

Abstract
The polo-like kinases (PLKs) are a family of serine/threonine kinases traditionally linked to cell cycle regulation. A structurally unique member of this family, PLK4, has been shown to regulate centriole duplication during the cell cycle via interactions with a variety of centrosomal proteins. Recent findings suggest that PLK4 is overexpressed in various human cancers and associated with poor cancer prognosis. Although several studies have shown that PLK4 inhibition may lead to cancer cell death, the underlying mechanisms are largely unknown. In this review, we discuss the structure, localization and function of PLK4, along with the functional significance of PLK4 in epithelial cancers and some preliminary work suggesting a role for PLK4 in the key cancer progression process epithelial-mesenchymal transition (EMT). We also discuss the potential of PLK4 as a druggable target for anti-cancer drug development based on critical analysis of the available data of PLK4 inhibitors in pre-clinical development and clinical trials. Overall, the emerging data suggests that PLK4 plays an essential role in epithelial cancers and should be further explored as a potential biomarker and/or therapeutic target. Continued detailed exploration of available and next-generation PLK4 inhibitors may provide a new dimension for novel cancer therapeutics following successful clinical trials.
Keywords: PLK4, Centriole, Centrosome Amplification, Cancer, EMT
Introduction
To remain healthy and duplicate efficiently, mammalian cells must undergo DNA replication and cell division in a strictly defined and tightly controlled manner, as any error in the normal choreography of cell cycle regulation may result in genomic instability or cell death (1). Genomic stability is in part maintained by centrosomes, which are membraneless organelles composed of two centrioles surrounded by a pericentriolar protein matrix. Chromosomal instability (CIN), characterized by a loss of proper rearrangement of chromosomes, is the predominant form of genomic instability. It is characterized by high rates of centrosome gain and loss. Errors in mitosis are attributable to CIN and include defects in the spindle assembly checkpoint, sister chromatid cohesion, and kinetochore-microtubule attachments (2). DNA damage and activation of the tumor suppressor TP53 can be consequences of these mitotic errors (3). Centrosomes function in the organization of microtubules, maintenance of cell shape, as well as polarity and motility. Each centrosome duplicates once per cell cycle, in a tightly regulated and coordinated process to ensure successful chromosomal segregation (4). Centrosome amplification (CA) refers to the presence of more than two centrosomes in any cell, resulting in genomic instability, one of the hallmark features of cancer cells (reviewed in (5)). However, the mechanisms associated with the prevalence of CA in cancer, and its therapeutic value are not fully understood.
The evolutionarily conserved polo-like kinase (PLK) family of serine-threonine kinases (PLKs 1–5) is involved in regulation of multiple cellular processes, including centrosome function (6–8). PLKs are characterized by an N-terminal catalytic domain and the presence of either one (PLK4) or two (PLKs 1–3, 5) highly conserved non-catalytic C-terminal polo boxes that comprise the polo-box domain (PBD; Figure 1), which are involved in subcellular localization, target binding, and cis-acting regulation of activity by binding to the catalytic domain (9). A structurally unique member of this family, PLK4, is intimately involved in the regulation of centrioles and centrosomes, as it localizes to centrioles, regulates centriole duplication during cell cycle progression (10), and autophosphorylates to promote its destruction to limit centriole duplication once per cell cycle (7). PLK4 plays an essential role in centriole duplication via interacting with the family of centriolar proteins (CEPs) (10) and other proteins. As centrosome aberrations are frequently observed in cancer, the central role of PLK4 in centriole replication suggests its possible involvement in tumorigenesis. Moreover, dysregulation of PLK4 causes loss of centrosome numerical integrity that promotes genomic instability and may sensitize cancer cells to targeted PLK4 inhibition. Additionally, studies have found increased PLK4 expression in several cancers and its involvement in tumorigenesis, cancer metastasis and the response to chemotherapy, suggesting that it may be a target for anti-cancer drug development.
Figure 1.
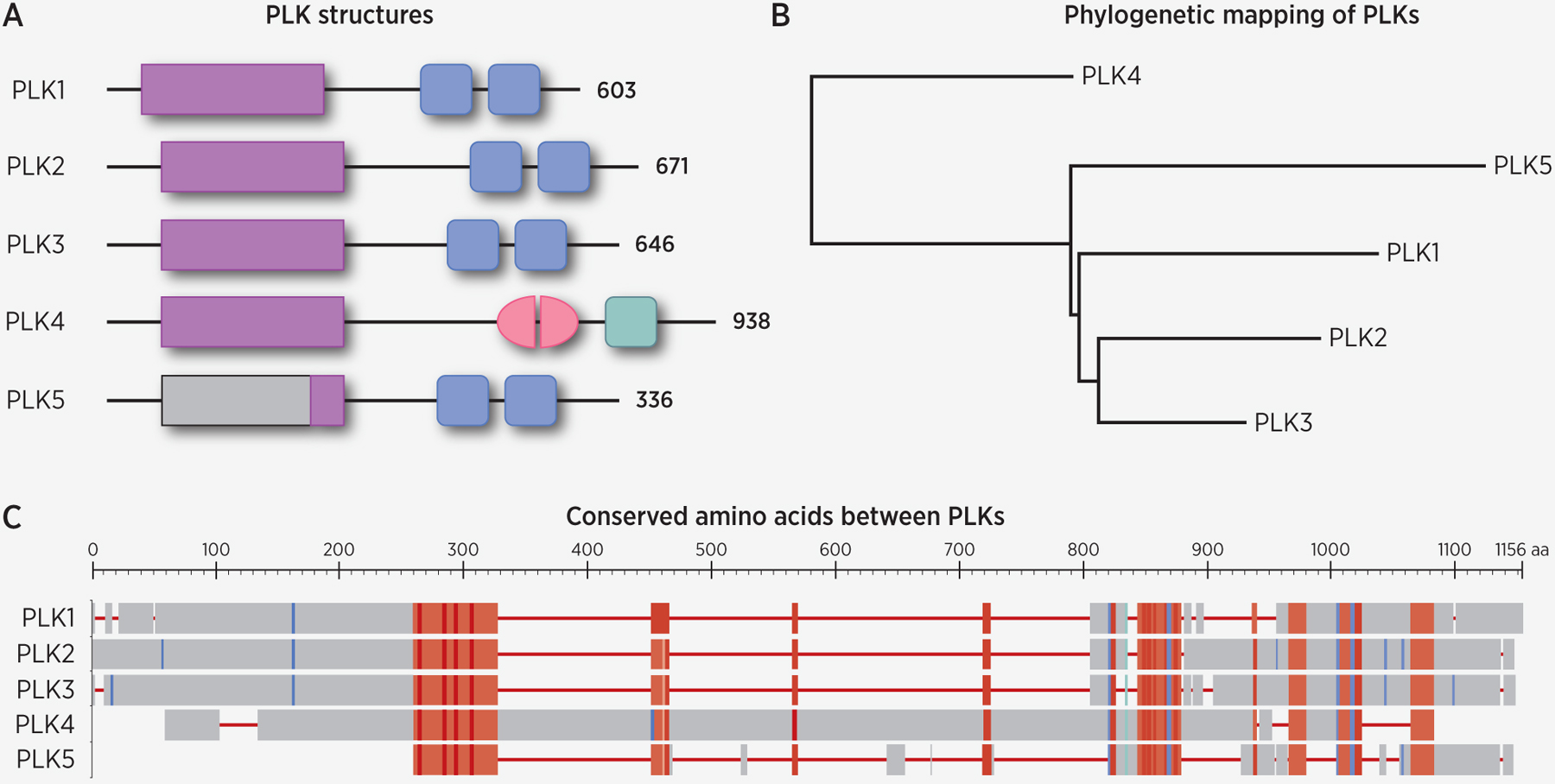
Comparisons of the mammalian polo-like kinases (PLKs). A, Mammalian PLKs have conserved kinase domains (purple rectangles) and polo box domains (PBD, blue/green squares). PLK4 is the most structurally divergent member of this family, as it contains only one PBD whose sequence is not fully homologous to the other PBDs, and one cryptic polo box (pink half-ovals). In humans, PLK5 contains an in-frame stop codon followed almost immediately by an in-frame start codon, resulting in a truncated kinase domain (purple rectangle). In all other mammals, the full PLK5 gene is encoded (light grey rectangle and line), although it shows no kinase activity. B, Phylogenetic analysis of PLK sequences show PLK4 as the most divergent, as determined using the COBALT Phylogenetic Tree widget (76). C, Alignment of PLK protein sequences using COBALT tool. Red areas are highly conserved, while blue areas are less conserved. Phylogenetic tree and alignment made using NCBI’s Constraint-based Multiple Alignment Tool and the following sequence IDs: NP_001177728.1, NP_001230008.1, NP_005021.2, NP_004064.2, and NP_001239155.1 (76).
