

Abstract
The role of peroxisome proliferator-activated receptor gamma (PPARγ) has been well characterized in the developmental process of adipogenesis, yet its aberrant expression patterns and functions in cancer subtypes are less understood. While PPARγ has been recently demonstrated to play non-cell-autonomous roles in promoting bladder urothelial carcinoma (UC) progression, underlying mechanisms of the cell-intrinsic oncogenic activity remain unknown. Here, we report robust expression and nuclear accumulation of PPARγ in 47% of UC patient samples, exceeding mRNA expression patterns published by The Cancer Genome Atlas. In vitro assays revealed for the first time that treatment of UC cells with PPARγ inverse agonist or PPARG knockout by CRISPR-Cas9 reduces proliferation, migration, and invasion of multiple established UC cell lines, most strongly in those characterized by PPARG genomic amplification or activating mutations of RXRA, the obligate heterodimer of PPARγ. Through genome-wide approaches including ChIP- and RNA-seq, we define a novel set of PPARγ-regulated genes in UC, including Sonic Hedgehog (SHH). Similar to PPARγ, genetic inhibition of SHH reduces proliferation and motility. Finally, we demonstrate the PPARγ dependency of UC tumors in vivo by genetic and pharmacological PPARγ inhibition in subcutaneous xenografts. Collectively, our data indicate that PPARγ promotes UC progression in a subset of patients, at least in part, through cell-autonomous mechanisms linked to SHH signaling.
Keywords: bladder urothelial carcinoma, PPARγ, CRISPR-Cas9, genomics, sonic hedgehog
Introduction
Bladder urothelial carcinoma (UC) is the 9th leading cause of cancer worldwide, with an estimated ~81,400 new cases in 2020 and ~17,980 annual deaths per year in the United States alone (1). Clinically, UC is stratified into non-muscle invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC), both contributing to disease morbidity and mortality (2). MIBC is currently treated by radical cystectomy preceded or followed by chemotherapy including methotrexate, vinblastine, doxorubicin, and cisplatin (3). Recently, large-scale genomic sequencing of hundreds of primary patient samples has revealed distinct clusters of MIBC, most broadly stratified into luminal and basal subcategories based on common transcriptional features (3, 4). The identification of molecular signatures and drivers of these programs will pave the way for improved targeted therapies, which are needed because there are currently few available to treat localized and metastatic UC.
One transcription factor reported to be a hallmark of luminal MIBC is the peroxisome proliferator-activated receptor gamma (PPARγ) (5). PPARγ is a member of the nuclear receptor family, which acts as molecular sensors to regulate gene expression based on environmental and metabolic cues (6). PPARγ binds DNA as a heterodimer with retinoid X receptor (RXR), and exists in complexes with co-repressor or co-activator proteins depending on its activation state. Associations with co-activator or co-repressor proteins facilitate chromatin remodeling and target gene activation or repression, respectively (7). During development, PPARγ and C/EBP transcription factors induce adipogenesis from mesenchymal progenitor cells (8). In differentiated post-mitotic adipocytes PPARγ regulates the expression of genes involved in lipid and glucose metabolism, as well as the expression of endocrine “adipokine” factors which affect whole-body energy homeostasis (9).
Long-term use of certain thiazolidinediones, synthetic agonists of PPARγ, Pioglitazone, but not rosiglitazone, used to manage blood glucose levels in type 2 diabetics, has been associated with increased risk of bladder cancer development (10–12). In a pre-clinical study employing a carcinogen-induced model of bladder cancer, rosiglitazone treatment increased incidence and size of UC in rats in a dose-dependent manner (13). Of note, rosiglitazone treatment alone did not induce UC tumors, suggesting a potential synergistic relationship between PPARγ activation and carcinogen exposure in UC development in vivo. Nevertheless, these observations have led our group and others to speculate that PPARγ might be an important factor in UC development and progression in a subset of patients. Indeed, multiple recent studies link PPARγ activity to UC, describing its ability to promote disease progression in both cell-autonomous (5, 14–16) and non-cell-autonomous manners (17). However, the direct molecular mechanisms by which PPARγ promotes tumor growth have not been described, nor have the downstream mediators of PPARγ-RXR transcriptional activity been defined. In this study, our objective was to further elucidate cell-intrinsic roles of PPARγ signaling in UC and identify candidate target genes responsible for UC progression based on genome-wide approaches. Importantly, our data demonstrated for the first time direct transcriptional regulation of the oncogene Sonic Hedgehog (SHH) by PPARγ. Moreover, we demonstrate a functional link between SHH and PPARγ dependent cell functions and thus providing insights into the tumorigenic nature and molecular mechanism involved in PPARγ dependent UC tumorigenesis.
