

Abstract
Intrinsic or acquired resistance to clinically approved CDK4/6 inhibitors has emerged as a major obstacle that hinders their utility beyond ER+ breast cancer. In this study, CDK4/6-dependent and -resistant models were employed to identify functional determinants of response to pharmacological CDK4/6 inhibitors. In all models tested, the activation of RB and inhibition of CDK2 activity emerged as determinants of sensitivity. While depleting CDK4 and 6 was sufficient to limit proliferation in specific resistance settings, RB loss rendered cells completely independent of these kinases. The main downstream target in this context was the activation status of CDK2, which was suppressed with CDK4/6 inhibition in an RB-dependent fashion. Protein levels of p27 were associated with plasticity/rigidity of the cell cycle and correlated with sensitivity to CDK4/6 inhibition. Exogenous overexpression and pharmacological induction of p27 via inhibition of SKP2 and targeting the MEK/ERK pathway enhanced the cytostatic effect of CDK4/6 inhibitors. Mice bearing ER+ xenografts displayed a durable ani-tumor response to palbociclib; however, over the course of treatment, few cells retained RB phosphorylation, which is associated with limited p27 protein levels as determined by multi-spectral imaging. Similarly, combination treatment of palbociclib with a MEK inhibitor in pancreatic cancer PDX models upregulated p27 and further enhanced the in vivo tumor response to palbociclib. Collectively, these results suggest that the cell cycle plasticity that enables tumor models to evade palbociclib-mediated activation of RB could be targeted using a clinically applicable CDK2 inhibitor.
Keywords: Cell-cycle plasticity, palbociclib, RB, CDK2 activity, P27, PF06873600
Introduction:
Cyclin dependent kinases 4 and 6 (CDK4/6) regulate cell-cycle initiation by phosphorylating the retinoblastoma tumor suppressor protein (RB) thereby inactivating its transcriptional repression function (1, 2). RB is further hyper-phosphorylated and inactivated by CDK2 that enhances the transition from G1 to S-phase of cell-cycle (3). The activity of CDK4/6 kinases are mainly governed by their binding partners, D-type cyclins, which are the transcriptional targets of different mitogenic signaling pathways (4, 5). The deregulation in cell-cycle machinery is one of the major mechanisms that transforms a normal cell to become cancerous (6). Such phenomenon can be driven by constitutive activation of upstream oncogenic signaling pathways, amplification of cyclin D1 and CDK4, or inactivation of a tumor suppressor protein, p16 that acts as an endogenous inhibitor of CDK4/6 (6–8).
Owing to their role in tumor biology, CDK4/6 has been considered as a promising molecular target to pharmacologically activate the RB pathway in different cancer types (9). Three selective CDK4/6 inhibitors, palbociclib, ribociclib and abemaciclib have been clinically approved for the treatment of estrogen receptor (ER) + breast cancer in combination with endocrine therapy (10–12). In multiple clinical studies, CDK4/6 inhibitors significantly increased the progression free survival (PFS) in patients with metastatic ER+ breast cancer (13, 14). However, development of acquired resistance to CDK4/6 inhibition limits the overall survival benefit. In other tumor settings CDK4/6 inhibition has had less of a positive impact, commonly due to intrinsic or rapidly acquiring resistance mechanisms (15). Hence to improve the clinical success of CDK4/6 inhibitors, combination treatment options will be required to target molecular mechanisms that drive resistance (16).
Studies have demonstrated that both normal and cancer cells bypass CDK4/6 pathway and continue to proliferate due to plasticity in their cell cycle machinery. In embryonic fibroblasts from mice devoid of CDK4 and CDK6 kinases, cell-cycle progression resumes under mitogenic stimuli because of the compensatory role of other CDKs (17, 18). On the other hand, in mammalian cancer cells, high levels of CDK4 activity overcomes the loss of CDK2 and promotes cell-cycle progression through the inactivation of RB pathway (19). In the context of pharmaceutical CDK4/6 inhibitors, there are myriad of molecular mechanisms that enable the cancer cells to bypass the negative cell-cycle regulation (20). Importantly, loss of RB and enhanced CDK2 activity due to amplification of cyclin E1 can promote resistance by driving cell-cycle progression even in the presence of pharmacological CDK4/6 inhibitors (21–23). However, the complexity in cell cycle machinery in different tumor models results in differing outcomes in many preclinical or clinical analyses and defining precise functional determinants of response to CDK4/6 inhibitors has remained a challenge (24, 25). Deepening the understanding of different cell-cycle regulatory factors that drive cell cycle plasticity would facilitate rational therapeutic approaches to achieve durable disease control.
Materials and Methods:
Cell culture and therapeutic agents:
Primary PDAC cells were grown in Keratinocyte SFM medium, supplemented with EGF (0.2 ng/mL), bovine pituitary extract (30 μg/mL) (Life Technologies, Carlsbad, CA) and 2% fetal bovine serum (FBS) on a collagen-coated (Millipore, Burlington, MA) tissue culture dishes. Select PDAC models were stably infected to express H2B-GFP as an independent measure for proliferation. MCF7 cells were maintained in DMEM medium containing 10% FBS and T47D and HCC1806 cells were maintained in RPMI media containing 10% FBS. Lung cancer cell lines, A549 and H1975 were kindly provided by Dr. Pamela Hershberger at RoswellPark Cancer Center and were grown in DMEM and RPMI media respectively containing 10% FBS. All the cells lines were grown at 37°C and 5% CO2 and were confirmed to be mycoplasma free. Cell-line authentication were performed using STR analysis. Palbociclib (IBRANCE) and PF06873600 were purchased from MedChemExpress (NJ, USA). Trametinib was purchased from Selleckchem (Houston, TX).
Plasmids and infection procedures:
Lentiviral overexpression vectors (pLX304) containing the ORFs clones for CCND1, V5 tag-CDK4 and CDKN1B were purchased from Cancer genetics and Genomics core; Roswell Park Cancer Center. Lentiviral infections were carried out in exponentially growing MCF7 and PDAC cells in the presence of Polybrene (Sigma Aldrich). The infected cells were selected using Blasticidin (5 μg/ml) and the protein overexpression was validated using western blotting.
CSII-EF lentiviral vector containing the cDNA for HDHB fused to mCHERRY was a gift from Dr. Steven Pruitt’s laboratory (Roswell Park Cancer Center). Lentiviral infection was performed on H2B-GFP labelled 1222 cells and the double positive clones containing both GFP and mCHERRY, cells were sorted using BD FACSAria II cell sorter.
Mice and xenografts:
NSG mice were maintained at University of Arizona and Roswell Park Cancer Center animal care facilities. All animal care, drug treatments and sacrifice were approved by the University of Arizona and Roswell Park Cancer Center Institutional Animal Care and Use Committee (IACUC) in accordance with the NIH guide for the care and use of laboratory animals. Mice were subcutaneously implanted with the early passage PDX tumor fragments. Early passaged MCF7 cells (1X 10^7 cells/mouse) and HCC1806 cells (5X10^6 cells/mouse) were subcutaneously injected into 8-10 weeks old female NSG mice. Mice were supplemented with estrogen pellet to promote the growth of MCF7 derived tumors. Treatment dosage and schedule are described in supplementary information.
RPPA and gene expression analysis:
The RPPA data for different breast cancer and pancreatic cancer cell lines were retrieved from the MD Anderson Cell Lines Project on the TCPA portal (26). Proteins with missing data across the cell lines were excluded for further analysis. The protein expression data for P21CIP1, p27 and cyclin E1 were compared between the breast and pancreatic cancer cell lines and were represented in a boxplot. The p-values were calculated based on student’s t-test. The IC50 values of palbociclib and abemaciclib in different breast cancer cell lines were retrieved from previously published studies (10, 27). Based on the IC50 values of palbociclib the cell lines were stratified as sensitive (IC50<0.2 μM) and resistant (IC50>0.2 μM) to palbociclib. For abemaciclib, IC50<0.38 is considered as sensitive cell lines and IC50>0.38 is considered as resistant. Heat map was generated to depict the differential expression of cell-cycle genes across the cell lines that were sorted based on their IC50 values of palbociclib. The gene expression data were retrieved from CCLE database. Correlation statistics were calculated based on the response to palbociclib and the expression of SKP2 and CCNE1 gene expression. Using the RPPA database correlation statistics were calculated based on the IC50 values and p27, cyclin E1 and P21CIP1 expressions in different breast cancer cell lines.
DepMAp Achilles :
The top 50 cell lines that are sensitive to CDK4, CDK6, CCNE1 and CDK2 based on their gene dependency scores were retrieved from the Project Achilles Portal (https://depmap.org/portal/achilles/) (28). From the top 50 cell lines that are sensitive to CCNE1 and CDK2 there were 28 cell lines sensitive to both CDK2 and CCNE1 (CDK2/CCNE1). The sensitivity of these cell lines to loss of CDK4 and CDK6 were analyzed. Similarly, from the top 50 cell lines that are sensitive to loss of CDK4 and CDK6, their corresponding sensitivity to CDK2 and/or CCNE1 was analyzed. To define differential sensitivity to other genes between the CDK4 or CDK6 (blue) and CCNE1/CDK2 (Orange) sensitive cell lines, the log fold change with a cut off of +/− 0.05 and p-value (0.05) were determined based on student t-test and a volcano plot was generated.
