

Abstract
The move from reading to writing the human genome offers new opportunities to improve human health. The United States National Institutes of Health (NIH) Somatic Cell Genome Editing (SCGE) Consortium aims to accelerate the development of safer and more-effective methods to edit the genomes of disease-relevant somatic cells in patients, even in tissues that are difficult to reach. Here we discuss the consortium’s plans to develop and benchmark approaches to induce and measure genome modifications, and to define downstream functional consequences of genome editing within human cells. Central to this effort is a rigorous and innovative approach that requires validation of the technology through third-party testing in small and large animals. New genome editors, delivery technologies and methods for tracking edited cells in vivo, as well as newly developed animal models and human biological systems, will be assembled—along with validated datasets—into an SCGE Toolkit, which will be disseminated widely to the biomedical research community. We visualize this toolkit—and the knowledge generated by its applications—as a means to accelerate the clinical development of new therapies for a wide range of conditions.
Subject terms: Targeted gene repair, Genetics research
This Perspective discusses how the Somatic Cell Genome Editing Consortium aims to accelerate the implementation of safe and effective genome-editing therapies in the clinic.
main
Genetic factors contribute to most categories of human disease, including those that are inherited, infectious and malignant. It has therefore been a long-standing goal of biomedical science to develop a means to modify genomes within patients to correct disease-causing mutations, disable the genomes of invading pathogens, arm immune cells to attack tumours and enable countless other therapeutic opportunities. In some instances, gene addition can have therapeutic value, and gene therapy—the field that develops this approach—is experiencing ever-increasing success1. In many other cases, however, the genome of the patient must be edited to achieve therapeutic benefit. Genome editing broadly encompasses diverse technologies that can make many different genomic alterations in different contexts, and the topic has been the subject of recent and comprehensive reviews2–4. Several concepts in genome editing (Fig. 1) are central to the goals and strategies of the SCGE Consortium, which we describe in this Perspective.
Fig. 1. Tools for editing the genomes of cells within the body.
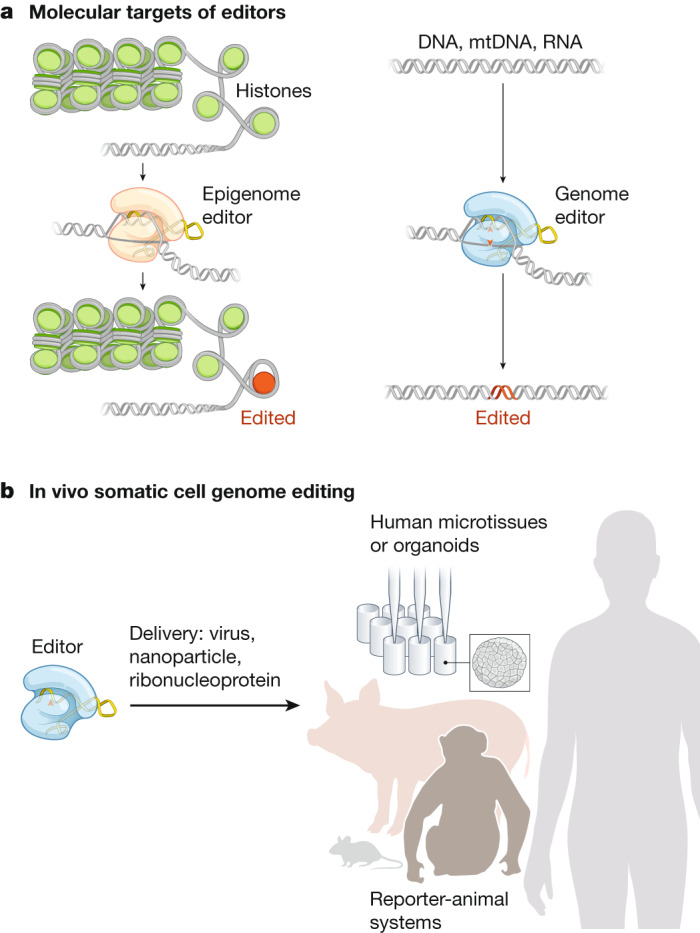
Activities of the SCGE Consortium converge on editing the genome of cells inside the human body. a, Targets of the genome editors (right) range from DNA within the nucleus of a cell to other nucleic acids elsewhere within a cell, such as DNA within the mitochondria (mtDNA) or RNA in the cytoplasm. Targets of epigenomic editors (left) produce targeted alteration of the chromatin structure—including remodelling, modification of the 3D structure and the direct modification of histones or DNA—without editing the DNA or RNA sequence. Approaches to editing cells outside of the body, as well as germline editing in embryos, are not directly supported by the SCGE Consortium, nor are strategies for gene augmentation through the addition of exogenous DNA. b, Interoperable tools assembled into an SCGE Toolkit will be disseminated to accelerate the translation of safe and effective genome-editing therapeutics into the clinic. Tools encompass several categories: newly developed genome editors, delivery technologies, reporter-animal systems, and human biological systems.
Over the past few decades, a steady progression of techniques and technologies that enable user-programmable genome editing has been introduced, tested, improved and implemented. These include homologous recombination, zinc-finger nucleases (ZFNs), meganucleases and transcription activator-like effector nucleases (TALENs)5–7. Most recently, engineered molecular machinery8–15 derived from bacterial immune pathways—known as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins (CRISPR–Cas systems)16—have revolutionized genome editing2–4, in part because their target sequences can be simply programmed with easily designed RNA guides. Despite these promising advances, challenges remain before the transformative potential of therapeutic genome editing can be fully realized. Here we outline the goals and strategies of the SCGE Consortium, which has been established by the United States NIH to accelerate the development of solutions to many of these challenges. The NIH has allocated around US$190 million over 6 years in support of the SCGE Consortium, which now includes 72 principal investigators from 38 institutions that are pursuing 45 distinct but well-integrated projects.
Until the past decade, the most prominent genome-editing platforms (ZFNs, meganucleases, TALENs and Cas9/Cas12a systems) relied almost exclusively on the realization17 that the repair of nuclease-induced breaks in the genome can be exploited to induce genome edits (Fig. 1a)—either gene knockouts (through insertions or deletions generated by non-homologous end joining (NHEJ) or microhomology-mediated end-joining) or precise correction through homology-directed repair (HDR)18. Some editing events involve the insertion of vector-derived, ‘cargo’ sequences into the genome: natural examples of recombinases and transposases that can accomplish this task have been investigated for decades, and some (for example, lentiviral vectors) are being applied for gene therapy and genome editing19. In addition, some platforms can be implemented in partially or completely nuclease-inactive forms, by tethering to other effector proteins20. These strategies include base editing21 (in which fused deaminase enzymes rewrite individual nucleotides without inducing double-strand breaks)22–24 and prime editing (in which a fused reverse transcriptase introduces edits templated by an extended guide RNA)25. Nuclease-inactive forms can also be fused to enzymes to alter chromatin without changing the DNA sequence26,27 (Fig. 1a). Of course, no platform is appropriate for all contexts, and factors critical to editing success include efficiency (the fraction of the intended loci that are edited), precision (the relative frequency of desired (for example, reversion of a pathogenic allele) versus undesired (for example, large deletions or translocations) modifications at the intended loci) and accuracy (how many off-target sites are unintentionally edited, and to what extent).
