

Abstract
Glycoproteins on the cell surface are essential for various cellular activities including cell-cell communication, cell-matrix interaction, and cell signaling. Alterations of glycosylation are correlated with many diseases such as cancer and infectious diseases. However, it is greatly challenging to systematically and site-specially analyze glycoproteins only located on cell surface because of the heterogeneity of glycans, the low abundance of many surface glycoproteins and the requirement of effective methods to separate surface glycoproteins. In this chapter, we briefly review existing mass spectrometry (MS)-based methods for global analysis of surface glycoproteins. Then we discuss an effective method integrating metabolic labeling, click and enzymatic reactions, and MS-based proteomics to comprehensively and site-specifically investigate cell surface N-glycoproteins. A detailed protocol for this method is also included. In combination with quantitative proteomics, we applied this method to quantify cell surface N-glycoproteins and study the relationship between cell invasiveness and N-sialoglycoproteins on the cell surface. Considering the importance of surface glycoproteins, this method can be extensively applied to advance glycoscience, which leads to a better understanding of the molecular mechanisms of human diseases, and the discovery of surface glycoproteins as biomarkers for disease detection.
Keywords: Cell surface, N-Glycoproteins, Metabolic labeling, Bioorthogonal chemistry, MS-based proteomics, Glycoprotein quantification
1. INTRODUCTION
Protein glycosylation is normally an enzymatic process, in which the anomeric hydroxyl group of a glycan is covalently bound to proteins. It is one of the most important protein modifications (Ohtsubo and Marth, 2006). Different side chains of the amino acid residues could be covalently attached to glycans and two types of the most common glycosylation are protein N- and O-glycosylation (Moremen et al., 2012). A growing number of evidence has indicated that glycosylation has fundamental significance for cells and is vital for protein folding and trafficking (Jayaprakash and Surolia, 2017). The interactions between glycans and proteins affect protein conformation and properties, which may regulate protein activity (Abu-Qarn et al., 2008). For protein N-glycosylation in eukaryotic cells, the initial N-glycan consisting of 14 monosaccharide residues (Glu3Man9GlcNAc2) is “en bloc” transferred to nascent polypeptides in the ER, followed by various glycan modifications with different enzymes in the ER and Golgi apparatus. This process determines the folding and trafficking of proteins (Aebi et al., 2010).
Glycoproteins located on the cell surface are of great importance to regulate many cellular activities, including cell-cell and cell-matrix interactions, cell signaling and cellular immune responses (Boscher et al., 2011; Rudd et al., 2001). Extensive studies have demonstrated that aberrant glycosylation is often correlated with diseases (Stowell et al., 2015; Veillon et al., 2018). Compared with normal cells, cancer cells display a wide range of aberrant glycosylation (Christiansen et al., 2014; Kim and Varki, 1997). Sialyation plays an important role in cellular adhesion and migration, and it increases (usually α2,6‑ and α2,3‑linked sialylation) in cancer cells because of the changed expression of glycosyltransferases (Dall’Olio and Chiricolo, 2001). Glycans on the cell surface also regulate different types of immune responses (Ogata et al., 1992). Changes in glycosylation on the cell surface disrupt the interactions between glycans and lectins, which helps cancer cells escape from the immune surveillance (Macauley et al., 2014; Rabinovich and Toscano, 2009). Therefore, characteristic alterations of glycosylation in many diseases can be used as biomarkers for diagnostic and prognostic purposes (Kailemia et al., 2017). Meanwhile, antibodies and inhibitors can be designed to specifically target the disease-associated glycan antigens for therapeutic treatment (Dalziel et al., 2014; Lavrsen et al., 2013).
Considering the critical roles of cell surface glycoproteins in various biological processes, it is an urgent task to systematically analyze them. However, the low abundance of many glycoproteins and the heterogeneity of glycans pose great challenges for global analysis of glycoproteins. It is even more challenging to specifically investigate glycoproteins only located on cell surface because it requires effective strategies to target cell surface glycoproteins. Antibody microarray has been employed to analyze cluster of differentiation (CD) molecules on the cell surface that are important for differentiation and classification of cells, and provided valuable information of cell surface protein expression (Belov et al., 2001). However, the drawbacks of antibody-based methods are that the availability and specificity of antibodies are sometimes problematic, and normally the throughput is low. Modern mass spectrometry (MS) has become a powerful tool to globally analyze proteins without antibody, and MS-based proteomics enables us to comprehensively identify and quantify protein post-translational modifications (PTMs) (Chen et al., 2018; Huang et al., 2015; Ruhaak et al., 2018; Simithy et al., 2017; Song et al., 2014; Wu et al., 2011; Xiao et al., 2018a; Xiao et al., 2019; Yuan et al., 2014; Zhang et al., 2003). Although MS can identify many peptides and proteins in bottom-up proteomics, in which proteins need to be proteolytically digested into peptides before MS analysis, the high-abundance intracellular proteins may mask surface glycoproteins during MS analysis. This results in the failure of identifying surface glycopeptides and glycoproteins. Moreover, MS cannot distinguish surface glycoproteins from intracellular ones. Therefore, specific separation and enrichment of surface glycoproteins is prerequisite prior to MS analysis.
In 2009, Wollscheid et al. developed an elegant method called cell surface capturing (CSC) to comprehensively investigate glycoproteins on the cell surface. They used a chemical method to oxidize glycans on the cell surface, and then enriched surface glycoproteins for MS analysis (Wollscheid et al., 2009). This method allowed them to identify and quantify surface glycoproteins. CSC has been applied to different samples including mouse and human cell lines, and primary cells (Gundry et al., 2009; Haverland et al., 2017). Bausch-Fluck et.al applied it to different types of human and mouse cells and built a MS-derived Cell Surface Protein Atlas (CSPA), which provides valuable information about surface glycoproteins from different species that forms the basis for classification of cell types and drug target screening (Bausch-Fluck et al., 2015). Recently, Leung et al. systemically studied the impact of azacitidine (AZA) on the surface proteome of acute myeloid leukemia (AML) cells (Leung et al., 2019). AML is one kind of disease involved with abnormal hematopoietic differentiation and epigenetic changes. By employing CSC, they identified the largest number of surface proteins in AML cells to date and provided a valuable resource for AML study and offered potential therapeutic strategies for AML by utilizing AZA.
Another nice approach to capture cell surface glycoproteins with high sensitivity is selective exoenzymatic labeling (SEEL) developed by Dr. Boons lab (Sun et al., 2016; Yu et al., 2016). For SEEL, a recombinant glycosyltransferase is used to specifically transfer a sugar analog to install a chemical reporter onto glycans on the cell surface that enables the enrichment of surface glycoprotein for MS analysis. Galactose oxidase (GAO), which is an enzyme and can specifically convert the hydroxyl group at C6 on galactose/N-acetylgalactosamine (Gal/GalNAc) to an aldehyde group, can be employed to tag surface glycoproteins, followed by the enrichment for MS analysis (Ramya et al., 2013). Since the reaction conditions are very mild, this method can eliminate the oxidative stress posed on cells and maintain the intact state of cells. Recently, by incorporating horseradish peroxidase (HRP) and methoxylamine, the efficiency of GAO-based method to capture cell surface glycoproteins has been dramatically improved (Sun et al., 2019). The method has also been used to systematically study the surface glycoprotein trafficking by combing with quantitative proteomics. However, the GAO-based method is biased for surface glycoproteins with glycans terminated with Gal/GalNAc because of the inherent specificity of GAO. In combination with the sialidase treatment to remove sialic acid, this method was demonstrated to be more effective to analyze surface glycoproteins (Sun et al., 2019). Despite the importance of surface glycoproteins, global analysis of surface proteins has been understudied compared to the whole proteomic analysis. In this book chapter, we discuss a chemoenzymatic method by integrating metabolic labeling, click and enzymatic reactions, and MS-based proteomics for global and site-specific analysis of surface glycoproteins.
