

Abstract
Purpose:
Cisplatin-based chemotherapy is a first-line treatment for muscle-invasive and metastatic urothelial cancer. Approximately 10% of bladder urothelial tumors have a somatic missense mutation in the nucleotide excision repair (NER) gene, ERCC2, which confers increased sensitivity to cisplatin-based chemotherapy. However, a significant subset of patients is ineligible to receive cisplatin-based therapy due to medical contraindications, and no NER-targeted approaches are available for platinum-ineligible or platinum-refractory ERCC2-mutant cases.
Experimental Design:
We used a series of NER-proficient and NER-deficient preclinical tumor models to test sensitivity to irofulven, an abandoned anticancer agent. In addition, we used available clinical and sequencing data from multiple urothelial tumor cohorts to develop and validate a composite mutational signature of ERCC2 deficiency and cisplatin sensitivity.
Results:
We identified a novel synthetic lethal relationship between tumor NER deficiency and sensitivity to irofulven. Irofulven specifically targets cells with inactivation of the transcription-coupled NER (TC-NER) pathway and leads to robust responses in vitro and in vivo, including in models with acquired cisplatin resistance, while having minimal effect on cells with intact NER. We also found that a composite mutational signature of ERCC2 deficiency was strongly associated with cisplatin response in patients and was also associated with cisplatin and irofulven sensitivity in preclinical models.
Conclusions:
Tumor NER deficiency confers sensitivity to irofulven, a previously abandoned anticancer agent, with minimal activity in NER-proficient cells. A composite mutational signature of NER deficiency may be useful in identifying patients likely to respond to NER-targeting agents, including cisplatin and irofulven.
Keywords: DNA repair, nucleotide excision repair, cancer genomics, biomarker discovery, synthetic lethality, irofulven, urothelial cancer
Introduction
Therapeutic approaches based on the principle of synthetic lethality are an attractive strategy for cancer treatment. Because DNA repair pathway aberrations are common in tumor cells, but are largely absent in normal cells, agents that target DNA repair–deficient cells may have a clinically exploitable therapeutic window. A successful example stemming from this principle is the development of PARP inhibitors for treatment of tumors with homologous recombination (HR) repair deficiency (1,2).
Nucleotide excision repair (NER) is a highly conserved DNA repair pathway that recognizes and repairs bulky intrastrand DNA adducts formed by genotoxic agents, such as UV radiation and platinum chemotherapies (3). NER is initiated through two separate branches of lesion recognition: transcription-coupled repair (TC-NER) is activated by RNA polymerase stalling at lesions in transcribed regions, while global genome repair (GG-NER) is able to recognize lesions throughout the genome. Following lesion recognition, TC-NER and GG-NER converge on a common NER pathway that excises and replaces the damaged DNA strand in an error-free manner.
Recently, sequencing and functional studies have revealed that NER pathway deficiency is present in a subset of tumors. Somatic missense mutations in ERCC2, a key NER gene that encodes the DNA helicase XPD, are present in approximately 10% of muscle-invasive bladder tumors (4). Although not strongly prognostic in patients treated without chemotherapy, ERCC2 mutations confer increased sensitivity to platinum-based chemotherapy (5–7), and mutations in ERCC2 and several other DNA repair genes (such as ATM, FANCC, and BRCA1/2) are being used as predictive biomarkers in several on-going clinical trials (8–10). Mutations in NER genes beyond ERCC2 also occur sporadically in bladder cancer and other tumor types, and it is possible that these events may also confer a therapeutically exploitable NER deficiency.
Cisplatin-based chemotherapy is a first-line treatment for muscle-invasive and metastatic urothelial cancer. Although 60–70% of patients have an initial response, resistance develops in the majority of patients (11). In addition, nearly half of all patients with urothelial cancer are ineligible for cisplatin-based chemotherapy due to medical comorbidities (12). Anti-PD-1/PD-L1 agents are approved for cisplatin-ineligible or cisplatin-resistant patients; however, only approximately 20%–30% of patients respond (13). Therefore, novel agents with activity in post-cisplatin and cisplatin-ineligible patient populations are needed.
Irofulven is a semisynthetic DNA alkylating agent that is a derivative of the fungal product, illudin S (14). Irofulven-mediated DNA damage is not recognized by mismatch repair (MMR) or GG-NER, but instead activates TC-NER when the damage is encountered by RNA polymerase. Therefore, irofulven has been shown to have approximately 100-fold increased cytotoxic activity in nontumor cell lines with a TC-NER or common NER pathway gene defect compared with the isogenic NER-proficient line (15,16). Although well-tolerated, irofulven showed only modest clinical benefit as a single agent in phase I/II clinical trials across a variety of tumor types (17–20). This apparent lack of efficacy may have occurred because NER deficiency is present in only a fraction of tumors, and these trials were conducted in biomarker-unselected populations.
Despite the potential clinical actionability of tumor NER deficiency, there are currently no functional or IHC assays available to reliably identify NER deficiency from clinical specimens. An alternative approach to identify tumor NER deficiency is through the use of next-generation sequencing (NGS)-based mutational signatures. Because DNA repair deficiency is frequently associated with specific mutational patterns (signatures), the presence of a mutational signature can indicate a specific DNA repair deficiency, even in the absence of an obvious DNA repair gene alteration (21,22). Such a strategy has been successfully employed to identify HR-deficient cancer cases for the prioritization of PARP inhibitor and platinum-based therapy in breast and ovarian cancer (23–25). For NER, a specific single-nucleotide variation (SNV)-based mutation signature (signature 5*) has been associated with ERCC2 mutations in bladder tumors (22), but the signature was not associated with platinum response in tumors that lacked an ERCC2 mutation. To improve predictive utility, SNV signatures can be combined with other non-SNV–based mutational features, such as short insertions/deletions (indels) or chromosomal rearrangements. These “composite” signatures may represent a more accurate measure of DNA repair deficiency, as has been demonstrated for HR deficiency (26).
Here, we identify a novel synthetic lethal relationship between NER deficiency and irofulven sensitivity. We show that inactivating mutations in genes of the TC-NER or common NER pathway are sufficient to drive irofulven sensitivity in vitro and in vivo, and we demonstrate that acquired cisplatin resistance does not induce cross-resistance to irofulven. We also define a composite mutational signature of ERCC2 deficiency in bladder cancer that is strongly associated with cisplatin sensitivity, including in cases that lack an ERCC2 mutation, and may, therefore, be a useful tool to predict sensitivity to NER-targeting agents, such as cisplatin and irofulven.
Materials and Methods
Patients and cohorts
In this study, 632 whole-genome (WGS) and whole-exome sequenced (WES) pretreatment samples were analyzed from four urothelial bladder tumor cohorts (Supp. Table 1).
The Cancer Genome Atlas (TCGA) cohort
The WGS normal and tumor bam files were downloaded from the International Cancer Genome Consortium data portal (https://dcc.icgc.org/). The WES normal and tumor bam files, as well as the vcf files generated by MuTect2, were downloaded from The Cancer Genome Atlas data portal (https://portal.gdc.cancer.gov/).
Dana-Farber Cancer Institute (DFCI)/Memorial Sloan Kettering Cancer Center (MSKCC) and Philadelphia cohorts
The normal and tumor bam files were downloaded from The Database of Genotypes and Phenotypes upon request (https://www.ncbi.nlm.nih.gov/gap/) using the phs000771 accession code.
Beijing Genomics Institute (BGI) cohort
The normal and tumor fastq files were downloaded from the Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/sra) using the SRA063495 accession code. The details of the alignment process are described in the Supplementary Methods.
The average coverage of the analyzed WGS and WES samples in each cohort is shown in Supp. Fig. 1–2.
