

Abstract
Chemical-induced liver cancer occurs in rodents through well characterized adverse outcome pathways (AOPs). We hypothesized that measurement of the 6 most common molecular initiating events (MIEs) in liver cancer AOPs in short-term assays using only gene expression will allow early identification of chemicals and their associated doses that are likely to be tumorigenic in the liver in two-year bioassays. We tested this hypothesis using transcript data from a rat liver microarray compendium consisting of 2013 comparisons of 146 chemicals administered at doses with previously established effects on rat liver tumor induction. Five MIEs were measured using previously characterized gene expression biomarkers composed of gene sets predictive for genotoxicity and activation of one or more xenobiotic receptors, (aryl hydrocarbon receptor (AhR), constitutive activated receptor (CAR), estrogen receptor (ER), and peroxisome proliferator-activated receptor α (PPARα)). Since chronic injury can be important in tumorigenesis, we also developed a biomarker for cytotoxicity that had a 96% balanced accuracy. Characterization of the genes in each biomarker set using the unsupervised TXG-MAP network model demonstrated that the genes were associated with distinct functional co-expression modules. Using the Toxicological Priority Index (ToxPi) to rank chemicals based on their ability to activate the MIEs showed that chemicals administered at tumorigenic doses clearly gave the highest ranked scores. Balanced accuracies using thresholds derived from either TG-GATES or DrugMatrix datasets to predict tumorigenicity in independent sets of chemicals were up to 93%. These results show that a MIE-directed approach using only gene expression biomarkers could be used in short-term assays to identify chemicals and their doses that cause tumors.
Keywords: adverse outcome pathway, constitutive activated receptor, transcript profiling, liver cancer, peroxisome proliferator-activated receptor α, aryl hydrocarbon receptor, genotoxicity, p53, estrogen receptor, cytotoxicity, ToxPi, key events, molecular initiating events
INTRODUCTION
Cancer imposes a tremendous burden on the United States economy due to the fact that it is the second leading cause of death in the US (CDC, 2017). For most chemicals currently in use, there is little, if anything, known about their potential to cause cancer in humans. These include ~ 30,000 chemicals in widespread commercial use in the United States and Canada (Muir et al., 2006), over 75,000 chemicals on the US EPA’s Toxic Substances Control Act Inventory (USEPA, 2004), and over 140,000 substances registered by the REACH (REACH, 2008). The 2-year rodent bioassay is currently the “gold-standard” to identify carcinogens, but due to the considerable resource expenditures required to assess a chemical in this manner (>800 rodents, histopathological analysis of more than 40 tissues, ~$2–4M USD), only ~1,500 chemicals in commercial use have been evaluated (Bucher et al., 2004; Gold et al., 2005; Waters et al., 2010). The situation demands new, resource-efficient methods that both identify carcinogenic potential of environmentally-relevant chemicals and pharmaceuticals as well as boundaries of exposure and human relevance of any risk identified in animal studies.
The last decade has witnessed the emergence of an evolving set of methods to identify human carcinogens that include in silico computational structure-activity relationships analysis (Benigni et al., 2007) to a range of in vitro or short-term in vivo approaches (Knudsen et al., 2015). A major drawback of many of these models is that they cannot recapitulate the complex biological changes that underlie the carcinogenesis process in the whole animal. Some but not all machine learning studies using whole-genome microarray data from short-term exposures in rats tends to bias outcomes toward reasonable balanced accuracies but with unacceptably high false negative rates (Ellinger-Ziegelbauer et al., 2008; Fielden et al., 2007; Gusenleitner et al., 2014; Nie et al., 2006; Uehara et al., 2008; Uehara et al., 2011; Yamada et al., 2013). In addition, the ability of these models for identification of hepatocarcinogens varies widely, and do not necessarily address the mechanistic pathway(s) through which the chemicals act. The latter information is a prerequisite to determine if potential mechanisms are rodent-specific or relevant to human risk (Cohen, 2010).
We have recently taken an adverse outcome pathway (AOP)-based approach to predicting rat liver cancer (Rooney et al., 2018a), the most common target organ for cancer in the rodent (Hill et al., 2017; Sistare et al., 2011). Pathway information organized by mode of action (MOA) (Boobis et al., 2006; USEPA, 2005) or more recently, by an AOP construct (Ankley et al., 2010; Edwards et al., 2016) plays a central role in compiling evidence of effects and determining human relevance for chemical carcinogens. A chemical-agnostic AOP starts with the interaction between a chemical and a molecular target (the molecular initiating event (MIE) followed by a series of downstream key events (KEs) that lead to an adverse outcome (AO). Overlay of chemical-specific information such as absorption, disposition, metabolism and excretion (ADME) and prediction of chemical concentrations at the site of the MIE permits the derivation of the corresponding MOA for risk assessment (Edwards, et al., 2016). In our earlier study, we hypothesized that measurement of the MIEs and KEs for rat liver cancer AOPs in short-term assays would allow early identification of chemicals, and their associated doses, that are likely to be tumorigenic in the liver in two-year bioassays. That hypothesis was predicated on the fact that while a number of mechanisms that lead to liver cancer have been described or hypothesized (Cohen, 2010; Yamada, 2018), most chemicals cause rodent liver cancer through only 6 major pathways. The MIEs for these include DNA damage, cytotoxicity and associated regenerative cell proliferation, and activation of one or more xenobiotic receptors (aryl hydrocarbon receptor (AhR), constitutive activated receptor (CAR), estrogen receptor (ER), and peroxisome proliferator-activated receptor α (PPARα)) (Figure 1). These MIEs and KEs were measured using a combination of gene expression biomarkers characterized as part of the study (AhR, CAR, ER, genotoxicity, PPARα), expression of individual genes (indirect measures of oxidative stress), clinical chemistry measurements (ALT and AST to measure cytotoxicity), and liver to body weights (an indirect measure of hepatocyte proliferation and hypertrophy). We employed the Toxicological Priority Index (ToxPi) to rank chemicals based on their ability to activate MIEs/KEs and found that chemicals administered at tumorigenic doses clearly gave the highest ranked scores. Depending on the time selected to make predictions, the accuracy ranged from 85–89% and was highest when endpoints were measured at 4d (Rooney, et al., 2018a).
Fig. 1. The network of major adverse outcome pathways leading to chemical-induced rodent liver tumors.
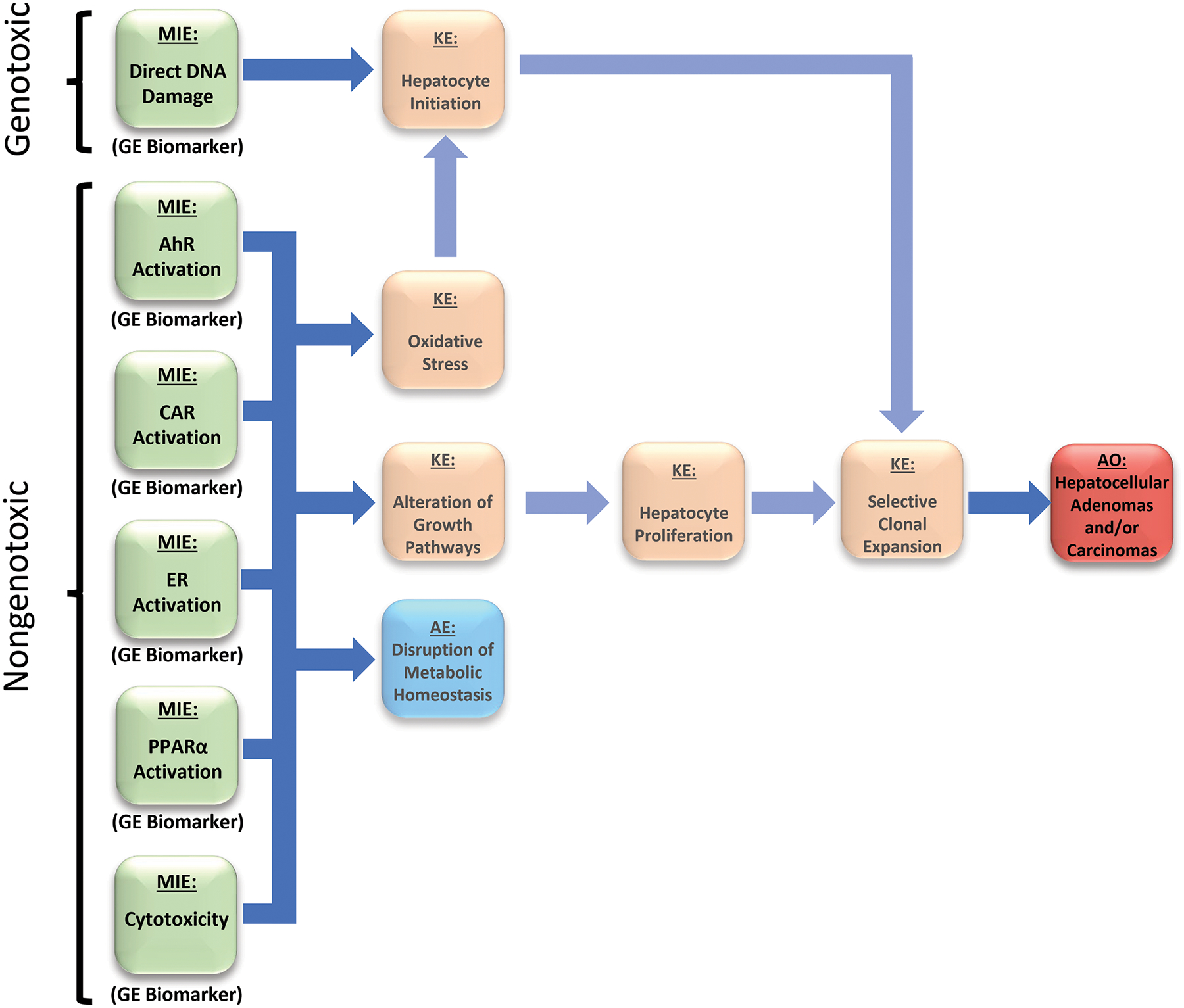
The major molecular initiating events (MIEs) and key events (KEs) in AOPs that lead to hepatocellular adenomas and carcinomas are shown. Genotoxic chemicals cause liver cancer through direct damage to DNA by the parent compound or its reactive metabolite(s); these chemicals can also induce other effects that contribute to liver cancer including cytotoxicity. Nongenotoxic compounds often activate one or more xenobiotic receptors, including the aryl hydrocarbon receptor (AhR), constitutive activated receptor (CAR), the estrogen receptor (ER), and the peroxisome proliferator-activated receptor α (PPARα) known to cause liver cancer through mechanisms that do not involve direct damage to DNA. These compounds generally may act at the promotion stage by altering hepatocyte fate, most notably through increases in cell proliferation (Budinsky, et al., 2014; Corton, et al., 2014; Elcombe, et al., 2014). Chemical-induced cytotoxicity can cause increases in hepatocyte death leading to compensatory regenerative hepatocyte proliferation. Activation of one or more of these five nongenotoxic MIEs can lead indirectly to DNA damage through increases in reactive oxygen species produced by increased and sustained activity of regulated enzymes. Promotion of initiated hepatocytes to preneoplastic foci precedes hepatocellular adenomas and carcinomas. Key events that are not the focus of the present study are faded out to highlight the goal of the project to predict liver cancer by measuring the activation of the 6 MIEs using only gene expression biomarkers.
We have refined our earlier hypothesis and in this study, we now propose that methods using only gene expression biomarkers could be used to accurately and precisely identify liver tumorigens in short-term assays. We tested our hypothesis using the microarray data mined from the TG-GATEs (Igarashi et al., 2015) and the DrugMatrix online databases (Svoboda D, 2019), both of which screened large cohorts of chemicals at multiple doses and times of exposure in the rat. Using a ToxPi analysis, we found that our set of 6 biomarkers displayed excellent predictive performance metrics. We also extended our investigation of these biomarker gene sets by investigating whether they were members of unsupervised co-expression gene sets (modules) using an open source analysis platform (Sutherland et al., 2019b) and weighted gene co-expression network analysis (WGCNA; (Sutherland et al., 2018)).
METHODS
Rationale for use of the MIEs in this study.
We performed an initial examination of the AOPs constructed for liver cancer found in the AOP Wiki (https://aopwiki.org) (2017). However, many relevant AOPs remain under development or are not yet fully endorsed. In addition, many of the KEs in the AOPs were not amenable to measurement using gene expression biomarkers from short-term studies (e.g., clonal expansion of preneoplastic foci). Therefore, we capitalized on our past experience using gene expression biomarkers to classify chemicals in the mouse liver (Oshida et al., 2015a; Oshida et al., 2015b; Oshida et al., 2015c) with an emphasis on the best known/characterized MIEs that are often involved in chemical-induced hepatocellular adenomas and carcinomas and that could be built using available microarray data in our rat liver compendium (Figure 1). Although the current state of the AOP Wiki has gaps for accepted MOAs for rodent liver carcinogenesis, we focused on MIEs for 6 well known MOAs for rat liver tumorigenesis including AhR (Budinsky et al., 2014), CAR (Elcombe et al., 2014), PPARα (Corton et al., 2014), ER (Yager et al., 1996), and cytotoxicity (Felter et al., 2017). The KE of direct DNA damage (genotoxicity) was also included in our analysis. We describe how each MIE was measured in the sections below. It should be emphasized that our objective in this study was not to construct new AOPs but to test the hypothesis that measurement of the most common and well characterized MIEs will allow identification of liver tumorigens using data from short-term exposures.
The TG-GATES study.
TG-GATES is a publicly-available database collaboration that reports microarray and pathology data across multiple-doses for subchronic in vivo rat studies, as well as in vitro exposures to human and rat primary hepatocytes. The published protocol for the TG-GATES experiments (Uehara, et al., 2011) uses 5 Sprague-Dawley rats per sampling point treated with compounds at each of 3 different dose levels (low, middle, high). The maximum tolerated dose of each compound was estimated from a preliminary 7-day repeated dosing study and was used as the highest dose level. In general, the ratio of the concentrations for the low, middle and high dose levels was set as 1:3:10, respectively (Igarashi, et al., 2015). For exposure timepoints of 24 h or less, rats were euthanized 3, 6, 9 and 24 h after dosing. In the remainder of the protocol, the animals were treated daily for 3, 7, 14 and 28 days and euthanized 24 h after the last dose (4, 8, 15 and 29d). Euthanasia was performed using ether anesthesia and exsanguination from the abdominal aorta, followed by immediate collection of tissue samples from the left lateral lobe of the liver. Microarray analysis was conducted on 3 of 5 samples from each group using Affymetrix Rat Genome RAE230 2.0 arrays (Affymetrix, Santa Clara, CA, USA) (Uehara, et al., 2011). In addition to microarray data, the database contains absolute liver and body weights, clinical chemistry results, and the results from pathological examination of tissues. All data are found in the TG-GATES database (http://toxico.nihbiohn.go.jp/english/) (accessed March 28, 2019).
The DrugMatrix study.
