

Abstract
Clinically, severe bacterial infection can cause septicemia and multiple organ dysfunction syndrome, especially liver injury. CD38 is closely related to many inflammatory pathways, but its role in liver injury caused by bacterial infection remains unclear. The purpose of this study is to discuss the specific role of CD38 in bacterial liver injury. Eight-week-old male C57BL/6 mice (WT, CD38−/− and CD38−/−TLR4mut) were used and stimulated with Escherichia coli (ATCC25922) or PBS, intraperitoneally. After 3 hours of bacterial stimulation, serum was collected to detect ALT and AST concentration, and liver tissue was harvested for hematoxylin and eosin staining and bacterial culture. The mRNA expressions of TLR4, NLRP3, IL-1β, IL-18, and GSDMD were quantitatively determined by RT-qPCR. The expressions of TLR4, MyD88, TRIF, NF-κB p65, NLRP3, GSDMD, and cytokines were detected by Western blot. The expression and localization of ERK1/2 were detected by immunohistochemistry and Western blot. The results showed that bacterial stimulation could upregulate the expression of inflammatory cytokines, leading to hepatic dysfunction. Moreover, bacterial stimulation of CD38-deficient mice can aggravate the inflammatory response, the expressions of TLR4, NF-κB, and ERK1/2 were significantly increased, and the biomarkers related to pyroptosis also manifested more obvious pyroptosis. However, TLR4 mutation significantly alleviated inflammation and pyroptosis in the liver caused by bacteria, on the basis of CD38 deficiency. Overall, CD38 knockout exacerbates bacteria-induced liver damage through TLR4-NLRP3-GSDMD-mediated pyroptosis.
1. Introduction
Clinically, severe bacterial infection can cause septicemia, and sepsis has been recognized as a global health priority by WHO because of the current estimates of 30 million episodes and six million deaths per year [1]. And the life-threatening organ dysfunction caused by dysregulated host response to infection is a thorny issue in sepsis [2], including liver injury, one of the main clinical manifestations and an independent risk factor for multiple organ dysfunction syndrome (MODS) and high mortality rate in patients with sepsis [3]. Hepatic dysfunction is considered to be one component of the MODS and usually associated with a poor prognosis in clinic during sepsis [4]. Moreover, the study has shown that liver injury due to sepsis was shown to be induced via oxidative damage, inflammatory response, and neutrophil infiltration [5] and hepatic pathology in sepsis mainly includes steatosis, cholangiolitis, intrahepatic cholestasis, periportal inflammation, and apoptosis [6].
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme in energy metabolism and electron transfer and regulates energy metabolism, mitochondrial function, apoptosis, necrosis, nuclear gene expression, and neurotransmitter release [7]. Cluster of differentiation 38 (CD38), a type II transmembrane protein, is the major ADP-ribosyl cyclase in mammals, which is a key NAD+-dependent enzyme [8]. CD38 is a multifunctional transmembrane protein that is widely expressed in immune cells [9], controls the innate immune response against infection [10, 11], and closely relates to inflammatory pathways such as the Toll-like receptor (TLR) pathway [12, 13] and mitogen-activated protein kinase (MAPK) pathway [13, 14]. CD38 also plays a critical role in many liver diseases such as glucagon-induced gluconeogenesis in hepatocytes [15] and lipopolysaccharide- (LPS-) induced acute damage of the liver [16]. And in inflammatory liver disease, the CD38/NAADP-mediated Ca2+ signaling pathway is a potential novel therapeutic target [17]. Moreover, it has been reported that CD38 is expressed in inflammatory cells [18], regulates the function of immunocytes [19], depresses the expressions of inflammatory cytokines, and inhibits the death of hepatocytes [16].
The innate immune system is considered to be the main defense against invading pathogens and maintaining homeostasis. The pathogens, damaged tissues, and cells may release pathogen-related molecular patterns (PAMPs) and injury-related molecular patterns (DAMPs). Then, pattern recognition receptors (PRRs) will recognize them and activate immune response. Among these receptors, NLR family protein 3 (NLRP3) is the most well-studied Nod-like receptor, because NLRs may form a protein complex called inflammasome [20]. The inflammasome is a multiprotein complex that is composed of NLRP3, apoptosis-associated speck-like protein (ASC), and serine protease caspase-1 (caspase-1), mainly mediating the host's immune response to microbial infection and cell damage [21]. The aggregation of the inflammasome leads to the cleavage of procaspase-1 and the formation of activated caspase-1. Cleaved caspase-1 promotes the transformation of prointerleukin-1 beta (pro-IL-1β) and prointerleukin-18 (pro-IL-18) into mature IL-1β and IL-18 [22]. In many immune responses, mature IL-1β is an effective proinflammatory mediator, which can recruit innate immune cells to the infection site and regulate acquired immune cells, while mature IL-18 can promote expression of interferon gamma (IFN-γ). At the same time, activated caspase-1 can cleave gasdermin-D (GSDMD) and induce proinflammatory cell death called pyroptosis [23, 24]. More and more studies have shown that NLRP3-meditated pyroptosis may cause vital injury in different organs affected by sepsis including liver injury [21, 25, 26]. And pyroptosis was found not only in immunocytes such as monocytes or macrophages but also in hepatocytes [21, 24]. Up to now, some mechanism studies [27, 28] have shown that inhibiting the assembly and activation of the inflammasome can improve the proinflammatory response in sepsis. However, the mechanism and effect of the NLRP3 inflammasome on the pathophysiology of sepsis need further study.
Recently, some study reported that Toll-like receptor 4 (TLR4) is a therapeutic target for the prevention and treatment of liver failure [29]. TLR4 is an important member of the Toll-like receptor family and can activate downstream intracellular signals such as nuclear factor-κB (NF-κB) and MAPK after LPS recognition [30, 31]. And the activated NF-κB and MAPKs pathways in turn lead to further inflammation responses, more production of proinflammatory cytokines, or immunosuppression [5, 32]. In addition, activated NF-κB can induce nitric oxide (NO) production and activate apoptotic protein to induce apoptosis and necrosis, and the final outcome is organ failure [5]. Furthermore, the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway, one of the MAPK superfamily pathways, is known to play an important role in the regulation of inflammation [33, 34] and apoptosis [35–37]. And it has been confirmed by researchers that the ERK1/2 signaling pathway plays a key role in liver injury [34, 36, 38].
Our previous studies have shown that TLR4 expression increases noticeably in CD38−/− mice when compared to wild-type (WT) mice. Therefore, in order to further explore the role of CD38 in the TLR4-related pyroptosis-meditated inflammatory signaling pathway and the potential role of CD38 in liver injury caused by bacterial infection, we injected E. coli into the abdominal cavity of mice (WT, CD38−/−, and CD38−/−TLR4mut) and then observed the pathological changes in liver tissues, expression variation of liver function indicator, cytokines, biomarkers of pyroptosis, and signaling pathways to reveal the effect of CD38 deficiency on bacterial infection-mediated sepsis liver injury.
2. Materials and Methods
2.1. Animals and Establishment of the Mouse Model
Wild-type mice (C57BL/6 of 4 weeks old) were purchased from the Laboratory Animal Center of Wuhan University, and C57BL/6 background CD38−/− and CD38−/−TLR4mut mice (B6.129P2-Ly96−/−) were kindly donated by professors Hongbo Xin and Keyu Deng from the Translational Medicine Research Institute of Nanchang University. All groups of mice were raised in SPF Animal Facility at the laboratory animal center of Nanchang University and kept well fed and watered. Eight-week-old male mice (20 ± 2 g) were selected for the experiment. All experiments were in conformance with the Management Ordinance of Laboratory Animals of Nanchang University and were approved by the Animal Protection Committee of Nanchang University.