2. COMPREHENSIVE ANALYSIS OF SURFACE N-GLYCOPROTEINS BY INTEGRATING METABOLIC LABELING, COPPER-FREE CLICK CHEMISTRY AND MS-BASED PROTEOMICS
2.1. Metabolic Labeling with Sugar Analogs and Bioorthogonal Chemistry
During the last decades, the rapid development of metabolic labeling with sugar analogs in glycobiology research has been achieved. Modified sugar analogs with a chemical reporter can be taken up into cells and incorporated into glycoconjugates (Mahal et al., 1997; Woo et al., 2015; Zhu et al., 2016). The sialic acid biosynthetic pathway in cells can tolerate unnatural sialic acid analogs with chemical reporters. For example, N-azidoacetylmannosamine (ManNAz) can be converted to sialic acid bearing an azido group in cells and then incorporated into glycoproteins through the biosynthetic machinery of a cell (Saxon and Bertozzi, 2000; Yang et al., 2011). In addition to the sialic acid biosynthetic pathway, both the GalNAc and N-acetylglucosamine (GlcNAc) salvage pathways can tolerate unnatural sugars bearing chemical reporters, including N-azidoacetylgalactosamine (GalNAz) and N-azidoacetylglucosamine (GlcNAz) (Hang et al., 2003; Vocadlo et al., 2003).
The chemical reporters incorporated into glycoproteins are critical for the following tagging and study of glycoproteins. The first requirement for the chemical reporters is that they cannot either exist in the biological systems or react with a plethora of chemical functionalities found in living cells, which ensures that the detection and/or enrichment of glycoproteins are specific and selective (Hubbard et al., 2011; Lim and Lin, 2010; Sletten and Bertozzi, 2009). In addition, ideally the chemical reporters can be recognized by biosynthetic machineries in cells without structural and biological perturbations in the biological systems. After being installed with the chemical reporters, glycoproteins can be tagged with fluorescence probes or other reagents with an affinity group for enrichment through a bioorthogonal reaction, which can take place in the presence of biomolecules or in living cells without the disruption of the biological systems (Dieterich et al., 2006). Normally, a bioorthogonal reaction needs to be fast and form a stable covalent linkage without generating toxic byproducts. Bertozzi lab employed the hydrazide/oxime condensation to target surface glycoproteins, in which carbonyls were used as the chemical reporters and under acidic conditions (pH 4–6) they reacted with hydrazines and alkoxyamines. They used N-levulinoyl mannosamine (ManLev), which bears a ketone functional group, to metabolically label cell surface glycoproteins, followed by being conjugated with molecules containing hydrazide under physiological conditions (Mahal et al., 1997). Azide has also been widely used as a chemical reporter in living systems because of its special properties including its absence from biological systems, relative stability and unique reactivity. A sugar analog containing azide was used to metabolically label cells and reacted with triarylphosphine through the Staudinger ligation for cell surface glycoproteins (Saxon and Bertozzi, 2000). However, the Staudinger ligation reaction is relatively slow and triarylphosphine is not very stable. Azide can also undergo reactions with alkynes through Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC), which proceeds faster than the Staudinger ligations under physiological conditions (Wang et al., 2003). Through metabolic labeling with ManNAz, glycans with the azide functionality on the cell surface can be tagged through CuAAC using tris(hydroxypropyltriazolyl)methylamine (THPTA) as the ligand (Hong et al., 2010). However, the copper ions used in CuAAC are toxic to cells and in order to lower the toxicity, decreasing copper concentration is a commonly used strategy, which leads to a significant decrease in the reaction rate. Strain-promoted azide–alkyne cycloaddition (SPAAC) was developed, which relies on the relatively high reactivity of a strained alkyne towards the azido group. The reaction can take place under physiological conditions without any catalysts (Agard et al., 2004). In order to further speed up the SPACC reactions, different types of cyclooctyne were developed and tested and the reaction rate was reported up to10−2 to 1 M−1 s−1 (Codelli et al., 2008). The reaction has been widely used in complex systems including live cells and animals (Ning et al., 2008).
Besides the hydrazide/oxime condensation, Staudinger ligations, CuAAC and SPAAC, many other types of bioorthogonal reactions have been developed and utilized to study biological processes, including metal-catalyzed olefin metathesis, the thiol-ene reaction and a light induced 1,3-dipolar cycloaddition reaction between tetrazoles and terminal alkenes (Lang and Chin, 2014; McKay and Finn, 2014). Inverse-electron demanding Diels–Alder reactions between highly strained alkenes or alkynes and tetrazines has recently become increasingly popular to label biomolecules in living systems because of its extraordinarily fast reaction rate (up to 105 M−1 s−1) (Taylor et al., 2011).
2.2. Combination of Metabolic Labeling and Bioorthogonal Reactions for Comprehensive Analysis of Surface N-Glycoproteins
Considering the importance of cell surface glycoproteins, we have worked on the development of effective methods to achieve their comprehensive analysis. As described above, it is extraordinarily challenging to globally analyze glycoproteins only located on the cell surface. With the rapid development in MS-based proteomics, site-specific analysis of glycosylation become possible. Site-specific analysis of protein modifications provides valuable information about modification sites, and similarly important, it offers solid evidence for the identification of modified peptides and proteins. In this book chapter, we describe a method integrating metabolic labeling, bioorthogonal chemistry, and MS-based proteomics to globally and site-specially identify and quantify surface N-glycoproteins (Chen et al., 2015; Smeekens et al., 2015; Xiao et al., 2016; Xiao et al., 2018b). A sugar analog including GalNAz, GlcNAz, or ManNAz, was fed to cells and then a chemical reporter, i.e. the azido group, can be incorporated into glycoproteins, including those located on the cell surface (Figure 1a and 1b). With the help of SPAAC, which takes place under physiological conditions and maintains the viability of cells, surface glycoproteins containing the azido group were conjugated with dibenzocyclooctyne (DBCO)-sulfo-biotin while cells were still alive. Due to the hydrophilic properties of DBCO-sulfo-biotin, it cannot penetrate the plasma membrane of cells and therefore only surface glycoproteins were tagged. After the removal of the remaining reagents, we lysed cells, extracted proteins and then digested them. Glycopeptides from surface glycoproteins contained the biotin tag, which allowed us to enrich them through NeutrAvidin beads based on the high affinity between biotin and avidin. One limitation of NeutrAvidin enrichment is the non-specific binding, which can be minimized by performing the enrichment at the peptide level instead of the protein level.
Figure 1.
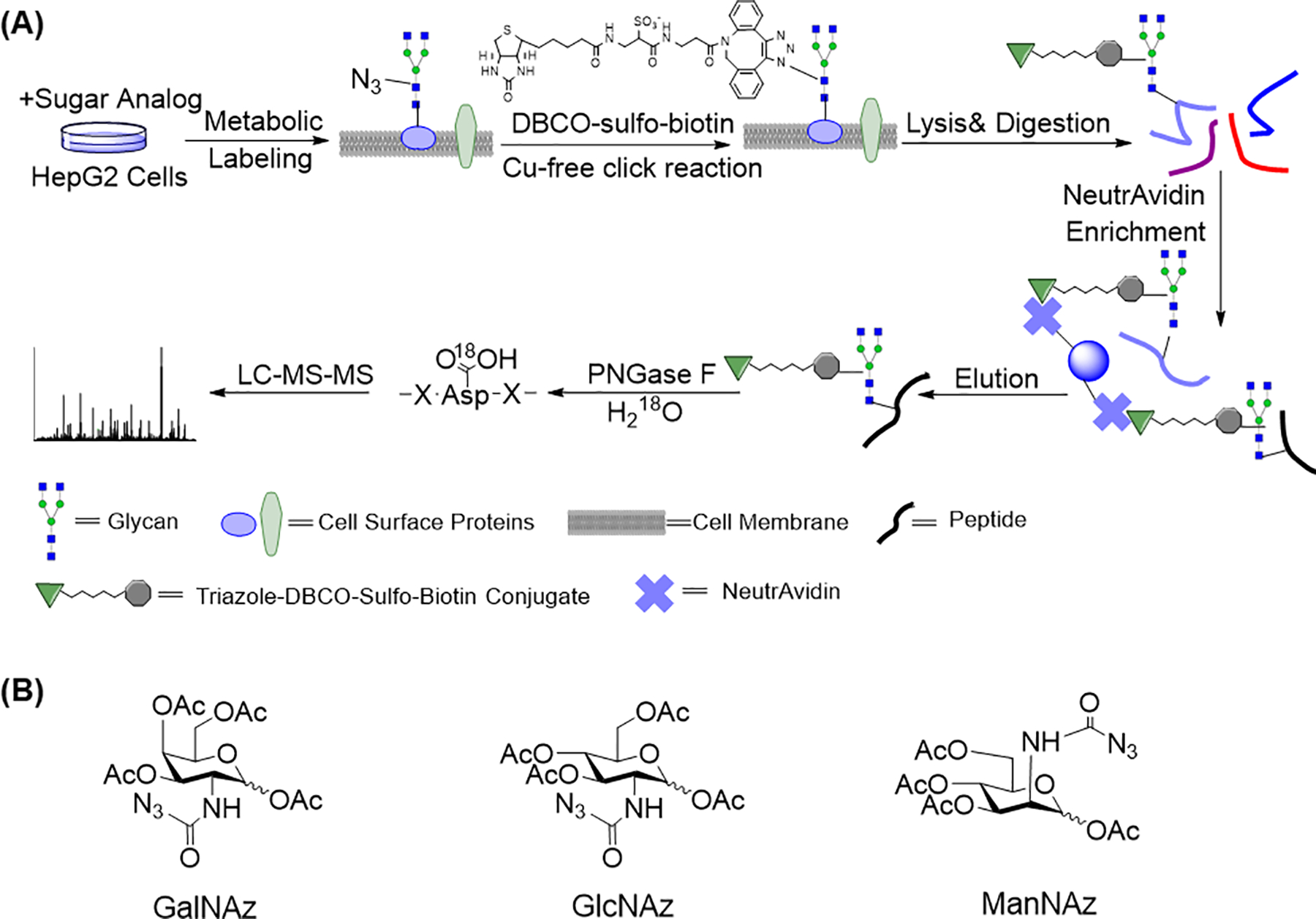
(a) Experimental procedure for the global analysis of the cell surface N-glycoproteome integrating metabolic labeling, copper-free click chemistry, and MS-based proteomics. (b) Different types of sugar analogs, GalNAz, GlcNAz, and ManNAz.
