

Abstract
Genetic mosaicism arises when a zygote harbors two or more distinct genotypes, typically due to de novo, somatic mutation during embryogenesis. The clinical manifestations largely depend on the differentiation status of the mutated cell; earlier mutations target pluripotent cells and generate more widespread disease affecting multiple organ systems. If gonadal tissue is spared—as in somatic genomic mosaicism—the mutation and its effects are limited to the proband, whereas mosaicism also affecting the gametes, such as germline or gonosomal mosaicism, is transmissible. Mosaicism is easily appreciated in cutaneous disorders, as phenotypically distinct mutant cells often give rise to lesions in patterns determined by the affected cell type. Genetic investigation of cutaneous mosaic disorders has identified pathways central to disease pathogenesis, revealing novel therapeutic targets. In this review, we discuss examples of cutaneous mosaicism, approaches to gene discovery in these disorders, and insights into molecular pathobiology that have potential for clinical translation.
INTRODUCTION
Genetic mosaicism describes an organism harboring two or more genetically distinct cells (90, 118). There are many potential causes of somatic mutation, which include UV radiation, viral integration, environmental genotoxic agents, errors occurring during DNA replication, and X chromosome inactivation. The great majority of somatic mutations occurs in noncoding DNA with little or no effect on cell viability or function (98), but if rare mutations with potentially larger effect occur during embryonic development or after birth and confer a growth or selective advantage, a mutant clone will expand and reach clinical significance (24, 127). More than 30% of genes are estimated to be indispensable to development in mammals, and many mutations in critical housekeeping genes or oncogenes cause embryonic lethality when arising de novo in a zygote and causing constitutional disease. Such mutations can be tolerated, however, in a limited, mosaic state (59, 114). Mutations arising after the establishment of immune tolerance can be limited by a T cell–mediated immune response against resulting neoantigens (24, 127). Thus, only those somatic mutations that permit viability and evade immune response lead to sustained mosaicism that can be visually appreciated.
Postnatal mutations can give rise to acquired benign seborrheic keratosis (56), nevi (51, 70, 74, 80, 81) and sporadic lobular capillary hemangiomas (53, 82, 84), but somatic genomic mosaicism is also a precursor to malignant transformation seen in basal cell carcinoma (18), squamous cell carcinoma (1), and melanoma (41, 59).Mutations occurring in embryonic development can affect cells of broad differentiation potential, giving rise to localized or generalized involvement of the organism, and are readily apparent in nature including the patterned coat color in calico or tortoiseshell cats (23, 89), the dual-colored eyes of heterochromia iridis (2), and the color variation in maize pericarp (108). In 1925, Calvin Bridges first observed genetic mosaicism in Drosophila melanogaster, after crossing wild-type males with Minute-n females harboring the X-linked dominant Minute gene mutation (which causes shorter thoracic bristles and a smaller, paler body) (20). Some of the daughters, which Bridges termed piebalds, exhibited spots on the thorax with normal bristles as well as mixed colors of the eyes, corresponding to random inactivation of the maternal X-linked dominant allele (20).
Postzygotic de novo mutations initiate all forms of genomic mosaicism, which is further classified on the basis of the presence or absence ofmutation in gonadal tissue, and hence, its heritability (16).Germline—or gonadal—mosaicism describes genetic heterogeneity within the gametes, permitting mutations to be inherited and expressed constitutionally by subsequent generations (9), whereas somatic mosaicism precludes mutation in gonadal tissues, restricting mutation to somatic cells and, consequently, the proband (57). Gonosomal mosaicism is the co-occurrence of both somatic and germlinemosaicism, where mutation is found in both somatic and gonadal tissue, and results from an early mutagenesis event (9, 116). When somatic mosaicism is present, the phenotypic consequence ofmosaicism is largely determined by cell lineage, gene function, and mutation timing (Figure 1) (16). Mutations earlier in embryogenesis likely affect pluripotent or multipotent cells, leading to multilineage effects observed in syndromic disorders such as epidermal nevus syndrome (58).
Figure 1. Mutation timing and end organ involvement.
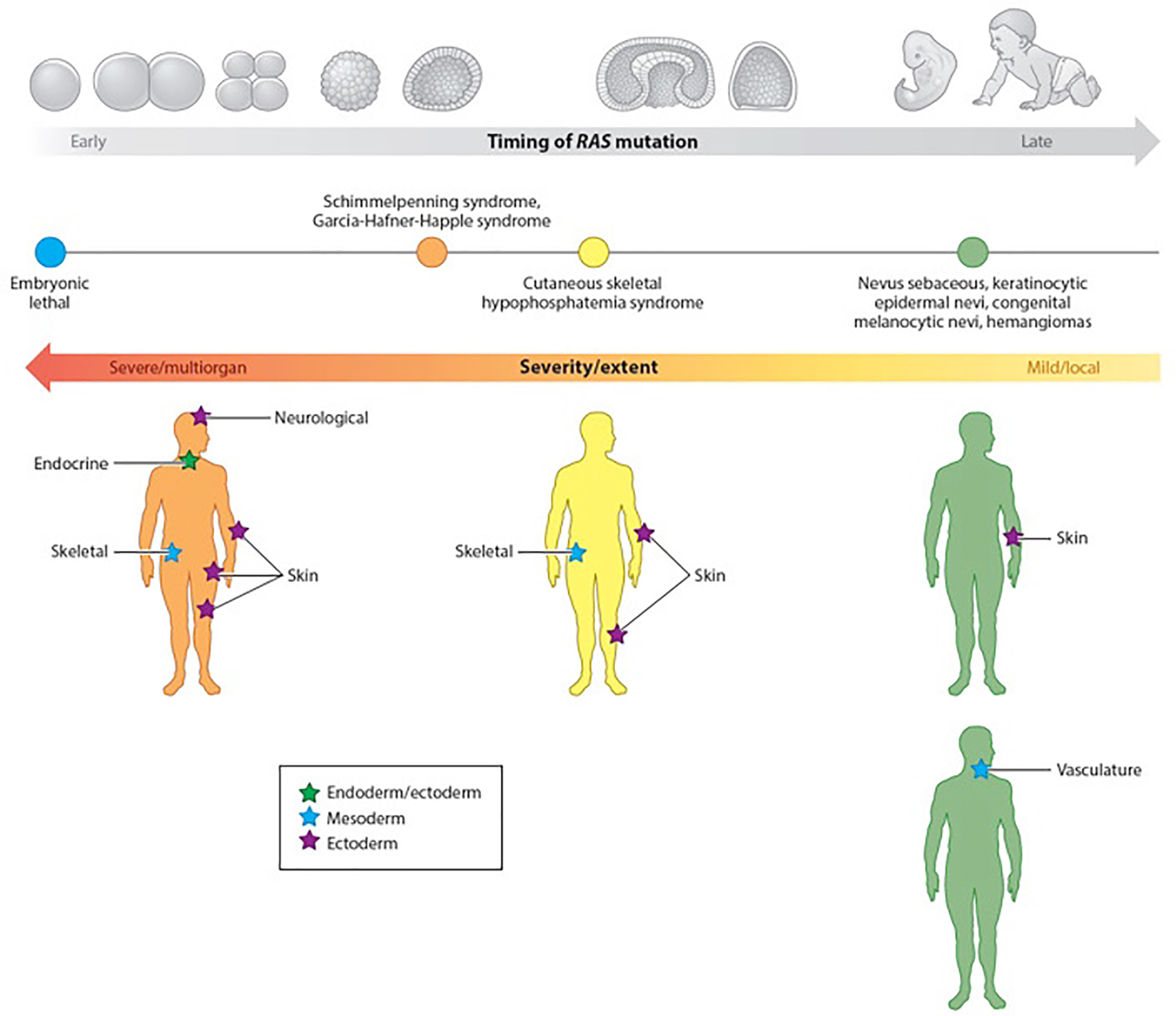
Clinical manifestation of cutaneous, genomic mosaic disorders largely depends on the timing of mutation. Activating RAS mutations demonstrate a pleiotropic phenotype, with the severity and extent of disease dependent on the timing of mutation and the corresponding potency of the affected cell. The spectrum of disease includes effects on the endoderm (green star), mesoderm (blue stars), and ectoderm (purple stars). Stochastic, postzygotic mutation of a cell early in development leads to multilineage disease affecting end organs of all germ layers, as found in Schimmelpenning syndrome and Garcia-Hafner-Happle syndrome, both of which demonstrate neurological, endocrine, skeletal and skin phenotypes. Disorders such as cutaneous skeletal hypophosphatemia syndrome and phacomatosis pigmentokeratotica, which have phenotypes in the skin and bone or keratinocyte and melanocyte, respectively, result from mutation later in development of a cell with less potency. Mutation occurring in cells that have fully committed to one lineage (e.g., keratinocyte, melanocyte, blood vessels) is reflected by a milder clinical phenotype, in which the RASopathy is nonsyndromic and may be isolated tumors or single lesions.