A standard Escherichia coli (E. coli) strain (ATCC25922) was used to establish mouse models of sepsis, which was kindly offered by the Laboratory of Jiangxi Provincial People's Hospital. A ring (2 μl) of resoluble bacterial solution was added into Columbia blood plate medium with three-zone scribing and then cultured in an incubator with atmosphere at 37°C for 12 hours. Intraperitoneal injection of 0.5 ml E. coli (3 × 108 cfu/ml) was used to induce sepsis according to a research by Shen et al. [39], and equal amounts of PBS were injected as a control. Three hours later, mice were sacrificed, and the blood and liver were collected for further analysis.
2.2. Serum Analysis
Blood was collected three hours after E. coli or PBS injection. Then, the supernatant was obtained after centrifugation at 10621g at 4°C for 10 minutes. And the concentrations of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were determined by using ELISA kits (Nanjing Jiancheng Bioengineering Institute, China) according to the instructions.
2.3. Bacterial Culture of Liver Tissue and Gram Staining
A total of 100 mg of liver tissue was taken into the homogenizer and then ground completely. And the homogenates (100 μl) were applied to agar plates and distributed evenly. The plates were cultured in a 5% CO2 incubator at 37°C for 24 hours and screened for bacterial colonies. A single colony was selected and transferred onto a slide. The slides were Gram stained and observed under the microscope.
2.4. Hematoxylin and Eosin Staining
Three hours after E. coli or PBS injection, the mice were killed and livers were taken out. Then, the tissues were fixed in 4% paraformaldehyde solution overnight and washed with 0.1 M phosphate buffer (pH 7.4) for 10 minutes 3 times. The liver tissues were rendered transparent with xylene and embedded after being dehydrated by successive washes in an ethanol gradient (60%, 75%, 85%, 95% twice, and 100% twice). Paraffin sections of 3 μm were prepared using a microtome. The sections were heated at 65°C for 2 hours for deparaffinization and hydrated by submersion in the following solutions: xylene for 5 minutes three times, 100% ethanol for 30 seconds twice, and 95% ethanol and 70% ethanol for 30 seconds. Hematoxylin and eosin staining (H&E) was performed, and the morphological changes were evaluated under a microscope.
2.5. RNA Extraction and Real-Time Quantitative PCR Assay
Total RNA was extracted from liver tissues (50-100 mg) with the TRizol reagent (Invitrogen, US). And then, the extracted RNA (1 μg) was treated with gDNA Eraser (Takara Biotech, Japan) at 42°C for 2 minutes to remove the genomic DNA. RT-PCR was performed using PrimeScript™ RT under the following conditions: 37°C for 15 minutes, 85°C for 5 seconds, and 4°C indefinitely for product preservation.
RT-qPCR was performed using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara Biotech, Japan) and the StepOne™ PLUS Real-Time PCR System (Applied Biosystems, New York, USA). We used primers for TLR4, NLRP3, IL-1β, IL-18, GSDMD, and GAPDH synthesized from TSINGKE Biotech (TSINGKE Biotech, Beijing, China) (TLR4 forward primer: 5′-CGCTTTCA CCTCTGCCTTCAC-3′; TLR4 reverse primer: 5′-TTGCCGTTCTTG TTCTTCTTC-3′; NLRP3 forward primer: 5′-GCCGTCTACGTCTTCTTCCTT TCC-3′; NLRP3 reverse primer: 5′-CATCCGCAGCCAGTGAACAGAG-3′; IL-1β forward primer: 5′-TTTTCCTCCTTGCCTCTGAT-3′; IL-1β reverse primer: 5′-GAGTGCTGCCTAATGTCCCC-3′; IL-18 forward primer: 5′-GACTCTTGC GTCAACTTCAAGG-3′; IL-18 reverse primer: 5′-CAGGCTGTCTTTTGTCAA CGA-3′; GSDMD forward primer: 5′-CCATCGGCCTTTGAGAAAGTG-3′; GSDMD reverse primer: 5′-ACACATGAATAACGGGGTTTCC-3′; GADPH forward primer: 5′-GAAGGTGGTGAAGCAGGCATC; and GADPH reverse primer: 5′-GTGGGAGTTGCTGTT GAAGTC). The PCR program was 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds, and 60°C for 30 seconds. We detected the threshold cycle (Ct) for all genes and determined their relative expression levels compared to GAPDH.
2.6. Immunohistochemical Staining
Sections of 3 μm were prepared using a microtome and baked in an oven at 65°C overnight and then dewaxed in xylene for 5 minutes three times, rehydrated in an ethanol gradient, and blocked by incubation in 0.3% H2O2 for 10 minutes. The slides were incubated with primary antibodies, anti-ERK1/2 rabbit mAb (1 : 250) (CST, USA) for 50 minutes at room temperature, and then with biotin-conjugated secondary antibodies for 25 minutes at room temperature after a thorough wash in PBS three times. The signal was detected with the help of diaminobenzidine (DAB). The samples were fixed on glass slides and observed under the microscope.
2.7. Western Blot
Western blot was performed to further detect the protein expressions of TLR4, TRIF, MyD88, NF-κB, IL-6, iNOS, BAX, NLRP3, ASC, caspase-1, IL-1β, IL-18, caspase-3, GSDMD, and ERK1/2 in livers from WT, CD38−/−, and CD38−/−TLR4mut mice (n = 3/group). The livers were ground up and lysed in the RIPA Lysis Buffer with phenylmethanesulfonyl fluoride. The equivalent protein sample was loaded into 10% SDS-PAGE gel and transferred onto polyvinylidene fluoride (PVDF) membranes. The primary antibodies anti-TLR4 rabbit mAb (1 : 500) (CST, USA), anti-TRIF rabbit mAb (1 : 1000) (Proteintech, USA), anti-MyD88 mouse mAb (1 : 2000) (Proteintech, USA), anti-NF-κB p65 rabbit mAb (1 : 1000) (CST, USA), anti-phospho-NF-κB p65 rabbit mAb (1 : 500) (CST, USA), anti-IL-6 mouse mAb (1 : 2000) (Proteintech, USA), anti-iNOS rabbit mAb (1 : 1000) (CST, USA), anti-BAX rabbit mAb (1 : 5000) (Proteintech, USA), anti-NLRP3 rabbit mAb (1 : 500) (Boster, China), anti-ASC rabbit mAb (1 : 500) (Affinity, China), anti-caspase-1 rabbit mAb (1 : 500) (Abcam, UK), anti-IL-1β rabbit mAb (1 : 1000) (CST, USA), anti-IL-18 rabbit mAb (1 : 1000) (Affinity, China), anti-caspase-3 rabbit mAb (1 : 500) (CST, USA), anti-GSDMD rabbit mAb (1 : 500) (Affinity, China), anti-ERK1/2 rabbit mAb (1 : 1000) (CST, USA), anti-phospho-ERK1/2 rabbit mAb (1 : 2000) (CST, USA), and anti-GAPDH rabbit mAb (1 : 5000) (Proteintech, USA) were used. The expressions of TLR4, TRIF, MyD88, NF-κB, IL-6, iNOS, BAX, NLRP3, ASC, caspase-1, IL-1β, IL-18, caspase-3, GSDMD, and ERK1/2 and GAPDH were visualized by the ECL assay (Sage creation) according to the manufacturer's instructions.