Adapted with permission from Xiao, H., Tang, G. X., & Wu, R. (2016). Site-specific quantification of surface N-glycoproteins in statin-treated liver cells. Analytical Chemistry, 88(6), 3324–3332. Copyright by the American Chemical Society.
High heterogeneity of glycans makes site-specific analysis of glycoproteins by MS extremely challenging. In order to enable site-specific analysis of surface N-glycoproteins, it is necessary to generate a universal tag on the glycosylation sites before MS analysis (Chen et al., 2014; Kuster and Mann, 1999). Peptide N-glycosidase F (PNGase F) was employed to remove the N-glycans on enriched surface glycopeptides, which converted asparagine (Asn) to aspartic acid (Asp). The reaction took place in heavy-oxygen water (H218O) and therefore a mass shift of +2.9883 Da on glycosylation sites (Asp) was generated, which can be readily identified by MS. This method can distinguish authentic glycosylation sites from those caused by chemical deamination on Asn that takes place in vivo and during sample preparation.
2.3. Identifications of Surface Glycopeptides with LC-MS/MS
Deglycosylated peptides can be analyzed by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS). The full MS of glycopeptides was recorded in the Orbitrap cell with high mass accuracy and high resolution, which increases the confidence of peptide identifications. Then peptides were fragmented using collision-induced dissociation (CID) and the fragments were recorded in a linear ion trap. In order to interpret the obtained tandem mass spectra, the observed fragmentation ions were matched against the predicted fragmentation ions from all the known protein sequences in the database (Eng et al., 1994). After obtaining peptide sequences, it is necessary to determine the confidence of glycosylation sites for site-specific analysis. The presence of the fragmentation ions generated from the tandem mass spectra were matched against the predicted fragmentation ions based on the theoretical sites, which would determine the possibility of the site localization (Beausoleil et al., 2006). It is well-known that the N-X-S/T (X is any amino acid residue except proline) motif is conserved for protein N-glycosylation and this motif was further utilized to control the confidence of glycosylation site identification. Eventually, the combination of metabolic labeling, copper-free click chemistry and MS-based proteomics enabled to comprehensively and site-specifically investigate N-glycoproteins only located on the cell surface.
3. PROTOCOL
Chemical Reagents and Materials
Dulbecco’s Modified Eagle Medium (DMEM), phosphate buffered saline (PBS), Benzonase® Nuclease, peptide-N-glycosidase F (PNGase F), dimethyl sulfoxide (DMSO), digitonin, sodium deoxycholate (SDC), penicillin-streptomycin, NaCl, HEPES, DTT, iodoacetamide, urea, ammonium bicarbonateare from Sigma-Aldrich; GalNAz, GlcNAz, ManNAz, DBCO-sulfo-biotin are purchased from Click Chemistry Tools; DMEM for SILAC, Pierce NeutrAvidin agarose beads, formic acid, trifluoroacetic acid, acetic acid are from Thermo Scientific; Fetal bovine serum (FBS) and dialyzed FBS are purchased from Corning and Atlanta Biologicals, respectively. Lys-C (mass spectrometry grade) and trypsin (sequence grade) are purchased from Wako and Promega. 13C615N2 L-lysine (+8 Da) and 13C6 L-arginine (+ 6 Da) are from Cambridge Isotopes Inc. Other chemicals are specified in the following protocol.
3.1. Cell Culture
-
3.1.1
Culture HepG2 (ATCC) in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C.
-
3.1.2
Wash cells with warm 10 mL PBS twice when the confluency of cells reaches about 50% in a T175 flask.
-
3.1.3
Add sugar analogs GalNAz, GlcNAz, or ManNAz (stock solution: 100 mM in DMSO) to the media to a final concentration of 100 μM. The sugar analogs need to be passed through a filter (0.2 μm, VWR) before being added to the media in order to remove possible microorganisms.
-
3.1.3
Transfer the prepared media to flasks and culture the cells for another 24 h before performing the copper-free click reaction.
3.2. Quantification Experiment Using Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
-
3.2.1
Prepare the SILAC media: for heavy media, add 87.5 mg heavy lysine (Lys-8), 42.0 mg heavy arginine (Arg-6), 100.0 mg proline (Sigma-Aldrich) and 50 mL dialyzed FBS to 450 mL SILAC media; for light media, add 73.0 mg lysine (Lys-0, Sigma-Aldrich), 34.7 mg arginine (Arg-0, Sigma-Aldrich), 100.0 mg proline and 50 mL dialyzed FBS to 450 mL SILAC media.
-
3.2.2
Grow MCF7 and MDA-MB-231 cells (ATCC) in light and heavy SILAC media in a humidified incubator at 37 °C supplemented with 5% CO2, respectively.
-
3.2.3
Culture cells for at least five generations before adding sugar analogs.
3.3. Cell Surface Glycoproteins Tagging Through Copper-Free Click Chemistry
-
3.3.1
Prepare the following solution: add DBCO-sulfo-biotin (stock solution: 40 mM in DMSO) to PBS to a final concentration of 100 μM.
-
3.3.2
Remove the media from cells and wash cells with warm 10 mL PBS twice.
-
3.3.3
Add 5 mL PBS containing DBCO-sulfo-biotin to cells in a T175 flask. Tilt the flask to ensure that the surface of cells is covered with the solution. Incubate the cells at 37 °C for 1 h.
-
3.3.4
After the copper-free click reaction, wash cells with 10 mL PBS twice and then harvest them in PBS by using cell scraper (Thermo Scientific).
-
3.3.5
Cells in PBS are pelleted through centrifuging at 500 g for 3 min and the cell pellet is washed with PBS one more time.
3.4. Cell Lysis
-
3.4.1
Prepare the following buffer: 150 mM NaCl (stock solution: 3 M), 50 mM HEPES (pH 7.4) (stock solution: 500 mM, pH 7.4), 25 μg/mL digitonin (stock solution: 250 μg/mL), 1 tablet/10 mL protease inhibitor (Roche).
-
3.4.2
Add 3 mL buffer to cells and pipette the solution up and down several times to ensure that cells are suspended in the solution. Put the cell mixture on ice for 10 min. The purpose of this step is to use digitonin to break the plasma membrane and remove cytosolic proteins.
-
3.4.3
Centrifuge the solution at 2000 g for 10 min. Then remove the supernatant and keep the pellet on ice.
-
3.4.4
Prepare the lysis buffer: 50 mM HEPES (pH 7.4), 150 mM NaCl, 0.5 % SDC, 20 units/mL benzonase, 1 tablet/10 mL protease inhibitor.
-
3.4.5
Add 3 mL lysis buffer to the cell pellet and vertex to suspend the pellet in solution. Incubate the solution on an end-over-end rotor at 4 °C for 45 min.
-
3.4.6
Centrifuge the lysis solution at 5000 g for 10 min and keep the supernatant. The protein concentrations were determined by the BCA protein assay.
3.5. Protein Reduction, Alkylation and Digestion
-
3.5.1
Add DTT (stock solution: 1 M) to the lysate to a final concentration of 5 mM and incubate the solution at 56 °C for 30 min. In order to minimize side reactions from alkylation, the solution needs to cool down to room temperature before adding iodoacetamide.
