are located within deep cartilage tissue. Therefore, liposomes are only particularly well suited for OA drug delivery to the cartilage surface (47, 48). Because of its small size and controllable surface charge, polyamidoamine (PAMAM) dendrimers can penetrate the cartilage efficiently (1). For example, Geiger et al. (1) conjugated insulin-like growth factor 1 to a cationic dendrimer, which could penetrate full-thickness bovine cartilage ex vivo. Unfortunately, thioetheramide-PC does not contain any reactive chemical groups for dendrimer conjugation. In addition, dendrimers have not translated into the clinic despite 40 years of research (49). Compared to the above NPs, phospholipid micelles we used here for loading sPLA2i have several advantages. First, sPLA2i-NPs consist almost solely of clinically used phospholipids and, thus, are readily translatable to clinical applications. Second, lipid-based sPLA2is can be incorporated into phospholipid micelles with high yield and without any conjugation or purification steps. Third, lipid-based sPLA2i-NPs are sPLA2 responsive. Fourth, the method for producing sPLA2i-NPs is highly reproducible, simple, and cost-effective because all components are included in starting materials before sPLA2i-NP formation, which would allow large-scale, good manufacturing practice (GMP) production of NPs, a necessary step for the initiation of future clinical trials. Therefore, the use of small size phospholipid micelles for loading lipid-based thioetheramide-PC is an appropriate vehicle/drug combination in the current specific settings because phospholipid micelles have a long history in clinical applications.
Inflammation plays a critical role in OA progression. Compared to rheumatoid RA, the prototypical inflammatory arthritis, the inflammation in OA is chronic and relatively low grade (50). Systemically blocking the activity of conventional inflammatory cytokines with biologic therapies approved for the treatment of RA, such as anti–tumor necrosis factor or anti–IL-1β therapies, provides no or minor benefit in generalized OA (50). These observations implicate that a new target or approach must be adopted for treating OA inflammation and resultant cartilage degeneration. Inflammation starts with the PLA2-induced hydrolysis of the membrane phospholipids, giving rise to the fatty acids, such as AA and lysophospholipids. Of all PLA2 types, sPLA2 is considered the most “inflammatory enzyme.” Past studies found that sPLA2 amount increases in human OA joints, including cartilage chondrocytes (20–23), but they did not further analyze the cause-effect relationship between sPLA2 up-regulation and OA progression. Our studies first confirmed the enhanced sPLA2 activity in both human and mouse OA cartilage. Then, we used an ex vivo bovine cartilage explant model to demonstrate that inhibiting sPLA2 protects chondrocytes against IL-1β insult via suppressing inflammation pathways. We used two in vivo mouse models of OA, which mimic different OA subtypes, to provide proof-of-principle evidence that intra-articular injections of sPLA2i-NPs are effective in attenuated OA progression. Notably, local delivery of sPLA2i-NPs, but not free sPLA2i or NP alone, greatly slowed the progression of cartilage degeneration, reduced synovial inflammation, prevented osteophyte formation, and relieved join pain. In addition to the articular cartilage, sPLA2i-NP could target other joint tissues, such as synovium and meniscus, to achieve its therapeutic effects. No signs of toxicity have been observed in the joints and other tissues, suggesting the safety of this nanomedicine. That to be said, because OA is a chronic disease, long-term treatment is required in the future to further prove its safety.
The development of sPLA2i-NP–based therapies for large animals or their translation into clinical settings requires further optimization and investigation. For example, in this work, we used thioetheramide-PC as sPLA2i due to its commercial availability and its ease of loading into phospholipid micelles. However, thioetheramide-PC is for research use only and has a relatively high IC50 (~2.5 μM). In the future, we will choose sPLA2is that have already been used in clinical trials and have a low IC50 value. There are some other variables that can be optimized to further improve the therapeutic efficacy of sPLA2i-NPs, including particle size, surface charge, hydrophobicity, and material compositions. A previous work has shown that weak-reversible binding between drug carriers and cartilage is key to allowing deep cartilage penetration (51, 52). If net surface charge of drug carriers is too high, then their diffusion rate could be slowed because of stronger binding interactions within the cartilage (53). In this work, zeta potential measurement in PBS showed a slight positive surface charge with 10 mol % DOTAP-doped sPLA2i-NPs. As expected, the DOTAP-doped sPLA2i-NPs penetrated into the deep zone of the articular cartilage. This is most likely due to a combination of the following features of NPs: favorable interactions with a slightly positive surface charge, some NPs with worm-like structures, and the modified spatial distribution of surface charge. It should be mentioned that the sPLA2i-NPs administration was initiated immediately after DMM surgery in this study. Future animal studies should incorporate injections at time points when early- or middle-stage OA symptoms appear. Given that sPLA2 level was markedly elevated in human knee cartilage at early, middle, and late stage of OA progression, it is reasonable to expect that the sPLA2i-NPs could also be used to slow cartilage degeneration and OA progression in cases where OA is already established. While our current engineered sPLA2i-NPs showed the therapeutic effects in a mouse model of OA, they may not be translated to large animals and humans with equal efficacy because the anatomy and physiology of small animals are significantly different from large animals and humans. Further optimization of this NP-based drug delivery system with better cartilage targeting, cartilage penetration and uptake, and joint retention, is needed in large animal models before translating this approach into clinical applications (54).