Cutaneous mosaic disorders offer a unique opportunity to investigate genetic mosaicism, as lesional skin is easily identified and mutant cells can be isolated via skin biopsy. Further, predictable embryonic dorsoventral migration patterns of ectodermal progenitors, which differentiate into the neural crest and cutaneous ectoderm, result in linear bands of involved tissue known as the lines of Blaschko in the setting of embryonic somatic mutation (17, 63). Somatic mosaicism can also give rise to proliferative/hamartomatous lesions such as congenital hemangiomas and nevi. Recently, a new system of classification of mosaic skin disorders based on genomic versus epigenetic etiology, and segmental versus nonsegmental distribution, was proposed (59). In this review, we discuss exemplary mosaic skin disorders, focusing on understanding pathogenesis and identification of potential targets for therapy. However, we must first review the types of genomicmosaicism found in genodermatoses and the current technologies employed to identify disease-causing mutations.
MENDELIAN OR MOSAIC?
Mosaicism is not independent fromMendelian inheritance, and the two are notmutually exclusive. The same genetic mutations underlying heritable disorders can occur in mosaic patterns, as in segmental neurofibromatosis type 1 (SNF1) due to somatic mutation in neurofibromin 1 (NF1) (91, 125). Unlike the generalized form of neurofibromatosis type 1 (NF1) caused by inherited autosomal dominant NF1 mutation, demonstrating disseminated neurofibromas, Lisch nodules of the iris, intellectual disability, and numerous café-au-lait spots, SNF1 presents with lesions occurring in a single, unilateral segment of the body, and these do not cross the midline (91, 105). A lack of systemic involvement or family history of disease is common in segmental disorders (47). Notably, the identical p.R1947X nonsense mutation in NF1 can lead to both SNF1 (32) and NF1 (142). Parents with SNF1 can also produce offspring with generalized NF1 in cases where gonosomal mosaicism underlies the segmental phenotype (32). Mutations that are only found in mosaic disease are likely lethal when constitutionally expressed even with widespread systemic involvement and suspected gonosomal mosaicism. This is likely the case for the activating p.E17K AKT1 mutation leading to the Proteus syndrome, which features segmental overgrowth of various tissues and organs, skin lesions including lipomas, nevi, and café-au-lait macules, and an increased susceptibility to developing tumors (88). To date, neither constitutional mutation nor intergenerational transmission has been reported, and the mutation is presumed to be embryonic lethal (21). Another example of obligate mosaic mutation is the activating GNAS1 mutations that lead to McCune-Albright syndrome (MAS) (139). Precocious puberty, which is part of the characteristic triad of MAS alongside café-au-lait spots and polyostotic fibrous dysplasia, results from large ovarian cysts secreting high levels of estrogen, and despite this confirmed presence of GNAS1 mutation in ovarian cells, the mutation is presumed to be lethal when constitutional (78,104). Other examples of presumed obligatory somatic mutations include activating mutations in IDH1 leading to Maffucci syndrome (3); PIK3CA mutations in congenital lipomatous overgrowth with vascular, epidermal, and skeletal anomalies (CLOVES) syndrome (76); KRAS mutations in Schimmelpenning syndrome (51); and activating GNAQ mutations underlying Sturge-Weber syndrome (115). Mendelian inheritance of other mutations may potentiate disease or alter its severity following postzygotic acquisition of a second mutation, according to Knudson’s two-hit model of disease pathogenesis (124). These second-hit scenarios can generate highly variable phenotypes of a dominantly inherited disorder, as seen in tuberous sclerosis complex (TSC), in which various non-nervous-system tumors can arise from second somatic mutations in TSC1, TSC2, and KRAS(102). These second hits may be loss of heterozygosity (LOH) of the wild-type allele leading to type 2 segmental mosaicism, whereby the areas with LOH display a more severe phenotype in the setting of milder, diffuse disease, as seen in Hailey-Hailey and Darier diseases (60, 99). This is in contrast to type 1 segmental mosaicism, whereby the mutation is confined to the affected tissue in an otherwise wild-type individual (Figure 2) (99).
Figure 2. Types of segmental mosaicism.
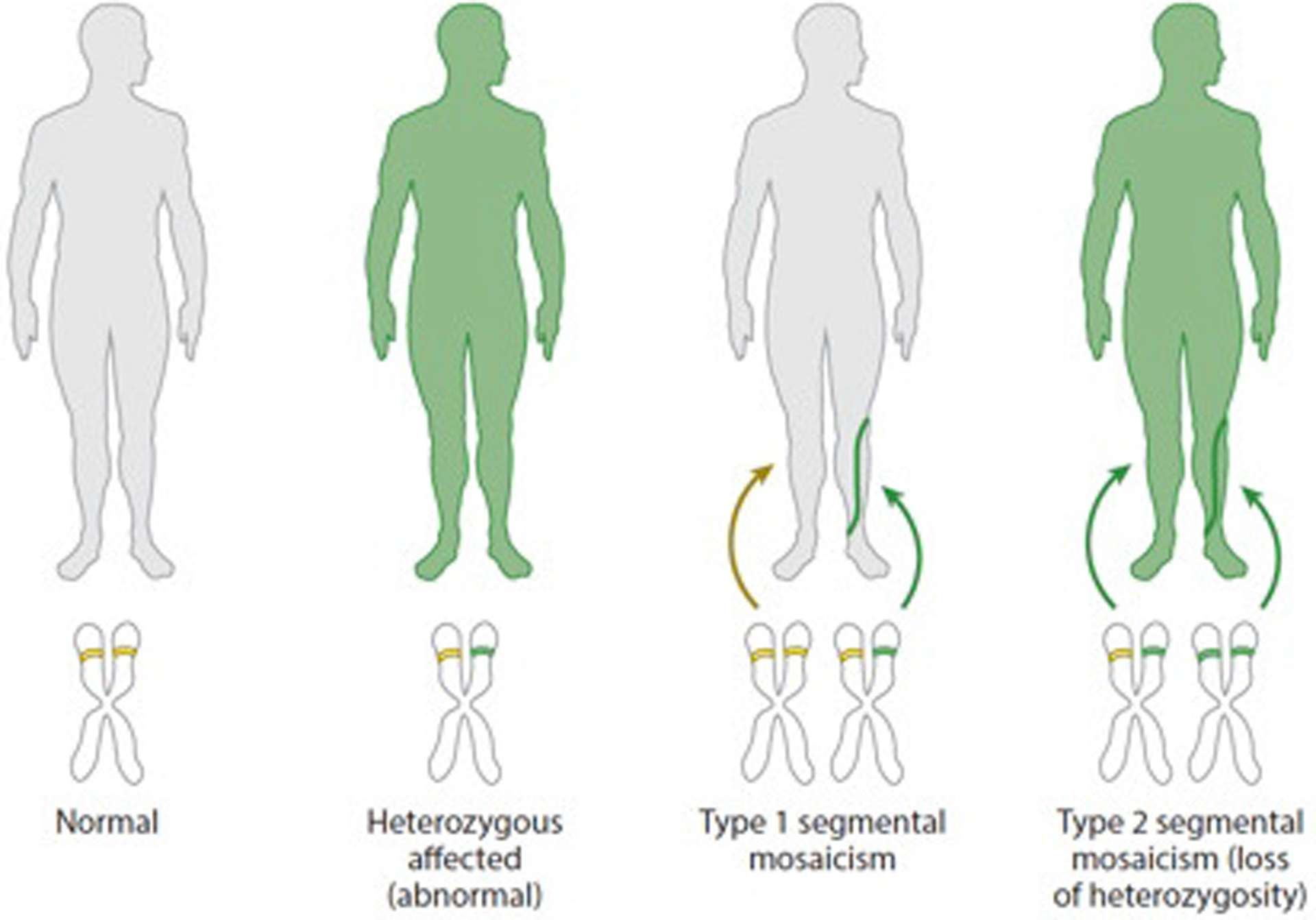
Segmental mosaicism refers to the Blaschko-linear patterning of cutaneous lesions that respect the midline: Single postzygotic mutations leading to segmental lesions reflect type 1 segmental mosaicism, by which the mutation is limited to lesional tissue in an otherwise wild-type individual. Type 2 segmental mosaicism occurs when a second mutagenesis event, such as loss of heterozygosity, occurs in individuals already carrying heritable mutations, leading to segments with more severe phenotype.