-
3.5.2
Add iodoacetamide to the lysate solution to a concentration of 14 mM. Use aluminum foil to cover the lysate and incubate on the end-over-end rotator at room temperature for 30 min.
-
3.5.3
Isolate proteins from the lysate solution through the methanol-chloroform protein precipitation method. For 1 mL of lysate solution, add 4 mL of methanol (EMD Millipore), 1 mL of chloroform (EMD Millipore), and 3 mL of deionized H2O. Mix and vortex after adding each solvent. Centrifuge the solution at 5000 g for 10 min and then remove the supernatant. Add the same volume of methanol to wash the pellet for one time and centrifuge at 5000 g for 10 min and remove the supernatant.
-
3.5.4
Prepare the digestion buffer: 50 mM HEPES (pH 8.6, stock solution: 500 mM pH 8.6), 1.6 M urea, 5% (v/v) ACN (EMD Millipore).
-
3.5.5
Add 3 mL digestion buffer to the protein pellet and sonicate for 15 seconds to suspend the protein pellet in the buffer. Add trypsin to the solution and the ratio of trypsin to proteins is 1:100 (w/w). Incubate at 37 °C with shaking overnight.
-
3.5.6
Quench the digestion by adding trifluoroacetic acid (TFA) until the pH is less than two. Centrifuge the solution at 5000 g for 10 min and keep the supernatant for peptide desalting.
3.6. Peptide Desalting
-
3.6.1
Purify peptides using a tC18 Sep-Pak cartridge (Waters). Here we use a 100 mg cartridge as an example. The maximum capacity is 5 % of the cartridge packing material weight.
-
3.6.2
Wash the cartridge with 3 mL ACN and then with 3 mL of 50 % ACN with 0.5 % acetic acid.
-
3.6.3
Equilibrate the cartridge with 3 mL of 0.1 % TFA and then load the sample.
-
3.6.4
Desalt the sample with 3 mL of 0.1 % TFA and then wash with 0.5 mL of 0.5 % acetic acid to remove TFA.
-
3.6.5
Elute peptides with 0.7 mL of 50 % ACN with 0.5 % acetic acid and then 0.3 mL of 75 % ACN with 0.5 % acetic acid.
-
6.6
Freeze the eluted peptides at −80 °C and lyophilize the sample using a speed vac overnight.
3.7. Glycopeptide Enrichment Using NeutrAvidin Beads
-
3.7.1
For ~3 mg peptides, get 100 μL NeutrAvidin slurry and wash the beads with 1 mL PBS for three times.
-
3.7.2
Dissolve peptides in 0.5 mL PBS and transfer the dissolved peptides to NeutrAvidin beads. After vertexing, incubate the mixture at 37 °C with shaking for 30 min.
-
3.7.3
Transfer the mixture to a spin column (Thermo Scientific) and wash the beads with 0.6 mL PBS for ten times. For each wash, vertex the spin column and then centrifuge at 2000 g for 2 min to remove the washing solution.
-
3.7.4
Elute glycopeptides by adding 200 μL of 8 M guanidine-HCl (pH 1.5, Sigma-Aldrich) and incubating at 56 °C for 2 min. The elution step is repeated for another two times. Combine the eluates for peptide desalting.
-
3.7.5
Desalt the gycopeptides using a 50 mg Sep-Pak cartridge. Freeze the eluted peptides at −80 °C and lyophilize the sample using a speed vac overnight.
3.8. Peptide Deglycosylation through the PNGase F Treatment
-
3.8.1
Dissolve lyophilized glycopeptide (entirely dry) in 40 μL 50 mM ammonium bicarbonate (NH4HCO3, pH = 9) in heavy-oxygen water (H218O) and then add 3 μL PNGase F (1 U/μL in H218O).
-
3.8.2
Incubate the reaction at 37 °C with shaking for 3 h. Quench the reaction by adding formic acid (FA) until the pH is less than 2, followed by stage tip.
3.9. Stage Tip
-
3.9.1
For each stage tip, load 50 μL methanol (HPLC grade, EMD Millipore), then centrifuge at 2500 rpm for 2 min. Remove the flow-through. It is necessary to make sure that there is no solvent left above the packing material.
-
3.9.2
Load 40 μL 80% ACN (LC-MS grade, EMD Millipore) with 1% acetic acid and then centrifuge at 2500 rpm for 2 min.
-
3.9.3
Equilibrate the stage tip with 40 μL 1% FA and centrifuge at 3000 rpm for 3 min.
-
3.9.4
Load the sample to stage tip and centrifuge at 2000 rpm for 10 min.
-
3.9.5
Load stage tip with 50 μL 1% FA and centrifuge at 3000 rpm for 3 min to desalt peptides.
-
3.9.6
Elute peptides with 20 %, 50% and 80% ACN with 1 % acetic acid, respectively. Collect each fraction in a vial used for mass spectrometric analysis.
-
3.9.7
Freeze the eluted peptides at −80 °C and lyophilize the sample using a speed vac for 20 min.
3.10. LC-MS/MS Analysis
-
3.10.1
Dissolve the dried peptides in 8 μL of 5% ACN with 4% FA. Vertex the sample to ensure peptides are completely dissolved.
-
3.10.2
Load 4 μL of each sample onto an in-house packed microcapillary column with C18 beads through a Dionex WPS-3000TPLRS autosampler. Peptides are separated by reversed-phase liquid chromatography using an UltiMate 3000 binary pump.
-
3.10.3
Thermo LTQ Orbitrap Elite is used to detect peptides with the following setup: resolution for one full MS scan in the Orbitrap: 60,000; automatic gain control (AGC) target: 106; MS/MS scanning in the LTQ: 20 for the most intense ions; normalized collision energy of CID: 40; isolation width (m/z) 2.0; maximum ion accumulation time for each full MS scan and each MS/MS scan: 1000 ms and 50 ms, respectively.
3.11. Database Searches, Data Filtering, and Glycosylation Site Localization
-
3.11.1
After converting the raw files into mzXML format, search all MS/MS spectra using SEQUEST algorithm (Eng et al., 1994), matching mass spectra against UniProt human (Homo sapiens) database containing all protein sequences with the following parameters: 10 ppm for precursor mass tolerance; 1.0 Da for product ion mass tolerance; fully digested with trypsin; up to three missed cleavages; fixed modification: carbamidomethylation of cysteine (+57.0214); variable modifications: oxidation of methionine (+15.9949), 18O tag of Asp (+2.9883), heavy lysine (+8.0142) and heavy arginine (+6.0201).
-
3.11.2
Employ the target-decoy method (Elias and Gygi, 2007) and linear discriminant analysis (LDA) to evaluate and control the false discovery rates (FDRs) of glycopeptide and glycoprotein identifications. Multiple parameters are integrated, such as Xcorr, ΔCn, and precursor mass error. The FDRs for both glycopeptides and glycoproteins are controlled to be less than 1%.
-
3.11.3
Use ModScore which indicates the possibility of the glycosylation site based on the comparison of fragmentation ions with theoretical ones to evaluate the confidence of site localizations (Beausoleil et al., 2006). It employs a probabilistic algorithm that considers the presence or absence of fragment ions generated in the tandem mass spectrum unique to each glycosylation site and calculates the possibility of the best site localization when compared with the theoretical fragments. Sites with a ModScore >19 (P < 0.01) are considered to be confidently localized.
-
3.11.4
For peptide quantification using SILAC, the ratio between signal and noise (S/N) for both heavy and light peptides need to be larger than 3. The S/N ratio of the light peptide needs to be larger than 10 when the S/N ratio of the heavy peptide counterpart is lower than 3, and vice versa. If the same peptides are quantified several times, their abundance change is the median ratio of those same peptides. For the glycosylation site quantification, quantified glycopeptide must follow two criteria below: it contains only one glycosylation site and the ModScore has to be larger than 19, which ensures that glycosylation site is well-localized.
-
3.11.5
Protein functional annotations can be obtained using the Database for Annotation, Visualization and Integrated Discovery (DAVID) and the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system.