Here we summarize recent progress on determination of the role and potential significance of PLK4 in cancer. Our review first describes the basic structure, localization and functions of PLK4 as well as its interactions with specific key proteins. Next, we discuss the roles of PLK4 in centrosome amplification and epithelial cancers. This is followed by an analysis of available data regarding links between PLK4 and epithelial-mesenchymal transition (EMT), responsible for cancer progression and metastasis. Finally, we discuss the recent progress on targeting PLK4 for cancer management focusing on current PLK4 inhibitors and recent pre-clinical and clinical developments.
Structure, Localization, Interactions and Function of PLK4
PLK4 is the most structurally divergent member of the PLK family, unlike the other members of this family, which have two PBDs, it contains three PBDs, out of which two are non-canonical and together form a cryptic polo-box (CPB) (Figure 1). PLK4 has been shown to interact with several proteins, both directly and indirectly (Table 1), to serve as a master regulator of centriole duplication. Through its CPB, PLK4 homodimerizes in an intermolecular manner, and it is responsible for binding to the partner CEP centrosomal proteins (CEPs 152 and 192) and other centriolar proteins to allow centriolar targeting of PLK4 to the proximal base of each parental centriole and downstream events (11,12). Additionally, PLK4 possesses three PEST sequences that are rich in proline (P), aspartate (D), glutamate (E), serine (S) and threonine (T) residues which are crucial in controlling the stability of the kinase (13). The initiation of centriole duplication relies not only on PLK4 but a core of components including CEP192, CEP152 and CEP135, the spindle assembly 6 homolog protein, SAS-6, the SCL/TAL1 interrupting locus protein, STIL, and the centrosome protein CPAP (14) (Table 1, Figure 2). The activity and levels of these core components in human cells must be tightly regulated to maintain the correct centriole number. Either overexpression or depletion of these proteins can result in the formation of multiple centrioles or a blockade in centriole formation (15).
Table 1.
Proteins directly or indirectly regulated by PLK4 that are essential to the centriole duplication process.
| Protein(s) | Interaction | Function | Reference(s) |
|---|---|---|---|
| CEP63 | Complexes with CEP152 |
|
(67,68) |
| CEP85 | Binds to STIL |
|
(24,69) |
| CEP131 | Phosphorylated by PLK4 |
|
(46,47) |
| CEP135 | Binds to CPAP |
|
(70) |
| CEP152 CEP192 | Bind to PLK4 |
|
(71) |
| SAS-6 | Binds to STIL after phosphorylation of STIL by PLK4 |
|
(21) |
| STIL | Binds to PLK4 |
|
(21,72) |
| CENPJ/CPAP | Interacts with centriole proteins and binds STIL |
|
(21,73) |
| FBXW5 | Phosphorylated by PLK4 to suppress ability to ubiquitylate SAS-6 |
|
(74,75) |
Figure 2.
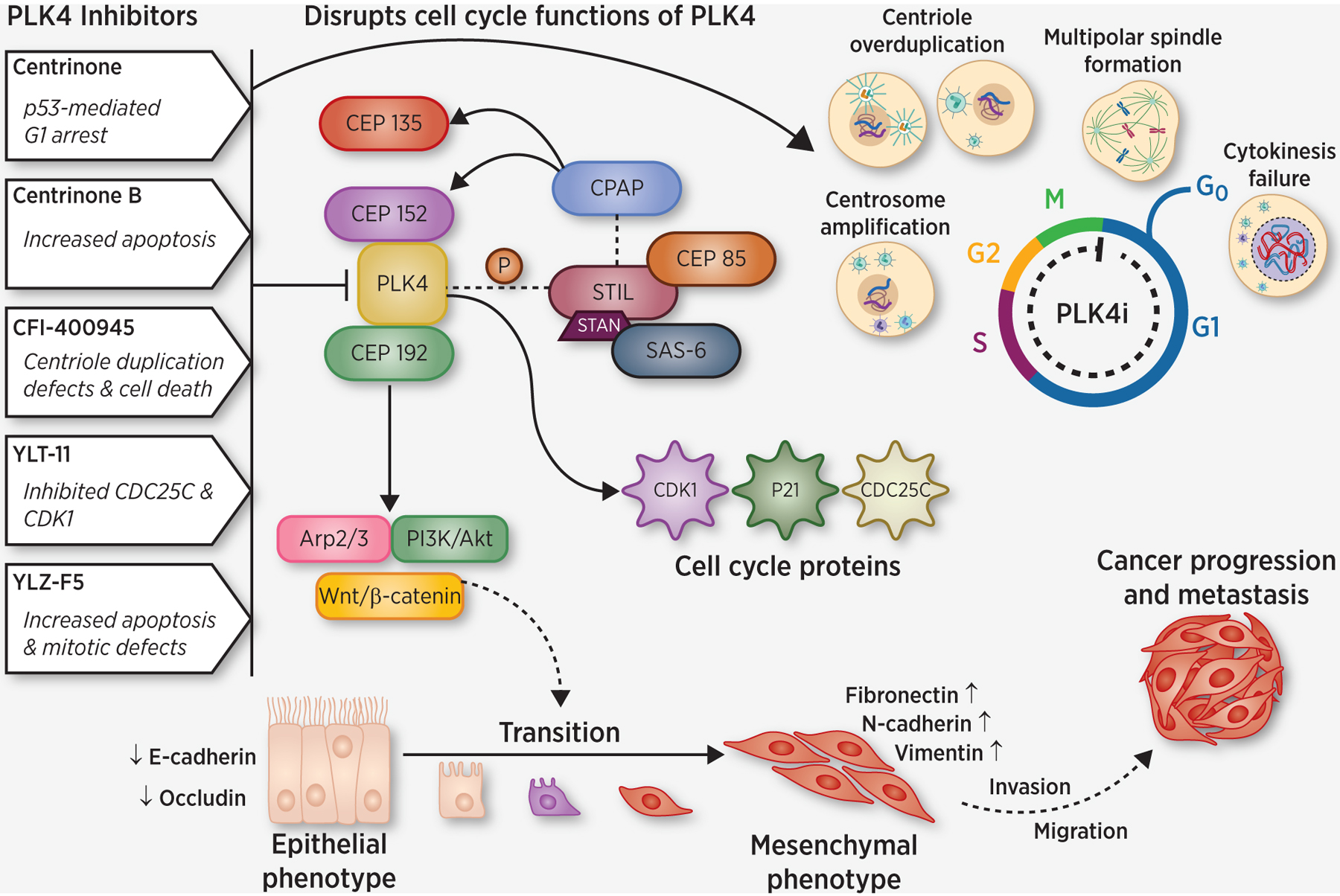
Schematic representation of PLK4 signaling in cancer with details of selected PLK4 inhibitors. Various PLK4 inhibitors along with their potential mechanisms to inhibit cancer progression are shown. PLK4 modulation, either by chemical/genetic inhibition, or overexpression, can disrupt cell cycle functions of PLK4, including induction of centrosome amplification through the simultaneous generation of multiple procentrioles adjoining each parental centriole during S phase of cell cycle leading to cancer progression. Various cellular proteins interact and complex with PLK4, leading to PLK4-mediated disruptions to cell cycle. Recent findings also suggest that PLK4 signaling is associated with epithelial-mesenchymal transition (EMT), which is well known to be linked to tumor invasion and metastasis. Visualization initially created with BioRender and professionally redrawn by the journal.