Materials and Methods
Primary Patient Samples and Genomics Data
Bladder urothelial carcinoma tissue array containing 60 cases of urothelial carcinoma plus adjacent normal bladder tissue was purchased from US Biomax, Inc. (cat. BC12011c). TCGA datasets (2, 18) were analyzed for PPARG and RXRA mutational burden, copy number variation, mRNA upregulation, and co-expression patterns using cBioPortal for Cancer Genomics (19, 20) and the Sanchez-Carbayo data set was analyzed for mRNA upregulation (Oncomine). PPARG expression levels of various cell lines were extracted from RNA-Seq data available on the Cancer Cell Line Encyclopedia. The CERES dependency score (Fig. 3H) is based on data from a cell depletion assay. A lower score indicates a higher likelihood that the gene of interest is essential in a given cell line. A score of 0 indicates a gene is not essential; correspondingly −1 is comparable to the median of all pan-essential genes (21).
Figure 3 – Genetic inhibition of PPARγ reduces growth and migratory capacities of several UC cell lines in vitro.
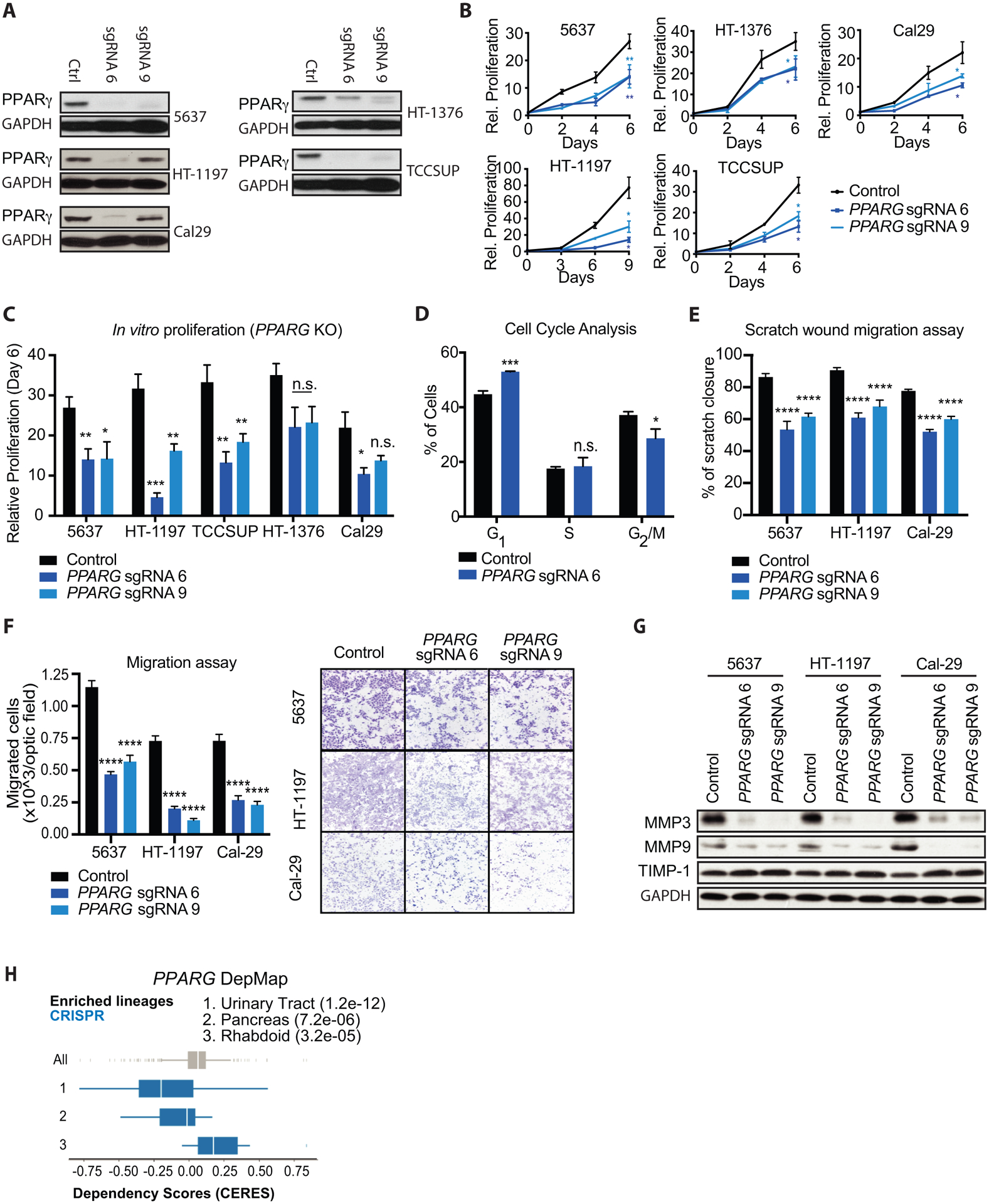
(A) Western blot for PPARγ expression in control, PPARG sgRNA 6 (sgRNA 6) and PPARG sgRNA 9 (sgRNA 9) showing knock out efficiency in different UC cell lines. GAPDH was used as loading control. (B) Growth curves of control, PPARG sgRNA 6, and