

GAT2 links metabolism to DNA methylation to orchestrate macrophage-dependent inflammation.
Abstract
Accumulating evidence shows that nervous system governs host immune responses; however, how γ-aminobutyric acid (GABA)ergic system shapes the function of innate immune cells is poorly defined. Here, we demonstrate that GABA transporter (GAT2) modulates the macrophage function. GAT2 deficiency lowers the production of interleukin-1β (IL-1β) in proinflammatory macrophages. Mechanistically, GAT2 deficiency boosts the betaine/S-adenosylmethionine (SAM)/hypoxanthine metabolic pathway to inhibit transcription factor KID3 expression through the increased DNA methylation in its promoter region. KID3 regulates oxidative phosphorylation (OXPHOS) via targeting the expression of OXPHOS-related genes and is also critical for NLRP3–ASC–caspase-1 complex formation. Likewise, GAT2 deficiency attenuates macrophage-mediated inflammatory responses in vivo, including lipopolysaccharide-induced sepsis, infection-induced pneumonia, and high-fat diet-induced obesity. Together, we propose that targeting GABAergic system (e.g., GABA transporter) could provide previously unidentified therapeutic opportunities for the macrophage-associated diseases.
INTRODUCTION
There is an increasing interest about the link between nervous system and immune system based on the discovery that neurons, neurotransmitters, and/or neuropeptides shape the function of immune cells (1, 2). The γ-aminobutyric acid (GABA)ergic system [e.g., GABA, GABA receptors, glutamic acid decarboxylase (GAD), and GABA transporters (GATs)] is a crucial family of the central nervous system. It has been well documented that various peripheral immune cells express GABAergic components, e.g., macrophages express GAD and GABA receptors, and T cells express GAD, GATs, and GABA receptors (3, 4). These findings indicate that GABAergic system may be closely associated with the activation and function of immune cells. Notably, more interest has been highlighted on the effect of GATs [i.e., GAT1 (SLC6A1), GAT2 (SLC6A13), GAT3 (SLC6A11), and GAT4/BGT1 (SLC6A12)] on T cells. For example, GAT1 deficiency enhances T cell–mediated responses (5, 6), and GAT2 deficiency promotes T helper 17 (TH17) cell differentiation during infection (4). Nonetheless, whether innate immune cells (macrophages in particular) express GATs and the roles of GATs in innate immune cell–mediated responses are still unknown.
Macrophages are of the principal innate immune cells and show high diversity and plasticity in response to microenvironmental variations, thereby exerting diverse functions (7, 8). For example, upon stimulation with proinflammatory signals, like lipopolysaccharide (LPS) [or plus interferon-γ (IFN-γ)], macrophages produce large amounts of proinflammatory cytokines [e.g., interleukin-1β (IL-1β)] and perform bactericidal and antitumor activities. However, defects in macrophage phenotypic switch, such as the prolonged activation of a proinflammatory property in macrophages, are associated with various macrophage-associated diseases, including sepsis, infection-induced inflammatory diseases, and high-fat diet (HFD)–induced obesity (9–11). Thus, there is an emerging field that attracts increasing attention to study the contributors to shape the polarization of macrophages, such as environmental signals, cellular signaling pathways, and cellular metabolism (12, 13). However, we are still lacking a complete understanding of the functions of GABAergic system, especially about GATs, in macrophage polarization and function.
Here, we have elucidated that GAT2 affects macrophage IL-1β production. Mechanistically, GAT2 deficiency boosts intracellular accumulation of hypoxanthine, which not only blocks NOD-like receptor family pyrin domain containing 3 (NLRP3)–apoptosis-associated speck-like protein (ASC)–caspase-1 complex formation but also enhances the expression of oxidative phosphorylation (OXPHOS)–related genes via reducing the expression of transcription factor (TF) KID3 through the methylation. We further provide proof-of-concept evidence that GAT2 deficiency attenuates macrophage-mediated inflammatory responses in vivo. Our findings provide evidence of targeting GABAergic system to direct macrophage-mediated inflammatory responses, which has implications for prevention of macrophage-associated diseases.
RESULTS
GAT2 is highly expressed in primary macrophages
Previous study has found that GATs highly shape the function of T cells (4, 14, 15); however, the roles of GATs in macrophages remain obscure. To uncover this, the expressions of GATs (GAT1-4) were detected in murine primary peritoneal exudate macrophages (PEMs). Notably, Slc6a13 (encoding GAT2) and Slc6a12 (encoding GAT4) were most strongly expressed in PEMs, while Slc6a1 and Slc6a11 could not be detected in PEMs (Fig. 1A). With immunoblotting analysis, LPS/IFN-γ stimulation inhibited GAT2 expression while promoted GAT4 expression in PEMs (Fig. 1B), indicating that GAT2/4 may affect the activation of macrophages. Considering that GAT2 transports GABA, while GAT4 for betaine and GABA (16), thus, we next focused on the GAT2.
Fig. 1. GAT2 deficiency lowers the secretion of IL-1β and inhibits the activation of NF-κB signaling and inflammasome in proinflammatory macrophages.
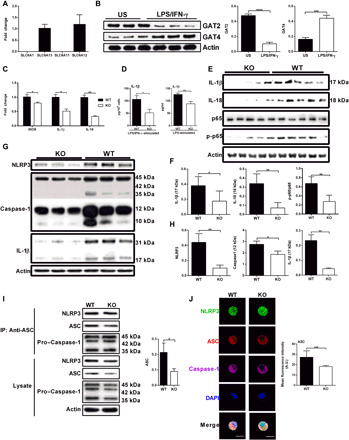
(A) Relative mRNA expressions of SLC6A1, SLC6A13, SLC6A11, and SLC6A12 in wild-type (WT) resting macrophages (n = 4). Results represent two independent experiments. Data are shown as means ± SEM. (B) Protein expressions of GAT2 and GAT4 in unstimulated (US) or LPS/IFN-γ–stimulated WT macrophages (n = 3). Results represent two independent experiments. (C) Relative mRNA expressions of inducible nitric oxide synthase (iNOS), IL-1β, and IL-18 in WT and knockout (KO) proinflammatory macrophages (n = 3). Results represent two independent experiments. Data are shown as means ± SEM. (D) The secretion of IL-1β from LPS or plus IFN-γ–stimulated WT and KO macrophages (n = 4). Results represent three independent experiments. (E to H) Protein expressions of IL-1β, IL-18, NLRP3, caspase-1, p65, and p-p65 in WT and KO proinflammatory macrophages (n = 3 to 5). Results represent two independent experiments. (I) Immunoblot analysis of ASC immunoprecipitates in WT and KO proinflammatory macrophages, probed for NLRP3, pro–caspase-1, and ASC (n = 3). Results represent two independent experiments. IP, immunoprecipitation. (J) Confocal microscopy of WT and KO M1 macrophages, immunostained for NLRP3 (green), ASC (red), and Caspase-1 (purple) (n = 3). Scale bars, 10 μm. Data were analyzed by unpaired t test and represented as means ± SD unless indicated.*P < 0.05, **P < 0.0 , ***P < 0.001, and ****P < 0.0001. A.U., arbitrary unit; DAPI, 4′,6-diamidino-2-phenylindole.
To explore the intrinsic roles of GAT2 in macrophage function, GAT2 deficiency [hereafter referred to as knockout (“KO”)] mice were constructed as shown previously (4). The percentages of F4/80+ cells in the peripheral blood mononuclear cells (PBMCs), spleens, and mesenteric lymph nodes (MLNs) were comparable between KO mice and the control [(hereafter referred to as wild type (“WT”)], except the bone marrow (fig. S1, A to D). Likewise, the number of macrophages in peritoneal cavity was comparable between KO mice and WT mice, even after eliciting with thioglycollate broth medium (Ftg) (fig. S1, E and F). As shown in fig. S1G, GAT2 was almost abolished in resting KO PEMs. However, there were no substantial difference on the mRNA expressions of macrophage markers [including F4/80, IL-1β, IL-6, IL-10, IL-18, and tumor necrosis factor–α (TNF-α)] (fig. S1H), IL-1β secretion (fig. S1I), and the activation of cellular nuclear factor κB (NF-κB) signaling pathway [phosphorylated IκB kinase α/β (IKKα/β) and IKBα/β, p65, and phosphorylated p65 in the cytoplasm and nucleus] (fig. S1, J and K). GAT2 deficiency reduced the intracellular protein abundance of NLRP3 inflammasome components (e.g., NLRP3, ASC, caspase-1, and IL-1β) in resting PEMs (fig. S1L). Collectively, macrophages express GAT2, which may be responsible for maintaining basal NLRP3 inflammasome activation in resting macrophages.
GAT2 deficiency dampens IL-1β secretion from proinflammatory macrophages
We then explored the influence of GAT2 deficiency on the function of activated macrophages. There were lower mRNA expressions of inducible nitric oxide synthase (iNOS), IL-1β, and IL-18 in LPS/IFN-γ–stimulated KO macrophages compared to the WT control (Fig. 1C). Besides, IL-1β (both LPS-induced and LPS/IFN-γ–induced) secretion (Fig. 1D) and the abundance of cleaved IL-1β and IL-18 were reduced in proinflammatory macrophages with GAT2 ablation (Fig. 1, E and F). Moreover, small interfering RNA (siRNA) was used to suppress endogenous GAT2, and we found that GAT2 knockdown considerably inhibited IL-1β secretion in LPS/IFN-γ–stimulated PEMs (fig. S2, A and B). To avoid cellular heterogeneity of conventional PEMs, we next chose ANA-1 cells to examine the function of GAT2. GAT2 silencing in ANA-1 cells also lowered the LPS/IFN-γ–induced IL-1β production (fig. S2C). GAT2 deficiency significantly lowered IL-6 production while had little effect on the production of TNF-α in LPS/IFN-γ–stimulated macrophages (fig. S2D). The inconsistent results of IL-1β and TNF-α indicate that GAT2 deficiency affects macrophage function in cytokine-specific manner.
Mammalian target of rapamycin (mTOR) signaling is involved in macrophage polarization and required for IL-1β production (17). We found that GAT2 deficiency had little effect on the phosphorylation of mTOR (fig. S2E) and 4EBP1 (fig. S2F) while highly enhanced the phosphorylation of S6K (as readout of mTORC1) and N-myc downstream regulated gene 1 (NDRG1) [as readout of serum- and glucocorticoid-induced kinase 1 (SGK1) activity that is a downstream node of mTORC2 signaling] (fig. S2F), indicating that the inhibition of LPS- (or plus IFN-γ)–induced IL-1β by GAT2 deficiency may through mTOR-independent pathways.
