

Abstract
Whole-slide histology images contain information that is valuable for clinical and basic science investigations of cancer but extracting quantitative measurements from these images is challenging for researchers who are not image analysis specialists. In this paper, we describe HistomicsML2, a software tool for learn-by-example training of machine learning classifiers for histologic patterns in whole-slide images. This tool improves training efficiency and classifier performance by guiding users to the most informative training examples for labeling and can be used to develop classifiers for prospective application or as a rapid annotation tool that is adaptable to different cancer types. HistomicsML2 runs as a containerized server application that provides web-based user interfaces for classifier training, validation, exporting inference results, and collaborative review, and that can be deployed on GPU servers or cloud platforms. We demonstrate the utility of this tool by using it to classify tumor-infiltrating lymphocytes in breast carcinoma and cutaneous melanoma.
Keywords: Digital pathology, computational pathology, machine-learning, immuno-oncology, biomarkers
INTRODUCTION
Whole-slide digital imaging of histologic sections is increasingly common in cancer research. Multiresolution whole slide images (WSIs) are a potentially rich source of data for clinical and basic science investigations, however, extracting quantitative information from WSIs presents many challenges for most investigators. Image analysis algorithms for detecting and classifying structures and patterns in WSIs are typically specific to a given tissue or dataset and are difficult for investigators to develop or to adapt to new applications. The scale and format of WSIs also make analysis with software tools like ImageJ (1) or CellProfiler (2) prohibitive.
The emergence of learning-based algorithms like convolutional neural networks (CNNs) has been transformational in biomedical image analysis. CNNs are largely powered by labeled data and have been demonstrated to be capable of detecting, classifying, and delineating histological structures in digital pathology applications provided adequate labeled datasets (3,4). Given their reliance on training data and the increasing commodification of deep learning algorithms and software, CNNs can be readily adapted to new applications and utilized by a broader community who are capable of generating annotations but who may lack experience developing conventional image analysis algorithms. Reaching these users in digital pathology applications requires addressing challenges in human-computer interaction, visualization, and high-performance computing.
We developed HistomicsML2, a server-based software platform for training and validation of CNN classifiers on WSI datasets. HistomicsML2 uses a learn-by-example approach to training, called active learning, that helps users identify the most informative examples in order to minimize effort and increase prediction accuracy. Active learning is a training paradigm where unlabeled examples are analyzed to identify the most impactful examples of labeling. The classifier uses an internal prediction confidence measure to rate unlabeled examples and presents poorly rated examples to the user for labeling. The labeled examples are used to update the classifier and the process is repeated again. This process creates a parsimonious training set where important patterns are represented without labeling redundant examples. Active learning has a long history as a technique for training machine learning algorithms but is challenging to implement in digital pathology due to demands on processing and visualizing WSI data (5,6). Superpixel segmentation is used to subdivide tissue regions into small patches that are then analyzed using a pre-trained CNN to extract features for classification. This combination of operations results in a fast and responsive algorithm that is scalable to WSI datasets and that is not application-specific. In this paper, we demonstrate how HistomicsML2 can be used to create accurate classifiers of tumor-infiltrating lymphocytes in triple-negative carcinoma of the breast and cutaneous melanoma, and show that active learning improves training efficiency.
HistomicsML2 builds on prior work that used conventional image analysis algorithms tailored to specific applications (7). In that paradigm, users had to develop bespoke image segmentation and feature extraction algorithms for their applications which are highly specific to a given tissue type and histologic pattern. This requirement has been eliminated in HistomicsML2 by using a CNN-based patch classification approach that is more robust than conventional image analysis methods and that has been demonstrated to perform well in a wide variety of applications. This avoids the need for users to provide their own bespoke algorithms and makes analysis capabilities accessible for cancer researchers. Several commercial and open-source tools exist for training machine learning classifiers with WSI datasets (8–11). QuPath, ASAP, and Boost are thick client applications that run on an end user’s workstation, desktop, or notebook computer. QuPath classification algorithms focus on individual cell nuclei and rely on explicit segmentation of nuclear boundaries using conventional image analysis methods. Orbit offers training of sophisticated convolutional network algorithms and can utilize GPU hardware acceleration or Apache Spark clusters to increase computing power beyond the limited client resources, but requires scripting by the end-user. Like HistomicsML2, Cytomine is a web-based application intended to run on a centralized server that facilitates collaboration and leverages the advanced hardware resources of server systems. Cytomine is also available as a Docker image but does not provide active learning training of algorithms.