2.8. Statistical Analysis
All data are depicted as the mean ± standard deviation (SD). A paired t-test was used to determine statistically significant differences between two groups, and one-way ANOVA followed by the Tukey post hoc test was used to estimate differences for multigroups. Differences were considered significant if the p value is <0.05. All analyses were made with the statistical software GraphPad Pro 5.0 (GraphPad, San Diego, CA, USA).
3. Results
3.1. E. coli Induces Inflammation Leading to Liver Injury in WT Mice
In order to induce liver injury, we injected 3 × 108 cfu/mlEscherichia coli (E. coli) into the abdominal cavity of WT mice, and the PBS was injected as the control. Three hours later, the liver was harvested for further study. H&E staining results showed that compared with the control group, after 3 hours of E. coli stimulation, obvious pathological changes can be observed in the liver of E. coli-injected mice, and the hepatocyte edema (yellow arrow), inflammatory cell infiltration (blue arrow), punctate necrosis (black arrow), and binucleate hepatocytes (green arrow) could be observed (Figures 1(a)–1(d)). And a study showed that the occurrence of binucleate hepatocytes activated TLR4-mediated signaling [40] and increases with the necro-inflammatory state, and it may be a reactive cell response to liver injury [41]. Furthermore, the liver suspension was taken for bacterial culture and Gram staining. There were scattered colonies in the stimulation group of E. coli (Figure 1(e)) and Gram-negative, rod-shaped bacteria in the bacterial staining (Figure 1(g)). No colony formation was observed in the normal control group (Figure 1(f)). Moreover, compared with the control group, the serum AST increased significantly after 3 hours of E. coli stimulation (Figure 1(h)). However, there was no significant difference in serum ALT between the two groups (Figure 1(i)). These data suggested that E. coli injected intraperitoneally can enter the liver tissues and cause pathological changes in the liver and damage to liver function.
Figure 1.

Escherichia coli can induce liver injury in mice. WT mice were injected with PBS or 3 × 108 cfu/mlE. coli intraperitoneally and sacrificed 3 hours later. Liver pathological injuries were observed with hematoxylin and eosin staining (a–d). The yellow arrows indicate edema, the blue arrows indicate inflammatory cell infiltration, the black arrows indicate punctate necrosis, and the green arrows indicate binucleate hepatocytes. Bacteria from the liver of PBS- or E. coli-stimulated WT mice were cultured in MH medium (e, f) overnight and identified by Gram staining (g). The serum AST (h) and ALT (i) concentrations were determined by using the detection kits. The mRNA of liver inflammatory cytokines TLR4 (j), NLRP3 (k), IL-1β (l), and IL-18 (m) of PBS- or 3 × 108 cfu/mlE. coli-stimulated mice were measured by RT-qPCR. Data are presented as means ± standard deviation. Statistical significance was determined by the paired t-test (n = 3, ∗p < 0.05, ∗∗p < 0.01).
In order to further explore the effect of E. coli stimulation on the liver, RNA was extracted from the liver for RT-qPCR. We found that in the liver tissue, compared with the control group, the TLR4 gene expression in the E. coli-stimulated group was significantly higher (Figure 1(j)). At the same time, the gene expression of proinflammatory cytokines NLRP3 (Figure 1(k)), IL-1β (Figure 1(l)), and IL-18 (Figure 1(m)) increased significantly. These results showed that E. coli can intrude into the liver, cause inflammation, and lead to liver damage.
3.2. CD38 Knockout Can Aggravate the Liver Damage Caused by E. coli
We found that E. coli can cause liver damage in WT mice. Further, we injected the equivalent amount of E. coli into the abdominal cavity of CD38−/− and CD38−/−TLR4mut mice to observe their response to bacteria. Three hours later, the liver and serum samples were collected for further study. Our results showed that in the liver of CD38-deficient mice, the expression levels of TLR4 mRNA and protein were significantly increased and CD38−/−TLR4mut mice showed lower expression levels of TLR4 (Figures 2(a) and 2(b)). And also, the results of serological analysis showed that the concentration of AST in serum of CD38-deficient mice was significantly higher than that of WT mice, while that of CD38−/−TLR4mut mice was decreased (Figure 2(c)). Nevertheless, there was no significant difference in serum ALT concentration among the three groups (Figure 2(d)). And the H&E staining results showed that compared with WT mice, CD38-deficient mice turned into having more severe pathological damage (Figures 2(e), 2(f), 2(h), and 2(i)), specifically more severe edema (yellow arrow), inflammatory cell infiltration (blue arrow), punctate necrosis (black arrow), and binucleate hepatocytes (green arrow). And in the liver of CD38−/−TLR4mut mice, the pathological changes of the above were obviously relieved (Figures 2(h)–2(k)). These results show that E. coli can cause more serious liver injury in CD38−/− mice than in WT mice, and the loss of CD38 will aggravate the damage of bacteria to the liver, but TLR4 mutation can reduce liver injury in septicemia aggravated by CD38 deletion.
Figure 2.
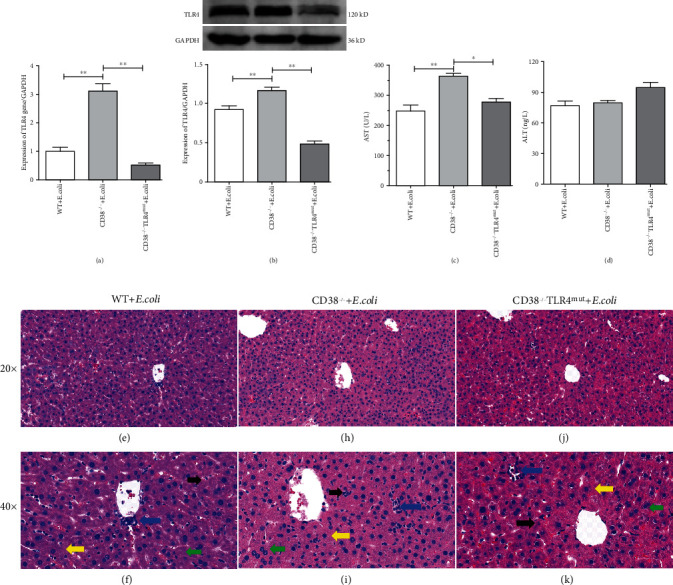
Escherichia coli can induce more serious liver injury in CD38−/− mice. The equivalent amount of E. coli suspension was injected intraperitoneally into WT, CD38−/−, and CD38−/−TLR4mut mice, and the liver and serum samples were harvested after 3 hours. The mRNA of TLR4 (a) in the liver was measured by RT-qPCR, and the expression of TLR4 (b) was detected by Western blot. The concentrations of AST (c) and ALT (d) in serum were determined by using the detection kits. And the liver samples were taken for hematoxylin and eosin staining (e, f, h, i, j, k). The yellow arrows indicate edema, the blue arrows indicate inflammatory cell infiltration, the black arrows indicate punctate necrosis, and the green arrows indicate binucleate hepatocytes. Data are presented as means ± standard deviation. Statistical significance was determined by one-way ANOVA (n = 3, ∗p < 0.05, ∗∗p < 0.01).