PPARG sgRNA 9 (PPARG KO) UC cell lines over the course of 6–9 days. Values are normalized to cell count of each cell line at the Day 0 timepoint. (C) Relative proliferation of PPARG KO to control cell lines at day 6 of the growth curves from panel (B). (D) Percentage of control and PPARG KO 5637 cells in each phase of the cell cycle, determined by flow cytometry staining of propidium iodide. (E) Scratch wound migration assays of control and PPARG KO UC cell lines. (F) Boyden chamber migration assays of control and PPARG KO cells, showing quantification (left) and representative micrographs (right). (G) Western blot for showing migration markers MMP3, MMP9 and TIMP1 in control, PPARG sgRNA 6 (sgRNA 6) and PPARG sgRNA 9 (sgRNA 9) UC cells. GAPDH was used as loading control. (H) DepMap analysis of PPARG CRISPR KO in Broad institute cell lines. A lower CERES score indicates that a given lineage is more sensitive to PPARG depletion. Data are presented as mean ± SEM. **** (p < 0.0001), *** (p < 0.001), ** (p < 0.01), * (p < 0.05), n.s. = not significant. Student’s two-tailed unpaired t-test for pairwise comparisons, one-way ANOVA for multiple comparisons, or two-way ANOVA for multiple comparisons involving two independent variables. Asterisks represent p values for comparison between control and PPARG sgRNA 6, and PPARG sgRNA 9 cells.