Mutation, copy number, and structural variant calling
Germline variants were called with HaplotypeCaller in key NER-related genes, while somatic point mutations and indels were called with MuTect2 (GATK, v3.8). Germline and somatic mutations in key NER-related genes in each cohort are presented in Supp. Fig. 3–7. The high fidelity of the reported variants was ensured by the application of additional hard filters in addition to the tools’ default filters (Supp. Table 2–3). Allele-specific copy-number profiles were estimated using Sequenza (27) (Supp. Fig. 8–9). Germline and somatic mutations were annotated using InterVar (28) and the genotypes of the samples were determined (Supp. Fig. 10–11). Structural variants were detected by BRASS (v6.0.0. http://github.com/cancerit/BRASS). Further details are available in the Supplementary Methods.
Mutational signatures
Somatic single-base substitution signatures were determined with the help of the deconstructSigs R package (29), using the Catalogue of Somatic Mutations in Cancer (COSMIC) signatures as a mutational process matrix (cancer.sanger.ac.uk/cosmic/signatures_v2). Doublet base substitutions and indels in each sample were classified into a 78-dimensional doublet base substitution and an 83-dimensional indel catalog, respectively, with the help of the ICAMS R package (30), and the previously described matrices of doublet base substitution and indel signatures were used (31) in a nonnegative least-squares problem to estimate the matrix of exposures to mutational processes. The extraction of rearrangement signatures was executed as described previously (32). The extracted single-base substitution signatures, doublet base substitution signatures, indel signatures and rearrangement signatures are shown in Supp. Fig. 12–19, and the cosine similarity between the single-base substitution signature profiles of each sample is presented in Supp. Fig. 20–21. Further details are provided in the Supplementary Methods.
Transcriptional strand bias
Transcriptional strand bias analysis was carried out using the MutationalPatterns R package (33), which identifies strand asymmetry in the reference frame of DNA transcription (34).
Cell lines and reagents
Bladder cancer cell lines were purchased from ATCC. The KE1 bladder cancer cell line is a derivative of KU19–19, in which an ERCC2 T484 mutation has been introduced by CRISPR/Cas9 gene editing as described previously (6). The MDA-MB-468 breast cancer cell line and its ERCC4-complemented derivative have been described previously (35). siRNAs targeting specific NER genes, as well as a nontargeting siRNA, were purchased from Integrated DNA Technologies Inc. (see Supp. Table 5 for sequences). Cells were seeded in 6-well plates and grown to 50% confluency. A transfection mixture containing siRNA diluted to a final concentration of 30 nmol/L in Opti-MEM containing Lipofectamine 3000 (Life Technologies) was then added to cells. After 48 hours, cells were trypsinized and realiquoted to separate wells and transfected with 15 nmol/L siRNA for 24 hours immediately, prior to performing viability assays, as described below. To create the SW1710 ERCC2 P463L-mutant cell line, a Cas9-encoding lentiviral vector, as well as a single-guide RNA (sgRNA) targeting the P463 codon were electroporated into SW1710 along with a donor template plasmid encoding the desired P463L codon change, as well as a GFP reporter cassette. After 48 hours, GFP-positive cells were isolated by cell sorting and expanded. The presence of the P463L mutation was validated by Sanger sequencing followed by RT-PCR and targeted NGS using the MSK-IMPACT assay. The sequences of sgRNA and PCR primers are listed in Supp. Table 6. Early-passage cells (≤6 passages) were used for all experiments and cell lines were tested monthly to confirm absence of Mycoplasma. For additional details, see Supplementary Methods.
Cell line mutational signatures
KU19–19 and KE1 cell populations were single-cell sorted, and individual cells were expanded to create multiple populations of cells. Each population of KU19–19 or KE1 cells was propagated separately in culture for 30 days while maintaining the population above 1×106 cells at all times. After 30 days, each population was single-cell sorted and individual clones were grown to approximately 1×106 cells. Cells were frozen, lysed, and genomic DNA was isolated. WGS was performed at the Broad Institute to an average depth of 30x. Somatic mutations were called using IsoMut (36), single-base substitution signatures, doublet base substitution signatures, and indel signatures were extracted as described in the Supplementary Methods.
In vitro drug sensitivity assays
Cells were seeded in either 96-well (5,000 cells/well) or 24-well (20,000 cells/well) plates. The following day, irofulven and cisplatin stock solutions were serially diluted in media and added to cells. After 48–72 hours, media were removed and CellTiter-Glo Reagent (Promega) was added. Plates were scanned using a Luminescence Microplate Reader (BioTek). Survival at each drug concentration was plotted as a percentage of survival in drug-free media with error bars representing the SD of at least three experiments.
For crystal violet imaging experiments, cells were seeded in 6-well plates (100,000–200,000 cells/well) with 15 nmol/L siRNA transfection mixture (as detailed above). At 24 hours, fresh media containing irofulven or PBS were added and the cells were incubated for an additional 72 hours. Cells were then fixed in formalin solution for 30 minutes and stained with crystal violet, prepared in equal volumes of methanol and water. Excess crystal violet was removed by washing with PBS, and plates were then dried and imaged.
Immunoblotting
Cells were lysed with ice-cold RIPA buffer supplemented with Protease and Phosphatase Inhibitors (Roche). Samples were then sonicated and protein concentrations were determined using the Bradford assay. Sample Buffer (Bio-Rad) was added and samples were then denatured at 90°C for 10 minutes. Samples were then loaded in NuPAGE Protein Gels (Thermo Fisher Scientific) and run at 90 V for 2–3 hours. The gels were then transferred to nitrocellulose membranes at 30 V overnight. Membranes were blocked for 30 minutes in 5% milk in TBS buffer. Sections of the membrane corresponding to the appropriate molecular weights were stained overnight in primary antibodies, prepared in 1% milk in TBST: XPF (1:700, clone D3G8C rabbit mAB, Cell Signaling Technology); cleaved caspase 3 (1:1000, clone D175, rabbit mAB, Cell Signaling Technology); total and cleaved PARP (1:1000, rabbit mAB, Cell Signaling Technology); phospho-H2AX Ser139 (1:1000, clone JBW301 mouse mAB, Millipore); β-tubulin (1:1000, mouse mAB, Santa Cruz Biotechnology); Rpb1 CTD (1:1000, clone 4H8 Mouse mAb, Cell Signaling Technology). A Li-COR Odyssey Infrared Imaging System was used for signal detection using IRDYE-conjugated secondary antibodies (LI- COR Biosciences).
Xenograft studies
Six-week-old female athymic nude mice, NU/J (stock no., 002019) were purchased from The Jackson Laboratory and housed at the Dana-Farber Cancer Institute Animal Resources Facility (Boston, MA). All animal experiments were performed in accordance with an Institutional Animal Care and Use Committee-approved protocol. At 7–10 weeks of age, mice were anesthetized with isoflurane and subcutaneously injected on the left flank with 3 million KE1 or 1 million KU19–19 cells mixed 1:1 with Matrigel (BD Biosciences) in PBS. Tumor size was measured with a digital caliper twice weekly and calculated using the formula: (L × W2) × 1/2. Drug treatments were administered when the average tumor volume reached a minimum of l00 mm3. Irofulven was prepared in PBS to a stock concentration of 200 μg/mL and was delivered intraperitoneally twice weekly at doses of 250 μg/kg, 500 μg/kg or 1 mg/kg for a total of 5 injections or until the tumor reached a prespecified protocol endpoint. Control mice were injected with PBS alone. At the end of the experiment, mice were sacrificed and tumors were excised for tumor weight measurements and imaging.
ERCC2mut composite mutational signature
We set out to build a statistical model for detecting mutational features associated with ERCC2 mutation status. The classification model was trained on the TCGA BLCA WES data. The information gained from the extraction of signatures of single-base substitutions, signatures of doublet base substitutions, signatures of indels, and transcriptional strand bias was used to build a logistic regression model. The training set consisted of 28 ERCC2 somatic mutants and 367 wild-type (WT) samples (Supp. Table 7). The data were log-transformed and standardized. The detailed description of the model-building process is available in the Supplementary Methods.
Survival analysis
Survival analysis is described in the Supplementary Methods.
Data availability
The cell line WGS bam files were deposited at the European Nucleotide Archive under the accession number PRJEB36417.