3.3. Expression of TLR4-NF-κB and Inflammatory Cytokines Increased in the Liver of CD38−/− Mice Infected with E. coli
In CD38-deficient mice, we observed more severe liver injury after E. coli challenge. Further, after 3 hours of E. coli challenge, we extracted the liver proteins of WT and CD38−/− and CD38−/−TLR4mut mice and detected the expression of TLR4-NF-κB and inflammatory cytokines to explore potential mechanisms. So Western blot was performed, and the results displayed that under bacterial stimulation, CD38-knockout mice showed more severe inflammatory response than WT mice. In CD38−/− mice, the expression of TLR4 (Figures 3(a) and 3(b)), MyD88 (Figures 3(a) and 3(c)), and TRIF (Figures 3(a) and 3(d)) was significantly higher than that in WT mice, and also, the expression of phosphorylated NF-κB p65 was significantly higher (Figures 3(a) and 3(e)). Furthermore, we also detected the protein levels of IL-6 (Figures 3(a) and 3(f)), iNOS (Figures 3(a) and 3(g)), and BAX (Figures 3(a) and 3(h)). As expected, compared with WT mice, the expression of these inflammatory proteins in the liver of CD38−/− mice increased significantly. However, the expression levels of these inflammatory proteins decreased significantly in the liver of CD38−/−TLR4mut mice. The above results manifested that CD38-deficient mice were more susceptible to E. coli, and more inflammatory proteins were expressed in the liver, resulting in more severe inflammatory reactions. And the inflammatory reactions upregulated the protein expression of downstream inflammatory factors such as IL-6, iNOS, and BAX through the TLR4-NF-κB p65 pathway.
Figure 3.
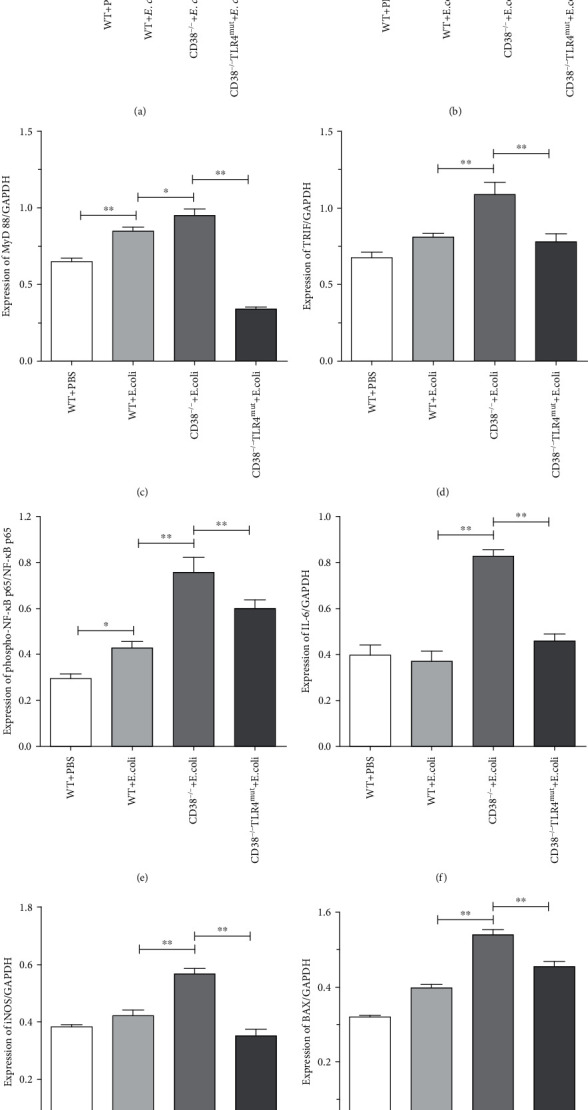
Expression of TLR4-NF-κB and inflammatory cytokines in the liver was detected by Western blot. Changes in expression of liver TLR4 and proinflammatory cytokines in WT, CD38−/−, and CD38−/−TLR4mut mice were detected at 3 hours after E. coli stimulation by Western blot. The expressions of TLR4, MyD88, TRIF, NF-κB p65 and phospho-NF-κB p65, IL-6, iNOS, and BAX were measured (a). And relative levels of TLR4 to GAPDH (b), MyD88 to GAPDH (c), TRIF to GAPDH (d), phospho-NF-κB p65 to NF-κB p65 (e), IL-6 to GAPDH (f), iNOS to GAPDH (g), and BAX to GAPDH (h) were analyzed by ImageJ software. Data are presented as means ± standard deviation. Statistical significance was determined by one-way ANOVA (n = 3, ∗p < 0.05, ∗∗p < 0.01).
3.4. Bacterial Stimulation Significantly Activated the TLR4-ERK Pathway in CD38−/− Mice
We found that CD38 deletion stimulated by E. coli can aggravate the inflammatory response of mice through the TLR4-NF-κB p65 pathway. Further, we want to explore whether MAPK, also an inflammatory-related signal pathway, is involved in the strong inflammatory response caused by E. coli in CD38−/− mice. After 3 hours of E. coli stimulation, the liver was taken for immunohistochemical staining. The results showed that compared with the WT PBS control group, bacterial stimulation could increase the nucleus expression of ERK1/2, which means that the phosphorylation level is increased, in liver tissue. And as expected, the expression of ERK1/2-positive cells in liver tissue of CD38−/− mice was significantly higher than that of WT mice (Figure 4(a)). In order to further verify the role of ERK1/2 in inflammatory mediators, we extracted the liver protein of mice for Western blot. The protein band showed that the phosphorylation level of ERK1/2 in the liver of CD38−/− mice was significantly higher than that of WT mice (Figures 4(b) and 4(c)). And interestingly, the results also showed that the activation level of ERK1/2 decreased significantly in CD38−/−TLR4mut mice. From the above results, we can know that CD38 deficiency aggravates the inflammatory response of the liver caused by bacterial invasion through the TLR4-ERK1/2 pathway.
Figure 4.
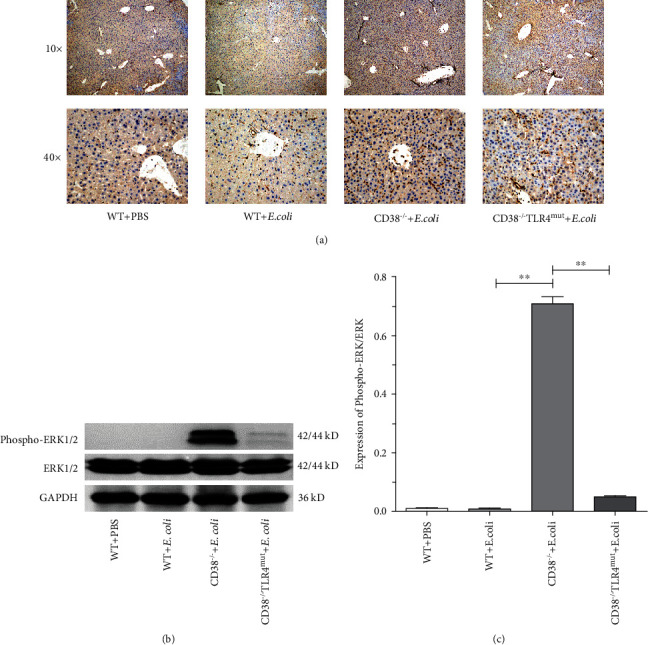
The expression of ERK1/2 was detected in mice induced by E. coli. After 3 hours of bacterial stimulation, liver tissues of WT CD38−/− and CD38−/−TLR4mut mice were taken out to detect the expression of ERK1/2. The liver tissue was stained by immunohistochemistry, and the pictures were obtained by using an Olympus electron microscope (a). The expression of ERK1/2 was detected by Western blot (b), and relative levels of phospho-ERK1/2 to ERK1/2 (c) were analyzed by ImageJ software. Data are presented as means ± standard deviation. Statistical significance was determined by one-way ANOVA (n = 3, ∗∗p < 0.01).