We then explored the NF-κB signaling and NLRP3 inflammasome, both are responsible for the production of IL-18 and IL-1β in proinflammatory macrophages (18). Notably, GAT2 deficiency inhibited the activation of NF-κB signaling (based on the lower ratio of p-p65/p65) in LPS/IFN-γ–stimulated macrophages (Fig. 1, E and F) and decreased the protein abundance of inflammasome components (e.g., NLRP3 and caspase-1) (Fig. 1, G and H). Furthermore, GAT2 deficiency selectively inhibited the mRNA expression of ASC (fig. S2G), suggesting that GAT2 deficiency dampens the activation of NLRP3 inflammasome at transcriptional level. Likewise, GAT2 silencing reduced the mRNA expression of ASC (fig. S2H). Immunoprecipitation and immunofluorescence analysis also showed that GAT2 deficiency in macrophages blocked NLRP3–ASC–caspase-1 complex formation, mainly lowering the abundance of ASC (Fig. 1, I and J). These results suggest that GAT2 deficiency negatively modulates macrophage NLRP3 inflammasome activation in the context of proinflammatory stimulation.
Considering that GAT2 expression was substantially increased in IL-4–stimulated WT macrophages (fig. S2I), we sought to determine whether GAT2 deficiency affects the activation of macrophage stimulated with IL-4. GAT2 deficiency reduced the mRNA expressions of Arg-1 and IL-10 (fig. S2J) and lowered the protein abundance of Arg-1 and CD206 (fig. S2K); however, it had little effect on IL-10 secretion (fig. S2L). Moreover, GAT2 deficiency had no effect on signal transducers and activators of transcription–6 activation, which is responsible for M2 macrophage polarization (fig. S2M) (19). Thus, whether GAT2 dictates IL-4–stimulated macrophage activation remains to uncover. Together, GAT2 deficiency dampens the function of proinflammatory macrophages, especially lowers the secretion of IL-1β through blocking NLRP3–ASC–caspase-1 complex formation.
GAT2 deficiency promotes OXPHOS in proinflammatory macrophages
To explore the nature of GAT2 ablation–induced the inhibition of IL-1β secretion, we performed RNA sequencing (RNA-seq) analysis on LPS/IFN-γ–stimulated macrophages with or without GAT2 deficiency. There was a significant difference about the transcriptomic profile between KO macrophages and WT macrophages (Fig. 2A). Further analysis demonstrated that GAT2 deficiency affected totally 959 genes (fold change > 2; P < 0.05; 355 up-regulated; 604 down-regulated) in LPS/IFN-γ–stimulated macrophages (Fig. 2B), enriching in immune system, signaling transduction, and metabolism (list hits > 5; P < 0.05) (Fig. 2C and fig. S3, A to C). For example, the NF-κB signaling pathway and NOD-like receptor signaling pathway were highly enriched (fig. S3, A to C), which were consistent with our previous results (Fig. 1, E to J).
Fig. 2. GAT2 deficiency alters the transcriptomic profile and enhances OXPHOS in proinflammatory macrophages.
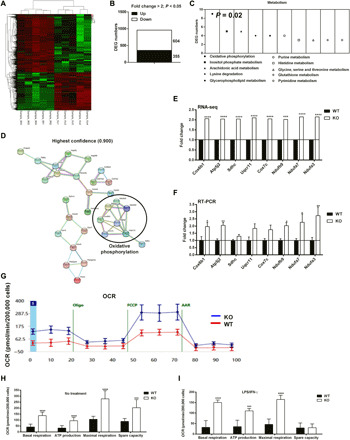
(A) Heatmap analysis of up-regulated (red) or down-regulated genes (green) in WT and KO proinflammatory macrophages (n = 5). (B) Histogram of differential expression genes (DEGs) (fold change > 2; P < 0.05) in WT and KO proinflammatory macrophages (n = 5). (C) Histogram of DEGs enriched in metabolism pathway in WT and KO proinflammatory macrophages (n = 5). (D) Schematic of the prediction of protein-protein interaction (confidence, 0.900). (E) The transcripts of the OXPHOS-related genes in WT and KO proinflammatory macrophages (n = 5). (F) Relative mRNA expressions of OXPHOS-related genes (Cox6b1, Atp5j2, Sdhc, Uqcr11c, Cox7c, Ndufb9, Ndufa7, and Ndufa3) in WT and KO proinflammatory macrophages (n = 5). Results represent two independent experiments. Data are shown as means ± SEM. (G to I) The basal respiration, ATP production, maximal respiration, and spare capacity of WT and KO resting/proinflammatory macrophages (n = 10). Results represent three independent experiments. Data were analyzed by unpaired t test and represented as means ± SD unless indicated. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone.
Notably, OXPHOS, which is negatively associated with the functions of proinflammatory macrophages (20), was the only significant pathway (P = 0.02) among the top 10 pathways in metabolism with Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis [differential expression genes (DEGs) > 3] (Fig. 2C). The prediction of protein-protein interaction of DEGs in metabolism indicated that most of DEGs were associated with OXPHOS (Fig. 2D), including Cox6b1, Atp5j2, Sdhc, Uqcr11, Cox7c, Ndufb9, Ndufa7, and Ndufa3 (Fig. 2E), and most of which (Cox6b1, Atp5j2, Ndufb9, Ndufa7, and Ndufa3) were validated by reverse transcription polymerase chain reaction (RT-PCR) analysis (Fig. 2F). These findings imply that GAT2 deficiency enhances OXPHOS in LPS/IFN-γ–stimulated macrophages.
We next assessed cellular bioenergetics with seahorse analysis and found that unstimulated KO macrophages became more “oxidative” as compared with unstimulated WT macrophages, on the basis of higher OCR (oxygen consumption rates; a measure of OXPHOS) in basal respiration, ATP production, maximal respiration, and spare capacity (Fig. 2, G and H). Notably, GAT2 deficiency enhanced OXPHOS in LPS/IFN-γ–stimulated macrophages (Fig. 2I) and seven in IL-4–stimulated macrophages (fig. S3D). However, the results of ECAR (extracellular acidification rate; a measure of glycolysis) were comparable between KO and WT macrophages (fig. S3, E to G). Collectively, GAT2 deficiency alters the transcriptomic profiles of LPS/IFN-γ–stimulated macrophages and especially switches proinflammatory macrophage cellular metabolic status toward to the OXPHOS.
GAT2 deficiency decreases IL-1β production through hypoxanthine
Since GAT2 deficiency in TH17 cells blocks GABA transport, resulting in higher levels of GABA in the medium (4); thus, we analyzed the concentrations of amino acids in the medium of LPS/IFN-γ–stimulated macrophages. There was no change about the concentrations of GABA and other amino acids (except decreased glutamate) in the medium (fig. S4, A to C), and no difference about the levels of intracellular GABA was detected both in resting and LPS/IFN-γ–stimulated macrophages (fig. S4, D and E), indicating that GAT2 deficiency affects macrophage IL-1β secretion may through GABA signaling independent way. Notably, GABA supplementation or bicuculline treatment (an inhibitor for GABA receptor) displayed little effect on IL-1β secretion from LPS/IFN-γ–stimulated WT macrophages (fig. S4F). Seemingly, the compensative increase of GAT4 expression in LPS/IFN-γ–stimulated KO macrophages (fig. S4G) may account for the comparable levels of GABA in WT/KO macrophages (fig. S4, A, D, and E). However, NNC05-2090 (an inhibitor for GAT4) treatment in LPS/IFN-γ–stimulated KO macrophages did not affect the IL-1β secretion (fig. S4H), ruling out the possibility that lower IL-1β secretion came from the increased GAT4 expression. Moreover, the involvement of decreased glutamate in IL-1β secretion from LPS/IFN-γ–stimulated KO macrophages was also ruled out on the basis of the results that glutamate supplementation had little effect on IL-1β secretion from both WT and KO proinflammatory macrophages (fig. S4I).
The cellular metabolism is highly associated with the functional output of macrophages (21). To explore the influence of GAT2 deficiency in cellular metabolic profiles in macrophages, we performed gas chromatography (GC)–3Q–mass spectrometry (MS) method (4) to identify the metabolites between WT macrophages with or without LPS/IFN-γ treatment, between WT and KO resting macrophages, as well as between LPS/IFN-γ–stimulated WT and KO macrophages. There was a significant difference about the cellular metabolites between unstimulated WT macrophages and LPS/IFN-γ–stimulated WT macrophages (Fig. 3A and fig. S5A), in which 12 metabolites were up-regulated, while 44 metabolites were down-regulated after LPS/IFN-γ stimulation (e.g., GABA and succinate). These metabolites were enriched in arginine and proline metabolism, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, riboflavin metabolism, and caffeine metabolism (Fig. 3A and fig. S5B). Likewise, there were also significant differences about the cellular metabolite profile between unstimulated WT and KO macrophages (Fig. 3B and fig. S5, C and D) and LPS/IFN-γ–stimulated WT/KO macrophages (Fig. 3C and fig. S5, E and F). Notably, tryosine, hypoxanthine, betaine, deoxyguanosine, citraconic acid, glyceric acid, and kynurenic acid were increased in LPS/IFN-γ–stimulated KO macrophages compared to those in WT control (Fig. 3C).
Fig. 3. GAT2 deficiency lowers IL-1β secretion through hypoxanthine.
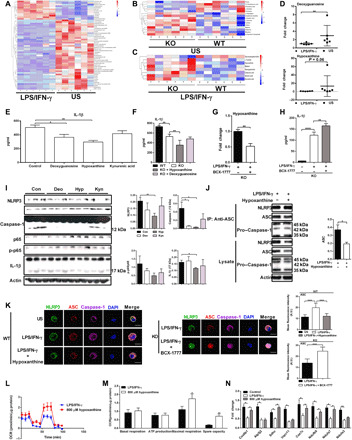
(A to C) Different metabolites in WT resting/proinflammatory macrophages (A), WT/KO resting macrophages (B), and WT/KO proinflammatory macrophages (C) (n = 6). (D) Intracellular deoxyguanosine and hypoxanthine in WT macrophages (n = 6). (E and F) IL-1β secretion from WT (E)/KO (F) proinflammatory macrophages with deoxyguanosine (5 μM), hypoxanthine (8 μM), and kynurenic acid (500 μM) supplementation (n = 3). (G and H) Intracellular hypoxanthine (G) and IL-1β secretion (H) in proinflammatory KO macrophages with 10 μM BCX-177 treatment (n = 6). (I) Protein abundance of inflammosome in WT proinflammatory macrophages treated as (E) (n = 3). (J) Immunoblot analysis of ASC immunoprecipitates in proinflammatory WT macrophages with hypoxanthine (800 μM) treatment (n = 3). (K) Confocal microscopy of proinflammatory WT macrophages with hypoxanthine (800 μM) treatment (left) and proinflammatory KO macrophages with BCX-1777 (10 μM) treatment (right) (n = 3). Scale bars, 10 μm. (L to N) The mitochondrial respiration [(L and M) n = 10] and relative mRNA expressions of OXPHOS-related genes [(N) n = 3] in WT proinflammatory macrophages with hypoxanthine (800 μM) supplementation. Results represent two (E, F, H to J, and L to N) or four [(G) means ± SEM] independent experiments. Data were analyzed by unpaired t test (D to G and J) or two-way analysis of variance (ANOVA) with Bonferroni correction (H, M, and N) and represented as means ± SD unless indicated. *P < 0.05, **P < 0.01, and ****P < 0.0001.
