

Abstract
Polo-like kinase 1 (PLK1) is an essential cell cycle regulator that is frequently overexpressed in various human cancers. To determine whether Plk1 overexpression drives tumorigenesis, we established transgenic mouse lines that ubiquitously express increased levels of Plk1. High Plk1 levels were a driving force for different types of spontaneous tumors. Increased Plk1 levels resulted in multiple defects in mitosis and cytokinesis, supernumerary centrosomes, and compromised cell cycle checkpoints, allowing accumulation of chromosomal instability (CIN) which resulted in aneuploidy and tumor formation. Clinically, higher expression of PLK1 positively associated with an increase in genome-wide copy number alterations in multiple human cancers. This study provides in vivo evidence that aberrant expression of PLK1 triggers CIN and tumorigenesis and highlights potential therapeutic opportunities for CIN-positive cancers.
Keywords: polo-like kinase 1 (PLK1), oncogene, chromosomal instability, cell cycle checkpoints, tumorigenesis
Introduction
Polo-like kinase 1 (PLK1) is a key regulator of mitotic events, including centrosome maturation, bipolar spindle formation, sister chromatid segregation, anaphase-promoting complex/cyclosome (APC/C) activation, and mitotic exit (1). PLK1 is also known to control many non-mitotic events, such as DNA replication, the DNA-damage response (DDR) and G2 DNA-damage checkpoint recovery, chromosome dynamics, and microtubule dynamics (1). Clinical evidence suggests that PLK1 has a pivotal role in human cancer development and could be a target for anticancer drug discovery (2,3). PLK1 has been reported to be highly expressed in a broad spectrum of malignant human tumors (2,3). High expression levels of PLK1 are often linked to high tumor grade and are correlated with a poorer patient prognosis (2), which strongly suggests an important role during tumor initiation and progression.
PLK1 has been proposed to control cancer development through several mechanisms including the canonical regulation of mitosis and cytokinesis, as well as modulation of transformation-regulating proteins. For instance, PLK1 knockdown in U2OS cells abrogates anchorage-independent growth (4). In addition, we and others have shown that enhanced expression of Plk1 in human prostate epithelial cells and NIH/3T3 cells leads to cellular transformation in vitro and promotes tumor formation in nude mice, which provides evidence that PLK1 is directly involved in neoplastic transformation (5,6). However, an in vivo animal model providing compelling evidence of PLK1 as a proto-oncogene and enabling elucidation of its oncogenic function is still lacking.
To address this, we established Plk1 transgenic mice that widely express Plk1 in a graded manner, Plk1TA/+ (heterozygous for the transgene) and Plk1TA/TA (homozygous for the transgene). We demonstrated that enhanced Plk1 expression is sufficient to drive the formation of spontaneous tumors (including lymphomas, carcinomas, and sarcomas) in multiple organs. We provide strong evidence that increased Plk1 expression promotes mitotic errors, cytokinesis failure, and cell cycle checkpoint weakening, which allows continued proliferation of cells harboring chromosomal instability (CIN) resulting in genomic chaos and cellular transformation. Furthermore, a meta-analysis of TCGA datasets from multiple types of human cancer revealed that high Plk1 expression is correlated with an increase in genome-wide copy number alterations (CNAs) and a poorer prognosis.
Materials and Methods
Plk1 transgenic mice
The Transgenic and Knockout Core at Mayo Clinic generated Plk1 transgenic mice as previously described (7). Mice were housed in pathogen-free barrier environments. All mouse experiments/procedures were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
For tumor susceptibility studies, mice were monitored daily. Moribund mice were euthanized and major organs were screened for tumors. Tumors were processed for histopathology by standard procedures.
Generation and culture of PMEFs
WT and Plk1 transgenic PMEFs were isolated from 13.5-day embryos and cultured in DMEM (10% fetal bovine serum [FBS], 1% penicillin-streptomycin, 1% non-essential amino acids). Cells were frozen at passage [P]1 and used for experiments between P1-P5. Cells were synchronized in G0 by serum starvation (0.1% FBS) for 24 hrs, then released in fresh media containing 10% FBS. The majority of cells were in G2 or M phase 24 hrs after release.
Histopathology
Specimens were collected from all organs exhibiting an abnormal appearance. Tissues were fixed in 10% formalin, processed, and embedded in paraffin. Tissue sections (5 μm) were stained with H&E using standard procedures. Tissue blocks were xylene deparaffinized, serially rehydrated in ethanol (100%, 95%, and 70%), and stained with hematoxylin. Specimens were then decolorized and counterstained with eosin. Staining was performed for the identification of liver carcinoma (AFP), differentiating B- versus T-cell lymphoid tumors (CD20 and CD3), and identification of apoptosis (cleaved-caspase 3), cell proliferation (Ki-67) and, ATM/Chk2 axis activation (pATM and pChk2). Sections were blocked in PBST using 10% goat serum, and incubated with primary antibody in a humidified chamber at 4℃ overnight. Biotin-conjugated secondary antibodies were diluted in PBST containing the corresponding blocking serum (2.5%). Sections were stained using DAB substrate solution (Vector laboratories).
Live cell imaging
Imaging was performed using an inverted microscope (Zeiss Cell Observer Spinning Disc confocal microscope) controlled by Zeiss software and equipped with a Yokogawa CSU-X1A spinning disc unit, 2 Photometrics Evolve 512 cooled emCCD cameras, and laser illumination system. For chromosome segregation analysis, mCherry-H2B–positive cells were seeded on glass-bottom dishes (μ-Dish, IBIDI), precoated with 0.2% gelatin (Sigma-Aldrich) and cultured in CO2-independent L15 medium (without phenol red, +10% FBS) (Life Technologies). Cells were recorded using a 40×/NA 1.30 oil immersion lens at 1 image every 3 min for 8 hrs. For long-term imaging, cells were imaged with a 10×/NA 0.3 phase 1 lens for 24 hrs at 1 image every 5 min. Analysis and quantification were performed with ImageJ software (NIH).
Immunostaining
Cells grown on coverslips were fixed for 15 min in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 for 5 min, and blocked in 3% BSA for 1 hr followed by primary antibodies overnight at 4℃. After 3×5 min PBS-0.1% Triton X-100 washes, coverslips were incubated with fluorescently conjugated Alexa Fluor 488 or 568 secondary antibodies (Life technologies) for 45 min at RT. All antibodies were diluted in 3% BSA in PBS. Cells were counterstained and mounted using Prolong gold mounting medium with DAPI (Molecular Probes). Images were taken using a Zeiss AxioImager A1 (upright) equipped with an Axiocam MRc color CCD camera and a 63x oil immersion lens. Analysis and quantification were performed with ImageJ software. For nuclear signal quantification of p53 and p21, cell nuclei were defined using DAPI staining and nuclear p53- and p21-integrated fluorescence intensity in cells was measured using imageJ software and analyzed using GraphPad Prism software.
Western blot
Cells were lysed with RIPA buffer (Santa Cruz, sc-24948) containing protease inhibitors (Cocktail, Roche). Protein lysate concentration was quantified using a Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA). Protein was resolved on denatured SDS-PAGE. The separated proteins were transferred to nitrocellulose membrane. The membrane was blocked in 3% BSA at room temperature (RT) for 1 hr, incubated with primary antibodies overnight at 4℃, washed 3 times for 5 min, and incubated with secondary antibody for 1 hr at RT. The membrane was then washed and developed using SuperSignal West Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Statistical analysis
All experiments were performed at least 3 times. Statistical tests were performed using GraphPad Prism. Quantitative data are presented as mean ± SEM, with p<0.05 indicating statistical significance. A two-tailed Student’s t test was used to compare differences between groups.
Results
Generation of Plk1 transgenic mice
To explore the role of PLK1 in tumorigenesis, we established Plk1 transgenic mice that express exogenous Plk1 in a wide variety of tissues and organs. A previously described protocol (7) was used to generate transgenic mice from embryonic stem (ES) cell clones carrying stably integrated exogenous DNA. The CAGGS (cytomegalovirus [CMV] early enhancer/chicken β-actin) promoter, which produces ubiquitous and robust gene expression in transgenic mice (8), was used to drive expression of a floxed β-geo-stop cassette (LSL), followed by the coding sequence of mouse Plk1 protein (Fig. 1A). EGFP was co-expressed from an internal ribosomal entry site (IRES) to serve as a reporter for Plk1 expression (Fig. 1A). The transgenic vector was introduced into 129SV/S ES cells by electroporation, and G418-resistant colonies were selected and expanded. Clones were subjected to initial screenings that included staining for β–galactosidase as a measure of CAGGS promoter activity and analysis of EGFP and Plk1 expression upon infection with adenovirus-expressing Cre, using fluorescence microscopy and western blotting. Southern blotting was used to confirm single-copy transgene integration in positive clones. Selected ES clones were injected into C57BL/6 blastocysts to generate chimeric males. Male chimeras from 2 independent ES clones produced transgenic offspring that were maintained by breeding hemizygous mice with C57BL/6 mice for several generations. There were no noticeable differences between the 2 LSL-Plk1 transgenic strains, thus the strain with the highest ubiquitous LacZ expression was used (Fig. 1B). This unique mouse strain provides a new in vivo approach to study the consequences of increased Plk1 expression.
Figure 1: Generation and characterization of Plk1 transgenic mice.