Genome editing of somatic cells can be carried out either ex vivo, followed by the re-introduction of edited cells into the patient, or in vivo, by delivering the editing machinery to tissues within the body. An important distinction is the editing of somatic tissues versus germline tissues: the latter has the potential to transmit genetic changes to future generations. The SCGE Consortium is strictly focused on somatic editing; germline editing is not only excluded as a goal but is also considered to be an unacceptable outcome that should be carefully prevented.
Existing capabilities and unmet needs
Genome-editing technologies have already demonstrated efficacy in diverse animal models of disease, including cancer, blood and metabolic disorders, inherited forms of blindness and deafness, and neuromuscular and neurological disease2,28–30. These successes have justified the move towards large animal models, in which signs of efficacy have also been found31–33. Early-stage clinical trials have shown that autologous edited cells can stably engraft and persist in humans34–36, and there have been early reports of the ex vivo editing of allogeneic T cells to fight cancer37 as well as the use of autologous haematopoietic stem cells to eliminate the need for blood transfusions in patients with sickle cell disease. However, ex vivo editing is logistically complex, expensive and hard to scale, given its requirement for substantial cell-manufacturing infrastructure. Therefore, in vivo approaches towards the editing of somatic cells are being developed29, and initial targets include cell types that are difficult to manufacture ex vivo (for example, in the retina (clinicaltrials.gov identifier NCT03872479) and in the liver (NCT03041324 and NCT04601051)). These studies highlight the great potential of genome editing to improve human health. However, they also underscore the need to address key limitations of these technologies. Specifically, in vivo editing still faces substantial hurdles in terms of efficacy and safety, especially in organ systems beyond the eye and the liver.
To achieve success in vivo, editors must be able to induce a range of edits to any target nucleic acid in the cell, including nuclear and mitochondrial DNA. Editors must be highly efficient but also safe, with acceptable levels of toxicity and minimal activation of innate immune responses. Adaptive immunity to either the editor38–41 or the delivery vehicle42,43 must also be managed, particularly in cases in which re-administration might be necessary to edit a desired proportion of a target cell population. Similarly, pre-existing immunity might need to be suppressed or circumvented in some cases44–47. A particularly daunting challenge is to develop delivery technologies that can ferry the editing machinery to numerous tissues in a safe and effective manner. We seek to better control the precise genomic changes that we intend to create at each targeted site, reduce the potential for unintended modifications at both targeted and non-targeted sites, and better understand the biological consequences of unintended editing events. These unmet needs are addressed by the initiatives of the SCGE Consortium, as elaborated below.
Despite the promise of changing any DNA sequence in the genome, the current programmable nucleases are most effective for gene knockout or for the excision of specific regions of genomic DNA. In fact, many gene-editing approaches for the treatment of diseases that are caused by mutations in a single gene—such as sickle cell disease, β thalassemia, Duchenne muscular dystrophy and Leber’s congenital amaurosis—are not intended to correct the inherited mutation or to restore the affected gene to a wild-type sequence. Instead, they are designed to knock out repressive genomic elements that will lead to the upregulation of compensatory factors48, to remove exons that will lead to the production of a partially functional gene product49–51, or to remove aberrant splice junctions32. The current inability to easily and accurately program specific sequences into the genome—given that HDR is largely ineffective in differentiated, post-mitotic cells18—is a fundamental obstacle to the broad use of genome editing in the treatment of genetic disease. Accordingly, new technologies that enable sequence-specific alterations—such as base editing22,23 and prime editing25—are also part of the SCGE Consortium’s portfolio of projects. In fact, base editing has already been used to correct pathogenic mutations, and in some cases has resulted in phenotypic rescue of the disease52–63.
Beyond new editing capabilities, there are numerous other technical limitations that must be overcome to advance the field. For example, there have been important advances in recent years in the prediction, characterization and validation of possible off-target editing64, building on foundational work with ZFNs65. Nonetheless, all of these methods are inherently incomplete, because it is not feasible to achieve non-destructive, whole-genome sequencing of every single edited cell. Similarly, most approaches are based on deep-sequencing technology, and are therefore limited by polymerase chain reaction biases, sensitivities, read lengths and the error rates of these methods. Moreover, off-target effects, unwanted events (for example, vector integrations, large deletions, rearrangements or translocations), genotoxicity and other adverse responses to genome editors might not be fully measurable in animal models. For these reasons, the development of methods to detect unwanted genomic events with increased predictive ability and sensitivity, as well as human cell and tissue systems such as organoids, are important components of the SCGE program.
The most substantial hurdle to the development of gene-editing therapies is the establishment of safe and effective delivery strategies. The genome-editing field can make use of four decades of innovation in the fields of gene therapy1 and nucleic acid therapeutics66, which have resulted in the development of numerous viral and non-viral delivery approaches. In fact, the recent regulatory approvals of gene therapies using both adeno-associated virus (AAV) and lentivirus vectors, as well as short interfering RNA (siRNA)-based and antisense-based drugs, provide lessons that are applicable to genome editors. However, many of the vectors that have been developed for gene therapy, which typically focuses on long-term expression to compensate for genetic defects, are not necessarily optimal for gene editing, which often requires transient delivery of editors. The most frequently used editors also introduce other challenges, including their large sizes (SpyCas9 and TALENs), their repetitive sequences and the need to deliver both components of a heterodimer (ZFNs and TALENs), and the requirement for delivery of a ribonucleoprotein complex (RNP; for example in CRISPR). Finally, the risk of on-target or off-target activity in inappropriate tissues underscores the need to ensure proper tissue targeting. Collectively, these challenges provide considerable opportunities for innovation.
Goals of the SCGE Consortium
After reviewing the state of the field in 2017 through a series of stakeholder workshops, the NIH Common Fund noted needs that spanned multiple clinical indications, genes and target tissues67. The consensus was that the field needed new genome editors, delivery systems and biological systems to measure the safety and efficacy of various genome-editing strategies. The Common Fund subsequently launched the SCGE Consortium in 2018, by assembling a collection of multidisciplinary teams working on individual projects designed to address these needs.
The overarching goal of the SCGE Consortium is to accelerate the translation of genome-editing technology to a wide range of tissues and diseases. One of the key challenges in the field is the comparison of various technologies using common metrics and standards. For instance, a retinal delivery system might produce on-target indels at a gene of interest, but it is unclear whether the same delivery system could correct a different gene in the lung. Developmental paths that enable the mixing and matching of various technologies and readouts are woven into the SCGE program. In one example, all new delivery technologies developed in the first three years of the program will be tested first in small animals (for example, mice) and then—if successful—in large animals such as pigs and non-human primates. The resulting third-party data will be shared with the larger research community and with the public. A key value of the SCGE Consortium is transparency, which enables others to access its research output and use its results and products to inform and accelerate their own disease-focused projects. Along with data, we aim to deliver a collection of tools, reagents, methods and best-practices that will be assembled into the SCGE Toolkit for Therapeutic Genome Editing (or SCGE Toolkit in short, Fig. 1b). Through these activities and deliverables, the SCGE Consortium seeks to have a lasting impact by reducing the time and cost required to develop new therapies.