IDENTIFYING GENETIC MOSAICISM IN SKIN DISORDERS
Genetic mosaicism is detected by comparing affected tissue to unaffected tissue from the same individual. The study of mosaicism was historically restricted to those disorders with visible clinical features that permitted collection of lesional tissue with sufficient purity (16, 45). Even among cutaneous mosaic disorders distinguished by clear phenotypic distinctions of texture, color, thickness, or other features, a low mutant allele fraction due to admixture with wild-type cells can prevent the detection of mutation (45, 131). Selection of the appropriate unaffected control tissue is also critical, and in cutaneous mosaic disorders, buccal swabs, saliva, or blood is commonly employed. Adjacent, phenotypically normal-looking skin may be a suitable control in cases where the mutation is limited to the affected tissue (type 1 segmental mosaicism). In cases where type 2 segmental mosaicism is suspected, comparison of SNP genotyping data between the segmental lesion and adjacent tissue or blood can help identify LOH (22) (see the section titled Microarray SNP Genotyping). Syndromic cutaneous disorders with widespread skin involvement may also have a subset of mutant cells in other tissues including saliva and blood, complicating identification of an appropriate normal control.
Laser Capture Microdissection
Isolation of pure mutant cell populations can be limited in routine skin biopsy by the presence of unaffected cells of the same or different lineages. For example, in a keratinocytic disorder, a punch biopsy is contaminated by dermal and subdermal fibroblasts, adipocytes, and endothelial cells. Furthermore, disorders accompanied by inflammation exhibit variable degrees of immune infiltration, which can further dilute the proportion of mutant cells (110). When histologic features or fluorescent markers can precisely delineate affected tissue, laser capture microdissection (LCM) can be employed to increase the fraction of affected cells sampled forDNAisolation (15, 96). LCM is a microscope-controlled technique using a focused laser beam to selectively isolate single cells or areas of tissue without compromising genetic content, and it was first successfully used for PCR-based mutation analysis in 1996 (40). Briefly, frozen or paraffin-embedded tissues are sectioned onto polyethylene naphthalate membrane-coated slides, which can be cut by the laser during capture (75). Dissected pieces of tissue fall directly into a lysis reservoir for immediate DNA extraction. Guiding the laser to areas of disease limits admixture from nearby cells, aiding the detection of low fraction mutations lying within complex tissues (34). Comparison of sequences from laser-captured lesions to control DNA can successfully identify mosaic mutations.
Mutation Detection
Early studies of mosaicism investigated chromosome integrity via cytogenetic analyses such as karyotyping and fluorescence in situ hybridization, which were employed to identify distinct karyotypes and aneuploidy within patient cells. These methods were successfully employed to identify genetic mechanisms in some of the earlier disorders to be solved, including the mixture of XY and XO sex-chromosome bone marrow cells in hermaphrodites (62) and trisomy 8 mosaicism syndrome (73). Advances in genotyping and sequencing technology, and the availability of large data repositories, have enabled the rapid detection and functional annotation of mutations underlying mosaic skin disorders.
Microarray SNP Genotyping
Mosaic chromosome-level abnormalities including small copy number variations require alternate identification strategies with higher resolution, and microarray-based techniques such as array comparative genomic hybridization and SNP genotyping have demonstrated marked sensitivity, detecting mosaicism at levels of <5% aneuploidy (31). Genome-wide analysis of SNPs across all chromosomes provides visualization of regions of LOH by plotting the B allele frequency and surveying for deviations from baseline that demonstrate loss of one haplotype. To differentiate between LOH resulting from deletion and copy-neutral LOH that can occur through recombination, the LogR ratio, which is the ratio of measured SNP signal intensity to the expected intensity of two alleles, is also measured. Targeted arrays permit assessment of genotypes in focused genomic regions. SNP genotyping was used to identify the copy-neutral LOH at the qter of chromosome 17 underlying the revertant mosaicism (RM) found in ichthyosis with confetti (25).
Whole Exome Sequencing
First employed to identify a disease-causing gene in an inherited disease in 2005, high-throughput sequencing platforms have permitted large-scale, low-cost DNA sequencing, accelerating the discovery of genetic mutations underlying human disorders and giving rise to the field of functional genomics (11). Although they comprise merely 1% of the human genome, the coding regions, also known as the exome, are estimated to host approximately 85% of disease-causing mutations. As such, limiting sequence analysis to the exome is a time-saving and cost-effective approach for identifying pathogenic mutations (27, 103). Whole exome sequencing (WES) platforms share a central workflow of hybridizing fragmented genomic DNA to exon-specific probes that isolates the exons and flanking sequences from nontargeted DNA. The resulting fragments are amplified for high-throughput sequencing (48, 94). Reads are aligned to the human genome and processed with custom or commercialized pipelines, such as the Genome Analysis Toolkit (GATK) Best Practices or VarScan, to then generate a variant call format (.vcf) file containing the proper headers and formatting for annotating mutations (37, 71).
Somatic variants underlying cutaneous mosaic disorders require paired WES, which includes adjacent sequencing and analysis of a controlDNA, most often blood (leukocytes) or a saliva/buccal swab (oral epithelial cells). Additional software such as MuTect/MuTect2 (Broad), Ingenuity Variant Analysis (QIAGEN), and Strelka can help automate the comparison of variants from lesional and control sequencing data and selectively identify the sites of difference by filtering for mutations unique to, or enriched in, the lesion (12, 28, 109). In cases where the causal mutation is intronic or lies within regions of transcriptional regulation, whole genome sequencing may be necessary (138). It is important to note that carefully selecting the source of control DNA is critical to ensure successful discovery of somatic mutations, as widespread mosaicism can affect end organs without an overt phenotype and lead to failed detection (35).
MOSAIC CUTANEOUS RASOPATHIES AND THE MAPK PATHWAY
Activating mutations in the Ras family of GTPases, including HRAS, KRAS, and NRAS, cause inherited and somatic mosaic disease as well as up to 30% of cancers (100, 101, 111). Like other G-proteins, Ras proteins exist in a binary “on” or “off ” state, characterized by their binding to guanosine triphosphate (GTP) or guanosine diphosphate (GDP), respectively (100). Following activation, Ras feeds two major signaling cascades: the Ras-Raf-MEK-ERK pathway and the PI3K-Akt pathway (111). Critical amino acid residues at codons 12 (glycine), 13 (glycine), and 61 (glutamine) mediate the hydrolysis of GTP to GDP necessary for proper inactivation of active Ras. Indeed, the missense mutations affecting these residues, which lead to a constitutively active GTP-bound Ras, account for the majority of identified mutations in cancer and RASopathies (55, 100). Most are considered embryonic lethal as they are not observed in a constitutional state, and widespread embryonic expression of KRAS p.G12D in amousemodel was demonstrated to be uniformly lethal (129). Missense mutations at other loci of KRAS, such as p.V14I and p.T58I, lead to only a mild reduction of GTPase activity and can constitute germline disorders like Noonan syndrome and cardiofaciocutaneous syndrome (36, 112). Similarly, the recurrent p.G12S HRAS mutation underlying the germline Costello syndrome was determined to be less potent than valine or aspartic acid substitutions at this position (4, 43). The attenuated molecular consequences of these alternate mutations evade the embryonic lethality characteristic of more strongly activating mutations.
NS and keratinocytic epidermal nevi (KEN) are examples of segmental mosaic RASopathies and appear linearly along Blaschko’s lines. Both can result from somatic activating mutations in one of the three Ras family members (51) and are benign; congenital lesions occur in 1–3 per 1,000 births (51). KEN are nonorganoid, appearing as lines and swirls of pigmentation frequently on the trunk or extremities, whereas NS are organoid nevi that appear as waxy plaques most commonly on the scalp, face, and neck (13). Secondary neoplasms like syringocystadenoma papilliferum (SCAP) can arise within these nevi and harbor the same RAS mutation as their associated nevi (51, 79, 97). Although the mutation is restricted to the epidermis in NS and KEN, somatic RAS mutations occurring earlier in embryogenesis can affect other cell lineages and end organs. Mutation in a multipotent progenitor cell giving rise to keratinocytic and melanocyte lineages causes phacomatosis pigmentokeratotica, a combination of NS and speckled lentiginous nevi occurring in the Blaschkoid pattern (52).Cutaneous skeletal hypophosphatemia syndrome (CSHS) is the clinical manifestation of RAS mutation affecting a progenitor cell prior to gastrulation, such that the phenotype affects not only the skin but also the mesoderm-derived skeleton and other organs. In CSHS, Ras activation in the bone leads to abnormally elevated levels of serum FGF23, causing skeletal dysplasia and rickets due to renal phosphate wasting (85).Earlier mutation targeting a cell with even greater potency leads to Schimmelpenning-Feuerstein-Mims (SFM) syndrome, which describes a more widespread constellation of symptoms involving the brain (seizures, mental retardation, structural defects), eyes (ocular colobomas), bone (osteomalacia, hypophosphatemic rickets), and skin (KEN and NS) (135, 137). In cases of SFM and CSHS, the identical RAS mutation can be isolated from the affected skin, skeleton, and other tissues, implicating a single mutant progenitor (42).