Code availability
There are no restrictions to access the custom code used for the analyses presented in this study. Information is available from the authors on request.
Results
Irofulven preferentially targets NER-deficient tumors
Given that tumor NER deficiency has been shown to drive sensitivity to platinum-based agents in several tumor contexts (6,37), we sought to determine if NER deficiency was also sufficient to confer sensitivity to other agents that create DNA damage typically repaired by the NER pathway. Irofulven is a DNA alkylating agent that creates lesions which do not distort the DNA helix and are thus not recognized by most DNA repair pathways, including the GG-NER pathway (Fig. 1A) (15). However, when present in transcribed regions of the genome, irofulven lesions block RNA polymerase and activate TC-NER (15).
Figure 1:

NER-deficient cell lines are sensitive to irofulven in vitro and in vivo. A. Chemical structure of irofulven. B. Cell viability assays demonstrate increased irofulven sensitivity in NER-deficient cell lines compared with their isogenic NER-proficient counterparts. C. Irofulven dose-response for KE1 (ERCC2 mutant, NER deficient) xenografts. The black arrow denotes the time of first irofulven injection. D. KE1 xenograft weights were significantly lower in 0.5 and 1.0 mg/kg irofulven-treated mice than in untreated mice. * p-value 0.01-0.05, ** p value 0.001< p<0.01, and *** p-value <0.001.
Given the activity of irofulven in TC-NER–deficient nontumor cells, we hypothesized that irofulven would also be toxic to tumor cells with loss of function of a TC-NER or common NER gene. KU19–19 is a bladder cancer cell line with WT ERCC2 and no other known NER pathway defects. We used CRISPR/Cas9 gene editing to introduce an inactivating mutation at the endogenous ERCC2 locus, and we have previously shown that the engineered ERCC2-mutated cell line (KE1) is NER deficient and has increased sensitivity to cisplatin in vitro and in vivo (6). To determine whether NER deficiency is also sufficient to drive sensitivity to irofulven, we compared irofulven sensitivity of KU19–19 and KE1 and found that the ERCC2-mutated, NER-deficient KE1 line exhibited significantly increased sensitivity to irofulven compared with the parental NER-proficient KU19–19 line (Fig. 1B, top).
To further investigate the relationship between ERCC2 mutations and irofulven sensitivity, we used similar CRISPR/Cas9 techniques to introduce a clinically observed ERCC2 mutation (P463L) in SW1710, another bladder cancer cell with no baseline NER dysfunction. The presence of the ERCC2 P463L mutation in the derivative cell line (SC14) was confirmed by NGS, and immunoblotting demonstrated the presence of a full-length ERCC2 gene product in the SC14 line (Supp. Fig. 22). The ERCC2-mutant SC14 line failed to resolve UV-induced DDB2 foci in an immunofluorescence NER reporter assay (6,38) (Supp. Fig. 23A), consistent with loss of NER capacity. As was observed for the NER-deficient KE1 bladder cancer line, the SC14 line also displayed significantly increased sensitivity to irofulven (Fig. 1B, middle) as well as cisplatin (Supp. Fig. 23B). Together, these data demonstrate that introduction of distinct, clinically relevant ERCC2 mutations in bladder cancer cell lines is sufficient to confer NER deficiency and drive marked sensitivity to irofulven.
Finally, we wished to determine whether NER deficiency conferred by alterations in genes other than ERCC2 is also sufficient to drive irofulven sensitivity. We recently showed that the breast cancer cell line, MDA-MB-468, is NER deficient due to epigenetic silencing of ERCC4 (XPF), and that NER activity and cisplatin sensitivity of the cell line could be rescued by reexpression of WT ERCC4 (35). We tested irofulven sensitivity of the NER-deficient parental MDA-MB-468 cell line, as well as its WT ERCC4-complemented counterpart, and again observed dramatically higher irofulven sensitivity in the NER-deficient compared with NER-proficient cell line (Fig. 1B, bottom), suggesting that NER loss mediated by dysfunction of an NER gene beyond ERCC2 can also drive irofulven sensitivity.
NER deficiency is sufficient to drive irofulven sensitivity in vivo
We next wished to test whether NER deficiency was sufficient to drive bladder tumor sensitivity to irofulven in vivo. We established NER-deficient KE1 xenografts and treated mice with increasing doses of irofulven twice weekly for 2.5 weeks (total of five doses). Irofulven was well-tolerated at all doses, and we observed a strong irofulven dose-response with near complete tumor regression observed at an irofulven dose of 1 mg/kg (Fig. 1C; Supp. Fig. 24). Post-treatment tumor weights were significantly lower in mice receiving 0.5 or 1 mg/kg irofulven compared with untreated mice (Fig. 1D). Conversely, as was observed in vitro, there was no response of the NER-proficient KU19–19 xenografts to irofulven (Supp. Fig. 25), confirming that irofulven preferentially targets NER-deficient tumor cells.
Irofulven specifically targets cancer cells with TC-NER or common NER pathway defects
To further investigate the specificity of the relationship between NER deficiency and irofulven sensitivity, we used siRNA to deplete genes in the TC-NER, GG-NER, or common NER pathways in the NER-proficient bladder cancer cell line KU19–19. Whereas depletion of the TC-NER gene, ERCC6, or the common NER gene, ERCC3, was sufficient to increase irofulven sensitivity, depletion of the GG-NER gene, DDB2, had minimal impact on irofulven sensitivity (Fig. 2A). These results are consistent with irofulven creating DNA lesions that are only recognized by the TC-NER pathway.
Figure 2:

Irofulven sensitivity is conferred by TC-NER or common NER gene defects and persists in cisplatin-resistant models. A. SiRNA-mediated depletion of the TC-NER gene, ERCC6, or the common NER gene, ERCC3, results in significantly higher irofulven sensitivity compared with depletion of the GG-NER gene, DDB2, as measured by crystal violet staining (top) or viability assay (bottom). B. Immunoblot analysis showing RNA polymerase (RBP1) levels following irofulven treatment in NER-proficient KU19-19 and NER-deficient KE1 cells (top) and immunoblot analysis showing accumulation of phospho-H2AX, cleaved PARP, and cleaved caspase in irofulven-treated KE1 cells. C. A cisplatin-resistant derivative of the NER-deficient MDA-MB-468 cell line demonstrates decreased sensitivity to cisplatin, but remains sensitive to irofulven. D. Immunoblot analysis showing that ERCC4 (XPF) protein expression remains absent in the cisplatin-resistant MDA-MB-468 line (lane 3), similar to the parental cisplatin-sensitive line (lane 2) and consistent with persistent NER deficiency. Lane 1 (positive control) shows an MDA-MB-468 line engineered to stably express WT ERCC4. * p-value 0.01-0.05, ** p value 0.001< p<0.01, and *** p-value <0.001.
The TC-NER pathway is normally activated by RNA polymerase stalling at DNA lesions. However, in cells with loss or dysfunction of a TC-NER or common NER gene, stalled RNA polymerase can undergo ubiquitination and proteasomal degradation to prevent toxic accumulation of stalled polymerase complexes (39). Following irofulven treatment, we observed a time-dependent loss of RNA polymerase in NER-deficient KE1 cells, but not in NER-proficient KU19–19 cells, consistent with an inability of KE1 cells to repair irofulven-mediated DNA damage via TC-NER (Fig. 2B, top). We also observed accumulation of the DNA damage marker, phospho-H2AX (γH2AX), as well cleaved PARP and cleaved caspase-3, in irofulven-treated NER-deficient cells, consistent with apoptotic cell death (Fig. 2B, bottom).