PLK4 levels in human cells are regulated by the SCF-Slimb/βTrCP-E3 ubiquitin ligase complex via a conserved phosphodegron motif commonly found in these substrates (7). Recently, it has been suggested that endoplasmic reticulum stress induces activating transcription factor 6 (ATF6) and CCAAT/enhancer-binding protein β (C/EBPβ) binding, which may inhibit transcription activity of the PLK4 gene (16). In either event, transcription of PLK4 is cell cycle-dependent, where transcript levels are undetectable in G0 and begin to rise in G1 with maximal levels detected during mitosis. Crucial to the regulation of PLK4 protein degradation is phosphorylation in the degron at two residues, Ser 293 and Thr 297. The degradation of PLK4, through autophosphorylation after homodimerization, is limited by its activity; thus, it is considered a suicide kinase (7). Mutations of the degron motif in PLK4 have given rise to stability of the protein along with centrosome amplification. As reviewed by Bose and Dalal (17), PLK4 is present as a ring around the mature centriole and coalesces into a single point, potentially through two mechanisms: one in which the stability of PLK4 is increased through formation of a complex with STIL and SAS-6 (17) and the other through self-assembly of PLK4 into a ring around the mother centriole through condensation-mediated self-organization (18). The complex formation is triggered by PLK4 phosphorylation of STIL within the STAN motif to activate STIL, leading to the centriolar recruitment of SAS-6, which marks the site for procentriole biogenesis and cartwheel assembly (a stack of ring-type symmetrical assemblies, providing the structural foundation for the procentriole) initiation, which is followed by centriole elongation (19,20). It has also been proposed that PLK4 phosphorylates STIL at an additional site, S428, to promote the binding of STIL to CPAP consequently linking the cartwheel to microtubules of the centriole wall (21).
PLK4, STIL and SAS-6 are all regulated by proteasomal degradation (22) since divergent expression or stabilization of these proteins can lead to supernumerary centrioles, while excessive centriole duplication can be curbed by their depletion (23). At the centriole, PLK4 can both inhibit nearby PLK4 molecules and undergo auto-activation, with its activity being further enhanced through the complex formation (18). More recently, it has been proposed that CEP85 is a regulator of PLK4 during early centriole duplication by associating with STIL during procentriole assembly (24). Although more work is needed to confirm the relationship, the phosphatase CDC25C and the RhoA guanine exchange factor (GEF), Ect2, are suggested to be mitotic substrates for PLK4. PLK4 co-localizes with CDC25C to the centrosome (25) and controls the association of Ect2 to the central spindle as it is dependent on the activity of PLK4 (13,26). One study investigated the subcellular distribution of autophosphorylated PLK4 and found it to localize to kinetochores, the central spindle midzone and midbody in different cell-cycle phases (27), which reveals the extent to which PLK4 may be involved past centriole duplication. While PLK4 has the ability to self-regulate, other proteins have proven to be required for this to occur, such as the centrosomal ubiquitin ligase TRIM37. It has been demonstrated that the expression levels of TRIM37 are indicative of the degree of response to PLK4 inhibition in certain cancers; elevated TRIM37 promotes cancer-specific sensitivity to PLK4 inhibition (28). This intricate control of the functions of PLK4 in the centrosomal makeup underscores the importance of this kinase in controlling the process of centriole and centrosome duplication, essential parts of maintaining the integrity of a healthy cell and its error-free division.
PLK4 in Centrosome Amplification
Centrosomes are comprised of two centrioles that are embedded in a matrix of proteins known as the pericentriolar material (PCM) which includes factors that nucleate and anchor microtubules, essential structures for multiple cellular functions. Indeed, the centrosome is known as the microtubule organizing center (MTOC) in most eukaryotic cells and is duplicated once per cell cycle in healthy cells to ensure equal dissemination of genetic material to the two daughter cells (29). Centrosome amplification (CA) is the acquisition of three or more centrosomes in one cell and has been reported in most human cancers, leading to its designation as a hallmark of cancer (5). When centriole duplication is deregulated, multipolar spindles may develop, leading to multipolar mitosis and promotion of genomic instability through aneuploidy (30). CIN is a hallmark of several human cancers, and several studies have shown that CA is one factor which contributes to CIN. Interestingly, about 75% of malignant breast tumors exhibit CA, albeit with wide variability for intratumoral cells (31). CA was also found to be strongly correlated with increasing tumor grade and stage of breast cancers, and was more pronounced in triple-negative and HER2 amplified subtypes (32). Triple-negative breast cancer (TNBC) has been found to possess extreme CA, which may be an underlying factor in its aggressive phenotype. Additionally, cells possessing supernumerary centrosomes in TNBC had a higher migration velocity than those with typical centrosomes (33). This supports the argument for a strong role of CA and CIN as potential cancer drivers, and PLK4, as a mediator of centrosome duplication, likely plays an important role in this process.
In mammalian cell lines, overexpression of PLK4 has been shown to induce centriole overduplication and form multiple pro-centrioles on a single mother centriole (34), while PLK4 knockdown has been shown to cause a loss of centrioles (35). Interestingly, use of the PLK4 small molecule inhibitor CFI-400945 was found to induce centriole overduplication at low concentrations, likely due to partial kinase activity, while increased inhibitor doses fully depleted centrioles and reduced centrosome number (36). Another research group discovered that elevated PLK4 expression led to CA and aneuploidy in vivo and accelerated the onset of intestinal tumors along with spontaneous tumorigenesis (34). They also evaluated the degree of aneuploidy and genome instability in tumors resulting from CA with PLK4 overexpression and found that all the tumors showed evidence of aneuploidy and chromosomal abnormalities. These findings indicate a central role for PLK4 in CA, but further work is required for a deeper understanding of the types of cancer that may exhibit these traits.