Data deposition:
RNA sequencing data are deposited in GEO: GSE113922 and GSE146788.
Detailed methods are described in supplementary information.
Results
Functional characterization of tumor models based on their response to CDK4/6 inhibition:
Using live-cell imaging we observed that three different patient-derived PDAC cell lines (1222, 226 and 3226) that harbor KRAS mutations (29, 30) displayed weak cytostatic response to palbociclib; whereas the proliferation of ER+ breast cancer models, MCF7 and T47D was robustly inhibited (Figures. 1A, Supplementary Figures S1A & S2A). Similar to palbociclib, another CDK4/6 inhibitor, abemaciclib, also showed a stronger cytostatic effect in ER+ breast cancer cells as compared to the PDAC models (Figure. 1A). The EC50 values of both palbociclib and abemaciclib in the PDAC cell lines were significantly higher than that of the ER+ breast cancer models (Figure. 1B). In lung adenocarcinoma models A549 and H1975, which are driven by KRAS and EGFR mutations respectively, palbociclib elicited a better control on the growth of A549 cells while H1975 cells continued proliferation, similar to the PDAC models (Supplementary Figure. S2A) (31, 32). The differential cytostatic effect of palbociclib among these cancer models further correlated with the inhibition of BrdU incorporation resulting in the most potent cell-cycle arrest in ER+ breast cancer models. (Figure. 1C). Consistent with 2D cell culture assays, palbociclib also yielded a more potent cytostatic response in limiting the organoid growth in ER+ breast cancer cells (T47D) as compared to the PDAC model (1222) (Figure. 1D, Supplementary Figure S2B). Biochemical analysis revealed that palbociclib treated cells resulted in suppression of endogenous RB phosphorylation in all tumor models; however, the most sensitive ER+ breast cancer cells showed the highest degree of dephosphorylation (Figure. 1E and Supplementary Figures S2C & S2D). Since CDK2 can potentially mediate RB phosphorylation, the effect of CDK4/6 inhibition on the enzymatic activity of CDK2 was determined using in vitro kinase assays (33). The ER+ breast cancer cells showed a robust inhibition of CDK2 kinase activity in the presence of palbociclib and abemaciclib whereas the PDAC models (1222 and 226) retained partial activity (Figures. 1F, 1G, 1H and Supplementary Figures S2E & S2F). Although abemaciclib is known to possess off-target activity against CDK2 kinase at higher concentration, the inhibitory effect observed in MCF7 and PDAC models is associated with CDK4/6 inhibition as confirmed by RB dephosphorylation and cyclin A downregulation (Supplementary Figure. S2G) (34). Consistent to the in vitro CDK2 kinase assays, the inhibitory effect of palbociclib on the intracellular CDK2 activity was more prominent in the ER+ breast cancer models as compared to the PDAC models as determined by its phosphorylation status (Supplementary Figure. S2H). Together, these data illustrate a spectrum of sensitivities to CDK4/6 inhibition that are not related to the presence of RB, but to the coordinated suppression of CDK2 activity.
Figure 1: Differential response of CDK4/6 inhibition in different models:
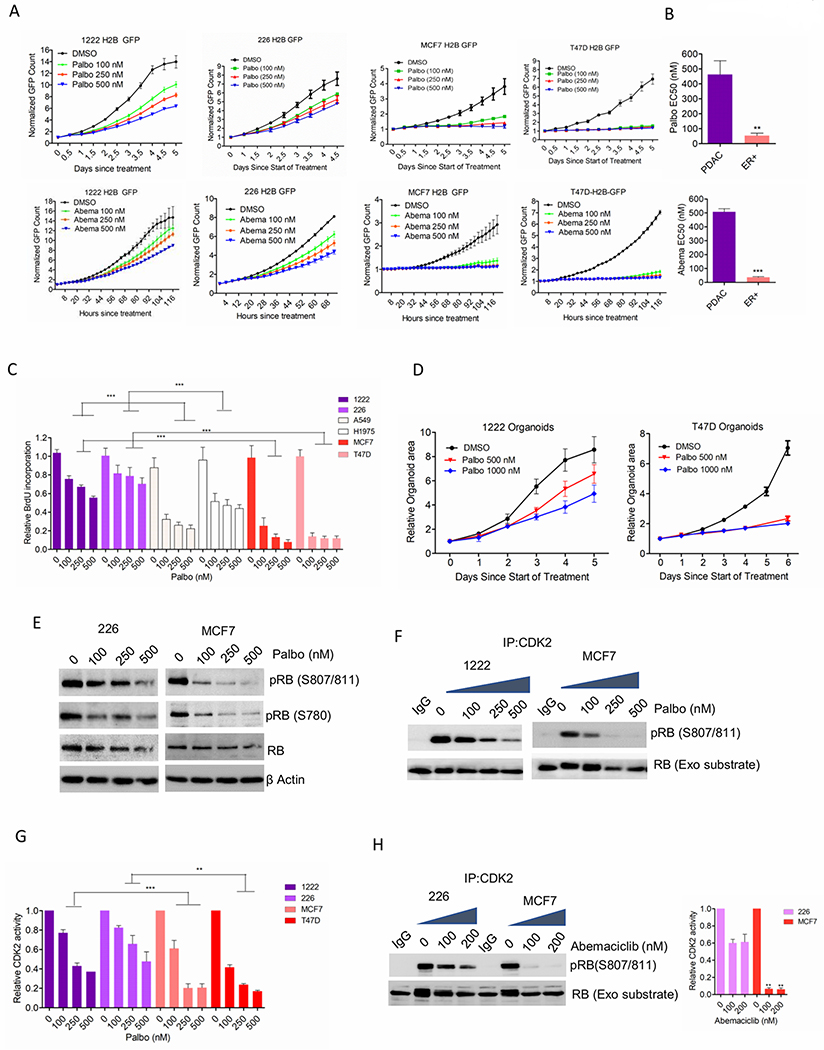
(A) Growth rates of 1222, 226, MCF7 and T47D cells, treated with palbociclib and abemaciclib. Bars indicate mean and SD. (B) Column graph represents the EC50 values of palbociclib and abemaciclib. Mean and SEM are shown (t-test). (C) BrdU incorporation on the indicated cell lines following 72 H exposure with palbociclib. Bars represent mean and SD (1-way ANOVA). (D) Growth of organoids derived from 1222 and T47D cells treated with palbociclib. Bars represent mean and SD. (E) Western blot analysis on MCF7 and 226 cells following 48 H exposure with palbociclib. (F) In vitro CDK2 kinase assays on MCF7 and 1222 cells treated with palbociclib up to 48 H. Kinase activity was evaluated based on the phosphorylation of an exogenous RB fragment as substrate at S807/811. (G) Densitometry analysis on RB phosphorylation. Column represents the mean and SEM (1-way ANOVA). (H) MCF7 and 226 cells treated were treated with up to 48 H and the CDK2 kinase activity was evaluated. Representative blot images and mean and SEM are shown (1-way ANOVA). Graphs represent 2 independent experiments with 3 replicates. (***p<0.001).
CDK4/6 regulate the proliferation of both ER+ breast cancer and PDAC models via RB dependent coupling to CDK2-activity:
To investigate whether the differential effect of palbociclib in ER+ breast cancer and PDAC cells is due to their differential addiction to CDK4 and CDK6, the endogenous expression of these kinases was depleted independently and concurrently using RNAi. In MCF7 cells, the inhibition of BrdU incorporation was more prominent following CDK4 knockdown as compared to CDK6 knockdown, indicating the dominant function of CDK4 in cell-cycle (Figure. 2A). However, the concurrent knockdowns yielded the most potent cell-cycle in both ER+ breast cancer and PDAC models (1222 and 226) (Figure. 2A). Biochemical analysis confirmed an enhanced inhibition of RB phosphorylation and cyclin A downregulation following the double knockdown in these models (Figure. 2B and Supplementary Figure S3A). Hence, the loss of CDK4 and CDK6 expressions have similar downstream effect on RB pathway in both ER+ breast cancer and PDAC cells, indicating that the resistance of PDAC is at least partly associated with the presence of target proteins CDK4 and CDK6. Hence, there could be a pool of CDK4/6 that is not particularly sensitive to pharmaceutical inhibition or that non-catalytic activities contribute to resistance.
Figure 2: Functional roles of CDK4 and CDK6 kinases.