3.5. CD38 Deficiency Aggravates Liver Injury in Septicemia Mediated by Pyroptosis
In the previous results, we found that CD38 deletion can lead to more severe sepsis related-liver injury, and there are studies reporting that in sepsis-related liver injury, pyroptosis activates immune cells and hugely induces inflammation [42]. Therefore, we investigated the role of pyroptosis in the liver injury of septicemia aggravated by CD38 deficiency. Western blot was performed to detect the levels of pyroptosis-related markers in liver tissues. And the results of Western blot showed that compared with the PBS control group, bacterial stimulation significantly increased the expression of ASC (Figures 5(a) and 5(c)), IL-1β (Figures 5(a) and 5(e)), and IL-18 (Figures 5(a) and 5(f)). In accordance with the expected results, we observed a significant increase in the expression levels of pyroptosis-related markers in the liver of CD38-deficient septicemia mice, such as NLRP3 (Figures 5(a) and 5(b)), cleaved caspase-1 (Figures 5(a) and 5(d)), IL-1β (Figures 5(a) and 5(e)), and IL-18 (Figures 5(a) and 5(f)), accompanied by the increase in cleaved caspase-3 (Figures 5(a) and 5(g)). Interestingly, these increased expression levels of pyroptosis-related markers caused by CD38 deletion can be reversed by TLR4 mutations. Taken together, these results suggested that CD38 deletion leads to more severe pyroptosis in bacteria-induced septic liver injury and is associated with the TLR4 signaling pathway.
Figure 5.
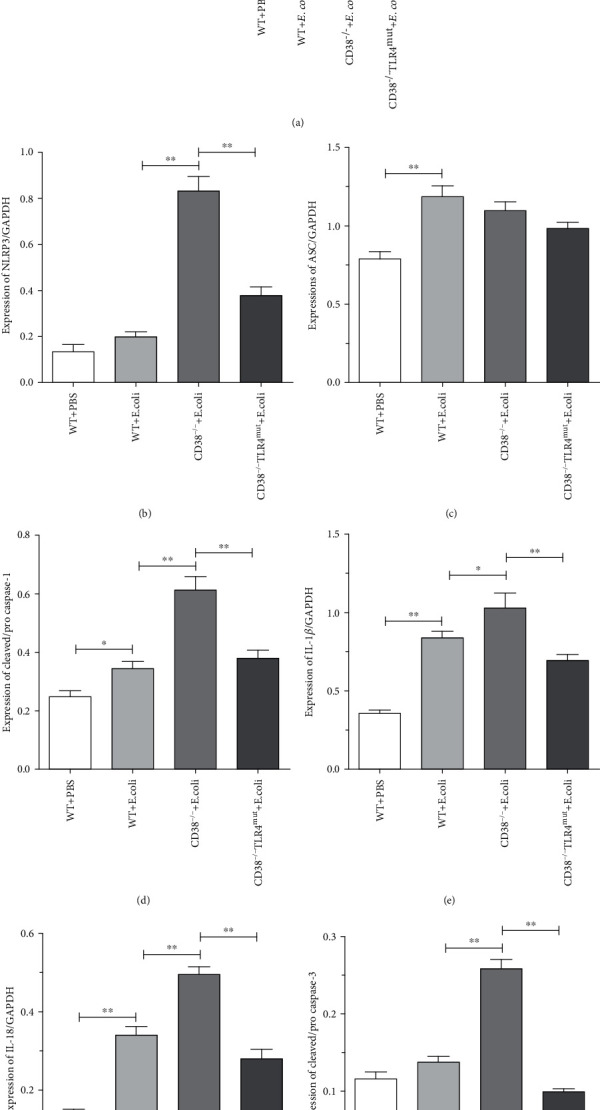
The expression levels of pyroptosis-related markers were detected by Western blot. The expressions of liver pyroptosis proteins in WT, CD38−/−, and CD38−/−TLR4mut mice were detected at 3 hours after E. coli stimulation by Western blot. The expressions of NLRP3, ASC, procaspase-1, cleaved caspase-1, IL-1β, IL-18, procaspase-3, and cleaved caspase-3 were measured (a). And relative levels of NLRP3 to GAPDH (b), ASC to GAPDH (c), cleaved to procaspase-1 (d), IL-1β to GAPDH (e), IL-18 to GAPDH (f), and cleaved to procaspase-3 (g) were analyzed by ImageJ software. Data are presented as means ± standard deviation. Statistical significance was determined by one-way ANOVA (n = 3, ∗p < 0.05, ∗∗p < 0.01).
3.6. CD38 Deficiency Aggravates GSDMD-Mediated Pyroptosis in the Liver of Sepsis Mice
More research reports suggest that GSDMD plays an important role in pyroptosis and is regarded as a pyroptosis executioner [43]. Similarly, we also detected the expression level of GSDMD to observe its role in pyroptosis. The results of RT-qPCR showed that under the stimulation of E. coli, the mRNA of GSDMD increased significantly in WT mice, and CD38 deficiency further increased GSDMD expression level significantly (5-fold to WT), but TLR4 mutation significantly decreased (Figure 6(a)). Recently, it is reported that the active form of GSDMD, termed GSDMD-N, was identified to mediate pyroptotic inflammatory cell death in several diseases [44]. Therefore, we decided to detect the protein expression levels of GSDMD and its activated cleavage C-terminal and N-terminal by Western blot. And the results showed that both of GSDMD-C (Figures 6(b) and 6(c)) and GSDMD-N (Figures 6(b) and 6(d)) fragments were overexpressed in CD38−/− mice than in other groups. And also, we found that the mutation of TLR4 in CD38-deficient mice decreased the expression level of cleaved GSDMD significantly. From these results, we can draw a conclusion that TLR4 mutation can mitigate CD38 deficiency which aggravates liver injury in sepsis by GSDMD-mediated pyroptosis.
Figure 6.
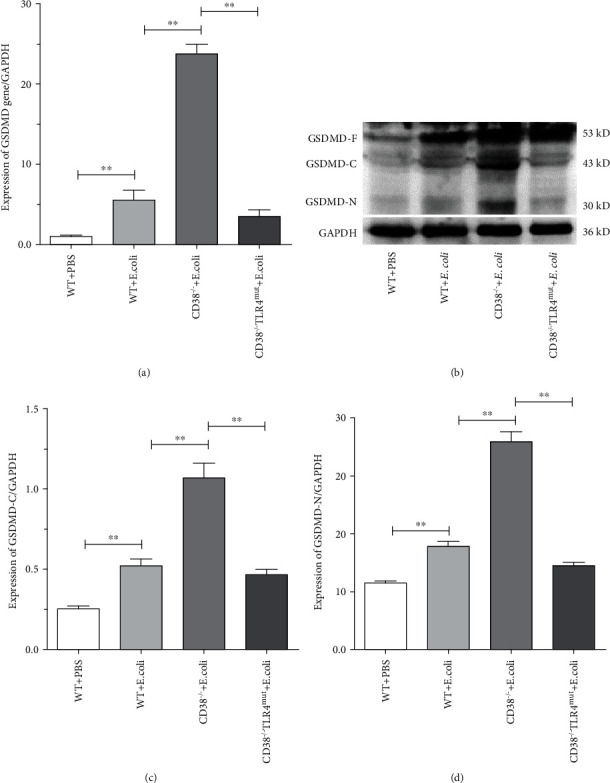
The expression of GSDMD was detected by Western blot. The expressions of GSDMD in the liver of WT, CD38−/−, and CD38−/−TLR4mut mice were detected at 3 hours after E. coli stimulation. The mRNA expression of GSDMD was measured (a) by RT-qPCR. And the expression of GSDMD was detected (b) by Western blot. And relative levels of GSDMD-C to GAPDH (c) and GSDMD-N to GAPDH (d) were analyzed by ImageJ software. Data are presented as means ± standard deviation. Statistical significance was determined by one-way ANOVA (n = 3, ∗∗p < 0.01).