PLK4 in Epithelial Cancers
Although the role of PLK4 has been studied in several cancers (37–40), to narrow our scope for this review, we have focused on epithelial cancers. Although PLK4 has not been found to be commonly mutated in human neoplasms, due to its frequently aberrant expression and its importance in regulating the centriole and centrosome duplication cycle and links to many other crucial proteins, researchers have focused on PLK4 as a potential cancer biomarker. For example, Li et al found increased mRNA expression of PLK4 in breast cancer tissues and an association of high PLK4 expression with lymph node and distant metastasis, suggesting it may be a potential prognostic factor of breast cancer (41). A similar trend was shown in non-small cell lung cancer in a study by Zhou et al (42). The authors found that increased PLK4 expression was associated with higher TNM stage, metastasis, and larger tumor size in clinical tumor specimens, as well as lower disease-free and overall survival in patients. Thus, PLK4 appears to be an independent predictive factor for poor prognosis in multiple epithelial cancers.
Interestingly, inhibition of PLK4 has been shown to impair centriole duplication and enhance genomic instability of cancer cells, resulting in cell death or cell cycle arrest (43). As discussed below, a few studies have evaluated the effect of PLK4 inhibition, via genetic manipulation or small molecule inhibitors, on cancer cells. In one study, breast cancer cells were found to have higher levels of PLK4, and inhibition reduced cell growth in vitro and tumor growth in vivo (36). Very recently, a group of researchers demonstrated that PLK4 inhibition resulted in mitotic catastrophe via centrosome depletion in human breast adenocarcinoma cells overexpressing TRIM37 (44). Further, Shinmura et al found that PLK4 is upregulated and associated with CA and CIN in a subset of studied human gastric cancers (45). More recently, a PLK4 genetic variant was found to be associated with a higher risk of liver cancer, and its overexpression increased cell proliferation, migration and invasion, while knockdown did the opposite (38). Lastly, the overexpression of CEP131, which is a substrate of PLK4 (46), was found to promote CA in colon cancer (47).
Further, it has been shown that supernumerary centrosomes are sufficient on their own to drive aneuploidy and the development of spontaneous tumors in multiple tissues, and that tumors arising from this mechanism exhibit frequent mitotic errors (34). In the skin, it was found that PLK4 overexpression led to CA and cutaneous barrier defects in developing mice, and cessation of PLK4 overexpression curbed the development of cancer (48). In TP53 knockout mice, concomitant PLK4 overexpression rescued skin barrier defects, but mice developed cancer even though the PLK4 overexpression was short term. In another impressive study, Levine et al used a mouse model of inducible PLK4 overexpression and found that the mice overexpressing PLK4 exhibited thickened epidermis and disrupted follicles, as well as progressive hair loss. These mice also developed spontaneous squamous cell carcinoma (SCC), even with transient overexpression of PLK4, and in the skin tissues, CA was found to be correlated with elevated PLK4 (34). Together, there is ample evidence suggesting that PLK4 is involved in cancer development or progression and may be a valid target to investigate the management and treatment of epithelial cancers.
PLK4 in Epithelial-to-Mesenchymal Transition (EMT)
Centrosome amplification has been reported to cause chromosomal instability and promote invasion of cancer cells through changes in cell polarity (49). Further, some of the centrosome/mitotic regulators causing centrosome amplification have been suggested to mediate cell invasion and EMT (50,51). EMT plays a key role in cancer progression when cancerous cells become invasive and metastatic by losing epithelial characteristics while acquiring mesenchymal characteristics (reviewed in (52)). Interestingly, along with the previously discussed research documenting the functional significance of PLK4 in various cancers, recent data suggest the involvement of PLK4 in EMT and metastasis of certain other cancer types. PLK4 has been shown to be overexpressed and associated with enhanced tumor size, lymph node metastasis and EMT in colorectal cancer (CRC) (53). Further, in vitro studies showed that PLK4 overexpression in CRC cells activated the Wnt/β-catenin pathway and modulated the expression of several EMT-associated proteins, including upregulation of N-cadherin and snail and downregulation of occludin (Figure 2). Additionally, PLK4 knockdown inhibited the Wnt/β-catenin pathway in CRC cells in vitro and suppressed the growth of xenograft tumors in nude mice (53). Together, these findings lead us to suggest a previously undefined mechanism associating PLK4 with EMT progression in colorectal cancer.
Similar results were found in neuroblastoma (NB), where PLK4 expression was remarkably increased in NB tissues and high PLK4 levels were negatively correlated with survival, suggesting PLK4 could have oncogenic functions in NB (54). Further, shRNA-mediated downregulation of PLK4 suppressed migration as well as invasion and promoted apoptosis in NB cells. Interestingly, downregulation of PLK4 in NB cells inhibited EMT, as the expression of epithelial marker E-cadherin was upregulated and expression of mesenchymal markers (N-cadherin, vimentin and Slug) were downregulated along with p-Akt. Further, LY294002, a specific inhibitor of the PI3K pathway, was shown to inhibit p-Akt and modulate EMT markers in PLK4-overexpressing NB cells, suggesting that PLK4 could be involved in regulation of the EMT process via the PI3K/Akt pathway. Moreover, the downregulation of PLK4 in SK-N-BE(2) neuroblastoma cells was found to dramatically suppress tumorigenesis and metastasis in nude mice in vivo (54). Overall, these results suggest that PLK4 promotes EMT through activation of the PI3K/Akt signaling pathway, and that PLK4 may serve as a potential therapeutic target for NB.
Evidence also exists for a role of PLK4 in promoting EMT in breast cancer. In one study, PLK4 was shown to promote cancer invasion and metastasis (55). In this study, the authors demonstrated that shRNA-mediated PLK4 knockdown inhibited cancer invasion and promoted an epithelial phenotype by increased expression of E-cadherin along with loss of expression of mesenchymal markers (fibronectin and N-cadherin) in poorly differentiated breast cancer cells. Further, utilizing murine xenografts of PLK4-depleted human breast cancer cells (MDA-MB-231), this study found that although there were minimal effects on primary tumor growth, a 25–45% reduction in PLK4 expression was able to completely inhibit invasive and metastatic progression of the breast cancer xenografts in vivo. Interestingly, the authors identified phosphorylation of actin related protein 2 (Arp2) at T237/T238 by PLK4 as a potential mediator of PLK4-related activity promoting migration and invasion (55). This study suggested that PLK4 inhibition may be a promising breast cancer therapy to suppress invasion and metastasis, while determination of Arp2 status in patients may be useful for the rational selection of patients for PLK4 inhibitor-based therapy. Overall, emerging data suggest an involvement of PLK4 in EMT, however, significantly more research is needed to validate these observations to put forth PLK4 as a key EMT player affecting the invasiveness and metastatic potential of certain cancers.
PLK4 as a Druggable Target
The important role of PLK4 in centrosome biology coupled with its demonstrated connection with cancer development and progression signaling provides a rationale for exploring PLK4 as a druggable target for cancer management. Although this field is still emerging, there are multiple agents currently in pre-clinical and clinical trials assessing the potential of PLK4 as a target for anti-cancer drug development. Below, we discuss some of these agents and their progress towards pre-clinical and clinical development. These important small molecule inhibitors are shown in Figure 2, along with their potential mechanisms of action. A brief account of PLK4 small molecule inhibitors at different stages of anti-cancer drug development is provided below.