We next studied the effect of these increased metabolites in IL-1β secretion from LPS/IFN-γ–stimulated macrophages by adding each of them to the medium during macrophage polarization. Hypoxanthine and deoxyguanosine were decreased in WT proinflammatory macrophages compared to WT resting macrophages (Fig. 3D), suggesting that hypoxanthine and deoxyguanosine might inhibit the function of proinflammatory macrophages. Deoxyguanosine and especially hypoxanthine decreased the IL-1β secretion from both LPS/IFN-γ–stimulated WT and KO macrophages (Fig. 3, E and F). Like GAT2 deficiency, hypoxanthine had little effect on TNF-α production (fig. S5, G and H). Moreover, the IL-1β secretion was not significantly influenced by other increased metabolites (e.g., kynurenic acid, citraconic acid, and betaine) (Fig. 3E and fig. S5, I and J). The levels of glycerophosphocholine were reduced in LPS/IFN-γ–stimulated KO macrophages (Fig. 3C), but glycerophosphocholine supplementation could not promote IL-1β secretion from both WT macrophages and KO macrophages (fig. S5, K and L). Notably, IRG1/ACOD1 (immune-regulated gene 1 or aconitate decarboxylase 1) mediates itaconate production and is important for regulating the proinflammatory state of M1 macrophages (22). Here, we found that GAT2 deficiency mildly inhibited IRG1 expression and itaconate production in LPS/IFN-γ–stimulated macrophages (fig. S5, M and N). Therefore, we envisioned that GAT2 deficiency inhibits IL-1β production in LPS/IFN-γ–stimulated macrophages through hypoxanthine.
To test our hypothesis, we used BCX-1777 [an inhibitor of the purine nucleoside phosphorylase (PNP) (23)] to suppress hypoxanthine biosynthesis (Fig. 3G). BCX-1777 treatment substantially increased IL-1β secretion from LPS/IFN-γ–stimulated KO macrophages (Fig. 3H). Furthermore, similar to GAT2 deficiency, hypoxanthine dampened the NLRP3–ASC–caspase-1 complex formation in WT macrophages (Fig. 3, I to K); however, inhibition of hypoxanthine biosynthesis highly enhanced inflammasome activation in KO macrophages (Fig. 3K). Like GAT2 deficiency, hypoxanthine inhibited the mRNA expressions of NLRP3 and ASC in LPS/IFN-γ–stimulated WT macrophages; however, BCX-1777 reversed these changes in KO macrophages (fig. S5O), suggesting that hypoxanthine also regulates the activation of inflammasome at transcriptional level.
We next asked whether hypoxanthine could also alter cellular bioenergetics in proinflammatory macrophages. In LPS/IFN-γ–stimulated WT macrophages, hypoxanthine (8 or 800 μM) treatment promoted OXPHOS (Fig. 3, L and M, and fig. S5, P and Q) but had little effect on glycolysis (fig. S5, R and S), which is similar to GAT2 deficiency (Fig. 2I and fig. S3G). Hypoxanthine supplementation also substantially increased the mRNA expressions of OXPHOS-related genes with dosage-dependent manner, including Cox6b1, Sdhc, Ndufa7, and Ndufb9 (Fig. 3N and fig. S5T). Collectively, GAT2 deficiency decreases IL-1β production in proinflammatory macrophages through hypoxanthine.
GAT2 deficiency decreases IL-1β secretion via betaine-hypoxanthine axis
To explore the underlying mechanism by which GAT2 deficiency promotes hypoxanthine accumulation in macrophages, we further analyzed the hypoxanthine metabolism in macrophages since there is a core purine metabolism in proinflammatory macrophages (24). There was a significant alteration about the hypoxanthine metabolism both in unstimulated macrophages and LPS/IFN-γ–stimulated macrophages after GAT2 deficiency, resulting in accumulation of inosine in unstimulated KO macrophages but hypoxanthine in LPS/IFN-γ–stimulated KO macrophages (Fig. 4A). Of note, GAT2 deficiency enhanced the enzyme activity of PNP in LPS/IFN-γ–stimulated macrophages (Fig. 4A), rather than resting macrophages (fig. S6A). Likewise, GAT2 deficiency increased mRNA level of PNP in LPS/IFN-γ–stimulated macrophages (fig. S6B). GAT2 deficiency also affected the enzyme activity and mRNA expressions of xanthine oxidase (XOD) and xanthine dehydrogenase (XDH) in LPS/IFN-γ–stimulated macrophages, which mediate the metabolism of hypoxanthine into xanthine and the production of reactive oxygen species (ROS) (Fig. 4A and fig. S6C) (25).
Fig. 4. GAT2 deficiency promotes betaine-hypoxanthine metabolism in proinflammatory macrophages.
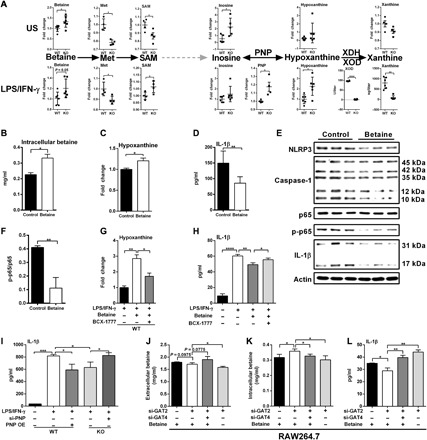
(A) Schematic of the generation of hypoxanthine from betaine. Metabolites in this metabolic axis analyzed with metabolomics (n = 4 to 6), and the activities of PNP and XOD also measured (n = 4). (B to D) Intracellular betaine (B), hypoxanthine (C) and IL-1β secretion (D) in WT proinflammatory macrophages with betaine delivery (1 mM; n = 3). (E and F) Protein abundance of interested proteins in WT proinflammatory macrophages after betaine delivery (n = 3). (G and H) Intracellular hypoxanthine [(G) n = 6] and IL-1β secretion [(H) n = 3)] in LPS/IFN-γ–activated WT macrophages with betaine delivery combined BCX-1777 (10 μM) treatment. (I) IL-1β secretion from WT resting macrophages, LPS/IFN-γ–activated WT macrophages with PNP overexpression, and LPS/IFN-γ–activated KO macrophages with PNP silencing (n = 3). (J to L) The levels of extracellular (J) or intracellular (K) betaine and IL-1β secretion (L) in LPS/IFN-γ–activated RAW264.7 cells with GAT2 and/or GAT4 silencing in the presence of 50 μM betaine (n = 3). Results represent two (H and J to L), three (B to D), or four (G) independent experiments. Data were analyzed by unpaired t test (A, B, C, D, F, and G) or two-way ANOVA with Bonferroni correction (G to L) and represented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Intriguingly, we observed that the intracellular betaine levels were increased in both unstimulated and LPS/IFN-γ–stimulated KO macrophages (Figs. 3, B and C, and 4A). This is not unexpected because LPS/IFN-γ–stimulated KO macrophages have higher expression of GAT4 (fig. S4G), which transports both GABA and betaine. GAT2 deficiency also increased GAT4 expression in IL-4–stimulated macrophages (fig. S6D), leading to intracellular accumulation of hypoxanthine (fig. S6E). Considering that GAT4 blockade and betaine supplementation during M1 macrophage polarization could not alter IL-1β secretion (figs. S4H and S5J), thus, we envisioned that GAT2 deficiency induces intracellular accumulation of betaine in resting macrophages by the compensated expression of GAT4, and betaine is converted into hypoxanthine by PNP to lower IL-1β production in LPS/IFN-γ–stimulated macrophages.
To test this, first, betaine was transfected into macrophages before LPS/IFN-γ stimulation. Betaine delivery increased intracellular levels of betaine and hypoxanthine and the activity of PNP (Fig. 4, B and C, and fig. S6, F to H). Notably, intracellular transfection with betaine extremely reduced the secretion of IL-1β from LPS/IFN-γ–stimulated WT macrophages (Fig. 4D) and the intracellular abundance of inflammasome components, p-p65 and IL-1β (Fig. 4, E and F). Then, we treated LPS/IFN-γ–stimulated WT macrophages with BCX-1777 after betaine transfection; BCX-1777 decreased the activity of PNP and intracellular level of hypoxanthine (Fig. 4G and fig. S6, H and I) but rescued the production of IL-1β (Fig. 4H). Similarly, PNP overexpression in WT macrophages lowered the IL-1β secretion, while PNP silencing in KO macrophages rescued IL-1β production (Fig. 4I and fig. S6J).
Subsequently, to support the importance of GAT4 in mediating intracellular accumulation of betaine in macrophages with GAT2 deficiency, we knocked down the GAT4 in KO bone marrow–derived macrophages (BMDMs) by means of siRNA delivery (fig. S6K). The GAT4 silencing in KO BMDMs remarkably rescued IL-1β production (fig. S6L). Subsequently, we knocked down the GAT2, GAT4, and GAT2 combined with GAT4 in RAW264.7 cells, respectively. It should be noted that RAW264.7 cells lack ASC expression (26); thus, the concentration of IL-1β in the culture medium measured by enzyme-linked immunosorbent assay (ELISA) is low (fig. S6M) and may reflect that of pro–IL-1β rather than the active form of IL-1β. Even so, we found that the change of extracellular IL-1β was consistent with those of the intracellular IL-1β by Western blot analysis (fig. S6, M and O). Therefore, we used ELISA for measuring the extracellular IL-1β in further experiment with RAW264.7 cells. To simulate the physiological situation in vivo, we supplemented ~50 μM betaine [20 to 70 μM in normal human plasma (27, 28); 50 to 100 μM in mouse plasma (29)] into medium during the whole transfection and/or polarization progress. The results also demonstrated the critical roles of GAT4 in the intracellular accumulation of betaine (Fig. 4, J and K) and reduced IL-1β production in GAT2-deficient macrophages (Fig. 4L and fig. S6P). Notably, GAT2 silencing without betaine supplementation had lower extracellular and/or intracellular level of betaine while higher IL-1β production in comparison to GAT2 silencing in the presence of betaine, highlighting the irreplaceable role of betaine in determining IL-1β production in LPS/IFN-γ–stimulated macrophages (Fig. 4, J to L). Together, our results highlight that GAT2 deficiency boosts betaine-hypoxanthine metabolic pathway to lower IL-1β production in proinflammatory macrophages.