An overview of the HistomicsML2 analysis pipeline is presented in Figure 1. Datasets are prepared for analysis by first performing a superpixel segmentation and feature extraction using the data generation Docker (Figure 1A). Superpixel segmentation is first performed at low magnification to generate a grid of small patches covering tissue regions. These superpixels adapt their boundaries to coincide with strong gradients like the edges of cell nuclei or tissue interfaces, producing an adaptive grid where the content of each patch is more homogeneous. A feature map describing each superpixel is generated in 3 steps: (1) A square patch centered on the superpixel is extracted at high magnification (2) A pre-trained VGG-16 CNN transforms this patch into a high-dimensional feature map (3) The size of this feature map is reduced using principal component analysis. The final step helps to improve speed during interactive classification by minimizing computation and storage. Extended details of our design and parameters used for experiments are presented in Methods and Figure S1A–D.
Figure 1. Overview of HistomicsML2 software for whole-slide image analysis.
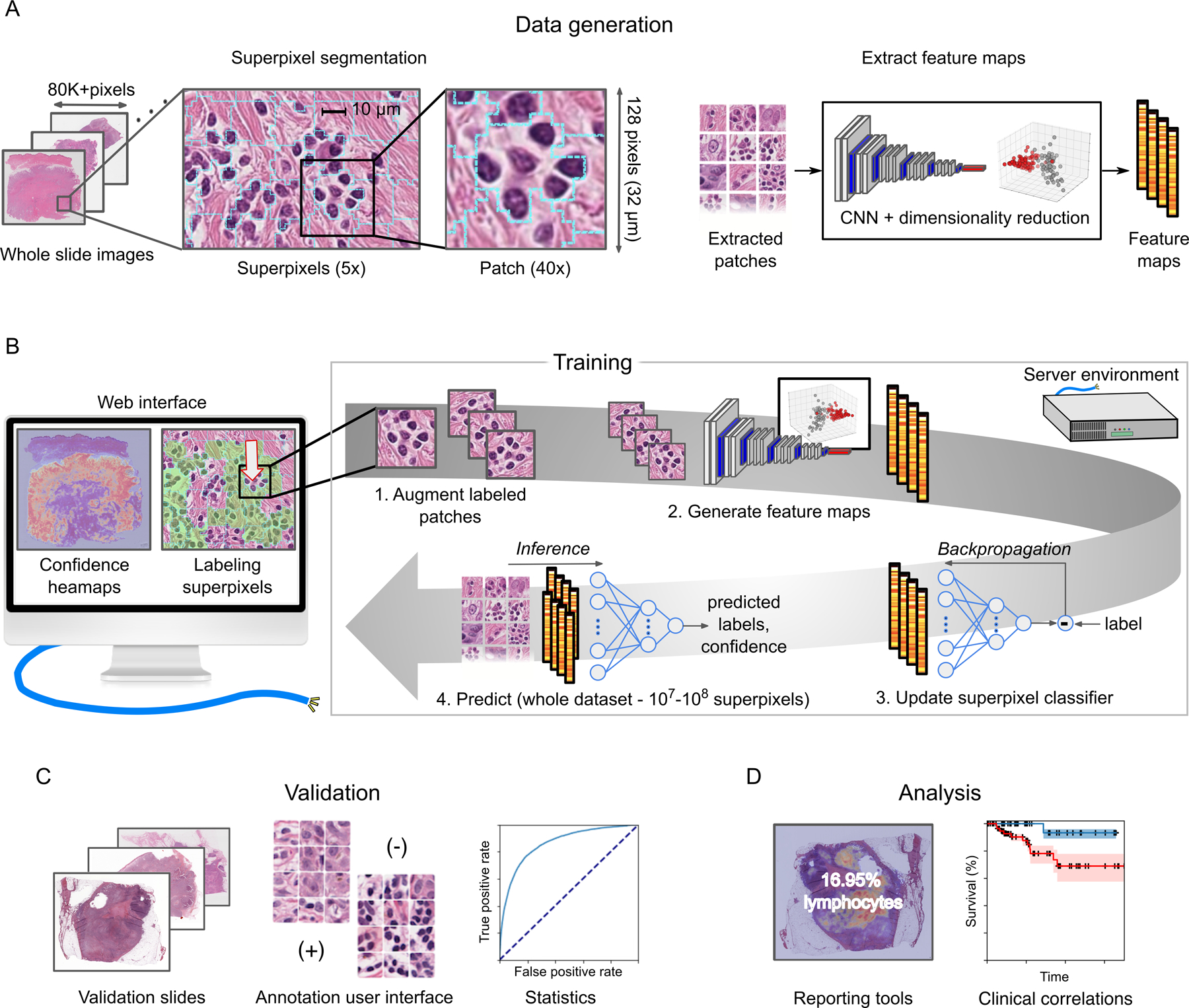
(A) Each WSI is analyzed at low magnification to divide the tissue into superpixel regions that adapt to gradients like cell nuclei borders or tissue interfaces. A patch is extracted from each superpixel at high-magnification and analyzed with a CNN to generate a feature map for machine learning. (B) A web interface communicating with a server is used to train a superpixel classifier. Zoomable WSI heatmaps guide users to regions that are enriched with superpixels having low prediction confidence. Each superpixel labeled by the user is augmented by rotation to increase the training set size, and feature maps from the labeled superpixels are used to update the classifier. The updated classifier is applied to the entire dataset to generate new predictions and confidence heatmaps, and the training process is repeated until the user is satisfied with the classifier predictions. (C) A validation interface helps users develop validation sets, to share and review these sets with others, and to calculate accuracy statistics for trained classifiers. (D) HistomicsML2 provides reporting tools to output summary statistics on WSI datasets. This data can be used to explore clinical or molecular correlations of histology.