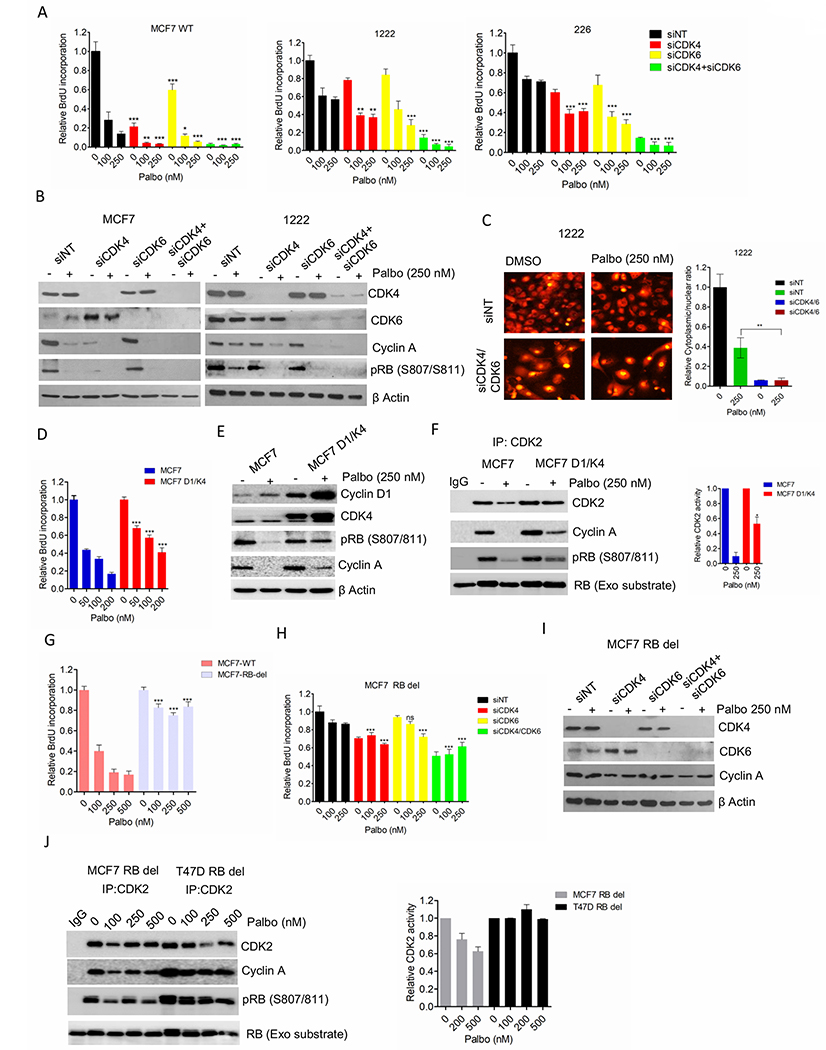
(A) BrdU incorporation in MCF7-WT, 1222 and 226 cell lines following the knockdowns of CDK4 and CDK6. Bars represent mean and SD (t-test) (B) Western blot analysis on MCF7 and 1222 cells following CDK4/6 knockdowns. (C) Representative images of 1222 cells stably expressing the CDK2 sensor following CDK4/6 concurrent knockdowns. Column graph indicates ratio of number of cells with cytoplasmic localization to number of cells with nuclear localization of the HDHB-mCHERYY. (D) BrdU incorporation in MCF7 and MCF7D1/K4 cells following 72 H exposure with palbociclib. Bars represent mean and SD (t-test) (E) Western blotting on MCF7 and MCF7D1/K4 cells following 48 H treatment with palbociclib (250 nM). (F) In vitro CDK2 kinase assays on MCF7 and MCF7D1/K4 cells treated with palbociclib (250 nM) up to 48 H. Representative blot images and mean and SEM are shown. (G) BrdU incorporation in MCF7-WT and MCF7-RB-del cells following 72 H exposure with palbociclib. Bars represent mean and SD (t-test). (H) BrdU incorporation in MCF7 RB depleted (MCF7 RB del) cell lines following the knockdowns of CDK4 and CDK6. Bars represent mean and SD (t-test). (I) Western blot analysis on MCF7 RB del following CDK4/6 knockdowns. (J) In vitro CDK2 kinase assays on MCF7 and T47D RB depleted cells treated with increasing concentrations of palbociclib up to 48 H. Representative blot images and mean and SEM are shown. Graphs represent 2 independent experiments with 3 replicates. (*p< 0.05, **p<0.01, ***p<0.001).
To interrogate intra-cellular CDK2 kinase activity in response to CDK4 and 6 dual knockdown, we employed a fluorescent-tagged CDK2 sensor (HDHB-mCHERRY) as described in a previous study (35). The phosphorylation of this sensor due to CDK2 activity as cells proliferate, drives its export from nuclear to cytoplasm. Hence, the ratio of cytoplasmic to nuclear fluorescence serves as a measure of CDK2 kinase activity. In PDAC models while palbociclib had a modest effect on the CDK2 activity, the depletion of CDK4/6 elicited a more pronounced impact that associated with a durable growth arrest (Figure. 2C and Supplementary Figure S3B). Conversely, overexpression of cyclin D1 and CDK4 in the palbociclib-sensitive MCF7 cells (MCF7 D1/K4) rendered them partially resistant to palbociclib as determined by BrdU incorporation (Figure. 2D). Furthermore, palbociclib-mediated RB dephosphorylation and suppression of cyclin A were rescued in the MCF7 D1/K4 cells, suggesting that this perturbation induces the ability to bypass pharmaceutical inhibition in ER+ breast cancer model (Figure. 2E). In accordance with the correlative analysis, MCF7 D1/K4 cells harbored active CDK2 kinase even in the presence of palbociclib (Figure. 2F).
To specifically dissect the role of RB in response to CDK4/6 inhibition on cell-cycle, RB knockout MCF7 cells were employed (Supplementary Figure. S3C). These cells were completely refractory to palbociclib as determined by BrdU incorporation, but unlike the PDAC models they were also resistant to CDK4/6 knockdown (Figures. 2G & 2H). Under these conditions the cells retained cyclin A expression (Figure. 2I). Like RB deficient ER+ breast cancer model, the pancreatic cancer cell line, 7310 that lacks RB displayed similar cellular response to CDK4/6 KD (Supplementary Figures S3D & S3E). Moreover, the RB deficient ER+ breast cancer models (MCF7 and T47D) cells maintained CDK2 kinase activity in the presence of palbociclib (Figure. 2J). These data indicate that RB acts as a central player in coupling the CDK4/6 kinase activity with CDK2 to mediate the cellular response to palbociclib in ER+ breast cancer, while in PDAC models there is a degree of plasticity in spite of the presence of RB.
Role of p27 expression in cell-cycle plasticity:
To determine additional determinants/biomarkers of response to CDK4/6 inhibition, we utilized DepMap data to stratify cell lines based on their degree of sensitivity to loss of CDK4 or 6 kinase (28). It was evident from our analysis that the dependency on CDK4 and CDK6 is mutually exclusive (Figure. 3A). Moreover, the cancer cells that are independent to both CDK4 and CDK6 were sensitive to loss of CDK2 and/or cyclin E1 (Figure. 3A). Analysis on the CDK4/6 resistant cell lines revealed the sensitivity to additional genes that are mainly involved in the regulation of CDK2 kinase activity (Figure. 3B). For example, SKP2, which is a negative regulator of p27 protein expression was associated with resistance to CDK4 and CDK6 kinases. To further validate this observation in response to CDK4/6 inhibitor, we interrogated the previously published IC50 values of palbociclib in different breast cancer models (10). Gene expression analysis on those models revealed that among several cell-cycle related genes the expression of RB regulated genes significantly correlated with response to palbociclib (Supplementary Figures S4A & S4B). Consistent to the Achilles data, the cell lines that are resistant to palbociclib (IC50>0.2 μM) harbored high levels of CCNE1 and SKP2 (Figure. 3C). Moreover, RPPA analysis indicated that low cyclin E1 and high p27 protein levels are the determinants of sensitivity to CDK4/6 inhibitors, suggesting the plausible role of SKP2 in regulating p27 at the protein level (Figures. 3D & S4C). In accordance with our computational analysis, p27 expression was relatively higher in the palbociclib-sensitive MCF7 cells as compared to the PDAC models (Figure. 3E). Additionally, p21, which also regulates CDK4 and CDK2 kinases was expressed more abundantly in MCF7 cells than the PDAC models (Figure 3E). Interestingly, MCF7 cells had higher cyclin D1 expression as compared to all the PDAC models due to its increased protein stability and CDK4 expression was modestly higher in MCF7 cells suggesting that the expression of these proteins cannot predict the response to palbociclib (Figure. 3E and Supplementary Figure S4D) (13, 36). Although loss of RB and amplification of cyclin E1 has been previously shown to be one of the mechanisms of resistance to palbociclib (23), PDAC models do not harbor such aberrations (Figure. 3E). RPPA analysis from different breast and pancreatic cancer cell lines further confirmed that PDAC models harbored significantly lower levels of cyclin E1, p21 and p27 protein expressions as compared to breast cancer models (Supplementary Figure. S4E) (26). Unlike p27 expression, no significant correlation was observed between p21 expression levels and IC50 of palbociclib (Supplementary Figure. S4F).
Figure 3: Functional roles of P27 in PDAC and ER+ breast cancer models.