4. DISCUSSION
4.1. Site-Specific Analysis of Surface N‑Glycoproteins with Different Sugar Analogs
An increase of surface glycoproteome coverage is essential to screen the desirable proteins for the classification of cells, discovery of biomarkers and treatment of diseases. GalNAz, GlcNAz, and ManNAz can all be used through the biosynthetic pathways inside a cell and then incorporated into glycoproteins. Each sugar analog enters a different biosynthetic pathway in cells and therefore labels glycoproteins on the basis of glycan structures and enzymes responsible for the glycan synthesis. Thus, it is expected that the labeling efficiency from these sugar analogs could be different, which can be reflected by the number of identified surface N-glycoproteins. Based on the biologically duplicate experiments, the result of GalNAz outperformed GlcNAz and ManNAz, and GlcNAz covered the fewest number of glycosylation sites and glycoproteins. The majority of the glycosylation sites and glycoproteins identified in the experiments with ManNAz and GlcNAz as the labeling reagents were covered in the experiment with GalNAz (Figure 2). The results clearly demonstrated that GalNAz resulted in the highest coverage of the surface N-glycoproteome among the parallel experiments (Xiao et al., 2016).
Figure 2.
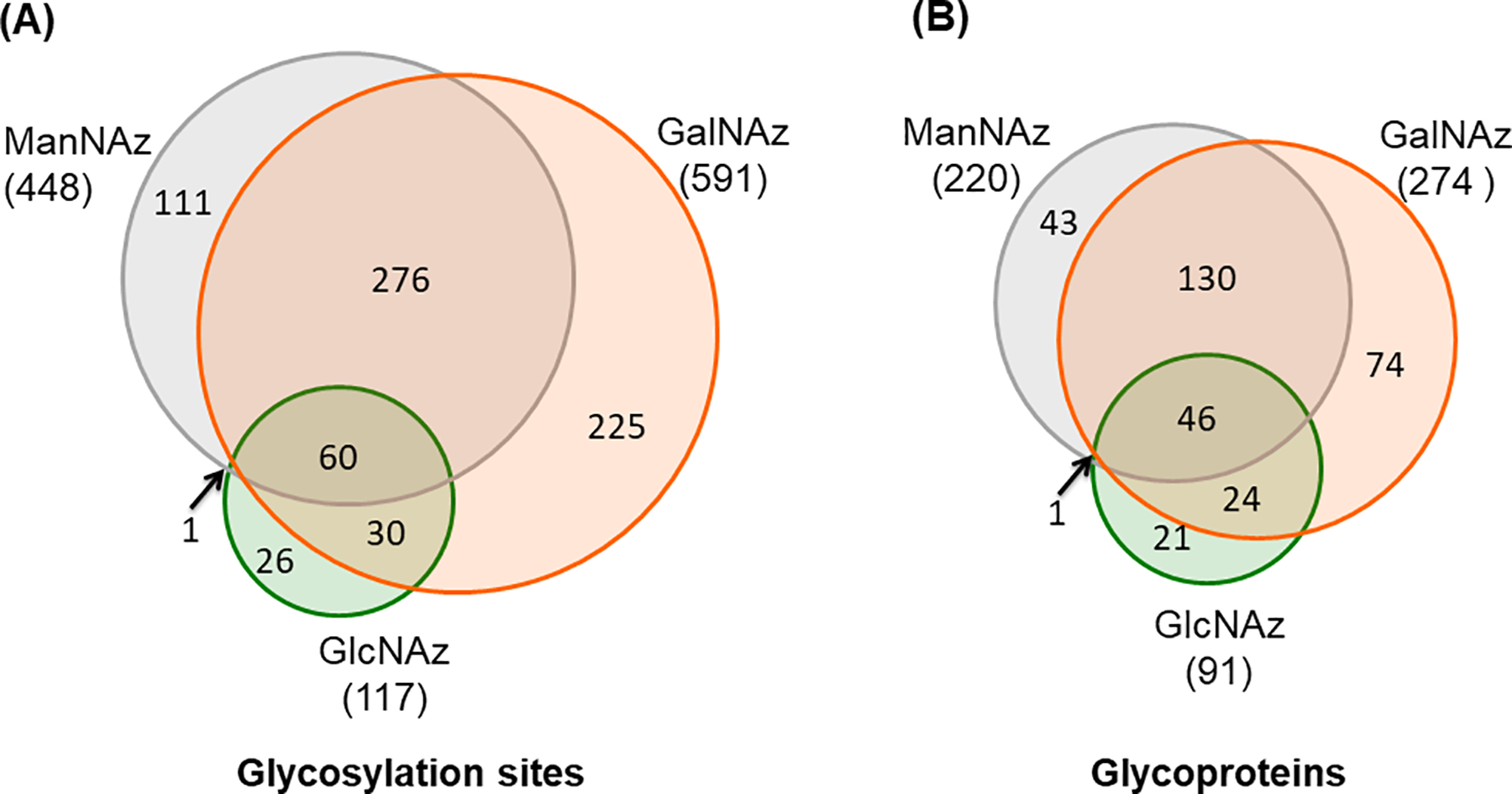
Identification of (a) surface N-glycosylation sites and (b) N-glycoproteins among GalNAz, GlcNAz, and ManNAz labeling experiments.
Adapted with permission from Xiao, H., Tang, G. X., & Wu, R. (2016). Site-specific quantification of surface N-glycoproteins in statin-treated liver cells. Analytical Chemistry, 88(6), 3324–3332. Copyright by the American Chemical Society.
In the process of globally identifying N-glycosylation sites on the cell surface, two potential issues need to be considered in order to ensure that the identified sites are authentic. The first one is non-specific binding from NeutrAvidin beads, which will undermine the hypothesis that only cell surface N-glycopeptides can be enriched and identified in the current method. A control experiment in which the step of copper-free click chemistry was omitted and the other steps remained the same. Compared to 886 unique glycopeptides identified in the GalNAz labeling experiment, only 20 unique glycopeptides were identified in the control experiment, which indicated that the non-specific binding from NeutrAvidin beads posed little impact on surface glycopeptide identification. Another issue is the chemical deamination of Asn, which leads to false positive identifications of surface glycopeptides. This issue can be solved by performing PNGase F treatment in H218O for only three hours. With this short period of treatment time and the nearly neutral reaction conditions, the effect from the chemical deamination on glycosylation site identification is minimal, and therefore can be ignored (Xiao et al., 2016).
4.2. Quantification of Cell Surface Glycoproteins in Combination with Quantitative Proteomics
Because the current method is effective to globally analyze cell surface N-glycoproteins, it can also be applied for quantitative analysis of surface N-glycoproteins, which will enhance our understanding of the molecular mechanism of diseases and the discovery of biomarkers for disease diagnosis and drug targets for disease treatment. Combing the current method with SILAC, we studied the surface N-glycoprotein abundance changes in cells with the statin treatment. Statin is an inhibitor of HMG-CoA reductase, an enzyme that controls the rate-limiting step in the mevalonate pathway of cholesterol synthesis. The inhibition of HMG-CoA reductase prevents the synthesis of one important molecule produced in this pathway-dolichol, which acts as a membrane anchor for the formation of the oligosaccharide Glc3Man9GlcNAc2 that can be transferred to the Asp residues and glycosylate proteins (Burda and Aebi, 1999). Therefore, quantification of surface N-glycoproteins on statin-treated cells leads to a better understanding of the relationship between N-glycosylation and the pleiotropic effects caused by statin. By using GalNAz as the sugar analog, the quantification results demonstrated that many N-glycosylation sites on surface proteins were down-regulated in statin-treated cells and it was also found that several N-glycosylation sites on proteins involved in Alzheimer’s disease pathway were down-regulated (Xiao et al., 2016).
4.3. Study on the Correlation between Surface N-Sialoglycoproteins and Cell Invasiveness
Sialic acid located at the terminus of glycans on surface glycoproteins carries a negative charge under physiological conditions, which may affect the cell surface properties and migration of cells. It is speculated that the increase of sialylation on cell surface glycoproteins is correlated with cancer development and metastasis. Therefore, in order to gain a better understanding of functions of surface N-sialoglycoproteins and cellular activities that they participate in, it is of great importance to achieve their comprehensive and site-specific analysis. Titanium dioxide, lectins and hydrophilic interaction chromatography (HILIC) were reported as the major ways to selectively enrich sialoglycoproteins (Larsen et al., 2007; Zhao et al., 2006). However, those methods are not suitable to specifically target sialoglycoproteins only located on cell surface.