MATERIALS AND METHODS
Materials
Thioetheramide-PC was purchased from Cayman Chemical (Ann Arbor, MI). HSPC, DSPE-PEG2000, DOTAP, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(Lissamine rhodamine B sulfonyl) (Rhod-PE), DSPE-PEG2000-Cy7, and NBD-PC were purchased from Avanti Polar Lipids Inc. sPLA2 enzyme from Naja mossambica was purchased from Sigma-Aldrich Co. Rabbit polyclonal antibody to sPLA2-IIa, p-P65, and p-P100 antibodies were purchased from Abcam. Bovine synovial fluid was obtained from Lampire Biological Laboratories Inc. All other chemicals were used as received. All of the buffer solutions were prepared with deionized water.
Synthesis of sPLA2i-loaded phospholipid NPs
sPLA2i-NPs were prepared by hydration of dry sPLA2i/lipid films. A mixture containing 10 mol % DOTAP/25 mol % thioetheramide-PC/65 mol % DSPE-PEG2000 was prepared in a round-bottom flask. The total amount of thioetheramide-PC was 0.25 mg. For the preparation of fluorescently labeled NPs, a small amount (1 mol %) of the fluorescent lipids, Rhod-PE, or DSPE-PEG2000-Cy7, was also added to the thioetheramide-PC/lipid mixture. The solvent was removed using a direct stream of nitrogen before vacuum desiccation for a minimum of 4 hours. NPs were formed by adding deionized water to the dried film and incubating in a 25°C water bath for 5 min and then votexing for another 3 min. The resulting solution was then centrifuged at 3000g for 5 min to remove the large aggregates. Last, the NPs were filtered through a 0.22-μm cellulose acetate membrane filter (Nalgene, Thermo Fisher Scientific) and stored in the dark at 4°C. Ctrl-NPs, including NPs without sPLA2i (10 mol % DOTAP/25 mol % HSPC/65 mol % DSPE-PEG2000) and NPs without DOTAP (10 mol % HSPC/25 mol % sPLA2i/65 mol % DSPE-PEG2000), were prepared using the similar procedures as above.
Synthesized sPLA2i-NPs were incubated in 0.1 M PBS (pH 7.4) and in bovine synovial fluid at 37°C for the stability study. The diameter and zeta potential of the NPs were measured with DLS (Zetasizer, Nano-ZS, Malvern). The morphology of the NPs was observed using a TEM (JOEL 1010) by the negative-staining technique. Phosphorus and sulfur concentration in the sample was quantified by inductively coupled plasma - optical emission spectrometry (ICP-OES) (SPECTRO GENESIS GmbH).
In vitro sPLA2 response study
To study in vitro sPLA2 response of sPLA2i-NPs, the NBD-incorporated liposomes were incubated with sPLA2i-NPs in the presence of sPLA2 enzyme, and the dequenching of NBD fluorescence signal was monitored. To prepare NBD liposomes, stock solutions of HSPC and NBD-PC in chloroform were mixed in the following molar ratios: HSPC/NBD-PC (80:20). The total amount of HSPC was 1 mg. The solvent was removed using a direct stream of nitrogen before vacuum desiccation for a minimum of 4 hours. A total of 0.2 ml of deionized water was then added to the dried lipid film and incubated in a 50°C water bath for 0.5 hours and then sonicated for another 30 min. The stock solution of NBD-incorporated liposomes was stored in the dark at 4°C.
Fluorescence measurements of the NBD-incorporated liposomes in 10 mM Hepes buffer (pH 7.4) [2 mM [CaCl2] and [HSPC] (0.022 mg ml−1)] were taken 10 min before the addition of the Ctrl-NPs, sPLA2i-NPs, or free sPLA2is at different sPLA2i concentrations: 0, 0.125, 0.25, 0.5, 1, and 2 μM. At a time of 20 min, sPLA2 enzyme was added at a final concentration of 0.05 U ml−1. The fluorescence intensity at 520 nm was measured on a SPEX FluoroMax-3 spectrofluorometer (HORIBA Jobin Yvon) using an excitation at 460 nm. The amount of NBD dequenched (% NBD dequenched) was calculated by means of Eq. 1
| (1) |
where I0 is the fluorescence intensity of the liposomal suspension containing NBD at the initial time, Ix is the fluorescence intensity at any given time, and It is the fluorescence intensity after addition of 20 μl of Triton X-100 (50 mM) to the suspension at the end of experiment.