Cell Culture, Plasmids, Lentiviral Production and Transduction
Human UC cell lines (5637, HT-1197, HT-1376, TCCSUP, T24) were purchased from the American Type Culture Collection (ATCC), with the exception of Cal29 (DSMZ, cat. ACC-515). Cell lines were authenticated by ATCC or DSMZ before distribution via morphology, karyotyping, and PCR based approaches. All cell lines were cultured in RPMI 1640 medium containing L-glutamine (ThermoFisher Scientific, cat. 11875–085) supplemented with 10% FBS (Gemini Bio-Products, cat. 900–108) and were tested regularly for mycoplasma contaminations using the Lonza MycoAlert assay. All cell lines were used for 6 weeks for 8–10 passages. Human single-guide RNAs (sgRNA) targeting PPARG #6 (cattacgaagacattccatt) and #9 (caactttgggatcagctccg) along with control gRNA targeting mouse Rosa26 locus (aagatgggcgggagtcttct) were cloned into LentiCRISPRv2 plasmid. Mature antisense human PPARG shRNA #2 sequence (clone ID: TRCN0000001672), SHH shRNA #4 sequence (Clone ID: TRCN0000033304) and shRNA #5 sequence (Clone ID: TRCN0000033305), along with scrambled (SCR) control were purchased from Dharmacon™ and cloned into a pLKO lentiviral plasmid. Lentivirus was prepared and cells were transduced as previously described (22).
Human SHH pre-packaged lentiviral particles (Hygromycin selective) were purchased from G&P Biosciences® (Cat. LTV7062). All experiments were performed with cells that survived puromycin and/ or hygromycin selection and displayed knockdown of PPARG, SHH and overexpression of SHH, as assayed by Western blot. Primary Bladder Epithelial Cells (A/T/N (ATCC® PCS-420–010™) were purchased from ATCC and processed immediately for protein and RNA sampling.
PPARγ Agonists and Inhibitors
Rosiglitazone (Sigma-Aldrich, cat. R2408) was used at a final concentration of 100 nM in all experiments. The PPARγ inverse-agonist T0070907 (Santa Cruz Biotechnology, cat. sc-203287) was used at a final concentration of 100 nM in all experiments. An equivalent volume of DMSO was used as a control for all experiments involving PPARγ pharmacological agents.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated using TRIzol reagent (ThermoFisher Scientific, cat. 15596026) and RNeasy mini kit (Qiagen, cat. 74104). Reverse transcription was performed using High-Capacity RNA-to-cDNA (Applied Biosystems, cat. 4387406). qRT-PCR was performed using ViiA7 Real-Time PCR system (Applied Biosystems) with TaqMan master mix (Life Technologies). TaqMan probes purchased from Thermo Fisher Scientific were used to quantitate expression of PPARG (cat. Hs01115513_m1), FABP4 (cat. Hs01086177_m1), PLIN2 (cat. Hs00605340_m1), SCD (cat. Hs01682761_m1), LPL (cat. Hs00173425_m1), PPARA (cat. Hs00947536_m1), SHH (cat. Hs00179843_m1), MMP3 (cat. Hs00968305_m1), MMP9 (Hs00957562_m1), TIMP1 (cat. Hs01092511_m1) and normalized to housekeeping gene TBP (Hs00427620_m1).
Western Blot and Immunohistochemistry
Western blot was performed as previously described (22). Primary antibodies were diluted 1:1000 in 5% w/v nonfat milk (except GAPDH, 1:10,000), and secondary antibodies were diluted 1:2000 in 5% w/v nonfat milk. PPARγ (cat. 2435), Shh (cat. 2207), MMP3 (Cat. 14351), MMP9 (Cat. 13667), TIMP1 (Cat. 8946), KRT5 (Cat. 25807), FOXA1 (Cat. 53528), GAPDH (cat. 2118), and anti-Rabbit IgG, HRP-linked (cat. 7074) antibodies were purchased from Cell Signaling Technology. KRT6 (Cat. MBS2526156) was purchased from MyBioSource
Human bladder cancer tissue array was processed for immunohistochemistry. Slide was baked in a dry oven for 20 minutes at 55°C, then deparaffinized by incubation in 100% xylene for 15 min, twice, followed by rehydration (100% ethanol (EtOH), 95% EtOH, 70% EtOH, 100% dH2O for 5 min each). Slide was then boiled in antigen unmasking solution (1M sodium citrate solution, pH 6.0) for 20 min, left to cool on bench for 20 min. Endogenous peroxidase activity was blocked with 1% H2O2/H2O solution for 30 minutes. Slide was washed in 1X TT buffer for 3 × 5 min, and incubated in blocking buffer (5% BSA, 2% goat serum diluted in 1X TT buffer) for 1 hr at RT. Samples were incubated overnight in PPARγ antibody (1:200) in blocking buffer at 4°C. Slide was washed in 1X TT buffer for 3 × 5 min, and incubated in secondary antibody (1:200 biotinylated goat anti-Rabbit, diluted in 1X TT) for 1 hr at RT. Slide was further processed using Vectastain Elite ABC kit (Fisher Scientific, cat. PK6100) according to the manufacturer’s instructions, and staining was visualized with DAB peroxidase substrate kit (Vector Laboratories, cat. SK4100). Slide was counterstained with hematoxylin and dehydrated before mounting and imaging. Where mentioned, densitometry was assessed using NIH Image J software package.