During training, users navigate the slides using superimposed heatmaps of classifier predictions, providing corrective feedback to iteratively improve the classifier (Figure 1B). These heatmaps represent the classification uncertainty of the individual superpixels and help users to focus their labeling in regions that are enriched with superpixels that confound the classifier, and to avoid labeling of redundant superpixels that are familiar to the classifier. Labeling is performed by clicking individual superpixels or by using a cursor paint tool for labeling larger areas. Each labeled superpixel is used to update a neural network (NN) that classifies the superpixels. A patch is first extracted from the labeled superpixel. Following extraction, rotated duplicates of this patch are generated to increase the available training data. Low dimensional feature vectors are extracted from these augmented patches and used to update the NN. Finally, the updated NN is applied to the entire dataset to generate new predicted superpixel labels and uncertainty heatmaps. As an alternative to the heatmap interface for training, HistomicsML2 provides an instance-based interface that presents a queue of low-confidence superpixels to the user for labeling (see Video 1). This interface does not require users to explore or navigate the WSIs to perform labeling.
Video 1.
The HistomicsML2 server application is deployed as a GPU-enabled Docker image that serves user interfaces for training, validation, inference and export of results (see Figure S2). A review interface presents superpixels labeled during training, organized by slide and class, and allows users to review and modify label assignments. This makes it possible for students or trainees to perform the bulk of labeling work in an experiment with periodic supervision from a more experienced investigator or pathologist. A validation user interface allows users to generate validation annotation sets and to validate trained classifiers using these annotations to measure accuracy and F1-score (Figure 1C). For results export, tables describing the abundance of inferred patterns (Figure 1D) in each slide can be generated from the user interface and used in correlative analyses with clinical outcomes or molecular endpoints and to generate visualizations like those presented in Figures 1 and 2. Mask images of inference results can also be exported, enabling additional customized post-processing, analysis of spatial patterns, or integration with other tools. In the case where HistomicsML is being used