Cell culture
Primary mouse chondrocytes were isolated from the distal femoral and proximal tibial epiphysis of mice (3 to 6 days old) via enzymatic digestion. Briefly, tissues were incubated with 0.25% trypsin (Invitrogen) for 1 hour, followed by 2-hour digestion with type I collagenase (300 U ml−1) (Worthington Biochemical). Dissociated cells were seeded in culture plates, and attached cells were considered primary mouse chondrocytes. They were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 containing 10% fetal bovine serum, streptomycin (100 μg ml−1), and penicillin (100 U ml−1). Primary mouse chondrocytes between passages 0 and 3 were used for MTT assay experiments.
MTT assay
Primary mouse chondrocytes (5000 cells per well) were seeded in 96-well plates and incubated overnight (37°C and 5% CO2) to allow the cells to attach to the surface of the wells. The sPLA2i-NPs were added to wells at five different sPLA2i concentrations ranging from 0.625 to 10 μg ml−1 (0.625, 1.25, 2.5, 5, and 10 μg ml−1), and the cell viabilities were determined according to the supplier’s instructions. After 24 hours of incubation, the medium containing NPs in each well was aspirated off and replaced with 100 μl of fresh medium and 10 μl of MTT reagent. The cells were incubated for 2 to 4 hours. Then, 100 μl of detergent reagent was added and left at room temperature in the dark for 4 hours. Last, the absorbance of formazan product was measured on a Tecan microplate reader (BioTek Instruments Inc.) at 570 nm. Cell viability was calculated using the following equation
| (2) |
Bovine cartilage explant harvest and culture
Young (1 to 2 weeks old) bovine knee joints were obtained from vendors (Lampire Biological Laboratories), and cartilage explants were harvested from the trochlear groove using biopsy punch and cultured with chemically defined medium [DMEM, 1% insulin-transferrin-selenous acid (ITS) + premix, l-proline (50 μg ml−1 ), 0.1 μM dexamethasone, 0.9 mM sodium pyruvate, and ascorbate 2-phosphate (50 μg ml−1)] in a 48-well plate.
Bovine cartilage explant penetration assay
Bovine cartilage explants (6 mm in diameter and 2 mm in thickness) were incubated with free rhodamine, rhodamine-labeled sPLA2i-NPs (DOTAP−), or sPLA2i-NPs (DOTAP+) in 500 μl of culture medium for 48, 96, 144, or 192 hours at 37°C and 5% CO2 under gentle agitation. Culture medium with free rhodamine, rhodamine-labeled sPLA2i-NPs (DOTAP−), or sPLA2i-NPs (DOTAP+) was replaced every other day. In all cases, the final rhodamine concentration in the culture medium was 10 μM. After incubation, cartilage explants were washed three times with PBS, fixed with 4% paraformaldehyde (PFA), dehydrated with 20% sucrose + 2% polyvinylpyrrolidone (PVP), followed by embedding with 30% sucrose + 2% PVP + 8% gelatin. Sections were mounted with 4′,6-diamidino-2-phenylindole (DAPI) Fluoromount-G mounting medium on glass slides and immediately observed under confocal microscope (Zeiss LSM 710). Quantitative analysis was performed on maximum intensity projections of Z-stack images taken from 100-μm-thick sections.
Bovine cartilage explant uptake assay
A total volume of 300 μl of free rhodamine, rhodamine-labeled sPLA2i-NPs (DOTAP−), or sPLA2i-NPs (DOTAP+) in culture medium was added to bovine cartilage explants (3 mm in diameter and 2 mm in thickness). The final rhodamine concentration in the culture medium was 10 μM. The explants were incubated for 24 hours at 37°C and 5% CO2 under gentle agitation. The explants were then removed from the medium, washed three times with PBS, and imaged by IVIS (Spectrum, PerkinElmer). Radiant efficiency within a fixed anatomical region of interest (ROI) was measured using Living Image software.
In vivo joint retention assay
The mouse knee joints retention assay was assessed by intra-articular injection of 10 μl of 10 μM free ICG or 10 μM Cy7-doped sPLA2i-NPs in healthy (3 months old) and OA (8 weeks after DMM surgery) mouse knees. The rat knee joints retention assay was assessed by intra-articular injection of 40 μl of 10 μM free ICG or 10 μM Cy7-doped sPLA2i-NPs in healthy rat knees (3 months old). An IVIS (Spectrum, PerkinElmer) was used to serially acquire fluorescence images within each joint over a period of 4 weeks. Using Living Image software, radiant efficiency within a fixed anatomical ROI was measured.