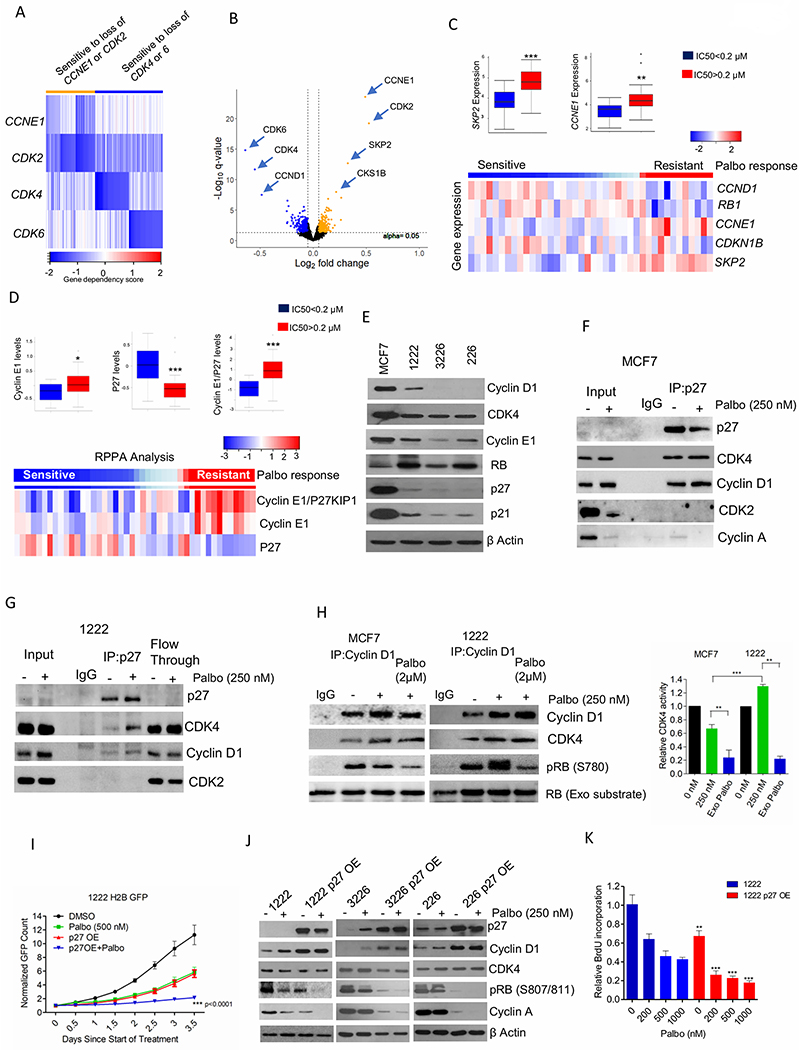
(A) Heat map represents the gene dependency score of different cancer cell lines that are sensitive to loss of CCNE1 (n=22), CDK2 (n=22) and to both CCNE1 and CDK2 (n=28) and their corresponding gene dependency scores to loss of CDK4 and CDK6 (Orange). Blue represents the top 50 cell lines that are only sensitive to loss of CDK4 and CDK6 independently and their corresponding gene dependency scores to loss of CCNE1 and/or CDK2. (B) Volcano plot indicates the differentially sensitive genes between the orange and blue groups, which are defined by a cut-off value +/− 0.05 for fold change and p-value 0.05 as determined by t-test. (C) Box plot representing the distribution of fold change of SKP2 and CCNE1 gene expression based on the IC50 values of palbociclib in different breast cancer cells. Heatmap indicates the distribution of fold change of the indicated genes based on the response to palbociclib. (D) Box plot and heat map representing the distribution of fold change of cyclin E1, P27 and cyclin E1/P27 based on the IC50 values of palbociclib in different breast cancer cells. (E) Western blot on the baseline expression of the indicated proteins from MCF7 and different PDAC cell lines. (F) Immunoprecipitation of P27 from MCF7 cells treated with palbociclib (250 nM) up to 48 H. Coimmunoprecipitated cyclin D1, CDK4, CDK2 and Cyclin A were determined by immunoblotting. (G) Immunoprecipitation of P27 from 1222 cells treated with palbociclib (250 nM) up to 48 H. Cyclin D1, CDK4 and CDK2 on the co-immunoprecipitated and flow through samples were determined by immunoblotting. (H) In vitro CDK4 kinase assay associated with cyclin D1 on MCF7 and 1222 cells treated with palbociclib (250 nM) up to 48 H. Exogenous palbociclib (2 μM) was added to the kinase reaction mix. Kinase activity was evaluated based on the phosphorylation of an exogenous RB fragment as substrate at S780. Representative blot images and mean and SD are shown. (I) Growth rate of 1222-WT and 1222 P27 OE cells treated with palbociclib (500 nM). Bars represent mean and SD (2-way ANOVA). (J) Immunoblot analysis on 1222, 226 and 3226 cell lines and their respective P27 OE cells in the presence of palbociclib (250 nM) up to 48 H. (K) BrdU incorporation of 1222 and 1222 P27 OE cells following 72 H exposure in the presence of palbociclib. Bars represent mean and SD (t-test). (*p< 0.05, **p<0.01, ***p<0.001).
To elucidate the functional role of differential p27 expression we investigated its association with different CDK binding partners using co-immunoprecipitation assays. In actively cycling MCF7 cells, p27 was associated predominantly with CDK4 rather than CDK2 (Figure. 3F). This implicates the non-catalytic function of CDK4 in sequestering p27 to ensure that CDK2 is catalytically active (37, 38) Even in the presence of palbociclib (250 nM), the entire pool of p27 was selectively bound with the CyclinD1/CDK4 complex, however, under this condition CDK2 is inactive due to limiting Cyclin A expression (Figure. 3F). In PDAC models that have low basal expression of p27, either in the absence or presence of palbociclib, p27 did not show a detectable interaction with CDK2, whereas only a modest interaction with Cyclin D1/CDK4 complex was observed (Figure. 3G). Western blotting from the residual lysate following p27 immunoprecipitation (flow through) retained large amounts of cyclin D1 and CDK4, indicating that in PDAC models a larger fraction of cyclin D1/CDK4 complex is free from p27 (Figure. 3G). The high p27 expression in MCF7 cells mediates assembly of cyclin D1/CDK4 complex because depletion of endogenous p27 expression in MCF7 cells using siRNA resulted in the dissociation of the cyclin D1/CDK4 complex (Supplementary Figure. S5A) (39, 40). Interestingly, such phenomenon was not observed in the PDAC model (1222), further confirming that only a very small fraction of cyclin D1 and CDK4 could form complex with p27 due to its low baseline expression (Supplementary Figure. S5A).
To examine how the differential association of p27 with Cyclin D1/CDK4 complex in MCF7 and PDAC cells could modulate the inhibitory effect of palbociclib, we determined the kinase activity of CDK4 associated with cyclin D1 in these cell lines. Although, palbociclib treated cells resulted in more cyclin D1/CDK4 complex in both MCF7 and PDAC models, the corresponding increasing in the kinase activity was selectively observed in the pancreatic cancer cell line (Figure. 3H). Interestingly, addition of exogenous palbociclib (2 μM) to the kinase reactions interfered with the CDK4 kinase activity in both the models confirming in vitro activity against each recovered kinase complex (Figure. 3H). This suggest that the differential levels of p27 between these models might contribute to the differential intra-cellular effect of palbociclib on the CDK4/6 kinase activity. Exogenous overexpression of p27 in the palbociclib partial-resistant PDAC models enhanced the cytostatic effect of palbociclib (Figure. 3I and Supplementary Figure S5B). Biochemical analysis revealed that p27 overexpression increased cyclin D1 protein levels without modulating the CDK4 expression in all the PDAC models and this is due to the increased protein stability of cyclin D1 (Figure 3J and Supplementary Figure S5C). Following the overexpression of p27, palbociclib elicited an enhanced effect on RB phosphorylation and downregulation of cyclin A, which resulted in an enhanced inhibition of BrdU incorporation (Figures 3J & 3K). Overall these data confirm the functional role of high levels of p27 in enhancing the cytostatic effect palbociclib.
Induction of p27 using pharmaceutical inhibitors:
Since SKP2 is a negative regulator of p27, we examined its function on p27 expression and cellular response to palbociclib in the resistant models. Depletion of SKP2 in PDAC models, 1222 and 226 significantly enhanced the anti-proliferative effect of palbociclib, which is accompanied by upregulation of p27 and dephosphorylation of RB (Figures 4A & B). Importantly, pharmacological inhibition of SKP2 by an NEDD8 inhibitor, pevonedistat resulted in a cooperative cytostatic effect in combination with palbociclib in PDAC models (Figure. 4C & D) (41). To investigate the involvement of other pathways in p27 regulation, a targeted drug screen using a panel of clinically approved and preclinically advanced agents was performed in PDAC cells (1222). Heatmap illustrating the relative growth rate of the cells in the presence of select classes of drugs in combination with vehicle (DMSO) and palbociclib (200 nM) indicates that the MEK inhibitors cooperate with palbociclib (Figure. 4E). Validation studies using different MEK inhibitors in combination with palbociclib resulted in a robust inhibition of cell proliferation and trametinib was identified as the most potent molecule (Figure. 4F). Prior studies have demonstrated that p27 is a downstream target for the RAS-driven MEK/ERK pathway, which results in its phosphorylation and subsequent degradation (42). In PDAC cells (226), KRAS knockdown resulted in the accumulation of p27 protein expression and further enhanced RB dephosphorylation and Cyclin A downregulation (Figure. 4G). Interestingly, trametinib and pimasertib resulted in the accumulation of p27 expression, and under such condition palbociclib possessed enhanced inhibitory effect on the downregulation of cyclin A and RB dephosphorylation (Figure. 4H). Since, MEK inhibitors are known to elicit their cytostatic effect through the inhibition of cyclin D1 transcription (43), we investigated how such phenomenon in concert with p27 upregulation could affect the cyclin D1/CDK4 complex in the presence of palbociclib. In the PDAC cell line (1222), trametinib limited the palbociclib-mediated accumulation of the cyclin D1/CDk4 complex (Figure. 4I). However, the residual cyclin D1/CDK4 retained p27 due to its increase in expression in the presence of Palbo/Tram and under such conditions the catalytic activity of this complex was prominently inhibited (Figures. 4I & J). Moreover, the high p27 expression in the Palbo/Tram treated cells also formed complex with CDK2, which in turn inhibited its kinase activity as determined by the nuclear localization of the HDHB-mCHERRY (Figures. 4K & L). Overall, these data indicate that the MEK inhibition cooperates with palbociclib and results in the dual inhibition of cyclin D1/CDK4 and CDK2 kinase activities thereby leading to a durable growth arrest. Additionally, cytostatic effect of Palbo/Tram treatment did not induce cell death as determined by PARP cleavage (Supplementary Figure. S5D).