Chromatin Immunoprecipitation (ChIP) and ChIP-seq
ChIP was performed with whole cell extracts isolated from 5637 parental cells using 2.5 μg PPARγ (Santa Cruz, cat. sc-7196), 2.5μg RXRα/β/ γ (Santa Cruz, cat. sc-774), or 2.5 μg Normal Rabbit IgG (Cell Signaling Technology, cat. 2729) antibodies for immunoprecipitation (IP). Briefly, confluent 10 cm dishes of cells were crosslinked with 1% formaldehyde for 15 min at RT, and quenched with 125 mM glycine for 5 min. Cells were harvested by scraping and pellets were resuspended in 200 μl SDS lysis buffer (50 mM HEPES/NaOH pH 7.5, 1% SDS, 10 mM EDTA, 1 mM PMSF, and Roche ULTRA protease inhibitor cocktail (cat. 05892791001)) on ice for 10 min. Sonication was performed using Bioruptor Pico (Diagenode, cat. B01060010) for 4 cycles of 30 seconds on, 30 seconds off, followed by centrifugation of lysates. 66 μl of sheared chromatin from each tube was then diluted 10X, with 5% saved as Input DNA and the rest prepared for either PPARγ, RXR, or IgG IP.
PPARγ IP, RXR IP, IgG IP and Input libraries were prepared in duplicate from two independent biological replicates. For ChIP-seq, sequencing data was mapped to the human genome (GRCh38) using STAR (23) with parameters appropriate for ungapped alignments. Peaks were called for each sample with input samples as background by HOMER (24). HOMER was also used for differential peak calling (PPARγ vs. Input and RXR vs. Input) and to annotate peaks to proximal genes as described in Ensembl v85 (http://www.ensembl.org/index.html). Motif enrichment analysis was performed using HOMER against the standard list of known motifs with consideration of lengths 8,10,12,15,18 bp. For bioinformatics analyses displayed in Figure 5A, “high-confidence sites” were defined by the following criteria: peak score ≥ 10 (≥ 1 read per million) and RXR peak called with strict overlap. Gene ontology was performed using Metascape (metascape.org).
Figure 5 – Identification of PPARγ target genes of interest from ChIP-seq and RNA-seq data.