for annotation, these masks can serve as ground truth for training other machine learning algorithms. Trained classifiers can also be exported which provides considerable flexibility in tool integration and offline analysis. The data generation tools are also provided as a GPU-enabled Docker image that can utilize multiple GPUs to accelerate the extraction of features from WSI datasets for producing HistomicsML2 datasets.
Figure 2. Prediction results on BRCA and SKCM.
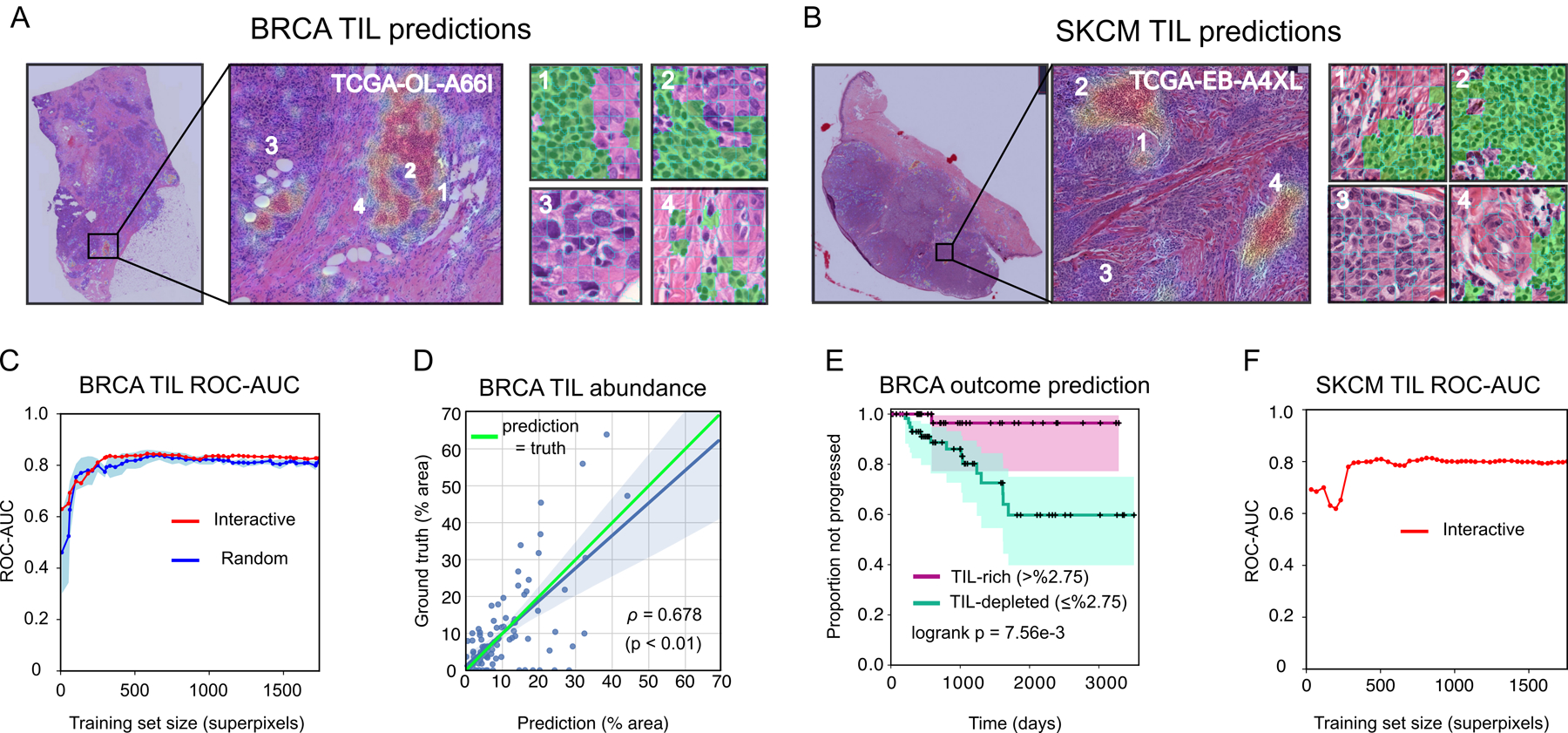
(A–B) TIL-density heatmaps and predicted TIL superpixels (green). Red heatmap areas indicate higher predicted TIL density. (C) ROC-AUC of BRCA-TIL classifier trained using active learning in HistomicsML2 (red). ROC-AUC of classifiers trained from randomly sampled superpixels shown for comparison (blue is average +/− 1 SD). (D) Correlation between predicted and ground truth abundance of TIL positive areas in triple-negative test slides. (E) Kaplan Meier plot of disease progression for TIL-rich and TIL-depleted triple-negative cases as stratified by HistomicsML2 predictions on WSIs. (F) SKCM-TIL classifier ROC-AUC.
MATERIALS AND METHODS
Data
Images and clinical outcomes data.
As an illustrative example, we applied HistomicsML2 to develop classifiers of tumor-infiltrating lymphocytes (TILs) in 1) Triple-negative breast carcinomas (BRCA) and 2) Primary cutaneous melanoma (SKCM). TIL quantification is an emerging diagnostic and predictive histological biomarker and has been shown to correlate with patient survival and therapeutic response in BRCA, SKCM, as well as a wide variety of solid tumors (12–14). Detailed guidelines for the manual scoring of TILs help reduce interobserver variability (15), yet computational assessment is hoped to improve reproducibility and pave the way for quantitative assessment with minimal bias (16).
These experiments used WSIs of hematoxylin and eosin-stained formalin-fixed paraffin embedded (FFPE) sections from The Cancer Genome Atlas (see Table S1). Classifier predictions were validated using ground truth annotations of tissue regions generated using the Digital Slide Archive (17) and a public dataset of breast cancer histopathology annotations dataset (18). Accuracy was measured at the individual pixel resolution on annotated regions of holdout WSIs not used in training. Pixel resolution accuracy accounts for discretization errors due to superpixel boundaries not conforming to ground truth. All clinical data used was derived from the TCGA Pan-Cancer resource (19).
Ground truth annotation data.