Spitz nevi and GCMN are melanocytic lesions caused by activating RAS mutations, predominantly in HRAS at glycine 13 and NRAS at glutamine 61, respectively (70, 107). Unlike NS and KEN, these lesions are nonsegmental: Spitz nevi are single point mosaic lesions occurring as a solitary benign tumor, whereas GCMN represent patchy mosaicism, often appearing in a coat-like pattern crossing the midline (59). Syndromic cases involving these melanocytic lesions are similar to the epidermal nevus syndromes, though GCMN syndrome can be associated with characteristic facies, including a wide forehead, orbital hypertelorism, a broad nasal tip, a prominent/long philtrum, and an everted lower lip (69). One of the first five cases of CSHS was a four-year-old girl with GCMN (85).
Recently, a subset of childhood hemangiomas was also identified as arising from somatic RAS mutation, adding to the spectrum of cutaneous RASopathies and nonsegmental mosaic tumors (53, 84). The most common tumors of infancy, hemangiomas affect 5–10% of all newborns, though most are benignGLUT-1-positive infantile hemangiomas that will spontaneously regress by three years of age (143). Activating RAS mutations are estimated to be responsible for up to 10% of childhood lobular capillary hemangiomas [also known as pyogenic granulomas (PGs)], as well as a small population of GLUT-1-negative, non-involuting congenital hemangiomas (NICHs).
Mutation in non-Ras members of the MAPK pathway, like BRAF, are frequently found in disorders classified as cutaneous RASopathies, including SCAP(79) and melanocytic nevi (74). The BRAF p.V600E mutation, which is the most common variant identified in mosaic disorders and cancer, dramatically increases the BRAF kinase activity, and, like activating RAS mutations, leads to increased phosphorylation of ERK1/2 to promote oncogenic MAPK activity. Alternatively, the discovery of novel mutations in disorders classified as RASopathies has helped to identify new proteins regulating the Ras-MAPK pathway. For example, activatingmutations of the Gaq family, including GNAQ, GNA11, and GNA14, were recently found to cause NICHs and PGs, the same group of vascular tumors arising from RAS and BRAF mutations (8, 82), and somatic mutation in GNAQ and GNA11 was also found to cause phacomatosis pigmentovascularis, a combination of melanocytic nevi and vascular birthmarks (123). Previously, mutations in GNAQ and GNA11, specifically at arginine 183 and glutamine 209, were associated with uveal melanoma and blue nevi (133, 134), whereas somatic GNAQ mutation at arginine 183 was associated with Sturge-Weber syndrome (115), a neurocutaneous disorder characterized by a congenital, extensive port-wine stain of the face involving the trigeminal nerve, leptomeningeal angioma, and variable neurological symptoms including seizures, hemiparesis, and glaucoma (120). The somatic mutations of Gaq family members occur at key residues (GNAQ and GNA11: arginine 183 and glutamine 209; GNA14: glutamine 205) located in their respective catalytic domains. The residues facilitate GTP hydrolysis, and their mutation leads to a constitutively active conformation of the Gaq proteins (8, 82).Gaq mutations were also recently found to specifically upregulate the MAPK pathway, but not the Akt-mTOR pathway (8, 53, 82, 84), and their expression in primary human melanocytes and umbilical vein endothelial cells was found to render the cells growth factor independent, suggestive of oncogenic transformation. Vascular anomalies caused by Gaq mutation can also be associated with disseminated nonsegmentalmosaicism, like the recently reported case of congenital hemangiomatosis with dozens of lesions all harboring the same GNA11 p.Q209P mutation (46).
Activation of Ras initiates a complex cascade of downstream signals involving diverse pathways including ERK-MAPK, and it is notable that mutations in GNAQ, GNA11, GNA14, BRAF, and RAS members give rise to similar vascular anomalies and, in the case of PGs, the same tumor. Therefore, it is intriguing to consider whether targeted inhibition of the MAPK pathway (e.g., MEK inhibitors) may prove effective against vascular lesions that are refractory to treatment. Furthermore, it is interesting to consider whether other G-protein disorders, likeMAS, that arise from a multilineage activating GNAS mutation leading to café-au-lait spots, polyostotic fibrous dysplasia, and precocious puberty may also respond to MAPK inhibition.
Recurrent FGFR1 p.N546K and p.K656E mutations were recently found to cause encephalocraniocutaneous lipomatosis, a neurocutaneous disorder featuring unilateral lipomatous cutaneous neoplasms devoid of hair, and both mutations were found to upregulate the MAPK pathway (14). Previously, other mosaic FGFR1 disorders, like dysembryoplastic neuroepithelial tumor (106) and pilocytic astrocytoma (65), were found to result from activating RAS or BRAF p.V600E mutations (64). Hence, like the G-protein disorders, cutaneous disorders resulting from activating mutations in FGFR1 may also respond to agents targeting the MAPK pathway.
EPIGENETIC MOSAIC SKIN DISORDERS
In the absence of postzygotic genetic mutation, alterations of gene expression can be induced by retrotransposon insertion, methylation/demethylation, and imprinting, which cause functional, rather than genomic, mosaicism. In such cases, a genomic mutation capable of causing an observable phenotype may be inherited, but its expression is mosaically modified. Random X-chromosome inactivation (XCI), which depends on the differential expression of the XIST RNA gene on the X-chromosome, occurs during female embryogenesis and can generate regions of healthy and diseased skin in the setting of X-linked inheritance, often occurring in the Blaschkoid pattern. X-linked epigenetic mosaic skin disorders can be largely divided into malelethal [X-linked dominant, e.g., incontinentia pigmenti (IP)], sublethal (e.g., Menkes disease), and nonlethal disorders (e.g., hypohidrotic ectodermal dysplasia) (59). IP, which is caused by genomic rearrangements within the X-linked NEMO gene encoding a regulatory component of the IκB kinase that activates the NF-κB pathway, features perinatal inflammatory vesicles that resolve into patterns of hyperpigmentation and dermal scarring alongside other highly variable abnormalities of the nails, teeth, eyes, and nervous system (117). Early cytolysis of cells selectively expressing the mutant X-chromosome leads to skewed XCI in female patients, and, in lesional tissue, activity of themutant allele interferes with NF-κB activation (117). Conradi-Hünermann-Happle syndrome is another example of an X-linked dominant, epigenetic cutaneous mosaic disorder, and it is due to differential expression of the X-linked EBP gene that encodes sterol isomerase, an enzyme critical for cholesterol biosynthesis (19). Affected skin expressing the EBP mutation is characterized by follicular atrophoderma and diffuse erythema with scaly keratotic lesions (44). Other associated systemic features can include alopecia, asymmetrical limb shortening, cataracts, and skeletal dysplasias (44). Rarely, survival of males with these traditionally X-linked lethal mosaic disorders has also been reported, bypassing in utero lethality via hypomorphic mutations, a 47,XXY karyotype (Klinefelter’s syndrome), or somatic mosaicism (6, 68).
Menkes disease is an X-linked recessive disorder due to mutation of ATP7A, a copper transporting P-type ATPase leading to copper deficiency, characterized by neurodegeneration, kinky hair, and connective tissue disease (128). Skewed XCI of the normal X-chromosome leads to a mild phenotype in the mosaic females compared with affected males (38, 92) who will rarely survive to adolescence (128). In nonlethal disorders like X-linked recessive hypohidrotic ectodermal dysplasia (also known as Christ-Siemens-Touraine syndrome), which demonstrates the triad of sparse hair (hypotrichosis), dental problems (hypodontia/anodontia), and the inability to sweat (hypohidrosis/anhidrosis) due to mutation in X-linked EDA/EDAR, females demonstrate abnormal skin temperature and sweating along the pattern of the lines of Blaschko (29). In fact, sweat testing has been suggested to identify female carriers (30).