In vivo biodistribution assay
In vivo biodistribution study was performed by intra-articular injection of 10 μl of PBS or 10 μM Cy7-doped sPLA2i-NPs in mouse knees (3 months old). At 24 hours or 1 month following injection, the mice were euthanized. The knee joints, blood, and major organs (heart, liver, spleen, lung, and kidney) were harvested. Knees were dissected to isolate the major joint components, including the surrounding tissues (quadriceps, patella, patellar ligament, synovium, and fat pad), femoral condyles, tibial plateau, and meniscus. All the major joint components, blood, and organs were imaged using the IVIS with the following imaging parameters: excitation, 745 nm; emission, 800 nm; exposure time, 1 s. The data were analyzed as described above.
Mouse femoral head explants penetration assay
Mouse femoral heads were collected from 8-week-old male mice and cultured for 48 hours in chemically defined medium [DMEM, streptomycin (100 μg ml−1) and penicillin (100 U ml−1), 1% ITS + premix, l-proline (40 μg ml−1), 0.1 μM dexamethasone, sodium pyruvate (100 μg ml−1), and ascorbate 2-phosphate (50 μg ml−1)] at 37°C and 5% CO2 in a 48-well plate. Following the culture, mouse femoral head explants were stimulated using recombinant mouse IL-1β (10 ng ml−1; PeproTech) for 2 days. On day 3, rhodamine-labeled sPLA2i-NPs were incubated with the IL-1β–stimulated femoral heads for 24 or 48 hours at 37°C with gentle agitation. The final rhodamine concentration in the culture medium was 10 μM. Following incubation, femoral heads were washed with PBS, fixed with 4% PFA, dehydrated with 30% sucrose, and embedded in Tissue-Tek optimum cutting temperature compound. The 6-μm-thick cyrosections were cut and mounted with DAPI Fluoromount-G mounting medium on the slides and imaged with a fluorescence microscope (Nikon, Eclipse 90i). Images were analyzed with ImageJ to quantify the penetration depth NP into the cartilage. Fluorescence intensity within each image was measured with ImageJ and normalized to the fluorescence of the outermost cartilage surface of the treated femoral head.
Mouse femoral head explants OA degradation assay
Mouse femoral heads were collected using the same procedure as described in mouse femoral head explants penetration assay. To test the therapeutic effects of sPLA2i-NPs, femoral head cartilage explants were divided into five groups to receive PBS (i.e., untreated), IL-1β, IL-1β and Ctrl-NP, IL-1β and sPLA2i, and IL-1β and sPLA2i-NP treatments, respectively, for 8 days. The final IL-1β and sPLA2i concentration in the culture medium was 10 ng ml−1 and 0.1 mg ml−1, respectively. The culture medium was replaced at days 2, 4, and 6. After an 8-day incubation, mouse femoral heads were then fixed with 4% PFA overnight, followed by decalcification in 0.5 M EDTA (pH 7.4) for 2 weeks and processed for 6-μm paraffin sections. Paraffin sections were used for Safranin O staining, and the Safranin O–positive area was quantified with ImageJ. OA severity was evaluated by Mankin score. Both quantifications were based on the section with the most severe loss of Safranin O staining and cartilage damage.
Quantitative reverse transcription polymerase chain reaction
Mouse femoral heads were collected and treated using the same procedure in femoral head explants degradation assay. Total RNA was harvested from the femoral head articular cartilage using TRI Reagent (Sigma-Aldrich Co.). TaqMan Reverse Transcription kit (Applied Biosystems) was used to reverse-transcribe mRNA into complementary DNA. Following this, polymerase chain reaction (PCR) was performed using a Power SYBR Green PCR Master Mix kit (Applied Biosystems). The primer sequences for the genes used in this study are listed in table S1.
Human articular cartilage samples
The human healthy and OA articular cartilage samples were prepared from the deidentified specimens obtained at the total arthroplasty of the knee joints and processed for paraffin sections. The serial sections were stained by Safranin O and Fast Green staining to evaluate OA grades as previously described (55). For sPLA2 staining, anti–sPLA2-IIA (1:800; Abcam, ab23705) antibody was used. We considered healthy, early OA, and middle OA cartilage as grades 0, 1 to 2, and 3 to 4 based on Osteoarthritis Research Society International score, respectively.
Animal care
In accordance with the standards for animal housing, mice with C57BL/6 background were group-housed in an atmosphere of 23° to 25°C with a 12-hour light/dark cycle and allowed free access to water and standard laboratory pellets. All work performed on animals was approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
To induce mouse OA that mimics chronic OA in human patients, male mice at 3 months of age were subjected to DMM surgery at right knees and sham surgery at left knees. Briefly, in DMM surgery, the joint capsule was opened immediately after anesthesia, and the medial meniscotibial ligament was cut to destabilize the meniscus without damaging other tissues. In sham surgery, the joint capsule was opened in the same fashion but without any further damage.