Priorities and strategies
Editing platforms
Both the discovery of new gene-editing tools and their engineering continue to advance rapidly. As such, we seek to discover new editors and build upon existing editors, in part by tuning them for increased precision (Fig. 2). Although the bulk of SCGE studies will focus on the CRISPR system that is already in widest use (SpyCas9), as well as on other established Cas9 and Cas12a homologues68–79, it is imperative to continue to identify and test new systems and related tools. For example, new CRISPR–Cas systems to which humans have not previously been exposed80—as well as gene editors that are based solely on nucleic acid analogues that do not require protein cofactors81—could serve to circumvent detection by the immune system and also facilitate delivery. By searching through microbial data obtained from uncultivated samples, we hope to identify new systems that can be harnessed for the manipulation of DNA—such as helicases, nucleases, transposases, or recombinases80,82–86. These new systems could provide resources with improved efficiencies, alternative targeting mechanisms, smaller cargoes for viral packaging or decreased immunogenicity. This approach is exemplified by the recent development of Cas12j, the smallest CRISPR–Cas genome editing system yet discovered, which was supported by the SCGE program87.
Fig. 2. New genome editors in development.

Major classes of genome editors include nucleases, base editors (BE), prime editors, PNAs, RNA editors and epigenome editors. The development of new editors involves mining metagenomic datasets and building upon existing editors, in part by tuning them for increased precision and accuracy. DNMT, DNA methyltransferase; Acr, anti-CRISPR protein; RT, reverse transcriptase; DN1S: dominant-negative mutant of tumour suppressor p53-binding protein 1, 53BP1; TALE-fusions, transcription activator-like effector-fusion with nucleases or cytidine deaminases (DddA).
In addition to the discovery of new CRISPR–Cas systems, we will continue to develop and improve engineered platforms—for example, base editing21—that efficiently edit genomes, including in post-mitotic cells and in mitochondrial DNA24. Well-established base editors can catalyse C-to-T transitions (cytosine base editors (CBEs))22, A-to-G transitions (adenine base editors (ABEs))23, or both88–90; very recently, C-to-G transversions in mammalian cells have also been enabled by base editing91,92. Ideally, programmed edits could change any nucleotide at any position in the genome; however, when using CRISPR–Cas effectors, editable bases are limited to regions that are near a compatible protospacer-adjacent motif sequence. Furthermore, editable sequences are restricted to a window that is a defined distance from the protospacer-adjacent motif. Through directed evolution, mining of natural variation or rational engineering, we aim to develop both broader targeting capabilities and increased specificity. Finally, we wish to eliminate limitations in changes to the targetable nucleotides. Prime editing, developed in part through the SCGE program, is an example of one such technology25.
Using CRISPR–Cas systems as ‘DNA cursors’ permits us to make edits not only to the DNA nucleotide sequence but also to the epigenetic marks that can alter gene-expression profiles and ultimately influence cellular function93. Like base editors, new CRISPR–Cas systems or variants that provide new binding sites can improve the accessibility of these new tools to all regions of the epigenome, and much has to be learned and developed to first understand and then to improve the specificity of epigenome editors. Such an approach extends the genome-engineering toolbox to apply to a much broader set of diseases, which can be addressed through changes in gene expression93,94 or through reprogramming cell phenotypes95. Epigenome-editing modalities have other potential advantages, including activating endogenous genes and networks for gain-of-function phenotypes, as well as tunability, reversibility and eliminating the possibility of off-target mutations or genotoxicity.
Although there is a considerable focus on CRISPR–Cas related systems within the SCGE Consortium, it is crucial to continue to explore alternative systems, in part because they could differ both in their potential for delivery and in the biological or immunological responses that they elicit. As one example, peptide nucleic acids (PNAs) are relatively small, synthetic molecules that recognize specific DNA sequences through triplex formation and subsequently induce editing96. The SCGE Consortium is developing improved methods for the production of PNAs, in addition to modifiers that could improve the function of PNAs for DNA editing, and a robust analysis of PNA function across many genetic loci97,98. Alternative systems could also target the many distinct mitochondrial genomes with human cells. These genomes are largely inaccessible to editing by systems that require guide RNAs or DNA donors, because of the current lack of reliable methods to transport these classes of molecules into mitochondria. The engineering of editors that target mitochondrial DNA could open up genome-editing therapies for the treatment of mitochondrial diseases, which affect 1 in approximately 5,000 people24,99. The discovery of DddA—an interbacterial toxin that catalyses the unprecedented deamination of cytidines within double-stranded DNA—led to the development of RNA-free DddA-derived CBEs (DdCBEs), which enabled the first purposeful sequence changes in mitochondrial DNA24. In addition to DdCBEs, other protein-based tools such as zinc fingers100–102 and TAL-like effectors103,104 are being fused to nucleases to control mitochondrial genome heteroplasmy.
Delivery systems
Regardless of the genome-editing system that is selected to edit a particular therapeutic locus, its translation to the clinic is currently limited by the capacity for the editing payload to reach the nuclei of target cells. This translational bottleneck presents multifaceted challenges that differ from one target tissue to the next. An ideal delivery platform would be capable of conveying the required macromolecular components across cellular boundaries and into the nucleus; able to induce therapeutically useful levels of editing; amenable to cost-efficient, reproducible and scalable production; specific for particular cell types; and consistent with acceptable thresholds of toxicity, genotoxicity and immunogenicity. Failure to satisfy any of these criteria could render candidate delivery strategies ineffective, inaccessible or unsafe. After decades of research effort dedicated to the therapeutic delivery of DNA or RNA, viral vectors and lipid nanoparticles have emerged as promising platforms105–107 through which to deliver genome-editing machinery. However, many existing platforms have practical limitations for clinical use, as highlighted by the modest supply of genetic therapies in spite of extensive academic and industrial efforts. For example, the clinical use of AAV as a vector for the delivery of DNA that encodes the components of an editor (for example, a Cas protein effector and its guide RNA) is hampered by manufacturing bottlenecks, limited target-tissue tropisms, insertional mutagenesis and the immunogenicity of viral proteins106. For CRISPR systems in particular, the restricted genome-packaging capacity can be another major issue106. Nanoparticles that consist of cationic and hydrophobic molecules, loaded with messenger RNA (mRNA) and guide RNA cargo, provide alternative strategies and can be just as effective as viral vectors in terms of editing efficiency108–110. However, the broad application of genome editing will require nanoparticles that can target the many different types of tissue in the body.
To address these needs, the SCGE Consortium is working on 20 distinct projects that will explore new methods for the delivery of genome-editing machinery to specific tissue types in vivo (Table 1). Existing viral vectors are being enhanced with improved tissue-targeting capacity, enabling high efficacy at lower doses. Similarly, nanoparticles are being augmented with molecules that drive cell-type-specific association, generating powerful homing systems that can be administered intravenously or locally. The delivery of pre-formed CRISPR RNPs has shown the capacity for editing of respiratory epithelial cells using amphiphilic cell-penetrating peptides111, retinal cells112 and neurons in the brain113, for which convection-enhanced delivery might augment tissue distribution. A hybrid approach will pair nanoparticles with an AAV that carries template DNA to facilitate HDR114. Virus-like particles constitute a chimeric strategy: virally derived carriers are packaged with pre-formed RNPs, potentially maintaining delivery efficiency without the prolonged expression of editing machinery that is potentially associated with increased genotoxicity and immunogenicity. Other promising strategies include the use of extracellular vesicles, ultrasound, amphiphilic cell-penetrating peptides or chemical modifications of RNA components105,107,115 to improve targeted in vivo delivery (Table 1).