Effective therapeutics for epigenetic mosaic skin disorders have not been identified. Although studies have revealed factors regulating the initiation of XCI like the X-inactivation center (Xic), dependable means of skewing XCI do not yet exist (7, 122). The utility of miRNAs, which are noncoding RNAs that act as posttranscriptional regulators of gene expression by targeting and cleaving mRNA, has been discussed in the treatment of human cancers, although both the delivery of miRNAs to vital layers of the skin and their potential cellular toxicity continue to be challenges (93). For mosaic epigenetic skin disorders, therapy is therefore limited to supportive care and symptom management.
REVERTANT MOSAICISM
Whereas mosaic pathogenic mutations lead to areas of lesional skin, RMappears on a background of completely affected skin, with genetic self-correction giving rise to patches of healthy skin. Several genetic mechanisms underlie RM. Back mutation site-specifically corrects the disease-causing mutation. Second-site mutations are compensatory secondary mutations that mitigate the effects of a disease-causing mutation, such as an insertion mutation via DNA polymerase slippage that restores a normal reading frame, or an enhancer or promoter mutation that silences a mutant gene. Finally, gene conversion or mitotic recombination can remove the mutant haplotype via nonreciprocal or reciprocal exchange of genetic information between sister chromatids, respectively, during repair of a double-strand break (39, 61, 66, 77). Although it is rare overall, RM is more common in some cutaneous disorders: Up to 36% of COL17A1 mutation-mediated epidermolysis bullosa (EB) patients and 33% of LAMB3 mutation-mediated EB patients demonstrate RM as patches of visibly normal skin with recovered expression of these genes (67, 95).Numerous revertant patches may occur within a patient over time via independent reversion events, and these areas may result from one or more distinct corrective mechanisms (25, 67).
Ichthyosis with confetti (IWC) is unique among the cutaneous disorders with RM given its striking phenotype and remarkable frequency of reversion: Individuals initially born with generalized erythema and scaling due to an autosomal dominant KRT10 or KRT1 mutation develop thousands of white, histologically and clinically normal skin areas, each representing an independent revertant clone arising via mitotic recombination leading to copy-neutral LOH that removes the mutant haplotype (25, 26, 86, 121). Other variable clinical features include ear malformations, mammillae hypoplasia, palmoplantar keratoderma, and hypertrichosis (54, 119). Unlike other keratinopathies, such as epidermolytic ichthyosis (EI), the mutations causing IWC have thus far been frameshift mutations targeting the tail domain of KRT10 or KRT1, often replacing their endogenous glycine-serine-rich tails with polyarginine or polyalanine tails leading to mislocalization of the keratin from the cytosol to the nucleus and/or nucleolus (25, 26, 86, 121). Notably, no cases of EI have been reported to demonstrate RM, and no reversion has been found in animal models of EI that faithfully recapitulate the disease phenotypes of generalized erythema, cutaneous blistering, hyperkeratosis, and palmoplantar keratoderma (5) (Figure 3). This further highlights the distinction of IWC mutations and implicates a potential role of keratin tail domains in genetic reversion (83).
Figure 3. Epidermolytic ichthyosis (EI) versus ichthyosis with confetti (IWC).
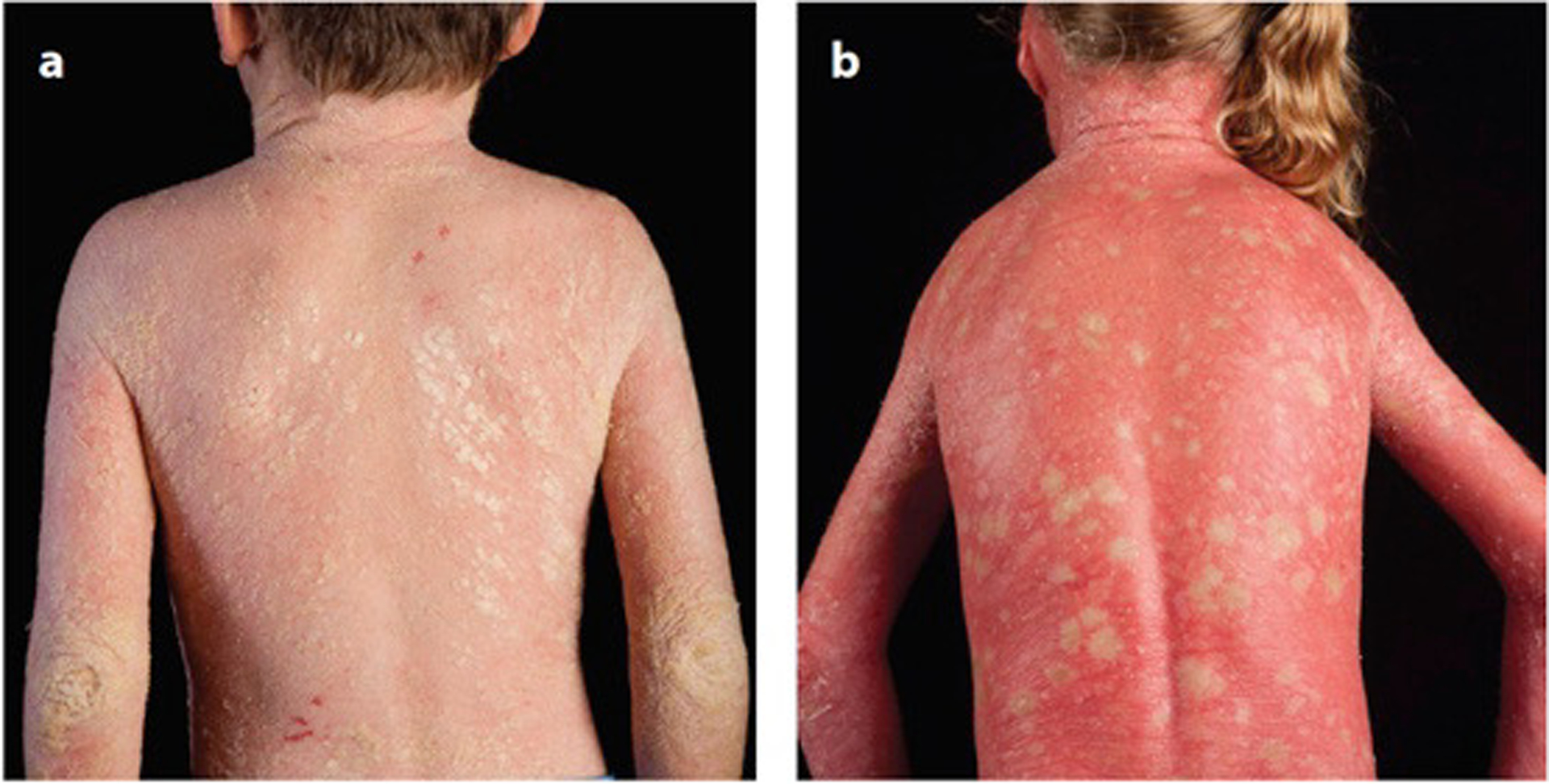
Age-matched patients with (a) EI due to KRT10 p.R156H mutation with extensive hyperkeratosis and plate-like scales and (b) IWC due to KRT10 mutation with erythroderma and confetti macules. Revertant mosaicism in IWC is demonstrated by the white macules of normal skin that are also histologically normal. To date, IWC has been associated with frameshift mutations targeting only the tail domains of KRT10 or KRT1, whereas mutations at other sites, such as the helical rod domains, lead to EI, which does not show clinical evidence of reversion.
In IWC, the size and number of revertant spots increase with age, suggesting that revertant keratinocytes have improved fitness over mutant cells. However, studies that have attempted to culture revertant basal stem cells isolated from biopsies of patients with revertant EB found mutant keratinocytes to be dominant, with revertant cells declining down to <1% of the population in subsequent passages (50). Autologous grafting of revertant skin to other areas of the body has had mixed results; functional repair of the graft has previously failed in COL17A1 mutant junctional EB (49), whereas punch grafting of split-thickness revertant biopsy specimens has been successful in an EB patient with LAMB3 mutation (49, 50). Alternatively, induced pluripotent stem cells (iPSCs) generated from revertant keratinocytes (132), or virus-associated editing of mutant iPSCs to wild type (113), have successfully been employed for transplantation in animal models, suggesting the potentials of stem cell therapy and genome editing in cutaneous disorders with RM. Finally, recessive dystrophic EB patients undergoing allogeneic bone marrow transplantation were reported to generate revertant patches with prolonged survival and COL7A1 expression in the basement membrane, and—with a large population of CD45 donor cells—in the skin. This finding suggests that hematopoietic stem cells can populate the skin and that such cells may be one potential source of revertant clones (126, 136). Indeed, bone marrow cells have previously been associated with the skin: Following cutaneous wounding in mice, bone marrow–derived cells were observed repopulating the skin (10), and some of these could potentially differentiate into keratinocytes (141).