Cisplatin resistance does not confer cross-resistance to irofulven
Platinum-based chemotherapy is a first-line treatment for numerous solid malignancies, including bladder cancer, but platinum resistance frequently arises and represents a challenging clinical problem. Although a variety of platinum resistance mechanisms have been characterized, restoration or upregulation of DNA repair is a common mechanism of resistance, particularly in HR-deficient breast and ovarian tumors (40). Mechanisms of platinum resistance in NER-deficient tumors are poorly understood, and we wished to determine whether acquired resistance to cisplatin would confer cross-resistance to irofulven through restoration of NER. To test this, we derived cisplatin resistance in the NER-deficient MDA-MB-468 cell line by gradual exposure to escalating doses of cisplatin. Although the cisplatin IC50 value was approximately 3.5-fold higher in the cisplatin resistant cell line, the cell line remained highly sensitive to irofulven (Fig. 2C). Immunoblotting showed persistent lack of ERCC4 expression in the cisplatin-resistant cell line, suggesting that cisplatin resistance in this model is driven by a mechanism other than restoration of NER (Fig. 2D). Taken together, these data suggest that irofulven may remain active in NER-deficient tumors that have become resistant to platinum-based therapy.
A composite mutational signature is strongly associated with ERCC2 mutations in multiple bladder cancer cohorts
Given the robust association between NER gene defects and irofulven sensitivity in our preclinical models, we wished to determine whether we could identify mutational features of bladder tumors that are associated with NER deficiency and sensitivity to NER-targeting agents, such as cisplatin and irofulven. Characteristic SNV-based mutational signatures are often present in tumors harboring specific DNA repair defects, such as HR- or MMR deficiency (21), and we previously identified an SNV-based mutational signature (“signature 5*”) that is enriched in ERCC2-mutated bladder tumors (22). However, DNA repair pathway alterations are also known to induce other types of genomic alterations, such as doublet base substitutions, short indels, and chromosomal rearrangements.
To further characterize the mutational features associated with ERCC2 mutations in treatment-naïve bladder tumors, we analyzed TCGA bladder cancer WGS and WES datasets (41) to identify multiple types of mutational features catalogued in cancer (31) (Supp. Fig. 12–19). TCGA database contains only 23 WGS cases; therefore, we used mutational features extracted from 412 WES samples to train a logistic regression-based classifier of ERCC2 mutation status (Supplementary Methods). In total, nine features contributed to the logistic regression-based ERCC2mut classifier: signature 2, signature 5, DBS4, ID2, ID8, ID10, TSB_ratio_CtoG, TSB_ratio_TtoA, and TSB_ratio_TtoG. (Fig. 3A; Supp. Table 8 and Supp. Fig. 26). Signature 2 and signature 5 were described previously to be associated with ERCC2 mutation status (22). DBS4 is dominated by GC>AA or TC>AA dinucleotide substitutions. ID2 is predominantly composed of deletions of T at long (≥5 bp) mononucleotide repeats and is found in many cancer types. ID8 is characterized by ≥5 bp deletions with short (≤2 bp) or no microhomologies and no repeats at deletion boundaries. ID10 predominantly consists of ≥5 bp insertions at one-unit repeats.
Figure 3:
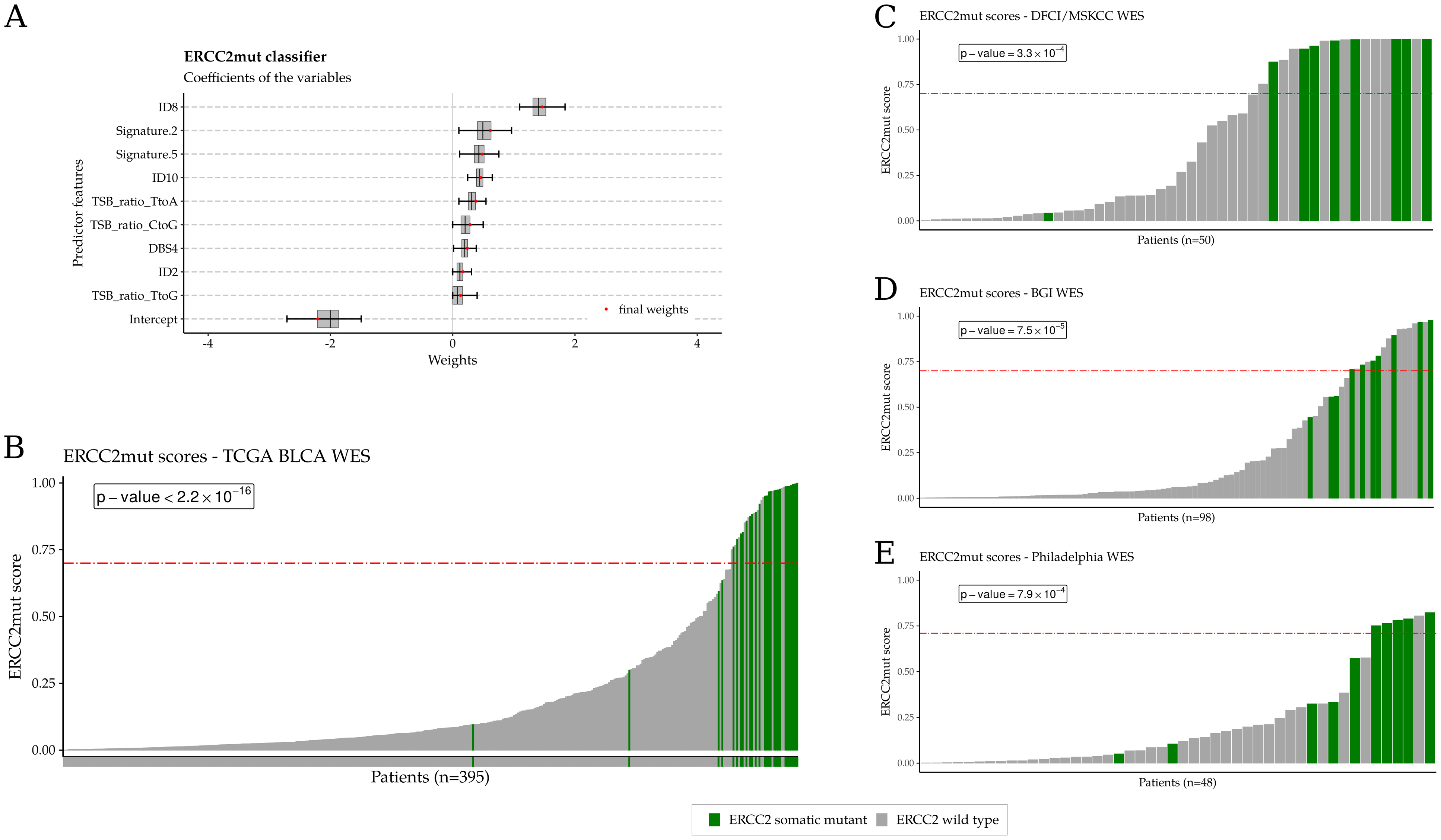
Development of the ERCC2mut logistic regression–based classifier and validation of the association between ERCC2mut scores and ERCC2 mutation status in three independent bladder cancer cohorts. Samples with an ERCC2 mutation are shown in green and WT ERCC2 samples are shown in gray. A. Nine mutational features were significantly associated with ERCC2 mutation status in TCGA bladder cancer WES cohort and were used to develop the composite ERCC2mut score. B. ERCC2mut signature scores are strongly associated with ERCC2 mutation status in TCGA bladder cancer cohort (p<2.2×10−16). A value of ≥0.70 maximally separates ERCC2-mutant from WT cases and is denoted by the red horizontal dash-dotted line. In each validation cohort, ERCC2 mutants are highly enriched among patients with a high ERCC2mut score (≥0.70). C: DFCI/MSK cohort (n=50), p=3.3×10−4. D: BGI cohort (n=98), p=7.5×10−5. E: Philadelphia cohort (n=48), p=7.9×10−4. P-values were calculated using Fisher exact test.