DrugMatrix is a publicly-available toxicogenomic reference database that contains microarray and clinical chemistry data from tissues of rats administered pharmaceutical agents, environmental chemicals, or other substances (Svoboda D, 2019). The DrugMatrix database was acquired by NTP in 2010 after development by Incyte Genomics, Inc. and Iconix Pharmaceuticals, Inc. The DrugMatrix database includes transcriptomic profiles from liver RNA samples hybridized to a number of types of microarrays. Most chemicals were administered to rats at two dose levels and multiple time points. All data are available at https://ntp.niehs.nih.gov/drugmatrix (accessed March 28, 2019). The following platforms were used in the DrugMatrix study: GPL5424 GE Healthcare/Amersham Biosciences CodeLink UniSet Rat I Bioarray, layout EXP5280X2–584; GPL5425 GE Healthcare/Amersham Biosciences CodeLink UniSet Rat I Bioarray, layout EXP5280X2–613; GPL5426 GE Healthcare/Amersham Biosciences CodeLink™ UniSet Rat I Bioarray, layout EXP5280X2–648. The microarray data used in the present study came from GSE8858 which used three platforms (GLP5424, GLP5425, GLP5426).
Identification of differentially expressed genes in BaseSpace Correlation Engine (BSCE) microarray datasets.
All of the statistically filtered gene lists used in our study were generated using BSCE standardized microarray analysis pipelines and are available in a searchable annotated format in the BSCE (Xu et al., 2013)(Xu et al., 2013)(Xu et al., 2013)database (https://www.illumina.com/informatics/research/biological-data-interpretation/nextbio.html) (accessed March 28, 2019). Raw microarray data available in Open TG-GATEs or available in Gene Expression Omnibus (e.g., for DrugMatrix study and additional smaller studies) were imported and analyzed by the BSCE analysis pipeline. Differentially expressed genes were identified as described in a previous publication (Kupershmidt et al., 2010). Lists of differentially expressed and statistically filtered genes are referred to as biosets.
Annotation of a rat liver gene expression compendium.
The annotation of the rat liver compendium was carried out in a manner similar to our studies in the mouse (Oshida, et al., 2015a; Oshida, et al., 2015b; Oshida, et al., 2015c) and as described in (Rooney, et al., 2018a). All biosets with gene expression from rat liver, rat primary hepatocytes, or rat hepatocyte-derived cell lines were annotated for study characteristics allowing a systematic assessment of the effect of chemicals and other factors on a number of transcription factors important in hepatocarcinogenesis. The list of descriptors provided for each of the biosets included study ID (i.e., source), name, classification of factor (e.g., chemical, diet, genotype, etc.), name of chemical or treatment, time and dose of exposure, sex, source of material (e.g., liver or hepatocyte), and microarray type.
Classification of hepatocarcinogenicity of chemical-dose pairs.
We evaluated chemicals that were examined in the TG-GATES and DrugMatrix studies for doses that have known effects in the rat liver for ability to induce hepatocellular adenomas or hepatocellular carcinomas (“liver tumorigens”) using information in the Carcinogenicity Potency Database (https://toxnet.nlm.nih.gov/cpdb/) (accessed March 28, 2019), Physicians’ Desk Reference (1997), or in Pharmapendium (https://www.elsevier.com/solutions/pharmapendium-clinical-data) (accessed March 28, 2019) (Wilson et al., 2005) (Wilson et al., 2005) (Wilson et al., 2005). The Carcinogenicity Potency Database is now found at ftp://anonftp.niehs.nih.gov/ntp-cebs/datatype/Carcinogenic_Potency_Database_CPDB/ (accessed March 9, 2020). Two dose levels were classified from the chronic cancer data: lowest dose that caused an increase in liver tumors and the highest dose that does not cause liver tumors. Annotations were made only when there was a clear response (positive or negative) at a given dose level in male or female rats of any strain; a total of 4739 chemical-dose-time comparisons were evaluated. In contrast to other studies (e.g., (Fielden, et al., 2007)),we did not make any assumptions about the ability of the chemicals to induce tumorigenesis, and thus our analysis resulted in fewer chemical-dose-time comparisons to which we could ascribe tumor induction. We were able to annotate 1421 chemical-dose-time comparisons. The chemicals evaluated in our study are listed in Supplemental File 1.
Analysis of ALT and AST measurements.
The TG-GATES database contains data for traditional measures from the animal studies. These data were used to calculate relative changes for all chemical-dose-time comparisons (a total of ~3200 each for alanine transaminase [ALT] and aspartate aminotransferase [AST]). Significant changes were identified by ANOVA for each chemical-time group across four doses (control and three doses) to determine significant effect of dose level at each endpoint. A Benjamini-Hochberg multiple test correction was applied to the ANOVA p-values (corrected p-value < 0.05) followed by a Tukey’s HSD post-hoc comparison between individual dose levels at each time point for each chemical.
Origin of the cytotoxicity biomarker genes.
In our previous study, we built and characterized six gene expression biomarkers used in our prediction models (AhR, CAR, ER, genotoxicity, PPARα, hepatocyte proliferation). Each of the biomarkers were built using a weight of evidence strategy similar to our previous studies in which we built and characterized biomarkers for AhR, CAR, PPARα, and STAT5b to predict effects in chemically-treated mouse livers (Oshida, et al., 2015a; Oshida, et al., 2015b; Oshida, et al., 2015c; Oshida et al., 2016). In our original study, we did not have a biomarker which could be used to predict cytotoxicity. In the present study, we used a previous analysis of cytotoxic chemicals in the rat by Glaab et al., as the basis for selection of 10 genes predictive of liver toxicity (Glaab WE, 2018). In their study (Glaab WE, 2018), tissue samples from short-term rat toxicity studies were used to develop, evaluate and qualify a set of gene expression signatures indicative of degeneration/necrosis. To do this, their research team used a reference set of toxicants for the rat that were known to cause degeneration/necrosis in one or more of 4 different tissues (liver, kidney, heart, and skeletal muscle). Tissue samples for all organs were collected from each animal at necropsy and processed for microscopic histopathologic assessment, with an additional adjacent tissue section removed for genomic analyses, allowing for direct comparison of the gene expression response to histopathology outcome. Glaab et al., first used their microarray data to identify approximately 400 genes with similar gene expression patterns across the 4 prioritized tissues (liver, kidney, heart, skeletal muscle), following exposure to the known toxicants; these genes were designated ‘universal’. They followed this with quantitative PCR targeted towards a subset of these universal genes.. The most consistent and robustly-responding transcripts were selected as markers for degeneration/necrosis, resulting in a final 12-gene set that accurately predicted degeneration/necrosis in the liver. Performance for their derived liver degeneration/necrosis marker signature on a test set of 34 chemicals (16 with positive histopathology findings) in Sprague Dawley rats was 88% sensitivity and 94% specificity and in Wistar rats was 94% sensitivity and 98% specificity (Glaab WE, 2018).
We attempted to incorporate the 12 genes identified by Glaab et al. in our current study, but we found that 2 of the 12 genes (Oldlr1, Serpine1) rarely responded with any significant change across the TG-GATES or DrugMatrix datasets and so were excluded from our current analysis. The remaining 10 genes (Anxa2, Bcl2a1a, Cdk1 (Cdc2a), Fcnb, Gpnmb, Pvr, S100a4, Spp1, Timp1, Tnfrsf12a) were each given an equal weight. This is in contrast to our other biomarkers in which each gene is weighted and ranked based on absolute value of the fold-change. The set of 10 genes were uploaded to BSCE in accordance with our previous publication (Rooney, et al., 2018a). It should be noted that there are no genes shared by the cytotoxicity biomarker and the other biomarkers used in our study including the previously characterized cell proliferation biomarker (Rooney, et al., 2018a).
Comparison of gene lists to the gene expression biomarkers.
The biomarker genes and associated fold-change values were uploaded into BSCE in which internal protocols rank the genes based on absolute fold-change. The biomarkers for AhR, CAR, PPARα, ER, and genotoxicity have been described (Rooney, et al., 2018a). Each ranked set of genes in the database is compared to all other ranked lists using the Running Fisher test, which calculates a correlation p-value for the overlapping genes between any two lists. This p-value is then transformed to a -Log(p-value), providing a simple scalar metric to evaluate correlation between any two gene lists. Thus, the higher the -Log(p-value), the lower the actual p-value, and the more significant the correlation between the data sets. A negative correlation is reported as a negative integer.
Determination of cytotoxicity biomarker balanced accuracy.
Biosets from microarray experiments in which cytotoxicity status was known were manually curated from the studies listed in Supplemental File 1. The bioset data used to determine the balanced accuracy for our study were extractedfrom the TG-GATES study because of the thorough ALT and AST measurements and histopathology annotations in TG-GATES associated with the microarray biosets. Chemicals were labeled as true positives if any dose or time of exposure led to 1) increases (≥ 2-fold) in ALT and/or AST, and 2) exhibited slight, moderate or severe increases in “necrosis” or “single cell necrosis” in over 33% of the samples. Chemicals were labeled as true negatives if there were no increases in either ALT or AST and no increases in “necrosis” or “single cell necrosis”. The final list of chemicals included 7 positives (colchicine, ethambutol, ethionamide, methapyrilene, monocrotaline, naphthyl isothiocyanate, thioacetamide) and 23 negatives (acarbose, adapin, ajmaline, bendazac, captopril, chloramphenicol, clofibrate, glibenclamide, meloxicam, nifedipine, nimesulide, nitrofurantoin, pemoline, phenacetin, phenylbutazone, ranitidine, rifampicin, sulindac, tamoxifen, tannic acid, tetracycline, theophylline, valproic acid). Each chemical was scored as true positive (TP), true negative (TN), false positive (FP) or false negative (FN) based on the highest -Log(p-value) for that chemical across the 24 dose-time comparisons. The equations used were: sensitivity (TP rate) = TP/(TP + FN); specificity (TN rate) = TN/(FP + TN); positive predictive value (PPV) = TP/(TP + FP); negative predictive value (NPV) = TN/(TN + FN); balanced or predictive accuracy = (sensitivity + specificity)/2; where FN = false negative and FP = false positive.
Co-Expression Analysis of biomarker genes using TXG-MAP.
The full list of genes in the biomarkers used in the study were analyzed for membership in co-expressed gene sets using TXG-MAP (Sutherland, et al., 2018). Weighted gene co-expression network analysis (WGCNA) was used to organize co-expressed sets of genes into gene networks or modules starting with the DrugMatrix rat liver microarray profiles. The resulting co-expression framework called the ‘toxicogenomic module associations with pathogenesis’ (the TXG-MAP) was integrated with standard pathology evaluation to successfully characterize mechanisms of drug-induced liver injury. Lists of the biomarker genes were uploaded into the TXG-MAP website (www.ctox.indianabiosciences.org).
Toxicological Priority Index (ToxPi) analysis.
The Toxicological Priority Index, or “ToxPi” framework (Reif et al., 2010), was used to generate and rank a liver tumorigen score for each comparison representing a chemical-dose-time based on the measured MIE responses. The ToxPi framework is a visualization tool used in this case to represent individual MIEs that are scaled and represented as “slices” that make up a circle or pie. For each slice, the distance from the origin is proportional to the normalized value of the data, and the width indicates the relative weight of that MIE. We predicted that chemical-dose pairs with the highest scores would be known tumorigens. A dimensionless ToxPi score was calculated using a linear scale for each comparison as normalized values between 0 and 1. All MIEs were weighted equally. Because the data contains only one component per slice, no bootstrapping step could be carried out. Scores were generated using ToxPi Software (v1.3) (Reif, et al., 2010). The -Log(p-value) scores were first filtered by removing any values < 0. ToxPi scores were exported and used to examine the relationships between scores and liver tumorigenicity.
Receiver-operator characteristic curves.
The ToxPi scores were used to develop a receiver-operator characteristic curve (ROC) and the area under the curves (AUC) were calculated to compare model performance and determine a capture threshold for tumorigens. The ROC curve is a mathematical method for evaluating inter-rater reliability or determining the critical value for clinical diagnostic assays against a dichotomous outcome. In our case, we applied this metric to the calculated ToxPi scores and the classifications of known tumorigens and non-tumorigens in the TG-GATES and DrugMatrix studies using a standardized data analysis program (Systat Software; San Jose, Ca). A paired correlation option was used for ROC area analysis using the DeLong, Delong and Clarke-Pearson method with the Systat software; there were no missing values in the data sets. Robustness of the ToxPi scoring system was determined by an area under the curve (AUC) closest to 1.0, as well as the false-positive and false-negative rate for the data set using a ToxPi cutoff at the point of balanced accuracy for each comparison. The balanced accuracy was evaluated using sensitivity/specificity analysis, and the optimal ToxPi cutoff was graphically determined using the point of intersection on a sensitivity and specificity versus cutoff plot.
Determination of predictive accuracy.
Predictive accuracy was determined either on an individual bioset level or by aggregating biosets that examine the same chemical. The tumorigenic prediction by chemical aggregate was scored as true positive (TP), true negative (TN), false positive (FP), or false negative (FN) based on whether the ToxPi value exceeded the ROC threshold. To be diagnosed as a FN, all biosets for a tumorigenic chemical had to be below the ToxPi threshold. If even one of the biosets for a non-tumorigenic chemical exceeded the ToxPi thresholds, it was diagnosed as a false positive. The calculations for predictive accuracy were the same as described above.
Additional methods.
Hierarchical clustering and visualization was carried out using the Cluster and TreeView programs from the Eisen lab (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) (assessed Sept. 10, 2019) or using the cluster program hclust with Ward.D2 in R.
RESULTS AND DISCUSSION
A MIE-driven strategy to identify chemicals that cause liver cancer.
The major MIEs and KEs in rodent liver cancer AOPs are shown in Figure 1. Activation of one or more of the MIEs including the xenobiotic receptors AhR and CAR, the lipid receptor PPARα, the steroid receptor ER, and cytotoxicity lead to downstream events that include cell proliferation, increases in oxidative stress, and indirect DNA damage. Also shown is the MIE of direct DNA damage induced through different genotoxic mechanisms. We hypothesized that the vast majority of rat liver tumorigens will activate one or more of the 6 MIEs and can be detected using gene expression biomarkers applied to transcript profile data from short-term exposures (≤ 1 month). We tested the hypothesis using two large microarray datasets, TG-GATES and DrugMatrix. The TG-GATES dataset included 133 chemicals at three doses and 8 time points (up to 29d) for a total of 3080 chemical-dose-time comparisons. Out of these, there were a total of 1066 chemical-dose-time comparisons representing 75 chemicals examined at doses with known liver tumorigenicity outcomes after ~2 years (18 positive and 57 negative). Gene expression was determined by Affymetrix arrays. The DrugMatrix dataset (GSE8858) included 339 chemicals conducted mostly at 3d and 5d for a total of 1659 chemical-dose-time comparisons. Out of these, there were a total of 355 chemical-dose-time comparisons representing 88 chemicals examined at doses with known liver tumor outcomes after ~2 years. Gene expression was determined by microarray platforms from GE Healthcare.
A 10 gene biomarker is predictive of cytotoxicity.
The cytotoxicity biomarker was comprised of 10 genes (Anxa2, Bcl2a1a, Cdk1, Fcnb, Gpnmb, Pvr, S100a4, Spp1, Timp1, Tnfrsf12a) reported in Glaab et al. to exhibit consistent increased expression changes after exposure to chemicals cytotoxic to the liver, which made them predictive for hepatic necrosis/degeneration (see Methods for a description) (Glaab WE, 2018). We used these 10 genes as the gene expression biomarker to predict cytotoxicity in the liver. Expression changes in the genes were first surveyed using the TG-GATES data for the prototypical cytotoxicant thioacetamide to determine if the gene expression patterns captured the dose- and time-dependent changes expected (Figure 2A, top). At the lowest dose (4.5 mg/kg/day), there were few changes in gene expression at any of the 8 time points. At the middle dose (15 mg/kg/day), a greater number of genes were increased, and at the highest dose (45 mg/kg/day), all time points exhibited increases in three or more of the genes which peaked after 1d of exposure.