Although clinical observations of RM in patients suggest expansion of revertant clones over time, this stands in contrast to the apparent reduced fitness observed to date in in vitro experiments. This paradox may result from the use of ex vivo culture, but specific mechanisms leading to the appearance of RM clones and their expansion over time remain unknown. Further, most genodermatoses do not exhibit clinically apparent reversion, and determining why only a subset of disorders undergo RM will be critical to understanding this phenomenon.
CONCLUSION
The discovery of novel mutations underlying cutaneous mosaic disorders continues at an unprecedented pace, supported by technological advances in genetic analysis. For example, just within the last two years, besides the identification of GNA14, GNA11, and GNAQ mutations causing childhood vascular tumors, somatic recurrent p. L412F mutation in SMO was found to cause Curry-Jones syndrome (130), NEK9 mutations were found to cause nevus comedonicus (80), and somatic PIK3CA and MAP3K3 mutations were found to cause venous malformations (33, 87). Functional investigation of these genetic mutations will enhance our understanding of epithelial biology and will identify novel therapeutic targets in the treatment of these disorders. Furthermore, the coexistence of wild-type and mutant cells, which defines mosaicism, has been shown to generate a unique cellular milieu involving cytokines and paracrine signaling pathways like the JAK-STAT system that modulates cell–cell competition, proliferation, and apoptosis (72, 140). Understanding the biology at this interface of mosaic-affected and adjacent normal tissue will provide new insights into cancer, signaling pathways, and other systemic disorders.
ACKNOWLEDGMENTS
This work was supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award and National Institutes of Health (NIH)/National Institute of Arthritis and Musculoskeletal and Skin Diseases grants K08AR056305, R01AR062111, and R01AR071491 to K.A.C. Y.H.L. is supported by a Doris Duke Charitable Foundation Clinical Mentorship Award and the Medical Scientist Training Program at Yale University (NIH/National Institute of General Medical Sciences T32GM007205).
REFERENCES CITED
- 1.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. 2011. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333:1154–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahuja YR. 1960. A human mosaic involving eye and hair color differences. Acta Genet. Med. Gemellol. (Roma) 9:427–31 [DOI] [PubMed] [Google Scholar]
- 3.Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, et al. 2011. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet 43:1262–65 [DOI] [PubMed] [Google Scholar]
- 4.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, et al. 2005. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet 37:1038–40 [DOI] [PubMed] [Google Scholar]
- 5.Arin MJ, Longley MA, Wang XJ, Roop DR. 2001. Focal activation of a mutant allele defines the role of stem cells in mosaic skin disorders. J. Cell Biol 152:645–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold AW, Bruckner-Tuderman L, Has C, Happle R. 2012. Conradi-Hünermann-Happle syndrome in males vs. MEND syndrome (male EBP disorder with neurological defects). Br. J. Dermatol 166:1309–13 [DOI] [PubMed] [Google Scholar]
- 7.Augui S, Nora EP, Heard E. 2011. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet 12:429–42 [DOI] [PubMed] [Google Scholar]
- 8.Ayturk UM, Couto JA, Hann S, Mulliken JB, Williams KL, et al. 2016. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am. J. Hum. Genet 98:789–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachoo S, Gibbons RJ. 1999. Germline and gonosomal mosaicism in the ATR-X syndrome. Eur. J. Hum. Genet 7:933–36 [DOI] [PubMed] [Google Scholar]
- 10.Badiavas EV, Abedi M, Butmarc J, Falanga V, Quesenberry P. 2003. Participation of bone marrow derived cells in cutaneous wound healing. J. Cell. Physiol 196:245–50 [DOI] [PubMed] [Google Scholar]
- 11.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, et al. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet 12:745–55 [DOI] [PubMed] [Google Scholar]
- 12.Bao R, Huang L, Andrade J, Tan W, Kibbe WA, et al. 2014. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform. 13:67–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baykal C, Yazganoglu KD. 2014. Clinical Atlas of Skin Tumors. New York: Springer [Google Scholar]
- 14.Bennett JT, Tan TY, Alcantara D, Tétrault M, Timms AE, et al. 2016. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am. J. Hum. Genet 98:579–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhattacherjee V, Mukhopadhyay P, Singh S, Roberts EA, Hackmiller RC, et al. 2004. Laser capture microdissection of fluorescently labeled embryonic cranial neural crest cells. Genesis 39:58–64 [DOI] [PubMed] [Google Scholar]
- 16.Biesecker LG, Spinner NB. 2013. A genomic view of mosaicism and human disease. Nat. Rev. Genet 14:307–20 [DOI] [PubMed] [Google Scholar]
- 17.Blaschko A 1901. Die Nervenverteilung in der Haut in ihrer Beziehung zu den Erkrankungen der Haut. Breslau, Ger.: W. Braumüller [Google Scholar]
- 18.Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, et al. 2016. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet 48:398–406 [DOI] [PubMed] [Google Scholar]
- 19.Braverman N, Lin P, Moebius FF, Obie C, Moser A, et al. 1999. Mutations in the gene encoding 3β-hydroxysteroid-Δ8,Δ7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat. Genet 22:291–94 [DOI] [PubMed] [Google Scholar]
- 20.Bridges CB. 1925. Elimination of chromosomes due to a mutant (Minute-n) in Drosophila melanogaster. PNAS 11:701–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. 2015. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 31:382–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castellanos E, Bielsa I, Carrato C, Rosas I, Solanes A, et al. 2015. Segmental neurofibromatosis type 2: discriminating two hit from four hit in a patient presenting multiple schwannomas confined to one limb. BMC Med. Genom 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Centerwall WR, Benirschke K. 1973. Male tortoiseshell and calico (T-C) cats. Animal models of sex chromosome mosaics, aneuploids, polyploids, and chimerics. J. Hered 64:272–78 [DOI] [PubMed] [Google Scholar]
- 24.Cheever MA, Disis ML, Bernhard H, Gralow JR, Hand SL, et al. 1995. Immunity to oncogenic proteins. Immunol. Rev 145:33–59 [DOI] [PubMed] [Google Scholar]
- 25.Choate KA, Lu Y, Zhou J, Choi M, Elias PM, et al. 2010. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science 330:94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choate KA, Lu Y, Zhou J, Elias PM, Zaidi S, et al. 2015. Frequent somatic reversion of KRT1 mutations in ichthyosis with confetti. J. Clin. Investig 125:1703–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, et al. 2009. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. PNAS 106:19096–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, et al. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31:213–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark RP, Goff MR, MacDermot KD. 1990. Identification of functioning sweat pores and visualization of skin temperature patterns in X-linked hypohidrotic ectodermal dysplasia by whole body thermography. Hum. Genet 86:7–13 [DOI] [PubMed] [Google Scholar]
- 30.Clarke A, Burn J. 1991. Sweat testing to identify female carriers of X linked hypohidrotic ectodermal dysplasia. J. Med. Genet 28:330–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, et al. 2010. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet 19:1263–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Consoli C, Moss C, Green S, Balderson D, Cooper DN, Upadhyaya M. 2005. Gonosomal mosaicism for a nonsense mutation (R1947X) in the NF1 gene in segmental neurofibromatosis type 1. J. Investig. Dermatol 125:463–66 [DOI] [PubMed] [Google Scholar]
- 33.Couto JA, Vivero MP, Kozakewich HP, Taghinia AH, Mulliken JB, et al. 2015. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am. J. Hum. Genet 96:480–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curran S, McKay JA, McLeod HL, Murray GI. 2000. Laser capture microscopy. Mol. Pathol 53:64–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De S 2011. Somatic mosaicism in healthy human tissues. Trends Genet. 27:217–23 [DOI] [PubMed] [Google Scholar]
- 36.Denayer E, Peeters H, Sevenants L, Derbent M, Fryns JP, Legius E. 2012. NRAS mutations in Noonan syndrome. Mol. Syndromol 3:34–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43:491–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desai V, Donsante A, Swoboda KJ, Martensen M, Thompson J, Kaler SG. 2011. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin. Genet 79:176–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellis NA, Lennon DJ, Proytcheva M, Alhadeff B, Henderson EE, German J. 1995. Somatic intragenic recombination within the mutated locus BLM can correct the high sister-chromatid exchange phenotype of Bloom syndrome cells. Am. J. Hum. Genet 57:1019–27 [PMC free article] [PubMed] [Google Scholar]