Hypoxanthine decreases IL-1β secretion via KID3
Then, we want to know the underlying mechanism by which hypoxanthine regulates inflammasome activation and OXPHS in macrophages, especially hypoxanthine-mediated expressions of OXPHOS-related genes. According to our previous observations that the mRNA encoding for Cox6b1, Ndufa7, and Ndufb9 are increased both in LPS/IFN-γ–stimulated KO and WT macrophages supplemented with hypoxanthine (Figs. 2F and 3N), thus, these genes are the upmost interest in the current study. In silico analysis shown that these genes are regulated by the TF KID3, CPBP, and PRRX2 (Fig. 5A). On the basis of the standard that resting macrophages, LPS/IFN-γ–stimulated KO macrophages and WT macrophages supplemented with hypoxanthine should have similar alteration in the expressions of TFs, compared to LPS/IFN-γ–stimulated WT macrophages, we took KID3 as the key TF (Fig. 5, B and C). Similarly, we also found that GAT2 silencing inhibited KID3 expression (fig. S7A). Of note, BCX-1777 treatment significantly increased KID3 expression while decreased Cox6b1, Ndufa7, and Ndufb9 expressions in LPS/IFN-γ–stimulated KO macrophages (Fig. 5D), suggesting that hypoxanthine inhibits the expression of KID3, which negatively regulates the expressions of these OXPHOS-related genes. In addition, PNP silencing increased KID3 expression while inhibited Ndufb9 expression; nevertheless, PNP overexpression showed contrary results (fig. S7, B and C). We also demonstrated that hypoxanthine treatment decreased the mRNA expressions of IL-1β and KID3 while increased the expression of Ndufb9 in LPS/IFN-γ–stimulated RAW264.7 cells (fig. S7D).
Fig. 5. Hypoxanthine enhances expressions of OXPHOS-related genes via KID3.
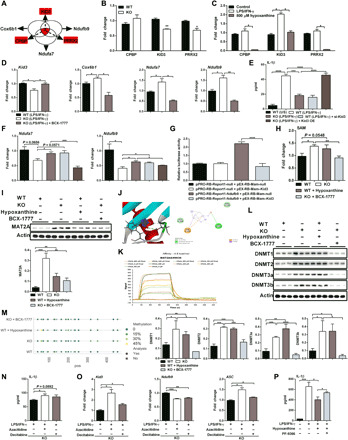
(A) Common TFs of Cox6b1, Ndufb9, and Ndufa7. (B and C) Gene expressions in proinflammatory macrophages [(B) n = 3] or in proinflammatory WT macrophages with hypoxanthine supplementation [(C) n = 3]. (D) Gene expressions in proinflammatory macrophages with BCX-1777 (10 μM) treatment (n = 3). (E and F) IL-1β secretion (E) and Ndufb9/Ndufa7 expression (F) in macrophages (n = 3). (G) Kid3 inhibits the luciferase activity of Ndufb9 promoter (n = 3 to 6). (H and I) Intracellular SAM (H) and MAT2A abundance (I) in macrophages (n = 3). (J) Molecular docking analysis of the complexes of MAT2A and hypoxanthine. (K) Hypoxanthine (H9636) directly interacts with MAT2A by SPR. (L) DNMTs abundance in macrophages (n = 3). (M) DNA total methylation level in promoter region of KID3 in macrophages (n = 3 and 4). (N and O) IL-1β secretion and gene expressions (O) from macrophages with azacitidine (500 nM) or decitabine (50 nM) (n = 3). (P) IL-1β secretion from WT macrophages with hypoxanthine combined PF-9366 (1 μM) (n = 3). Results represent two (B and C), three (G and H), four (D), or five (E and F) independent experiments. Data are shown as means ± SD (E, G, H, P, L, and N) or means ± SEM. Data were analyzed by unpaired t test (B) or two-way ANOVA with Bonferroni correction (C, D to I, L, and N to P). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To further validate the roles of KID3 in regulating mRNA expressions of these OXPHOS-related genes, we transfected WT macrophages and KO macrophages with si-Kid3 and Kid3-containing plasmid to inhibit and enhance the expression of KID3, respectively (fig. S7E). Like GAT2 deficiency or hypoxanthine supplementation, KID3 inhibition reduced production and expression of IL-1β (Fig. 5E and fig. S7F) and promoted the expressions of both Ndufa7 and Ndufb9 in LPS/IFN-γ–stimulated WT macrophages (Fig. 5F). Similar to PNP inhibition in KO macrophages, KID3 overexpression increased production and expression of IL-1β in KO macrophages (Fig. 5E and fig. S7F) and decreased the expressions of both Ndufa7 and Ndufb9 in LPS/IFN-γ–stimulated KO macrophages (Fig. 5F). These results indicate that the expressions of Ndufa7 and Ndufb9 are negatively regulated by KID3. Unexpectedly, KID3 silencing reduced the mRNA expressions of NLRP3 and ASC, while KID3 overexpression promoted the mRNA expressions of NLRP3 and ASC (fig. S7G), suggesting that these two genes might be positively controlled by KID3. To identify the role of KID3 in determination of gene expression, we conducted dual luciferase report system assay and found that Ndufb9 was negatively modulated by KID3 in a direct way (Fig. 5G), suggesting that KID3 affects OXPHOS by directly targeting the expression of OXPHOS-related genes. Collectively, hypoxanthine inhibits the expression of KID3, which regulates the expressions of genes associated with OXPHOS and inflammasome.
Hypoxanthine inhibits KID3 through methylation
Next, we sought to reveal the underlying mechanism by which KID3 is inhibited in LPS/IFN-γ–stimulated KO macrophages or hypoxanthine supplementation. We focused on the methylation of KID3 because betaine metabolism is accompanied by the production of SAM (functions as the main methyl donor), which could be metabolized into hypoxanthine (24, 30). Notably, GAT2 silencing and GAT2 deficiency improved the intracellular level of SAM in LPS/IFN-γ–stimulated macrophages (Figs. 4A and 5H and fig. S7H). Hypoxanthine increased the intracellular level of SAM in LPS/IFN-γ–stimulated WT macrophages, while BCX-1777 blocked the accumulation of SAM in LPS/IFN-γ–stimulated KO macrophages (Fig. 5H). These results indicated that hypoxanthine positively regulates the accumulation of SAM in proinflammatory macrophages. Of note, we found that the relative abundance of MAT2A, the main enzyme that favors in the SAM de novo synthesis from methionine in mammalian cells (31), was much higher in LPS/IFN-γ–stimulated KO macrophages or WT macrophages with hypoxanthine supplementation but lower in LPS/IFN-γ–stimulated KO macrophages with BCX-1777 treatment (Fig. 5I). Likewise, PNP overexpression in WT macrophages and PNP silencing in KO macrophages affected the MAT2A expression (fig. S7I).
To better understand how hypoxanthine specifically improves the enzymatic activity of MAT2A, we performed in silico docking analysis between MAT2A as receptor and hypoxanthine as the ligand. Intriguingly, we found that hypoxanthine could bind to MAT2A through van der Waals with two amino acid residues (Ser206 and Arg249), hydrogen bonds with Ser247, and orthophosphate (Pi)-cation/sigma with Phe250 of MAT2A (Fig. 5J). It has been well demonstrated that adenosine occupies the active site of MAT2A through several hydrogen bonds with Ser247 and Arg249, as well as π-π stacking between the purine base of adenine and Phe250 of MAT2A (32). Adenosine and hypoxanthine have identical chemical properties (fig. S7J), implying that hypoxanthine could also occupy the active site and hydrogen bond to MAT2A, thereby enhance its enzymatic activity. To firmly validate our hypothesis, we conducted surface plasmon resonance (SPR) assay about hypoxanthine and human MAT2A protein [since no available commercial recombinant murine MAT2A protein, and the MAT2A is highly conserved between mouse and human (fig. S7K)]. The results showed that hypoxanthine directly interacted with MAT2A in a dose-dependent manner (Fig. 5K).
These data suggested that the DNA hypermethylation may be involved in the reduced expression of KID3 after GAT2 deficiency or hypoxanthine supplementation. This hypothesis had been demonstrated by the following evidence: (i) The relative abundance of DNA methyltransferases (i.e., DNMT1, DNMT2, DNMT3a, and DNMT3b) were much higher both in LPS/IFN-γ–stimulated KO macrophages and WT macrophages with hypoxanthine supplementation, but lower in LPS/IFN-γ–stimulated KO macrophages with BCX-1777 treatment (Fig. 5L). (ii) The accumulation of hypoxanthine could enhance DNA total methylation level of KID3 (Fig. 5M). (iii) The specific DNA methyltransferase inhibitors azacitidine and decitabine (33) both highly increased KID3 and ASC expression and IL-1β production (Fig. 5, N and O) while inhibited Ndufb9 expression in LPS/IFN-γ–stimulated KO macrophages (Fig. 5O). (iv) The blockage of MAT2A by PF-9366 (31) reversed the inhibitory effect of hypoxanthine on IL-1β production in WT proinflammatory macrophages (Fig. 5P). In summary, these data illustrate that GAT2 deficiency promotes hypoxanthine accumulation, which inhibits KID3 expression via the enhanced methylation.
GAT2 deficiency attenuates LPS-induced sepsis, Pasteurella multocida–induced pneumonia, and HFD-induced obesity in mice
We then asked whether GAT2 deficiency suppresses the inflammatory responses in vivo. We first investigated the effect of GAT2 deficiency on LPS-induced sepsis (a model that largely reflects the inflammatory functions of monocytes/macrophages). The survival rate of KO mice after intraperitoneal injection of LPS was significantly increased (fig. S8A). GAT2 deficiency inhibited mRNA expressions and abundance of inflammatory cytokines (especially IL-1β and TNF-α) in the jejunum and liver of mice (fig. S8, B and C). GAT2 deficiency significantly lowered numbers of macrophages (marked by F4/80) in the lung and liver of mice under LPS challenge (fig. S8D). After deletion of macrophages using clodronate-loaded liposome, the survival rates of WT and KO mice were comparable under LPS challenge (fig. S8E).
We next addressed the suppressive role of GAT2 deficiency in inflammation in vivo using a model of P. multocida (PmCQ2)–infected pneumonia. Likewise, the KO mice were less sensitive to PmCQ2 infection in overall survival assays (Fig. 6A). GAT2 deficiency substantially reduced mRNA expressions of IL-1β and TNF-α in the lung of mice (Fig. 6B) and attenuated infection-induced pneumonia (Fig. 6C) and decreased levels of IL-6, IL-1β, and TNF-α in the lung and serum (Fig. 6, D and E). Our previous study reported that PmCQ2 infection triggers inflammatory responses in mouse lung depending on macrophages and neutrophils (34). Here, GAT2 deficiency significantly lowered the numbers of macrophages (marked by CD68), rather than neutrophils (marked by Ly6G) in the lung of mice (Fig. 6, F and G). To verify that GAT2 deficiency inhibits host inflammatory responses by regulating the functions of alveolar macrophages, the alveolar macrophages in vivo were deleted using clodronate-loaded liposome. In this model, GAT2 deficiency had no effect on the numbers of alveolar macrophages (Fig. 6H) and mRNA expressions and secretion of IL-1β in the lung (Fig. 6I) and serum (Fig. 6J) of mice.