Pre-clinical development
Centrinone
Centrinone is a reversible and selective PLK4 inhibitor [inhibition constant (Ki) = 0.16 nM in vitro] developed on a template of the pan-Aurora kinase inhibitor VX-680, which was also found to inhibit PLK4 (56). This compound was modified via a methoxy substituent at the C5 position to target the relatively unique hinge methionine (Met 91) in PLK4. Optimization and characterization of several analog inhibitors produced two highly selective PLK4 inhibitors with robust cellular activity, one of which was named centrinone. This compound exhibited >1000-fold selectivity for PLK4 over Aurora A and/or B in vitro and did not affect cellular Aurora A and/or B substrate phosphorylation at concentrations that deplete centrosomes. This small molecule blocks centriole duplication and depletes centrosomes at nanomolar concentrations by preventing centriole assembly.
In one recent study, centrinone was found to induce cell death in both TP53-wild type and mutant Ewing’s sarcoma cells, which was associated with G2/M cell cycle arrest and DNA fragmentation (57). In in vitro experiments in HeLa cells, centrinone treatment was found to cause a TP53-mediated cell cycle arrest in the G1 phase suggesting a decrease in proliferation rate. However, some other cancer cell lines continued to proliferate even after centrosome loss due to centrinone treatment, suggesting an intrinsic set point for centrosome number. Further, centrinone treatment was shown to block centriole duplication, leading to progressive loss of centrosomes as cells divide. Most cell lines with cancer-associated mutations (frequently in TP53) continued to proliferate after centrinone-mediated centrosome removal, albeit at a reduced rate because of an increase in mitotic errors (56). In contrast to cancer-derived cell lines, retinal pigmented epithelial cells (RPE1) and three primary cell cultures exhibited irreversible G1 arrest after centrinone-induced centrosome loss. Overall, this study suggested that in these tissues, centrinone-mediated centrosome depletion may be insufficient for cancer therapy alone and for best effect centrinone may need to be combined with other targeted therapies (56).
Centrinone-B
As the second selective PLK4 inhibitor developed alongside centrinone, centrinone-B was also found to have >1000-fold selectivity over Aurora A/B (Ki = 0.6 nM in vitro and centrosome depletion at 500 nM) (56). A study from our laboratory has shown that treatment with centrinone-B resulted in significant decrease in cell viability and an increase in apoptosis in TP53 wild-type human melanoma cell lines (40), suggesting a pro-proliferative function of PLK4 in melanoma. The effects of centrinone-B on melanoma cells were found to be due to selective PLK4 inhibition, as exogenous expression of the centrinone-B–resistant mutant (PLK4G95L) partially reversed the centrinone-B–mediated inhibition of melanoma cell proliferation. This study also suggested that PLK4 is required for centriole overduplication in melanoma, as inhibition of PLK4 with centrinone-B reduced CA in melanoma cell lines. Further, to test the hypothesis that CA could be used as a biomarker for sensitivity to PLK4 inhibition, this study also correlated centriole numbers with sensitivity to centrinone-B in the melanoma cell lines. Additionally, this study utilized inducible PLK4 overexpression in MCF10A and RPE1 immortalized, non-transformed human cell lines to model CA. These cells were treated with various concentrations of centrinone-B followed by an assessment of cell viability. However, no significant correlation was observed between CA and sensitivity to PLK4 inhibition by centrinone-B, suggesting that CA is not an independent predictive biomarker of sensitivity to PLK4 inhibition (40). Although more research is needed in this and other cancers, centrinone-B appears to be a selective small-molecule PLK4 inhibitor with significant anti-proliferative effects against melanoma cells.
YLT-11
YLT-11 is a small-molecule PLK4 inhibitor characterized by an (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine skeleton (58). Lei et al evaluated antineoplastic activity and the possible mechanism of YLT-11 against human breast cancer in vitro and in vivo (59). In this study, YLT-11 was shown to fit well into the ATP pocket of PLK4, suggesting its potential as a novel PLK4 inhibitor. Further, YLT-11 was shown to selectively inhibit PLK4 enzymatic activity >200-fold over other PLK family members (PLK1, PLK2, and PLK3) with an IC50 of 22 nM. It was also found that at low concentrations (≤0.25 μM), YLT-11 showed an increase in centriole number while at high concentrations (≥0.5 μM), a decrease in centriole number was observed. This reduction might be attributable to the level of inhibition of PLK4 activity i.e. full inhibition versus partial inhibition. Interestingly, YLT-11 significantly decreased the viability of a panel of human breast cancer cell lines, including TNBC cell lines, with a weak inhibitory activity on the proliferation of normal mammary cells. YLT-11 has been shown to cause mitotic exit without proper segregation of sister chromatids, leading to cytokinesis failure. Although it is not clear, the effects of YLT-11 could be resulting from inhibition of both PLK4 and Aurora B. With the encouraging in vitro data, further investigation into the anti-cancer effects of YLT-11 was conducted in breast xenograft models. At an oral dose of 90 mg/kg, YLT-11 showed a 60% reduction in tumor growth in TNBC xenograft model, without any appreciable toxicity to animals. Further, this in vivo study showed that YLT-11 inhibited tumor cell proliferation and induced apoptosis, mainly associated with the inhibition of cell cycle-associated proteins such as CDC25C and CDK1 as well as an increase in the level of P21. Overall, this study suggested that YLT-11, a selective PLK4 inhibitor, has an appreciable anti-proliferative response in vitro as well as anti-tumor activity in vivo in human breast cancer xenograft without any significant toxicity (59), and needs to be further assessed in detail.
YLZ-F5
YLZ-F5 is another PLK4 small-molecule inhibitor formulated as a derivative of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine. Very recent in vitro pre-clinical evaluation (60) found that YLZ-F5 inhibited ovarian cancer cell proliferation and colony formation ability, as well as induced apoptosis by activating caspases 3 and 9. Moreover, YLZ-F5 inhibited PLK4 phosphorylation and caused aberrant centriole duplication and promoted the accumulation of ovarian cancer cells with mitotic defects (>4n DNA content). YLZ-F5 also markedly inhibited the migration of ovarian cancer cells (60). Overall, these results suggest that YLZ-F5 is a PLK4 inhibitor with considerable anti-proliferative activity in vitro. However, these observations need to be further validated in in vivo models and other cancers.
CFI-400945
CFI-400945 is a first-in-class, orally available, potent PLK4 inhibitor. This compound is currently licensed by Treadwell Therapeutics, but was identified through an academic drug discovery program at the Campbell Family Institute (CFI) for breast cancer research at the University Health Network in Toronto, Canada. CFI-400945 is an optimized, indolinone-derived, selective ATP-competitive kinase inhibitor that has been shown to selectively inhibit PLK4 over other members of PLK family, with an IC50 value of 2.8 ± 1.4 nM. However, some other protein kinases have also been shown to be inhibited by CFI-400945 at a much higher concentration (IC50 >100 nM) (36). Further, CFI-400945 has also been shown to inhibit members of the aurora kinase (AURK) and tropomyosin receptor kinase (TRK) families, albeit at higher concentrations than PLK4 (61).