To induce mouse OA that is noninvasive and mimics PTOA in human patients, male mice (2 months old) were subjected to mechanic loading at the right knees and sham loading at left knees. Briefly, under anesthesia, the right tibiae were positioned with the knee downward in deep flexion between custom-made cups and subjected to axial compressive loads with a peak force of 6 or 9 N with a 0.5-N preload force to maintain the limb in position between loading cycles. Cyclic loads were applied for 0.34 s with a rise and fall time each of 0.17 s and a baseline hold time of 10 s between cycles for 60 cycles. The uninjured left knees were used as controls.
NP administration
For treatment, NPs were administrated using sterile techniques: The right knees were kept in a flexed position, and a total volume of 10 μl of PBS, sPLA2i (0.25 mg ml−1), Ctrl-NPs, or sPLA2i-NPs (sPLA2i, 0.25 mg ml−1) was injected intra-articularly with a 30-gauge needle. For DMM surgical OA model, the first injection was performed immediately after surgery. Injections were then repeated every 2 weeks for 2 or 4 months. In total, there are four injections for the 2-month group and eight injections for 4-month group. For load-induced OA model, intra-articular injections were performed immediately and at 48 hours after loading. There are two injections in total for each mouse in this model.
After 2-month treatment for DMM mice, some major organs (kidney, liver, lung, heart, and spleen) and blood were collected. Tissue sections were stained with hematoxylin and eosin (H&E) to assess the effects of different treatments on mouse organ morphology. The blood indexes were measured after receiving 2-month treatment of PBS, sPLA2i, Ctrl-NPs, or sPLA2i-NPs.
Histology
After euthanasia, mouse knee joints were harvested and fixed in 4% PFA overnight, followed by decalcification in 0.5 M EDTA (pH 7.4) for 4 weeks before paraffin embedding. A serial of 6-μm-thick sagittal sections (about 100) were cut across the entire medial compartment of the joint until ACL junction. To measure the thicknesses of articular cartilage and chondrocyte numbers, three sections from each knee, corresponding to one of four (sections 20 to 30), two of four (sections 45 to 55), and three of four (sections 70 to 80) regions of the entire section set, were stained with Safranin O/Fast Green and quantified using BIOQUANT software. The final measurement is an average of these three sections. We defined uncalcified cartilage as the area from articular surface to the tide mark and calcified cartilage as the area from tide mark to cement line. The method to measure Mankin score was described previously (56). Briefly, two sections within every consecutive six sections in the entire section set for each knee were stained with Safranin O/Fast Green and scored by two blinded observers. Each knee received a single score representing the maximal score of its sections.
Synovitis score grading was carried out in 6-μm paraffin sections of sagittal mouse knee sections stained with H&E. The following basic morphological parameters of synovitis were included: (i) hyperplasia/enlargement of synovial lining layer, (ii) degree of inflammatory infiltration, and (iii) activation of resident cells/synovial stroma, including fibroblasts, endothelial cells, histiocytes, macrophages, and multinucleated giant cells. All parameters are graded from 0 (absent), 1 (slight), 2 (moderate) to 3 (strong positive).
Cartilage injury length was measured in Safranin O/Fast Green–stained paraffin sections. From the serial Safranin O–stained sections in each sample, we selected one section with the widest cartilage lesion that featured by focal loss of Safranin O staining, minor fissuring of the articular cartilage, and atrophy of articular chondrocytes. According to these histological changes, it is possible to identify the demarcation between the normal and injured cartilage tissue, and the length of the cartilage lesion range was measured.
Paraffin sections were used for IHC and TUNEL assay. For mouse samples, after appropriate antigen retrieval, slides were incubated with primary antibodies, such as rabbit anti–sPLA2-IIA (1:100; Abcam, ab23705), rabbit anti-p-P65 (1:50: Abcam, ab86299), and rabbit anti-p-P100 (1:50; Abcam, ab194919) at 4°C overnight, followed by binding with biotinylated secondary antibodies and DAB (3,3′-diaminobenzidine) color development. The TUNEL assay was carried out according to the manufacturer’s instructions (Millipore, s7101).
OA pain analysis
The knee joint pain after DMM surgery or loading injury was evaluated in mice weekly before and after surgery using von Frey filaments as described previously (57). An individual mouse was placed on a wire-mesh platform (Excellent Technology Co.) under a 4 cm by 3 cm by 7 cm cage to restrict their move. Mice were trained to be accustomed to this condition every day starting from 7 days before the test. During the test, a set of von Frey fibers (Stoelting Touch Test Sensory Evaluator Kit #2 to #9, ranging from 0.015- to 1.3-g force) were applied to the plantar surface of the hind paw until the fibers bowed and then held for 3 s. The threshold force required to elicit withdrawal of the paw (median, 50% withdrawal) was determined five times on each hind paw with sequential measurements separated by at least 5 min.