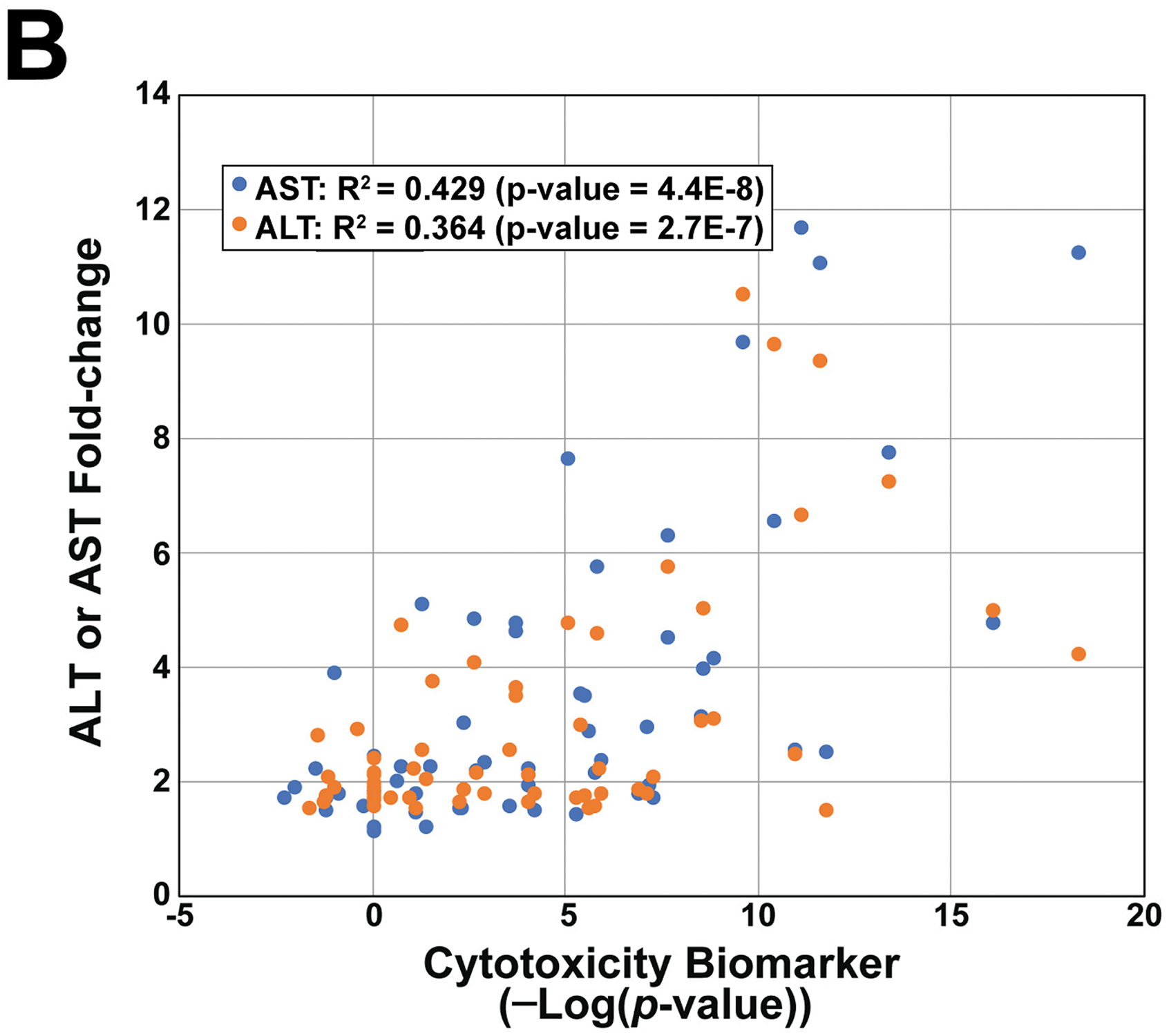
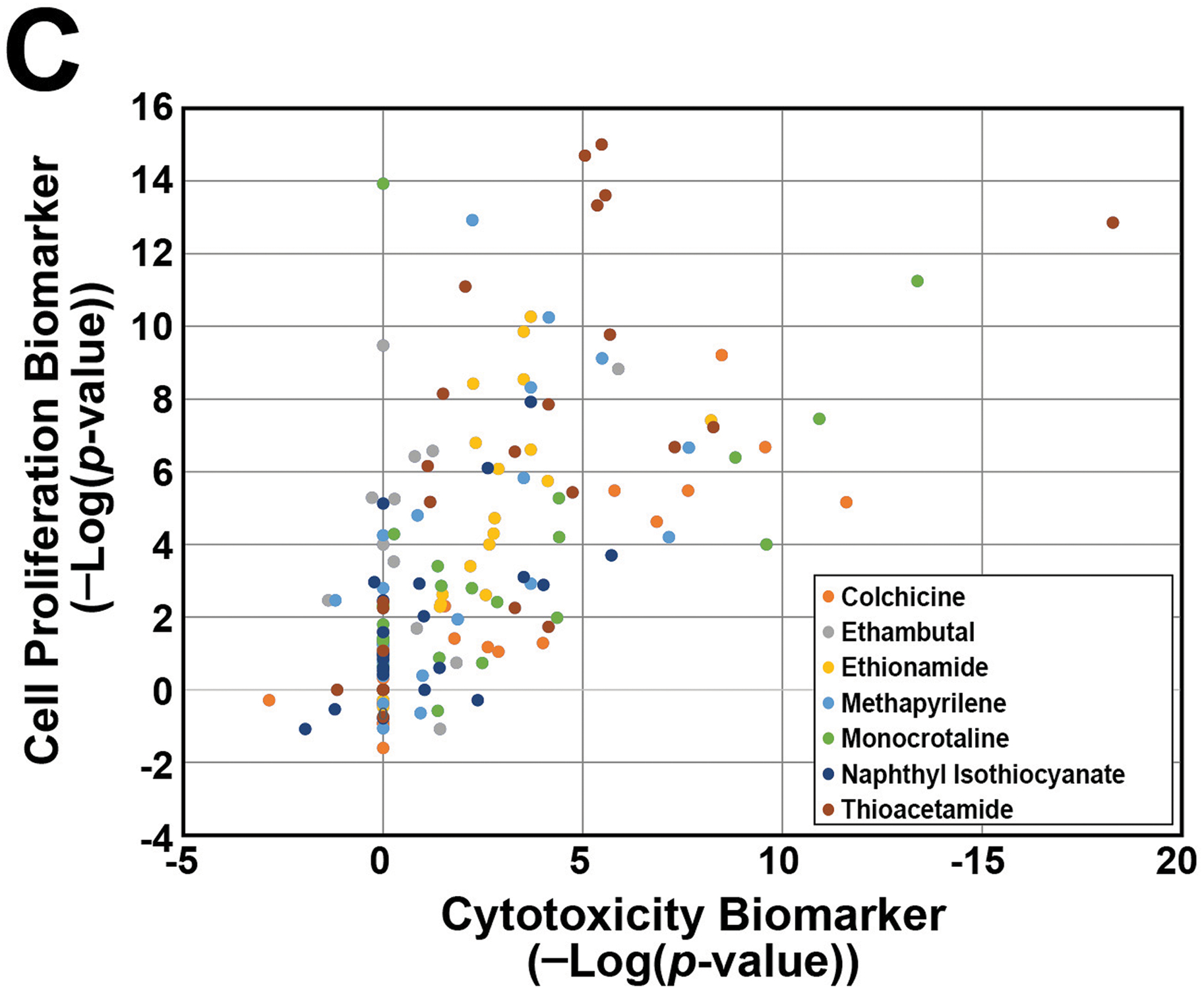
Fig. 2. Characterization of a cytotoxicity biomarker.
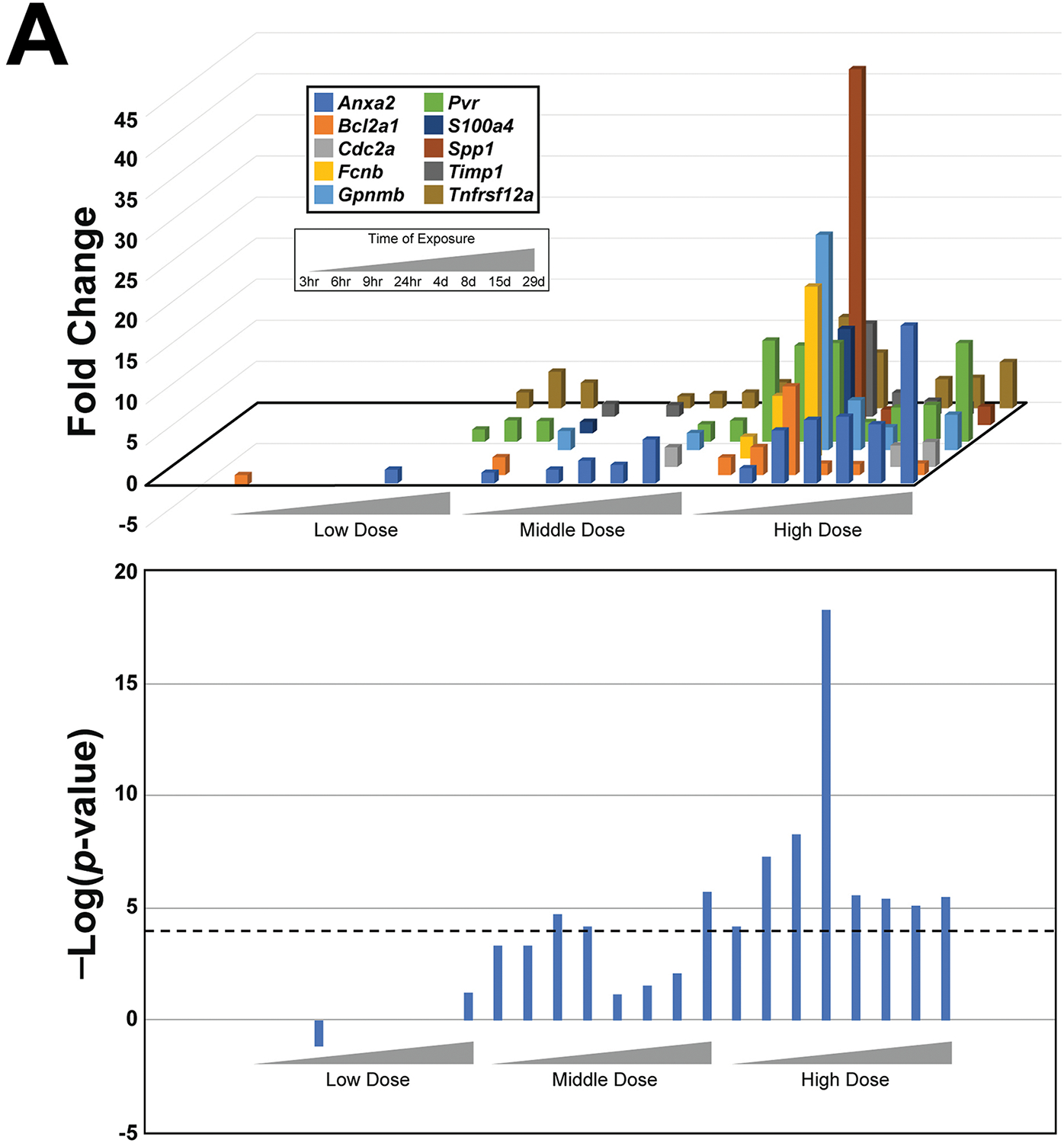
A. Activation of cytotoxicity biomarker genes by thioacetamide. (Top) The expression changes of the 10 genes in the cytotoxicity biomarker are shown across increasing time and dose. Only significant changes in gene expression are shown (p-value < 0.05). (Bottom) The significance of the correlation (in -Log(p-value)s) between the cytotoxicity biomarker and the gene expression changes of each time-dose pair is shown. The cutoff of -Log(p-value) = 4 is shown.
B. Relationship between alterations of ALT/AST and the cytotoxicity biomarker. Biosets in the TG-GATES study were filtered for those with significant increases in ALT or AST and then compared to the biomarker -Log(p-value) for that same treatment.
C. Relationship between correlations with the cytotoxicity and cell proliferation biomarkers for biosets from treatments with 7 cytotoxic chemicals. The transcript profiles of treatment groups in the TG-GATES study selected as described in the Methods were compared to the two biomarkers.
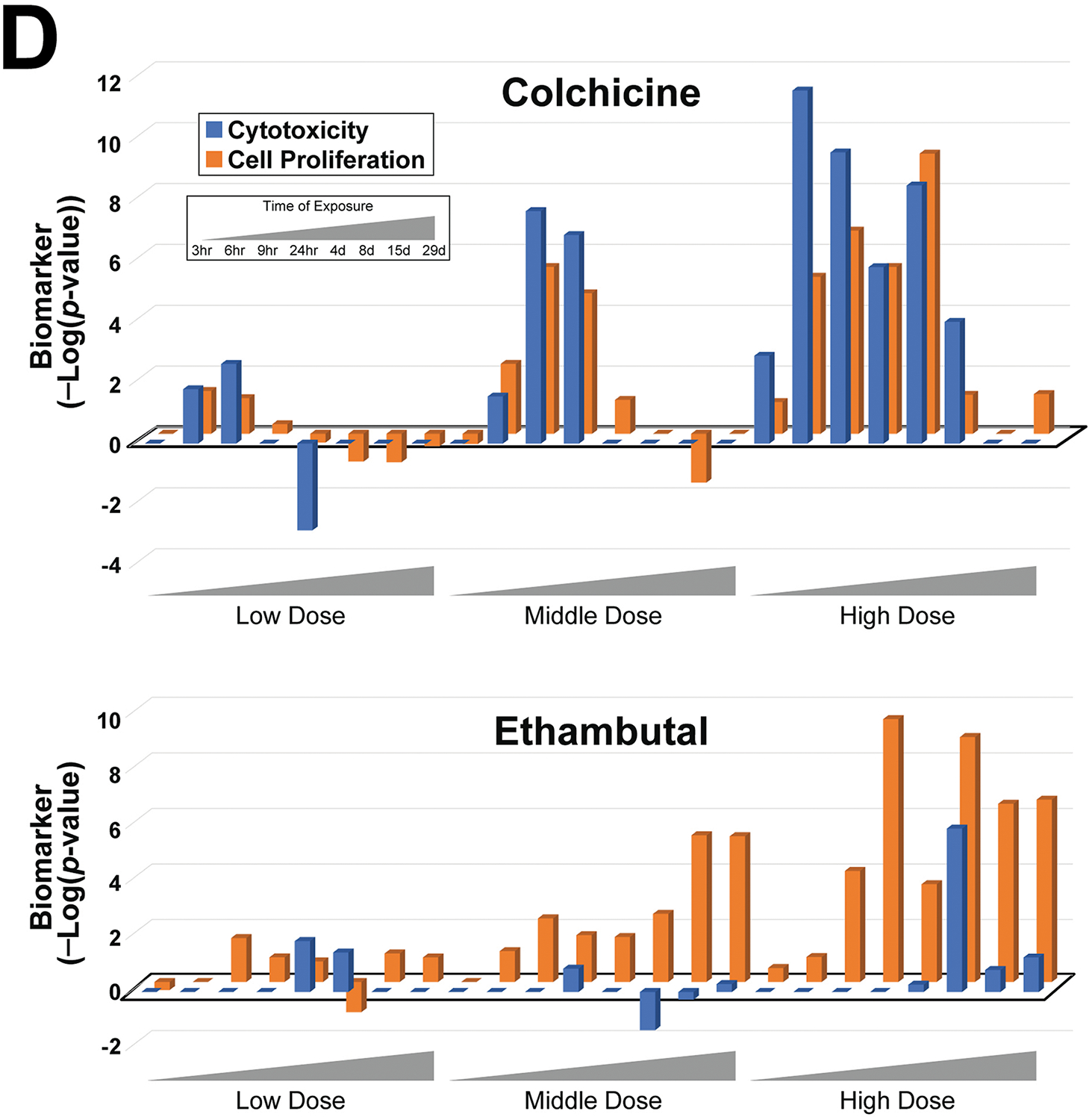
D. Correlations between the transcript profiles from exposure to colchicine (top) or ethambutal (bottom) and either the cytotoxicity or cell proliferation biomarkers across time and dose. The results of a linear regression analysis of each chemical are as follows: colchicine (R2 = 0.800, p-value = 4.13E-09) and ethambutal (R2 = 0.159, p-value = 0.054).