In vitro studies in U2OS osteosarcoma cells and MDA-MB-468 and MDA-MB-231 breast cancer cell lines demonstrated that CFI-400945 treatment resulted in significant decrease in centriole numbers, leading to centriole duplication defects and cell death. Interestingly, like YLT-11, CFI-400945 was also found to produce bimodal effects on centriole number, with increased centriole numbers at lower doses while suppressing centrosome duplication at higher doses. In CFI-400945, this phenomenon was attributed to the self-regulation pattern of PLK4, as at low concentrations of CFI-400945, partial inhibition of PLK4 was not enough for its degradation, though it was sufficient to cause substrate phosphorylation leading to an increase in PLK4 levels and centriole number. In contrast, higher concentrations of CFI-400945 completely blocked PLK4 activity, thereby inhibiting centriole duplication (36). In additional studies, CFI-400945 has been shown to impair proliferation, survival, migration, and invasion, as well as induce apoptosis, senescence, and polyploidy of malignant rhabdoid tumor (MRT), atypical teratoid rhabdoid tumors (AT/RT) and medulloblastoma (MB) tumor cells while sparing non-tumor human fibroblasts (62,63). In a very recent study, CFI-400945 was also found to elicit anti-proliferative activity as well as apoptosis-type cell death and polyploidy in Ewing’s sarcoma cells (57).
Several in vivo studies with CFI-400945 have been performed in multiple tumor xenograft models. CFI-400945 treatment was found to be efficacious in reducing tumors in pancreatic (64), breast (36), lung (65), and AT/RT (63) xenograft models. Lohse et al have shown that CFI-400945 treatment reduced tumor growth and prolonged survival in four out of six human pancreatic cancer xenograft models studied (64). Kawakami et al demonstrated that CFI-400945 treatment blocked proliferation and induced polyploidy, apoptosis, and mitotic aberrations in lung cancer cells and inhibited tumor growth in a xenograft model of lung cancer (65). Further, Mason et al evaluated the efficacy of CFI-400945 in PTEN wild-type and PTEN null HCT116 colorectal cancer cells xenografts. In this study, CFI-400945 showed higher activity in the PTEN null xenografts compared to the PTEN wild-type xenografts. However, in breast tumor xenografts bearing MDA-MB-468 (PTEN null) or MDA-MB-231 (PTEN wild-type), the high dose of CFI-400945 (9.4 mg/kg) showed comparable antitumor activity, while the low dose of CFI-400945 (3 mg/kg) showed higher activity in the MDA-MB-468 tumor xenografts compared to the MDA-MB-231 tumor xenografts (36). In the same study, CFI-400945 efficacy was also evaluated in patient-derived xenografts of PTEN null high-grade invasive ductal carcinoma. A marked tumor regression was observed following 77 days of an intermittent dosing schedule of CFI-400945, with complete disappearance of palpable tumors in 3 of 5 animals. In addition, CFI-400945 was well tolerated at the doses and schedules examined, with no significant decrease in body weight, normal behavior of experimental animals, and favorable pharmacokinetic (PK) and pharmacodynamic (PD) characteristics (36). In another study, Sredni et al reported that treatment with CFI-400945 significantly reduced tumor growth of intracranial AT/RT xenografts and extended the survival of treated animals. Further, analysis of brain tumors obtained from pre-symptomatic mice at the end of the study showed significantly increased nuclear diameter and nuclear area, consistent with CFI-400945 pharmacodynamics (63). Although some of the effects found in these studies may be due in part to the inhibition of AURKs, the fact that PLK4 is affected at lower doses than other PLKs or AURKs suggests that there is a strong argument for the further evaluation of CFI-400945, a multi-kinase inhibitor with a defined selectivity for PLK4, in a clinical setting, even for advanced tumors.
Clinical development of PLK4 inhibitors
Phase 1 and 2 Clinical Trials
Following the successful in vitro and in vivo pre-clinical studies, CFI-400945 was evaluated in clinical trials for solid tumors (66). A phase 1 clinical trial was designed to evaluate the safety, PKs, PDs, recommended phase 2 dose (RP2D), and preliminary clinical antitumor activity of CFI-400945 administered orally to patients with advanced solid tumors. The experimental design consisted of forty-three patients treated in dose escalation from 3 to 96 mg/day, and nine treated in 64 mg dose expansion, which was established as the RP2D. It is important to note that there was no biomarker selection in this study and the study population was heavily pretreated. However, despite this, CFI-400945 was generally well tolerated, with neutropenia as the most common high-grade treatment-related adverse event at ≥64 mg (66). The half-life of CFI-400945 was reported as 9 h, with Cmax achieved 2–4 h following dosing. Favorable PK profiles were achieved with daily dosing (66). Although the overall observed efficacy of CFI-400945 was modest in this study of advanced solid tumors, the favorable PK characteristics, together with the absence of toxicities other than myelosuppression, supports further investigation and development of CFI-400945 as a potential therapeutic agent against cancer.
Another phase 1 clinical trial is currently recruiting patients to evaluate the safety and tolerability of CFI-400945 to determine the best dose (maximum tolerated dose or RP2D) in patients with relapsed or refractory acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) (NCT03187288). In addition, two phase 2 studies have been initiated with CFI-400945 in prostate cancer (NCT03385655) and advanced and metastatic breast cancer (NCT03624543). These studies are aimed at evaluating the efficacy of CFI-400945 in disease-specific cohorts and its effects on biomarkers of sensitivity in relevant populations.
Combinatorial Drug Trials
Based on promising pre-clinical in vivo data demonstrating combination activity of CFI-400945 with immune checkpoint blockade with no overlapping toxicities, a phase 2 study of CFI-400945 in combination with Durvalumab (immune checkpoint blockade) has been initiated in patients with advanced/metastatic TNBC by Canadian Cancer Trials Group. This study is aimed at evaluating the objective response rate (RECIST 1.1) of CFI-400945 given with durvalumab. Secondary objectives of this study include (i) to evaluate the disease control rate (defined as complete remission, partial remission, or stable disease >16 weeks in duration) of CFI-400945 given with durvalumab, (ii) to determine the immune-related response rate (iRECIST) of CFI-400945 given with durvalumab, (iii) to establish the safety and tolerability of CFI-400945 given orally in combination with durvalumab and (iv) to confirm the RP2D in patients with metastatic TNBC and to assess the pharmacodynamic and immune effects of CFI-400945 combined with durvalumab.
Conclusions
As integral kinases involved in regulating numerous facets of the cell cycle, the PLKs play essential roles in maintaining appropriate levels of genetic material and orchestrating the coordination of critical events as cells divide. PLK4, a structurally divergent member of this family, is critical in maintaining centrosomal integrity and regulating centriole duplication. Researching the involvement of PLK4 in genomic instability, in tandem with centrosome amplification, could not only aid in understanding the underlying mechanisms of PLK4 but also be useful as additional prognostic markers for a variety of cancers. PLK4 is overexpressed in many epithelial cancers and has been suggested as a potential therapeutic target. However, at this time, there is not much more known about the exact role of PLK4 in both epithelial and non-epithelial cancers, and closure of this gap in research may allow for the use of PLK4-targeting and molecular mechanism-based co-targeting strategies in anti-cancer studies.