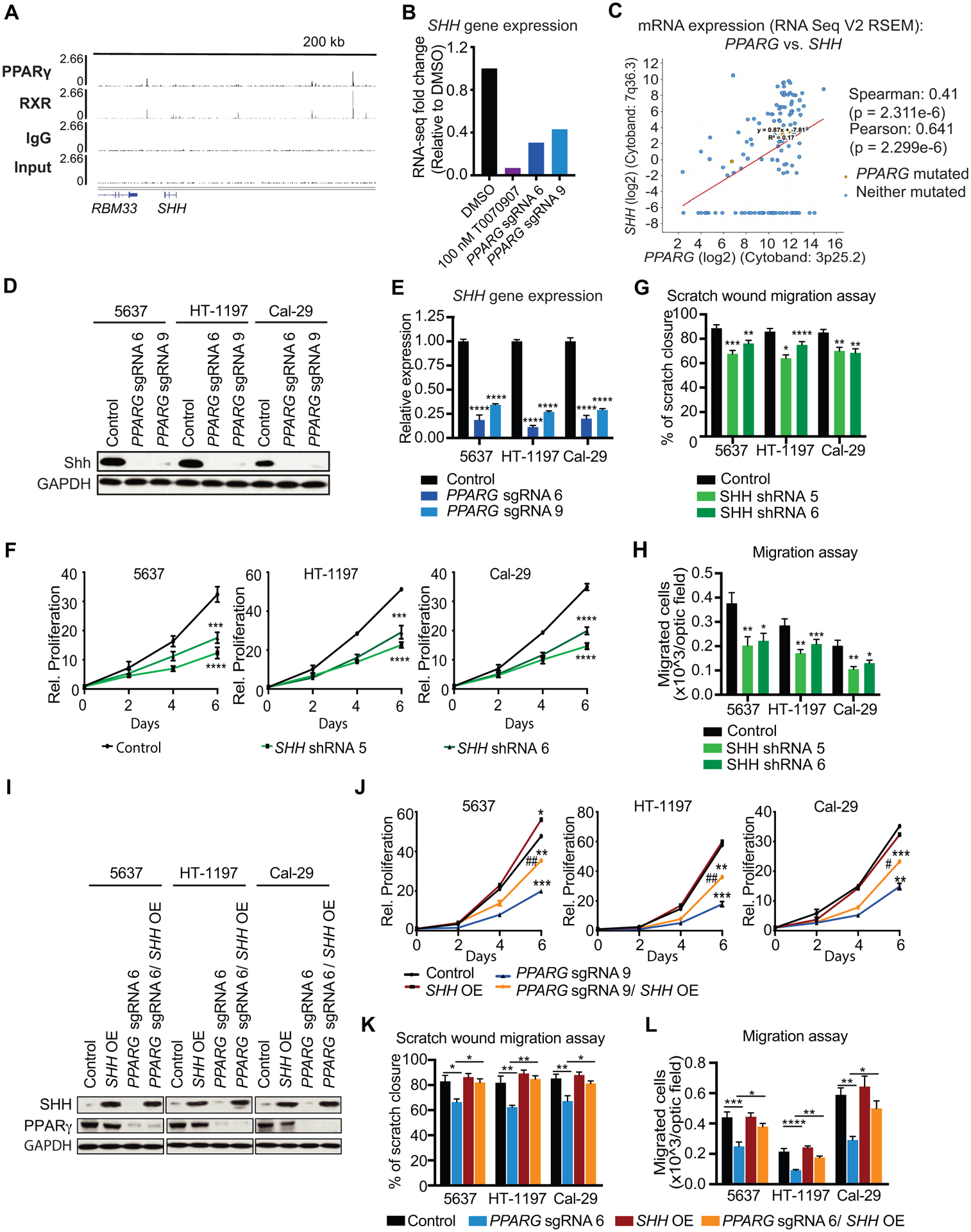
(A) ChIP-seq tracks of PPARγ, RXR, IgG and Input, showing PPARγ-RXR occupancy within 200 kb of SHH. (B) SHH gene expression data from RNA-seq in 100 nM T0070907, PPARG sgRNA 6, and PPARG sgRNA 9, relative to DMSO. (C) Co-expression data for PPARG and SHH in UC tumors from the TCGA provisional dataset (cBioPortal). (D) Western blot analysis for PPARγ and SHH of control, PPARG sgRNA 6, and sgRNA 9 in UC cell lines. GAPDH is used as loading control. (E) SHH mRNA expression in control, PPARG sgRNA 6, and PPARG sgRNA 9 UC cells. (F) Growth curves of control, SHH shRNA 5, and SHH shRNA 6 cells over the course of 6 days. Values are normalized to cell count of each line at the Day 0 timepoint. (G) Scratch wound migration assay of control, SHH shRNA 5, and SHH shRNA 6 UC cell lines. (H) Boyden chamber migration assay of control, SHH shRNA 5, and SHH shRNA 6 cells. (I) Western blot analysis for PPARγ and SHH of control, PPARG sgRNA 6, SHH OE, and PPARG sgRNA 6/ SHH OE in UC cell lines. GAPDH is used as loading control. (J) Growth curves of control, PPARG sgRNA 6, SHH OE, and PPARG sgRNA 6/ SHH OE cells over the course of 6 days. Values are normalized to cell count of each line at the Day 0 timepoint. (K) Scratch wound migration assay control, PPARG sgRNA 6, SHH OE, and PPARG sgRNA 6/ SHH OE cells. (L) Boyden chamber migration assay of control, PPARG sgRNA 6, SHH OE, and PPARG sgRNA 6/ SHH OE cells. Data are presented as mean ± SEM. **** (p < 0.0001), **** (p < 0.0001), *** (p < 0.001), * (p < 0.05), One-way ANOVA for multiple comparisons, or two-way ANOVA for multiple comparisons involving two independent variables. Asterisks represent p values for comparison between control and PPARG KO or SHH KD cells except for multiple comparisons.