Annotation data for the BRCA experiments was obtained from a public repository of pathologist generated and reviewed annotations (18). This dataset contains freehand annotations of lymphocytic infiltration, tumor, stromal, and necrotic tissue regions performed within rectangular regions in WSIs from 149 triple-negative carcinomas. Ground truth annotations for the SKCM experiments were generated using the Digital Slide Archive (17) and similar annotation protocols as those published in the BRCA annotation dataset (16). Large rectangular regions of interest (ROIs) ranging from 2.4 – 11.2 mm2 were selected for annotation and within each ROI, areas containing lymphocytic infiltration of the tumor-associated stroma were manually delineated using a free-hand polygon tool. For the purpose of this work, we considered all small mononuclear cells with lymphocyte-like morphology to be TILs. Dense purely plasma cell infiltrates were not included in TIL regions, but admixtures of lymphocytes and plasma cells were counted as lymphocytic infiltrates. Artifacts, normal epidermal or dermal structures, and nearby lymphoid aggregates were excluded from the regions of interest. The manual polygonal boundaries were stored on a Mongo database and queried and converted to masks for analysis (pixel values encode region membership). This procedure follows the protocol described in the BRCA annotation dataset (18).
Software
Superpixel segmentation.
Tissue areas in WSIs were masked using the HistomicsTK simple_mask function (https://digitalslidearchive.github.io/HistomicsTK/histomicstk.segmentation.html) to define the regions for data extraction and analysis. Masked tissue areas were color normalized using the Reinhard function from the preprocessing module of HistomicsTK (https://digitalslidearchive.github.io/HistomicsTK/histomicstk.preprocessing.html). Normalized color images were then segmented with the scikit-image SLIC superpixel algorithm at 40X objective creating roughly 64 × 64 pixel sized superpixels. Square patches sized 128 × 128 pixels are extracted from the superpixel centroids and resized to 224 × 224 to be used as the input of the pre-trained VGG-16 model. These steps were performed on 8192 × 8192 tiled versions of the WSI. The objective magnifications for superpixel analysis and patch extraction as well as the superpixel parameters are fully adjustable.
Generating convolutional feature maps.
The VGG-16 pre-trained CNN was used to extract feature maps from the color-normalized patches (https://keras.io). This network consists of 13 convolutional layers, 5 max-pooling layers, and 3 fully-connected layers. We truncated this network to extract 4096-dimensional feature maps after the first fully-connected layer. These vectors are reduced to 64-dimensional feature maps using PCA to improve the storage footprint of datasets and responsiveness of the training interface and to reduce inference time when applying trained classifiers to new datasets.
HistomicsML2 can use a variety of magnifications for superpixel segmentation and feature extraction, although for most applications we recommend performing superpixel segmentation at 5–20X objective where feature extraction should be performed at 20–40X objectives. Reducing these objectives can significantly help with the time required to generate data and may be possible in some applications where the patterns of interest cover large areas and have simple boundaries.
Machine learning.
HistomicsML2 uses a multi-layer neural network for superpixel classification and interactive learning. Keras version 2.2.2 and Tensorflow version 1.12 were used to implement the machine learning algorithms in HistomicsML2. In our experiments, this neural network was configured with three layers (containing 64–32-1 neurons). The hidden layer uses a rectified linear activation function and a dropout fraction of 30%, and the output layer uses a sigmoidal activation for class prediction. This network was optimized using cross-entropy loss with the Adam optimizer. All of these parameters are adjustable by editing the networks.py source file.
Training.
The HistomicsML2 BRCA classifier was trained over 49 iterations to label a total of 1741 superpixels. The HistomicsML2 SKCM classifier was trained over 56 iterations to label the 2009 superpixels. A superpixel was considered TIL-positive if it contains one or more complete TIL nuclei. The model was initially primed by choosing 4 positive and 4 negative superpixels. After priming, each training cycle consisted of the following steps: 1. Choose a slide for analysis; 2. Use the uncertainty heatmap to navigate to a field of view with low prediction confidence; 3. View predicted superpixel labels in a high-magnification field of view and correct a roughly-equal number of false-positive and false-negative superpixels; 4. Repeat step 3 for two more fields of view in variable geographic locations, until the total number of corrected superpixels ranges between 30–60; 5. Retrain the network; 6. Navigate to the next slide; 7. Repeat the process, making sure that all training slides are eventually represented in the labeled training set. Accuracy on the testing slides was not evaluated during the training process.
For the BRCA dataset, a series of 10 classifiers was generated as a comparison to evaluate the benefit of active learning training in HistomicsML2. Each training slide in the BRCA dataset has an associated ground truth annotation8, and so we formed 10 training sets by sampling superpixels from these images and using the ground-truth annotations for labeling. These training sets were formulated to identically match the number of superpixels and slides where these superpixels were labeled during each iteration of HistomicsML2 classifier training. Superpixels were randomly sampled from annotation regions and were labeled as positive if 75% or more of their pixels were annotated as TIL.
Simulation.