ERCC2 mutants were highly enriched among cases with high ERCC2mut classifier scores (Fig. 3B; p < 2.2×10−16, Fisher exact test) in the training data set. The composite ERCC2mut classifier more accurately identified ERCC2-mutant cases than was previously possible using COSMIC 5 signature alone (22): the AUCROC and AUCPRC were 0.97 and 0.77, respectively, compared with 0.88 and 0.37 with the COSMIC 5 signature (Supp. Fig. 27). We identified a threshold score of ≥0.70 that minimized the cost of misclassification between ERCC2-mutant and WT cases in TCGA WES cohort (Supp. Fig. 28). The ERCC2mut classifier was also applied to the subset of TCGA cases with WGS data available (n=23) and there was a strong correlation between scores calculated using WGS and WES (r=0.87, Supp. Fig. 29).
To validate the ability of the composite ERCC2mut classifier to discriminate between ERCC2-mutant and WT bladder tumors, we analyzed WES data from three additional independent bladder cancer cohorts (5,7,42). The DFCI/MSKCC (5) (n=50) and Philadelphia (7) (n=48) cohorts are comprised of pretreatment tumors from patients with muscle-invasive bladder cancer who received neoadjuvant cisplatin-based chemotherapy followed by radical cystectomy, while the BGI cohort (42) (n=98) included patients with muscle-invasive bladder cancer or nonmuscle-invasive bladder cancer. In each cohort, ERCC2-mutant cases were significantly associated with an ERCC2mut classifier score ≥0.70 (p = 3.3×10−4 in the DFCI/MSKCC cohort, p = 7.5×10−5 in the BGI cohort, and p = 7.9×10−4 in the Philadelphia cohort; Fisher exact test; Fig. 3C–E, Supp. Fig. 30).
The composite mutational signature score correlates with cisplatin response in cases that lack an ERCC2 mutation
ERCC2 mutations are associated with complete pathologic response and improved survival in patients with muscle-invasive bladder cancer treated with neoadjuvant cisplatin-based chemotherapy (5,7). However, not all patients with complete response harbor an ERCC2 mutation; therefore, a method to identify potential cisplatin responders among patients who lack an ERCC2 mutation could inform therapy selection. We hypothesized that tumors that are cisplatin sensitive, but lack an ERCC2 mutation, may harbor other genetic or epigenetic alterations that confer functional NER deficiency and increased activity of the ERCC2mut composite signature.
Patients in the DFCI/MSKCC and Philadelphia cohorts were treated with cisplatin-based chemotherapy followed by radical cystectomy, and cisplatin responders were defined as patients with no residual invasive disease on pathologic examination of the cystectomy specimen. We found that the ERCC2mut signature scores were strongly associated with cisplatin response in both the DFCI/MSKCC and Philadelphia cohorts (p = 2.1×10−4 and p = 0.003, respectively; Fisher exact test; Fig. 4A, B). For example, in the DFCI/MSKCC cohort, 15 of 17 cases (88%) with an ERCC2mut signature score ≥0.70 had a complete response versus only 10 of 33 (30%) cases with a score <0.70 (Fig. 4A). In the Philadelphia cohort, all six cases (100%) with an ERCC2mut signature score ≥0.70 had a complete response versus only 14 of 42 (33%) cases with score <0.70 (Fig. 4B). In both cohorts, overall survival (OS) was significantly longer in patients with a score ≥0.70 (Supp. Fig. 31–32).
Figure 4:
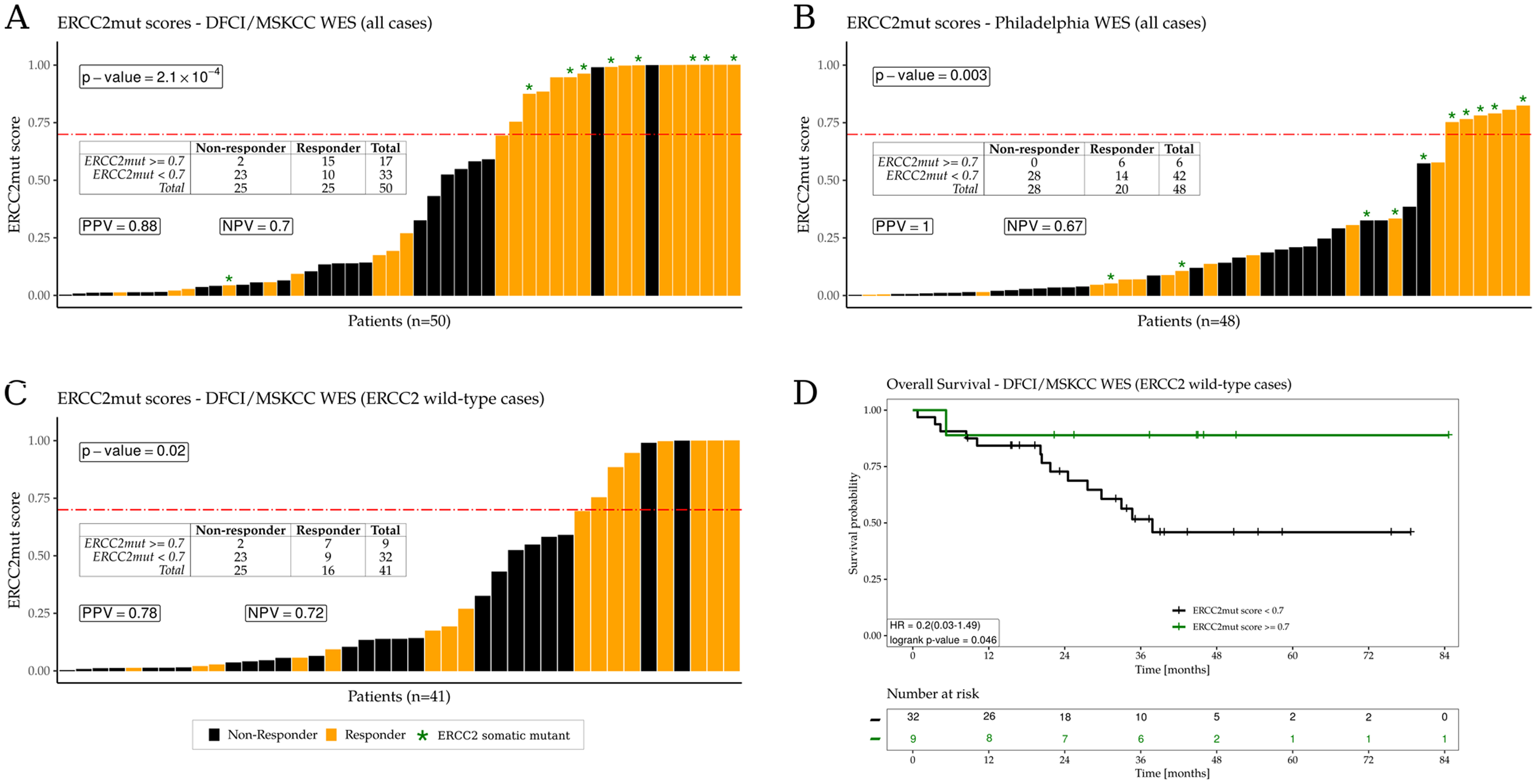
ERCC2mut signature scores are associated with cisplatin response, including among WT ERCC2 cases. Cisplatin responders are colored in orange and nonresponders are in black. ERCC2 mutant cases are denoted by green asterisks. A. ERCC2mut signature scores for all cases in the DFCI/MSKCC cohort (n=50). B. ERCC2mut signature scores for all cases in the Philadelphia cohort (n=48). C. ERCC2mut signature scores for WT ERCC2 cases in the DFCI/MSKCC cohort (n=41); high ERCC2mut signature scores (≥0.70) are significantly associated with cisplatin response (p=0.02). D. OS for patients with WT ERCC2 tumors in the DFCI/MSKCC cohort. OS was significantly longer for WT ERCC2 patients with ERCC2mut signature scores ≥0.70 (p=0.046). Positive predictive value (PPV) and negative predictive value (NPV) of the ERCC2mut composite signature were calculated for each cohort. P-values were calculated by the Fisher exact test.