Combing SILAC and the method discussed in this book chapter, we can quantitatively compare the different expression of surface N-sialoglycoproteins between two breast cancer cells, MCF7 and MDA-MB-231 cells with low and high invasiveness, respectively (Figure 3a) (Chen et al., 2015). In order to specifically study N-sialoglycoproteins, ManNAz was employed for the metabolic labeling of sialoglycoproteins, including those on the cell surface. Four hundred and thirty-nine N-sialoglycosylation sites were identified on the surface of MDA-MB-231 cells and 237 sites were identified in MCF7 cells (Figure 3b). There are different types of membrane proteins, including type I and type II membrane proteins, which extends the N- or C-terminus in the extracellular region, respectively. Of all the N-sialoglycoproteins identified in both types of cells, the vast majority were membrane proteins based on the information from Uniprot and the prediction results from SecretomeP and Phobius (Figure 3c) (Bendtsen et al., 2004; Käll et al., 2004). The location of all the glycosylation sites identified on type I and type II membrane proteins was in the extracellular region (Figure 3d). These results demonstrated that the current method is very specific for surface N-sialoglycoprotein analysis. Among quantified N-sialoglycopeptides, over 40% (179 out of 406) from 99 proteins were up-regulated by over two-fold (Figure 4a). Gene Ontology analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) was performed to determine biological processes those up-regulated N-sialoglycoproteins are involved in (Huang et al., 2008). Proteins correlated with cell adhesion were the most highly enriched, and proteins belonging to the categories of integrin-mediated signaling, cell motion and cell-matrix adhesion were also overrepresented (Figure 4b). These results indicated that surface protein sialylation was closely correlated to cell migration and invasiveness.
Figure 3.
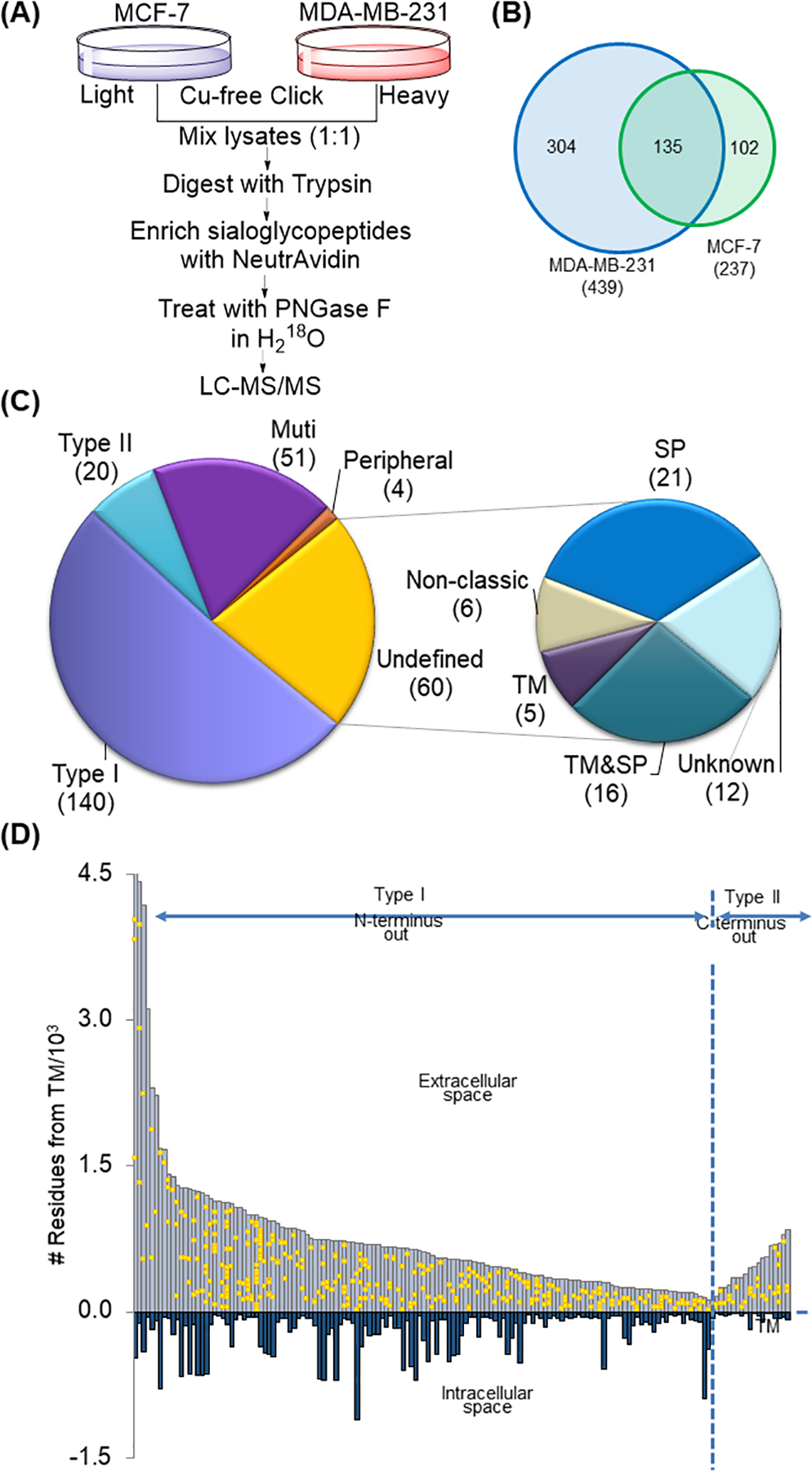
(a) Experimental procedure for the quantification of the surface N-sialoglycoproteome between MCF7 and MDA-MB-231cells. (b) Identification of N-sialoglycosylation sites from MCF7 and MDA-MB-231 cells. (c) Classification of surface proteins identified from MCF7 and MDA-MB-231 cells. (d) The distribution of site location of identified N-sialoglycosylation sites from type I and type II membrane proteins.
Adapted with permission from Chen, W., Smeekens, J. M., & Wu, R. (2015). Systematic and site-specific analysis of N-sialoglycosylated proteins on the cell surface by integrating click chemistry and MS-based proteomics. Chemical Science, 6(8), 4681–4689. Copyright by the Royal Society of Chemistry.
Figure 4.
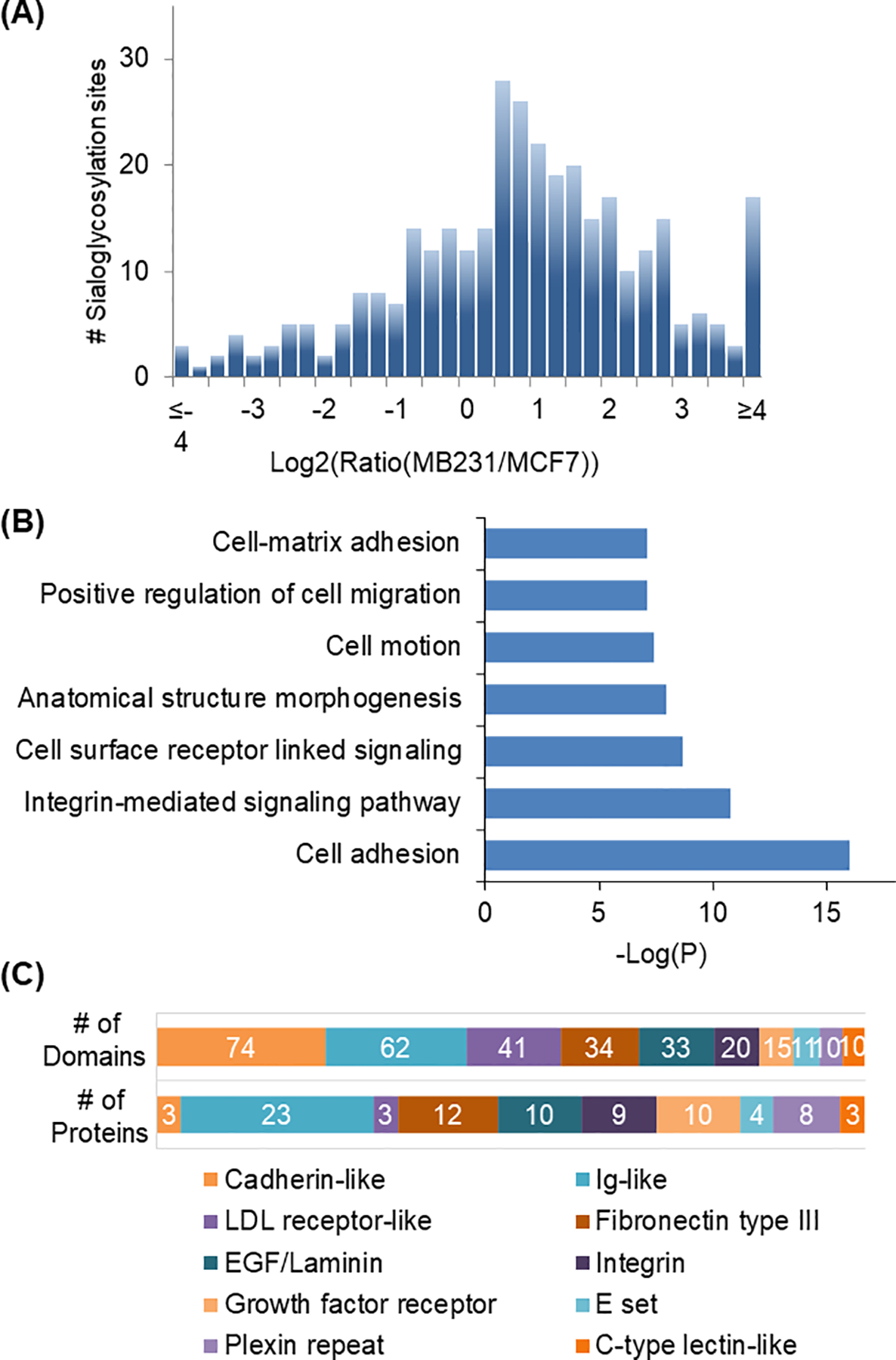
(a) Abundance distribution of quantified N-sialoglycosylation sites between MCF7 and MDA-MB-231 cells. (b) Protein clustering of up-regulated N-sialoglycoproteins based on biological processes. (c) Domain analysis of up-regulated N-sialoglycoproteins.