We performed simulations to measure the impact of different feature extraction and dimensionality reduction methods on classifier performance, and to evaluate tumor classification in breast cancer. These simulations used ground truth annotations from our breast cancer dataset described above with training and testing slide assignments from Figure S1A–D. In each simulated training cycle 10 examples from each class were selected and added to the training set. Each cycle performed 10 epochs of training updates. Samples were selected without replacement either randomly or from the top quartile of uncertain samples to simulate active learning. The resnet18 network was evaluated as a relatively simpler alternative network to VGG-16 for feature extraction. Dimensionality reduction was evaluated on VGG-16 extracted features by reducing them to 1024 dimensions, or by retaining 95% of the variance during PCA. We also compare resnet18 with VGG-16 in TIL and tumor classification and evaluate random versus active learning using resnet18 features. Classifiers trained on the full 96,800 training samples were used as an upper-bound reference of performance.
Reproducibility.
During training HistomicsML2 records all metadata associated with labeled samples needed to reconstruct a session including the epoch a sample was labeled in, sequence of labeling, prediction confidence (used in active learning) and how the sample was labeled (heatmap or instance interface). In addition to this information, a serialized prediction model is maintained during training and can be exported and used outside of HistomicsML2.
Data analysis
Validation.
Slides in the BRCA and SKCM datasets were assigned to non-overlapping training and testing sets so that no single case/sample has slides represented in both sets to avoid data leakage. The BRCA dataset was split into training (45 slides) and testing (10 slides) sets. The SKCM dataset was split into training (40 slides) and testing (10 slides) sets. HistomicsML2 classifiers were trained on the training slides and their performance was evaluated on the testing slides. Since HistomicsML2 trains classifiers iteratively, classifier performance was evaluated at every iteration to analyze sensitivity to training set size.
The prediction accuracy of trained classifiers was measured on a pixel-wise basis. Superpixel predictions were mapped by filling each superpixel with the predicted class or class-probability. These prediction images were compared to ground truth annotations, measuring error on a pixel-wise basis to account for the fact that superpixel boundaries do not conform perfectly to ground truth annotations. Accuracy, receiver operating characteristic area under the curve (AUC), and Matthews correlation coefficient (MCC) were measured by a pixel-wise comparison of HistomicsML2 predictions for superpixels with masks from the ground truth annotations. Accuracy is defined as:
where P and N are the number of predicted positive and negative pixels respectively, TP is the number of true positive pixel predictions, TN is the number of true negative pixel predictions, FN is the number of false negative pixel predictions, and FP is the number of false positive pixel predictions.
Breast cancer data analysis.
We applied the trained HistomicsML2 classifier to an additional 96 slides to analyze TIL abundance and to evaluate the prognostic accuracy of HistomicsML2 predictions. We combined these 96 slides with the 10 testing slides to form an analysis set. These analysis slides have associated ground truth annotations8 and do not overlap with cases in the training set.
Percent area occupied by TIL regions was calculated on a pixel-wise basis within the ROI for each analysis set slide for both the ground-truth annotations and the HistomicsML2 predictions. The correlation between predicted and actual percentages was calculated using Pearson correlation.
To evaluate prognostics accuracy, we used predicted TIL percentages calculated over WSIs. The median TIL percentage on training slides (2.75%) was used as a threshold to define TIL-rich (>2.75% abundance by area) and TIL-depleted (≤2.75%) categories. Analysis slides were placed in these categories using predicted TIL percentages, and a Kaplan Meier analysis was performed to compare these two groups. The logrank test was also used for hypothesis testing to compare survival distributions of the TIL-rich and TIL-depleted analysis cases.
Software
All source code, documentation, and data are available on Github (https://github.com/CancerDataScience/HistomicsML2) ReadtheDocs (https://histomicsml2.readthedocs.io/en/latest/index.html) and Synapse (https://www.synapse.org/#!Synapse:syn22293065). Containerized versions of software for data generation and the HistomicsML2 server have been published on Docker hub (https://cloud.docker.com/u/cancerdatascience/repository/docker/cancerdatascience/histomicsml).
Hardware
Experiments were performed using a two-socket server with 2 × 16 Intel Xeon cores, 256 GB memory, 54 TB network mounted storage, and two NVIDIA Tesla P100 GPUs.