We next restricted our analysis to patients without an ERCC2 mutation because this is the subset of patients for whom cisplatin response is currently most difficult to predict. Among the 41 WT ERCC2 cases in the DFCI/MSKCC cohort, only 16 (39%) were cisplatin responders (Fig. 4C). However, there was a significant enrichment of cisplatin responders among the cases with high ERCC2mut signature scores, 7 of the 9 (78%) WT ERCC2 patients with a score ≥0.70 were responders versus only 9 of the 32 (28%) WT ERCC2 patients with an ERCC2mut score <0.70 (p = 0.02). Therefore, WT ERCC2 cases with a high ERCC2mut signature score were nearly three times as likely to have complete response to cisplatin as WT ERCC2 patients with a low score. In addition, WT ERCC2 patients with ERCC2mut score ≥0.70 had significantly better survival than WT ERCC2 patients with lower scores (p=0.046; Fig. 4D). ERCC2mut scores were lower in the formalin-fixed, paraffin-embedded (FFPE)-derived Philadelphia cohort than in the other cohorts (which were derived from fresh frozen tissue), and there was only one WT ERCC2 case with a score ≥0.70 (Fig. 4B; Supp. Fig. 33). Together, these data demonstrate that the composite ERCC2mut signature is associated with cisplatin response, including in bladder tumors that lack an ERCC2 mutation. This result suggests that the ERCC2mut composite signature may reflect tumor NER deficiency conferred by mechanisms beyond ERCC2 mutation, and may, therefore, provide additional predictive power to prioritize patients for NER-targeting agents, such as cisplatin or irofulven.
An NER gene defect is sufficient to induce the composite mutational signature
To further investigate the relationship among NER pathway function, sensitivity to NER-directed therapy, and the ERCC2mut composite mutational signature, we tested if ERCC2 inactivation was sufficient to generate the composite mutational signature. KU19–19 is a bladder cancer cell line with no known NER pathway alterations (Supp. Fig. 34), while KE1 is an ERCC2-mutated derivative of KU19-19 that is NER deficient and displays increased sensitivity to cisplatin and irofulven (Fig. 1B) (6). For each cell line, separate clonal populations were propagated in parallel for 30 days and single cells were isolated, expanded to approximately 1×106 cells, and harvested for genomic DNA isolation (Fig. 5A). WGS was performed from clonal ‘parental’ (P0) populations, as well as from two independent ‘postpropagation’ clonal populations. Mutations were called as described previously (36) and ERCC2mut scores were determined for each sample (Supplementary Methods). Among the postpropagation populations, the NER-deficient KE1 cell line clones had significantly higher composite mutational signature scores than NER-proficient KU19–19 clones (Fig. 5B). These data demonstrate that NER deficiency created by loss of an NER gene is sufficient to induce the composite mutational signature and further supports a direct link among NER deficiency, the composite mutational signature, and sensitivity to cisplatin and irofulven.
Figure 5:
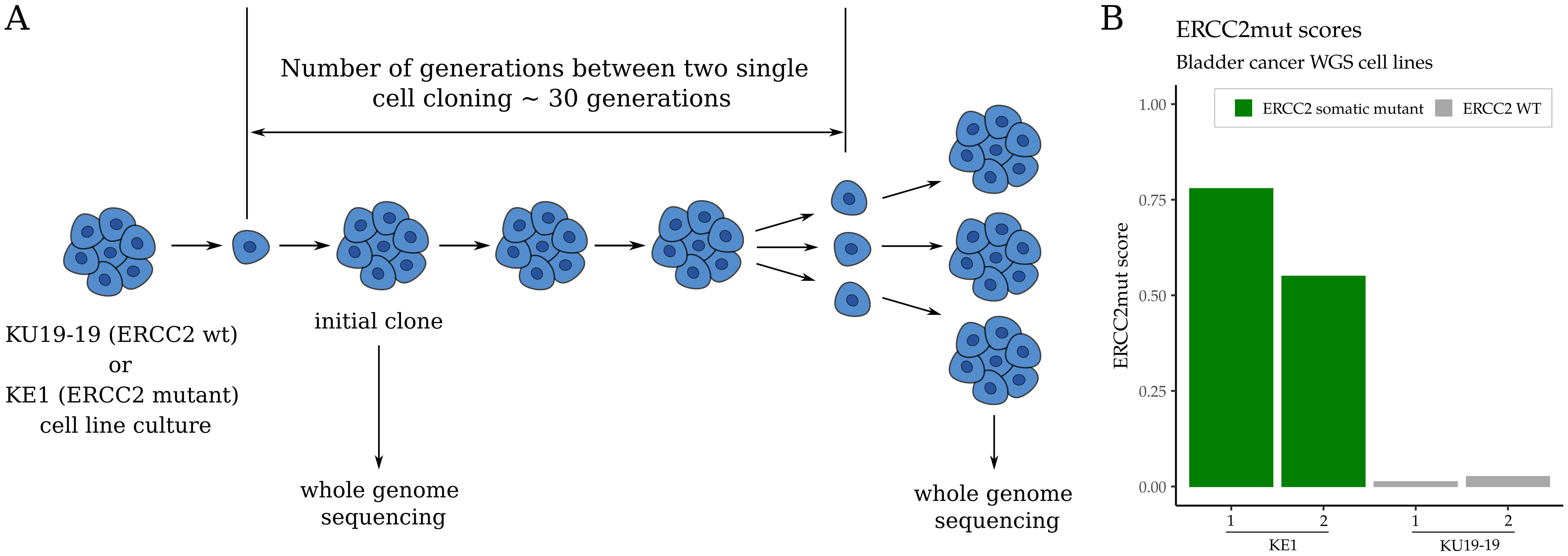
NER deficiency drives the ERCC2mut composite mutational signature. A. Separate clonal populations of NER-proficient KU19–19 or NER-deficient KE1 cells were cultured in parallel for approximately 30 generations and single cells were then isolated, expanded, and harvested. B. WGS and mutational signature analysis revealed significantly higher ERCC2mut scores in the NER-deficient KE1 samples compared with the NER-proficient KU19–19 samples.
Discussion
Tumor DNA repair deficiency is of particular interest in clinical oncology because it can potentially be targeted using a synthetic lethal–based strategy. However, the success of the synthetic lethal approach is dependent on accurate identification of the relevant DNA repair pathway deficiency in clinical tumor specimens, as well as on the availability of a therapeutic agent that is specifically active in DNA repair–deficient cells. Here, we characterize a novel synthetic lethal relationship between tumor NER deficiency and the abandoned anticancer drug, irofulven, and we show that NER-deficient tumors have a unique mutational signature that may expand identification of patients likely to respond to NER-targeted agents, such as cisplatin or irofulven.
Cisplatin-based chemotherapy is a preferred treatment approach for patients with muscle-invasive or metastatic bladder cancer. Mutations in ERCC2 or other DNA repair genes are present in approximately 20% of bladder tumors and are associated with cisplatin response rates of 80–100% (7,9). However, despite the demonstrated activity of cisplatin in patients with tumor NER deficiency, 30–50% of patients with bladder cancer are unable to receive cisplatin due to renal dysfunction or other medical comorbidities (43). For these patients, other agents that target tumor NER deficiency represent an attractive therapeutic strategy. On the basis of its mechanism of action and on its reported activity in NER-deficient nontumor models, we elected to investigate irofulven, a semisynthetic alkylating agent previously tested as an anticancer agent in a number of phase I/II trials (17–20). Whereas irofulven response rates were very modest (10–15%) in unselected patient populations, our functional analyses showed that NER-deficient tumor cells are profoundly sensitive to irofulven both in vitro and in vivo. Similar NER-dependent irofulven sensitivity was also observed in additional cell line and patient-derived cancer models, including breast cancer models harboring ERCC3 mutations (44). Because irofulven creates DNA lesions that are only recognized by the TC-NER pathway when transcribing RNA polymerase encounters the lesion, we found that inactivation of TC-NER or common NER genes, but not GG-NER–specific genes, was sufficient to induce irofulven sensitivity. These findings are consistent with a synthetic lethal mechanism of irofulven-induced tumor cell killing that is analogous to the synthetic lethal relationship that exists between HR deficiency and PARP inhibitor sensitivity in other tumor settings. Unfortunately, no tumors from irofulven-treated patients with bladder cancer are currently available to validate the relationship between NER gene mutation status and clinical irofulven sensitivity; however, a prospective clinical trial of irofulven is planned for patients with advanced urothelial tumors harboring predicted NER dysfunction as defined by NER gene defects (with or without LOH) or the presence of a high ERCC2mut signature score.