Figure 4: Pharmacological induction of p27 in PDAC models.
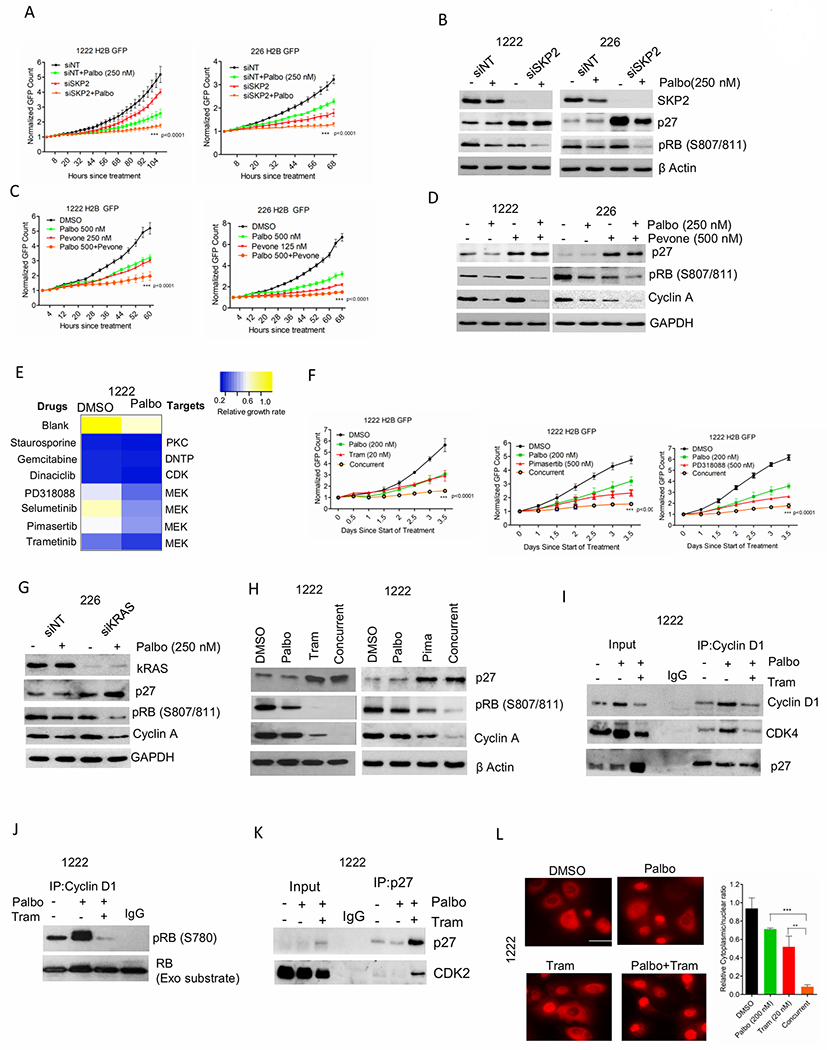
(A) Growth of 1222 and 226 cells following SKP2 knockdown. Bars represent mean and SD (2-way ANOVA). (B) Western blotting on 1222 and 226 cells following SKP2 knockdown. (C) Growth of 1222 and 226 cells that were treated with palbociclib (250 nM) in combination with SKP2 inhibitor, pevonedistat (500 nM). Graphs represent mean and SD (2-way ANOVA). (D) Western blotting on 1222 and 226 cells following combination treatment with palbociclib and pevonedistat. (E) Heat map showing the changes in relative growth rate of 1222 cells to a panel of targeted therapies in combination with DMSO and palbociclib (200 nM). (F) Growth of 1222 cells treated with palbociclib (200 nM) in combination with different MEK inhibitors, trametinib (Tram) (20 nM), pimasertib (500 nM) and PD318088 (500 nM). Graph represents mean and SD (2-way ANOVA). (G) Western blot on 226 cells following kRAS knockdown. (H) Western blot on 1222 cells following the combination treatment with palbociclib (200 nM) and trametinib (20 nM) and pimasertib (Pima) (500 nM). (I) Immunoprecipitation of cyclin D1 from 1222 cells treated with palbociclib (200 nM) +/− trametinib (20 nM) up to 48 H. Coimmunoprecipitated P27 and CDK4 were determined by immunoblotting. (J) In vitro CDK4 kinase assay associated on 1222 cells treated with palbociclib (200 nM) +/− trametinib (20 nM) up to 48 H. (K) Immunoprecipitation of P27 from 1222 cells treated with palbociclib (200 nM) +/− trametinib (20 nM) up to 48 H. Coimmunoprecipitated P27 and CDK2 were determined by immunoblotting. (L) Representative images of 1222 cells stably expressing the CDK2 sensor following 48 H exposure with palbociclib (200 nM) +/− trametinib (20 nM). Column graph indicates ratio of number of cells with cytoplasmic localization to number of cells with nuclear localization of the HDHB-mCHERYY protein at the indicated conditions. Bars represent the mean and SD (t-test). Graphs represent 2 independent experiments with 3 replicates. (*p< 0.05, **p<0.01, ***p<0.001).
In vivo evidence of cell cycle plasticity in response to palbociclib:
To investigate whether the in vitro observations apply in the tumor environment in vivo, we employed ER+ xenografts and PDAC PDX models to match the cell line data. Mice harboring MCF7 xenografts were treated with palbociclib (100 mg/kg) that resulted in a robust inhibition of tumor growth over the course of 21 days (Figures. 5A & 5B) and suppressed cell proliferation as determined by ki67 staining. (Figure. 5C). To delineate the in vivo mechanism of palbociclib, multi-spectral staining was employed in excised tumor tissue. The phosphorylation of RB was significantly inhibited in the palbociclib-treated tumor as compared to vehicle treatment, indicating the downstream effect of CDK4/6 inhibition (Figure. 5D). The phosphorylation of histone H3 (pHH3) was also inhibited, confirming that palbociclib prevented the cells from entering into mitosis to stop cell-division (Figure. 5D). Although a pronounced anti-tumor response was observed, there were subset of tumor cells that escaped the inhibitory effect of palbociclib (Figure. 5D). Interestingly those populations of pRB positive cells displayed a low expression of nuclear p27, whereas palbociclib-mediated RB dephosphorylation is largely observed in cells that have p27, mainly localized within the nucleus (Figure. 5D). The relationship between p27 and the phosphorylation status of RB was analyzed from different regions of the tumor. In the vehicle treated group where tumor cells are actively proliferating, the expression of p27 was surprisingly positively correlated with pRB suggesting that in this context p27 does not possess an intrinsic CDK inhibitory function (Fisher’s Exact test p<0.0001) (Figure. 5E). In contrast, palbociclib treatment resulted in a negative correlation between nuclear p27 and the phosphorylation of RB (Fisher’s Exact test p<0.0001). Tumor cells with nuclear p27 harbored lower fraction of pRB positive cells while cells with cytoplasmic p27 had the higher fraction (Figure. 5E). In addition, the fraction of pRB positive cells with the nuclear p27 was significantly lower in the palbociclib treated group as compared to that of the vehicle treated group (Figure. 5F). Overall, these data imply that nuclear p27 expression in the presence of palbociclib exerts an inhibitory effect to cease tumor proliferation. To further define whether MEK inhibition increases p27 expression in vivo and enhance the anti-cancer effect of palbociclib, a PDAC PDX model (3226 PDX) was treated with palbociclib in combination with trametinib. The in vivo response of PDAC xenografts to palbociclib was not as potent as the MCF7 xenografts suggesting that the intrinsic cell-cycle plasticity in PDAC models was also observed in an in vivo setting (Supplementary Figure. S5E) (30). Moreover, immunohistochemical staining revealed that the baseline p27 expression in PDAC tumors was very low and the concurrent treatment of trametinib and palbociclib (Palbo/Tram) resulted in increase in its nuclear expression (Figure. 5G). Under such condition, a robust decrease in the ki67 levels were observed, indicating an anti-tumor response in the presence of palbociclib and trametinib (Figure. 5G). Multispectral staining further confirmed that the Palbo/Tram treatment resulted in a significant decrease in RB dephosphorylation and inhibition of pHH3, suggesting a profound cell-cycle arrest at G1-phase in the tumor cells (Figure. 5H). Overall, these data demonstrate that the anti-cancer effect of palbociclib is more potent in cells that harbor high p27 expression.
Figure 5: In vivo function of P27.