Table 1.
Delivery systems under development
| Delivery system | Target tissue | Administration | Cargo classa | PI(s)b |
|---|---|---|---|---|
| Viral: AAV | Brain | Intravenous | DNA | B. E. Deverman |
| Viral: AAV | Endothelium | Intravenous | DNA | G. Bao, W. R. Lagor |
| Viral: adenovirus | Endothelium | Intravenous | DNA | D. T. Curiel |
| Viral: AAV | Brain, skeletal muscle | Intravenous | DNA | A. Asokan, C. Gersbach |
| Non-viral: engineered guide RNAs | Brain | Local injection | RNP | E. J. Sontheimer, A. Khvorova, J. K. Watts, S. A. Wolfe |
| Non-viral: polymeric NP | Bone marrow, lung | Intravenous | mRNA, PNA | W. M. Saltzman, P. M. Glazer |
| Non-viral: polymeric NP | Brain | Local injection, intravenous | RNP | S. Gong, M. Emborg, J. E. Levine, S. Roy, K. Saha |
| Non-viral: polymeric NP | Brain | CED, intravenous | RNP | J. Zhou |
| Non-viral: cell-targeted NP | HSPCs | Intravenous | mRNA | J. Dahlman, P. J. Santangelo |
| Non-viral: liposomal NP | Inner ear | Local injection | mRNA, RNP | Z. Chen, D. R. Liu, Q. Xu |
| Non-viral: extracellular vesicles | Bone marrow | Intravenous | mRNA, RNP | I. Ghiran |
| Non-viral: PEGylated particles | Brain | CED | RNP | K. S. Bankiewicz, N. Murthy |
| Non-viral: ultrasound | Brain | Intravenous | DNA, RNP | K. W. Leong |
| Non-viral: amphiphilic peptides | Lung epithelium | Nasal instillation | RNP | P. McCray |
| Non-viral: engineered RNP | Immune cells | Intravenous | RNP | R. Wilson, J. A. Doudna |
| Non-viral: engineered RNP, VLP | HSPCs | Intravenous | RNP | E. Chaikof |
| Non-viral: engineered capsids | Intestinal cell types | Oral, intravenous | DNA, mRNA, RNP | K. Lam, R. H. Cheng |
| Non-viral: VLP | T cells | Intravenous | RNP | G. Yi |
| Non-viral: VLP | Lung, gastrointestinal tract | Intravenous | RNP | J. C. Tilton, M. Drumm, C. Flask, Z. Wang |
| Hybrid: NP and AAV | Lung epithelium | Inhalation/intratracheal | DNA, mRNA | G. Gao, D. G. Anderson, W. Xue |
CED, convection-enhanced delivery; HSPCs, haematopoietic stem and progenitor cells; NP, nanoparticle; VLP, virus-like particle.
a‘Cargo’ refers to the molecular form(s) of genome-editing enzyme component(s): DNA encoding protein and guide RNA, mRNA encoding protein co-delivered with guide RNA, a RNP complex or a PNA.
bThe lead principal investigator (PI) of the project is listed first. Additional PIs follow, listed alphabetically by last name.
Testing in animals
Animal models provide essential validation of delivery systems within a living organism. Such models also serve as a proving ground for new therapeutics and a detection system for adverse events, including toxicity and immunogenicity. Target-indication-specific in-animal efficacy and safety studies are currently treated as essential by regulatory authorities in the United States and the European Union for nearly all genome-editing therapeutics that are being advanced to the clinic. One goal of the SCGE program is to generate in vivo reporter systems that are broadly applicable to many delivery systems and editing technologies, independent of the target cell or tissue type, or the specific disease to be corrected. These reporters should have the ability to detect and quantify genome editing in the intended target tissue, as well as editing events that result from non-specific delivery to other tissues throughout the body.
Small- and large-animal testing centres (SATCs and LATCs, respectively) within the SCGE Consortium centralize expertise with animal models (Table 2) to aid investigators in assessing the efficiency, specificity and safety of new delivery formulations in both wild-type and reporter-animal models. For example, the two SATCs are developing mouse reporter systems because mice are an ideal tool for the preliminary testing of new delivery formulations given their small size, low costs and well-established utility. Large animals are required for preclinical determination of safety, efficiency, dosing and reagent distribution, and as alternatives to mouse models when mice do not adequately recapitulate human responses. Engineered nucleases have enabled efficient and accurate genetic modification of large animals, such as non-human primates and pigs. Three research teams in the SCGE Consortium are developing large animal in vivo reporter systems: one group is dedicated to pigs and two others are dedicated to non-human primates, specifically marmosets and rhesus monkeys. The role of the LATCs is to assess the efficiency and safety of in vivo genome editing and delivery technologies, initially in wild-type animals. When the research teams that create and evaluate the reporter animals have accomplished their goals, they will provide the reporter animals to the LATCs to conduct independent validation and to establish large cohorts for the testing of genome editors.
Table 2.
Animal testing systems under development
| Organism | Editing events detected | Primary readout | Secondary readout | Editors | PIsa |
|---|---|---|---|---|---|
| Mouse | NHEJ, HDR, off-target cutting | Fluorescent signal in situ | Luciferase | SpyCas9, SauCas9, Cas12a | J. D. Heaney, M. E. Dickinson, W. R. Lagor |
| Mouse | NHEJ, HDR, base editing, PNA | Fluorescent signal in situ | Luciferase, NaI symporter | SpyCas9, SauCas9, Cas12a, Nme2Cas9, CjeCas9, ABE, CBE, PNA | S. A. Murray, C. M. Lutz |
| Pig | NHEJ, HDR | Fluorescent signal | NaI symporter | SpyCas9, SauCas9, Cas12a, ABE | D. F. Carlson; K. D. Wells, R.S. Prather |
| Macaque | NHEJ, HDR, C base editing | Fluorescent signal | Luciferase | SpyCas9, SauCas9, Cas12a, CBE | J. D. Hennebold; A. F. Tarantal, D. J. Segal |
| Marmoset | NHEJ | Akaluciferase | Fluorescence | SpyCas9, SauCas9, Nme2Cas9, Cas12a, ABE | G. Feng; A. F. Tarantal, D. J. Segal |
NaI, sodium iodide.
aThe lead PI of the project is listed first. Additional PIs follow, listed alphabetically by last name. Reporter Development and Testing Center teams are separated by semicolons.
The reporter-animal models are designed to faithfully activate in all cells and tissues in response to a specific gene-editing event. Fluorescent proteins provide a simple and robust means to detect activity at the single-cell level in situ, enabling identification of the specific cell types that are targeted. Reporters can be designed to detect different types of editing activity, often with a multi-functional arrangement to enable user flexibility. This includes nuclease activity through the detection of NHEJ-mediated repair events, as well as HDR of an inactivated reporter protein. The capacity to detect the activity of multiple nucleases (for example, SpyCas9, Cas12a and others) is highly desirable to enable comparative studies. Embodying these principles, SCGE reporter systems (Table 2) are primarily designed as improved variations of the Ai9 system50,116 or have a ‘traffic-light reporter’ design117,118. Other models will detect the activities of other types of editors, including ABEs and CBEs21 and PNA-based editing systems96–98. Additional reporter cassettes, such as Akaluciferase119 or sodium iodide symporters120, will be included to permit longitudinal detection by distinct imaging platforms. Importantly, all new reporter animals created as part of the SCGE program will be available for distribution to the wider biomedical community.