Adapted with permission from Chen, W., Smeekens, J. M., & Wu, R. (2015). Systematic and site-specific analysis of N-sialoglycosylated proteins on the cell surface by integrating click chemistry and MS-based proteomics. Chemical Science, 6(8), 4681–4689. Copyright by the Royal Society of Chemistry.
Protein domains represent the functions of a given protein sequence and the investigation of their relationship with N-glycosylation sites will provide valuable information about the correlation between protein functions and N-glycosylation (Milac et al., 2004). Among all the up-regulated N-sialoglycoproteins, over sixty types of protein domains were identified and the most frequent domain was the cadherin-like domain, which acts as one of the major adhesion domains that regulate cell-cell adhesion (Figure 4c). About 25% of the up-regulated proteins contained the immunoglobulin (Ig)-like domain, which is one of the most widespread domains involved in binding with different types of ligands, ranging from small molecules to hormones, and to macromolecules. Fibronectin type III domain related to cell adhesion and migration was found in twelve of the identified up-regulated N-sialoglycoproteins. Integrin, EGF-like, and growth factor receptor domains also frequently appeared among these up-regulated N-sialoglycoproteins. The domain analysis further indicated that sialylation may regulate the functions of proteins, and then promote the migration of cancer cells.
5. SUMMARY
Effective methods are critically important to tackle the mystery of glycoproteins located on the cell surface, which will facilitate our understanding of their biological functions and reveal the molecular mechanisms of disease. In this book chapter, we describe the challenges to analyze surface glycoproteins and briefly review existing methods, including CSC, SEEL, and GAO-based techniques, to systematically investigate cell surface N-glycoproteins. We mainly focus on an effective method integrating metabolic labeling, click and enzymatic reactions, and MS-based proteomics for global and site-specific analysis of surface glycoproteins. In parallel experiments for the comparison of different sugar analogs, GalNAz was found to be the most effective for surface N-glycoprotein analysis. This analog was then employed to systematically quantify surface N-glycoprotein changes in cells with the statin treatment. By utilizing ManNAz that specifically labels sialylated glycoproteins, systematic quantification of surface N-sialoglycosylated proteins between MCF7 and MDA-MB-231 cells with different invasiveness was performed in combination with quantitative proteomics. The results provide valuable information about the correlation between cell invasiveness and protein N-sialoglycosylation on the cell surface. This method can be extensively applied to decipher glycoprotein functions and various biological processes regulated by surface glycoproteins.
ACKNOWLEDGMENT
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R01GM118803).
REFERENCES
- Abu-Qarn M, Eichler J, and Sharon N (2008). Not just for eukarya anymore: protein glycosylation in Bacteria and Archaea. Current Opinion in Structural Biology 18, 544–550. [DOI] [PubMed] [Google Scholar]
- Aebi M, Bernasconi R, Clerc S, and Molinari M (2010). N-glycan structures: recognition and processing in the ER. Trends in Biochemical Sciences 35, 74–82. [DOI] [PubMed] [Google Scholar]
- Agard NJ, Prescher JA, and Bertozzi CR (2004). A strain-promoted [3 + 2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. Journal of the American Chemical Society 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A, Moest H, Omasits U, Gundry RL, Yoon C, et al. (2015). A mass spectrometric-derived cell surface protein atlas. PLoS One 10, e0121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villen J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature Biotechnology 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Belov L, de la Vega O, dos Remedios CG, Mulligan SP, and Christopherson RI (2001). Immunophenotyping of leukemias using a cluster of differentiation antibody microarray. Cancer Research 61, 4483–4489. [PubMed] [Google Scholar]
- Bendtsen JD, Jensen LJ, Blom N, von Heijne G, and Brunak S (2004). Feature-based prediction of non-classical and leaderless protein secretion. Protein Engineering, Design and Selection 17, 349–356. [DOI] [PubMed] [Google Scholar]
- Boscher C, Dennis JW, and Nabi IR (2011). Glycosylation, galectins and cellular signaling. Current Opinion in Cell Biology 23, 383–392. [DOI] [PubMed] [Google Scholar]
- Burda P, and Aebi M (1999). The dolichol pathway of N-linked glycosylation. Biochimica et Biophysica Acta (BBA) - General Subjects 1426, 239–257. [DOI] [PubMed] [Google Scholar]
- Chen W, Smeekens JM, and Wu R (2014). Comprehensive analysis of protein N-glycosylation sites by combining chemical deglycosylation with LC-MS. Journal of Proteome Research 13, 1466–1473. [DOI] [PubMed] [Google Scholar]
- Chen W, Smeekens JM, and Wu R (2015). Systematic and site-specific analysis of N-sialoglycosylated proteins on the cell surface by integrating click chemistry and MS-based proteomics. Chemical Science 6, 4681–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZW, Yu Q, Hao L, Liu FB, Johnson J, Tian ZC, Kao WJ, Xu W, and Li LJ (2018). Site-specific characterization and quantitation of N-glycopeptides in PKM2 knockout breast cancer cells using DiLeu isobaric tags enabled by electron-transfer/higher-energy collision dissociation (EThcD). Analyst 143, 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, and Packer NH (2014). Cell surface protein glycosylation in cancer. PROTEOMICS 14, 525–546. [DOI] [PubMed] [Google Scholar]
- Codelli JA, Baskin JM, Agard NJ, and Bertozzi CR (2008). Second-generation difluorinated cyclooctynes for copper-free click chemistry. Journal of the American Chemical Society 130, 11486–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall’Olio F, and Chiricolo M (2001). Sialyltransferases in cancer. Glycoconjugate Journal 18, 841–850. [DOI] [PubMed] [Google Scholar]
- Dalziel M, Crispin M, Scanlan CN, Zitzmann N, and Dwek RA (2014). Emerging principles for the therapeutic exploitation of glycosylation. Science 343, 1235681. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, and Schuman EM (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proceedings of the National Academy of Sciences 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Gundry RL, Raginski K, Tarasova Y, Tchernyshyov I, Bausch-Fluck D, Elliott ST, Boheler KR, Van Eyk JE, and Wollscheid B (2009). The mouse C2C12 myoblast cell surface N-linked glycoproteome. Molecular & Cellular Proteomics 8, 2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Yu C, Kato DL, and Bertozzi CR (2003). A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proceedings of the National Academy of Sciences 100, 14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverland NA, Waas M, Ntai I, Keppel T, Gundry RL, and Kelleher NL (2017). Cell surface proteomics of n-linked glycoproteins for typing of human lymphocytes. PROTEOMICS 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V, Steinmetz NF, Manchester M, and Finn MG (2010). Labeling live cells by copper-catalyzed alkyne–azide click chemistry. Bioconjugate Chemistry 21, 1912–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44. [DOI] [PubMed] [Google Scholar]
- Huang H, Lin S, Garcia BA, and Zhao Y (2015). Quantitative proteomic analysis of histone modifications. Chemical Reviews 115, 2376–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SC, Boyce M, McVaugh CT, Peehl DM, and Bertozzi CR (2011). Cell surface glycoproteomic analysis of prostate cancer-derived PC-3 cells. Bioorganic & Medicinal Chemistry Letters 21, 4945–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaprakash NG, and Surolia A (2017). Role of glycosylation in nucleating protein folding and stability. Biochemical Journal 474, 2333. [DOI] [PubMed] [Google Scholar]
- Kailemia MJ, Park D, and Lebrilla CB (2017). Glycans and glycoproteins as specific biomarkers for cancer. Analytical and Bioanalytical Chemistry 409, 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, and Sonnhammer ELL (2004). A combined transmembrane topology and signal peptide prediction method. Journal of Molecular Biology 338, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kim YJ, and Varki A (1997). Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconjugate Journal 14, 569–576. [DOI] [PubMed] [Google Scholar]
- Kuster B, and Mann M (1999). O-18-labeling of N-glycosylation sites to improve the identification of gel-separated glycoproteins using peptide mass mapping and database searching. Analytical Chemistry 71, 1431–1440. [DOI] [PubMed] [Google Scholar]