Fig. 6. GAT2 deficiency alleviates PmCQ2 infection in mice.
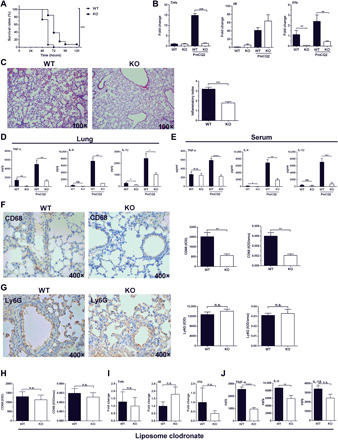
(A) GAT2 deficiency improves the survival rate of mice (n = 10). (B) Relative mRNA expressions of TNF-α, IL-6, and IL-1β in lungs of mice (n = 3). Data are shown as means ± SEM. (C) The inflammation in the lung was analyzed with hematoxylin and eosin staining (400×) (n = 20 to 30). Data are shown as means ± SEM. (D and E) The levels of TNF-α, IL-6, and IL-1β in lungs (D) and serum (E) of mice (n = 6). n.s., not significant. (F) The expressions of CD68 in lungs of mice were determined by immunohistochemistry (100×) (n = 6). IOD, integrated optical density. Data are shown as means ± SEM. (G) The expressions of Ly6G in lungs of mice were determined by immunohistochemistry (400×) (n = 6). Data are shown as means ± SEM. (H) The expressions of CD68 in lungs of mice with liposome clodronate treatment were determined by immunohistochemistry (100×) (n = 6). Data are shown as means ± SEM. (I) Relative mRNA expressions of TNF-α, IL-6, and IL-1β in the lungs of mice with liposome clodronate treatment (n = 5). Data are shown as means ± SEM. (J) Levels of TNF-α and IL-1β in the serum of mice with liposome clodronate treatment (n = 6 to 8). Data were analyzed by unpaired t test and represented as means ± SD unless indicated. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The above two models indicate that GAT2 deficiency could mitigate macrophage-mediated acute inflammation in vivo; however, whether GAT2 deficiency affects macrophage-mediated chronic inflammation (e.g., obesity) is still unknown. To uncover this, we fed mice with a HFD for 12 weeks. As expected, HFD-treated KO mice had a lower body weight, liver weight, and adipose tissue weight than those in HFD-treated WT mice (Fig. 7, A and B). GAT2 deficiency prevented insulin resistance, although no significantly effect on glucose intolerance was observed in mice after HFD treatment (fig. S8, F and G). GAT2 depletion reduced the production of inflammatory cytokines in adipose tissues and serum (Fig. 7C and fig. S8H), suggesting a suppressive action of GAT2 deficiency on metabolic syndrome–related inflammation in vivo. In addition, GAT2 deficiency remarkably reduced the numbers of macrophages in adipose tissues (Fig. 7D), especially the ratio of iNOS+ (M1-like):CD206+ (M2-like) macrophages (Fig. 7E). Of note, in our experimental models, GAT2 deficiency decreased TNF-α level, which is not consistent with the finding in vitro, which is probably due to the contribution of IL-1β to TNF-α production in vivo (35). Collectively, these results indicate that GAT2 deficiency ameliorates macrophage-mediated inflammatory responses in vivo.
Fig. 7. GAT2 deficiency attenuates HFD-induced obesity in mice.
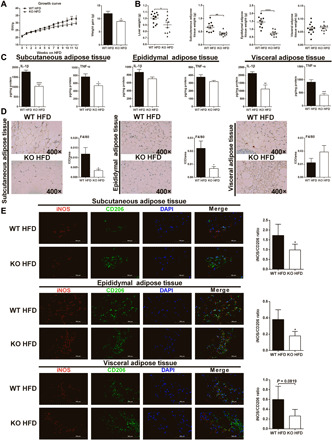
(A) GAT2 deficiency slows the increase of body weight of HFD-fed mice (n = 10 to 12). BW, body weight. (B) GAT2 deficiency reduces the weight of liver and adipose tissues of HFD-fed mice (n = 10 to 12). (C) The levels of IL-1β and TNF-α in adipose tissues (n = 10 to 12). (D) The expressions of F4/80 in adipose tissues of mice were determined by immunohistochemistry (400×) (n = 6). Data are shown as means ± SEM. (E) Immunofluorescence analysis of adipose tissues (400×), immunostained for iNOS (red) and CD206 (green). Scale bars, 100 μm (n = 6). Data were analyzed by unpaired t test and represented as means ± SD unless indicated. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
DISCUSSION
Mounting evidence shows the cross-talk between the nervous system with various immune cells to regulate host homeostasis and immunity. Intriguingly, both central and enteric nervous system can physiologically contribute to the inflammatory responses through different mediators (e.g., neurotransmitters) at homeostasis and during pathogen infection (36, 37). However, whether GABAergic system has implications for controlling macrophage-mediated inflammatory responses are not well characterized. This study reported that GAT2 affects IL-1β production in macrophages. During pathogen infection, sympathetic neurons could polarize muscularis macrophages toward a tissue-protective phenotype (38); nevertheless, the mechanism whereby sympathetic neurons alter macrophage polarization is not clear. We provided evidence that GAT2 deficiency inhibited M1 macrophage polarization by suppressing inflammasome activation and enhancing OXPHOS capacity. In addition, previous study showed that the activation of GABAergic system in macrophages enhances autophagy activation, phagosomal maturation, and antimicrobial responses against mycobacterial infection (3).
It is noteworthy that macrophages arise from monocytes and that monocyte migration could be markedly suppressed by GABAB receptor agonist (39); thus, it is interesting to investigate whether GABA signaling is decisive for the development and maturity of macrophages. Moreover, other GABAergic components, including GABAAR, GAD65, and GABA, were also observed in mouse macrophages (3, 15). The impacts of these GABAergic components in macrophage function need further study. As recently reviewed (40), the activation of macrophages increases the levels of the GABAergic components and GABA could induce inward currents (although smaller in amplitude and slower to rise and decay) in macrophages. These findings suggest that GABAergic signaling links to the multifarious functions of macrophages despite the underlying mechanisms remain elusive.
Neutrophils express GABAAR and GABABR, and the latter of which can stimulate neutrophilic chemotaxis (41). Dendritic cells (DCs) could express GABAAR, GAD65, and GAT4 and secrete GABA (42–44). GABA has stimulatory effects on DCs (e.g., chemotactic responses, motility, and transmigration) while inhibits the antigen presentation function of DCs (44). Moreover, a recent fascinating study has demonstrated that mast cells could express GAT1 (45). These findings support the importance of GABAergic signaling in modulating the biology of innate immune cells. Considering that GATs are the pivotal components of GABAergic system and most of the studies have centered on the GATs in T cells thus far, future additional works should be focused on GATs in the modulation of innate immune cell functions (e.g., mast cells). Notably, similar to GABA, glutamate (excitatory amino acid), aspartate (excitatory amino acid), and glycine (inhibitory amino acid) can also function as the amino acid neurotransmitters; hence, it is meaningful to further investigate the cross-talk between the nervous system (e.g., glutamatergic and glycinergic signaling) and the immune system.
A purine nucleotide cycle is active in M1 macrophages, which could supply tricarboxylic acid (TCA) cycle intermediates (e.g., fumarate) to synchronize mitochondrial activity with glycolysis by balancing electron transfer into mitochondria (24). However, we did not find any alterations of TCA cycle intermediates from LPS/IFN-γ–stimulated KO macrophages in comparison to the WT control, indicating that GAT2 deficiency switches cellular bioenergetics of proinflammatory macrophages independent of TCA cycle intermediates. Our data demonstrated that GAT2 deficiency boosts the levels of intracellular purine metabolites (especially hypoxanthine) to function as the pivotal metabolite to enhance OXPHOS by promoting Ndufb9 expression in LPS/IFN-γ–stimulated macrophages. Mechanistically, we confirmed that GAT2 deficiency increases PNP-mediated intracellular hypoxanthine level by improving betaine level involving the compensatory increase of GAT4 expression, which serves as the betaine transporter in the cells (15). These data highlight the importance of GABA transporters in directing immune cell function. Moreover, hypoxanthine can funnel into the salvage pathway for purine nucleotides (46), whether hypoxanthine can support the proliferation of macrophages is still unknown. Likewise, inosine (the metabolic precursor of hypoxanthine) could serve as an alternative carbon source for CD8+ T cell function under glucose restriction (47), whether hypoxanthine could exploit the similar effects in macrophages warrants further investigation.
Masses of C2H2 zinc finger family (the largest class of TFs in mammals) members are key transcriptional regulators involved in immune cell development and function (48–50). Unexpectedly, we noticed that KID3 (also termed as Zfp354c, one of the C2H2 zinc finger protein) displays discrepant roles in targeting Ndufb9 and ASC to regulate their expression. The possible reasons may be as follows: (i) Other factors that share large amino acid sequence similarity and/or DNA sequence identity with KID3 could also bind to GC-rich regulatory regions of Ndufb9 and ASC. (ii) KID3 cooperates with epigenetic control mechanisms (e.g., acetylation and methylation) to exert different transcriptional activities. Virtually, emerging evidence propose that epigenetic regulation drives immune responses and that SAM supports proinflammatory macrophages (24, 30). We suggested that the occurrence of the enhanced methylation evidenced by GAT2 deficiency or hypoxanthine supplementation facilitates betaine to generate maximum SAM. Now, we concluded that GAT2 deficiency increases DNA total methylation in the promoter region of KID3 to silence its expression to augment OXPHOS-related gene (chiefly Ndufb9) expression, thereby lowering IL-1β production in LPS/IFN-γ–stimulated macrophages. However, other underlying mechanisms (e.g., cofactors and signaling) connecting to KID3 in orchestrating proinflammatory macrophage function (especially in guiding ASC activation) are not fully elucidated.
It is mentionable that GAT2 deficiency had no effects on IKK and IκB while significantly inhibiting p65 phosphorylation, which is probably due to the fact that GAT2 deficiency reduces the activity of preexisting NF-κB nuclear complexes, inducing transcriptional inactivation (51). ASC is an adapter protein that is required for the formation of the inflammasome, contributing to macrophage activation (52–54). Upon inflammasome activation, ASC forms the ASC speck, a supramolecular aggregate of ASC dimers, which serves as another platform for the activation of Caspase-1 (55, 56). Moreover, phosphorylation of ASC is critical for speck formation and caspase-1 activation (26). Now, hypoxanthine highly reduces the mRNA expression of ASC, dampening the formation of NLRP3–ASC–caspase-1 complex. However, it is interesting to know whether hypoxanthine affects the NLRP3 inflammasome activation through protein posttranslation modification.