E. Comparison of correlations to the cytotoxicity biomarker for the 7 true positive chemicals. The cytotoxicity biomarker was compared to each of the indicated chem-time comparisons across the highest dose used. The chemicals were ordered to facilitate observing the maximum correlation with the chemicals. There were no values for the 29d timepoint for ethionamide and monocrotaline.
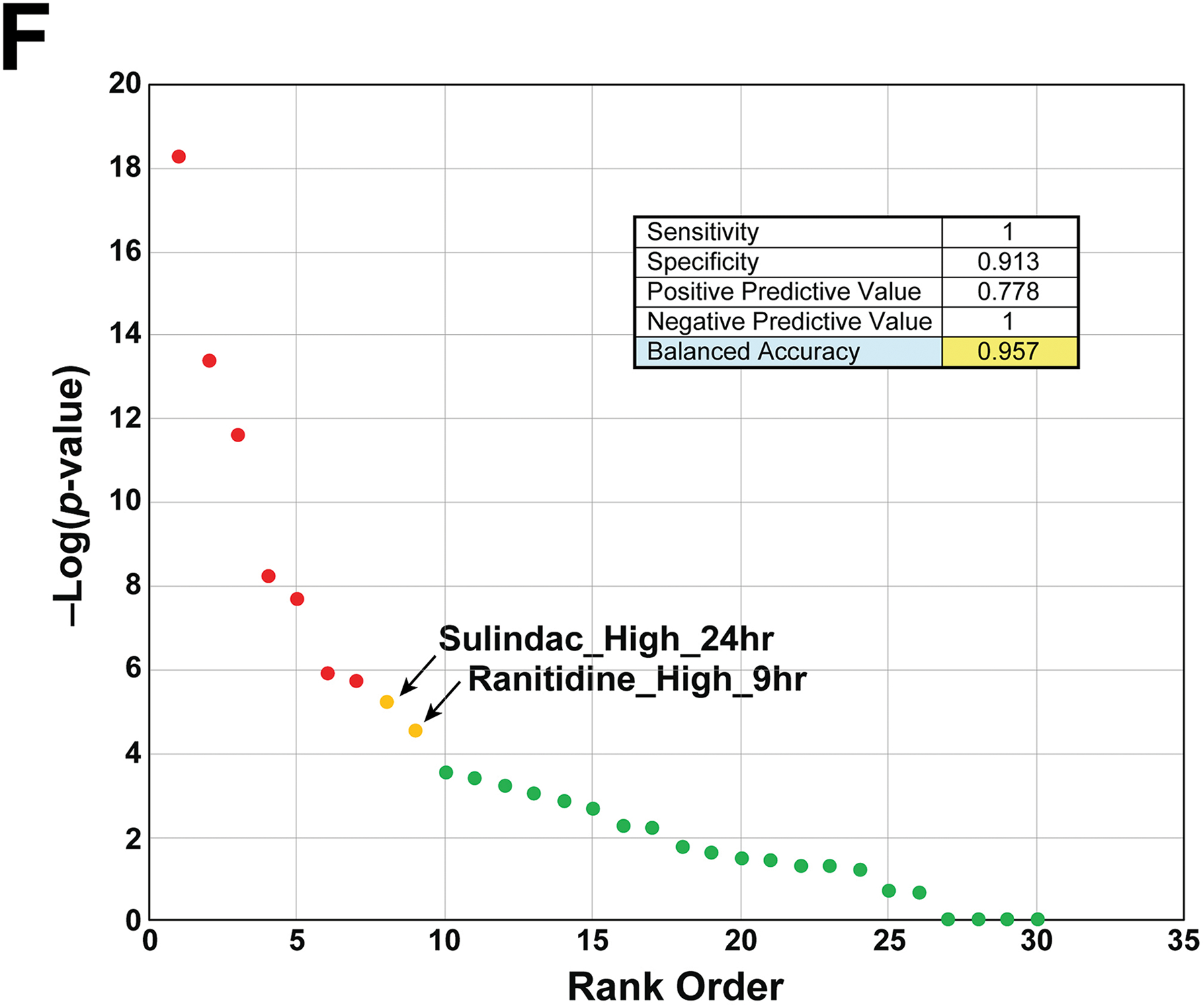
F. Predictive accuracy of the cytotoxicity biomarker. The biomarker was compared to transcript profiles of chemicals that are known positives (7) or negatives (23) for cytotoxicity. See Methods for criteria for selection of chemicals. The -Log(p-value) for each chemical represents the highest value within all of the time-dose pairs found in the TG-GATES study. A summary of the prediction sensitivity and specificity of the cytotoxicity biomarker is shown. The two false positives are shown that caused significant correlation to the biomarker.
The expression of the 10 genes in each of the 24 transcript profiles for thioacetamide were compared to the cytotoxicity biomarker using a Running Fisher test, resulting in a correlation p-value for the overlapping genes. This p-value was then transformed to a -Log(p-value), providing a simple scalar metric to evaluate correlation. The higher the -Log(p-value), the lower the p-value, and the more significant the degree of correlation between the biomarker and the gene expression changes. A negative correlation is reported as a negative integer. The -Log(p-value)s of the correlations between the cytotoxicity biomarker and the expression of the 10 genes in each of the comparisons are shown in Figure 2A (bottom). At the low dose, no dose-time pairs achieved significance (-Log(p-value) ≥ 4), whereas at the middle dose significance was achieved at two early time points (9h, 24h) and at the end of the study (29d). All time points at the high dose were significant. The increases in the correlation to the cytotoxicity biomarker peaked at 24h in parallel with the maximal increases in the expression of the genes. At 24h, there were increases in alanine transaminase (ALT) (4.2-fold) and aspartate aminotransferase (AST) (11.2-fold), commonly used to provide evidence of liver damage (data not shown). Previous studies of thioacetamide showed that a dose of 50 mg/kg (high dose in the TG-GATES study is 45 mg/kg) induced ALT levels in male SD rats that peaked at 36h (Mangipudy et al., 1995), similar to the findings reported here.
We determined if there was a relationship between increases in cytotoxicity as assessed by the biomarker and increases in ALT and AST across the TG-GATES chemicals. Figure 2B shows that for many exposure conditions, the cytotoxicity -Log(p-value)s increase with increased ALT or AST levels. Linear regression analysis gave a R2 = 0.429 (p-value = 4.4E-8) for AST compared to the biomarker and R2 = 0.364 (p-value = 2.66E-7) for ALT compared to the biomarker. There were a number of biosets which did not achieve significance for the biomarker but resulted in increases in ALT or AST, and there were biosets in which the biomarker achieved the threshold but there were no increases in ALT or AST. These could be due to temporal differences in gene expression changes and release of ALT and AST from damaged hepatocytes after chemical exposure.
Cytotoxicity leads to hepatocyte death that is followed by regenerative cell proliferation to replace lost cells (Felter et al., 2018). We predicted that there would be increases in a cell proliferation response when there were increases in chemical-induced cytotoxicity. Although no cell proliferation data was generated as part of the TG-GATES or DrugMatrix studies (e.g., histochemical evaluation of Ki67 or PCNA protein expression), we had previously characterized a biomarker of cell proliferation genes derived from microarray comparisons under conditions known to have increases in either mitogen-induced or regenerative cell proliferation. This cell proliferation biomarker consisting of only cell cycle genes showed a significant positive relationship with increases in liver weight to body weight ratios (Rooney, et al., 2018a). To determine if relationships exist between responses detected by the cytotoxicity and cell proliferation biomarkers, we focused on the responses to 7 chemicals identified as cytotoxic agents in the TG-GATES study based on both histopathology (“necrosis”, “single cell necrosis”) and increases in ALT and/or AST levels (see Methods). The gene expression profiles representing the dose-time pairs of the 7 chemicals were compared to the cytotoxicity and cell proliferation biomarkers. Figure 2C shows that for most of the dose-time comparisons, there were increases in the correlation to both biomarkers (R2 = 0.381; p-value = 7.9E-19), indicating that both cytotoxicity and cell proliferation genes were regulated in the same samples. Two examples of chemicals with differences in biomarker responses are shown in Figure 2D. Colchicine showed striking parallel increases in both biomarkers across time and dose. Increases occurred early and were no longer seen after 7d. In contrast, ethambutal exhibited cell proliferation at the middle dose in the absence of cytotoxicity, and at the high dose, cytotoxicity only occurred at 7d, indicating cell proliferation was induced through another mechanism. The other 5 chemicals (ethionamide, methapyrilene, monocrotaline, 1-naphyl isothiocyanate, thioacetamide) exhibited mostly parallel increases in both biomarkers (Figure S1), indicating the cytotoxicity and cell proliferation responses were mechanistically linked.
To determine the balanced accuracy of the biomarker, an analysis was performed on biosets from chemicals known to be positive or negative for cytotoxicity. (The list of chemicals used in the analysis is found in Supplemental File 1.) Given the transient nature of the cytotoxicity response in rat liver (e.g., as observed for thioacetamide and colchicine above), chemicals were evaluated across all time points and doses, and scored using the highest -Log(p-value) of any time-dose for that chemical. This approach was warranted due to differences in the peak of the significance of the cytotoxicity biomarker response to the different dose-time pairs of the cytotoxic chemicals. A comparison of the -Log(p-value)s across the 8 time points for the highest dose of each chemical is shown in Figure 2E. The peak of correlation ranged between 6h (colchicine) and 29d (methapyrilene) with intermediate peaks of 24h (thioacetamide), 8d (ethambutol, monocrotaline) and 15d (ethionamide, napthyl isothiocyanate) observed. Criteria for classification included a -Log(p-value) ≥ 4 that was used with other biomarkers in past studies (Oshida, et al., 2015c; Rooney, et al., 2018a). The final number of chemicals evaluated were 7 positives and 23 negatives. The biomarker had 100% sensitivity and 91% specificity, giving a balanced accuracy of 96% (Figure 2F). The two false positive chemicals were sulindac and ranitidine both of which had only one of the 24 possible dose-time comparisons reach significance and both were at the highest dose (Sulindac_High_24 hr, Ranitidine_High_9 hr). We repeated the analysis with either the 4 acute time points (3h – 24h) or the 4 multiple dose time points (4d – 29d). Using only the acute time points, ethambutol, monocrotaline and napthyl isothiocyanate were not identified while using the later time points only ethionamide was not predicted (data not shown). This analysis indicates that examination of time points later than 24h would be more useful for prediction of cytotoxicity. Overall, these results indicate that the cytotoxicity biomarker can identify chemicals that cause cytotoxicity in the rat liver.
Identification of cytotoxic agents in a rat liver microarray compendium
The cytotoxicity biomarker was also used to identify chemicals in a rat liver microarray compendium that exhibit transcript profiles indicative of cytotoxicity. There were 407 chemicals represented by 4745 biosets from the TG-GATES and DrugMatrix studies that were examined. Profiles were compared to the biomarker using the Running Fisher test and sorted by the resultant -Log(p-value)s. Biosets that had a positive correlation to the biomarker (Figure 3, top, left side), exhibited greater numbers of the 10 genes increased in expression. There were 178 biosets representing 95 chemicals that exhibited significant positive correlation (-Log(p-value) ≥ 4) to the biomarker. The top 10 ranking biosets are shown and include some prototypical liver cytotoxic agents. Figure 3 (top, right side) shows a much smaller group of biosets that exhibited significant negative correlation (-Log(p-value) ≤ −4) to the biomarker. There were 13 biosets representing 11 chemicals that exhibited negative correlation to the biomarker. These included acetazolamide, caffeine, ibufenac, and theophylline. The underlying basis for the suppression of the biomarker genes by these chemicals awaits further investigation. The entire dataset is provided in Supplemental File 1.
Fig. 3. Screening for cytotoxicants in a rat liver microarray compendium.
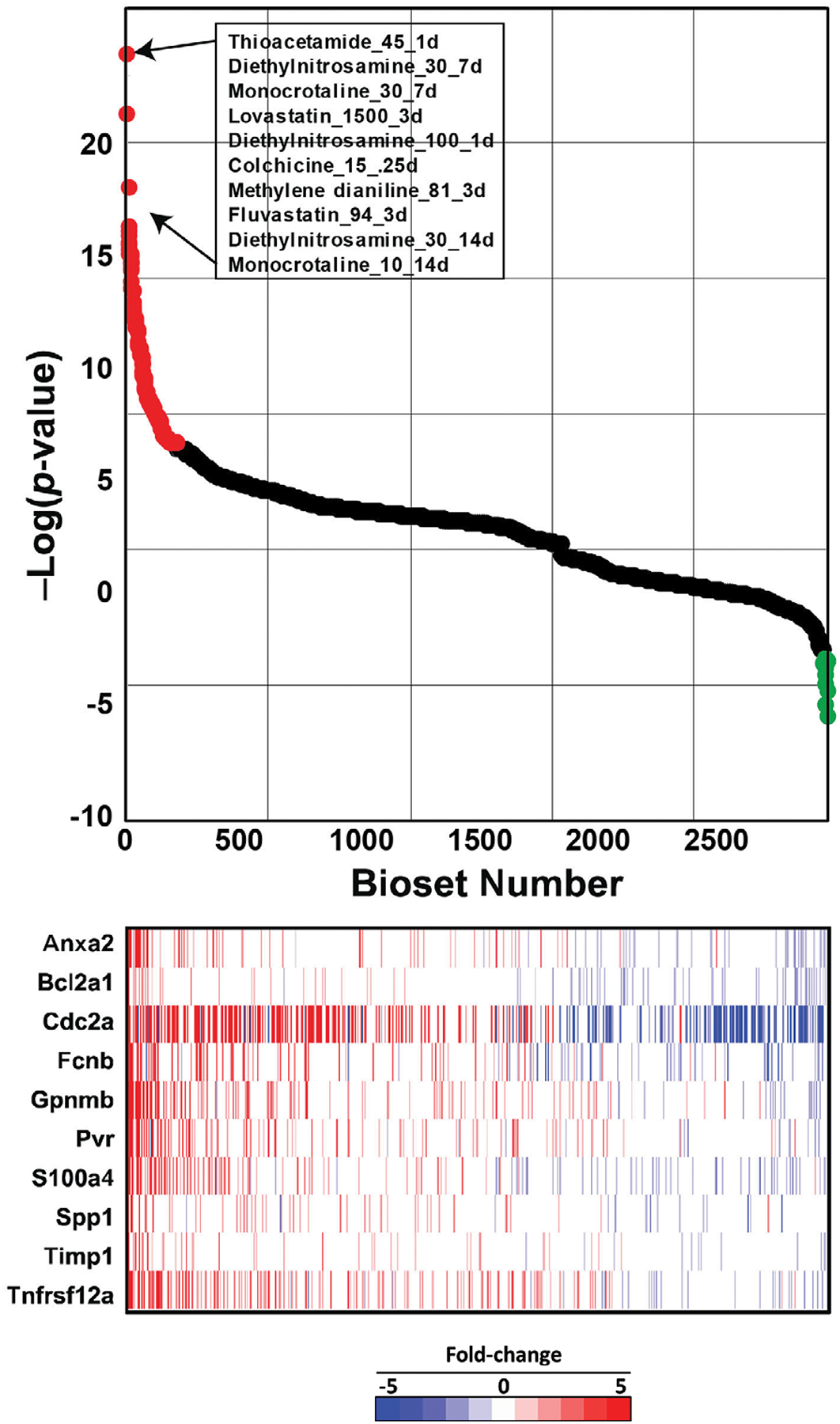
All 4745 transcript profiles representing chemical exposures in the microarray compendium were compared to the cytotoxicity biomarker using the Running Fisher test. (Top) The series of points represents the rank ordered -Log(p-value)s of the biosets compared to the biomarker. Biosets with positive correlation are on the left, and if the -Log(p-value) ≥ 4, they are indicated in red. The biosets with negative correlations are on the right and if the -Log(p-value) ≤ −4, they are indicated in green. Biosets with a -Log(p-value) = 0 are not shown. The top 10 most positively correlated biosets are shown and are represented as dose in mg/kg/day and time of exposure in days. (Bottom) The heat map shows the expression of the 10 biomarker genes in the corresponding biosets.