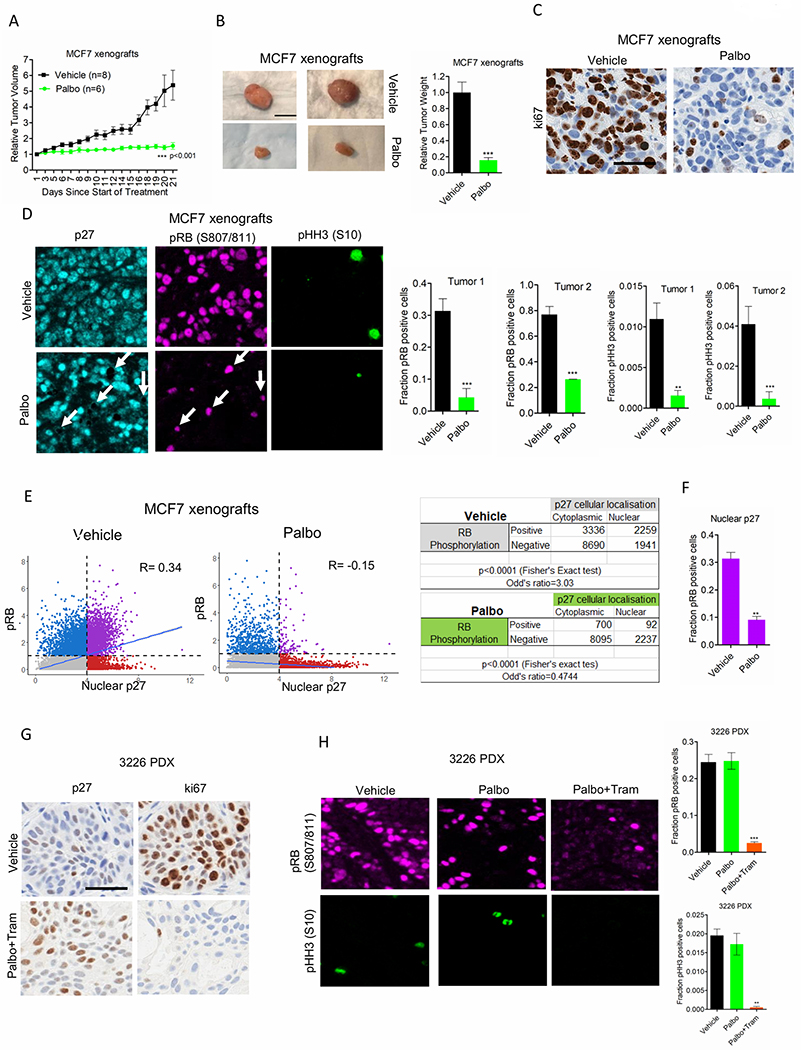
(A) Mice bearing MCF7 xenografts were randomized for treatment with Vehicle (n=8) and palbociclib (n=6). Graph represents mean and SEM (2-way ANOVA). (B) Representative images of tumors that were excised from mice at the end of treatments. Column graph represents the relative tumor weight from the palbociclib treated group with mean and SEM (t test). (C) Tumors from the vehicle and palbociclib treated mice were stained for ki67. Representative images are shown (scale bar = 100 μm). (D) Representative images of multispectral staining on the tumor tissues from vehicle and palbociclib treated mice. P27 (cyan), pRB(S807/811) (pink) and pHH3 (S10) (green). Arrows indicate the pRB positive cells in the palbociclib treated tumor tissue and the corresponding P27 localization. Graphs represent the fraction of pRB and pHH3 positive cells from the vehicle (n=2) and palbociclib (n=2) treated mice. Mean and SD are shown (t-test). (E) The correlation between pRB positive cells and nuclear localization of P27 from the vehicle and palbociclib treated tumor at 3 different region of interest (ROI) were determined using Fisher exact test. (F) Graph represents the fraction of pRB positive cells that harbor nuclear localization of P27 from the vehicle and palbociclib treated groups. Mean and SD are shown (t-test). (G) Tumors excised from the mice bearing 3226 PDX, treated with vehicle and palbociclib in combination with trametinib (palbo/tram) were stained for ki67 and p27. Representative images are shown (scale bar = 100 μm). (H) Representative images of multispectral staining on the tumor tissues from vehicle and palbo/tram treated mice. pRB(S807/811) (pink) and pHH3 (S10) (green). Graphs represent the fraction of pRB and pHH3 positive cells from the vehicle and palbo/tram treated mice. Mean and SD are shown (t-test). (**p<0.01, ***p<0.001).
Targeting CDK2 activity as an approach to rescue CDK4/6 inhibitor resistance:
Multiple mechanisms that limit the efficacy of CDK4/6 inhibitors (ie. RB loss, enhanced cyclin D1/CDK4 kinase and low p27 expression) coalesce to impact on CDK2 activity. Thus, targeting CDK2 kinase activity could be an alternate therapeutic approach on models that are partially or completely refractory to CDK4/6 inhibition. CDK2 proved to be a valid target because depletion of this protein resulted in cell-cycle arrest at G1 phase, which is independent of RB status (Supplementary Figure. S6A). We employed a small molecule inhibitor, PF06873600, which is known to target both CDK4 (Ki = 1.37 nM) and CDK2 (ki=0.12 nM) kinases and is currently being used in a phase I clinical trial (NCT03519178) (44). However, in our study we demonstrate that PF06873600 also inhibits CDK1 kinase activity with similar potency as it inhibits CDK2 (Supplementary Figure. S6B). To compare the cellular efficacies of PF068736 and palbociclib on multiple signaling pathways, we performed RNA sequencing on both MCF7-WT and MCF7 RB knockout cells. Gene expression analysis revealed that, following palbociclib and PF06873600 treatments the downregulated genes were significantly enriched in pathways that regulate cell-cycle progression (Figure. 6A). However, palbociclib is more potent in driving the repression of these genes (Supplementary Figure. S6C and Supplementary Table S1). The same sets of genes were not downregulated in the RB deficient MCF7 cells following the treatment with either of the drugs, indicating the activity of PF06873600 in driving transcriptional repression of cell cycle genes is RB-dependent (Figure. 6A and Supplementary Figure S6C, Supplementary Table S1). Interestingly, PF06873600 is very potent in inhibiting the proliferation of both MCF7-WT and MCF7 RB deleted cells (Figure. 6B). Similarly, in HCC1806 cell line, which is an RB proficient triple negative breast cancer model completely refractory to palbociclib, PF06873600 effectively ceased proliferation (Figure. 6C and Supplementary Figure S6D). Furthermore, PF06873600 resulted in the inhibition of BrdU incorporation in an RB independent manner and the overexpression of cyclin D1/CDK4 in MCF7 cells could not ameliorate this effect (Figure. 6D). Interestingly, the potent activity of PF06873600 against multiple CDKs and cell proliferation did not induce cytotoxic effects, such as induced by the pan-cdk inhibitor dinaciclib (Supplementary Figure. S7A). These data suggest that the cytotoxic effects associated with pharmacological inhibitors likely reflect inhibition of CDK molecules that regulate transcription, not cell cycle. PF06873600 treatment resulted in a prominent change in cell morphology, leading to flat and enlarged cells, a phenomenon resembling senescence (Figure. 6E) (45). Consistent with this notion, prolonged inhibition of cell growth by PF06873600 increased senescence associated β-galactosidase (SA- β-Gal) activity a marker for cellular senescence in MCF7 cells irrespective of their molecular perturbations (MCF7-WT, RB-del, D1/K4) (Supplementary Figure. S7B). Based on the DNA content it was evident that PF06873600 resulted in the accumulation of 4N cells suggesting that cell-cycle was blocked after DNA replication, which is distinct from CDK4/6 inhibition and CDK2 knockdown that prevent G1 to S phase transition (Figure. 6F and Supplementary Figure S7C) (46). Moreover, CDK2 inhibition by PF06873600 did not alter the p27 levels in MCF7 cells, which was upregulated by depleting CDK2 (Supplementary Figure. S7D). Biochemical analysis revealed that PF-06873600 resulted in the accumulation of cyclin A, Cyclin B1 and stable expression of CDK1 selectively in MCF7 RB depleted cells whereas it decreased the expression of these proteins in MCF7-WT cells (Supplementary Figure. S7E). Although PF-06873600 increased the population of cells with 4N DNA content in MCF7-WT model, the transcriptional repression of CCNA2, CCNB1 and CDK1 genes resulted in the downregulation of their target proteins (Figure. 6G and Supplementary Figure S7E). Similar to ER+ breast cancer models, the PDAC cells (1222 and 226) that harbor intrinsic cell-cycle plasticity in the presence of CDK4/6 inhibition also yielded a durable cytostatic effect associated with BrdU incorporation (Figure. 6H and Supplementary Figure S7F). The RB deficient PDAC line, 7310 cells was also sensitive to PF06873600 inhibition and the biochemical analysis matched with the MCF7 RB depleted cells (Supplementary Figure. S7G). Based on gene expression and cell-cycle analysis the impact of PF06873600 on the RB proficient PDAC models led to the downregulation of the E2F target genes and accumulation of cells with 4N DNA content (Figure. 6I and Supplementary Figure S7H, Supplementary Table S2).
Figure 6: Cellular response to CDK2 inhibitor, PF06873600.