- 40.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, et al. 1996. Laser capture microdissection. Science 274:998–1001 [DOI] [PubMed] [Google Scholar]
- 41.Evans DG, Wallace AJ, Wu CL, Trueman L, Ramsden RT, Strachan T. 1998. Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am. J. Hum. Genet 63:727–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farschtschi S, Mautner VF, Hollants S, Hagel C, Spaepen M, et al. 2015. Keratinocytic epidermal nevus syndrome with Schwann cell proliferation, lipomatous tumour and mosaic KRAS mutation. BMC Med. Genet 16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, Wigler M. 1984. Analysis of the transforming potential of the human H-ras gene by random mutagenesis. PNAS 81:4008–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feldmeyer L, Mevorah B, Grzeschik KH, Huber M, Hohl D. 2006. Clinical variation in X-linked dominant chondrodysplasia punctata (X-linked dominant ichthyosis). Br. J. Dermatol 154:766–69 [DOI] [PubMed] [Google Scholar]
- 45.Flaherty P, Natsoulis G, Muralidharan O, Winters M, Buenrostro J, et al. 2012. Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res. 40:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Funk T, Lim Y, Kulungowski AM, Prok L, Crombleholme TM, et al. 2016. Symptomatic congenital hemangioma and congenital hemangiomatosis associated with a somatic activating mutation in GNA11. JAMA Dermatol. 152:1015–20 [DOI] [PubMed] [Google Scholar]
- 47.Gabhane SK, Kotwal MN, Bobhate SK. 2010. Segmental neurofibromatosis: a report of 3 cases. Indian J. Dermatol 55:105–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, et al. 2013. Sequencing studies in human genetics: design and interpretation. Nat. Rev. Genet 14:460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gostynski A, Deviaene FC, Pasmooij AM, Pas HH, Jonkman MF. 2009. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br. J. Dermatol 161:444–47 [DOI] [PubMed] [Google Scholar]
- 50.Gostynski A, Pasmooij AM, Jonkman MF. 2014. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J. Am. Acad. Dermatol 70:98–101 [DOI] [PubMed] [Google Scholar]
- 51.Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, et al. 2012. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet 44:783–87 [DOI] [PubMed] [Google Scholar]
- 52.Groesser L, Herschberger E, Sagrera A, Shwayder T, Flux K, et al. 2013. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J. Investig. Dermatol 133:1998–2003 [DOI] [PubMed] [Google Scholar]
- 53.Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. 2015. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J. Investig. Dermatol 136:481–86 [DOI] [PubMed] [Google Scholar]
- 54.Guerra L, Diociaiuti A, El Hachem M, Castiglia D, Zambruno G. 2015. Ichthyosis with confetti: clinics, molecular genetics and management. Orphanet J. Rare Dis 10:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hafner C, Groesser L. 2013. Mosaic RASopathies. Cell Cycle 12:43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hafner C, López-Knowles E, Luis NM, Toll A, Baselga E, et al. 2007. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. PNAS 104:13450–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall JG. 1988. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am. J. Hum. Genet 43:355–63 [PMC free article] [PubMed] [Google Scholar]
- 58.Halvorsen M, Petrovski S, Shellhaas R, Tang Y, Crandall L, et al. 2016. Mosaic mutations in early-onset genetic diseases. Genet. Med 18:746–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Happle R 2016. The categories of cutaneous mosaicism: a proposed classification. Am. J. Med. Genet. A 170A:452–59 [DOI] [PubMed] [Google Scholar]
- 60.Happle R, Itin PH, Brun AM. 1999. Type 2 segmental Darier disease. Eur. J. Dermatol 9:449–51 [PubMed] [Google Scholar]
- 61.Hastings PJ. 2010. Mechanisms of ectopic gene conversion. Genes 1:427–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirschhorn K, Decker WH, Cooper HL. 1960. Human intersex with chromosome mosaicism of type XY/XO. Report of a case. N. Engl. J. Med 263:1044–48 [DOI] [PubMed] [Google Scholar]
- 63.Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461:1071–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janzarik WG, Kratz CP, Loges NT, Olbrich H, Klein C, et al. 2007. Further evidence for a somatic KRAS mutation in a pilocytic astrocytoma. Neuropediatrics 38:61–63 [DOI] [PubMed] [Google Scholar]
- 65.Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, et al. 2013. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet 45:927–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jonkman MF, Castellanos Nuijts M, van Essen AJ. 2003. Natural repair mechanisms in correcting pathogenic mutations in inherited skin disorders. Clin. Exp. Dermatol 28:625–31 [DOI] [PubMed] [Google Scholar]
- 67.Jonkman MF, Pasmooij AM. 2009. Revertant mosaicism---patchwork in the skin. N. Engl. J. Med 360:1680–82 [DOI] [PubMed] [Google Scholar]
- 68.Kenwrick S, Woffendin H, Jakins T, Shuttleworth SG, Mayer E, et al. 2001. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am. J. Hum. Genet 69:1210–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kinsler VA, Shaw AC, Merks JH, Hennekam RC. 2012. The face in congenital melanocytic nevus syndrome. Am. J. Med. Genet. A 158A:1014–19 [DOI] [PubMed] [Google Scholar]
- 70.Kinsler VA, Thomas AC, Ishida M, Bulstrode NW, Loughlin S, et al. 2013. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J. Investig. Dermatol 133:2229–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, et al. 2009. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25:2283–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kolahgar G, Suijkerbuijk SJ, Kucinski I, Poirier EZ, Mansour S, et al. 2015. Cell competition modifies adult stem cell and tissue population dynamics in a JAK-STAT-dependent manner. Dev. Cell 34:297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korgaonkar S, Vundinti BR. 2015. Trisomy 8 mosaicism in a boy with dysmorphic features. Indian Pediatr. 52:812–13 [PubMed] [Google Scholar]
- 74.Kumar R, Angelini S, Snellman E, Hemminki K. 2004. BRAF mutations are common somatic events in melanocytic nevi. J. Investig. Dermatol 122:342–48 [DOI] [PubMed] [Google Scholar]
- 75.Kummari E, Guo-Ross SX, Eells JB. 2015. Laser capture microdissection---a demonstration of the isolation of individual dopamine neurons and the entire ventral tegmental area. J. Vis. Exp (96):e52336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, et al. 2012. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet 90:1108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lai-Cheong JE, McGrath JA, Uitto J. 2011. Revertant mosaicism in skin: natural gene therapy. Trends Mol. Med 17:140–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lavoue V, Morcel K, Bouchard P, Sultan C, Massart C, et al. 2008. Restoration of ovulation after unilateral ovariectomy in a woman with McCune-Albright syndrome: a case report. Eur. J. Endocrinol 158:131–34 [DOI] [PubMed] [Google Scholar]
- 79.Levinsohn JL, Sugarman JL, Bilguvar K, McNiff JM, Yale Cent. Mendel. Genom., Choate KA. 2015. Somatic V600E BRAF mutation in linear and sporadic syringocystadenoma papilliferum. J. Investig. Dermatol 135:2536–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levinsohn JL, Sugarman JL, Yale Cent. Mendel. Genom., McNiff JM, Antaya RJ, Choate KA. 2016. Somatic mutations in NEK9 cause nevus comedonicus. Am. J. Hum. Genet 98:1030–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levinsohn JL, Tian LC, Boyden LM, McNiff JM, Narayan D, et al. 2013. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J. Investig. Dermatol 133:827–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim YH, Bacchiocchi A, Qiu J, Straub R, Bruckner A, et al. 2016. GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am. J. Hum. Genet 99:443–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim YH, Choate KA. 2016. Expanding the mutation spectrum of ichthyosis with confetti. J. Investig. Dermatol 136:1941–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lim YH, Douglas SR, Ko CJ, Antaya RJ, McNiff JM, et al. 2015. Somatic activating RAS mutations cause vascular tumors including pyogenic granuloma. J. Investig. Dermatol 135:1698–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, et al. 2014. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum. Mol. Genet 23:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lim YH, Qiu J, Saraceni C, Burrall BA, Choate KA. 2016. Genetic reversion via mitotic recombination in ichthyosis with confetti due to a KRT10 polyalanine frameshift mutation. J. Investig. Dermatol 136:1725–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Limaye N, Kangas J, Mendola A, Godfraind C, Schlögel MJ, et al. 2015. Somatic activating PIK3CA mutations cause venous malformation. Am. J. Hum. Genet 97:914–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, et al. 2011. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med 365:611–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lyon MF. 1962. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet 14:135–48 [PMC free article] [PubMed] [Google Scholar]