We have delineated the influences of systemic knockout of GAT2 in macrophage functions; additional work will be highly helpful by exploring the macrophage functions with Slc6a13fl/fl-LysMcre mice, in which GAT2 was deleted specifically in macrophages (although we have conducted macrophage-deleting experiments). Moreover, although we have validated that GAT2 deficiency enhances betaine/SAM/hypoxanthine metabolic pathway in proinflammatory macrophages, it is critical to perform a stable isotope-based metabolomic approach to identify the metabolic route of betaine. This study lacks a stable isotope-based metabolomic approach to identify the metabolic route of betaine (e.g., usage of [13C] betaine or [15N] betaine) to hypoxanthine for following reasons: (i) Only the methyl group (CH3) from betaine attaches to the methionine sulfur atom in SAM, which then physically latches on to certain areas along the DNA strand to inactive genes (called “methylation”) and cannot participate in the downstream metabolism. (ii) All carbon atoms and nitrogen atoms in hypoxanthine entirely originate from adenosine.
Collectively, we have elucidated that GAT2 affects macrophage IL-1β production. Mechanistically, GAT2 deficiency boosts intracellular accumulation of hypoxanthine, which not only blocks NLRP3–ASC–Caspase-1 complex formation but also enhances the expression of OXPHOS-related genes via reducing expression of TF KID3 through the methylation (Fig. 8). We postulate that metabolic licensing of DNA methylation provides another layer of control that serves to fine-tune the immune cell activation. Our study expands the role of the nervous system in host innate immune responses by showing that GAT2 tailors IL-1β production in proinflammatory macrophages. Thus, manipulation of GABAergic system could be a strategy to tackle macrophage-associated diseases.
Fig. 8. Mechanisms whereby GAT2 deficiency lowers IL-1β production in proinflammatory macrophages.
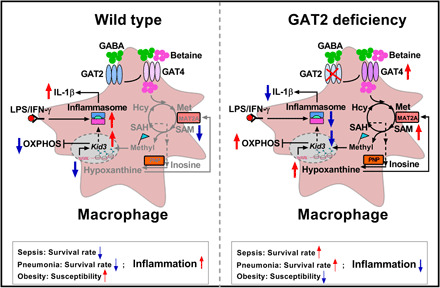
Depletion of GAT2 activates MAT2A/PNP-mediated betaine/SAM/hypoxanthine signaling through increased GAT4 expression to dampen NLRP3–ASC–caspase-1 complex formation and boost OXPHOS-related gene expression (chiefly Ndufb9) by targeting KID3 via methylation, lowering IL-1β in proinflammatory macrophages. Notably, GAT2 deficiency attenuates macrophage-mediated inflammatory responses in vivo, including LPS-induced sepsis, infection-induced pneumonia, and HFD-induced obesity.
MATERIALS AND METHODS
Bacterial strains
Bovine P. multocida serotype A strain CQ2 (PmCQ2) was generally grown in Martin’s broth agar containing 5% horse serum at 37°C for 24 hours.
Cell lines
RAW264.7 cells and ANA-1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and kept in a 5% carbon dioxide (CO2) incubator at 37°C. Cells were polarized for 12 hours toward proinflammatory macrophages with LPS (1 μg/ml) plus IFN-γ (20 ng/ml) or left unstimulated and used as resting macrophages.
Mice
All animal care and experimental procedures used in this study were approved by the Laboratory Animal Ethical Commission of the South China Agricultural University. GAT2−/− mice (KO) have previously been described (4). All experiments were performed using littermate control mice (WT). Six- to 8-week-old mice were used for all experiments and were age- and gender-matched for individual experiments.
Murine primary PEMs
Murine primary PEMs were prepared as described in previous studies with some minor modifications. Briefly, 2 to 4 days after the injection of 4% thioglycolate, macrophages were flushed from peritoneal cavity with phosphate-buffered saline (PBS) and cultured in complete DMEM. After adherent purification, PEMs were polarized for 12 hours toward proinflammatory macrophages with LPS (1 μg/ml) or plus IFN-γ (20 ng/ml) or toward anti-inflammatory macrophages with IL-4 (20 ng/ml) or left unstimulated and used as resting macrophages.
Bone marrow–derived macrophages
Murine BMDMs were prepared as described in previous study with some minor modifications (57). Briefly, murine bone marrow cells were flushed from tibias and femurs with iced-cold RPMI 1640 and cultured in complete RPMI 1640 medium supplemented with macrophage colony–stimulating factor (10 ng/ml) to generate BMDMs. BMDMs were polarized for 24 hours toward proinflammatory macrophages with LPS (100 ng/ml) plus IFN-γ (20 ng/ml) or left unstimulated and used as resting macrophages.
Deletion of macrophages in mice
Macrophage deletion was performed as previously described with some minor adjustments (34, 58). Briefly, the mice were completely anesthetized and intraperitoneally or intratracheally administered 200 μl of empty liposomes or liposome clorophosphate.
In vivo LPS challenge
Mice with or without clodronate-loaded liposome treatment were injected intraperitoneally with LPS (100 mg/kg body weight) for the sepsis model. Survival rates were measured in all groups after injection. Mice were sacrificed, and the serum, jejunum, and liver were collected for further experiments.
In vivo PmCQ2 infection
Mice with or without clodronate-loaded liposome treatment were infected by an intraperitoneal injection with PmCQ2 at a dose of 2.2 × 106 colony-forming units/ml according to the previous study (34). Survival rates were investigated in all groups after infection. Mice were killed to collect the serum and lung, and the samples were stored at −80°C until processing.
HFD-induced obesity model
Six-week-old male mice were maintained on HFD for 12 weeks. For glucose tolerance test, mice were injected intraperitoneally with glucose (2 g/kg body weight) after 16-hour fasting. Blood glucose was determined by Accu-Chek glucose meters (Roche) at different time points. For insulin tolerance test, mice were injected with recombinant human insulin (0.75 U/kg body weight) after 4-hour fasting, and blood glucose was also determined at different time points. Moreover, serum and adipose tissues were sampled for further analysis.
Reverse transcription polymerase chain reaction
The total RNA of the samples was extracted using TRIzol (Invitrogen, USA) based on the manufacturer’s instructions. RNA was reverse transcribed to complementary DNA by using the Primer Script TM RT reagent Kit (Takara, Qingdao, China) under the manufacturer’s recommendations. RT-PCR was conducted via SYBR Green on the Quant Studio 6 Real-Time PCR System (Thermo Fisher Scientific, America). Fold change was assessed using the 2−∆∆Ct method using β-actin as a reference gene.
Western blotting
Immunoblotting analysis was performed following the standard procedures. The tissues and cells were washed with iced-cold PBS and lysed in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Beyotime). Protein concentration was quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime), and an equal amount of protein was subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred onto polyvinylidene difluoride membranes, followed by incubations with primary and secondary antibodies, and visualized using chemiluminescent reagent. The optical density of the signals on the film was quantified by ImageJ software.
Detection of cytokines and mediators
Culture supernatants from cell culture and serum and supernatants from homogenized tissue or cell samples were collected, and levels of cytokines (e.g., IL-1β, IL-6, and TNF-α) and mediators (e.g., ROS and xanthine) were measured by commercial kits following the protocols.
Coimmunoprecipitation
PEMs were treated as indicated and collected, then lysed with lysate on ice for 1 hour, and followed by centrifugation at 12,000g at 4°C for 30 min. Subsequently, the supernatants were harvested and incubated with the appropriate antibody at 4°C overnight and precipitated with protein A/G agarose beads (Santa Cruz Biotechnology) for another 4 hours at 4°C. The beads were washed with the lysate for three times and centrifuged at 1000g at 4°C. The immunoprecipitated proteins were separated by SDS-PAGE, and Western blotting was conducted with the indicated antibodies.
Immunofluorescence
Immunofluorescence analysis was conducted according to the standard protocols. The fixed, permeabilized, and sliced samples were blocked by 5% bovine serum albumin (BSA), followed by incubation with primary and secondary antibodies. Stained cells and tissues were viewed using a confocal fluorescence microscope (Zeiss).
Flow cytometry
Cells isolated from mouse bone marrow, PBMC, spleen, and mesenteric lymph node were resuspended, and surface staining (fluorescein isothiocyanate–F4/80) was performed in accordance with the manufacturer’s instructions. F4/80+ cells were analyzed by a fluorescence-activated cell sorting (FACS) Calibur (BD Biosciences).
In silico docking analysis
Molecular docking between MAT2A and hypoxanthine was conducted using the AutoDock Vina tool without the incorporation of water molecules. Interactions between hypoxanthine and the residues of the binding sites in MAT2A are shown using a two-dimensional diagram by Discovery Studio 4.5.
SPR (OpenSPR) assay
Binding experiments was performed via an SPR (OpenSPR) localized SPR (LSPR) biosensor (Nicoya Lifesciences). Briefly, the COOH sensor chip (Nicoya, #SEN-AU-100-10-COOH) was activated by injecting 200 μl of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS) (1:1). Subsequently, MAT2A was diluted in immobilization buffer [10 mM acetate (pH 3.5)] and immobilized on the chip at a flow rate of 20 μl/min for 4 min. The free carboxyl groups were deactivated using 200 μl of blocking buffer (Nicoya) followed by achieving a stable baseline. Then, the buffer-matched hypoxanthine (0, 100, 200, 400, and 800 μM) was successively injected into the flow cell at a rate of 20 μl/min. Following a 4-min interaction time, the dissociation was recorded for an additional 3 min. The data were analyzed using TraceDrawer 1.8 (Ridgeview Instruments AB, Sweden).
RNA sequencing
PEMs were polarized into proinflammatory macrophages and collected. RNA extraction, quality verification, library preparation, and sequencing were performed by NovoGene (Beijing). Briefly, clean data were obtained by filtering low-quality reads from raw data and mapped to the reference genome. Differential expression analysis was performed with DESeq2.GO, and KEGG pathway enrichment analysis of the DEGs (FC > 2; P < 0.05) were performed.
Seahorse assay
Real-time measurements of OCR and ECAR were performed using an XFe24 Extracellular Flux Analyzer as per the manufacturer’s instructions (Seahorse Agilent). PEMs (2 × 105 cells per well) were plated in XFe24 plates and polarized as described above toward proinflammatory macrophages with or without hypoxanthine treatment or toward anti-inflammatory macrophages or left unstimulated and used as resting macrophages. Subsequently, macrophages were washed and incubated for 1 hour in XF assay medium [nonbuffered DMEM (pH 7.4) + 10 mM glucose + 2 mM l-glutamine] in a non-CO2 incubator at 37°C. OCR was measured under basal conditions and after the addition of the following drugs:100 μM oligomycin, 100 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone, and 50 μM rotenone plus antimycin A. ECAR was assayed under basal conditions and after the addition of the following drugs:1 mM glucose, 1 mM oligomycin, and 1 mM 2-deoxyglucose.