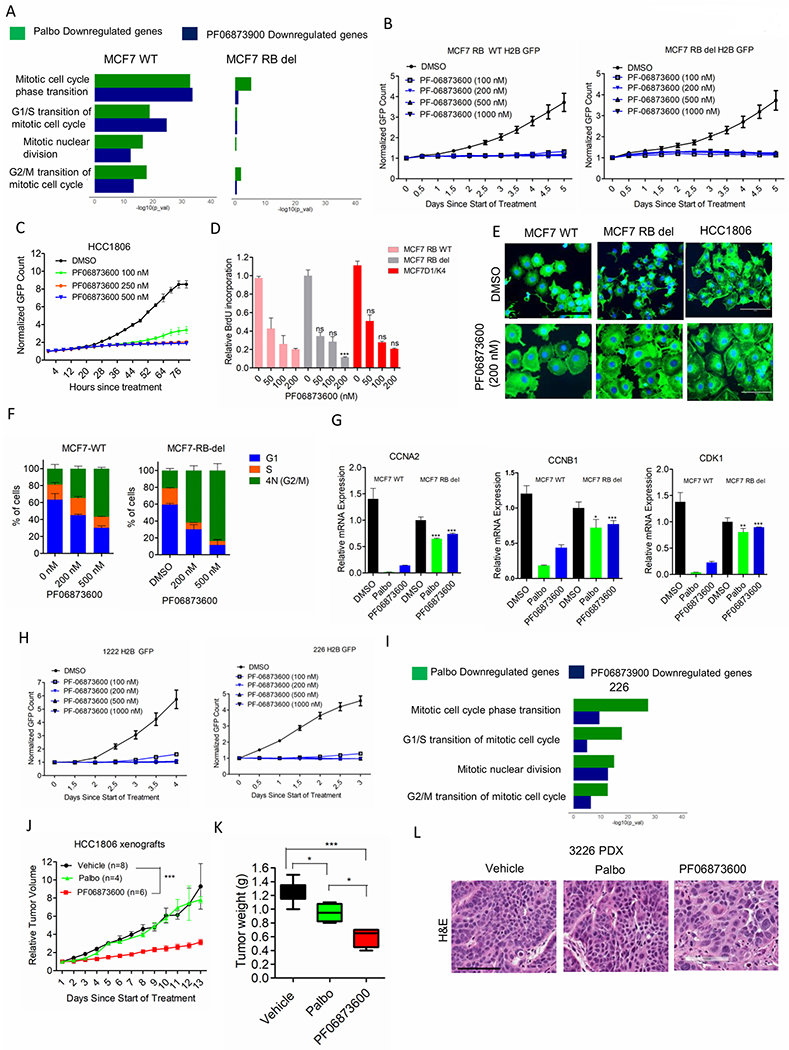
(A) ENRICHR analysis to display the top gene ontology sets that were significantly suppressed based on RNAseQ analysis on MCF7-WT and MCF7-RB-del cells treated with palbociclib (250 nM) (green) and PF06873600 (200 nM) (blue) up to 48 H. (B) Growth of MCF7-WT and MCF7-RB-del cells treated with different concentrations of PF06873600. Graphs indicate the mean and standard deviation (SD). (C) Growth of HCC1806 cells treated with different concentrations of PF06873600 up to the indicated number of days. Graphs represent mean and SD. (D) BrdU incorporation in MCF7-WT, MCF7-RB-del and MCF7D1/K4 cells treated with different concentrations of PF06873600 up to 72 H. Bars represent mean and SD (t-test). (E) Phalloidin staining on MCF7-WT, MCF7-RB-del and HCC1806 cells following 48 H exposure with PF06873600 (200 nM). DAPI was used to stain for the nuclei. (F) Cell-cycle profile based on PI staining in MCF7-WT and MCF7-EB-del cells following 48 H exposure with PF06873600. Error bars represent mean and SEM from 2 independent experiments. (G) Relative mRNA expression in MCF7-WT and MCF7-RB-del cells treated with palbociclib (250 nM) (green) and PF06873600 (200 nM) (blue) up to 48 H. Mean and SD are shown. (t-test). (H) Growth of 1222 and 226 cells treated with PF06873600 Mean and SD are shown. (I) ENRICHR analysis to display the top gene ontology sets that were significantly suppressed based on RNAseQ analysis on 226 cells treated with palbociclib (250 nM) (green) and PF06873600 (200 nM) (blue) up to 48 H. (J) Mice bearing HCC1806 xenografts were randomized for treatment with Vehicle (n=8), palbociclib (n=4) and PF06873600 (n=6). Data show the mean and SEM (2-way ANOVA). (K) Box plot represents the weights of the tumors that were excised from mice treated with vehicle, palbociclib and PF06873600. Data show the mean and SEM (t-test). (L) H&E staining on tumor tissues excised from 3226 PDX treated with the vehicle, palbociclib and PF06873600 up to 6 days. Representative images are shown (scale bar = 100 μm). (*p< 0.05, **p<0.01, ***p<0.001).
To evaluate the in vivo efficacy of PF06873600 on tumor progression, we utilized the palbociclib-resistant HCC1806 xenografts. PF06873600 significantly delayed tumor growth as compared to the vehicle and palbociclib treated groups (Figures 6J & K). Histological analysis on tumor tissues from HCC1806 xenografts and 3226 PDX revealed that PF06873600 prominently alters the tumor morphology, resulting in fewer and more enlarged cells, which is consistent with the cell-culture results (Figure. 6L and Supplementary Figure S8A). Administration of PF06873600 was well tolerated in mice since no significant change in the body weight was observed among the treatment groups (Supplementary Figure. S8B). Moreover, PF06873600 had no major impact on the histo-architecture of different organs such a s gut, liver, and kidney (Supplementary Figure. S8C). Together, these findings suggest that dual inhibition of CDK4/6 and CDK2 would be a potent approach to achieve durable disease control and improved therapeutic efficacy.
Discussion:
Identifying the functional determinants of response or resistance to molecularly targeted therapies is vital to improve clinical outcomes. CDK4/6 inhibitors are successful treatment options for ER+ breast cancer patients, but the therapeutic limitations are mainly due to the development of acquired resistance mechanisms (21, 23). Multiple preclinical and clinical studies have indicated that loss of RB, which is a preferred CDK4/6 substrate is a mechanism that contributes to resistance to CDK4/6 inhibitors (47–49). However, multiple perturbations can limit the effectiveness of CDK4/6 inhibitors in the presence of RB, and as such to predict the response of cancers to CDK4/6 inhibitors has not progressed to the point of clinically actionable biomarkers (24, 25).
Genetic analyses in mice that lack both CDK4 and CDK6 have shown that most cell types undergo proliferation, indicating that the underlying ability to attain CDK4/6 independence could contribute to resistance to pharmaceutical inhibitors (17, 18). In ER+ breast cancer cells that respond potently to CDK4/6 inhibition by palbociclib, we believe there are two molecular responses that contribute to efficacy. First, RB is completely dephosphorylated or activated to configure robust cell-cycle arrest via the transcriptional repression of essential cell-cycle genes. Second, CDK2 activity is potently inhibited. This event in essence imparts the function of CDK4/6 inhibitors to further block the activities of other CDKs in select settings. In resistant models like PDAC, we have shown here there is a response relative to RB phosphorylation, but it is limited and downstream genes like cyclin A continue to be expressed. Furthermore, work with RB-deficient models further illustrates that the coupling of CDK4/6 to CDK2 is dependent on RB. Thus, there exists a form of cell-cycle plasticity by maintaining an active CDK2 kinase even in the presence of CDK4/6 inhibition. This phenomenon is operable in PDAC highlighting the necessity for combinatorial treatment approaches using different molecularly targeted drugs along with CDK4/6 inhibitors (30, 50). Additionally, recent studies have combined CDK4/6 inhibitors with chemotherapy agents in a sequential manner to prevent cell-cycle re-entry at least in part through the suppression of CDK2 activity (46, 51). Although these studies primarily focus to reverse cell-cycle plasticity in PDAC models by improving the response to CDK4/6 inhibition, the intrinsic mechanism that drives resistance still remains unaddressed.
Mixed results have emerged relative to the role of CDK4/6 and its associated D-type cyclins as markers of sensitivity or resistance to CDK4/6 inhibitors (52, 53). Genetic analyses of tumor cell lines indicated that signatures of cyclin D1 deregulation can be predictive of response; however, in several settings it has been shown that depletion of cyclin D1 cooperates with CDK4/6 inhibitors and as shown here enforced expression can yield resistance (21, 30, 54). Although, the genetic alterations that initiate tumorigenesis in ER+ breast cancer and PDAC models are distinct, veritably all oncogenic signaling pathways coalesce to dysregulate cell-cycle machinery due aberrant CDK4 or 6 kinase activity. Consistent with this, both breast cancer and PDAC models depend on CDK4/6 for their proliferation. However, a differential sensitivity to pharmaceutical inhibitors is observed between these models, indicating that either the CDK4/6 inhibitors cannot inhibit the total pool of kinase, or the CDK4/6-cyclin D1 complexes can function non-catalytically in a tissue specific manner. This study sheds light on the differential regulation of CDK4 and CDK2 kinases in ER+ breast cancer and PDAC models to identify potential markers of resistance or sensitivity to CDK4/6 inhibitors.