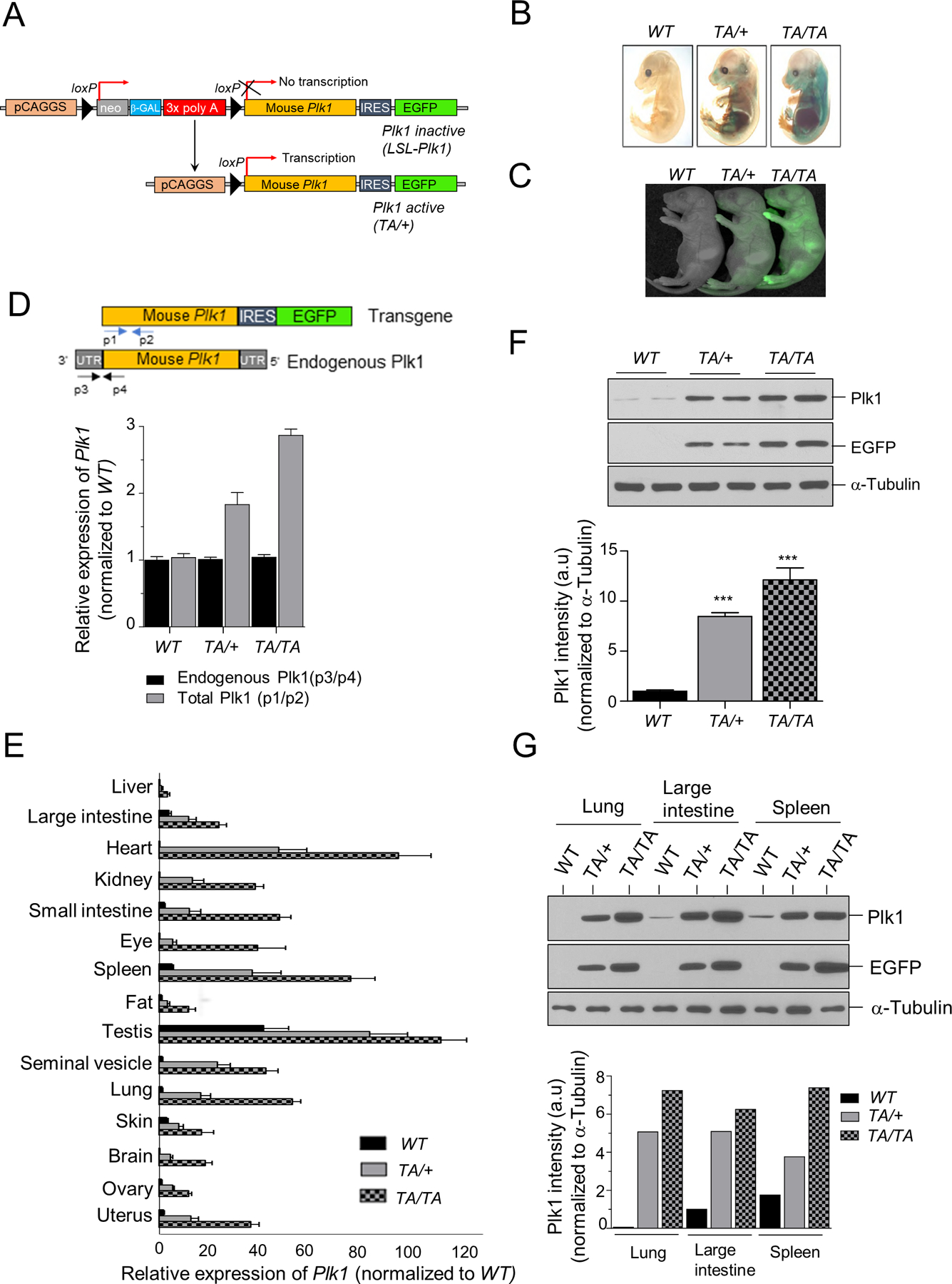
(A) Schematic of Plk1 transgenic mice generation. (B) β-galactosidase staining of WT: wild-type; TA/+: Plk1TA/+; TA/TA: Plk1TA/TA E13.5 mouse embryos. (C) EGFP fluorescence from 1d-old pups of the indicated genotypes. (D-E) qRT-PCR for Plk1 transcripts in PMEFs (D); tissues from transgenic mice (E) of indicated genotypes. Blue and black arrows show primer positions for analysis of the total p1/p2) and endogenous (p3/p4) Plk1 transcript, respectively. (F-G) Western blot (WB) analysis of PMEF cell extracts (F); tissue extracts (G) from transgenic mice. Plk1 and EGFP WB with α-Tubulin loading control. Bottom panel: Plk1 protein quantification of this WB.
Hprt-Cre transgenic mice were crossed with LSL-Plk1 mice for ubiquitous activation of the Plk1 transgene (Fig. 1A). Their offspring are referred to as Plk1TA/+ (heterozygous for the activated Plk1 transgene). Plk1TA/+ mice were intercrossed to generate cohorts of wild-type (WT), Plk1TA/+, and Plk1TA/TA (homozygous for the transgene) mice. As expected, a graded expression of the transgene was found in the pups of the indicated genotypes (Fig. 1C).
Plk1 expression was assessed by quantitative RT-PCR (qRT-PCR) on primary mouse embryonic fibroblasts (PMEFs) derived from these transgenic mice. Total (endogenous and transgenic) Plk1 transcript levels showed 2- and 3-fold increases in Plk1TA/+ and Plk1TA/TA PMEFs, respectively, when compared to WT PMEFs (Fig. 1D). Plk1 protein expression increases were verified by western blot (Fig. 1F). These experiments were also preformed using various tissues from WT and transgenic mice (Fig. 1E/G). Plk1 transcript and protein levels considerably increased in a graded manner in tissues from WT to Plk1TA/+ and Plk1TA/TA mice, although the magnitude varied per tissue (Fig. 1E/G). Together, these data indicate that Plk1TA/+ and Plk1TA/TA mice have graded increases of Plk1 expression across multiple tissues.
Increased Plk1 expression causes malignant transformation and spontaneous tumor formation
Several studies have demonstrated that PLK1 plays an important role during oncogenic transformation (9–11). We examined the ability of WT and Plk1 transgenic MEFs to grow in soft agar, a property that frequently correlates with tumorigenicity in animals. Because of the limited lifespan of PMEFs, we used spontaneously immortalized (P22) WT and Plk1 transgenic MEFs. WT MEFs cells formed few, if any, colonies in soft agar, whereas Plk1 transgenic MEF cells showed robust colony formation after 4 weeks, indicating that Plk1 overexpression in MEFs promotes oncogenic transformation (Supplementary Fig. S1A).
We then investigated whether increased Plk1 expression causally predisposes mice to spontaneous tumorigenesis. Mice cohorts were monitored for up to 24 months or until death. Plk1 transgenic mice had a shorter lifespan than their WT littermates (p< 0.0001), with a significant difference between Plk1TA/+ and Plk1TA/TA mice (p = 0.0013), suggesting that survival is inversely associated with increased Plk1 expression (Fig. 2A). Both Plk1TA/+ and Plk1TA/TA cohorts had marked increases in tumor incidence (54.6% and 76%, respectively) compared to WT littermates (4.7%) (p <0.001) (Fig. 2B). Histological analysis of Plk1TA/+ and Plk1TA/TA mice showed a wide spectrum of tumors, with B-cell lymphoma (CD19+, CD20+, and CD3-) being the most predominant and diffuse neoplasm that infiltrated lymph nodes, liver, spleen, lung, kidney, colon, and intestines (Fig. 2B/C and Supplementary Fig. S1B/C). Sarcomas of the intestine, liver, spleen, skin, and muscle were found in these mice, but to a lesser extent (Fig. 2B and Supplementary Fig. S1D). Carcinomas of the liver, lung, and kidney were also detected in transgenic mice; with liver carcinoma being the second most frequently induced neoplasm (Fig. 2B/C and Supplementary Fig. S1E). Of note, “giant” malignant polyploid or multinucleated cells were detected in these tumors (Fig. 2C). Plk1 transgenic mice frequently developed multiple concurrent tumors (Supplementary Fig. S1F), suggesting potent oncogenic activity. Taken together, these results provide strong evidence that increased Plk1 expression drives aggressive malignancies in various tissues.
Figure 2: Plk1 overexpression promotes spontaneous tumorigenesis.
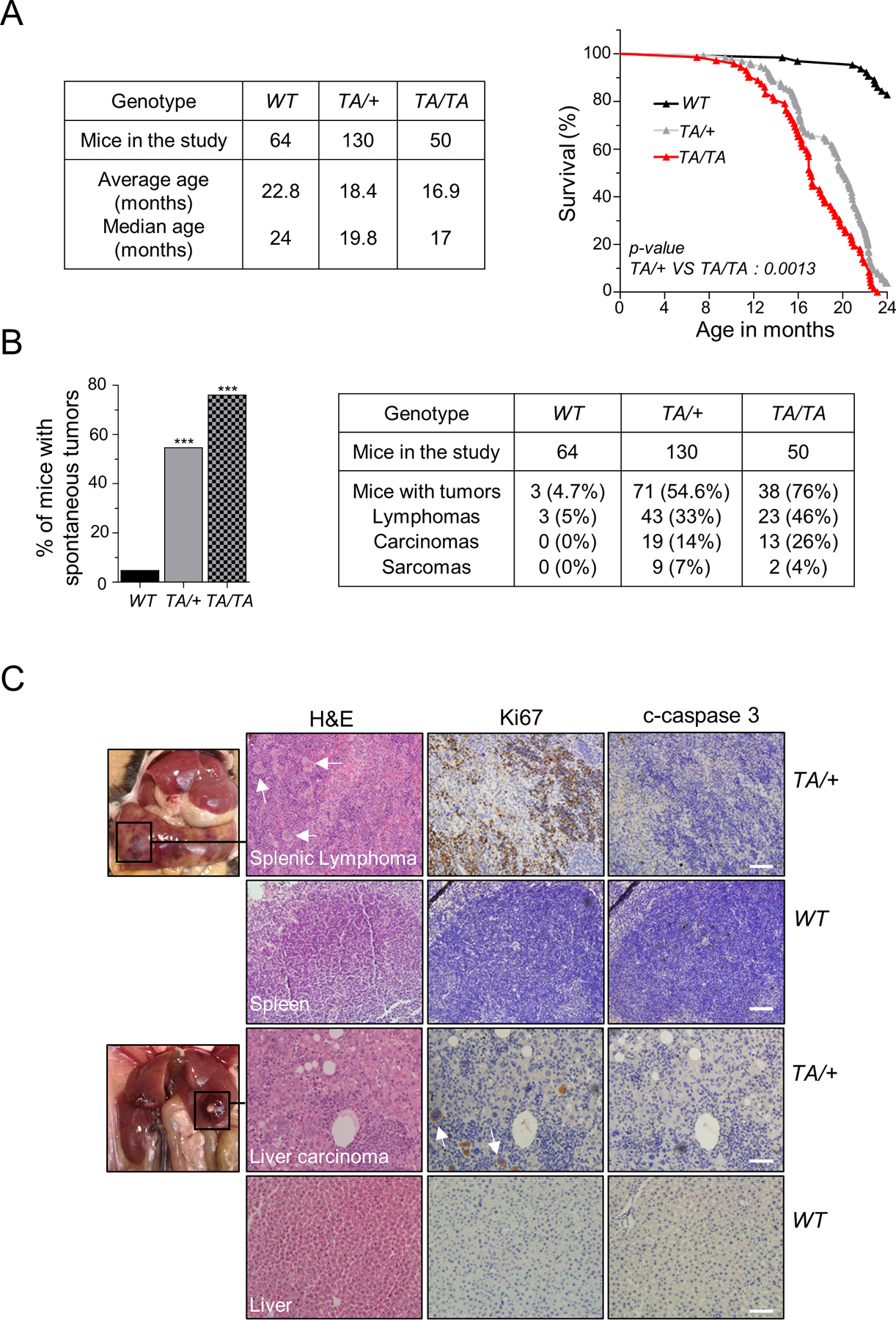
(A) (Left) Mouse cohort information, between 0 to 24 months. (Right) Survival curves for WT, TA/+, and TA/TA mice (TA/+ vs TA/TA: p = 0.0013, log-rank test). (B) (Left) Spontaneous tumor incidence in WT, TA/+, and TA/TA mice. (***: p < 0.001, Chi-squared test) (Right) Distinct neoplasm incidence in transgenic mice. (C) Examples of spontaneous tumors from TA/+ mice and corresponding immunohistochemistry (IHC) staining of Ki67 and cleaved-caspase 3 (c-caspase 3), and H&E in paraffin-embedded tissues. Below: Staining of respective WT tissue. Scale bar: 100 µm. White arrows indicate neoplastic polyploid cells.