Along with the development of new model organisms, new non-invasive methods are needed to measure editing-associated outcomes. The SCGE Consortium is developing techniques for in vivo cell tracking using advanced imaging methods, including total-body positron emission tomography (PET) imaging121, magnetic particle imaging (MPI)122 and chemical exchange saturation transfer magnetic resonance imaging (CEST MRI)123, as outlined in Table 3. Ongoing projects will enable quantitative tracking of the locations and the fates of genome-edited cells after in vivo implantation124–127 or administration using cell labelling (MPI/MRI)128 and reporter gene (MRI/PET)129–132 approaches. Additionally, tracking the delivery and transduction of gene-editing cargo using AAV capsid proteins as endogenous CEST MRI contrast mechanisms is being examined. Each of these methods can be performed alongside existing, standard, non-invasive imaging to assess maladaptive responses to treatment. Together, these tools will provide a powerful combination of methods to quantify and link the delivery of gene-editing technology or gene-edited cells with subsequent biological outcomes.
Table 3.
In vivo cell monitoring and in vitro human biological systems under development
| In vivo cell monitoring | ||
|---|---|---|
| Cell and tissue target | Reporter and/or contrast mechanism | PI(s) |
| hiPS cells in CNS | Tri-modal: iron oxide nanoparticle labelling and tracking for MRI + MPI and 18F-DCFPyL for PET | J. W. M. Bulte |
| Cardiac and hepatic tissues | AAV2 capsid as an endogenous contrast agent Genetically encoded reporter: lysine-rich protein | M. Vandsburger |
| CAR-T cells | Genetically encoded reporter genes MRI: OATP1B3 PET: NaI symporter | J. A. Ronald |
| Whole body, muscle and liver | Genetically encoded reporter: HSV-sr39tk Probe: 18F-FHBG | A. F. Tarantal, D. J. Segal |
| Human biological systems | ||
| Tissue | Cell source | PI(s) |
| Brain | WTC11 hiPS cells | T. C. McDevitt |
| Heart | WTC11 hiPS cells | J.T. Hinson; T.C. McDevitt |
| Liver | WA09 hES cells, WTC11 hiPS cells | S. Kiani; T. C. McDevitt |
| Haematopoietic | Primary T cells | S. Q. Tsai |
| Eye | WA09 hES cells | K. Saha, D. M. Gamm, S. Roy, M. C. Skala |
| Muscle | hiPS cells, primary myoblasts, primary immune cells | C. A. Gersbach, N. Bursac, G. A. Truskey |
| Kidney | WTC11 hiPS cells, BJFF hiPS cells, WA09 hES cells | B. S. Freedman; R. Morizane, J. A. Lewis, V. Sabbisetti |
CNS, central nervous system; 18F-DCFPyL, 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid; 18F-FHBG, 9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine substrate for mutant herpes simplex virus type 1 thymidine kinase (HSV1-sr39TK); OATP1B3, human organic anion transporter polypeptide 1B3; CAR, chimeric antigen receptor; hES cells, human embryonic stem cells; hiPS cells, human induced pluripotent stem cells.
aThe lead PI is listed first. Additional PIs follow, listed alphabetically by last name. Teams are separated by semicolons.
Testing in human biological systems
The development of human biological systems to detect and minimize unintended biological effects of genome editing is a major focus of the SCGE Consortium. Although substantial progress has been made regarding methods for defining the genome-wide off-target mutations induced by genome editors65,133–140, as well as unintended outcomes (such as large deletions and rearrangements) at the on-target site, the interpretation of potential biological consequences associated with these mutations within human cells remains a major challenge141. Additionally, other effects of the editors or of the delivery components themselves—including the potential to stimulate immune responses38,39,43–45,142—have not been fully characterized. The SCGE Consortium is working to develop human cell-based and organoid platforms to define the unintended biological effects of editing (Table 3).
Projects use human primary cells when possible. For example, a primary T cell platform will define some of the unintended biological effects of genome editors. T cells are readily amenable to sensitive, unbiased methods for defining the genome-wide activities of editors143, and unique genomic rearrangements that establish a diverse T cell receptor repertoire can serve as cellular barcodes, facilitating single-cell analysis. Ex vivo screens for a T cell adaptive immune response to editors can also be readily implemented. When primary cells from the relevant target tissue are limiting, bioengineers within the SCGE Consortium will use self-renewing human cells to construct functional three-dimensional organoids or microphysiological systems. These platforms can bring together multiple cell types and extracellular matrices in a tissue-like architecture, providing an in vitro mimic of complex human tissues. Relevant functional assays with these systems can be defined, such as force generation from skeletal muscle, contraction of cardiac tissue and phototransduction in retinal organoids144,145. These systems can be scaled up to enable studies at higher throughput than would be feasible in animal models, and can also facilitate deeper molecular characterization of the various outcomes after editing different human cell types within a tissue. An ultimate aim of these studies is to produce assays that are relevant to regulatory science, to better evaluate various genome-editing strategies. Previous studies involving immunodeficient mice and edited T cells have been broadly enabling for many immune-cell-therapy products, and any new biological system that is developed by the SCGE Consortium, once established, could be similarly enabling for studies aiming towards investigational new drug filings in that cell and gene-therapy space.
Integration of SCGE technologies
The initiatives described above—new editing platforms, delivery technologies, in vivo reporter systems and human biological systems—are expected to recombine and synergize in multiple ways, both planned and spontaneous. One prominent example, arising from the ever-growing recognition of the need for greater reproducibility during clinical translation146, is an explicit requirement for independent validation for all of the delivery technologies in development. Each delivery project involves multiple phases: an initial phase that establishes proof-of-principle within each laboratory that is developing a delivery technology; an intermediate phase that involves testing at a SATC, performed by SATC personnel; and finally, for those technologies that meet pre-defined SATC efficacy milestones, scale-up of the delivery system and testing at a LATC. Large-animal testing is contingent upon successful independent validation of the technology by an SATC—that is, outside of the laboratory that developed the delivery system in question. In another example, investigators that are developing new editors might choose to apply newly developed delivery systems to enable testing in vivo; delivery groups might use human biological systems to assess performance and adverse consequences before commencing with animal tests; and human biological systems could be developed to assess the editing precision of new editing platforms. Such cross-team, integrated projects are nurtured through internal calls within the SCGE Consortium to discuss collaborative proposals each year.
Standards and the SCGE Toolkit
Although the above activities will generate a wide array of data and tools, the maximal impact will be achieved only when SCGE technologies use common standards and are interoperable. Data and resource standards and shared lexicons are imperative for the development of new technologies, and will be particularly critical for translating genome-editing systems into approved therapies147,148. To ensure the highest-quality data, interoperability of tools and reproducibility, the SCGE Consortium’s Dissemination and Coordinating Center (DCC) serves as a communication hub, facilitates collaboration among consortium members and builds platforms to enable the sharing of SCGE program resources and data, including through the SCGE Toolkit. Furthermore, to contribute to standards development, the SCGE Consortium is interfacing with the Food and Drug Administration, the National Institute of Standards and Technology (NIST) and the Defence Advanced Research Projects Agency. In particular, the SCGE Consortium is a member of the NIST Genome Editing Consortium.