Using broad-based approaches, it is clear that regulatory networks that control CDK2 are important for the acquisition of resistance. This analysis defined p27 as well as regulatory networks that control its stability (ie. SKP2). The functional role of p27 in regulating the kinase activities of CDK2 and CDK4/6 is more complex, and it remains controversial. In breast cancer models p27 acts as a scaffold and facilitates the assembly of Cyclin D1/CDK4 to form a stable trimeric complex (55). Recent studies have reported that palbociclib binds the monomeric CDK4 and prevents its interaction with cyclin D1 thereby limiting kinase activity (39). Additionally, the trimeric complex in palbociclib-treated MCF7 cells harbors a non-phosphorylated p27 that renders this complex to exist in an inactive state (40). Hence the outcome of such a phenomenon in MCF7 cells in the presence of palbociclib is lower CDK4 activity, which is sufficient to block CDK2 through RB activation. On the other hand, in PDAC models, the low abundance of p27 limits its ability to assemble the entire pool of cyclin D1 and CDK4 and failed to exert a negative impact on the kinase activity in the presence of palbociclib. Therefore, our data implicates that the protein levels of p27 are associated with sensitivity to CDK4/6 inhibitors in vitro cell-culture and in individual cells within a tumor microenvironment. Pharmacological induction of p27 using a MEK inhibitor, trametinib enhances the cytostatic effect of palbociclib and reverses cell-cycle plasticity in PDAC models. MEK inhibition has been previously shown to attenuate the palbociclib-mediated upregulation of cyclin D1 to induce a potent cytostatic effect in PDAC models (50). However, in this study we provide additional mechanistic evidence that the increase in stochiometric ratio of p27 to cyclin D1 and assembly of p27/CDK2 complex by palbo/tram combination treatment leads to dual inhibition of both CDK4 and CDK2 kinase activities respectively to achieve robust cell-cycle arrest.
From our study it emerges that control over CDK2 activity represents the pivotal event in the response to CDK4/6 inhibition. Thus, targeting the function of CDK2 using a clinically applicable agent, PF06873600, exerted potent anti-proliferative effect in all the cancer models irrespective of cell-cycle plasticity. The impact of CDK2 activity on cell-cycle progression depends upon its binding partners, cyclin E1 and cyclin A because depletion of CCNE1 and CCNA2 blocked cell-cycle at G1 and G2/M phases respectively (Supplementary Figure. S8D). Interestingly, the cytostatic effect of PF06873600 in different tumor models is mediated through the inhibition of cyclin A/CDK2 activity because this drug mimics the effect of CCNA2 knockdown. This feature of PF06873600 explains that the CDK2 kinase activity in cell-cycle progression is predominantly regulated through Cyclin A binding. Hence, the use of a targeted agent to directly block CDK2 activity, which is a critical factor in cell-cycle plasticity serves as an alternate approach to enforce a durable cell-cycle exit.
Together, the study herein indicates the intersection between multiple factors in mediating the response to CDK4/6 inhibition. Notably RB, p27, and CDK2 activity are all involved, and are undoubtedly responsive to cyclin levels (e.g. Cyclin E) that can shift the overall balance of CDK2 activity in the cell. Although CCNE1 gene expression is associated with resistance to CDK4/6 inhibition in ER+ breast cancer based on preclinical studies, the clinical evidence to support this notion is very elusive (24, 25). Hence, the analysis of protein levels will be particularly important since much of the cell cycle machinery is under significant post-transcriptional regulation. Another critical factor to be considered is the complex interplay of several regulatory elements that determine the activities of these proteins will also need to be addressed for predictive value. Thus, multi-marker protein analyses will likely be crucial to accurately predict response vs. resistance with CDK4/6 inhibitors and other cytostatic acting agents in a clinical setting.
Supplementary Material
Statement of Significance.
This work provides a mechanistic insight towards understanding the functional roles of multiple cell-cycle regulators that drive plasticity and sensitivity to CDK4/6 inhibition.
Acknowledgements:
The author thank all members of the laboratory group and colleagues in the discussion and preparation of the manuscript. Dr. Pamela Hershberger kindly provided the A549 and H1975 cell lines that were used in this study. Dr. Steven Pruitt provided the lentiviral vector for CDK2 sensor. Multi-spectral imaging was performed in the in the Center for Personalized Medicine. Drug screening was performed through Small Molecule Screening facility at Roswell Park Cancer Center. We would like to thank Dr. Sandra Sexton, Facility director of Laboratory Animal Resources at Roswell Park for assisting us with xenografts. The research was supported by a grant to AKW and ESK from National Cancer Institute (NCI).
Competing Interests: Dr. Knudsen and Dr. Witkiewicz have received research funding from Eli Lilly, Novartis and Pfizer over the last 5 years. There is no current research support from these entities and the study was written in the absence of input from any pharmaceutical company.
References:
- 1.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. [DOI] [PubMed] [Google Scholar]
- 2.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagano M, Theodoras AM, Tam SW, Draetta GF. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994;8:1627–39. [DOI] [PubMed] [Google Scholar]
- 5.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–97. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 7.Knudsen ES, Pruitt SC, Hershberger PA, Witkiewicz AK, Goodrich DW. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer. 2019;5:308–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10:2497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20:3379–83. [DOI] [PubMed] [Google Scholar]
- 10.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin Cancer Res. 2017;23:3251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corona SP, Generali D. Abemaciclib: a CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des Devel Ther. 2018;12:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. [DOI] [PubMed] [Google Scholar]
- 14.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425–39. [DOI] [PubMed] [Google Scholar]
- 15.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6:740–53. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez-Fernandez M, Malumbres M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell. 2020;37:514–29. [DOI] [PubMed] [Google Scholar]
- 17.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. [DOI] [PubMed] [Google Scholar]
- 18.Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S, et al. Mice thrive without Cdk4 and Cdk2. Mol Oncol. 2007;1:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–45. [DOI] [PubMed] [Google Scholar]
- 20.Knudsen ES, Witkiewicz AK. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer. 2017;3:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016;76:2301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget. 2016;7:68012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guarducci C, Bonechi M, Benelli M, Biagioni C, Boccalini G, Romagnoli D, et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer. 2018;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol. 2019;37:1169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Dieras V, et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin Cancer Res. 2020;26:110–21. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, et al. Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell. 2017;31:225–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien N, Conklin D, Beckmann R, Luo T, Chau K, Thomas J, et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol Cancer Ther. 2018;17:897–907. [DOI] [PubMed] [Google Scholar]
- 28.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knudsen ES, Balaji U, Mannakee B, Vail P, Eslinger C, Moxom C, et al. Pancreatic cancer cell lines as patient-derived avatars: genetic characterisation and functional utility. Gut. 2018;67:508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knudsen ES, Kumarasamy V, Ruiz A, Sivinski J, Chung S, Grant A, et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R, et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat. 2009;30:1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–20. [DOI] [PubMed] [Google Scholar]
- 34.Hafner M, Mills CE, Subramanian K, Chen C, Chung M, Boswell SA, et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol. 2019;26:1067–80 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21:995–1001. [DOI] [PubMed] [Google Scholar]
- 37.Soos TJ, Kiyokawa H, Yan JS, Rubin MS, Giordano A, DeBlasio A, et al. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–46. [PubMed] [Google Scholar]
- 38.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. [DOI] [PubMed] [Google Scholar]
- 39.Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel P, Tsiperson V, Gottesman SRS, Somma J, Blain SW. Dual Inhibition of CDK4 and CDK2 via Targeting p27 Tyrosine Phosphorylation Induces a Potent and Durable Response in Breast Cancer Cells. Mol Cancer Res. 2018;16:361–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li GZ, Okada T, Kim YM, Agaram NP, Sanchez-Vega F, Shen Y, et al. Rb and p53-Deficient Myxofibrosarcoma and Undifferentiated Pleomorphic Sarcoma Require Skp2 for Survival. Cancer Res. 2020;80:2461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res. 2005;65:4870–80. [DOI] [PubMed] [Google Scholar]
- 43.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behenna DC (SJC, CA, US), Ping Chen (San Diego, CA, US), Kevin Daniel Freeman-cook (Carlsbad, CA, US), Robert Louis Hoffman (San Diego, CA, US), Mehran Jalaie (San Diego, CA, US), Asako Nagata (San Diego, CA, US), Sajiv Krishnan Nair (Vista, CA, US), Sacha Ninkovic (La Jolla, CA, US), Martha Alicia Ornelas (San Diego, CA, US), Cynthia Louise Palmer (La Mesa, CA, US), Eugene Yuanjin Rui (San Diego, CA, US), inventor; Pfizer Inc. (New York, NY, US), assignee. CDK2/4/6 Inhibitors. United States: 2018. [Google Scholar]
- 45.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumarasamy V, Ruiz A, Nambiar R, Witkiewicz AK, Knudsen ES. Chemotherapy impacts on the cellular response to CDK4/6 inhibition: distinct mechanisms of interaction and efficacy in models of pancreatic cancer. Oncogene. 2020;39:1831–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018;29:640–5. [DOI] [PubMed] [Google Scholar]
- 48.Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014;16:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569:560–4. [DOI] [PubMed] [Google Scholar]
- 50.Knudsen ES, Kumarasamy V, Chung S, Ruiz A, Vail P, Tzetzo S, et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salvador-Barbero B, Alvarez-Fernandez M, Zapatero-Solana E, El Bakkali A, Menendez MDC, Lopez-Casas PP, et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell. 2020;37:340–53 e6. [DOI] [PubMed] [Google Scholar]
- 52.Zhang YX, Sicinska E, Czaplinski JT, Remillard SP, Moss S, Wang Y, et al. Antiproliferative effects of CDK4/6 inhibition in CDK4-amplified human liposarcoma in vitro and in vivo. Mol Cancer Ther. 2014;13:2184–93. [DOI] [PubMed] [Google Scholar]
- 53.Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013;31:2024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108:1744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.