4. Discussion
The role of CD38 in promoting or inhibiting inflammation in the inflammatory response has been controversial, and it plays different roles in the inflammation of different organs. In a mouse model of colitis, CD38 deficiency relieves colonic inflammatory symptoms [18]. Blocking the CD38 pathway protected the hippocampus from apoptosis, oxidative stress, and ultrastructural morphology damage in a septic rat [45], and also, the hearts, livers, and kidneys of septic rats were protected from sepsis-induced damage [8]. On the contrary, accumulated evidence indicates that CD38-knockout (KO) mice are more susceptible to pathogenic bacterial infection [9, 46, 47] and CD38 deficiency can aggravate the inflammatory response [12]. And a research by Lischke et al. showed that CD38-KO mice were highly susceptible to Listeria monocytogenes infection and absence of CD38 caused alterations of the migration pattern of neutrophils and inflammatory monocytes to sites of infection and more accumulation of cells in the liver [10]. CD38 overexpression protects against LPS/GalN-induced acute liver injury [17], and CD38-KO mice demonstrated significant increases in serum ALT and AST [16]. In accordance with the results of the latter, we infected WT and CD38−/− mice with E. coli. Compared with WT, CD38−/− mice showed to be more susceptible to E. coli causing more severe liver injury, more inflammatory cell infiltration, increased expression of inflammatory cytokines, and upregulated expression of the apoptosis gene.
It has been reported that CD38 deletion can activate NF-κB and the expression of downstream inflammatory factors to aggravate the inflammatory response [12], and this pathway is related to TLR4 [48]. Takayashiki et al. showed that increased expression of TLR4 enhances endotoxin-induced hepatic failure [49], and our previous studies have shown that CD38 deficiency enhances TLR4 expression in the kidneys of LPS-induced septic mice [50]. In line with these, in this study, we also found that CD38 deletion can upregulate TLR4 expression, induce phosphorylation of downstream NF-κB p65, and activate the expression of downstream inflammatory factors and apoptosis gene in an E. coli-induced liver injury model, and TLR4 mutation significantly rescues the liver injury from inflammatory response aggravated by CD38 deficiency.
Recently, growing evidence suggests that the NLRP3 inflammasome activation is an important regulator of pyroptosis, which plays various roles in the development of liver diseases [26]. There is a research reporting that experiments in cell cultures, mice, and human samples show that a specific form of cell death, called pyroptosis, leads to the release of complex inflammatory particles, the NLRP3 inflammasome, from inside hepatocytes into the extracellular space, and from there, they are taken up by other cells and thereby mediate inflammatory signals [24]. Chen et al. demonstrated that the hepatic cell pyroptosis increased in a time-dependent manner with the highest rate at 24 h after CLP-induced sepsis and also the severity of liver pyroptosis was correlated with the liver damage [51]. Inhibiting the liver pyroptosis by NLRP3 and caspase-1 inhibitors could reduce the degree of septic acute liver injury [28] and knockdown of NLRP3 or GSDMD significantly restored LPS/D-Gal-induced acute liver injury and lethality [52]. Moreover, our previous study [53] showed that CD38 deletion upregulates TLR4 expression, and also, there is a paper [54] reporting that CD38 can induce inflammasome-mediated activation of caspase-1 by activating NLRP3 in head and neck squamous cell carcinoma (HNSCC). And in this study, we observed severe pyroptosis and serious damage of the liver in septicemia-related liver injury in CD38-deficient mice, whereas both pyroptosis and damage of the liver can be significantly reversed in TLR4 mutant mice, accompanied by the decrease in NLRP3 and GSDMD. Therefore, we concluded that the CD38-regulated TLR4-NLRP3-GSDMD pathway plays an important role in liver injury of septicemia and its mechanism needs further study.
As early as 2001, Guha and Mackman had reported that LPS stimulates the activation of various MAPK pathways with the signaling receptor TLR4, and specific inhibitors of the ERK1/2 and p38 MAPK pathways blocked nuclear NF-κB activity and the transactivation activity of NF-κB p65 [55]. And in particular, studies have demonstrated that the ERK1/2 signaling pathway participates in the mitochondrial dysfunction, oxidative stress, cell apoptosis, and inflammation [37, 56]. Recently, there are research reports that in an acetaminophen- (APAP-) induced liver injury mouse model, ERK1/2 activity was markedly increased in the APAP group than in the control group, and no significant difference was observed in the hepatic JNK and p38 protein expression and phosphorylation [34, 38], indicating that ERK1/2 is more related to liver injury than another two members of MAPKs. Similarly, our results showed that ERK1/2 nuclear translocation and phosphorylation levels in the lesion liver of CD38−/− mice were significantly higher than those of WT mice after E. coli stimulation. In addition, we also observed that NF-κB p65 activation increased and the expression of inflammatory factors and apoptosis genes was significantly upregulated. These results strongly suggested that ERK1/2 is involved in the occurrence and development of bacterial liver injury and that CD38 deletion can activate the ERK1/2 signaling pathway to aggravate liver injury. The same as the current data, a report showed that the level of ERK1/2 phosphorylation in liver and lung tissue increased significantly compared to that in sham in polymicrobial sepsis; activated ERK1/2 can induce the activation of NF-κB and a series of proinflammatory cytokine gene expressions, leading to systemic inflammatory response and even multiple organ dysfunction in sepsis [57].
In conclusion, the current study suggested that E. coli can enter the liver tissue and cause bacterial liver injury, which is manifested by histopathological changes, liver function damage, intrahepatic inflammation, and pyroptosis increase. Importantly, the above-mentioned reactions are more serious in the liver of CD38−/− mice. At the same time, TLR4-NF-κB p65, ERK1/2 phosphorylation, and pyroptosis-related marker proteins are significantly increased, aggravating the liver damage, while TLR4 mutation can significantly reduce the above reaction and improve the liver injury caused by bacteria. Our results showed that CD38 deficiency causes severe bacterial liver injury through TLR4-NLRP3-GSDMD-mediated pyroptosis. This study also shows that CD38-deficient host is susceptible to bacteria, which will aggravate the damage caused by bacterial invasion.
5. Conclusions
Based on the results of this study, we concluded that CD38 deficiency could increase E. coli-induced inflammatory response and activate TLR4-NLRP3-GSDMD-mediated pyroptosis, aggravating liver injury in septic mice. Thus, TLR4 inhibitors or CD38 activators may serve as potential drugs to attenuate E. coli-induced liver injury in septicemia.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) (grant numbers 31960165 and 81760288).