The SCGE Toolkit (Fig. 1b) will be generated to develop the infrastructure and data to promote collaborations among the different projects within the SCGE Consortium, and to create a platform for investigators (and eventually, the broader scientific community and the public) to access data generated by the program. To ensure data integration and functional mining tools, standardized data formats and vocabularies are being developed and will be made available through the SCGE Consortium website. There will be several components of the SCGE Toolkit, including a public Resource Portal to provide both consortium members and other investigators with a single stop for information on existing data repositories, public tools and algorithms used in genome-editing research. Investigators within the SCGE Consortium will submit data to these existing resources when available. As these components are tested, validated and used together in experimental procedures, they will be integrated into a centralized database for both the SCGE Consortium and the public, facilitating the comparison of results across experiments and enabling researchers to further refine experimental designs for genome-editing research. Because much of the ongoing clinical development of genome editing is occurring within industry, the SCGE Consortium seeks to contribute broadly accessible data, tools, systems and assays that could enable a more open-access approach for clinical development.
Outlook
New opportunities for the clinical translation of genome-editing technologies are arising from a deeper understanding of the human genome and from rapidly advancing bioengineering capabilities. The SCGE Consortium aims to develop new technologies and adapt existing tools to take immediate advantage of these opportunities, define and mitigate safety risks, and extend therapeutic genome editing into the most challenging somatic tissue contexts. Previous large-scale projects149–155 advanced the frontiers of genomics not only by producing new knowledge, but also by developing a common framework that ensured reproducibility, applied common standards and established the interoperability of distinct technologies. Inspired by these efforts, the SCGE program is designed to advance the field of genome editing towards a broadened spectrum of human therapeutic applications.
Supplementary information
Grouped membership list of the five SCGE initiatives.
Acknowledgements
We acknowledge ArtforScience for support with initial figure ideas.
Author contributions
All authors contributed to writing the manuscript.
Data availability
Data from the consortium will be deposited in public databases, and also made available online through the SCGE Toolkit (https://scge.mcw.edu/toolkit), as described in the SCGE Data Sharing Policy (available at https://scge.mcw.edu/policies/; periodically updated and amended by SCGE Consortium members).
Competing interests
K.S. receives sponsored research support from Spotlight Therapeutics. E.J.S. is a co-founder and advisor of Intellia Therapeutics. C.A.G. is a co-founder of Tune Therapeutics, Element Genomics and Locus Biosciences, and an advisor to Sarepta Therapeutics, Levo Therapeutics and Iveric Bio. D.R.L. is a co-founder or founder and scientific advisor of Editas Medicine, Pairwise Plants, Beam Therapeutics and Prime Medicine. S.Q.T. is a member of the scientific advisory board of Kromatid, Inc. D.G.A. is a co-founder of CRISPR Therapeutics, Sigilon, Verseau, VasoRx and Orna, and a consultant for Translate Bio and Obsidian Therapeutics. A.A. is a co-founder of StrideBio and TorqueBio, and an advisor to AstraZeneca, Sarepta Therapeutics, Gemini Therapeutics and BridgeBio. J.F.B. is a co-founder of Metagenomi. J.W.M.B. receives research support from NovaDip Biosciences, Philips Healthcare and Weinberg Medical Physics. D.F.C. and J.C. are employees and shareholders in Recombinetics, Inc. J.E.D. is a co-founder of Guide Therapeutics. K.J.C. is a co-founder of Recombinetics, Inc. and LifEngine Technologies. B.E.D. is on the scientific advisory board of Tevard Biosciences. J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, Intellia Therapeutics and Mammoth Biosciences; a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Algen Biotechnologies, Felix Biosciences and Inari; a Director at Johnson & Johnson; and has research projects sponsored by Biogen, Pfizer, AppleTree Partners and Roche. S.C.E. is co-founder and chief executive officer of LifEngine Technologies, co-founder of LEAH Labs and chief scientific officer of Mettaforge Therapeutics. D.M.G. is a co-founder and chief scientific advisor for Opsis Therapeutics and holds equity in this company, and also receives sponsored research support from Ascidian Therapeutics. G.G. is a scientific co-founder of Voyager Therapeutics, Adrenas Therapeutics and Aspa Therapeutics. P.M.G. is a co-founder of Cybrexa Therapeutics and Delmab Bio and an advisor to pHLIP, Inc. P.B.M. is on the scientific advisory board and receives support for sponsored research from Spirovant Sciences, Inc. S.K. is the chief scientific officer and co-founder of SafeGen Therapeutics. W.M.S. is a co-founder of and receives research support from Stradefy Biosciences. J.K.W. is a consultant for Flagship Pioneering.
Footnotes
Peer review information Nature thanks Fyodor Urnov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Krishanu Saha, Erik J. Sontheimer
A full list of members and their affiliations appears in the Supplementary Information
Contributor Information
Krishanu Saha, Email: ksaha@wisc.edu.
Erik J. Sontheimer, Email: erik.sontheimer@umassmed.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-021-03191-1.
References
- 1.High KA, Roncarolo MG. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 2.Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–236. doi: 10.1038/s41586-020-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pickar-Oliver A, Gersbach CA. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019;20:490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 5.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 6.Smith J, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JC, et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 8.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho SW, Kim S, Kim JM, Kim J-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 13.Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarova KS, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020;18:67–83. doi: 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl Acad. Sci. USA. 1994;91:6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh CD, Richardson CD, Corn JE. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019;21:1468–1478. doi: 10.1038/s41556-019-0425-z. [DOI] [PubMed] [Google Scholar]
- 19.Naldini L, Trono D, Verma IM. Lentiviral vectors, two decades later. Science. 2016;353:1101–1102. doi: 10.1126/science.aah6192. [DOI] [PubMed] [Google Scholar]
- 20.Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaudelli NM, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mok BY, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anzalone AV, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kearns NA, et al. Functional annotation of native enhancers with a Cas9–histone demethylase fusion. Nat. Methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilton IB, et al. Epigenome editing by a CRISPR–Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol. Ther. 2016;24:430–446. doi: 10.1038/mt.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porteus MH. A new class of medicines through DNA editing. N. Engl. J. Med. 2019;380:947–959. doi: 10.1056/NEJMra1800729. [DOI] [PubMed] [Google Scholar]
- 30.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amoasii L, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeder ML, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 33.Moretti A, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L, et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N. Engl. J. Med. 2019;381:1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 36.Stadtmauer EA, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qasim W, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 38.Chew WL, et al. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther. 2015;26:432–442. doi: 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson CE, et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018;36:717–725. doi: 10.1038/nbt.4182. [DOI] [PubMed] [Google Scholar]
- 42.Sedic M, et al. Safety evaluation of lipid nanoparticle-formulated modified mRNA in the Sprague-Dawley rat and cynomolgus monkey. Vet. Pathol. 2018;55:341–354. doi: 10.1177/0300985817738095. [DOI] [PubMed] [Google Scholar]