The genes in the biomarkers overlap with a number of functional modules in a rat liver co-expression network.
In our previous study, pathways enriched with biomarker genes were consistent with the known functions of the MIEs being predicted (Rooney, et al., 2018a). One of the goals in the present study was to determine if gene expression biomarkers not only allow classification of carcinogens, but also to validate that building biomarkers based on literature knowledge of MIEs provides a more robust pathway-based context. Therefore, we determined the correlation between the 6 biomarkers and canonical pathway/gene ontology (GO) using TXG-MAP module scores for 3528 TG-GATEs rat liver experiments from the CTox web application (Sutherland et al., 2019a) (Table 1), similar to the approach we took in a recent study (Podtelezhnikov, 2020). Four of the biomarkers (genotoxicity, CAR, PPARα, cytotoxicity) exhibited high correlation vs. pathways/GO and/or TXG-MAP modules, indicating that treatments having high biomarker scores also have high pathway/module scores. For pathways/GO terms, biological themes were consistent with the biomarkers (e.g., BIOCARTA p53 pathway is the pathway most correlated with the genotoxicity biomarker). TXG-MAP modules are associated with histologically-defined outcomes; module 42m (rank 1 for hypertrophy), and 18m (ranks 1, 5, and 18 for necrosis, fibrosis and biliary hyperplasia, respectively) were most similar to the CAR and cytotoxicity biomarkers. Thus, the pathology associations are consistent with pathogenic mechanisms associated with that AOP, e.g. hypertrophy in the case of the prototypical CAR activator, phenobarbital, and the expected tissue injury in the case of the cytotoxicity bioset. The ER and AhR biomarkers had lower similarity compared to pathways/modules, suggesting that these perturbations result in multifactorial effects not well represented by single pathways or modules. Similar results are obtained when identifying TXG-MAP modules that include genes from the 6 biomarkers: >30% of biomarker genes are present in one (or closely-related) modules for cytotoxicity, genotoxicity, CAR and PPARα, whereas genes for AhR and ER are distributed across several modules with low correlation vs. biomarkers (Table S1, Figure S2).
Table 1.
Most similar canonical pathways, GO terms and TXG-MAP modules compared to the six gene expression biomarkers
| Biomarker | Most similar gene set | Pearson R1 |
|---|---|---|
| Canonical pathways / GO terms | ||
| Cytotox biomarker | GO cellular response to hydrogen peroxide | 0.71 |
| AhR Biomarker | REACTOME Genes involved in Xenobiotics | 0.48 |
| CAR Biomarker | KEGG Glutathione metabolism | 0.75 |
| ER Biomarker | BIOCARTA Nuclear Receptors in Lipid Metabolism and Toxicity | 0.44 |
| Genotoxicity Biomarker | BIOCARTA p53 Signaling Pathway | 0.56 |
| PPARα Biomarker | GO fatty acid catabolic process | 0.94 |
| TXG MAP modules (enriched GO term) | ||
| Cytotox biomarker | Module 18m (cell adhesion) | 0.64 |
| AhR Biomarker | Module 108 (insulin secretion) | 0.50 |
| CAR Biomarker | Module 42m (glutathione metabolic process) | 0.71 |
| ER Biomarker | Module 206 (prostate gland development) | 0.53 |
| Genotoxicity Biomarker | Module 205 (cellular response to DNA damage) | 0.69 |
| PPARα Biomarker | Module 17m (fatty acid metabolic process) | 0.82 |
Pearson R obtained by comparing biomarker scores for 3528 treatments from TG-GATEs (compound, dose and time) vs. the most similar canonical pathway / GO term or TXG-MAP module, as obtained from the CTox application (Sutherland, et al., 2019a).
Assessment of the six molecular initiating events in the rat liver
We hypothesized that chemicals that are tumorigenic would activate one or more of the 6 MIEs. Biomarker scores were visualized across the chemicals in the TG-GATES and DrugMatrix datasets relative to liver tumor classification. Figure 4A shows the activation of the MIEs across 4 time points (4, 8, 15, and 29d; 393 total biosets) of the TG-GATES study clustered by one-dimensional hierarchical clustering. Most of the chem-dose pairs did not activate any of the MIEs and were associated with doses that were nontumorigenic. In general, the chem-dose comparisons that were tumorigenic exhibited strong signals for one or more of the MIEs. There were clusters of biosets in which there was activation of each of the biomarkers associated with tumorigenic doses (lower part of Figure 4A). There were many chem-dose pairs in which there was no tumor incidence data available that clustered with treatments that were tumorigenic.
Figure 4. Activation of liver cancer MIEs across the TG-GATES dataset.
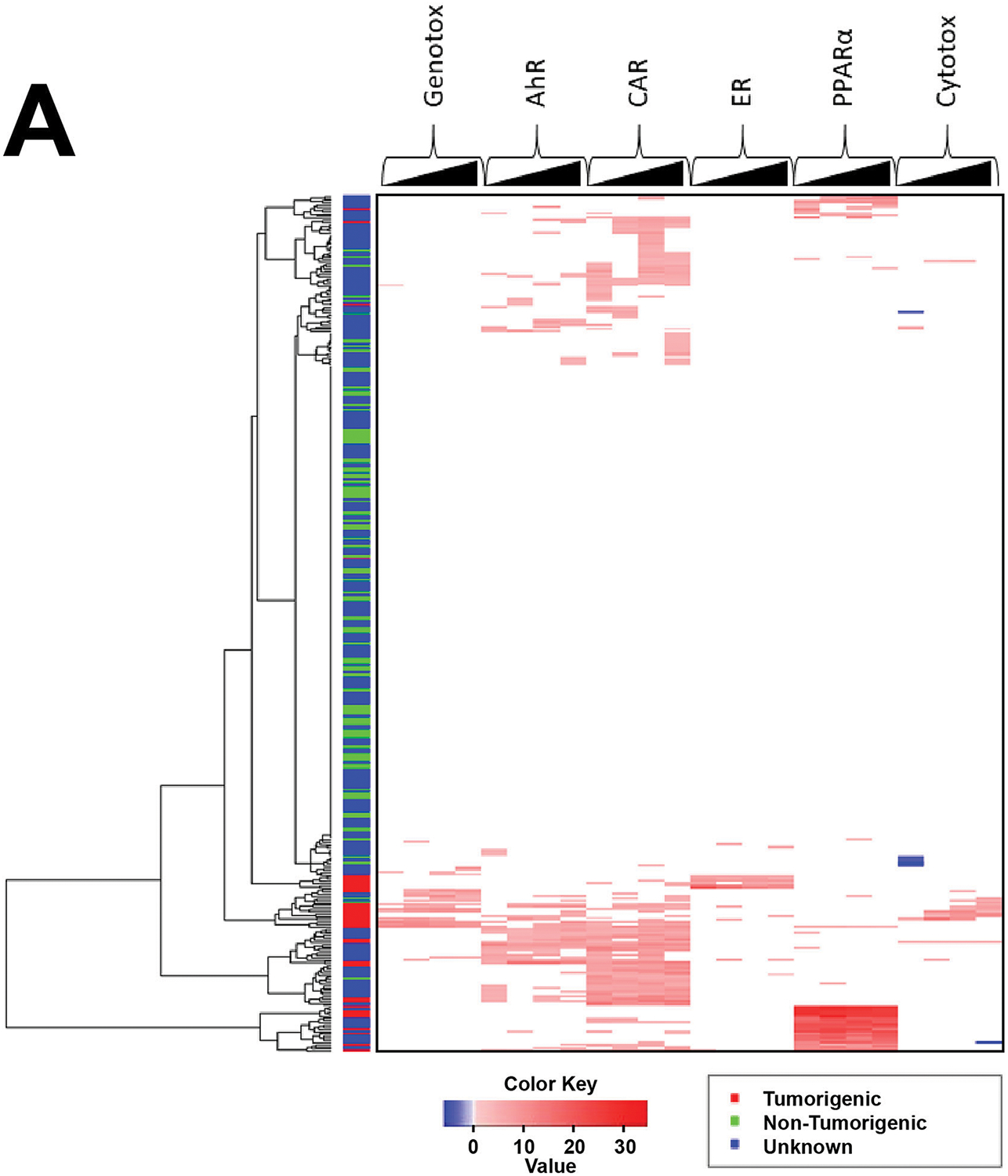
A. Heat map showing the modulation of the 6 MIEs across the chemical-dose pairs analyzed in the TG-GATES study. The changes are shown for each MIE for the 4 time points from 4d to 29d. Only biomarker values |−Log(p-value)s| ≥ 4 are shown. All 393 chemical-dose pairs were clustered using one-dimensional hierarchical clustering. The liver tumorigenicity of each chemical-dose pair is shown on the left. Red, tumorigenic; green, nontumorigenic; blue, not known.
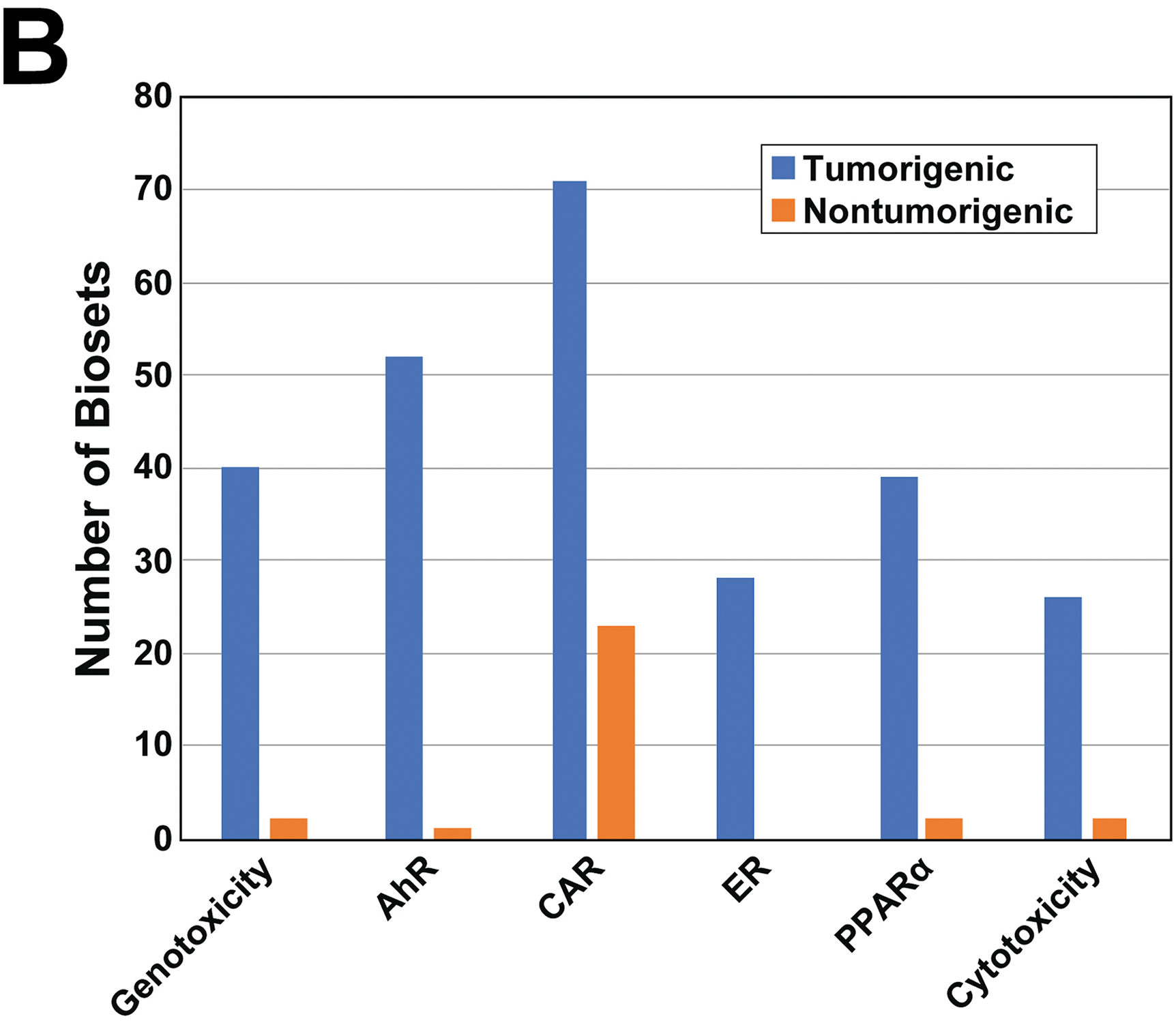
B. Activation of the 6 biomarkers after chemical treatment in the TG-GATES dataset. The number of tumorigenic and nontumorigenic biosets that had -Log(p-value) ≥ 4 for each biomarker are shown. The total number of biosets analyzed was 562.