Emerging data also suggests an involvement of PLK4 in the EMT process. However, a definite research gap exists at this point, and further research should be done to determine if PLK4 may be a key EMT player affecting tumor invasiveness and metastasis. If research supports this idea, the existence of PLK4-targeting compounds may offer a quick turnaround for metastatic cancer treatment and/or prevention. Although these molecular mechanisms have not been fully explored, several PLK4-targeting compounds have been developed and additional research is being conducted to 1) determine the clinical usefulness of the existing PLK4 inhibitors, 2) develop the next generation of more efficient and specific PLK4 inhibitors, and 3) identify other combinatorial partners of PLK4 inhibitors. Overall, PLK4 appears to have promise as a potential therapeutic target. However, further detailed mechanistic pre-clinical, as well as clinical studies, are needed for a bench-to-bedside development of PLK4-based therapeutic strategies.
Acknowledgements
This work was partially supported by funding from the National Institutes of Health [grant numbers R01AR059130] and the Department of Veterans Affairs [grant numbers I01CX001441, 1I01BX004221, IK6BX003780] to NA.
Footnotes
Declaration of Competing Interest
The authors declare that there are no conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 2.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol 2010;20:R285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine MS, Holland AJ. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev 2018;32:620–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bornens M The centrosome in cells and organisms. Science 2012;335:422–6. [DOI] [PubMed] [Google Scholar]
- 5.Chan JY. A Clinical Overview of Centrosome Amplification in Human Cancers. Int J Biol Sci 2011;7:1122–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004;5:429–40. [DOI] [PubMed] [Google Scholar]
- 7.Cunha-Ferreira I, Bento I, Pimenta-Marques A, Jana SC, Lince-Faria M, Duarte P, et al. Regulation of Autophosphorylation Controls PLK4 Self-Destruction and Centriole Number. Curr Biol 2013;23:2245–54. [DOI] [PubMed] [Google Scholar]
- 8.Driscoll DL, Chakravarty A, Bowman D, Shinde V, Lasky K, Shi J, et al. Plk1 inhibition causes post-mitotic DNA damage and senescence in a range of human tumor cell lines. PLoS One 2014;9:e111060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KS, Grenfell TZ, Yarm FR, Erikson RL. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc Natl Acad Sci U S A 1998;95:9301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 2005;7:1140–6. [DOI] [PubMed] [Google Scholar]
- 11.Levine MS, Holland AJ. Polo-like kinase 4 shapes up. Structure 2014;22:1071–3. [DOI] [PubMed] [Google Scholar]
- 12.Wei Z, Kim TS, Ahn JI, Meng L, Chen Y, Ryu EK, et al. Requirement of the Cep57-Cep63 Interaction for Proper Cep152 Recruitment and Centriole Duplication. Mol Cell Biol 2020;40:e00535–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sillibourne JE, Bornens M. Polo-like kinase 4: the odd one out of the family. Cell Div 2010;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganser A, Carlo-Stella C, Greher J, Volkers B, Hoelzer D. Effect of recombinant interferons alpha and gamma on human bone marrow-derived megakaryocytic progenitor cells. Blood 1987;70:1173–9. [PubMed] [Google Scholar]
- 15.Arquint C, Gabryjonczyk AM, Imseng S, Bohm R, Sauer E, Hiller S, et al. STIL binding to Polo-box 3 of PLK4 regulates centriole duplication. Elife 2015;4:e07888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen T, Li Y, Chen Z, Liang S, Qiu Y, Zhu L, et al. Activating transcription factor 6 (ATF6) negatively regulates Polo-like kinase 4 expression via recruiting C/EBPbeta to the upstream-promoter during ER stress. Biochim Biophys Acta Gene Regul Mech 2020;1863:194488. [DOI] [PubMed] [Google Scholar]
- 17.Bose A, Dalal SN. Centrosome Amplification and Tumorigenesis: Cause or Effect? The Golgi Apparatus and Centriole: Functions, Interactions and Role in Disease. Cham: Springer International Publishing; 2019. p 413–40. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto S, Kitagawa D. Self-organization of Plk4 regulates symmetry breaking in centriole duplication. Nat Commun 2019;10:1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nigg EA, Holland AJ. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 2018;19:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLamarrah TA, Speed SK, Ryniawec JM, Buster DW, Fagerstrom CJ, Galletta BJ, et al. A molecular mechanism for the procentriole recruitment of Ana2. J Cell Biol 2020;219:e201905172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moyer TC, Holland AJ. PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. Elife 2019;8:e46054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arquint C, Cubizolles F, Morand A, Schmidt A, Nigg EA. The SKP1-Cullin-F-box E3 ligase betaTrCP and CDK2 cooperate to control STIL abundance and centriole number. Open Biol 2018;8:170253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zitouni S, Francia ME, Leal F, Montenegro Gouveia S, Nabais C, Duarte P, et al. CDK1 Prevents Unscheduled PLK4-STIL Complex Assembly in Centriole Biogenesis. Curr Biol 2016;26:1127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Gupta GD, Barnabas DD, Agircan FG, Mehmood S, Wu D, et al. Direct binding of CEP85 to STIL ensures robust PLK4 activation and efficient centriole assembly. Nat Commun 2018;9:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonni S, Ganuelas ML, Petrinac S, Hudson JW. Human Plk4 phosphorylates Cdc25C. Cell Cycle 2008;7:545–7. [DOI] [PubMed] [Google Scholar]
- 26.Rosario CO, Ko MA, Haffani YZ, Gladdy RA, Paderova J, Pollett A, et al. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc Natl Acad Sci U S A 2010;107:6888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Press MF, Xie B, Davenport S, Zhou Y, Guzman R, Nolan GP, et al. Role for polo-like kinase 4 in mediation of cytokinesis. Proc Natl Acad Sci U S A 2019;116:11309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meitinger F, Ohta M, Lee KY, Watanabe S, Davis RL, Anzola JV, et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020;585:440–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luders J The amorphous pericentriolar cloud takes shape. Nat Cell Biol 2012;14:1126–8. [DOI] [PubMed] [Google Scholar]
- 30.Slevin LK, Nye J, Pinkerton DC, Buster DW, Rogers GC, Slep KC. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure 2012;20:1905–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogden A, Rida PCG, Aneja R. Centrosome amplification: a suspect in breast cancer and racial disparities. Endocr Relat Cancer 2017;24:T47–T64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, Burkard ME. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016;16:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pannu V, Mittal K, Cantuaria G, Reid MD, Li X, Donthamsetty S, et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget 2015;6:10487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev Cell 2017;40:313–22 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 2005;15:2199–207. [DOI] [PubMed] [Google Scholar]
- 36.Mason JM, Lin DC, Wei X, Che Y, Yao Y, Kiarash R, et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014;26:163–76. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Ren D, Sun Y, Xu C, Wang C, Cheng R, et al. Inhibition of PLK4 might enhance the anti-tumour effect of bortezomib on glioblastoma via PTEN/PI3K/AKT/mTOR signalling pathway. J Cell Mol Med 2020;24:3931–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng L, Zhou Y, Ju S, Han J, Song C, Kong J, et al. A cis-eQTL genetic variant in PLK4 confers high risk of hepatocellular carcinoma. Cancer Med 2019;8:6476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maniswami RR, Prashanth S, Karanth AV, Koushik S, Govindaraj H, Mullangi R, et al. PLK4: a link between centriole biogenesis and cancer. Expert Opin Ther Targets 2018;22:59–73. [DOI] [PubMed] [Google Scholar]