Increased Plk1 expression leads to CIN
PMEFs are well-defined, fairly homogeneous, and easy to derive and maintain and have been widely used to study the physiological consequences of selective gene alterations in many branches of cell biology since the early 1970s (12–15). We examined whether increased Plk1 expression induces CIN, a characteristic of most human cancers (16) that alters nuclear morphology (17). We stained WT and Plk1 transgenic PMEFs with DAPI to examine variations in nuclear size and shape. We observed that transgenic PMEFs exhibited a graded increase in nuclear size compared to WT PMEFs, which was sustained over time (comparison of P1 and P5 PMEFss) (Fig. 3A). Since Plk1 transgenic mice frequently develop carcinomas, pulmonary alveolar epithelial cells (PAEpiCs) from 2-month-old mice were used to validate our observations (Supplementary Fig. S2A/B). Similar alterations were detected in the Plk1 transgenic PAEpiCs (Supplementary Fig. S2C). Strikingly, Plk1 transgenic PMEFs and PAEpiCs presented with nuclear protrusions, also called “buds” (Fig. 3A and Supplementary Fig. S2C). Nuclear bud presence and micronuclei frequency are widely used to assess CIN (17). Plk1 PMEFs and PAEpiCs showed a significant increase in micronuclei frequency compared to WT cells (Fig. 3B and Supplementary Fig. S2D). Interestingly, 16% of Plk1TA/+ and 36% of Plk1TA/TA PMEFs were multinucleated compared to just 4% of WT PMEFs (Fig. 3C). In agreement, FACS analyses showed an accumulation of polyploid cells in Plk1 PMEFs (Fig. 3D). This trend was also observed in Plk1 PAEpiCs (Supplementary Fig. S2D), indicating that elevated Plk1 expression promotes an increase in genomic content.
Figure 3: Overexpression of Plk1 induces aneuploidy.
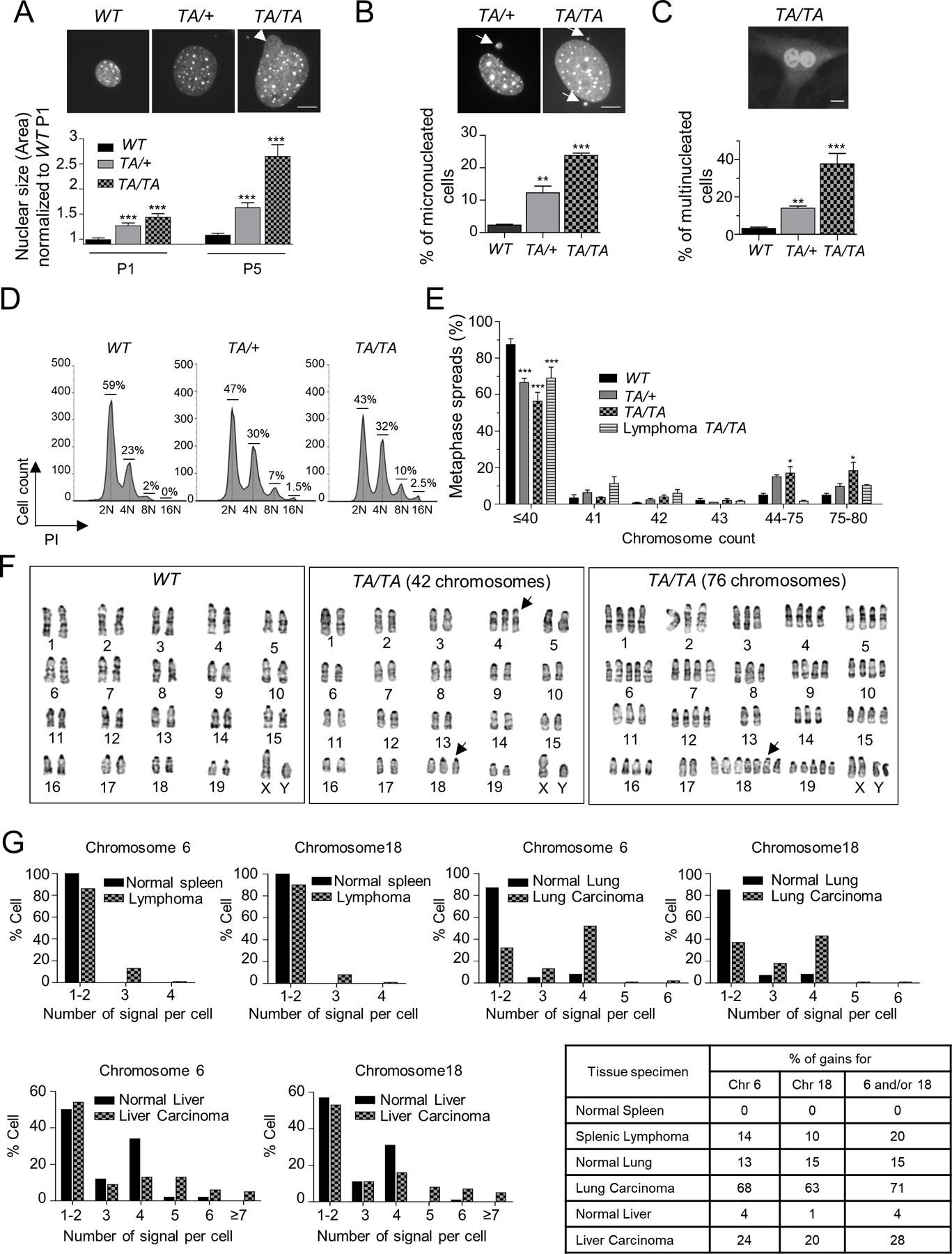
(A, B) (Top) Representative WT, TA/+ and TA/TA PMEFs stained with DAPI. (Bottom) (A) Nuclear size of WT, TA/+ and TA/TA P1 and P5 PMEFs, normalized to P1 WT PMEFs (B) Percentage of micronucleated WT, TA/+ and TA/TA PMEFs. Fifty cells/genotype, repeated 3x. (Mean ± SEM, **: p <0.01; ***: p< 0.001). Scale bar: 10 µm. Arrow head: nuclear buds; arrow: micronuclei. (C) (Top) Representative binucleated TA/TA PMEFs stained with DAPI and Plk1. (Bottom) Percentage of multinucleated cells per genotype. Fifty cells/genotype, repeated 3x. (Mean ± SEM, **: p < 0.01; *** p < 0.001). Scale bar: 10 μm. (D) DNA content in asynchronous WT, TA/+ and TA/TA PMEFs via flow cytometry. (E) Chromosome counts of WT, TA/+ and TA/TA PMEFs, and TA/TA lymphomas metaphase spreads. One hundred metaphases/genotype; repeated 3x. (Mean ± SEM, WT vs TA/+ or TA/TA, *: p < 0. 05; *** p < 0.001, t-test). (F) WT and TA/TA PMEFs karyograms. (Left) Representative GTG-banding of chromosomes from WT cells, showing a typical mouse complement of 40 chromosomes. (Middle) Representative metaphase spread from TA/TA PMEFs showing a cell with a total of 42 chromosomes (additional chromosome 4 and 18 [highlighted by arrows]). (Right) Representative TA/TA PMEF metaphase spread showing a cell with a near-tetraploid complement (76 chromosomes), including 7 copies of chromosome 18. (G) Interphase FISH analysis of 5-µm paraffin sections of a splenic lymphoma and lung and liver carcinomas from Plk1 transgenic mice and their respective WT tissues, hybridized to both peri-centromeric chromosome 6 and 18 probes. 100 cells/section. Percent gain in lung and spleen specimens represent gains of ≥1 signal in each cell; in liver, gains of >2 signals due to the physiological hepatocyte tetraploidy.
To further investigate if increased Plk1 expression drives chromosomal aneuploidy, chromosome counts were performed on metaphase spreads derived from WT and transgenic PMEFs. Strikingly, 34% of Plk1TA/+ and of 43% Plk1TA/TA metaphase spreads showed whole chromosome gains compared to just 12% in WT metaphase spreads (Fig. 3E). Moreover, 12% of Plk1TA/+ and 19% of Plk1TA/TA PMEFs were near-tetraploid (76–80 chromosomes) (Fig. 3E). Giemsa (G)-banding of Plk1TA/+ and Plk1TA/TA metaphase cells revealed an overall increase in chromosome copy number (Fig. 3F). Of note, chromosome 18, which contains genetic factors that enhance susceptibility to testicular germ cell tumors and promote ES cell derivation (18), was the chromosome most often present in extra copies in Plk1 PMEFs (Fig. 3F). A significant increase in the gain of whole chromosome(s) was also observed in Plk1 PAEpiCs (22% for Plk1TA/+ and 27% for Plk1TA/TA) compared to WT metaphase spreads (5%) (Supplementary Fig. 3E).