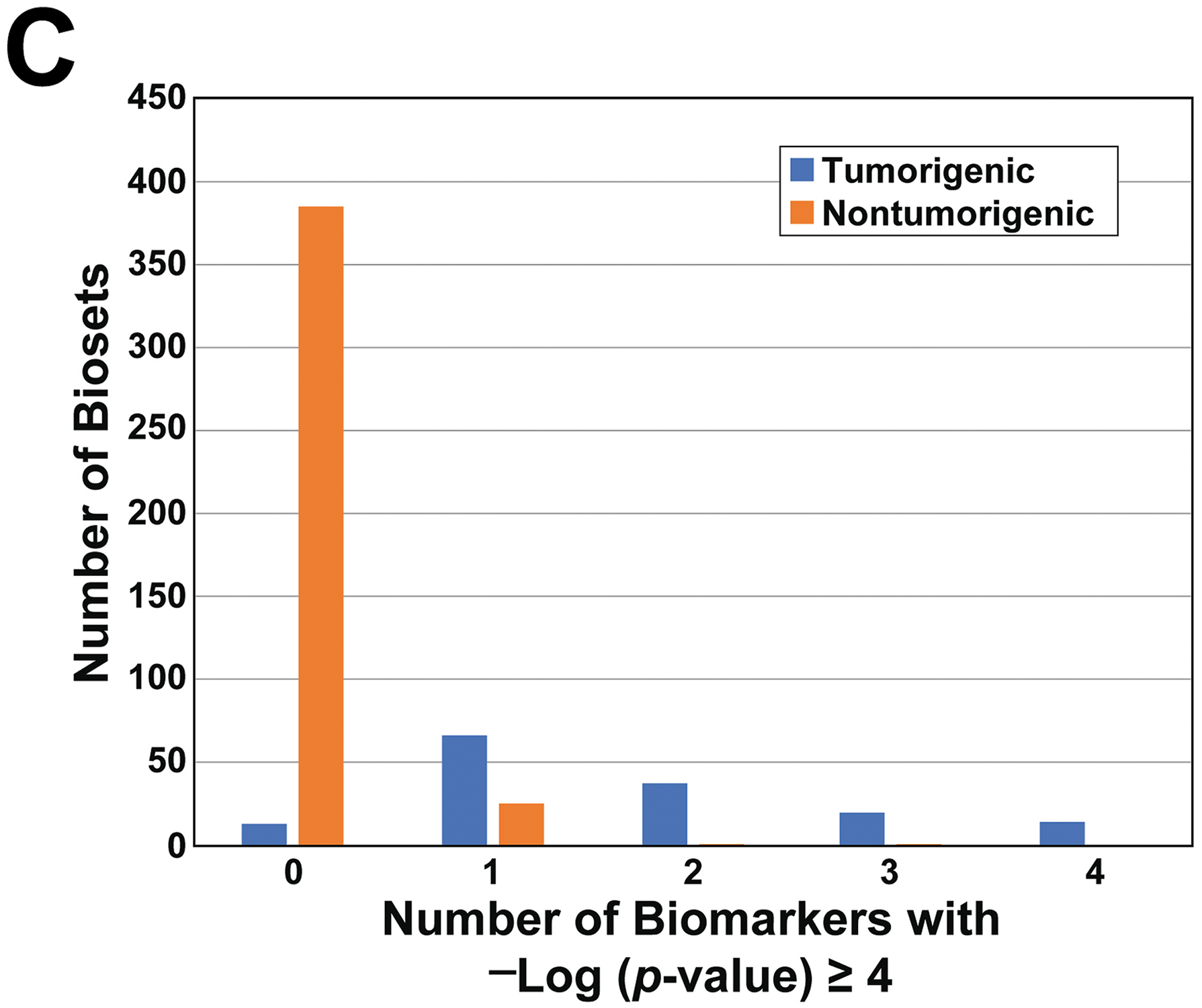
C. Relationship between tumorigenicity and number of MIEs activated. The number of tumorigenic and nontumorigenic biosets that activated 0 to 4 biomarkers are shown.
We also examined the chemicals that were in the DrugMatrix dataset. Figure S3 shows the activation of the MIEs across the comparisons. Most of the biosets from tumorigenic chem-dose pairs resulted in activation of one or more of the MIEs. Out of the 151 tumorigenic biosets, there were 19 representing 7 tumorigenic chemicals (carbimazole, carbon tetrachloride, diethylnitrosamine, fenofibrate, hydrazine, lovastatin, tamoxifen) that did not activate any of the MIEs. Eleven of the 19 biosets were from exposures of 24hrs or less. However, other time-dose comparisons for the same chemicals did activate one or more of the MIEs.
Using the assembled information, we determined how often each of the MIEs were activated in the chemical-dose-time conditions that were tumorigenic or nontumorigenic using a cutoff of -Log(p-value) ≥ 4 for activation of the biomarkers across the 562 comparisons in the dataset. Figure 4B shows that CAR was most frequently activated in the TG-GATES dataset followed by AhR > Genotoxicity > PPARα > ER > Cytotoxicity. While most of the MIEs were rarely activated in the nontumorigenic treatments, CAR was activated in over 20 comparisons. In the DrugMatrix dataset, the MIEs were perturbed in a similar order (CAR > AhR > PPARα > ER > Cytotoxicity > Genotoxicity; Figure S4) for the tumorigenic chemicals and again, out of all of the MIEs, CAR was activated the most in the nontumorigenic group. Any differences between TG-GATES and DrugMatrix may be due to differences in the sets of chemicals examined in each collection.
We also asked the question: for tumorigenic exposures, how many MIEs are simultaneously activated? For the TG-GATES dataset, most of the tumorigenic chemicals activated more than one MIE. While there were 66 biosets in which one MIE was activated, there were 71 total biosets that activated 2 or more MIEs (Figure 4C). In the nontumorigenic group, there were 25 biosets in which one of the MIEs was activated but activation of more than one MIE was rarely observed (2 biosets total). The DrugMatrix dataset exhibited similar trends; most of the tumorigenic biosets activated more than one MIE (Figure S5). These findings indicate that activation of more than one MIE is a common feature of tumorigenic chemicals.
Do any chemicals cause liver tumors independent of the 6 MIEs?
The entire microarray compendium was examined to determine if there were any tumorigenic chemicals that do not activate one or more of the MIEs. One set of chemicals that have been thought to cause liver cancer through a mechanism independent of the AOPs examined in our study are the statins (Cohen, 2010; Yamada, 2018). Statins work by inhibiting the rate-limiting enzyme in the cholesterol synthetic pathway, HMG-CoA reductase resulting in decreases in circulating cholesterol. There were five statins (atorvastatin, fluvastatin, lovastatin, pravastatin, simvastatin) examined. If the statins were working through a unique mechanism, it could be postulated that there would be no consistent activation of any of the MIEs. The MIEs were examined in exposures greater than 1d in biosets from either the DrugMatrix (Figure S6A) or TG-GATES (Figure S6B) studies. Under tumorigenic conditions in the DrugMatrix study, lovastatin, pravastatin, and simvastatin consistently activated AhR and CAR. Lovastatin activated cytotoxicity at 3d and genotoxicity at 3d and 5d at the highest dose. PPARα was rarely and only weakly activated. In contrast, simvastatin in the TG-GATES study primarily activated PPARα. A search of the literature indicated that CAR activation after statin exposure has not been examined in the rat liver. However, using trans-activation assays, fluvastatin and simvastatin were found to activate rat CAR (Kobayashi et al., 2005). Additionally, simvastatin induction of some genes was found to be PPARα-dependent in the mouse liver (Landrier et al., 2004). While discrepancies between the responses of simvastatin in the two studies remain, it appears that the statins examined under tumorigenic conditions cause increases in one or more of the 6 MIEs that could explain their tumorigenic effects. Thus, it does not appear that the statins cause liver tumors through a mechanism independent of the AOPs examined in the present study.
Out of the 38 tumorigenic chemicals examined, only acetamide and ethionine exhibited inconsistent or no activation of the MIEs. Acetamide is detected in some common foods, not considered to be genotoxic, and caused hepatocellular carcinomas after 52 weeks of exposure in female (888 mg/kg) and male (710 mg/kg) rats (CPD; https://toxnet.nlm.nih.gov/cpdb/chempages/ACETAMIDE.html; accessed June 6, 2019). Acetamide was examined in GSE53082 (Romer et al., 2014) at 3000 mg/kg/day for 14d. Despite the massive dose used, there were only 288 genes altered. Besides the lack of activity of the 6 MIEs, there was also no induction of cell proliferation assessed by the cell proliferation biomarker (data not shown). In a recent study, acetamide was examined at 30, 100, 300, and 1000 mg/kg/day for 7d in male rats (Nault et al., 2020). None of the doses exhibited significant correlation with any of the 6 biomarkers (data not shown). The evidence indicates that acetamide causes cancer through a mechanism independent of the 6 AOPs.
Ethionine is not considered to be genotoxic; hepatocellular carcinomas were increased in male rats after 34w at the only dose tested (65.4 mg/kg). In the TG-GATES study, ethionine was examined at 25, 80, and 250 mg/kg. Genotoxicity (1d at 250 mg/kg), AhR (7d at 80 mg/kg) or CAR (28d at 250 mg/kg) were activated in only one each of the 24 comparisons and near the threshold of significance. At the highest dose, cell proliferation assessed using our biomarker was activated at 7, 14, and 28d (data not shown). In another study (GSE31307; (Lee et al., 2013)), ethionine activated AhR at 200 mg/kg at 14d, again near the threshold of significance. In that study, the ethionine treatment group clearly separated from the other nongenotoxic and genotoxic chemicals examined by principle component analysis indicating a novel set of genes were perturbed. The authors speculated that ethionine causes hepatotoxicity through increases in oxidative stress. Overall, the evidence is weak that ethionine may act through one or more of the 6 MIEs. With the exception of acetamide and ethionine, the data does not support any of the tumorigenic chemicals acting independently of the 6 MIEs.
There is evidence that ethionine causes cancer through a mechanism that involves methyl deficiency and hypomethylation of DNA. Ethionine is structurally-related to methionine (an ethyl group is in place of the methyl group). Supplementation of ethionine-containing diets with methyl group donors including methionine reduced ethionine-induced carcinogenesis in rats (Farber, 1963). Previous experiments of rodent methyl deficiency showed that diets lacking methionine lead to a number of effects associated with hepatocarcinogenesis including rapid fat accumulation in the liver, lipid peroxidation, necrotic and apoptotic cell death, increased cell proliferation, depletion of intracellular methyl group pools, an imbalance of deoxynucleotide pool resulting in uracil incorporation into DNA, DNA strand breakage, and increased genome-wide and gene-specific hypomethylation (Christman et al., 1993; James et al., 2003; Wainfan et al., 1992). DNA from animals fed ethionine, like that of rats on a methyl-restricted diet was hypomethylated (Shivapurkar et al., 1984). Thus, the evidence points to a mechanism for ethionine-induced liver tumors that involves mechanisms similar to that of a methyl deficient diet.
To provide insights into the mechanism of ethionine-induced liver cancer, we determined whether a common set of regulated genes could be identified that are linked to pathways associated with cancer. We built a gene expression biomarker from the 14 biosets in our compendium derived from three studies of ethionine-treated rats. Genes were selected if they were altered in the same direction in 13 or 14 out of the 14 biosets. Figure S7A shows the consistent expression of the resulting 106 genes across the 14 biosets. The biomarker was compared to the gene lists derived from the TG-GATES and DrugMatrix studies using the Running Fisher test and then separated by tumorigenic class. Figure S7B shows the range of -Log(p-value)s across the tumorigenic and nontumorigenic comparisons. There was a striking enrichment of highly correlated biosets in the tumorigenic group. The top most correlated biosets were from exposures to prototypical cytotoxic agents at tumorigenic doses. Given that there is evidence that thioacetamide (Huang et al., 1999), methapyrilene (Ozden et al., 2015), carbon tetrachloride (Varela-Moreiras et al., 1995), and acetamidofluorene (Yerokun et al., 1994) exposures led to alterations in methyl pools and DNA hypomethylation in the rat liver, it is possible that the genes in the ethionine biomarker represent a core set of pathways that underly fundamental changes in hepatocyte growth related to changes in the methyl pool. Comparison of the biomarker to canonical pathways from Broad MSigDB showed overlaps with genes involved in solute carrier (SLC)-mediated transmembrane transport, the 55S mitochondrial ribosome, and the spliceosome or mRNA splicing (-Log(p-value) > 5; data not shown). Further work is needed to better understand how ethionine is inducing liver tumors.
The 6 biomarkers predict liver tumorigenicity.
ToxPi was used to score each chemical-dose-time comparison based on the ability to induce the MIEs. The ToxPi scores were based on the correlations (represented as -Log(p-value)s) between each microarray comparison and the 6 biomarkers. ToxPi scores are shown for the TG-GATES study (Figure 5, top). The tumorigenic treatments had scores that were mostly higher than those from treatments that were nontumorigenic. The heatmap (Figure 5, bottom) shows that the highest scores reflect higher correlations to a greater number of biomarkers. Many of the top scores in the nontumorigenic group exhibited strong correlations to the CAR biomarker. This observation is consistent with the results in Figure 4B showing that activation of CAR does not necessarily lead to liver cancer. The differences in the scores between tumorigenic and nontumorigenic groups were broken out by time of treatment (Figure S8A). The top ToxPi score for the nontumorigenic chemicals was relatively consistent across the time points. ToxPi scores were also generated for biosets from the DrugMatrix study. Like the TG-GATES study, the tumorigenic treatments had scores that were mostly higher than those from the nontumorigenic treatments, although there was clearly a greater overlap between the two classes compared to the TG-GATES study (Figure S8B).
Figure 5. Distribution of ToxPi scores across tumorigenic and nontumorigenic chemicals.
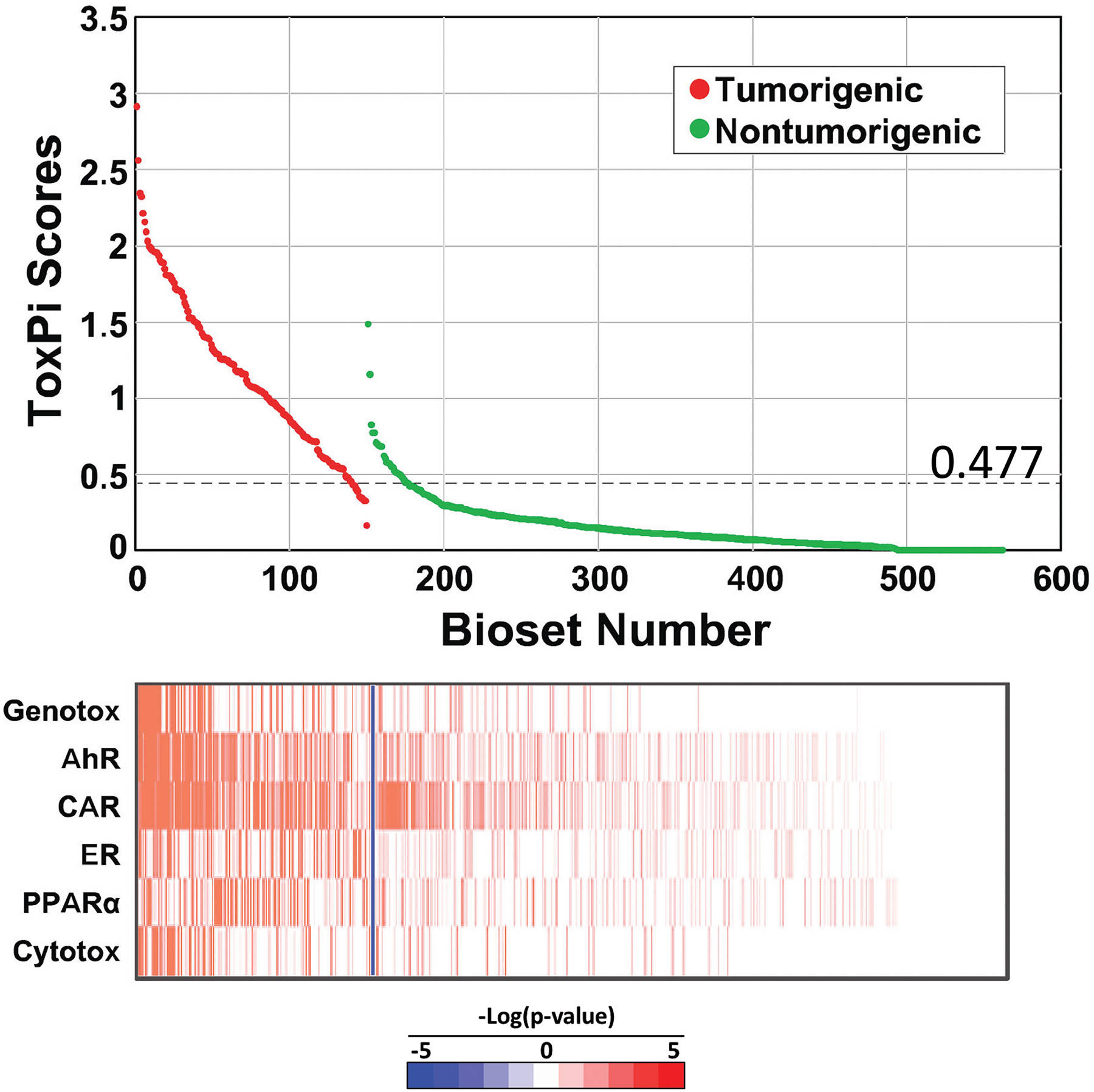
Distribution of ToxPi scores between tumorigenic and nontumorigenic chem-dose-times in the TG-GATES study. ToxPi scores were derived from the -Log(p-value)s of the 6 biomarkers compared to each bioset. (Top) Distribution of scores. The optimal ROC threshold is indicated. (Bottom) Heatmap of -Log(p-value)s of the biomarkers. The blue line separates the tumorigenic and nontumorigenic exposure conditions.