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Authors' Contributions
Huiqing Zhang and Yuna Du contributed equally to this work. YG, ZW, HL, and ZL carried out the experiments, and LZ, YC, ZX, and RL participated in the project design, coordinated the experiments, and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Minasyan H. Sepsis: mechanisms of bacterial injury to the patient. Scandinavian Journal of Trauma, Resuscitation and Emergency Medicine. 2019;27(1):p. 19. doi: 10.1186/s13049-019-0596-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhodes A., Evans L. E., Alhazzani W., et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Medicine. 2017;43(3):304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 3.Dou J., Zhou Y., Cui Y., Chen M., Wang C., Zhang Y. AST-to-platelet ratio index as potential early-warning biomarker for sepsis-associated liver injury in children: a database study. Frontiers in Pediatrics. 2019;7:p. 331. doi: 10.3389/fped.2019.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nesseler N., Launey Y., Aninat C., Morel F., Malledant Y., Seguin P. Clinical review: the liver in sepsis. Critical Care. 2012;16(5):p. 235. doi: 10.1186/cc11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ismail Hassan F., Didari T., Khan F., Niaz K., Mojtahedzadeh M., Abdollahi M. A review on the protective effects of metformin in sepsis-induced organ failure. Cell Journal. 2020;21(4):363–370. doi: 10.22074/cellj.2020.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garofalo A. M., Lorente-Ros M., Goncalvez G., et al. Histopathological changes of organ dysfunction in sepsis. Intensive Care Medicine Experimental. 2019;7(Suppl 1):p. 45. doi: 10.1186/s40635-019-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch-Nolte F., Fischer S., Haag F., Ziegler M. Compartmentation of NAD+-dependent signalling. FEBS Letters. 2011;585(11):1651–1656. doi: 10.1016/j.febslet.2011.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Peng Q. Y., Ai M. L., Zhang L. N., Zou Y., Ma X. H., Ai Y. H. Blocking NAD+/CD38/cADPR/Ca2+ pathway in sepsis prevents organ damage. The Journal of Surgical Research. 2016;201(2):480–489. doi: 10.1016/j.jss.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 9.Glaria E., Valledor A. F. Roles of CD38 in the immune response to infection. Cells. 2020;9(1) doi: 10.3390/cells9010228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lischke T., Heesch K., Schumacher V., et al. CD38 controls the innate immune response against Listeria monocytogenes. Infection and Immunity. 2013;81(11):4091–4099. doi: 10.1128/IAI.00340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estrada-Figueroa L. A., RamÍRez-JimÉNez Y., Osorio-Trujillo C., et al. Absence of CD38 delays arrival of neutrophils to the liver and innate immune response development during hepatic amoebiasis by Entamoeba histolytica. Parasite Immunology. 2011;33(12):661–668. doi: 10.1111/j.1365-3024.2011.01333.x. [DOI] [PubMed] [Google Scholar]
- 12.Qian Y., Chen C., Ma L., et al. CD38 deficiency promotes inflammatory response through activating Sirt1/NF-κB-mediated inhibition of TLR2 expression in macrophages. Mediators of Inflammation. 2018;2018:13. doi: 10.1155/2018/8736949.8736949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartiala P., Hytonen J., Yrjanainen H., et al. TLR2 utilization of Borrelia does not induce p38- and IFN-β autocrine loop-dependent expression of CD38, resulting in poor migration and weak IL-12 secretion of dendritic cells. Journal of Immunology. 2010;184(10):5732–5742. doi: 10.4049/jimmunol.0803944. [DOI] [PubMed] [Google Scholar]
- 14.Benkisser-Petersen M., Buchner M., Dörffel A., et al. Spleen tyrosine kinase is involved in the CD38 signal transduction pathway in chronic lymphocytic leukemia. PLoS One. 2016;11(12, article e0169159) doi: 10.1371/journal.pone.0169159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rah S. Y., Kim U. H. CD38-mediated Ca2+ signaling contributes to glucagon-induced hepatic gluconeogenesis. Scientific Reports. 2015;5(1) doi: 10.1038/srep10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y., Liang L., Shi G. CD38 protein reduces LPS/D-galactosamine-induced acute damage of liver tissues via down-regulating inflammatory cytokine expressions. Xi bao yu fen zi mian yi xue za zhi = Chinese journal of cellular and molecular immunology. 2014;30(10):1009–1012. [PubMed] [Google Scholar]
- 17.Rah S. Y., Lee Y. H., Kim U. H. NAADP-mediated Ca2+ signaling promotes autophagy and protects against LPS-induced liver injury. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 2017;31(7):3126–3137. doi: 10.1096/fj.201601290R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider M., Schumacher V., Lischke T., et al. CD38 is expressed on inflammatory cells of the intestine and promotes intestinal inflammation. PLoS One. 2015;10(5, article e0126007) doi: 10.1371/journal.pone.0126007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burlock B., Richardson G., García-Rodríguez S., Guerrero S., Zubiaur M., Sancho J. The role of CD38 on the function of regulatory B cells in a murine model of lupus. International Journal of Molecular Sciences. 2018;19(10):p. 2906. doi: 10.3390/ijms19102906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel S. Inflammasomes, the cardinal pathology mediators are activated by pathogens, allergens and mutagens: a critical review with focus on NLRP3. Biomedicine & Pharmacotherapy. 2017;92:819–825. doi: 10.1016/j.biopha.2017.05.126. [DOI] [PubMed] [Google Scholar]
- 21.Wree A., Eguchi A., McGeough M. D., et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang Y., Tian S., Pan Y., et al. Pyroptosis: a new frontier in cancer. Biomedicine & Pharmacotherapy. 2020;121:p. 109595. doi: 10.1016/j.biopha.2019.109595. [DOI] [PubMed] [Google Scholar]
- 23.Awad F., Assrawi E., Louvrier C., et al. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacology & Therapeutics. 2018;187:133–149. doi: 10.1016/j.pharmthera.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Gaul S., Leszczynska A., Alegre F., et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. Journal of Hepatology. 2021;74(1):156–167. doi: 10.1016/j.jhep.2020.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danielski L. G., Giustina A. D., Bonfante S., Barichello T., Petronilho F. The NLRP3 inflammasome and its role in sepsis development. Inflammation. 2020;43(1):24–31. doi: 10.1007/s10753-019-01124-9. [DOI] [PubMed] [Google Scholar]
- 26.Al Mamun A., Wu Y., Jia C., et al. Role of pyroptosis in liver diseases. International Immunopharmacology. 2020;84:p. 106489. doi: 10.1016/j.intimp.2020.106489. [DOI] [PubMed] [Google Scholar]
- 27.Mangan M. S. J., Olhava E. J., Roush W. R., Seidel H. M., Glick G. D., Latz E. Erratum: targeting the NLRP3 inflammasome in inflammatory diseases. Nature Reviews. Drug Discovery. 2018;17(9):p. 688. doi: 10.1038/nrd.2018.149. [DOI] [PubMed] [Google Scholar]