We next sought to analyze aneuploidy in tumors originating from Plk1 transgenic mice. Chromosome counts were performed on metaphase spreads of lymphomas from 4 different Plk1 transgenic mice. Interestingly, 25% of these cells had gains of one or more chromosomes, with 10% having a near-tetraploid complement (Fig. 3E). We further analyzed chromosome copy number gains in splenic lymphoma and lung and liver carcinomas that arose from Plk1 transgenic mice, along with their respective WT tissues. Interphase FISH analysis was performed with pericentromic probes for chromosome 18 (as it was frequently found in extra copies in PMEFs) and chromosome 6 (due to the presence of numerous oncogenes [e.g., CCND2, KRAS, BRAF] and transcription factors [e.g., FOXM1]) (Fig. 3G). Few, if any, gains for chromosomes 6 and 18 were detected in cells from the normal spleen and lung tissue (Fig. 3G). In contrast, the splenic lymphoma and lung tumor samples showed a significant increase in both chromosomes (Fig. 3G). Of note, hepatocytes can exist as a mixed population of diploid and tetraploid cells (19). However, we found that 20% to 24% of the liver cancer cells had 5 or more signals for at least one of the probes evaluated, compared to just 4% of normal hepatocytes. These results provide strong evidence that increased Plk1 expression drives aneuploidy.
Increased Plk1 expression promotes mitotic errors, cytokinesis failure, and centrosome amplification
Since PLK1 coordinates several crucial steps in cell division (1), we analyzed potential mitotic defects that might promote aneuploidy. WT and Plk1 PMEFs were infected with a lentivirus encoding H2B–mCherry, allowing for visualization of chromosomes. Enhanced Plk1 expression led to prolonged mitotic progression, particularly, from metaphase to cytokinesis (~10 min in WT, ~28 min for Plk1TA/+, and ~33 min for Plk1TA/TA) (Fig. 4A/C). Strikingly, 21% of Plk1TA/+PMEFs and 29% of Plk1TA/TA PMEFs, failed to complete cytokinesis, resulting in the formation of multinucleated cells (Fig. 4B/C). Furthermore, 72% of Plk1TA/+and 85% of Plk1TA/TA PMEFs showed mitotic defects, which included chromosomal misalignments and mis-segregations, compared to 15% of WT cells (Fig. 4B/C; Video 1). Chromatin bridges, formed due to chromosome mis-segregation in Plk1 PMEFs, failed to be resolved and were found at the cytokinesis abscission sites, resulting in a significant delay in cytokinesis (Fig. 4C; Videos 1, 2). Eventually, some of the bridges broke during cytokinesis and gave rise to micronuclei associated with the daughter cells (Fig. 4C; Video 1). Alternatively, some cells underwent abscission failure, followed by furrow regression and polyploid cell formation (Fig. 4B/C; Video 2). These results strongly suggest that increased Plk1 expression triggers numerous mitotic aberrations that contribute to the CIN and the complex karyotypes observed in Plk1 cells.
Figure 4: Plk1-overexpressing cells display numerous mitotic aberrations, cytokinesis defects, and centrosome amplification.
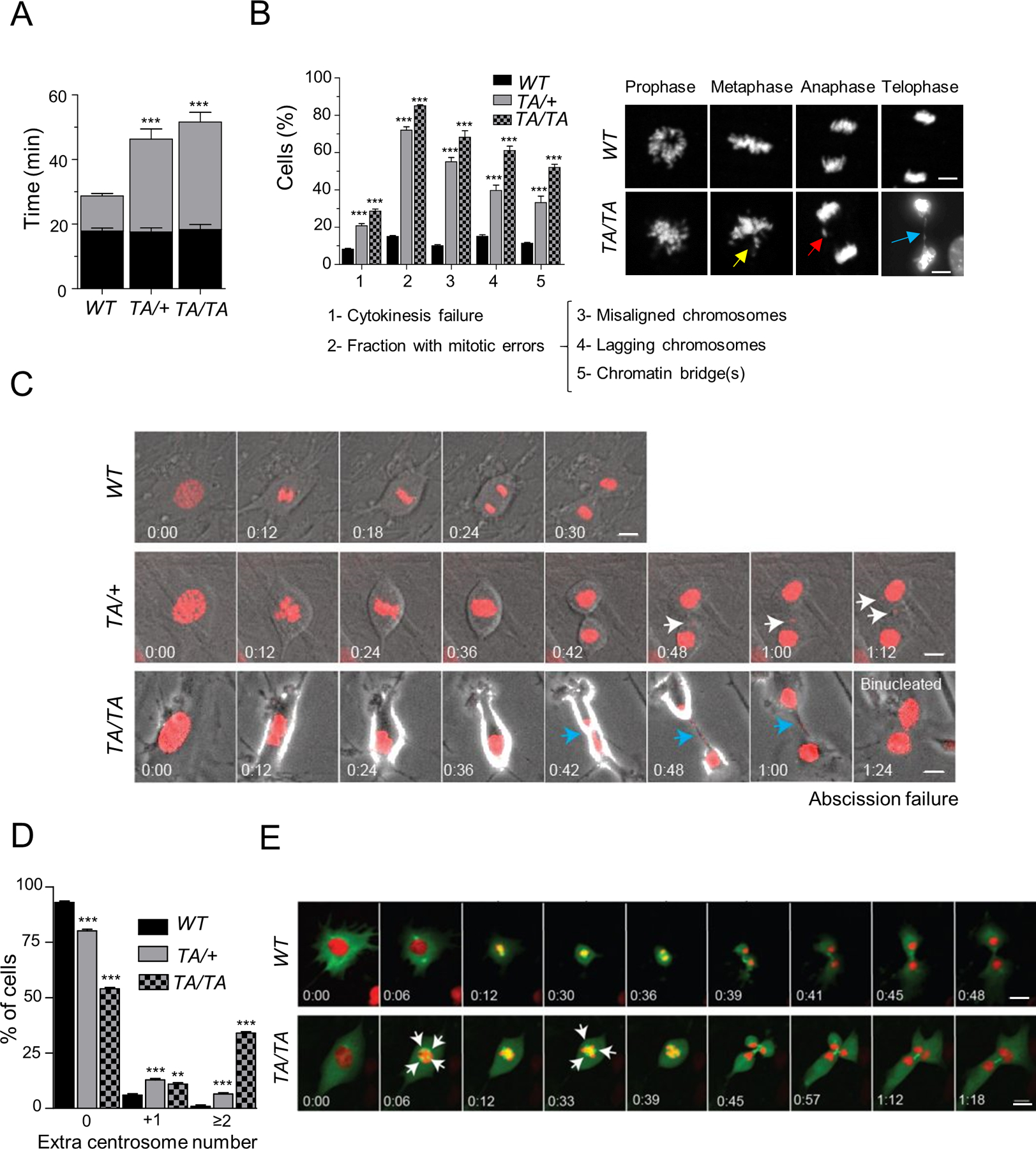
(A) Mitotic duration between prophase to metaphase (P → M) and metaphase to cytokinesis (M → C) in WT, TA/+ and TA/TA PMEFs expressing H2B-mcherry. Fifty cells/genotype, repeated 3x. (Mean ± SEM, ***: p < 0.001). (B) (Left) Percentage of cell division defects in WT, TA/+ and TA/TA PMEFs. Fifty cells/genotype, repeated 3x. (Mean ± SEM, WT vs TA/+ or TA/TA ***: p < 0.001). (Right) Representative PMEFs in each phase. Arrows – yellow: misaligned chromosome; red: lagging chromosome; blue: chromatin bridge. Scale, 10 µm. (C) Time-lapse images of PMEFs expressing H2B-mCherry monitored (3 min/image) as cells enter mitosis. Arrows – white: micronuclei; blue: chromatin bridge. Scale, 10 µm. (D) Centrosome count of G2 PMEF cells immunostained for γ-Tubulin. One hundred metaphases/genotype, repeated 3x. (Mean ± SEM, WT vs TA/+ or TA/TA, **: p < 0.01; *** p < 0.001, t-test). (E) Time-lapse images (3 min/image) of WT and TA/TA PMEFs co-expressing H2B-mCherry and EGFP-α-Tubulin as cells enter mitosis. White arrow - extra centrosome and multipolar spindle. Scale bar: 10 μm.
Cytokinesis failure is usually associated with centrosome amplification and the formation of multipolar spindles, leading to chromosome mis-segregation and rearrangements (20). Thus, we investigated whether enhanced Plk1 expression promotes centrosome amplification, which, in turn, contributes to chromosome mis-segregation. Centrosome counts were performed in G2 PMEFs immunostained for γ-tubulin (a centrosome marker) and phospho-histone H3 (a G2/M cell marker). Approximately 20% of Plk1TA/+ PMEFs and 45% of Plk1TA/TA PMEFs showed centrosome amplifications, compared to just 6% of WT PMEFs (Fig. 4D). A more dramatic increase was observed in transgenic PAEpiCs (35% of Plk1TA/+ and 69% of Plk1TA/TA cells) compared to their WT counterparts (20%) (Supplementary Fig. S3A/B). We then examined whether cells with centrosome amplifications undergo multipolar divisions or cluster their centrosomes (20).We simultaneously monitored chromosome segregation and mitotic spindle in PMEFs expressing both H2B–mCherry and EGFP-α-Tubulin, by live imaging (Fig. 4E; Video 3). A significant fraction of Plk1 transgenic PMEFs assembled multipolar spindles and underwent asymmetric multipolar division, leading to misaligned chromosomes in metaphase, lagging chromosomes in anaphase, and multinucleated cell formation (Fig. 4E; Video 3).
Plk1 drives uncontrolled proliferation of cells with CIN
Higher eukaryotes have developed multiple mechanisms to eliminate mitotic-incompetent cells, preventing the proliferation of cells with chromosomal abnormalities (21). Thus, we examined whether Plk1 PMEFs that harbor extensive CIN were able to activate senescence and/or apoptotic pathways. We assessed the senescence pathway in Plk1 PMEFs using a senescence-associated (SA) β−galactosidase assay. Compared to WT, no increase in SA β-galactosidase-positive Plk1 PMEFs was observed (Fig. 5A). Only ~30% of Plk1 PMEFs entered senescence in response to genotoxic stimuli (0.5 μΜ doxorubicin for 24 hrs) compared to 79% of WT PMEFs, suggesting that Plk1 suppressed the senescence pathway (Fig. 5A). We next assessed the apoptotic fraction of WT and Plk1 PMEFs using Annexin V (+) staining by flow cytometry. Only 6% of P5 Plk1 PMEFs underwent apoptosis compared to 14% of P5 WT PMEFs (Fig. 5B). In addition, when cells were challenged with doxorubicin to stimulate apoptosis, measured by poly (ADP-ribose) polymerase-1 (c-PARP-1) cleavage and caspase-3 (c-caspase 3), caspase-dependent apoptosis was induced in WT, but not Plk1 PMEFs (Fig. 5C). Collectively, these results suggest that Plk1 is associated with a “protective” response and inhibits cell death pathways.