Micro–computed tomography analysis
The distal femur of mouse knee joints was scanned at a 6-μm isotropic voxel size with a micro–computed tomography (micro-CT) 35 scanner (Scanco Medical AG, Brüttisellen, Switzerland). All images were smoothened by a Gaussian filter (sigma = 1.2, support = 2.0). Measurement of SBP thickness was described previously (58). Briefly, sagittal images were contoured for the SBP, followed by generating a 3D color map of thickness for the entire SBP along with a scale bar. This map was then converted to a grayscale thickness map. The ROI was circled, and the average SBP thickness within ROI is calculated by average gray value/255 × maximum scale bar value. Coronal images were also contoured for the osteophyte, followed by 3D reconstruction and volume calculation.
Statistical analysis
Data are expressed as means ± SEM and analyzed by t tests, one-way analysis of variance (ANOVA) with Dunnett’s or Turkey’s post test, and two-way ANOVA with Bonferroni’s or Turkey’s post test for multiple comparisons using Prism 8 software (GraphPad Software, San Diego, CA). For assays using primary chondrocytes and bovine cartilage explants, experiments were repeated independently at least three times, and representative data were shown here. Values of P < 0.05 were considered statistically significant.
Acknowledgments
Funding: This study was supported by NIH grants NIH/NIAMS R01AR066098, R21AR074570, and R01AG067698 (to L.Q.), P30AR069619 (to Penn Center for Musculoskeletal Disorders), R01NS100892 (to Z.C.), and 2019 Health Research Formula Fund from the Pennsylvania Department of Health (to Z.C.). Author contributions: L.Q. and Z.C. designed the study. Y.W., L.L., and T.G. performed animal experiments. L.L., A.A., and L.Y. made NP formulations. Y.W., L.L., L.Y., and T.G. performed the histology and imaging analysis. Y.W. and T.G. performed cell culture and quantitative reverse transcription PCR experiments. B.J. performed the ICP-OES measurement. T.Y. and A.T. provided technical or material support and consultation. L.Q. and Z.C. wrote the manuscript. A.T. reviewed and revised the manuscript. L.Q. and Z.C. approved the final version. Competing interests: Z.C., L.Q., A.T., and Y.W. are inventors on a patent related to this work filed by the University of Pennsylvania (no. 63/043,025., filed 23 June 2020). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe6374/DC1
REFERENCES AND NOTES
- 1.Geiger B. C., Wang S., Padera R. F. Jr., Grodzinsky A. J., Hammond P. T., Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 10, eaat8800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kou L., Xiao S., Sun R., Bao S., Yao Q., Chen R., Biomaterial-engineered intra-articular drug delivery systems for osteoarthritis therapy. Drug Deliv. 26, 870–885 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karsdal M. A., Michaelis M., Ladel C., Siebuhr A. S., Bihlet A. R., Andersen J. R., Guehring H., Christiansen C., Bay-Jensen A. C., Kraus V. B., Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: Lessons learned from failures and opportunities for the future. Osteoarthr. Cartil. 24, 2013–2021 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Sokolove J., Lepus C. M., Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 5, 77–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martel-Pelletier J., Pathophysiology of osteoarthritis. Osteoarthr. Cartil. 12 Suppl. A, S31–S33 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Lin J. B., Poh S., Panitch A., Controlled release of anti-inflammatory peptides from reducible thermosensitive nanoparticles suppresses cartilage inflammation. Nanomedicine 12, 2095–2100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daghestani H. N., Pieper C. F., Kraus V. B., Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee osteoarthritis. Arthritis Rheumatol. 67, 956–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi K. E., Management of neuropathic pain. J. Am. Osteopath. Assoc. 105, S12–S19 (2005). [PubMed] [Google Scholar]
- 9.Burke J. E., Dennis E. A., Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 50 Suppl, S237–S242 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z., Liu N., Huang J., Lu P. H., Xu X. M., Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J. Neurochem. 116, 1057–1065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toborek M., Malecki A., Garrido R., Mattson M. P., Hennig B., Young B., Arachidonic acid-induced oxidative injury to cultured spinal cord neurons. J. Neurochem. 73, 684–692 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Carabaza A., Cabre F., Garcia A. M., Gomez M., Sanchez J., Mauleon D., Carganico G., Inhibition of phospholipase A2 purified from human herniated disc. Biochem. Pharmacol. 45, 783–786 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Biyani A., Andersson G. B., Low back pain: Pathophysiology and management. J. Am. Acad. Orthop. Surg. 12, 106–115 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Franson R. C., Saal J. S., Saal J. A., Human disc phospholipase A2 is inflammatory. Spine 17, S129–S132 (1992). [DOI] [PubMed] [Google Scholar]