- 90.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, et al. 2015. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348:880–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miller RM, Sparkes RS. 1977. Segmental neurofibromatosis. Arch. Dermatol 113:837–38 [PubMed] [Google Scholar]
- 92.Møller LB, Lenartowicz M, Zabot MT, Josiane A, Burglen L, et al. 2012. Clinical expression of Menkes disease in females with normal karyotype. Orphanet J. Rare Dis 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Monroig PD, Calin GA. 2013. MicroRNA and epigenetics: diagnostic and therapeutic opportunities. Curr. Pathobiol. Rep 1:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, et al. 2014. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 15:256–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pasmooij AM, Garcia M, Escamez MJ, Nijenhuis AM, Azon A, et al. 2010. Revertant mosaicism due to a second-site mutation in COL7A1 in a patient with recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol 130:2407–11 [DOI] [PubMed] [Google Scholar]
- 96.Percoco G, Benard M, Ramdani Y, Lati E, Lefeuvre L, et al. 2012. Isolation of human epidermal layers by laser capture microdissection: application to the analysis of gene expression by quantitative real-time PCR. Exp. Dermatol 21:531–34 [DOI] [PubMed] [Google Scholar]
- 97.Pewitt JD, Burns EK, Chan LS. 2015. Eruptive syringocystadenoma papilliferum, keratoacanthoma, and verruca vulgaris in a keratinocytic epidermal nevus on the leg. SKINmed 13:395–97 [PubMed] [Google Scholar]
- 98.Piraino SW, Furney SJ. 2015. Beyond the exome: the role of non-coding somatic mutations in cancer. Ann. Oncol 27:240–48 [DOI] [PubMed] [Google Scholar]
- 99.Poblete-Gutiérrez P, Wiederholt T, König A, Jugert FK, Marquardt Y, et al. 2004. Allelic loss underlies type 2 segmental Hailey-Hailey disease, providing molecular confirmation of a novel genetic concept. J. Clin. Investig 114:1467–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res. 72:2457–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. 2011. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11:761–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, et al. 2010. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 20:1096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rabbani B, Tekin M, Mahdieh N. 2014. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet 59:5–15 [DOI] [PubMed] [Google Scholar]
- 104.Rey RA, Venara M, Coutant R, Trabut JB, Rouleau S, et al. 2006. Unexpected mosaicism of R201H-GNAS1 mutant-bearing cells in the testes underlie macro-orchidism without sexual precocity in McCune-Albright syndrome. Hum. Mol. Genet 15:3538–43 [DOI] [PubMed] [Google Scholar]
- 105.Riccardi VM. 1982. Neurofibromatosis: clinical heterogeneity. Curr. Probl. Cancer 7:1–34 [DOI] [PubMed] [Google Scholar]
- 106.Rivera B, Gayden T, Carrot-Zhang J, Nadaf J, Boshari T, et al. 2016. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 131:847–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarin KY, McNiff JM, Kwok S, Kim J, Khavari PA. 2014. Activating HRAS mutation in nevus spilus. J. Investig. Dermatol 134:1766–68 [DOI] [PubMed] [Google Scholar]
- 108.Sastry GR, Cooper HB Jr., Brink RA. 1965. Paramutation and somatic mosaicism in maize. Genetics 52:407–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28:1811–17 [DOI] [PubMed] [Google Scholar]
- 110.Scheimberg I, Harper JI, Malone M, Lake BD. 1996. Inherited ichthyoses: a review of the histology of the skin. Pediatr. Pathol. Lab. Med 16:359–78 [DOI] [PubMed] [Google Scholar]
- 111.Schubbert S, Shannon K, Bollag G. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7:295–308 [DOI] [PubMed] [Google Scholar]
- 112.Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, et al. 2006. Germline KRAS mutations cause Noonan syndrome. Nat. Genet 38:331–36 [DOI] [PubMed] [Google Scholar]
- 113.Sebastiano V, Zhen HH, Haddad B, Bashkirova E, Melo SP, et al. 2014. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med 6:264ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shamseldin HE, Tulbah M, Kurdi W, Nemer M, Alsahan N, et al. 2015. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, et al. 2013. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med 368:1971–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Siegel DH, Sybert VP. 2006. Mosaicism in genetic skin disorders. Pediatr. Dermatol 23:87–92 [DOI] [PubMed] [Google Scholar]
- 117.Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, et al. 2000. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. Nature 405:466–72 [DOI] [PubMed] [Google Scholar]
- 118.Snyder RD, Green JW. 2001. A review of the genotoxicity of marketed pharmaceuticals. Mutat. Res 488:151–69 [DOI] [PubMed] [Google Scholar]
- 119.Spoerri I, Brena M, De Mesmaeker J, Schlipf N, Fischer J, et al. 2015. The phenotypic and genotypic spectra of ichthyosis with confetti plus novel genetic variation in the 3′ end of KRT10: from disease to a syndrome. JAMA Dermatol. 151:64–69 [DOI] [PubMed] [Google Scholar]
- 120.Sudarsanam A, Ardern-Holmes SL. 2014. Sturge-Weber syndrome: from the past to the present. Eur. J. Paediatr. Neurol 18:257–66 [DOI] [PubMed] [Google Scholar]
- 121.Suzuki S, Nomura T, Miyauchi T, Takeda M, Nakamura H, et al. 2016. Revertant mosaicism in ichthyosis with confetti caused by a frameshift mutation in KRT1. J. Investig. Dermatol 136:2093–95 [DOI] [PubMed] [Google Scholar]
- 122.Tan K, An L, Miao K, Ren L, Hou Z, et al. 2016. Impaired imprinted X chromosome inactivation is responsible for the skewed sex ratio following in vitro fertilization. PNAS 113:3197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thomas AC, Zeng Z, Rivière JB, O’Shaughnessy R, Al-Olabi L, et al. 2016. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J. Investig. Dermatol 136:770–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ting TW, Shahdadpuri R, Jamuar SS. 2015. Mosaicism in traditional Mendelian diseases. Curr. Genet. Med. Rep 3:101–9 [Google Scholar]
- 125.Tinschert S, Naumann I, Stegmann E, Buske A, Kaufmann D, et al. 2000. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur. J. Hum. Genet 8:455–59 [DOI] [PubMed] [Google Scholar]
- 126.Tolar J, Wagner JE. 2013. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet 382:1214–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, et al. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tumer Z, Moller LB. 2010. Menkes disease. Eur. J. Hum. Genet 18:511–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, et al. 2004. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5:375–87 [DOI] [PubMed] [Google Scholar]
- 130.Twigg SR, Hufnagel RB, Miller KA, Zhou Y, McGowan SJ, et al. 2016. A recurrent mosaic mutation in SMO, encoding the hedgehog signal transducer smoothened, is the major cause of Curry-Jones syndrome. Am. J. Hum. Genet 98:1256–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Uchiyama Y, Nakashima M, Watanabe S, Miyajima M, Taguri M, et al. 2016. Ultra-sensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge-Weber syndrome. Sci. Rep 6:22985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Umegaki-Arao N, Pasmooij AM, Itoh M, Cerise JE, Guo Z, et al. 2014. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci. Transl. Med 6:264ra164. [DOI] [PubMed] [Google Scholar]
- 133.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, et al. 2009. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457:599–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, et al. 2010. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med 363:2191–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.van Steensel MA. 2015. Neurocutaneous manifestations of genetic mosaicism. J. Pediatr. Genet 4:144–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, et al. 2010. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N. Engl. J. Med 363:629–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang SM, Hsieh YJ, Chang KM, Tsai HL, Chen CP. 2014. Schimmelpenning syndrome: a case report and literature review. Pediatr. Neonatol 55:487–90 [DOI] [PubMed] [Google Scholar]
- 138.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. 2014. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet 46:1160–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. 1991. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med 325:1688–95 [DOI] [PubMed] [Google Scholar]
- 140.Wu M, Pastor-Pareja JC, Xu T. 2010. Interaction between RasV12 and scribbled clones induces tumour growth and invasion. Nature 463:545–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu Y, Zhao RC, Tredget EE. 2010. Concise review: bone marrow-derived stem/progenitor cells in cutaneous repair and regeneration. Stem Cells 28:905–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang Q, Huang C, Yang X, Feng Y, Wang Q, Liu M. 2008. The R1947X mutation of NF1 causing autosomal dominant neurofibromatosis type 1 in a Chinese family. J. Genet. Genom 35:73–76 [DOI] [PubMed] [Google Scholar]
- 143.Zheng JW, Zhang L, Zhou Q, Mai HM, Wang YA, et al. 2013. A practical guide to treatment of infantile hemangiomas of the head and neck. Int. J. Clin. Exp. Med 6:851–60 [PMC free article] [PubMed] [Google Scholar]