Figure 5: Plk1 overexpression promotes proliferation of giant polyploidy and micronucleated cells.
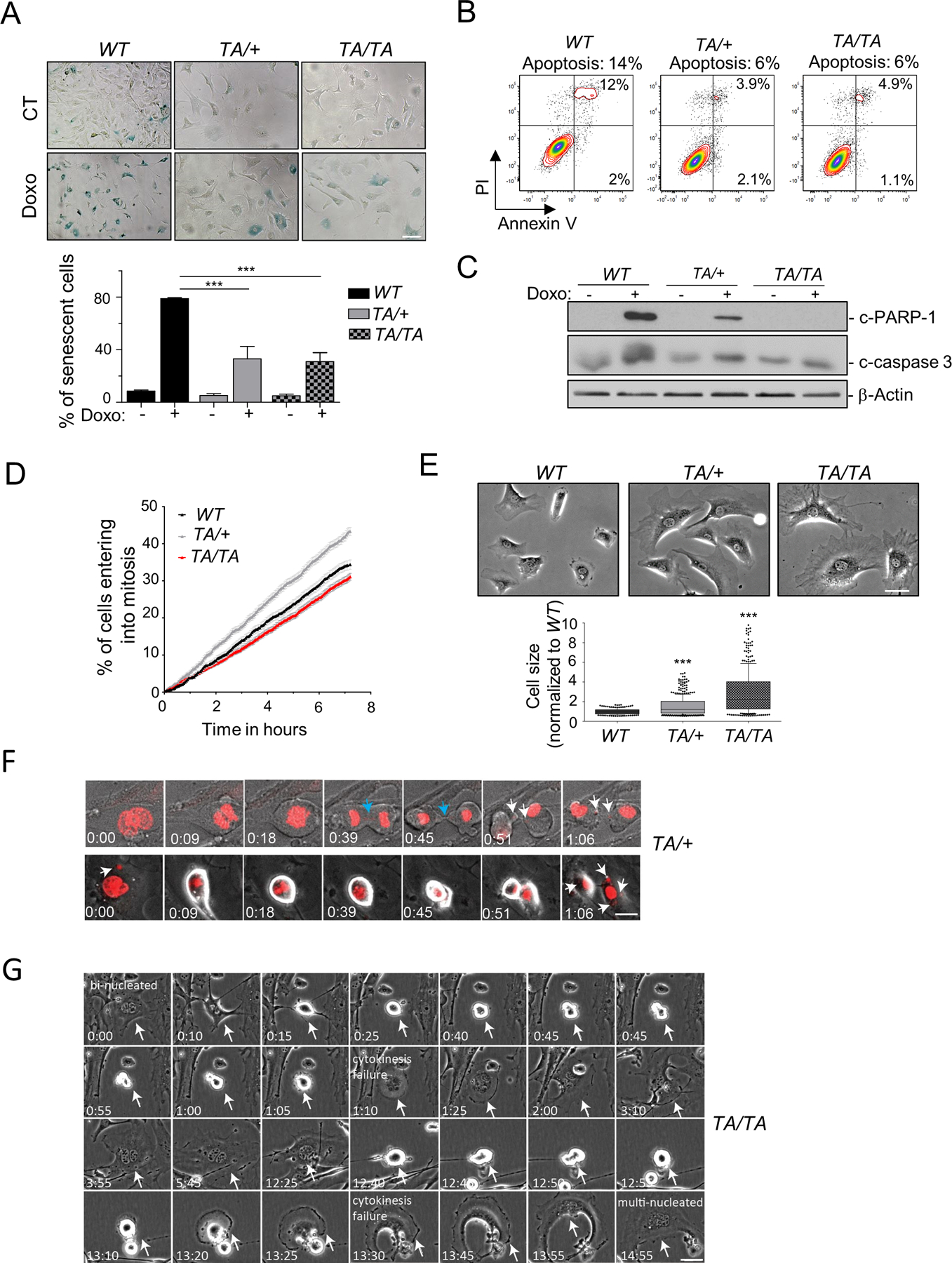
(A) (Top) Representative senescence-associated (SA) β-galactosidase activity in unperturbed PMEFs (P5) and 48 hrs post-treatment with doxorubicin (Doxo) (0.5μM, 8 hrs). Scale bar: 20 μm. (Bottom) Quantification of SA β-galactosidase positive cells. 200 cells/genotype, repeated 3x. (Mean ± SEM, ***: p < 0.001, t-test). (B) Unperturbed WT and Plk1 transgenic PMEFs (P5) were stained with Annexin V and PI and analyzed by flow cytometry. Numbers indicate percentage of apoptotic cells in the total population. (C) WB of untreated or Doxo (0.5μM, 8 hrs) treated WT and Plk1 transgenic PMEFs. Loading control: β-Actin. (D) Entry into mitosis (defined by nuclear envelope break down) of asynchronous PMEFs of indicated genotypes recorded by phase-contrast video-microscopy (5 min/image). Graph represents cumulative percentage of cells that have entered mitosis over time, normalized to cell density. 200 cells/genotype, repeated 3x. (Mean ± SEM). (E) (Top) Representative phase images of WT and Plk1 transgenic PMEFs. (Bottom) Cell size quantification, normalized to WT. 200 cells/genotype, repeated 3x (Mean ± SEM, ***: p < 0.001, t-test). Scale bar: 20 μm. (F) Representative images of binucleated and micronucleated TA/+ PMEFs expressing H2B-mCherry monitored during mitotic entry progression. Arrows – white: micronuclei; blue: chromatin bridge. Scale. Scale bar: 10 μm. (G) Representative binucleated TA/TA PMEFs monitored during 2 cell divisions. White arrow: binucleated cell. Scale bar: 10 μm.
We then assessed whether Plk1 transgenic PMEFs progress through active cell cycle or are arrested, by examining the cumulative number of cells entering mitosis within 8 hrs by live-cell imaging (22). No significant differences between WT and Plk1 PMEFs were observed, indicating that the genomic chaos elicited by increased Plk1 expression did not halt cell cycle progression and, instead, cells continued to proliferate (Fig. 5D). Strikingly, in contrast to WT cells, a substantial fraction of micronucleated cells and “giant” multinucleated Plk1 PMEFs were capable of progressing through active cell cycle (Fig. 5E/F; Videos 4A and B). Two outcomes were observed: (1) multinucleated or micronucleated Plk1 PMEFs were able to divide into 2 progeny cells with increased micronuclei as a result of cleavage of chromatin bridges (Fig. 5F; Video 4A), or (2) a portion of these cells continued to double their genomic content due to cytokinesis failure (Video 4B). To examine whether these “giant” progeny cells underwent G1 arrest or progressed through a second round of division, we monitored the multinucleated Plk1 PMEFs over an extended time (24 hrs). Multinucleated Plk1 PMEFs were still able to progress through a second round of division, resulting in genomic chaos (Fig. 5G; Video 5). Consistent with this observation, the multinucleated Plk1 PAEpiCs were also able to progress through a second round of division (Supplementary Fig.S4).
Increased Plk1 expression compromises cell cycle checkpoints
Cell cycle checkpoints function at key cell cycle transitions and are an essential surveillance mechanism to guard against improper cell division (21). The spindle assembly checkpoint (SAC) monitors the attachment to and tension on sister kinetochores by spindle microtubules, preventing premature chromosomal segregation and mitotic exit (23). It has been reported that PLK1 ensures efficient SAC activation by regulating critical components including haspin, Aurora B, monopolar spindle 1 (MPS1), budding uninhibited by benzimidazoles 1 (BUB1), and BUB1-related 1 (BUBR1) (24). SAC activity was assessed by live-cell imaging measuring the duration of mitotic arrest in PMEFs challenged with the spindle poison nocodazole before entry into mitosis. The duration of nocodazole-induced mitotic arrest was reduced in Plk1 PMEFs compared to WT PMEFs (3 hrs in Plk1 PMEFs vs. 5 hrs in WT cells), suggesting that high Plk1 levels weakened SAC signaling (Fig. 6A). The maintenance of levels of SAC effectors including MAD1&2, BUBR1, BUB1, and CDC20 (PLK1 targets) represents an important regulatory mechanism of SAC activity in prometaphase (25). We examined whether enhanced Plk1 expression impairs BUBR1 and/or CDC20 expression after nocodazole treatment. Indeed, BUBR1 and CDC20 protein levels were decreased in Plk1TA/TA PMEFs and, to a lesser extent, in Plk1TA/+ PMEFs, compared to WT PMEFs upon nocodazole treatment (Fig. 6B). Thus, increased Plk1 expression compromises SAC signaling, leading to chromosomal segregation defects and, therefore, CIN.
Figure 6: Plk1 overexpression impairs the cell cycle checkpoints.