RESULTS
Predictions from trained classifiers on testing images are presented in Figures 2A and 2B. The ROC-AUC of the BRCA-TIL classifier trained with HistomicsML2 is 0.828 with 1741 superpixels labeled in 49 iterations (88.9% accuracy, 0.409 MCC). After 11 training iterations with 332 superpixels labeled, the HistomicsML2 classifier has ROC 0.837 (89.3% accuracy, 0.366 MCC) (Figure 2C, Table S2). To compare active learning to conventional algorithm training we simulated five BRCA-TIL classifiers by randomly selecting labeled superpixels from ground truth annotations of the training slides. These classifiers were trained without active learning and had generally lower ROC-AUC values and slower convergence (median ROC-AUC 0.796 +/− 0.028 at 12 iterations). These random classifiers eventually converge to have similar performance as the HistomicsML2 classifiers given the larger training set. We found agreement between the percentage of tissue corresponding to TILs with the percentages from ground truth annotations in testing images (Pearson ρ=0.68, see Figure 2D, Table S3). We also assessed the prognostic value of TIL percentages predicted by our HistomicsML2 classifier on testing WSIs from our triple-negative BRCA cohort (See Figure 2E, Table S4). Stratification of cases into TIL-rich and TIL-depleted using HistomicsML2 predictions accurately predicts disease progression risk in this cohort (logrank p=7.56e-3). The SKCM-TIL classifier converged to 0.800 ROC-AUC (92.4% accuracy, 0.370 MCC) (see Figure 2F). Analyses of clinical outcomes and comparisons to random classifiers were not possible for the SKCM-TIL predictions due to lack of ground-truth annotations for the training dataset.
To evaluate the impact of different dimensionality reduction and feature extraction and methods we performed classifier training simulations using the breast cancer ground truth dataset for TIL and tumor classification (see Figure S3A–E, Table S5). Greater reduction of VGG-16 features through PCA has a significant positive impact on TIL classification. VGG-16 features were not effective in classifying tumor, and dimensionality reduction did not improve performance. We also evaluated features from a resnet18 pre-trained network in TIL and tumor classification as a relatively simpler alternative to VGG-16 features. The resnet18 features excelled in tumor classification (>0.70 PR-AUC) but were inferior to VGG-16 features in TIL classification. Finally, we validated the utility of active learning in tumor classification, where we observed that an active learning tumor classifier had faster convergence, greater accuracy, and less variability than a classifier trained with randomly selected samples.
We performed experiments to compare the execution times of training updates with and without GPU acceleration (see Figure S4, Table S6). Using the same network from our experiments, we varied the number of labeled samples in the training set from 200 to 1000, and measured the time required to update the network with and without a single GPU for acceleration. With GPU acceleration training updates do not exceed 2.5 seconds. Without GPU acceleration training times can exceed 40 seconds with training sets of 1000 superpixels.
DISCUSSION
HistomicsML2 is a server application that is built for analyzing WSI data. Web-based user interfaces enable interactive training of classifiers, review and editing of training sets, and export of quantitative measurements of histology that can be compared to clinical outcomes or molecular endpoints. HistomicsML can be used to develop predictive models for prospective applications, or as a tool to rapidly annotate a collection of slides. Containerization using Docker simplifies deployment by bundling all software dependencies and improves the ease of cloud platform deployments. Documentation is provided (https://histomicsml2.readthedocs.io/en/latest/index.html) as a guide for users, and also describes the design of HistomicsML2 for developers who want to extend or modify this tool using our source repository (https://github.com/CancerDataScience/HistomicsML2). While this software was designed and optimized to minimize computational requirements, a GPU equipped server can accelerate data-generation and enhance the responsiveness of the system during interactive training (see Figure S3A–E). These resources are widely available on cloud platforms if on-premises resources are not available.
Our experiments demonstrate that HistomicsML2 can assist users in analyzing patterns in WSI datasets, yielding valuable quantitative measurements of cancer histology. This system can be used to develop predictive models that are applied prospectively to new cases not seen in training, or as an annotation tool to rapidly annotate patterns and yield measurements for a collection of slides. The success of CNNs in analyzing a wide variety of histologic patterns motivated our use of CNNs for feature extraction. Quality and abundance of training data is the primary determinant in CNN performance, and so our tool seeks to make it easy for investigators to produce parsimonious training sets using active learning. Similarly, the superpixel segmentation used in HistomicsML2 has been used to delineate tissue components in a variety of contexts and is not application-specific. Our design also permits advanced users to substitute custom feature extraction pipelines and to use HistomicsML2 as an interface to their algorithms by following our documented data formats.
Our experiments used popular “commodity” feature extraction networks to develop classifiers of TILs in breast cancer and cutaneous melanoma. We demonstrate that for triple-negative BRCA patients, quantitative TIL measurements are effective in stratifying clinical outcomes, and that training with active learning accelerates convergence and improves TIL classification accuracy. We also observed clear benefits of active learning training in simulations of tumor classification in breast cancer. In studies of cancer, these capabilities could be used to detect tumor regions and to describe components of the tumor microenvironments, to analyze spatial distribution patterns of histologic features, or to quantify immunohistochemical staining.