Despite initial responses in a subset of patients, most patients with metastatic bladder cancer who receive cisplatin-based chemotherapy ultimately develop cisplatin-resistant disease. Although several anti-PD-1/PD-L1 are approved in this setting, only a fraction of patients respond and median OS for platinum-refractory bladder cancer remains less than one year. Whereas mechanisms of resistance to platinum drugs and PARP inhibitors in HR-deficient tumors have been extensively studied, mechanisms of cisplatin resistance in NER-deficient tumors are not known. We found that acquired cisplatin resistance in an NER-deficient tumor model failed to restore NER activity and, therefore, conferred minimal cross-resistance to irofulven, suggesting that irofulven may be an effective treatment for patients with tumor NER deficiency who progress on cisplatin-based chemotherapy. Given that upregulation of alternative DNA repair pathways is common in tumors with loss of a specific DNA repair pathway, such as NER or HR, combining irofulven with other DNA damage response–targeting agents, such as PARP inhibitors, may also be a rational strategy to explore (44).
In addition to identifying a drug that specifically targets a DNA repair–deficient state, successful implementation of a synthetic lethal therapeutic strategy also relies on accurate identification of tumors harboring the targetable DNA repair deficiency. Most commonly, this is accomplished by sequencing the DNA repair genes of interest, and putative damaging germline or somatic NER pathway alterations are present in up to 10% of patients with bladder cancer and other tumor types (44). However, relying exclusively on this approach to identify tumor DNA repair deficiency has several shortcomings, including (1) uncertainty regarding the biological impact of novel mutations (i.e., variants of unknown significance), (2) the potential for secondary genetic events to modulate the effect of an observed mutation (for example, REV7 loss offsetting the impact of BRCA1 loss (45)), and (3) loss of DNA repair pathway activity via an epigenetic mechanism or through alteration of a gene not previously implicated in the pathway. Mutational signature-based approaches have the potential to overcome many of these challenges because they detect the genomic consequences of DNA repair pathway deficiency, rather than the underlying cause. Furthermore, integrative analyses of multiple types of mutation events (SNVs, indels, rearrangements) may increase the ability of mutational signatures to identify clinically relevant DNA repair deficiency, as has been demonstrated for detection of HR deficiency in breast tumors (46). Here, we describe a composite mutational signature of ERCC2 inactivation in bladder cancer. In multiple independent cohorts, this signature was strongly associated with ERCC2 mutation status, and importantly, the signature was also associated with cisplatin response in cases that lack an ERCC2 mutation. In fact, WT ERCC2 cases with a high ERCC2mut score were nearly three times more likely to have complete response to cisplatin-based chemotherapy than WT ERCC2 cases with a low signature score. Interestingly, several of the WT ERCC2 cisplatin responders with a high ERCC2mut signature score harbored a predicted deleterious mutation in another NER gene, including two patients with truncating ERCC6 mutations (Supp. Fig. 35). NER deficiency may also arise via epigenetic mechanisms, and further integrative genomic analyses will be required to understand the contribution of epigenetic processes to NER deficiency. We identified an optimal threshold signature score of ≥0.70 for bladder tumors sequenced from fresh frozen tissue; however, validation is required in prospectively collected cohorts, and additional work will be needed to define the optimal threshold for FFPE bladder tumor cohorts and to determine whether the signature can be reliably identified from less than WES data.
In summary, we identified a novel synthetic lethal relationship between tumor NER deficiency and sensitivity to the previously discarded anticancer agent, irofulven, and we show that NER deficiency is sufficient to drive sensitivity to irofulven in cisplatin-sensitive and cisplatin-resistant tumor models. Furthermore, we developed and validated a composite mutational signature of ERCC2 deficiency that is strongly associated with NER deficiency and irofulven sensitivity in preclinical models and also correlates with clinical cisplatin response, including in cases that lack an ERCC2 mutation.
Supplementary Material
Translational relevance.
ERCC2 mutations are present in approximately 10% of bladder tumors and confer increased sensitivity to cisplatin-based chemotherapy. However, up to half of patients with advanced bladder cancer are ineligible to receive cisplatin-based chemotherapy due to medical comorbidities. We identified a synthetic lethal relationship between bladder tumor NER deficiency and sensitivity to irofulven, a discarded anticancer agent that creates DNA lesions which can only be repaired by the NER pathway. We show that NER-deficient tumors, including those with acquired cisplatin resistance, are sensitive to irofulven, suggesting that irofulven may provide a novel treatment approach for cisplatin-ineligible or cisplatin-resistant patients with tumor NER deficiency. In addition, we developed and validated a composite mutational signature of tumor ERCC2 deficiency that is strongly associated with cisplatin response, including in cases that lack an ERCC2 mutation. This signature may be useful in identifying patients likely to benefit from cisplatin or irofulven.
Acknowledgments
Results shown here are based, in part, from data generated by The Cancer Genome Atlas Research Network: http://cancergenome.nih.gov/ and the International Cancer Genome Consortium: https://icgc.org/. This work was supported by the Research and Technology Innovation Fund (KTIA_NAP_13-2014-0021 to Z. Szallasi), Breast Cancer Research Foundation (BCRF-17-156 to Z. Szallasi), the Novo Nordisk Foundation Interdisciplinary Synergy Programme grant (NNF15OC0016584 to Z. Szallasi), the Novo Nordisk Foundation Center for Protein Research core grant (NNF14CC0001 to S. Brunak), Department of Defense through the Prostate Cancer Research Program (W81XWH-18-2-0056 to Z. Szallasi), Det Frie Forskningsråd Sundhed og Sygdom (7016-00345B to Z. Szallasi), Burroughs-Wellcome Fund (to K.W. Mouw), Dana-Farber Whole Foods Golf Fund (to M.-E. Taplin), the NCI (5K08CA219504 to K.W. Mouw and R01 CA227388 to E.M. Van Allen), Fox Chase Cancer Center NCI Core grant (P30CA00692 to E.R. Plimack), and the Velux Foundations (00018310 to Z. Sztupinszki and J. Börcsök).
Footnotes
Conflict of interest
J. Börcsök, M. Diossy, Z. Sztupinszki, K.W. Mouw and Z. Szallasi are listed as co-inventors on a provisional patent owned by Children’s Hospital Boston to quantify the ERCC2 mutational signature in cancer biopsies. K.W. Mouw - Consulting or Advisory Role: EMD Serono, Pfizer. Research Funding: Pfizer. J.E. Rosenberg - Honoraria: UpToDate, Bristol-Myers Squibb, AstraZeneca, Medscape, Vindico, Peerview, Chugai Pharma, Research To Practice, Intellisphere, Clinical Care Options, Clinical Mind. Consulting or Advisory Role: Lilly, Merck, Agensys, Genentech, Sanofi, AstraZeneca/MedImmune, Bristol-Myers Squibb, EMD Serono, Seattle Genetics, Bayer, Inovio Pharmaceuticals, BioClin Therapeutics, QED Therapeutics, Adicet Bio, Sensei Biotherapeutics, Fortress Biotech, Pharmacyclics, Western Oncolytics, GlaxoSmithKline, Janssen Oncology, Astellas Pharma. Research Funding: Oncogenex (Inst), Agensys (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Viralytics (Inst), Genentech (Inst), Incyte (Inst), Seattle Genetics (Inst), Bayer (Inst), AstraZeneca (Inst), QED Therapeutics (Inst), Astellas Pharma (Inst), Jounce Therapeutics (Inst). Patents, Royalties, Other Intellectual Property: Predictor of platinum sensitivity (Inst). Travel, Accommodations, Expenses: Genentech, Bristol-Myers Squibb. E.M. Van Allen - Advisory/Consulting: Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Manifold Bio, Monte Rosa, Janssen. Research support: Novartis, BMS. Equity: Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Manifold Bio, Monte Rosa, Microsoft. Travel reimbursement: Roche/Genentech. Patents: Institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation. E.R. Plimack - Clinical Research Support/Data Safety Monitoring Board: Astellas Pharma Inc., AstraZeneca Pharmaceuticals LP, Bristol-Myers Squibb Company, Genentech Inc., Infinity Pharmaceuticals Inc., Merck & Co. Inc., Pfizer Inc. Scientific Advisory Boards, Consultant, or Expert Witness: Genentech Inc., Merck & Co. Inc., Seattle Genetics Inc. D.B. Solit has consulted with/received honoraria from Pfizer, Loxo Oncology, Lilly Oncology, Illumina, Vivideon Oncology and Q.E.D. Therapeutics. J.H. Hoffman-Censits - Medical writing services: Roche Genentech. Consultant: AstraZeneca. Research support: Foundation Medicine.