- 28.Qu J., Yuan Z., Wang G., Wang X., Li K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. International Immunopharmacology. 2019;70:147–155. doi: 10.1016/j.intimp.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Engelmann C., Sheikh M., Sharma S., et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. Journal of Hepatology. 2020;73(1):102–112. doi: 10.1016/j.jhep.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Joh E. H., Gu W., Kim D. H. Echinocystic acid ameliorates lung inflammation in mice and alveolar macrophages by inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways. Biochemical Pharmacology. 2012;84(3):331–340. doi: 10.1016/j.bcp.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 31.Wang H., Wei X., Wei X., et al. 4-Hydroxybenzo[d]oxazol-2(3H)-one ameliorates LPS/D-GalN-induced acute liver injury by inhibiting TLR4/NF-κB and MAPK signaling pathways in mice. International Immunopharmacology. 2020;83:p. 106445. doi: 10.1016/j.intimp.2020.106445. [DOI] [PubMed] [Google Scholar]
- 32.Qi M., Zheng L., Qi Y., et al. Corrigendum to "Dioscin attenuates renal ischemia/reperfusion injury by inhibiting theTLR4/MyD88 signaling pathway via up-regulation of HSP70" [Pharmacol. Res. 100 (2015) 341-352] Pharmacological Research. 2019;150:p. 104449. doi: 10.1016/j.phrs.2019.104449. [DOI] [PubMed] [Google Scholar]
- 33.Yu S. M., Kim S. J. The thymoquinone-induced production of reactive oxygen species promotes dedifferentiation through the ERK pathway and inflammation through the p38 and PI3K pathways in rabbit articular chondrocytes. International Journal of Molecular Medicine. 2015;35(2):325–332. doi: 10.3892/ijmm.2014.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao C. C., Day Y. J., Lee H. C., Liou J. T., Chou A. H., Liu F. C. ERK signaling pathway plays a key role in baicalin protection against acetaminophen-induced liver injury. The American Journal of Chinese Medicine. 2017;45(1):105–121. doi: 10.1142/S0192415X17500082. [DOI] [PubMed] [Google Scholar]
- 35.Sun Y., Liu W. Z., Liu T., Feng X., Yang N., Zhou H. F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. Journal of Receptor and Signal Transduction Research. 2015;35(6):600–604. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- 36.Duan T., Hu T., Wu C., et al. PINK1/Parkin-mediated mitophagy is involved in NaAsO2-induced apoptosis of human hepatic cells through activation of ERK signaling. Toxicology in Vitro. 2020;66:p. 104857. doi: 10.1016/j.tiv.2020.104857. [DOI] [PubMed] [Google Scholar]
- 37.Liu P., Yang J., Chen Z. Y., Zhang P., Shi G. J. Mitochondrial protein UCP1 mediates liver injury induced by LPS through EKR signaling pathway. European Review for Medical and Pharmacological Sciences. 2017;21(16):3674–3679. [PubMed] [Google Scholar]
- 38.Lee H. C., Yu H. P., Liao C. C., Chou A. H., Liu F. C. Escin protects against acetaminophen-induced liver injury in mice via attenuating inflammatory response and inhibiting ERK signaling pathway. American Journal of Translational Research. 2019;11(8):5170–5182. [PMC free article] [PubMed] [Google Scholar]
- 39.Shen W. C., Wang X., Qin W. T., Qiu X. F., Sun B. W. Exogenous carbon monoxide suppresses Escherichia coli vitality and improves survival in an Escherichia coli-induced murine sepsis model. Acta Pharmacologica Sinica. 2014;35(12):1566–1576. doi: 10.1038/aps.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon L. J., Barnes M., Tang H., Pritchard M. T., Nagy L. E. Kupffer cells in the liver. Comprehensive Physiology. 2013;3(2):785–797. doi: 10.1002/cphy.c120026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grizzi F., Chiriva-Internati M. Human binucleate hepatocytes: are they a defence during chronic liver diseases? Medical Hypotheses. 2007;69(2):258–261. doi: 10.1016/j.mehy.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 42.Shen L., Li L., Yan J. Research progress of pyroptosis in sepsis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2019;31(4):498–500. doi: 10.3760/cma.j.issn.2095-4352.2019.04.026. [DOI] [PubMed] [Google Scholar]
- 43.Shi J., Gao W., Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends in Biochemical Sciences. 2017;42(4):245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Li Y., Xia W., Wu M., et al. Activation of GSDMD contributes to acute kidney injury induced by cisplatin. American Journal of Physiology. Renal Physiology. 2020;318(1):F96–F106. doi: 10.1152/ajprenal.00351.2019. [DOI] [PubMed] [Google Scholar]
- 45.Peng Q. Y., Wang Y. M., Chen C. X., et al. Inhibiting the CD38/cADPR pathway protected rats against sepsis associated brain injury. Brain Research. 2018;1678:56–63. doi: 10.1016/j.brainres.2017.09.029. [DOI] [PubMed] [Google Scholar]
- 46.Viegas M. S., do Carmo A., Silva T., et al. CD38 plays a role in effective containment of mycobacteria within granulomata and polarization of Th1 immune responses against Mycobacterium avium. Microbes and Infection. 2007;9(7):847–854. doi: 10.1016/j.micinf.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Partida-Sánchez S., Cockayne D. A., Monard S., et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nature Medicine. 2001;7(11):1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 48.Zi S. F., Li J. H., Liu L., et al. Dexmedetomidine-mediated protection against septic liver injury depends on TLR4/MyD88/NF-κB signaling downregulation partly via cholinergic anti-inflammatory mechanisms. International Immunopharmacology. 2019;76:p. 105898. doi: 10.1016/j.intimp.2019.105898. [DOI] [PubMed] [Google Scholar]
- 49.Takayashiki T., Yoshidome H., Kimura F., et al. Increased expression of toll-like receptor 4 enhances endotoxin-induced hepatic failure in partially hepatectomized mice. Journal of Hepatology. 2004;41(4):621–628. doi: 10.1016/j.jhep.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 50.Li Q., Wu C., Liu Z., et al. Increased TLR4 expression aggravates sepsis by promoting IFN-γ expression in CD38−/− mice. Journal of Immunology Research. 2019;2019:12. doi: 10.1155/2019/3737890.3737890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y. L., Xu G., Liang X., et al. Inhibition of hepatic cells pyroptosis attenuates CLP-induced acute liver injury. American Journal of Translational Research. 2016;8(12):5685–5695. [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao T., Cui Y., Ji H., Yan L., Pei D., Qu S. Baicalein attenuates acute liver injury by blocking NLRP3 inflammasome. Biochemical and Biophysical Research Communications. 2021;534:212–218. doi: 10.1016/j.bbrc.2020.11.109. [DOI] [PubMed] [Google Scholar]
- 53.Fu Q., Wu J., Zhou X. Y., et al. NLRP3/caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation. 2019;42(1):306–318. doi: 10.1007/s10753-018-0894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M. J., Gao W., Liu S., et al. CD38 triggers inflammasome-mediated pyroptotic cell death in head and neck squamous cell carcinoma. American Journal of Cancer Research. 2020;10(9):2895–2908. [PMC free article] [PubMed] [Google Scholar]
- 55.Guha M., Mackman N. LPS induction of gene expression in human monocytes. Cellular Signalling. 2001;13(2):85–94. doi: 10.1016/S0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 56.Nowak G. Protein kinase C-α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. The Journal of Biological Chemistry. 2002;277(45):43377–43388. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ang S.-F., Moochhala S. M., MacAry P. A., Bhatia M. Hydrogen sulfide and neurogenic inflammation in polymicrobial sepsis: involvement of substance P and ERK-NF-κB signaling. PLoS One. 2011;6(9, article e24535) doi: 10.1371/journal.pone.0024535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.