Amino acid analysis
The levels of amino acids in supernatants from cell culture and lysed cells were analyzed with isotope dilution liquid chromatography–MS (LC-MS) methods.
Metabolite profiling analysis
PEMs (1 × 107 cells per well) were plated in six-well plates and polarized as described above toward proinflammatory macrophages or left unstimulated and used as resting macrophages. Macrophages were then used to perform metabolomics assay at Shanghai Biotree Biotech Co. Ltd. Briefly, for each sample, 1000 μl of extract solution (acetonitrile:methanol:water = 2:2:1) was added. The samples were homogenized at 35 Hz for 4 min and sonicated for 5 min with three cycles in iced water bath followed by incubating at −40°C for 1 hour and centrifuging at 12,000g for 15 min at 4°C. The supernatant (800 μl) was transferred to a fresh tube and dried in a vacuum concentrator at 37°C. Subsequently, the dried samples were reconstituted in 100 μl of 50% acetonitrile by sonication on ice for 10 min. The constitution was then centrifuged at 12,000g for 15 min at 4°C, and 75 μl of supernatant was transferred to a fresh glass vial for LC-MS analysis.
Measuring the enzyme activity of PNP
For measuring the enzyme activity of PNP, the cells were washed with iced-cold PBS and collected; then, the cells were repeatedly frozen and thawed for several times [note that the freeze-thaw process was initiated with 30 min of freezing process at a −80°C refrigerator until completely frozen, followed by a thawing process under room temperature (25°C)]. Last, the enzyme activity of PNP was measured according to the manufacturer’s recommendation (Andy gene, AD3407Mo).
Gene silencing or overexpression
Gene silencing or overexpression in macrophages was achieved via lipofection using the Lipofectamine 3000 transfection reagent according to the manufacturer’s protocol.
Intracellular transfection of betaine
PEMs were prepared as described above. For betaine delivery, first, macrophages were cultured in 1× basic DMEM containing a FuGENE HD Transfection reagent (5 μl/ml) with or without 1 mM betaine and kept in a 5% CO2 incubator at 37°C for 45 min. Then, the above medium was replaced with the normal complete DMEM, and macrophages were incubated at 37°C in 5% CO2 for 24 hours. Last, macrophages were stimulated as described above.
Luciferase reporter assay
The promoter of mouse Ndufb9 was produced by PCR-based amplification and subcloned into the pPRO-RB-Report1 to form luciferase reporter plasmids. PEMs (2 × 104 cells per well) were plated in 96-well plates and cotransfected with 50 ng of the luciferase reporter plasmid and 50 ng of pEX-RB-Mam-m-Kid3 plasmid or the control vector. After 36 to 48 hours, the luciferase activity was determined by the Dual-Glo Luciferase Assay System in accordance with the manufacturer’s instructions.
MassARRAY methylation assay
PEMs (2 × 106 cells per well) were plated in six-well plates and polarized as described above toward proinflammatory macrophages and treated as indicated in the legend. Then, the DNA samples of macrophages were isolated and used to perform methylation assay at Shanghai OE Biotech. Co. Ltd. Briefly, the potential CpG islands in the KID3 promoter region were predicated by the website (www.ebi.ac.uk). The primer sets for the methylation analysis of the KID3 promoter region were designed by EpiDesigner software (http://epidesigner.com; primer: 5′-aggaagagagTAGGTATAATGAAGGGTTAAGGGGT-3′; 5′-agtaatacgactcactatagggagaaggct AATCAAAAAAAACCTCAAACATACC-3′). Subsequently, the quantitative gene-specific methylation analysis was conducted on the Sequenom MassARRAY platform, including bisulfite treatment of DNA, amplification of DNA, Shrimp Alkaline Phosphatase (SAP) treatment, RNA transcription, base-specific cleavage by ribonuclease A, and matrix-assisted laser desorption/ionization–time-of-flight analysis. Last, the methylation data of individual units were generated and analyzed by EpiTYPER software (SEQUENOM).
Histopathological examination
Mouse lung samples were fixed in 4% paraformaldehyde (PFA) at 4°C overnight and dehydrated and embedded into paraffin. Prepared sections (5 μm) were stained with hematoxylin and eosin using standardized procedures. Histopathological scoring was performed by a pathologist blinded to all groups according to the previous study (59).
Immunohistochemistry
Briefly, mouse lung, liver, and adipose tissues were collected and washed with iced-cold PBS and fixed in 4% PFA at 4°C overnight. The fixed samples were dehydrated in graded ethanol, embedded in paraffin, sliced and blocked by 2% BSA, followed by incubation with appropriate primary and secondary antibodies. The positive areas of the sample were measured using ImageJ software.
Statistical analysis
All statistical analyses were performed using Prism 6.0 software (GraphPad), and results are represented as means ± SEM or SD. Survival rates of mice were evaluated using Kaplan-Meier analysis (Prism 6.0). Data between two groups were analyzed by unpaired t test (Prism 6.0) if the data were in Gaussian distribution and had equal variance, by unpaired t test with Welch’s correction (Prism 6.0) if the data were in Gaussian distribution but with unequal variance, or by nonparametric test (Mann-Whitney U test, Prism 6.0) if the data were not normally distributed. Data among more than two groups were analyzed by the one-way analysis of variance (ANOVA) followed by Dunnett multiple comparisons (Prism 6.0) if the data were in Gaussian distribution and had equal variance or analyzed by Kruskal-Wallis followed by Dunn’s multiple comparisons (Prism 6.0) if the data were not normally distributed. The Gaussian distribution of data was analyzed by D’Agostino-Pearson omnibus normality test (Prism 6.0) and Kolmogorov-Smirnov test (Prism 6.0). The variance of data was analyzed by homogeneity of variance test (SPSS 22.0) or Brown-Forsythe test (Prism 6.0). Differences with P < 0.05 were considered significant.
Acknowledgments
Funding: This work was supported by National Natural Science Foundation of China (31922079 and 31872365) and Guangdong Basic and Applied Basic Research Foundation (2019B1515210002). Author contributions: Y.X. and W.R. designed the research and analyzed data. Y.X., F.H., and B.T. performed sorting cells, Western blot analysis, immunoprecipitation, immunofluorescence, and animal experiments. Y.X., F.H., X.W., Siyuan Chen, and Shuai Chen performed the detection of RT-PCR and ELISA. Y.L. and M.Q. helped with the FACS analysis and LSPR analysis, respectively. Y.P. and Y.Y. supervised initial experiments. Y.X. and W.R. wrote the manuscript. W.R. supervised the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. RNA-seq data generated in this paper are available under the accession number SRP151076.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe9274/DC1
REFERENCES AND NOTES
- 1.Lai N. Y., Musser M. A., Pinho-Ribeiro F. A., Baral P., Jacobson A., Ma P., Potts D. E., Chen Z., Paik D., Soualhi S., Yan Y., Misra A., Goldstein K., Lagomarsino V. N., Nordstrom A., Sivanathan K. N., Wallrapp A., Kuchroo V. K., Nowarski R., Starnbach M. N., Shi H., Surana N. K., An D., Wu C., Huh J. R., Rao M., Chiu I. M., Gut-Innervating nociceptor neurons regulate peyer’s patch microfold cells and SFB levels to mediate salmonella host defense. Cell 180, 33–49.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarret A., Jackson R., Duizer C., Healy M. E., Zhao J., Rone J. M., Bielecki P., Sefik E., Roulis M., Rice T., Sivanathan K. N., Zhou T., Solis A. G., Honcharova-Biletska H., Vélez K., Hartner S., Low J. S., Qu R., de Zoete M. R., Palm N. W., Ring A. M., Weber A., Moor A. E., Kluger Y., Nowarski R., Flavell R. A., Enteric nervous system-derived IL-18 orchestrates mucosal barrier immunity. Cell 180, 813–814 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Kim J. K., Kim Y. S., Lee H.-M., Jin H. S., Neupane C., Kim S., Lee S.-H., Min J.-J., Sasai M., Jeong J.-H., Choe S.-K., Kim J.-M., Yamamoto M., Choy H. E., Park J. B., Jo E.-K., GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections. Nat. Commun. 9, 4184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren W., Liao Y., Ding X., Jiang Y., Yan J., Xia Y., Tan B., Lin Z., Duan J., Jia X., Yang G., Deng J., Zhu C., Hardwidge P. R., Li J., Zhu G., Yin Y., Slc6a13 deficiency promotes Th17 responses during intestinal bacterial infection. Mucosal Immunol. 12, 531–544 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Dionisio L., De Rosa M. J., Bouzat C., Del Carmen Esandi M., An intrinsic GABAergic system in human lymphocytes. Neuropharmacology 60, 513–519 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Wang Y., Feng D., Liu G., Luo Q., Xu Y., Lin S., Fei J., Xu L., γ-aminobutyric acid transporter 1 negatively regulates T cell-mediated immune responses and ameliorates autoimmune inflammation in the CNS. J. Immunol. 181, 8226–8236 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Locati M., Curtale G., Mantovani A., Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weichhart T., Hengstschläger M., Linke M., Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 15, 599–614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martínez-García J. J., Martínez-Banaclocha H., Angosto-Bazarra D., de Torre-Minguela C., Baroja-Mazo A., Alarcón-Vila C., Martínez-Alarcón L., Amores-Iniesta J., Martín-Sánchez F., Ercole G. A., Martínez C. M., González-Lisorge A., Fernández-Pacheco J., Martínez-Gil P., Adriouch S., Koch-Nolte F., Luján J., Acosta-Villegas F., Parrilla P., García-Palenciano C., Pelegrin P., P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 10, 2711 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langston P. K., Nambu A., Jung J., Shibata M., Aksoylar H. I., Lei J., Xu P., Doan M. T., Jiang H., MacArthur M. R., Gao X., Kong Y., Chouchani E. T., Locasale J. W., Snyder N. W., Horng T., Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 20, 1186–1195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaitin D. A., Adlung L., Thaiss C. A., Weiner A., Li B., Descamps H., Lundgren P., Bleriot C., Liu Z., Deczkowska A., Keren-Shaul H., David E., Zmora N., Eldar S. M., Lubezky N., Shibolet O., Hill D. A., Lazar M. A., Colonna M., Ginhoux F., Shapiro H., Elinav E., Amit I., Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caputa G., Castoldi A., Pearce E. J., Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 20, 793–801 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Ji L., Zhao X., Zhang B., Kang L., Song W., Zhao B., Xie W., Chen L., Hu X., Slc6a8-mediated creatine uptake and accumulation reprogram macrophage polarization via regulating cytokine responses. Immunity 51, 272–284.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Luo Q., Xu Y., Feng D., Fei J., Cheng Q., Xu L., γ-aminobutyric acid transporter 1 negatively regulates T cell activation and survival through protein kinase C-dependent signaling pathways. J. Immunol. 183, 3488–3495 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Bhat R., Axtell R., Mitra A., Miranda M., Lock C., Tsien R. W., Steinman L., Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. U.S.A. 107, 2580–2585 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalby N. O., Inhibition of γ-aminobutyric acid uptake: Anatomy, physiology and effects against epileptic seizures. Eur. J. Pharmacol. 479, 127–137 (2003). [DOI] [PubMed] [Google Scholar]