RNA-seq
Total RNA was isolated from four technical replicates of 5637 control, 100 nM T0070907-treated, PPARG sgRNA 6, and PPARG sgRNA 9 cell lines as described above. NEBNext Ultra II RNA Library Prep Kit for Ilumina was used for preparing ~250–300 bp insert cDNA, non-directional libraries according to manufacturer’s instructions at Novogene Corporation (Sacramento, CA). Libraries were sequenced using the Ilumina platform with paired-end 150 bp sequencing at 20M raw reads/sample at Novogene. Raw sequence files (fastq) for 16 samples were mapped using Salmon (25) against the human transcripts described in GENCODE (version 28, built on the human genome GRCh38.p12). Transcript counts were summarized to the gene level using tximport (26), and normalized and tested for differential expression using DESeq2 (27). Statistics for each contrast of interest were exported for further analysis. Genes used for gene ontology met the following criteria: significantly up- or down-regulated by inverse-agonist or CRISPR treatment at least 1.5-fold with padj < 0.05. Gene ontology was performed using Metascape (metascape.org).
Annexin V-PI Apoptosis Assay
5,000 cells of each cell line were plated in triplicate on 6-well plates. Cells were treated for 8 days with media containing DMSO or 100 nM T0070907, and then prepared using the FITC–Annexin V, PI Kit (BD Biosciences, cat. 556547) according to the manufacturer’s instructions. Flow cytometry was performed using the BD Accuri C6 instrument, with viable cells represented as the double-negative population.
Proliferation Assay
25,000 cells of tested conditions were plated in triplicate on 6-well plates. The following day (Day 0), cells were trypsinized and counted using the Countess Automated Cell Counter (Invitrogen, cat. C10281), as per the manufacturer’s instructions with Trypan blue. Cells were then counted again at the indicated timepoints, and fresh media was added every two days.
Clonogenic Growth Assay
5,000 cells of each cell line were plated in triplicate on 6-well plates. The next day, media containing either 100 nM T0070907 or an equivalent volume of DMSO was added to the wells in triplicate. UC cells were treated with DMSO or T0070907 for 9 days (with the exception of T24, 5 days) with fresh medium supplied every 2 days. At the end, cells were washed in PBS and stained with crystal violet solution (0.5% crystal violet in 20% methanol) for 10 minutes. Plates were then washed twice with PBS and allowed to dry. Quantitation was performed by adding 500 μL methanol to dry wells and incubating at RT with rocking for 20 minutes. Absorbance was read using a spectrophotometer at 570 nm, and data are represented as fold change relative to DMSO readings.