- 15.Korovessis P. G., Phospholipase A2 activity in herniated lumbar discs. Spine 24, 99 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Kawakami M., Tamaki T., Hayashi N., Hashizume H., Nishi H., Possible mechanism of painful radiculopathy in lumbar disc herniation. Clin. Orthop. Relat. Res. , 241–251 (1998). [PubMed] [Google Scholar]
- 17.Murakami M., Nakatani Y., Atsumi G., Inoue K., Kudo I., Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 17, 225–283 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Leistad L., Feuerherm A. J., Faxvaag A., Johansen B., Multiple phospholipase A2 enzymes participate in the inflammatory process in osteoarthritic cartilage. Scand. J. Rheumatol. 40, 308–316 (2011). [DOI] [PubMed] [Google Scholar]
- 19.van Rossum G. S. A. T., Drummen G. P. C., Verkleij A. J., Post J. A., Boonstra J., Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: A p42/44MAPK-dependent and phosphorylation-independent mechanism. Biochim. Biophys. Acta 1636, 183–195 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Jamal O. S., Conaghan P. G., Cunningham A. M., Brooks P. M., Munro V. F., Scott K. F., Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Ann. Rheum. Dis. 57, 550–558 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruzanski W., Bogoch E., Wloch M., Vadas P., The role of phospholipase A2 in the physiopathology of osteoarthritis. J. Rheumatol. Suppl. 27, 117–119 (1991). [PubMed] [Google Scholar]
- 22.Pruzanski W., Bogoch E., Stefanski E., Wloch M., Vadas P., Enzymatic activity and distribution of phospholipase A2 in human cartilage. Life Sci. 48, 2457–2462 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Zhang W., Sun G., Aitken D., Likhodii S., Liu M., Martin G., Furey A., Randell E., Rahman P., Jones G., Zhai G., Lysophosphatidylcholines to phosphatidylcholines ratio predicts advanced knee osteoarthritis. Rheumatology (Oxford) 55, 1566–1574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quach N. D., Arnold R. D., Cummings B. S., Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem. Pharmacol. 90, 338–348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiDomenico C. D., Lintz M., Bonassar L. J., Molecular transport in articular cartilage – What have we learned from the past 50 years? Nat. Rev. Rheumatol. 14, 393–403 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Evans C. H., Kraus V. B., Setton L. A., Progress in intra-articular therapy. Nat. Rev. Rheumatol. 10, 11–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S., Adjei I. M., Brown S. B., Liseth O., Sharma B., Manganese dioxide nanoparticles protect cartilage from inflammation-induced oxidative stress. Biomaterials 224, 119467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McHugh J., Promising drug delivery system. Nat. Rev. Rheumatol. 15, 64 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Yan H., Duan X., Pan H., Holguin N., Rai M. F., Akk A., Springer L. E., Wickline S. A., Sandell L. J., Pham C. T. N., Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc. Natl. Acad. Sci. U.S.A. 113, E6199–E6208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Q., Yan L., Chiorazzo M., Delikatny E. J., Tsourkas A., Cheng Z., PLA2-responsive and SPIO-loaded phospholipid micelles. Chem. Commun. (Camb.) 51, 12313–12315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mwangi T. K., Berke I. M., Nieves E. H., Bell R. D., Adams S. B., Setton L. A., Intra-articular clearance of labeled dextrans from naive and arthritic rat knee joints. J. Control. Release 283, 76–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Partain B. D., Unni M., Rinaldi C., Allen K. D., The clearance and biodistribution of magnetic composite nanoparticles in healthy and osteoarthritic rat knees. J. Control. Release 321, 259–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J., Liang Q., Zuscik M., Shen J., Chen D., Xu H., Wang Y. J., Chen Y., Wood R. W., Li J., Boyce B. F., Xing L., Distribution and alteration of lymphatic vessels in knee joints of normal and osteoarthritic mice. Arthritis Rheumatol. 66, 657–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia H., Ma X., Tong W., Doyran B., Sun Z., Wang L., Zhang X., Zhou Y., Badar F., Chandra A., Lu X. L., Xia Y., Han L., Enomoto-Iwamoto M., Qin L., EGFR signaling is critical for maintaining the superficial layer of articular cartilage and preventing osteoarthritis initiation. Proc. Natl. Acad. Sci. U.S.A. 113, 14360–14365 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J. P., Fahmi H., Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Scanzello C. R., Goldring S. R., The role of synovitis in osteoarthritis pathogenesis. Bone 51, 249–257 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenham C. Y. J., Conaghan P. G., The role of synovitis in osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2, 349–359 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malfait A. M., Ritchie J., Gil A. S., Austin J.-S., Hartke J., Qin W., Tortorella M. D., Mogil J. S., ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthr. Cartil. 18, 572–580 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Nikolaou A., Kokotou M. G., Vasilakaki S., Kokotos G., Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 941–956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradley J. D., Dmitrienko A. A., Kivitz A. J., Gluck O. S., Weaver A. L., Wiesenhutter C., Myers S. L., Sides G. D., A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 32, 417–423 (2005). [PubMed] [Google Scholar]