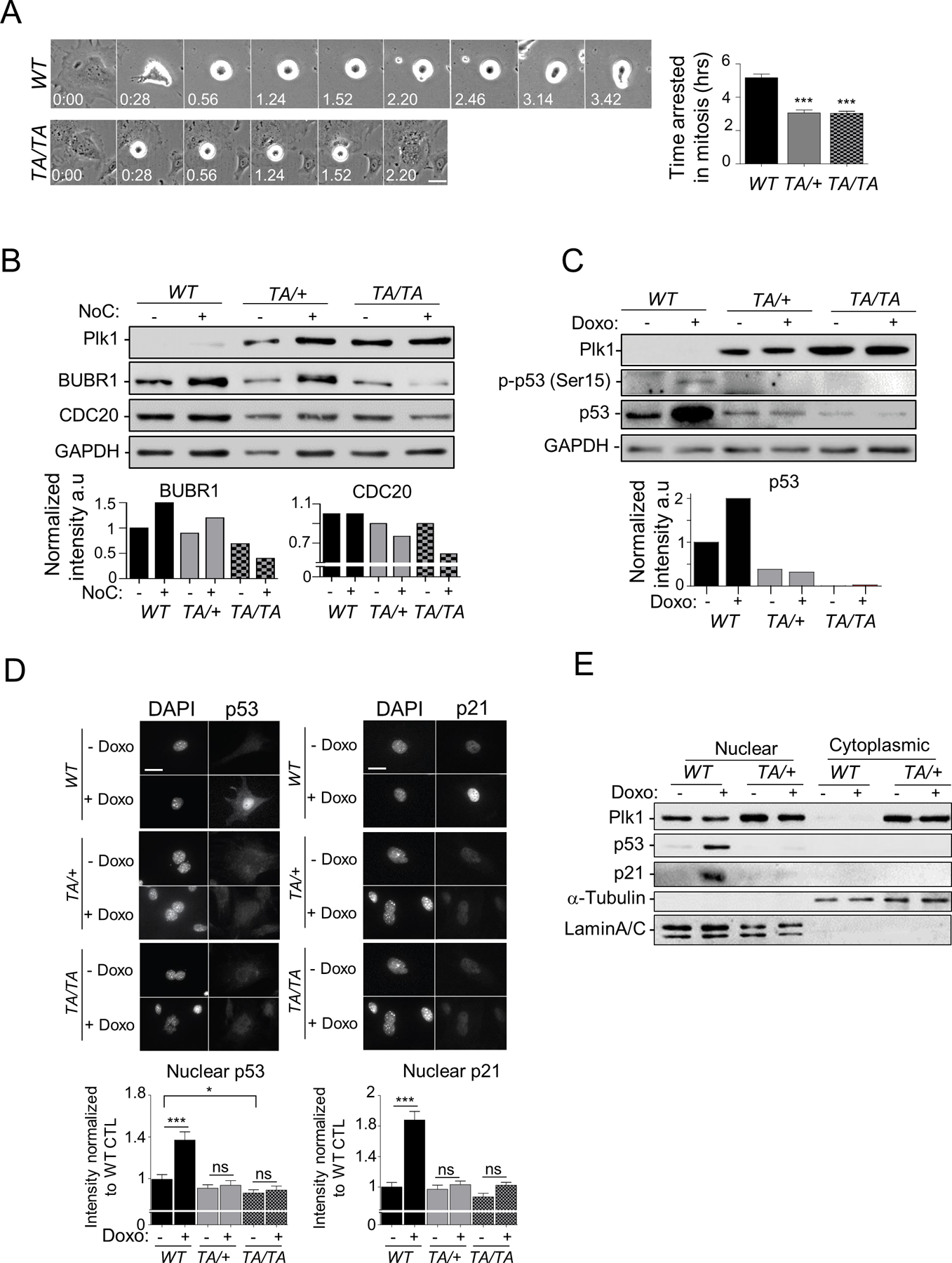
(A) Synchronized G2 WT and Plk1 transgenic PMEFs were treated with nocodazole (NoC) (200 ng/ml) and tracked through mitotic entry and progression by phase-contrast video-microscopy (4 min/image). (Left) Time-lapse images of WT and TA/TA PMEFs arrested in mitosis by NoC treatment. (Right) Duration of mitotic arrest induced by NoC treatment in PMEFs. Fifty cells/genotype; repeated 3x. (Mean ± SEM, WT vs TA/+ or WT vs TA/TA ***: p < 0.001 t-test). (B) (Top) WB of untreated or Noc (200 ng/ml, 6 hours) treated WT and Plk1 transgenic PMEFs, Loading control: GAPDH. (Bottom) Quantification of BUBR1 and CDC20 expression levels normalized to GAPDH (C) (Top) Analysis of p53 expression and activity (p-p53 [Ser15]) in untreated or doxorubicin (Doxo) (0.5 mM for 2 hrs) treated WT and Plk1 transgenic PMEFs synchronized in G2. (Bottom) Quantification of p53 expression level normalized to GAPDH (D) (Top) Representative images of cells treated as in (C), and immunostained for p53 or p21. (Bottom) Quantification of p53 and p21 nuclear intensity using ImageJ software. 50 cells/genotype, repeated 3x. (Mean ± SEM, *: p < 0.05; ***: p < 0.001). Scale, 10 µm. (E) Nuclear and cytoplasmic p53 and p21 levels analyzed by WB from synchronous G2 PMEFs of indicated genotype treated as in (A). Loading and fractionation controls: α-Tubulin and Lamin A/C.
See Supplementary Fig.S5/56/7.
DNA-damage checkpoints are regulatory pathways that trigger cell cycle arrest in response to DNA damage, thereby enabling repair prior to mitotic entry (26). Although harboring CIN, Plk1 PMEFs continue to proliferate (Fig. 5), which prompted us to investigate if DNA-damage checkpoints are impaired in these cells. In mammalian cells, ataxia-telangiectasia mutated (ATM) kinase is one of the most upstream DDR kinases, which orchestrates the propagation of the DNA-damage signal through its direct transductors, such as Chk2 (checkpoint kinase 2) and p53 (27). Thus, we sought to investigate whether increased of Plk1 expression triggers the activation of ATM/Chk2 signaling in nonmalignant and malignant tissues. As expected, lung tissues from Plk1 transgenic mice showed strong pSer 1981-ATM (mouse ortholog S1987) staining when compared to tissues from WT mice, indicating ATM activation (Supplementary Fig. S5A). However, Chk2 activation was compromised in tissues derived from Plk1 transgenic mice (Supplementary Fig. S5A), suggesting checkpoint silencing of the ATM/Chk2 axis. ATM activation was also observed in tumors derived from Plk1 transgenic mice (Supplementary Fig. S5A). Similar results were obtained with Plk1 PAEpiCs (Supplementary Fig. S5B/C).
In eukaryotic cells, p53 governs most of the molecular events in the DDR by regulating the expression of a large number of target genes, leading to cell cycle arrest, DNA repair, or apoptosis (28). Previous studies showed that PLK1 is an essential regulator of checkpoint recovery from DNA damage-induced G2 arrest, in part, by directly and indirectly controling p53’s activity (29). We investigated whether increased Plk1 expression modulates p53 signaling in Plk1 PMEFs. We first analyzed p53 levels and activity (phospho-p53 [Ser15]) by Western blot in WT and Plk1 PMEFs synchronized in G2 (when PLK1 expression is high (1)). Surprisingly, Plk1 transgenic cells that harbored CIN had lower p53 basal expression and activity levels compared to WT cells (Fig. 6C), suggesting that Plk1 may, directly or indirectly, affect p53 protein levels. Exon sequencing of p53 in 19 samples (2 independent sets of WT, Plk1TA/+, and Plk1TA/TA PMEFs and immortalized MEFs; 1 WT and 1 Plk1TA/+ lung sample; 1 Plk1TA/TA lung tumor sample; 2 independent sets of WT spleens and 2 Plk1TA/TA splenic lymphoma) confirmed no detectable tumor-associated mutations.
We performed qRT-PCR for 5 of p53’s canonical targets (p21, Mdm2, Puma, Bax, and GADD45α). As expected, Plk1 PMEFs had lower p53 target gene expression compared to WT cells (Supplementary Fig. S6). We then examined the ability of these cells to activate p53 in response to genotoxic stimuli (Fig. 6C and Supplementary Fig. S6). As expected, WT cells showed a dramatic increase in both the total and active form of p53 as well as its target gene expression upon treatment with doxorubicin (Fig. 6C and Supplementary Fig. S6). This response was not observed in Plk1 transgenic cells (Fig. 6C and Supplementary Fig. S6). Similar results were obtained in PAEpiCs (Supplementary Fig. S7A/B). Upon DNA damage, p53 translocates to the nucleus, where it acts as a nuclear transcription factor, initiating the transcription of direct target genes implicated in cell cycle arrest, DNA repair, apoptosis, or senescence (30). To determine whether Plk1 also modulates p53’s subcellular localization, we performed immunofluorescence (IF) to visualize the subcellular localization of endogenous p53 in PMEFs under the same conditions as those in Fig. 6A. Interestingly, Plk1TA/TA PMEFs exhibited a reduction in baseline levels of nuclear p53 compared to WT PMEFs (Fig. 6D); doxorubicin treatment induced an increase in p53 protein levels and its nuclear translocation in WT PMEFs, but not in Plk1 PMEFs (Fig. 6D). Subcellular fractionation assays further confirmed these results (Fig. 6E). In many cell types, p53-mediated growth inhibition is contingent upon the induction of its downstream target the cyclin-dependent kinase inhibitor p21(28). A dramatic increase in nuclear p21 was induced by doxorubicin in WT, but not in Plk1 PMEFs (Fig. 6D/E). Of note, Plk1 PMEFs showed a dramatic increase in Plk1 expression in both cytoplasmic and nuclear compartments, compared to PLK1’s predominant nuclear localization in WT cells (Fig. 6E). Early studies demonstrated that PLK1 executes its cell-cycle regulatory functions in the nucleus (31). Interestingly, cytoplasmic accumulation of PLK1 has been reported in multiple cancers, including bladder, breast, rectal, and lung, and was associated with the aggressiveness of the tumor (32–35), which suggest that PLK1 may have distinct oncogenic functions (acting on specific effectors) in the cytoplasm. A deeper understanding of PLK1’s compartmentalized-functions will be needed to further establish the role of PLK1 in cancer development.
These results indicate that increased Plk1 expression compromises DDR signaling, providing an explanation for why chromosomally unstable Plk1 transgenic cells continue to proliferate and accumulate CIN, leading to malignant transformation and cancer development.
Plk1 expression is positively correlated with genome-wide CNAs in human tumors
Given our observation that increased Plk1 expression drives the formation of aneuploid tumors in Plk1 transgenic mice (Fig. 3G), we sought to explore the relationship between PLK1 and CIN in human cancers using a combination of publicly available datasets and bioinformatics approaches. We selected 6 cancer types, including prostate adenocarcinoma [PRAD], liver hepatocellular carcinoma [LIHC], lung squamous cell carcinoma [LUSC], lung adenocarcinoma [LUAD], uterine corpus endometrial carcinoma [UCEC], and sarcoma [SARC], all of which showed significant increases in PLK1 expression compared to the normal tissues (Supplementary Fig. S8, (2)). Datasets for both genome-wide CNAs and PLK1 expression levels from a large collection of human tumor specimens were downloaded through the public UCSC Xena platform Portal, which gathers datasets from TCGA, Pan-Cancer Analysis of Whole Genomes (PCAWG), the International Cancer Genome Consortium (ICGC), and the Genomic Data Commons (GDC) (36). Segment counts were summed for each sample according to segment mean data generated based on hg19 reference with germline probes removed. A total of 2298 tumor specimens (492 PRAD, 364 LIHC, 498 LUSC, 511 LUAD, 176 UCEC, and 257 SARC) were analyzed. Pearson correlation analysis revealed that PLK1 expression was significantly and positively correlated with genome-wide copy number in all 6 tumor types (Fig. 7A, p < 0.0001), supporting our observations that increased Plk1 expression drives aneuploid tumor formation in mice.