- Lang K, and Chin JW (2014). Bioorthogonal reactions for labeling proteins. ACS Chemical Biology 9, 16–20. [DOI] [PubMed] [Google Scholar]
- Larsen MR, Jensen SS, Jakobsen LA, and Heegaard NHH (2007). Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Molecular & Cellular Proteomics 6, 1778. [DOI] [PubMed] [Google Scholar]
- Lavrsen K, Madsen CB, Rasch MG, Woetmann A, Ødum N, Mandel U, Clausen H, Pedersen AE, and Wandall HH (2013). Aberrantly glycosylated MUC1 is expressed on the surface of breast cancer cells and a target for antibody-dependent cell-mediated cytotoxicity. Glycoconjugate Journal 30, 227–236. [DOI] [PubMed] [Google Scholar]
- Leung KK, Nguyen A, Shi T, Tang L, Ni X, Escoubet L, MacBeth KJ, DiMartino J, and Wells JA (2019). Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proceedings of the National Academy of Sciences 116, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RKV, and Lin Q (2010). Bioorthogonal chemistry: recent progress and future directions. Chemical Communications 46, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Crocker PR, and Paulson JC (2014). Siglec-mediated regulation of immune cell function in disease. Nature Reviews Immunology 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Yarema KJ, and Bertozzi CR (1997). Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125. [DOI] [PubMed] [Google Scholar]
- McKay Craig S., and Finn MG (2014). Click chemistry in complex mixtures: bioorthogonal bioconjugation. Chemistry & Biology 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milac A-L, Petrescu A-J, Wormald MR, Dwek RA, and Petrescu SM (2004). Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14, 103–114. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, and Nairn AV (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nature Reviews Molecular Cell Biology 13, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X, Guo J, Wolfert MA, and Boons G-J (2008). Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angewandte Chemie International Edition 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata S, Maimonis PJ, and Itzkowitz SH (1992). Mucins bearing the cancer-associated sialosyl-Tn antigen mediate inhibition of natural killer cell cytotoxicity. Cancer Research 52, 4741. [PubMed] [Google Scholar]
- Ohtsubo K, and Marth JD (2006). Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, and Toscano MA (2009). Turning ‘sweet’ on immunity: galectin–glycan interactions in immune tolerance and inflammation. Nature Reviews Immunology 9, 338–352. [DOI] [PubMed] [Google Scholar]
- Ramya TN, Weerapana E, Cravatt BF, and Paulson JC (2013). Glycoproteomics enabled by tagging sialic acid- or galactose-terminated glycans. Glycobiology 23, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd PM, Elliott T, Cresswell P, Wilson IA, and Dwek RA (2001). Glycosylation and the immune system. Science 291, 2370. [DOI] [PubMed] [Google Scholar]
- Ruhaak LR, Xu GG, Li QY, Goonatilleke E, and Lebrilla CB (2018). Mass spectrometry approaches to glycomic and glycoproteomic analyses. Chemical Reviews 118, 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E, and Bertozzi CR (2000). Cell surface engineering by a modified staudinger reaction. Science 287, 2007. [DOI] [PubMed] [Google Scholar]
- Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, et al. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nature Communications 8, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten EM, and Bertozzi CR (2009). Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angewandte Chemie International Edition 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens JM, Chen WX, and Wu RH (2015). Mass spectrometric analysis of the cell surface N-glycoproteome by combining metabolic labeling and click chemistry. Journal of the American Society for Mass Spectrometry 26, 604–614. [DOI] [PubMed] [Google Scholar]
- Song E, Mayampurath A, Yu CY, Tang HX, and Mechref Y (2014). Glycoproteomics: identifying the glycosylation of prostate specific antigen at normal and high isoelectric points by LC-MS/MS. Journal of Proteome Research 13, 5570–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell SR, Ju T, and Cummings RD (2015). Protein glycosylation in cancer. Annual Review of Pathology 10, 473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Suttapitugsakul S, and Wu R (2019). Enzymatic tagging of glycoproteins on the cell surface for their global and site-specific analysis with mass spectrometry. Analytical Chemistry 91, 4195–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun TT, Yu SH, Zhao P, Meng L, Moremen KW, Wells L, Steet R, and Boons GJ (2016). One-step Selective Exoenzymatic Labeling (SEEL) strategy for the biotinylation and identification of glycoproteins of living cells. Journal of the American Chemical Society 138, 11575–11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MT, Blackman ML, Dmitrenko O, and Fox JM (2011). Design and synthesis of highly reactive dienophiles for the tetrazine–trans-cyclooctene ligation. Journal of the American Chemical Society 133, 9646–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, and Mechref Y (2018). Glycosylation Changes in Brain Cancer. Acs Chemical Neuroscience 9, 51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vocadlo DJ, Hang HC, Kim E-J, Hanover JA, and Bertozzi CR (2003). A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proceedings of the National Academy of Sciences 100, 9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, and Finn MG (2003). Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. Journal of the American Chemical Society 125, 3192–3193. [DOI] [PubMed] [Google Scholar]
- Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R, Bibel M, Schiess R, Aebersold R, and Watts JD (2009). Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nature Biotechnology 27, 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, and Bertozzi CR (2015). Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nature Methods 12, 561-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, and Gygi SP (2011). A large-scale method to measure absolute protein phosphorylation stoichiometries. Nature Methods 8, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Chen W, Smeekens JM, and Wu R (2018a). An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycoproteins. Nature Communications 9, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Sun F, Suttapitugsakul S, and Wu R (2019). Global and site-specific analysis of protein glycosylation in complex biological systems with Mass Spectrometry. Mass Spectrometry Reviews, DOI: 10.1002/mas.21586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Tang GX, and Wu R (2016). Site-specific quantification of surface N-glycoproteins in statin-treated liver cells. Analytical Chemistry 88, 3324–3332. [DOI] [PubMed] [Google Scholar]
- Xiao HP, Suttapitugsakul S, Sun FX, and Wu RH (2018b). Mass spectrometry-based chemical and enzymatic methods for global analysis of protein glycosylation. Accounts of Chemical Research 51, 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LF, Nyalwidhe JO, Guo SQ, Drake RR, and Semmes OJ (2011). Targeted identification of metastasis-associated cell-surface sialoglycoproteins in prostate cancer. Molecular & Cellular Proteomics 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S-H, Zhao P, Sun T, Gao Z, Moremen KW, Boons G-J, Wells L, and Steet R (2016). Selective exo-enzymatic labeling detects increased cell surface sialoglycoprotein expression upon megakaryocytic differentiation. Journal of Biological Chemistry 291, 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZF, Arnaudo AM, and Garcia BA (2014). Mass spectrometric analysis of histone proteoforms. In Annual Review of Analytical Chemistry, Vol 7, Cooks RG, and Pemberton JE, eds. (Palo Alto: Annual Reviews), pp. 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, and Aebersold R (2003). Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nature Biotechnology 21, 660–666. [DOI] [PubMed] [Google Scholar]
- Zhao J, Simeone DM, Heidt D, Anderson MA, and Lubman DM (2006). Comparative serum glycoproteomics using lectin selected sialic acid glycoproteins with mass spectrometric analysis: application to pancreatic cancer serum. Journal of Proteome Research 5, 1792–1802. [DOI] [PubMed] [Google Scholar]
- Zhu YT, Wu J, and Chen X (2016). Metabolic Labeling and Imaging of N-Linked Glycans in Arabidopsis Thaliana. Angewandte Chemie-International Edition 55, 9301–9305. [DOI] [PubMed] [Google Scholar]