References
- 1.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstantinopoulos PA, Matulonis UA. PARP Inhibitors in Ovarian Cancer: A Trailblazing and Transformative Journey. Clin Cancer Res. 2018;24:4062–5. [DOI] [PubMed] [Google Scholar]
- 3.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–81. [DOI] [PubMed] [Google Scholar]
- 4.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540–556.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Damish AW, Frazier Z, Liu D, Reznichenko E, Kamburov A, et al. ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin Cancer Res. 2019;25:977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu D, Plimack ER, Hoffman-Censits J, Garraway LA, Bellmunt J, Van Allen E, et al. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol. 2016;2:1094–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grivas P DNA Damage Response Gene Alterations in Urothelial Cancer: Ready for Practice? Clin Cancer Res. 2019;25:907–9. [DOI] [PubMed] [Google Scholar]
- 9.Plimack ER, Dunbrack RL, Brennan TA, Andrake MD, Zhou Y, Serebriiskii IG, et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur Urol. 2015;68:959–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teo MY, Bambury RM, Zabor EC, Jordan E, Al-Ahmadie H, Boyd ME, et al. DNA Damage Response and Repair Gene Alterations Are Associated with Improved Survival in Patients with Platinum-Treated Advanced Urothelial Carcinoma. Clin Cancer Res. 2017;23:3610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussain SA, James ND. The systemic treatment of advanced and metastatic bladder cancer. Lancet Oncol. 2003;4:489–97. [DOI] [PubMed] [Google Scholar]
- 12.Sonpavde G, Watson D, Tourtellott M, Cowey CL, Hellerstedt B, Hutson TE, et al. Administration of cisplatin-based chemotherapy for advanced urothelial carcinoma in the community. Clin Genitourin Cancer. 2012;10:1–5. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Gracia JL, Loriot Y, Rosenberg JE, Powles T, Necchi A, Hussain SA, et al. Atezolizumab in Platinum-treated Locally Advanced or Metastatic Urothelial Carcinoma: Outcomes by Prior Number of Regimens. Eur Urol. 2018;73:462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacDonald JR, Muscoplat CC, Dexter DL, Mangold GL, Chen SF, Kelner MJ, et al. Preclinical antitumor activity of 6-hydroxymethylacylfulvene, a semisynthetic derivative of the mushroom toxin illudin S. Cancer Res. 1997;57:279–83. [PubMed] [Google Scholar]
- 15.Jaspers NGJ, Raams A, Kelner MJ, Ng JMY, Yamashita YM, Takeda S, et al. Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair (Amst). 2002;1:1027–38. [DOI] [PubMed] [Google Scholar]
- 16.Koeppel F, Poindessous V, Lazar V, Raymond E, Sarasin A, Larsen AK. Irofulven cytotoxicity depends on transcription-coupled nucleotide excision repair and is correlated with XPG expression in solid tumor cells. Clin Cancer Res. 2004;10:5604–13. [DOI] [PubMed] [Google Scholar]
- 17.Seiden MV, Gordon AN, Bodurka DC, Matulonis UA, Penson RT, Reed E, et al. A phase II study of irofulven in women with recurrent and heavily pretreated ovarian cancer. Gynecol Oncol. 2006;101:55–61. [DOI] [PubMed] [Google Scholar]
- 18.Schilder RJ, Blessing JA, Shahin MS, Miller DS, Tewari KS, Muller CY, et al. A phase 2 evaluation of irofulven as second-line treatment of recurrent or persistent intermediately platinum-sensitive ovarian or primary peritoneal cancer: a Gynecologic Oncology Group trial. Int J Gynecol Cancer. 2010;20:1137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senzer N, Arsenau J, Richards D, Berman B, MacDonald JR, Smith S. Irofulven demonstrates clinical activity against metastatic hormone-refractory prostate cancer in a phase 2 single-agent trial. Am J Clin Oncol. 2005;28:36–42. [DOI] [PubMed] [Google Scholar]
- 20.Yeo W, Boyer M, Chung HC, Ong SYK, Lim R, Zee B, et al. Irofulven as first line therapy in recurrent or metastatic gastric cancer: a phase II multicenter study by the Cancer Therapeutics Research Group (CTRG). Cancer Chemother Pharmacol. 2007;59:295–300. [DOI] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016;48:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20:636–48. [DOI] [PubMed] [Google Scholar]
- 24.Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin Cancer Res. 2017;23:7521–30. [DOI] [PubMed] [Google Scholar]
- 25.Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med. 2019;381:2416–28. [DOI] [PubMed] [Google Scholar]
- 26.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015;26:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100:267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boot A, Huang MN, Ng AWT, Ho S-C, Lim JQ, Kawakami Y, et al. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018;28:654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, et al. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell. 2016;164:538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajkumar-Calkins AS, Szalat R, Dreze M, Khan I, Frazier Z, Reznichenkov E, et al. Functional profiling of nucleotide Excision repair in breast cancer. DNA Repair (Amst). 2019;82:102697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pipek O, Ribli D, Molnár J, Póti Á, Krzystanek M, Bodor A, et al. Fast and accurate mutation detection in whole genome sequences of multiple isogenic samples with IsoMut. BMC Bioinformatics. 2017;18:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ceccaldi R, O’Connor KW, Mouw KW, Li AY, Matulonis UA, D’Andrea AD, et al. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015;75:628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dreze M, Calkins AS, Gálicza J, Echelman DJ, Schnorenberg MR, Fell GL, et al. Monitoring repair of UV-induced 6–4-photoproducts with a purified DDB2 protein complex. PLoS ONE. 2014;9:e85896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tufegdzic Vidakovic A, Harreman M, Dirac-Svejstrup AB, Boeing S, Roy A, Encheva V, et al. Analysis of RNA polymerase II ubiquitylation and proteasomal degradation. Methods. 2019;159–160:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45:1459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galsky MD, Hahn NM, Rosenberg J, Sonpavde G, Hutson T, Oh WK, et al. Treatment of patients with metastatic urothelial cancer “unfit” for Cisplatin-based chemotherapy. J Clin Oncol. 2011;29:2432–8. [DOI] [PubMed] [Google Scholar]
- 44.Topka S, Steinsnyder Z, Ravichandran V, Tkachuk K, Kemel Y, Bandlamudi C, et al. Targeting Germline and Tumor Associated Nucleotide Excision Repair Defects in Cancer. Clin Cancer Res 2020. November 16;clincanres.3322.2020. doi: 10.1158/1078-0432.CCR-20-3322. Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–4. [Google Scholar] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staaf J, Glodzik D, Bosch A, Vallon-Christersson J, Reuterswärd C, Häkkinen J, et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat Med. 2019;25:1526–33. [Google Scholar] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cell line WGS bam files were deposited at the European Nucleotide Archive under the accession number PRJEB36417.