Figure 7: PLK1 overexpression is associated with increased genome-wide copy numbers and poorer prognosis in human cancers.
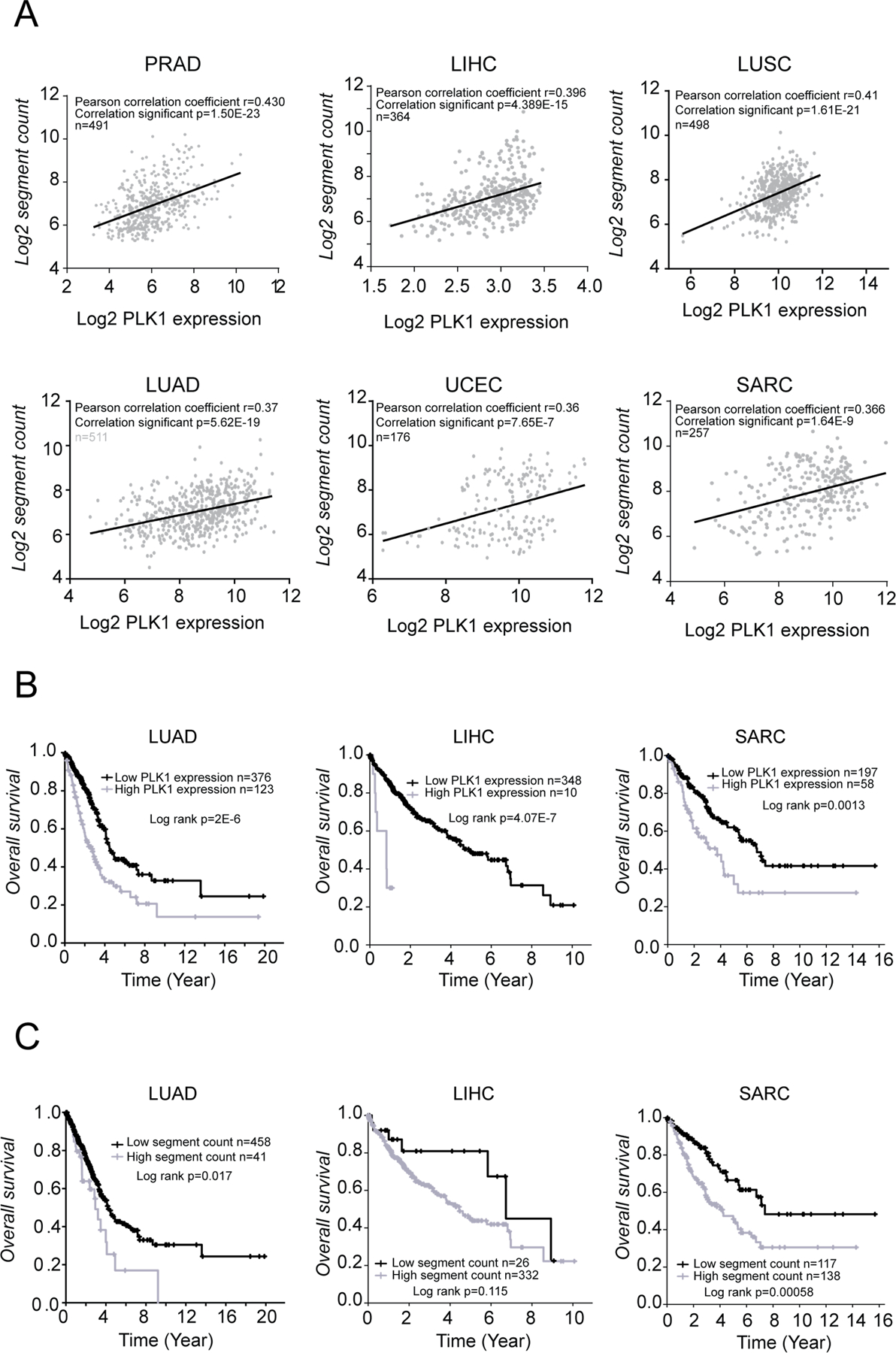
(A) Correlations between CNAs and PLK1 expression levels using TCGA cohort datasets. (B) Association between PLK1 expression and prognosis. Mean PLK1 expression levels was used to divide patients into high/low expression groups. (C) Association between genome segmentation counts and prognosis. Patients were divided into high/low segment count groups, using optimal segment count mean. Optimal split cutoff was defined using Cutoff finder.
We next assessed the prognostic value of increased PLK1 expression in human cancers. Overall survival (OS) was examined for cancer types with available clinical follow-up, which included LUAD, LIHC, and SARC. Kaplan-Meier analysis revealed that individuals with high expression levels of PLK1 had a significantly worse prognosis than those with low expression level of PLK1 (p < 0.01; Fig. 7B). Patients with LUAD and SARC with high CNAs also had a poorer prognostic value than those with low CNA levels (p < 0.05; Fig. 7C).
Discussion
The list of proto-oncogenes and tumor suppressors is continually expanding (37). Increased understanding of their specific roles during tumorigenesis and targeting these pathways has led to numerous treatment breakthroughs. PLK1 is frequently overexpressed in various human cancers, and its expression level is positively correlated with increased tumor aggressiveness and poorer prognosis, highlighting PLK1’s close association with human cancer and oncogenic potential. However, direct evidence to substantiate the claim of PLK1 as a potent proto-oncogene has been lacking. Using a new PLK1 transgenic mouse model, we provide direct evidence that Plk1 has potent oncogenic properties, driving malignant transformation and spontaneous tumorigenesis. Our results show that increased Plk1 expression drives CIN by promoting the formation of supernumerary centrosomes and numerous mitotic errors, including chromosome misalignment, chromosome mis-segregation, and cytokinesis failure, leading to genomic chaos and the formation of giant multinucleated cells and micronucleated cells. Increased Plk1 expression overrides cell cycle checkpoints, which allows cells to tolerate CIN and continue to proliferate. Furthermore, meta-analysis of publicly available genome-wide CNAs and gene expression datasets revealed that higher Plk1 expression is significantly correlated with an increase in genome-wide segment copy number and is associated with poorer prognosis in multiple human cancers, clinical evidence that further substantiates the role of PLK1 overexpression in CIN and tumor development. Therefore, this study provides the first evidence that PLK1 is a potent proto-oncogene in vivo.
Two other Plk1 transgenic mouse models have recently been established (38,39). In Li Z. et al’s study, the CMV promoter was used to drive expression of Cre recombinase that in turn activated the Plk1 transgene (38), in contrast to the CAGGS-driven Cre expression in our transgenic model. It has been well documented that the CMV promoter is much less efficient than the CAGGS promoter (40), which may contribute to the insufficient expression of transgenic Plk1 and the subtle tumor phenotype in that model (Fig. 1 and (38)). De Cárcer and his colleagues established inducible (Tet-on) knock-in Plk1 transgenic mice (39). Of note, approximately 50% of the control mice developed tumors by 20 months of age, suggesting that their genetic background predisposes them to tumor development, which may contribute to the minimal increase in tumor incidence observed when Plk1 is overexpressed, as reported in Apcmin/+ mice (41). Paradoxically, they reported that Plk1 overexpression suppresses the development of Kras-induced or Her2-induced mammary gland tumors (39). Both Kras and Her2 are potent proto-oncogenes, and, in combination with increased Plk1 expression levels, are likely to activate a powerful senescence response, known as oncogene-induced senescence (42,43), which prevents cellular transformation and tumorigenesis, reminiscent of the phenotypes reported in their studies (39). This observation highlights the oncogenic function of PLK1 in the early stages of tumor development and also underscores the need for further assessment of Plk1 function between and within species to ensure that the models evaluated are reflective of human function. Additionally, several groups have reported that PLK1 is closely associated with breast cancer development. PLK1 is strongly overexpressed in breast cancer, particularly in triple-negative breast cancer (TNBC), and is associated with poorer clinical outcomes (44,45). Inhibition of PLK1 using RNAi or pharmacological inhibitors triggered apoptosis in TNBC cell lines (44,45). In our transgenic model, the magnitude of enhanced Plk1 expression mirrors what is observed in human tumors (2). Our finding that ubiquitously enhanced expression of Plk1 drives spontaneous tumorigenesis in multiple tissues is consistent with the overwhelming clinical reports showing that PLK1 is overexpressed in a variety of cancers and often correlates with cellular proliferation, metastasis, and poor prognosis (2,9,10). Our model provides substantive evidence that PLK1 functions as a proto-oncogene, and when overexpressed, becomes a potent oncogene.
We observed supernumerary centrosomes in Plk1 transgenic cells, which resulted in multipolar mitotic spindle formation, chromosome misalignment and mis-segregation, and cytokinesis defects (Fig. 4D/E and Supplementary Fig. S3). Supernumerary centrosomes result from centrosome overduplication or cytokinesis failure (20). PLK1 has reportedly been involved in centriole duplication, centrosome separation and maturation, mitotic spindle assembly (1), and centrosome amplification (32). In this study, we showed chromosomal instability in Plk1 transgenic cells, as well as in tumors, as evidenced from increased frequencies of somatic cell chromosomal aberrations, micronuclei, and multinuclei (Fig. 3 and Supplementary Fig. S2). In addition to centrosome regulation, PLK1 plays a critical role in the formation of stable microtubule-kinetochore attachments, which are required for proper alignment of chromosomes, along with the dissociation of cohesion during chromosome segregation (1). Therefore, increased Plk1 expression may interfere with these key events, leading to mitotic aberrations and resultant CIN (Fig. 4).
Our data demonstrate that Plk1’s role as a potent proto-oncogene stems from its ability to enable cells to tolerate genomic chaos and proliferate, by overriding cell cycle checkpoints, including SAC and the DNA-damage checkpoint. We found that ATM is activated in the context of Plk1 overexpression, mainly owing to CIN induction (Supplementary Fig. S4A). However, activation of its downstream effector Chk2 was compromised. It has been reported that PLK1 deactivates Chk2 through phosphorylation of its FHA domain, which reduces its ability to bind to other proteins, as part of the mechanism of checkpoint inactivation (26,46,47). Likewise, the budding yeast polo-like kinase Cdc5 is essential for DNA damage checkpoint silencing in the presence of persisting double-strand breaks (48). In addition, Plk1 overexpression negatively regulates p53 expression and activity, which, in turn, contributes to compromised DDR and senescence pathways (Fig. 6C/D/E and Supplementary Fig. S5 and S6). PLK1 was also linked to inactivation of the ATR/Chk1 pathway directly through regulation of BRCA1 and Chk1, or indirectly via phosphorylation of the checkpoint mediator claspin (26). Given that PLK1 interacts with several components of DNA-damage pathways, it is not surprising that Plk1 overexpression causes robust inactivation of genomic surveillance mechanisms, allowing aneuploid cells to continue to proliferate.