ToxPi scores from different training sets were used to predict liver tumorigenesis in three test sets of independent chemicals using a DeLong, Delong and Clarke-Pearson receiver operating curve (ROC) analysis. The TG-GATES dataset was divided into a training set consisting of 38 chemicals including 9 that were tumorigenic and a test set consisting of 37 independent chemicals including 9 that were tumorigenic. In the first test, the ToxPi scores from the training set were used to predict tumorigenesis in the test set. Tests were carried out by individual biosets of chem-dose-time comparisons or by chemical (see Methods for description). The optimal ROC threshold for the training set occurred at a ToxPi score of 0.477. Applying this threshold to the test set resulted in 8 false negatives, 6 false positives, 90% sensitivity, 97% specificity, and a balanced accuracy of 93% (Table 2).
Table 2.
Predictive accuracies using the 6 biomarkers.
| Unit of Prediction | Training Set | Test Set | ToxPi Threshold Used | Total Number of Biosets or Chemicals Examined | TP | TN | FP | FN | Sensitivity | Specificity | PPV | NPV | Balanced Accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bioset | TG-GATES Training | TG-GATES Test | 0.4765 | 270 | 70 | 186 | 6 | 8 | 0.897 | 0.969 | 0.921 | 0.959 | 0.933 |
| Chemical | TG-GATES Training | TG-GATES Test | 0.4765 | 37 | 9 | 24 | 4 | 0 | 1.000 | 0.857 | 0.692 | 1.000 | 0.929 |
| Bioset | TG-GATES All | DrugMatrix GE Healthcare | 0.4765 | 179 | 74 | 55 | 44 | 6 | 0.925 | 0.556 | 0.627 | 0.902 | 0.740 |
| Chemical | TG-GATES All | DrugMatrix GE Healthcare | 0.4765 | 88 | 35 | 24 | 29 | 0 | 1.000 | 0.453 | 0.547 | 1.000 | 0.726 |
| Bioset | DrugMatrix GE Healthcare | TG-GATES All | 0.6329 | 562 | 119 | 402 | 10 | 31 | 0.793 | 0.976 | 0.922 | 0.928 | 0.885 |
| Chemical | DrugMatrix GE Healthcare | TG-GATES All | 0.6329 | 75 | 17 | 51 | 6 | 1 | 0.944 | 0.895 | 0.739 | 0.981 | 0.920 |
Unit refers to the comparisons used to determine the balanced accuracies. ToxPi thresholds were calculated using receiver-operating curves as described in the Methods.
In the second test, ToxPi values derived from the TG-GATES training dataset were used to identify tumorigenic chemical-dose combinations in the DrugMatrix chemicals. Applying the threshold of 0.477 to the DrugMatrix set resulted in 6 false negatives, 44 false positives, 93% sensitivity, 56% specificity, and a balanced accuracy of 74%. The balanced accuracy on a chemical basis was similar (73%).
ToxPi values derived from the DrugMatrix dataset were used to predict the TG-GATES chemicals. The optimal ROC threshold occurred at a ToxPi score of 0.633, higher than that derived using the TG-GATES data. Applying this threshold to the TG-GATES dataset resulted in a large number of false negatives (31), 10 false positives, 79% sensitivity, 98% specificity, and a balanced accuracy of 88%. On a chemical basis, the threshold derived from the DrugMatrix dataset performed well resulting in 94% sensitivity, 89% specificity, and a balanced accuracy of 92%. Thus, our method performs well when using the threshold derived from the TG-GATES training set applied to the test set and when the DrugMatrix threshold was applied to test the TG-GATES dataset.
Summary
In this study, we developed a novel MIE-driven strategy for prediction of chemicals that cause liver cancer in rodents. The strategy utilized a set of 6 gene expression biomarkers, 5 of which were characterized previously with balanced accuracies between 92% and 98%. A sixth biomarker that predicts cytotoxicity is described in the present study and was found to be accurate at identifying chemicals that cause hepatocyte damage (96% balanced accuracy). Furthermore, the biomarker was able to identify chemical exposures in which a number of hallmarks of cytotoxicity were simultaneously occuring including pathological changes (necrosis, single cell necrosis), increases in ALT and/or AST, and increases in the expression of cell cycle genes that are part of a previously characterized cell proliferation biomarker, as a response to replace hepatocytes by induction of regenerative hyperplasia.
The biomarkers allowed us to address a number of incompletely answered questions about chemical carcinogenesis in the rat liver. We found that in our datasets CAR is activated more often than other MIEs, but that activation does not necessarily lead to liver tumors as many treatments that were not tumorigenic resulted in CAR activation. These results indicate that CAR must be strongly activated for liver tumor induction to occur. We also found that most tumorigenic exposure conditions activate more than one MIE and in some cases up to 4 out of the 6. Thus, to label a tumorigenic chemical as an activator of a single MIE may, for the majority of chemicals, be too simplistic. Given that many of the exposure conditions were carried out at relatively high doses which may lead to multiple pathways being activated, a dose-response analysis of the biomarker genes would be useful to determine what MIE is activated at the lowest tumorigenic dose.
We were also able to determine if there were any tumorigenic chemicals that did not activate one or more of the 6 MIEs. Out of the 38 tumorigenic chemicals examined, we could find only two chemicals (acetamide, ethionine) that did not consistently induce one or more of the MIEs. Thus, our methods were able to identify 95% of all liver tumorigens. For ethionine, we found a core set of genes that overlapped with those induced by a set of cytotoxic agents. All of these chemicals deplete methyl pools resulting in hypomethylation and for ethionine could represent a new AOP that is not covered by the 6 MIEs. Our results indicate that the vast majority of chemicals that comprise our compendium cause liver tumors through one or more of the 6 MIEs, but that one chemical may cause cancer through an incompletely characterized AOP involving depletion of methyl pools. Further work is needed to determine if environmentally-relevant chemicals not included in the compendium, also cause liver tumors through these or other MIEs.
Prediction of the MIE and thus the AOP through which a compound alters early indicators of cancer risk will facilitate hazard assessment during early (preclinical) testing and will allow prioritization of chemicals with respect to an understanding of their potential to cause cancer in chronic bioassays. Chemicals could then be assessed relative to whether the AOP has human relevance. Our MIE-driven strategy is flexible in that as additional MIEs are characterized they can be included in the ToxPi analysis. Our approach is complementary to the approach by Podtelezhnikov et al (Podtelezhnikov, 2020) who used seed genes and a co-expression strategy. Both approaches demonstrate overlap with an unsupervised approach to identifying MIE of interest (Sutherland, et al., 2018). While only microarray information was used to identify liver tumorigens in the present study, ToxPi could be used to incorporate additional data types including liver to body weights and clinical chemistry measures as in our previous study which together were very predictive to identify liver tumorigens (Rooney, et al., 2018a). Our strategy provides a scientific basis for applying the methods to other tissues that are the targets of rat carcinogens.
The biomarkers that we have characterized in mice (Oshida, et al., 2015a; Oshida, et al., 2015b; Oshida, et al., 2015c; Oshida, et al., 2016; Rooney, 2018; Rooney et al., 2018b), rats (Rooney, et al., 2018a), and humans (Rooney et al., 2018c; Ryan et al., 2016) have the potential to be widely used to identify the underlying mechanisms underlying toxicity. For example, using the BSCE environment, our set of mouse biomarkers were used by another group to identify CAR as the major MIE that leads to sedaxane-induced liver tumors (Peffer et al., 2018). While we have provided the biomarker genes and associated fold-change values as part of this and past studies (Rooney, et al., 2018a), there is no publicly-available version of the Running Fisher test and supporting platform to compare large sets of gene lists to each other. Efforts should be made to create and test a publicly available version of the Running Fisher test that can recapitulate the ability to make predictions of MIE activation that accurately replicate prior results. The Running Fisher test would then need to be embedded into a publicly available interface for comparison of gene lists. An example of how this interface might look comes from a web tool that allows comparison of uploaded microarray data with the TGx-DNA Damage Induction (DDI) biomarker used as an indicator of DNA damage in human cells (Jackson et al., 2017). Each uploaded gene list is compared to the expression of the 64 TGx-DDI biomarker genes across the 28 reference treatments used to create the biomarker. The analysis uses a number of methods for comparison and gives a summary classification probability of whether the treatment causes DNA damage. In principle, a platform containing a set of comparison tools including the Running Fisher test would allow simultaneous comparison of any gene list generated by chemical exposure to a battery of characterized biomarkers. Having information about the AOPs activated by short-term exposures to a chemical including any biological thresholds breached that would be predicted to cause liver cancer (Hill et al., 2020) could assist chemical and pharmaceutical industries to identify human-irrelevant AOPs that support waivers for conducting a full 2 year bioassay. From the perspective of the EPA, the approach has the potential to substantially reduce the number of animals used in chemical testing and help move us towards the EPA Administrator’s goal of complete elimination of animal testing by 2035 (AR, 2019).
Supplementary Material
Supplemental File 1. Excel document.
Contains 1) information about the chemicals from the TG-GATES and GSE8858 datasets evaluated in our study including liver tumor response and alteration of biomarkers, 2) biosets from microarray experiments in which cytotoxicity status was known, and 3) identification of putative cytotoxic chemical exposure conditions in the rat liver compendium using the cytotoxicity biomarker.
Supplemental File 2. Word document.
Contains
1) Fig. S1. Relationships between the cytotoxicity biomarker and cell proliferation biomarker correlations for cytotoxic chemicals.
2) Fig S2. Representation of genes from biomarker signatures on the TXG-MAP.
3) Fig. S3. Activation of liver cancer MIEs across the DrugMatrix dataset.
4) Fig. S4. Activation of the 6 biomarkers across the DrugMatrix dataset.
5) Fig. S5. Relationship between tumorigenicity and number of MIEs activated in the DrugMatrix dataset.
6) Fig. S6. Statins activate one or more of the 6 MIEs.
7) Fig. S7. The ethionine biomarker exhibits greater correlation to tumorigenic chemical exposure conditions.
8) Fig. S8. Distribution of ToxPi scores across time of exposure.
Acknowledgements
We thank the TG-GATES consortium for making their data public and Drs. Hisham El-Masri and Urmilla Kodavanti for critical review of this manuscript. Funding for this study came from the U.S. EPA Office of Research and Development.
Footnotes
Publisher's Disclaimer: Disclaimer: The information in this document has been funded in part by the U.S. Environmental Protection Agency. It has been subjected to review by the National Health and Environmental Effects Research Laboratory and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- (1997). The Physicians’ Desk Reference, 51 ed. Thomson PDR. [Google Scholar]
- (2017). AOP Wiki. Available at: https://aopwiki.org. Accessed September 1, 2017.
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, et al. (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environmental toxicology and chemistry / SETAC 29(3), 730–41. [DOI] [PubMed] [Google Scholar]
- AR W (2019). Directive to prioritize efforts to reduce animal testing. https://www.epa.gov/sites/production/files/2019-09/documents/image2019-09-09-231249.pdf.
- Benigni R, Netzeva TI, Benfenati E, Bossa C, Franke R, Helma C, Hulzebos E, Marchant C, Richard A, Woo YT, et al. (2007). The expanding role of predictive toxicology: an update on the (Q)SAR models for mutagens and carcinogens. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 25(1), 53–97. [DOI] [PubMed] [Google Scholar]
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, and Farland W (2006). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Critical reviews in toxicology 36(10), 781–92. [DOI] [PubMed] [Google Scholar]
- Bucher JR, and Portier C (2004). Human carcinogenic risk evaluation, Part V: The national toxicology program vision for assessing the human carcinogenic hazard of chemicals. Toxicological sciences : an official journal of the Society of Toxicology 82(2), 363–6. [DOI] [PubMed] [Google Scholar]
- Budinsky RA, Schrenk D, Simon T, Van den Berg M, Reichard JF, Silkworth JB, Aylward LL, Brix A, Gasiewicz T, Kaminski N, et al. (2014). Mode of action and dose-response framework analysis for receptor-mediated toxicity: The aryl hydrocarbon receptor as a case study. Critical reviews in toxicology 44(1), 83–119. [DOI] [PubMed] [Google Scholar]
- CDC (2017). National Center for Health Statistics: Deaths and Mortality. Available at: https://www.cdc.gov/nchs/fastats/deaths.htm. Accessed May 17, 2017.