There are important limitations and areas for future work. While we examined TIL classification in BRCA and SKCM, we did not evaluate performance in other tissue types or for other histologic patterns. Although our tool allows users to specify parameters of the trainable portion of the neural network like number of layers, dropout fraction, and optimization algorithm, we only evaluated a single network architecture in our experiments. The flexibility of the superpixel segmentation also does not address all possible use cases in cancer histology and is best suited to tissue region and larger scale patterns. It is unlikely to permit analysis of individual cells, as many cells would be subdivided by superpixel boundaries, and the significant increase in the number of superpixels would increase computational demand. Our analyses also did not perform quantitative assessments of user experience or multi-user studies to account for interobserver variability. We have taken steps to simplify setup, data generation, and data import, but the scale of data involved and implementation as a server application requires support from a bioinformatician or information technology staff.
In future work, we plan to explore more advanced active learning strategies for helping users with training in multiclass problems and to improve training efficiency by selecting better samples for labeling. More advanced feature extraction methods can also improve accuracy and there are a number of developments in unsupervised and self-supervised techniques that we plan to investigate. Ideally the network parameters could be determined adaptively during training for users who lack expertise in tuning algorithms. This remains an open problem since in active learning the selection of optimal training samples is specific to a network architecture. Future software development efforts will pursue enhancements to further simplify data generation, to develop deployment scripts for popular cloud platforms, and to optimize system design to take advantage of the elastic scalability that exists in the cloud.
Supplementary Material
SIGNIFICANCE.
An interactive machine learning tool for analyzing digital pathology images enables cancer researchers to apply this tool to measure histologic patterns for clinical and basic science studies.
ACKNOWLEDGEMENTS
This research was supported in part by the National Institutes of Health National Cancer Institute (NCI) grant U01CA220401 that supported Mohamed Amgad, David A Gutman, and Lee AD Cooper, National Institutes of Health National Cancer Institute (NCI) grant U24CA19436201 that supported Sanghoon Lee, Pooya Mobadersany, David A Gutman and Lee AD Cooper, and National Institutes of Health National Library of Medicine grant KLM011576A that supported Lee AD Cooper.
FINANCIAL SUPPORT
This research was supported in part by the National Institutes of Health National Cancer Institute (NCI) grants U01CA220401 and U24CA19436201 and National Institutes of Health National Library of Medicine grant KLM011576A.
Footnotes
DISCLOSURE
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones TR, Kang IH, Wheeler DB, Lindquist RA, Papallo A, Sabatini DM, et al. CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 2008;9:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehteshami Bejnordi B, Veta M, Johannes van Diest P, van Ginneken B, Karssemeijer N, Litjens G, et al. Diagnostic Assessment of Deep Learning Algorithms for Detection of Lymph Node Metastases in Women With Breast Cancer. JAMA 2017;318:2199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Litjens G, Sanchez CI, Timofeeva N, Hermsen M, Nagtegaal I, Kovacs I, et al. Deep learning as a tool for increased accuracy and efficiency of histopathological diagnosis. Sci Rep 2016;6:26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Settles B From theories to queries: Active learning in practice. In Active Learning and Experimental Design workshop In conjunction with AISTATS 2010; 1–18
- 6.Kutsuna N, Higaki T, Matsunaga S, Otsuki T, Yamaguchi M, Fujii H, et al. Active learning framework with iterative clustering for bioimage classification. Nat Commun 2012;3:1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalisnik M, Amgad M, Lee S, Halani SH, Velazquez Vega JE, Brat DJ, et al. Interactive phenotyping of large-scale histology imaging data with HistomicsML. Sci Rep 2017;7:14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stritt M, Stalder AK, Vezzali E. Orbit Image Analysis: An open-source whole slide image analysis tool. PLoS Comput Biol 2020;16:e1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marée R Open Practices and Resources for Collaborative Digital Pathology. Frontiers in Medicine 2019;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marée R, Rollus L, Stévens B, Hoyoux R, Louppe G, Vandaele R, et al. Collaborative analysis of multi-gigapixel imaging data using Cytomine. Bioinformatics 2016;32:1395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 2015;26:259–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method From the International Immunooncology Biomarkers Working Group: Part 1: Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma In Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv Anat Pathol 2017;24:235–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv Anat Pathol 2017;24:311–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kos Z, Roblin E, Kim RS. et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. npj Breast Cancer 2020;6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amgad M, Stovgaard ES, Balslev E, et al. Report on computational assessment of Tumor Infiltrating Lymphocytes from the International Immuno-Oncology Biomarker Working Group. npj Breast Cancer 2020;6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutman DA, Khalilia M, Lee S, Nalisnik M, Mullen Z, Beezley J, et al. The Digital Slide Archive: A Software Platform for Management, Integration, and Analysis of Histology for Cancer Research. Cancer Res 2017;77:e75–e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173:400–16 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.