Our results indicate that PLK1 has a profound effect on chromosomal stability. Plk1 transgenic PMEFs, PAEpiCs, and tumors display extensive CIN, mainly chromosomal gains and micronuclei accumulation, suggesting that PLK1 may be one of the upstream drivers of intratumor heterogeneity and cancer genome evolution. CIN offers the opportunity to select for cells with advantageous karyotypes (e.g., increased cell proliferation, metastatic potential, and/or drug resistance) that drive cancer development and progression.
Numerous studies have reported that PLK1 overexpression is associated with poor prognosis in a variety of human cancers. Additionally, studies have shown that PLK1 is closely associated with chemoresistance and PLK1 inhibition can overcome the drug resistance to anticancer drugs, such as gemcitabine (49), docetaxel (50), and doxorubicin (51). In this context, PLK1-induced CIN is likely one of the major underlying mechanisms. An elegant study by Strebhardt’s group reported that PLK1 inhibition compromises SAC and increases CIN (52), which suggests that PLK1 is an important cell cycle regulator whose expression and activity needs to be tightly regulated and that the precise level of its enzymatic activity is important for the correct execution of its cellular functions.
In conclusion, using our newly-generated Plk1 transgenic mice, we provide solid evidence that increased Plk1 expression drives spontaneous tumorigenesis in various types of tissues, which is strongly supported by clinical studies in humans. This oncogenic activity relies on PLK1’s induction of CIN and the overriding of cell cycle checkpoints, which leads to malignant transformation and tumor development. These findings not only establish PLK1 as a potent proto-oncogene and a CIN gene, but also guide the development of more effective and targeted treatment regimens across the landscape of PLK1-overexpressing and CIN-positive cancers.
Supplementary Material
Significance:
These findings establish roles for PLK1 as a potent proto-oncogene and a CIN gene and provide insights for the development of effective treatment regimens across PLK1-overexpressing and CIN-positive cancers.
Acknowledgements
This work was supported by grants from the American Cancer Society (ACS Research Scholar Grant 127626-RSG-15–005-01-CCG to Z.F.), and the National Institutes of Health (NIH R01 CA191002 to Z.F.). The authors are very grateful to the Transgenic and Knockout Core at Mayo Clinic for help with the generation of the Plk1 transgenic mice. Histological staining was in part performed by the Virginia Commonwealth University Cancer Mouse Models Core Laboratory, supported, in part, with funding to the Massey Cancer Center from NIH-NCI Cancer Center Support Grant P30 CA016059. Microscopy was performed at the VCU Massey Cancer Center Microscopy Core Facility and supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. We thank Heidi Sankala for editorial assistance with the manuscript.
Footnotes
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
- 1.Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Liu Z, Sun Q, Wang X. PLK1, A Potential Target for Cancer Therapy. Transl Oncol, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer, 2006. [DOI] [PubMed] [Google Scholar]
- 4.van Vugt MATM, van de Weerdt BCM, Vader G, Janssen H, Calafat J, Klompmaker R, et al. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Ivanov AI, Fisher PB, Fu Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, et al. Malignant Transformation of Mammalian Cells Initiated by Constitutive Expression of thePolo-like Kinase1. Biochem Biophys Res Commun, 1997. [DOI] [PubMed] [Google Scholar]
- 7.van Ree J, Zhou W, Li M, van Deursen JM. Transgenesis in Mouse Embryonic Stem Cells. Methods Mol Biol, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, et al. Mouse in red: Red fluorescent protein expression in mouse ES cells, embryos, and adult animals. genesis, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Fu Z, Wen D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers (Basel), 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai XP, Chen LD, Song H Bin, Zhang CX, Yuan ZW, Xiang ZX. PLK1 promotes epithelial-mesenchymal transition and metastasis of gastric carcinoma cells. Am J Transl Res, 2016. [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong SB, Im JH, Yoon J-H, Bui QT, Lim SC, Song JM, et al. Essential Role of Polo-like Kinase 1 (Plk1) Oncogene in Tumor Growth and Metastasis of Tamoxifen-Resistant Breast Cancer. Mol Cancer Ther, 2018. [DOI] [PubMed] [Google Scholar]
- 12.Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garfield AS. Derivation of primary mouse embryonic fibroblast (PMEF) cultures. Methods Mol Biol, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Vargas-Rondón N, Villegas VE, Rondón-Lagos M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers (Basel), 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luzhna L, Kathiria P, Kovalchuk O. Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Front Genet, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson PD, Nelson VR, Tesar PJ, Nadeau JH. Genetic factors on mouse chromosome 18 affecting susceptibility to testicular germ cell tumors and permissiveness to embryonic stem cell derivation. Cancer Res, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson PD, Delgado ER, Alencastro F, Leek MP, Roy N, Weirich MP, et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godinho SA, Pellman D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. B Biol. Sci, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Gheghiani L, Loew D, Lombard B, Mansfeld J, Gavet O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep, 2017. [DOI] [PubMed] [Google Scholar]
- 23.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol, 2007. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor A, Maffini S, Rainey MD, Kaczmarczyk A, Gaboriau D, Musacchio A, et al. Requirement for PLK1 kinase activity in the maintenance of a robust spindle assembly checkpoint. Biol Open, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumura S, Toyoshima F, Nishida E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Shaltieì IA, Krennin L, Bruinsma W, Medema RH. The same, only different – DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci, 2015. [DOI] [PubMed] [Google Scholar]
- 27.Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyun SY, Hwan HI, Jang YJ. Polo-like kinase-1 in DNA damage response, BMB Rep, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’brate A, Giannakakou P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Bruinsma W, Aprelia M, Kool J, Macurek L, Lindqvist A, Medema RH. Spatial Separation of Plk1 Phosphorylation and Activity. Front Oncol, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto Y, Matsuyama H, Kawauchi S, Matsumoto H, Nagao K, Ohmi C, et al. Overexpression of Polo-Like Kinase 1 (PLK1) and Chromosomal Instability in Bladder Cancer. Oncology, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Donizy P, Halon A, Surowiak P, Kaczorowski M, Kozyra C, Matkowski R. Augmented expression of polo-like kinase 1 is a strong predictor of shorter cancer-specific overall survival in early stage breast cancer at 15-year follow-up. Oncol Lett, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tut TG, Lim SHS, Dissanayake IU, Descallar J, Chua W, Ng W, et al. Upregulated polo-like kinase 1 expression correlates with inferior survival outcomes in rectal cancer. PLoS One, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Bossche J, Deben C, de Beeck KO, Deschoolmeester V, Hermans C, Pauw I De, et al. Towards prognostic profiling of non-small cell lung cancer: New perspectives on the relevance of polo-like kinase 1 expression, the TP53 mutation status and hypoxia. J Cancer, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldman M, Craft B, Kamath A, Brooks A, Zhu J, Haussler D. The UCSC Xena Platform for cancer genomics data visualization and interpretation. bioRxiv, 2018. [Google Scholar]
- 37.Lee EYHP, Muller WJ. Oncogenes and tumor suppressor genes. Cold Spring Harb Perspect Biol, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Liu J, Li J, Kong Y, Sandusky G, Rao X, et al. Polo-like kinase 1 (Plk1) overexpression enhances ionizing radiation-induced cancer formation in mice. J Biol Chem, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Cárcer G, Venkateswaran SV, Salgueiro L, El Bakkali A, Somogyi K, Rowald K, et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat Commun, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen C, Krohn J, Bhattacharya S, Davies B. A comparison of exogenous promoter activity at the ROSA26 locus using a ΦiC31 integrase mediated cassette exchange approach in mouse ES cells. PLoS One, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoevenaar WHM, Janssen A, Quirindongo AI, Ma H, Klaasen SJ, Teixeira A, et al. Degree and site of chromosomal instability define its oncogenic potential. Nat Commun, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Ding J, Meng L. Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cisowski J, Sayin VI, Liu M, Karlsson C, Bergo MO. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene, 2016. [DOI] [PubMed] [Google Scholar]
- 44.Ueda A, Oikawa K, Fujita K, Ishikawa A, Sato E, Ishikawa T, et al. Therapeutic potential of PLK1 inhibition in triple-negative breast cancer. Lab Investig, 2019. [DOI] [PubMed] [Google Scholar]
- 45.Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, et al. Polo-like Kinase 1: A Potential Therapeutic Option in Combination with Conventional Chemotherapy for the Management of Patients with Triple-Negative Breast Cancer. Cancer Res, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Vugt MATM, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong S-E, et al. A Mitotic Phosphorylation Feedback Network Connects Cdk1, Plk1, 53BP1, and Chk2 to Inactivate the G2/M DNA Damage Checkpoint. Lichten M, editor. PLoS Biol, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell,1997. [DOI] [PubMed] [Google Scholar]
- 49.Song B, Liu XS, Rice SJ, Kuang S, Elzey BD, Konieczny SF, et al. Plk1 phosphorylation of Orc2 and Hbo1 contributes to gemcitabine resistance in pancreatic cancer. Mol Cancer Ther, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al Nakouzi N, Cotteret S, Commo F, Gaudin C, Rajpar S, Dessen P, et al. Targeting CDC25C, PLK1 and CHK1 to overcome Docetaxel resistance induced by loss of LZTS1 in prostate cancer. Oncotarget, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sero V, Tavanti E, Vella S, Hattinger CM, Fanelli M, Michelacci F, et al. Targeting polo-like kinase 1 by NMS-P937 in osteosarcoma cell lines inhibits tumor cell growth and partially overcomes drug resistance. Invest New Drugs, 2014. [DOI] [PubMed] [Google Scholar]
- 52.Raab M, Sanhaji M, Matthess Y, Hörlin A, Lorenz I, Dötsch C, et al. PLK1 has tumor-suppressive potential in APC- truncated colon cancer cells, Nat Commun, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.