- 40.Denu RA, Shabbir M, Nihal M, Singh CK, Longley BJ, Burkard ME, et al. Centriole Overduplication is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma. Mol Cancer Res 2018;16:517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, Dai K, Wang C, Song Y, Gu F, Liu F, et al. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J Cancer 2016;7:1125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Q, Fan G, Dong Y. Polo-like kinase 4 correlates with greater tumor size, lymph node metastasis and confers poor survival in non-small cell lung cancer. J Clin Lab Anal 2020;34:e23152. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell 2015;60:524–36. [DOI] [PubMed] [Google Scholar]
- 44.Yeow ZY, Lambrus BG, Marlow R, Zhan KH, Durin MA, Evans LT, et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature 2020;585:447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shinmura K, Kurabe N, Goto M, Yamada H, Natsume H, Konno H, et al. PLK4 overexpression and its effect on centrosome regulation and chromosome stability in human gastric cancer. Mol Biol Rep 2014;41:6635–44. [DOI] [PubMed] [Google Scholar]
- 46.Denu RA, Sass MM, Johnson JM, Potts GK, Choudhary A, Coon JJ, et al. Polo-like kinase 4 maintains centriolar satellite integrity by phosphorylation of centrosomal protein 131 (CEP131). J Biol Chem 2019;294:6531–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim DH, Ahn JS, Han HJ, Kim H-M, Hwang J, Lee KH, et al. Cep131 overexpression promotes centrosome amplification and colon cancer progression by regulating Plk4 stability. Cell Death Dis 2019;10:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, et al. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol 2016;18:100–10. [DOI] [PubMed] [Google Scholar]
- 49.Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, et al. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 2014;510:167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jusino S, Saavedra HI. Role of E2Fs and mitotic regulators controlled by E2Fs in the epithelial to mesenchymal transition. Exp Biol Med (Maywood) 2019;244:1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu L, et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis 2008;29:1930–7. [DOI] [PubMed] [Google Scholar]
- 52.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer 2018;18:128–34. [DOI] [PubMed] [Google Scholar]
- 53.Liao Z, Zhang H, Fan P, Huang Q, Dong K, Qi Y, et al. High PLK4 expression promotes tumor progression and induces epithelialmesenchymal transition by regulating the Wnt/betacatenin signaling pathway in colorectal cancer. Int J Oncol 2019;54:479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian X, Zhou D, Chen L, Tian Y, Zhong B, Cao Y, et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis 2018;9:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kazazian K, Go C, Wu H, Brashavitskaya O, Xu R, Dennis JW, et al. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res 2017;77:434–47. [DOI] [PubMed] [Google Scholar]
- 56.Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015;348:1155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerschner-Morales SL, Kuhne M, Becker S, Beck JF, Sonnemann J. Anticancer effects of the PLK4 inhibitors CFI-400945 and centrinone in Ewing’s sarcoma cells. J Cancer Res Clin Oncol 2020;146:2871–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z L Q Wei W Xiong L Shi Y Yan G Gao C Ye T Wang N Yu L Synthesis and biological evaluation of (e)-4-(3-arylvinyl-1h-indazol-6-yl)pyrimidin-2-amine derivatives as plk4 inhibitors for the treatment of breast cancer. RSC Adv 2017;7 27737–46. [Google Scholar]
- 59.Lei Q, Xiong L, Xia Y, Feng Z, Gao T, Wei W, et al. YLT-11, a novel PLK4 inhibitor, inhibits human breast cancer growth via inducing maladjusted centriole duplication and mitotic defect. Cell Death Dis 2018;9:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Y, Liu Z, Qu Y, Zeng J, Yang M, Li X, et al. YLZ-F5, a novel polo-like kinase 4 inhibitor, inhibits human ovarian cancer cell growth by inducing apoptosis and mitotic defects. Cancer Chemother Pharmacol 2020;86:33–43. [DOI] [PubMed] [Google Scholar]
- 61.Sampson PB, Liu Y, Forrest B, Cumming G, Li SW, Patel NK, et al. The discovery of Polo-like kinase 4 inhibitors: identification of (1R,2S).2-(3-((E).4-(((cis).2,6-dimethylmorpholino)methyl)styryl). 1H.indazol-6-yl)-5’-methoxyspiro[cyclopropane-1,3’-indolin]-2’-one (CFI-400945) as a potent, orally active antitumor agent. J Med Chem 2015;58:147–69. [PubMed] [Google Scholar]
- 62.Sredni ST, Suzuki M, Yang JP, Topczewski J, Bailey AW, Gokirmak T, et al. A functional screening of the kinome identifies the Polo-like kinase 4 as a potential therapeutic target for malignant rhabdoid tumors, and possibly, other embryonal tumors of the brain. Pediatr Blood Cancer 2017;64:e26551. [DOI] [PubMed] [Google Scholar]
- 63.Sredni ST, Bailey AW, Suri A, Hashizume R, He X, Louis N, et al. Inhibition of polo-like kinase 4 (PLK4): a new therapeutic option for rhabdoid tumors and pediatric medulloblastoma. Oncotarget 2017;8:111190–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lohse I, Mason J, Cao PM, Pintilie M, Bray M, Hedley DW. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017;8:3064–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawakami M, Mustachio LM, Zheng L, Chen Y, Rodriguez-Canales J, Mino B, et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc Natl Acad Sci U S A 2018;115:1913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Veitch ZW, Cescon DW, Denny T, Yonemoto LM, Fletcher G, Brokx R, et al. Safety and tolerability of CFI-400945, a first-in-class, selective PLK4 inhibitor in advanced solid tumours: a phase 1 dose-escalation trial. Br J Cancer 2019;121:318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown NJ, Marjanovic M, Luders J, Stracker TH, Costanzo V. Cep63 and cep152 cooperate to ensure centriole duplication. PLoS One 2013;8:e69986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar A, Rajendran V, Sethumadhavan R, Purohit R. CEP proteins: the knights of centrosome dynasty. Protoplasma 2013;250:965–83. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Kim J, Philip R, Sridhar V, Chandrashekhar M, Moffat J, et al. Direct interaction between CEP85 and STIL mediates PLK4-driven directed cell migration. J Cell Sci 2020;133(8):jcs238352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin YC, Chang CW, Hsu WB, Tang CJ, Lin YN, Chou EJ, et al. Human microcephaly protein CEP135 binds to hSAS-6 and CPAP, and is required for centriole assembly. EMBO J 2013;32:1141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD, Nigg EA. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci 2013;126:3223–33. [DOI] [PubMed] [Google Scholar]
- 72.Breslow DK, Holland AJ. Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu Rev Biochem 2019;88:691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, et al. Control of centriole length by CPAP and CP110. Curr Biol 2009;19:1005–11. [DOI] [PubMed] [Google Scholar]
- 74.Pagan J, Pagano M. FBXW5 controls centrosome number. Nat Cell Biol 2011;13:888–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Puklowski A, Homsi Y, Keller D, May M, Chauhan S, Kossatz U, et al. The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat Cell Biol 2011;13:1004–9. [DOI] [PubMed] [Google Scholar]
- 76.Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007;23:1073–9. [DOI] [PubMed] [Google Scholar]