- 43.Moreno AM, et al. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 2019;3:806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charlesworth CT, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner DL, et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 46.Ferdosi SR, et al. Multifunctional CRISPR–Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019;10:1842. doi: 10.1038/s41467-019-09693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li A, et al. AAV-CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol. Ther. 2020;28:1432–1441. doi: 10.1016/j.ymthe.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson CE, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabebordbar M, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amoasii L, et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, et al. Highly efficient RNA-guided base editing in rabbit. Nat. Commun. 2018;9:2717. doi: 10.1038/s41467-018-05232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zafra MP, et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–893. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villiger L, et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 55.Li Q, et al. CRISPR–Cas9-mediated base-editing screening in mice identifies DND1 amino acids that are critical for primordial germ cell development. Nat. Cell Biol. 2018;20:1315–1325. doi: 10.1038/s41556-018-0202-4. [DOI] [PubMed] [Google Scholar]
- 56.Yeh W-H, Chiang H, Rees HA, Edge ASB, Liu DR. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018;9:2184. doi: 10.1038/s41467-018-04580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng Y, et al. Correction of the Marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol. Ther. 2018;26:2631–2637. doi: 10.1016/j.ymthe.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryu S-M, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 59.Levy JM, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeh W-H, et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 2020;12:eaay9101. doi: 10.1126/scitranslmed.aay9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng J, et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020;26:535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song C-Q, et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020;4:125–130. doi: 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sürün D, et al. Efficient generation and correction of mutations in human iPS cells utilizing mRNAs of CRISPR base editors and prime editors. Genes (Basel) 2020;11:511. doi: 10.3390/genes11050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim D, Luk K, Wolfe SA, Kim J-S. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu. Rev. Biochem. 2019;88:191–220. doi: 10.1146/annurev-biochem-013118-111730. [DOI] [PubMed] [Google Scholar]
- 65.Gabriel R, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011;29:816. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 66.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perry ME, Valdes KM, Wilder E, Austin CP, Brooks PJ. Genome editing to ‘re-write’ wrongs. Nat. Rev. Drug Discov. 2018;17:689–690. doi: 10.1038/nrd.2018.91. [DOI] [PubMed] [Google Scholar]
- 68.Esvelt KM, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hou Z, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl Acad. Sci. USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim E, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017;8:14500. doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Agudelo D, et al. Versatile and robust genome editing with Streptococcus thermophilus CRISPR1–Cas9. Genome Res. 2020;30:107–117. doi: 10.1101/gr.255414.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edraki A, et al. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol. Cell. 2019;73:714–726.e4. doi: 10.1016/j.molcel.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirano H, et al. Structure and engineering of Francisella novicida Cas9. Cell. 2016;164:950–961. doi: 10.1016/j.cell.2016.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrington LB, et al. A thermostable Cas9 with increased lifetime in human plasma. Nat. Commun. 2017;8:1424. doi: 10.1038/s41467-017-01408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatterjee P, Jakimo N, Jacobson JM. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018;4:eaau0766. doi: 10.1126/sciadv.aau0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chatterjee P, et al. A Cas9 with PAM recognition for adenine dinucleotides. Nat. Commun. 2020;11:2474. doi: 10.1038/s41467-020-16117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu Z, et al. A compact Cas9 ortholog from Staphylococcus auricularis (SauriCas9) expands the DNA targeting scope. PLoS Biol. 2020;18:e3000686. doi: 10.1371/journal.pbio.3000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burstein D, et al. New CRISPR–Cas systems from uncultivated microbes. Nature. 2017;542:237–241. doi: 10.1038/nature21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM. Nanotechnology for delivery of peptide nucleic acids (PNAs) J. Control. Release. 2016;240:302–311. doi: 10.1016/j.jconrel.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan WX, et al. Functionally diverse type V CRISPR–Cas systems. Science. 2019;363:88–91. doi: 10.1126/science.aav7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu J-J, et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019;566:218–223. doi: 10.1038/s41586-019-0908-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dolan AE, et al. Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type I CRISPR–Cas. Mol. Cell. 2019;74:936–950.e5. doi: 10.1016/j.molcel.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Strecker J, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53. doi: 10.1126/science.aax9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klompe SE, Vo PLH, Halpin-Healy TS, Sternberg SH. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature. 2019;571:219–225. doi: 10.1038/s41586-019-1323-z. [DOI] [PubMed] [Google Scholar]
- 87.Pausch P, et al. CRISPR–CasΦ from huge phages is a hypercompact genome editor. Science. 2020;369:333–337. doi: 10.1126/science.abb1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sakata RC, et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020;38:865–869. doi: 10.1038/s41587-020-0509-0. [DOI] [PubMed] [Google Scholar]
- 89.Grünewald J, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020;38:861–864. doi: 10.1038/s41587-020-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang X, et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020;38:856–860. doi: 10.1038/s41587-020-0527-y. [DOI] [PubMed] [Google Scholar]
- 91.Kurt IC, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021;39:41–46. doi: 10.1038/s41587-020-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao D, et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021;39:35–40. doi: 10.1038/s41587-020-0592-2. [DOI] [PubMed] [Google Scholar]
- 93.Holtzman L, Gersbach CA. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 2018;19:43–71. doi: 10.1146/annurev-genom-083117-021632. [DOI] [PubMed] [Google Scholar]
- 94.Zeitler B, et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019;25:1131–1142. doi: 10.1038/s41591-019-0478-3. [DOI] [PubMed] [Google Scholar]
- 95.Black JB, Gersbach CA. Synthetic transcription factors for cell fate reprogramming. Curr. Opin. Genet. Dev. 2018;52:13–21. doi: 10.1016/j.gde.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 96.Economos NG, et al. Peptide nucleic acids and gene editing: perspectives on structure and repair. Molecules. 2020;25:735. doi: 10.3390/molecules25030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McNeer NA, et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat. Commun. 2015;6:6952. doi: 10.1038/ncomms7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bahal R, et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016;7:13304. doi: 10.1038/ncomms13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gammage PA, Moraes CT, Minczuk M. Mitochondrial genome engineering: the revolution may not be CRISPR-ized. Trends Genet. 2018;34:101–110. doi: 10.1016/j.tig.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Minczuk M, Kolasinska-Zwierz P, Murphy MP, Papworth MA. Construction and testing of engineered zinc-finger proteins for sequence-specific modification of mtDNA. Nat. Protoc. 2010;5:342–356. doi: 10.1038/nprot.2009.245. [DOI] [PubMed] [Google Scholar]
- 101.Gammage PA, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018;24:1691–1695. doi: 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bacman SR, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018;24:1696–1700. doi: 10.1038/s41591-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hashimoto M, et al. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther. 2015;23:1592–1599. doi: 10.1038/mt.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Campbell, J. M. et al. Engineering targeted deletions in the mitochondrial genome. Preprint at 10.1101/287342 (2018).