- 17.O’Neill L. A., Hardie D. G., Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Rathinam V. A., Fitzgerald K. A., Inflammasome complexes: Emerging mechanisms and effector functions. Cell 165, 792–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu T., Gan S., Zhu Q., Dai D., Li N., Wang H., Chen X., Hou D., Wang Y., Pan Q., Xu J., Zhang X., Liu J., Pei S., Peng C., Wu P., Romano S., Mao C., Huang M., Zhu X., Shen K., Qin J., Xiao Y., Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 10, 4353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill L. A. J., Kishton R. J., Rathmell J., A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray P. J., Rathmell J., Pearce E., SnapShot: Immunometabolism. Cell Metab. 22, 190–190.e1 (2015). [DOI] [PubMed] [Google Scholar]
- 22.O’Neill L. A. J., Artyomov M. N., Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 19, 273–281 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Fan K.-Q., Li Y.-Y., Wang H.-L., Mao X.-T., Guo J.-X., Wang F., Huang L.-J., Li Y.-N., Ma X.-Y., Gao Z.-J., Chen W., Qian D.-D., Xue W.-J., Cao Q., Zhang L., Shen L., Zhang L., Tong C., Zhong J.-Y., Lu W., Lu L., Ren K.-M., Zhong G., Wang Y., Tang M., Feng X.-H., Chai R.-J., Jin J., Stress-induced metabolic disorder in peripheral CD4+ T cells leads to anxiety-like behavior. Cell 179, 864–879.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Cader M. Z., Pereira de Almeida Rodrigues R., West J. A., Sewell G. W., Md-Ibrahim M. N., Reikine S., Sirago G., Unger L. W., Iglesias-Romero A. B., Ramshorn K., Haag L.-M., Saveljeva S., Ebel J.-F., Rosenstiel P., Kaneider N. C., Lee J. C., Lawley T. D., Bradley A., Dougan G., Modis Y., Griffin J. L., Kaser A., FAMIN is a multifunctional purine enzyme enabling the purine nucleotide cycle. Cell 180, 278–295.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusano T., Ehirchiou D., Matsumura T., Chobaz V., Nasi S., Castelblanco M., So A., Lavanchy C., Acha-Orbea H., Nishino T., Okamoto K., Busso N., Targeted knock-in mice expressing the oxidase-fixed form of xanthine oxidoreductase favor tumor growth. Nat. Commun. 10, 4904 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hara H., Tsuchiya K., Kawamura I., Fang R., Hernandez-Cuellar E., Shen Y., Mizuguchi J., Schweighoffer E., Tybulewicz V., Mitsuyama M., Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang W. H. W., Wang Z., Levison B. S., Koeth R. A., Britt E. B., Fu X., Wu Y., Hazen S. L., Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig S. A., Betaine in human nutrition. Am. J. Clin. Nutr. 80, 539–549 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Imbard A., Benoist J.-F., Esse R., Gupta S., Lebon S., de Vriese A. S., de Baulny H. O., Kruger W., Schiff M., Blom H. J., High homocysteine induces betaine depletion. Biosci. Rep. 35, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu W., Wang Z., Zhang K., Chi Z., Xu T., Jiang D., Chen S., Li W., Yang X., Zhang X., Wu Y., Wang D., One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell 75, 1147–1160.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Quinlan C. L., Kaiser S. E., Bolaños B., Nowlin D., Grantner R., Karlicek-Bryant S., Feng J. L., Jenkinson S., Freeman-Cook K., Dann S. G., Wang X., Wells P. A., Fantin V. R., Stewart A. E., Grant S. K., Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 13, 785–792 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Murray B., Antonyuk S. V., Marina A., Lu S. C., Mato J. M., Hasnain S. S., Rojas A. L., Crystallography captures catalytic steps in human methionine adenosyltransferase enzymes. Proc. Natl. Acad. Sci. U.S.A. 113, 2104–2109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flotho C., Claus R., Batz C., Schneider M., Sandrock I., Ihde S., Plass C., Niemeyer C. M., Lübbert M., The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 23, 1019–1028 (2009). [DOI] [PubMed] [Google Scholar]
- 34.He F., Yin Z., Wu C., Xia Y., Wu M., Li P., Zhang H., Yin Y., Li N., Zhu G., Ren W., Peng Y., L-Serine Lowers the Inflammatory Responses during Pasteurella multocida Infection. Infect. Immun. 87, e00677-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., Haneklaus M., Caffrey B. E., Pierce K., Walmsley S., Beasley F. C., Cummins E., Nizet V., Whyte M., Taylor C. T., Lin H., Masters S. L., Gottlieb E., Kelly V. P., Clish C., Auron P. E., Xavier R. J., O’Neill L. A. J., Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birchenough G. M., Nyström E. E., Johansson M. E., Hansson G. C., A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science (New York, N.Y.) 352, 1535–1542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labed S. A., Wani K. A., Jagadeesan S., Hakkim A., Najibi M., Irazoqui J. E., Intestinal epithelial Wnt signaling mediates acetylcholine-triggered host defense against infection. Immunity 48, 963–978.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabanyi I., Muller P. A., Feighery L., Oliveira T. Y., Costa-Pinto F. A., Mucida D., Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164, 378–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang I., Jo K., Shin K. C., Kim J. I., Ji Y., Park Y. J., Park J., Jeon Y. G., Ka S., Suk S., Noh H. L., Choe S. S., Alfadda A. A., Kim J. K., Kim S., Kim J. B., GABA-stimulated adipose-derived stem cells suppress subcutaneous adipose inflammation in obesity. Proc. Natl. Acad. Sci. U.S.A. 116, 11936–11945 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prud’homme G. J., Glinka Y., Wang Q. H., Immunological GABAergic interactions and therapeutic applications in autoimmune diseases. Autoimmun. Rev. 14, 1048–1056 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Rane M. J., Gozal D., Butt W., Gozal E., Pierce W. M. Jr., Guo S. Z., Wu R., Goldbart A. D., Thongboonkerd V., McLeish K. R., Klein J. B., Gamma-amino butyric acid type B receptors stimulate neutrophil chemotaxis during ischemia-reperfusion. J. Immunol. 174, 7242–7249 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Kanatani S., Fuks J. M., Olafsson E. B., Westermark L., Chambers B., Varas-Godoy M., Uhlén P., Barragan A., Voltage-dependent calcium channel signaling mediates GABAA receptor-induced migratory activation of dendritic cells infected by Toxoplasma gondii. PLOS Pathog. 13, e1006739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barragan A., Weidner J. M., Jin Z., Korpi E. R., Birnir B., GABAergic signalling in the immune system. Acta Physiol (Oxford) 213, 819–827 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Fuks J. M., Arrighi R. B. G., Weidner J. M., Kumar Mendu S., Jin Z., Wallin R. P. A., Rethi B., Birnir B., Barragan A., GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLOS Pathog. 8, e1003051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plum T., Wang X., Rettel M., Krijgsveld J., Feyerabend T. B., Rodewald H.-R., Human mast cell proteome reveals unique lineage, putative functions, and structural basis for cell ablation. Immunity 52, 404–416.e405 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Bzowska A., Kulikowska E., Shugar D., Purine nucleoside phosphorylases: Properties, functions, and clinical aspects. Pharmacol. Ther. 88, 349–425 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Wang T., Gnanaprakasam J. N. R., Chen X., Kang S., Xu X., Sun H., Liu L., Rodgers H., Miller E., Cassel T. A., Sun Q., Vicente-Muñoz S., Warmoes M. O., Lin P., Piedra-Quintero Z. L., Guerau-de-Arellano M., Cassady K. A., Zheng S. G., Yang J., Lane A. N., Song X., Fan T. W. M., Wang R., Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat. Metab. 2, 635–647 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo J., Xue Z., Ma R., Yi W., Hui Z., Guo Y., Yao Y., Cao W., Wang J., Ju Z., Lu L., Wang L., The transcription factor Zfp281 sustains CD4(+) T lymphocyte activation through directly repressing Ctla-4 transcription. Cell. Mol. Immunol. 17, 1222–1232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brayer K. J., Segal D. J., Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 50, 111–131 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Razin S. V., Borunova V. V., Maksimenko O. G., Kantidze O. L., Cys2His2 zinc finger protein family: Classification, functions, and major members. Biochem. Biokhimiia 77, 217–226 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Sitcheran R., Gupta P., Fisher P. B., Baldwin A. S., Positive and negative regulation of EAAT2 by NF-kappaB: A role for N-myc in TNFalpha-controlled repression. EMBO J. 24, 510–520 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X., Eagen W. J., Lee J. C., Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc. Natl. Acad. Sci. U.S.A. 117, 3174–3184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratsimandresy R. A., Chu L. H., Khare S., de Almeida L., Gangopadhyay A., Indramohan M., Misharin A. V., Greaves D. R., Perlman H., Dorfleutner A., Stehlik C., The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 8, 15556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stutz A., Kolbe C. C., Stahl R., Horvath G. L., Franklin B. S., van Ray O., Brinkschulte R., Geyer M., Meissner F., Latz E., NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 214, 1725–1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang R., Uchiyama R., Sakai S., Hara H., Tsutsui H., Suda T., Mitsuyama M., Kawamura I., Tsuchiya K., ASC and NLRP3 maintain innate immune homeostasis in the airway through an inflammasome-independent mechanism. Mucosal Immunol. 12, 1092–1103 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Guo C., Xie S., Chi Z., Zhang J., Liu Y., Zhang L., Zheng M., Zhang X., Xia D., Ke Y., Lu L., Wang D., Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 802–816 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Weischenfeldt J., Porse B., Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008, pdb.prot5080 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Brown R. L., Sequeira R. P., Clarke T. B., The microbiota protects against respiratory infection via GM-CSF signaling. Nat. Commun. 8, 1512 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van den Boogaard F. E., van’t Veer C., Roelofs J. J. T. H., Meijers J. C. M., Schultz M. J., Broze G. J. Jr., van der Poll T., Endogenous tissue factor pathway inhibitor has a limited effect on host defence in murine pneumococcal pneumonia. Thromb. Haemost. 114, 115–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/15/eabe9274/DC1