- 41.Coulthard L. G., Costello J., Robinson B., Shiels I. A., Taylor S. M., Woodruff T. M., Comparative efficacy of a secretory phospholipase A2 inhibitor with conventional anti-inflammatory agents in a rat model of antigen-induced arthritis. Arthritis Res. Ther. 13, R42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruzanski W., Phospholipase A2: Quo vadis? J. Rheumatol. 32, 400–402 (2005). [PubMed] [Google Scholar]
- 43.Prasad L. K., O’Mary H., Cui Z., Nanomedicine delivers promising treatments for rheumatoid arthritis. Nanomedicine (Lond.) 10, 2063–2074 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He T., Zhang C., Vedadghavami A., Mehta S., Clark H. A., Porter R. M., Bajpayee A. G., Multi-arm avidin nano-construct for intra-cartilage delivery of small molecule drugs. J. Control. Release 318, 109–123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook Sangar M. L., Girard E. J., Hopping G., Yin C., Pakiam F., Brusniak M. Y., Nguyen E., Ruff R., Gewe M. M., Byrnes-Blake K., Nairn N. W., Miller D. M., Mehlin C., Strand A. D., Mhyre A. J., Correnti C. E., Strong R. K., Simon J. A., Olson J. M., A potent peptide-steroid conjugate accumulates in cartilage and reverses arthritis without evidence of systemic corticosteroid exposure. Sci. Transl. Med. 12, eaay1041 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Ai X., Duan Y., Zhang Q., Sun D., Fang R. H., Liu-Bryan R., Gao W., Zhang L., Cartilage-targeting ultrasmall lipid-polymer hybrid nanoparticles for the prevention of cartilage degradation. Bioeng. Transl. Med. 6, e10187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bajpayee A. G., Grodzinsky A. J., Cartilage-targeting drug delivery: Can electrostatic interactions help? Nat. Rev. Rheumatol. 13, 183–193 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Kavanaugh T. E., Werfel T. A., Cho H., Hasty K. A., Duvall C. L., Particle-based technologies for osteoarthritis detection and therapy. Drug Deliv. Transl. Res. 6, 132–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Svenson S., The dendrimer paradox–High medical expectations but poor clinical translation. Chem. Soc. Rev. 44, 4131–4144 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Robinson W. H., Lepus C. M., Wang Q., Raghu H., Mao R., Lindstrom T. M., Sokolove J., Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 580–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bajpayee A. G., Scheu M., Grodzinsky A. J., Porter R. M., Electrostatic interactions enable rapid penetration, enhanced uptake and retention of intra-articular injected avidin in rat knee joints. J. Orthop. Res. 32, 1044–1051 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Vedadghavami A., Wagner E. K., Mehta S., He T., Zhang C., Bajpayee A. G., Cartilage penetrating cationic peptide carriers for applications in drug delivery to avascular negatively charged tissues. Acta Biomater. 93, 258–269 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vedadghavami A., Zhang C., Bajpayee A. G., Overcoming negatively charged tissue barriers: Drug delivery using cationic peptides and proteins. Nano Today 34, 100898 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajpayee A. G., Scheu M., Grodzinsky A. J., Porter R. M., A rabbit model demonstrates the influence of cartilage thickness on intra-articular drug delivery and retention within cartilage. J. Orthop. Res. 33, 660–667 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Pritzker K. P. H., Gay S., Jimenez S. A., Ostergaard K., Pelletier J.-P., Revell P. A., Salter D., van den Berg W. B., Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 14, 13–29 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Aigner T., Cook J. L., Gerwin N., Glasson S. S., Laverty S., Little C. B., McIlwraith W., Kraus V. B., Histopathology atlas of animal model systems - Overview of guiding principles. Osteoarthr. Cartil. 18 Suppl. 3, S2–S6 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Piel M. J., Kroin J. S., Im H. J., Assessment of knee joint pain in experimental rodent models of osteoarthritis. Methods Mol. Biol. 1226, 175–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jia H., Ma X., Wei Y., Tong W., Tower R. J., Chandra A., Wang L., Sun Z., Yang Z., Badar F., Zhang K., Tseng W. J., Kramer I., Kneissel M., Xia Y., Liu X. S., Wang J. H. C., Han L., Enomoto-Iwamoto M., Qin L., Loading-induced reduction in sclerostin as a mechanism of subchondral bone plate sclerosis in mouse knee joints during late-stage osteoarthritis. Arthritis Rheumatol. 70, 230–241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe6374/DC1