- 105.Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017;16:387–399. doi: 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- 106.Wang D, Zhang F, Gao G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020;181:136–150. doi: 10.1016/j.cell.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van Haasteren J, Li J, Scheideler OJ, Murthy N, Schaffer DV. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020;38:845–855. doi: 10.1038/s41587-020-0565-5. [DOI] [PubMed] [Google Scholar]
- 108.Finn JD, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 109.Wei T, Cheng Q, Min Y-L, Olson EN, Siegwart DJ. Systemic nanoparticle delivery of CRISPR–Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11:3232. doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yin H, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krishnamurthy S, et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019;10:4906. doi: 10.1038/s41467-019-12922-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen G, et al. A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Nat. Nanotechnol. 2019;14:974–980. doi: 10.1038/s41565-019-0539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Staahl BT, et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yin H, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moon SB, Kim DY, Ko J-H, Kim J-S, Kim Y-S. Improving CRISPR genome editing by engineering guide RNAs. Trends Biotechnol. 2019;37:870–881. doi: 10.1016/j.tibtech.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 116.Wu W, et al. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proc. Natl Acad. Sci. USA. 2017;114:1660–1665. doi: 10.1073/pnas.1614775114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chu VT, et al. Increasing the efficiency of homology-directed repair for CRISPR–Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 118.Certo MT, et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods. 2011;8:671–676. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Iwano S, et al. Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science. 2018;359:935–939. doi: 10.1126/science.aaq1067. [DOI] [PubMed] [Google Scholar]
- 120.Penheiter AR, Russell SJ, Carlson SK. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr. Gene Ther. 2012;12:33–47. doi: 10.2174/156652312799789235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Minn I, et al. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci. Adv. 2019;5:eaaw5096. doi: 10.1126/sciadv.aaw5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bulte JWM. Superparamagnetic iron oxides as MPI tracers: A primer and review of early applications. Adv. Drug Deliv. Rev. 2019;138:293–301. doi: 10.1016/j.addr.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pumphrey AL, et al. Cardiac chemical exchange saturation transfer MR imaging tracking of cell survival or rejection in mouse models of cell therapy. Radiology. 2017;282:131–138. doi: 10.1148/radiol.2016152766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang J, Lee CCI, Sutcliffe JL, Cherry SR, Tarantal AF. Radiolabeling rhesus monkey CD34+ hematopoietic and mesenchymal stem cells with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for microPET imaging. Mol. Imaging. 2008;7:1–11. [PubMed] [Google Scholar]
- 125.Tarantal AF, et al. Radiolabeling and in vivo imaging of transplanted renal lineages differentiated from human embryonic stem cells in fetal rhesus monkeys. Mol. Imaging Biol. 2012;14:197–204. doi: 10.1007/s11307-011-0487-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tarantal AF, Lee CCI, Kukis DL, Cherry SR. Radiolabeling human peripheral blood stem cells for positron emission tomography (PET) imaging in young rhesus monkeys. PLoS ONE. 2013;8:e77148. doi: 10.1371/journal.pone.0077148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tarantal AF, Lee CCI, Martinez ML, Asokan A, Samulski RJ. Systemic and persistent muscle gene expression in rhesus monkeys with a liver de-targeted adeno-associated virus vector. Hum. Gene Ther. 2017;28:385–391. doi: 10.1089/hum.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bulte JWM, et al. Quantitative “hot spot” imaging of transplanted stem cells using superparamagnetic tracers and magnetic particle imaging (MPI) Tomography. 2015;1:91–97. doi: 10.18383/j.tom.2015.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tarantal AF, Lee CCI, Jimenez DF, Cherry SR. Fetal gene transfer using lentiviral vectors: in vivo detection of gene expression by microPET and optical imaging in fetal and infant monkeys. Hum. Gene Ther. 2006;17:1254–1261. doi: 10.1089/hum.2006.17.1254. [DOI] [PubMed] [Google Scholar]
- 130.Tarantal AF, Lee CCI. Long-term luciferase expression monitored by bioluminescence imaging after adeno-associated virus-mediated fetal gene delivery in rhesus monkeys (Macaca mulatta) Hum. Gene Ther. 2010;21:143–148. doi: 10.1089/hum.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meier S, et al. Non-invasive detection of adeno-associated viral gene transfer using a genetically encoded CEST-MRI reporter gene in the murine heart. Sci. Rep. 2018;8:4638. doi: 10.1038/s41598-018-22993-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nyström NN, et al. Longitudinal visualization of viable cancer cell intratumoral distribution in mouse models using Oatp1a1-enhanced magnetic resonance imaging. Invest. Radiol. 2019;54:302–311. doi: 10.1097/RLI.0000000000000542. [DOI] [PubMed] [Google Scholar]
- 133.Crosetto N, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods. 2013;10:361–365. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim D, et al. Digenome-seq: genome-wide profiling of CRISPR–Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243, 1, 243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 136.Tsai SQ, et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cameron P, et al. Mapping the genomic landscape of CRISPR–Cas9 cleavage. Nat. Methods. 2017;14:600–606. doi: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 138.Lazzarotto CR, et al. Defining CRISPR–Cas9 genome-wide nuclease activities with CIRCLE-seq. Nat. Protoc. 2018;13:2615–2642. doi: 10.1038/s41596-018-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wienert B, et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. 2019;364:286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmid-Burgk JL, et al. Highly parallel profiling of Cas9 variant specificity. Mol. Cell. 2020;78:794–800.e8. doi: 10.1016/j.molcel.2020.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cheng Y, Tsai SQ. Illuminating the genome-wide activity of genome editors for safe and effective therapeutics. Genome Biol. 2018;19:226. doi: 10.1186/s13059-018-1610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Simhadri VL, et al. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 2018;10:105–112. doi: 10.1016/j.omtm.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lazzarotto CR, et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Truskey GA. Development and application of human skeletal muscle microphysiological systems. Lab Chip. 2018;18:3061–3073. doi: 10.1039/c8lc00553b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang J, et al. Engineered skeletal muscles for disease modeling and drug discovery. Biomaterials. 2019;221:119416. doi: 10.1016/j.biomaterials.2019.119416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Freedman LP, Cockburn IM, Simcoe TS. The economics of reproducibility in preclinical research. PLoS Biol. 2015;13:e1002165. doi: 10.1371/journal.pbio.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Plant AL, Locascio LE, May WE, Gallagher PD. Improved reproducibility by assuring confidence in measurements in biomedical research. Nat. Methods. 2014;11:895–898. doi: 10.1038/nmeth.3076. [DOI] [PubMed] [Google Scholar]
- 148.Plant AL, et al. How measurement science can improve confidence in research results. PLoS Biol. 2018;16:e2004299. doi: 10.1371/journal.pbio.2004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.The ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 150.Brown JB, Celniker SE. Lessons from modENCODE. Annu. Rev. Genomics Hum. Genet. 2015;16:31–53. doi: 10.1146/annurev-genom-090413-025448. [DOI] [PubMed] [Google Scholar]
- 151.Stunnenberg HG, Hirst M. The International Human Epigenome Consortium: a blueprint for scientific collaboration and discovery. Cell. 2016;167:1145–1149. doi: 10.1016/j.cell.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 152.Dekker J, et al. The 4D nucleome project. Nature. 2017;549:219–226. doi: 10.1038/nature23884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Warren CR, Jaquish CE, Cowan CA. The NextGen Genetic Association Studies Consortium: a foray into in vitro population genetics. Cell Stem Cell. 2017;20:431–433. doi: 10.1016/j.stem.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 154.HuBMAP Consortium The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature. 2019;574:187–192. doi: 10.1038/s41586-019-1629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Collins F, Galas D. A new five-year plan for the U.S. Human Genome Project. Science. 1993;262:43–46. doi: 10.1126/science.8211127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Grouped membership list of the five SCGE initiatives.
Data Availability Statement
Data from the consortium will be deposited in public databases, and also made available online through the SCGE Toolkit (https://scge.mcw.edu/toolkit), as described in the SCGE Data Sharing Policy (available at https://scge.mcw.edu/policies/; periodically updated and amended by SCGE Consortium members).