- Christman JK, Sheikhnejad G, Dizik M, Abileah S, and Wainfan E (1993). Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis 14(4), 551–7. [DOI] [PubMed] [Google Scholar]
- Cohen SM (2010). Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays: the two-year bioassay is no longer necessary. Toxicologic pathology 38(3), 487–501. [DOI] [PubMed] [Google Scholar]
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, and Klaunig JE (2014). Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Critical reviews in toxicology 44(1), 1–49. [DOI] [PubMed] [Google Scholar]
- Edwards SW, Tan YM, Villeneuve DL, Meek ME, and McQueen CA (2016). Adverse Outcome Pathways-Organizing Toxicological Information to Improve Decision Making. The Journal of pharmacology and experimental therapeutics 356(1), 170–81. [DOI] [PubMed] [Google Scholar]
- Elcombe CR, Peffer RC, Wolf DC, Bailey J, Bars R, Bell D, Cattley RC, Ferguson SS, Geter D, Goetz A, et al. (2014). Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Critical reviews in toxicology 44(1), 64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H, Gmuender H, Bandenburg A, and Ahr HJ (2008). Prediction of a carcinogenic potential of rat hepatocarcinogens using toxicogenomics analysis of short-term in vivo studies. Mutation research 637(1–2), 23–39. [DOI] [PubMed] [Google Scholar]
- Farber E (1963). ETHIONINE CARCINOGENESIS. Advances in cancer research 7, 383–474. [DOI] [PubMed] [Google Scholar]
- Felter SP, Foreman JE, Boobis A, Corton JC, Doi AM, Flowers L, Goodman J, Haber LT, Jacobs A, Klaunig JE, et al. (2017). Human relevance of rodent liver tumors: Key insights from a Toxicology Forum workshop on nongenotoxic modes of action. Regulatory toxicology and pharmacology : RTP 92, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felter SP, Foreman JE, Boobis A, Corton JC, Doi AM, Flowers L, Goodman J, Haber LT, Jacobs A, Klaunig JE, et al. (2018). Human relevance of rodent liver tumors: Key insights from a Toxicology Forum workshop on nongenotoxic modes of action. Regulatory toxicology and pharmacology : RTP 92, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielden MR, Brennan R, and Gollub J (2007). A gene expression biomarker provides early prediction and mechanistic assessment of hepatic tumor induction by nongenotoxic chemicals. Toxicological sciences : an official journal of the Society of Toxicology 99(1), 90–100. [DOI] [PubMed] [Google Scholar]
- Glaab WE, H. D., He YD, Gerhold DL, Baily WJ, Beare C, Erdos Z, Lane P, Michna L, Muniappa N, Lawrence JW, Sina JF, Skopek TR, Sistare FD (2018). Universal toxicity gene signatures for early identification of drug-induced tissue injuries in rats. The Toxicologist, 1265. [DOI] [PubMed] [Google Scholar]
- Gold LS, Manley NB, Slone TH, Rohrbach L, and Garfinkel GB (2005). Supplement to the Carcinogenic Potency Database (CPDB): results of animal bioassays published in the general literature through 1997 and by the National Toxicology Program in 1997–1998. Toxicological sciences : an official journal of the Society of Toxicology 85(2), 747–808. [DOI] [PubMed] [Google Scholar]
- Gusenleitner D, Auerbach SS, Melia T, Gomez HF, Sherr DH, and Monti S (2014). Genomic models of short-term exposure accurately predict long-term chemical carcinogenicity and identify putative mechanisms of action. PloS one 9(7), e102579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill T 3rd, Nelms MD, Edwards SW, Martin M, Judson R, Corton JC, and Wood CE (2017). Editor’s Highlight: Negative Predictors of Carcinogenicity for Environmental Chemicals. Toxicological sciences : an official journal of the Society of Toxicology 155(1), 157–169. [DOI] [PubMed] [Google Scholar]
- Hill T 3rd, Rooney J, Abedini I, El-Masri H, Sistare FD, Wood CE, and Corton JC (2020). Gene Expression Thresholds Predictive of Rat Liver Tumorigens in Short-Term Assays. Submitted. [Google Scholar]
- Huang ZZ, Mato JM, Kanel G, and Lu SC (1999). Differential effect of thioacetamide on hepatic methionine adenosyltransferase expression in the rat. Hepatology (Baltimore, Md.) 29(5), 1471–8. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Nakatsu N, Yamashita T, Ono A, Ohno Y, Urushidani T, and Yamada H (2015). Open TG-GATEs: a large-scale toxicogenomics database. Nucleic acids research 43(Database issue), D921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MA, Yang L, Lea I, Rashid A, Kuo B, Williams A, Lyn Yauk C, and Fostel J (2017). The TGx-28.65 biomarker online application for analysis of transcriptomics data to identify DNA damage-inducing chemicals in human cell cultures. Environmental and molecular mutagenesis 58(7), 529–535. [DOI] [PubMed] [Google Scholar]
- James SJ, Pogribny IP, Pogribna M, Miller BJ, Jernigan S, and Melnyk S (2003). Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. The Journal of nutrition 133(11 Suppl 1), 3740s–3747s. [DOI] [PubMed] [Google Scholar]
- Knudsen TB, Keller DA, Sander M, Carney EW, Doerrer NG, Eaton DL, Fitzpatrick SC, Hastings KL, Mendrick DL, Tice RR, et al. (2015). FutureTox II: in vitro data and in silico models for predictive toxicology. Toxicological sciences : an official journal of the Society of Toxicology 143(2), 256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka Y, Iwazaki N, Nakajo I, Hosokawa M, Negishi M, and Chiba K (2005). Identification of HMG-CoA reductase inhibitors as activators for human, mouse and rat constitutive androstane receptor. Drug metabolism and disposition: the biological fate of chemicals 33(7), 924–9. [DOI] [PubMed] [Google Scholar]
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, et al. (2010). Ontology-based meta-analysis of global collections of high-throughput public data. PloS one 5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrier JF, Thomas C, Grober J, Duez H, Percevault F, Souidi M, Linard C, Staels B, and Besnard P (2004). Statin induction of liver fatty acid-binding protein (L-FABP) gene expression is peroxisome proliferator-activated receptor-alpha-dependent. The Journal of biological chemistry 279(44), 45512–8. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Yum YN, Kim SC, Kim Y, Lim J, Lee WJ, Koo KH, Kim JH, Kim JE, Lee WS, et al. (2013). Distinguishing between genotoxic and non-genotoxic hepatocarcinogens by gene expression profiling and bioinformatic pathway analysis. Scientific reports 3, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangipudy RS, Chanda S, and Mehendale HM (1995). Tissue repair response as a function of dose in thioacetamide hepatotoxicity. Environ Health Perspect 103(3), 260–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir DC, and Howard PH (2006). Are there other persistent organic pollutants? A challenge for environmental chemists. Environmental science & technology 40(23), 7157–66. [DOI] [PubMed] [Google Scholar]
- Nault R, Bals B, Teymouri F, Black MB, Andersen ME, McMullen PD, Krishnan S, Kuravadi N, Paul N, Kumar S, et al. (2020). A toxicogenomic approach for the risk assessment of the food contaminant acetamide. Toxicology and applied pharmacology 388, 114872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie AY, McMillian M, Parker JB, Leone A, Bryant S, Yieh L, Bittner A, Nelson J, Carmen A, Wan J, et al. (2006). Predictive toxicogenomics approaches reveal underlying molecular mechanisms of nongenotoxic carcinogenicity. Molecular carcinogenesis 45(12), 914–33. [DOI] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Jones C, Moore T, Hester S, Nesnow S, Auerbach S, Geter DR, Aleksunes LM, Thomas RS, et al. (2015a). Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nuclear receptor signaling 13, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Gonzalez FJ, Aleksunes LM, Klaassen CD, and Corton JC (2015b). Screening a mouse liver gene expression compendium identifies modulators of the aryl hydrocarbon receptor (AhR). Toxicology 336, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Rosen M, Abbott B, Lau C, Guo G, Aleksunes LM, Klaassen C, et al. (2015c). Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) in a mouse liver gene expression compendium. PloS one 10(2), e0112655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Waxman DJ, and Corton JC (2016). Disruption of STAT5b-Regulated Sexual Dimorphism of the Liver Transcriptome by Diverse Factors Is a Common Event. PloS one 11(3), e0148308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozden S, Turgut Kara N, Sezerman OU, Durasi IM, Chen T, Demirel G, Alpertunga B, Chipman JK, and Mally A (2015). Assessment of global and gene-specific DNA methylation in rat liver and kidney in response to non-genotoxic carcinogen exposure. Toxicology and applied pharmacology 289(2), 203–12. [DOI] [PubMed] [Google Scholar]
- Peffer RC, Cowie DE, Currie RA, and Minnema DJ (2018). Sedaxane-Use of Nuclear Receptor Transactivation Assays, Toxicogenomics, and Toxicokinetics as Part of a Mode of Action Framework for Rodent Liver Tumors. Toxicological sciences : an official journal of the Society of Toxicology 162(2), 582–598. [DOI] [PubMed] [Google Scholar]
- Podtelezhnikov AA, Monroe JJ, Aslamkhan AG, Pearson K, Qin C, Tamburino AM, Loboda AP, Glaab WE, Sistare FD, & Tanis KQ (2020). Quantitative Transcriptional Biomarkers of Xenobiotic Receptor Activation in Rat Liver for the Early Assessment of Drug Safety Liabilities. Toxicological sciences : an official journal of the Society of Toxicology 175(1), 98–112. [DOI] [PubMed] [Google Scholar]
- REACH (2008). European Chemical Agency -- List of Pre-registered Substances. Available at: http://apps.echa.europa.eu/preregistered/pre-registered-sub.aspx. Accessed May 17, 2017.
- Reif DM, Martin MT, Tan SW, Houck KA, Judson RS, Richard AM, Knudsen TB, Dix DJ, and Kavlock RJ (2010). Endocrine profiling and prioritization of environmental chemicals using ToxCast data. Environ Health Perspect 118(12), 1714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer M, Eichner J, Metzger U, Templin MF, Plummer S, Ellinger-Ziegelbauer H, and Zell A (2014). Cross-platform toxicogenomics for the prediction of non-genotoxic hepatocarcinogenesis in rat. PloS one 9(5), e97640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney J, Chorley B, Corton JC (2018). A Gene Expression Biomarker Identifies Chemicals and Other Factors in the Mouse Liver That Modulate Sterol Regulatory Element Binding Protein (SREBP) Highlighting Differences in Targeted Regulation of Cholesterogenic and Lipogenic Genes. Submitted. [Google Scholar]
- Rooney J, Hill T 3rd, Qin C, Sistare FD, and Corton JC (2018a). Adverse outcome pathway-driven identification of rat liver tumorigens in short-term assays. Toxicology and applied pharmacology 356, 99–113. [DOI] [PubMed] [Google Scholar]
- Rooney J, Oshida K, Vasani N, Vallanat B, Ryan N, Chorley BN, Wang X, Bell DA, Wu KC, Aleksunes LM, et al. (2018b). Activation of Nrf2 in the liver is associated with stress resistance mediated by suppression of the growth hormone-regulated STAT5b transcription factor. PloS one 13(8), e0200004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JP, Chorley B, Kleinstreuer N, and Corton JC (2018c). Identification of Androgen Receptor Modulators in a Prostate Cancer Cell Line Microarray Compendium. Toxicological sciences : an official journal of the Society of Toxicology 166(1), 146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan N, Chorley B, Tice RR, Judson R, and Corton JC (2016). Moving Toward Integrating Gene Expression Profiling Into High-Throughput Testing: A Gene Expression Biomarker Accurately Predicts Estrogen Receptor alpha Modulation in a Microarray Compendium. Toxicological sciences : an official journal of the Society of Toxicology 151(1), 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivapurkar N, Wilson MJ, and Poirier LA (1984). Hypomethylation of DNA in ethionine-fed rats. Carcinogenesis 5(8), 989–92. [DOI] [PubMed] [Google Scholar]
- Sistare FD, Morton D, Alden C, Christensen J, Keller D, Jonghe SD, Storer RD, Reddy MV, Kraynak A, Trela B, et al. (2011). An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: support for a proposal to modify current regulatory guidelines. Toxicologic pathology 39(4), 716–44. [DOI] [PubMed] [Google Scholar]
- Sutherland JJ, Stevens JL, Johnson K, Elango N, Webster YW, Mills BJ, and Robertson DH (2019a). A novel open access web portal for integrating mechanistic and toxicogenomic study results. Toxicological sciences : an official journal of the Society of Toxicology doi: 10.1093/toxsci/kfz101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland JJ, Stevens JL, Johnson K, Elango N, Webster YW, Mills BJ, and Robertson DH (2019b). A Novel Open Access Web Portal for Integrating Mechanistic and Toxicogenomic Study Results. Toxicological sciences : an official journal of the Society of Toxicology 170(2), 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland JJ, Webster YW, Willy JA, Searfoss GH, Goldstein KM, Irizarry AR, Hall DG, and Stevens JL (2018). Toxicogenomic module associations with pathogenesis: a network-based approach to understanding drug toxicity. The pharmacogenomics journal 18(3), 377–390. [DOI] [PubMed] [Google Scholar]
- Svoboda D, S. T., Auerbach SS (2019). An overview of the National Toxicology Program’s Toxicogenomic Applications: DrugMatrix and ToxFX. Advances in Computational Toxicology: Methodologies and Applications in Regulatory Sciences Chapter 8. [Google Scholar]
- Uehara T, Hirode M, Ono A, Kiyosawa N, Omura K, Shimizu T, Mizukawa Y, Miyagishima T, Nagao T, and Urushidani T (2008). A toxicogenomics approach for early assessment of potential non-genotoxic hepatocarcinogenicity of chemicals in rats. Toxicology 250(1), 15–26. [DOI] [PubMed] [Google Scholar]
- Uehara T, Minowa Y, Morikawa Y, Kondo C, Maruyama T, Kato I, Nakatsu N, Igarashi Y, Ono A, Hayashi H, et al. (2011). Prediction model of potential hepatocarcinogenicity of rat hepatocarcinogens using a large-scale toxicogenomics database. Toxicology and applied pharmacology 255(3), 297–306. [DOI] [PubMed] [Google Scholar]
- USEPA (2004). What is the TSCA Chemical Substance Inventory? Available at: https://www.epa.gov/tsca-inventory/about-tsca-chemical-substance-inventory. Accessed May 17 2017.
- USEPA (2005). Guidlelines for Carcinogen Risk Assessment. 166. [Google Scholar]
- Varela-Moreiras G, Alonso-Aperte E, Rubio M, Gasso M, Deulofeu R, Alvarez L, Caballeria J, Rodes J, and Mato JM (1995). Carbon tetrachloride-induced hepatic injury is associated with global DNA hypomethylation and homocysteinemia: effect of S-adenosylmethionine treatment. Hepatology (Baltimore, Md.) 22(4 Pt 1), 1310–5. [PubMed] [Google Scholar]
- Wainfan E, and Poirier LA (1992). Methyl groups in carcinogenesis: effects on DNA methylation and gene expression. Cancer research 52(7 Suppl), 2071s–2077s. [PubMed] [Google Scholar]
- Waters MD, Jackson M, and Lea I (2010). Characterizing and predicting carcinogenicity and mode of action using conventional and toxicogenomics methods. Mutation research 705(3), 184–200. [DOI] [PubMed] [Google Scholar]
- Wilson CL, and Miller CJ (2005). Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics (Oxford, England) 21(18), 3683–5. [DOI] [PubMed] [Google Scholar]
- Xu X, So JS, Park JG, and Lee AH (2013). Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Seminars in liver disease 33(4), 301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager JD, and Liehr JG (1996). Molecular mechanisms of estrogen carcinogenesis. Annual review of pharmacology and toxicology 36, 203–32. [DOI] [PubMed] [Google Scholar]
- Yamada F, Sumida K, Uehara T, Morikawa Y, Yamada H, Urushidani T, and Ohno Y (2013). Toxicogenomics discrimination of potential hepatocarcinogenicity of non-genotoxic compounds in rat liver. Journal of applied toxicology : JAT 33(11), 1284–93. [DOI] [PubMed] [Google Scholar]
- Yamada T (2018). Case examples of an evaluation of the human relevance of the pyrethroids/pyrethrins-induced liver tumours in rodents based on the mode of action. Toxicology research 7(4), 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerokun T, Lyn-Cook BD, and Ringer DP (1994). Hypomethylation of the rat aryl sulfotransferase IV gene and amplification of a DNA sequence during multistage 2-acetylaminofluorene hepatocarcinogenesis. Chemico-biological interactions 92(1–3), 363–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1. Excel document.
Contains 1) information about the chemicals from the TG-GATES and GSE8858 datasets evaluated in our study including liver tumor response and alteration of biomarkers, 2) biosets from microarray experiments in which cytotoxicity status was known, and 3) identification of putative cytotoxic chemical exposure conditions in the rat liver compendium using the cytotoxicity biomarker.
Supplemental File 2. Word document.
Contains
1) Fig. S1. Relationships between the cytotoxicity biomarker and cell proliferation biomarker correlations for cytotoxic chemicals.
2) Fig S2. Representation of genes from biomarker signatures on the TXG-MAP.
3) Fig. S3. Activation of liver cancer MIEs across the DrugMatrix dataset.
4) Fig. S4. Activation of the 6 biomarkers across the DrugMatrix dataset.
5) Fig. S5. Relationship between tumorigenicity and number of MIEs activated in the DrugMatrix dataset.
6) Fig. S6. Statins activate one or more of the 6 MIEs.
7) Fig. S7. The ethionine biomarker exhibits greater correlation to tumorigenic chemical exposure conditions.
8) Fig. S8. Distribution of ToxPi scores across time of exposure.