Cell Cycle Analysis
5,000 cells of the 5637 cell line were plated on 6-well plates. The following day, cells were serum starved for 24 hours to synchronize the population, after which media containing DMSO and 100 nM T0070907 were added. Cells were harvested after 5 days of drug treatment, fixed using ice-cold 70% ethanol, and stored at −20°C for 2 hours. Following centrifugation and removal of ethanol, cells were washed twice in FACS buffer (PBS containing 2% FBS and 1 mM EDTA). For dual Ki67/PI staining to determine non-proliferative (G0 population): samples were resuspended in 200 μL FACS buffer and stained for 30 minutes at RT in the dark with Ki67-FITC monoclonal antibody (SolA15) (Life Technologies, cat. 11-5698-82) at 1:100. Cells were then washed twice with FACS buffer and resuspended in 500 μL PI staining solution (PBS containing 50 μg/mL PI, 100 μg/mL RNAse A and 2 mM MgCl2). Cells were strained through 40 μm filters and incubated at RT in the dark for 20 minutes before analysis by flow cytometry. For analysis of the cell cycle phases, cells were stained with PI antibody only.
EdU incorporation assay
Cells were labeled with 10 mM EdU over 4 hr, collected by trypsinization and fixed (4% PFA). The incorporated EdU was detected by a ‘click-It reaction’ with Alexa Fluor 488 according to manufacturer’s instructions (ThermoFisher Scientific). Data were recorded by flow cytometry, and resultant data were analyzed with the FlowJo 10.6.2 software (https://www.flowjo.com).
Adipogenesis Assay
250,000 NIH-3T3 cells (ATCC) were infected with MSCV-retroviral empty vector or vector containing PPARG cDNA resistant to shRNA #2. When cells were confluent, induction medium was added to the plates for 48 hours (DMEM containing 10% FBS supplemented with 125 nM indomethacin, 0.5 mM isobutylmethylxanthine (IBMX), 1 mM dexamethasone, 20 nM insulin, and 1 μM rosiglitazone). After 48 hours, medium was changed to DMEM containing 10% FBS supplemented with 20 nM insulin and 1 μM rosiglitazone. Medium was changed every 2 days until plates were harvested on day 10 after induction and stained with oil red O to visualize adipogenesis. Oil red O staining was performed as previously described (22).
Boyden Chamber Migration and invasion Assay:
100,000 cells were plated in the upper chamber of semi-permeable inserts for 24-well plates (6.5 mm Transwell® with 8.0 μm Pore Polycarbonate Membrane Insert. Corning Cat. 3422) in RPMI 1640 medium containing L-glutamine, supplemented with 10% FBS. For the Invasion assay, 40 μl of Matrigel (Corning Cat. 354234) and solidified in a 37 °C incubator for 15–30 minutes to form a thin gel layer. 100,000 cells were seeded on top of the Matrigel coating.
30 min after seeding, growth medium in the upper chamber was carefully replaced by serum free RPMI 1640 medium. After 20 hours incubation at 37°C with 5% CO2, cells on the upper side of the membrane were removed with a cotton tipped applicator. Migrated cells were fixed in cold methanol and stained with crystal violet. The membranes were removed from the insert using a scalpel and mounted on a glass slide. Bright-field images were taken with 10X objectives on a Leica DM 5000B microscope. Images were analyzed using NIH Image J software package.
Scratch Wound Assay:
100,000 cells of each experimental condition were plated in 24 well plates and grown to confluence. These cells were treated overnight with 10 μg/ml Mitomycin C. A straight scratch was made using a P200 pipette tip. The cells were then washed with PBS three times, and initial gap sizes at T0 were photographed. The cultures were further incubated for 8 hours (37 °C and 5% CO2) in RPMI 1640 medium containing L-glutamine, supplemented with 10% FBS, and photographed again (T8). Gap areas were measured with NIH Image J software package and results are expressed in % wound closure = ratio: [gap area at T0 minus gap area at T8] / gap area T0.
Mouse experiments
Subcutaneous xenograft experiments were performed using male 6–7 week old BALB/c Nude Mouse (CAnN.Cg-Foxn1nu/Crl), purchased from Jackson Laboratory (Bar Harbor, ME). 1×106 5637 WT/ Ctl vector/ or PPARG sgRNA 6 cells, suspended in PBS were mixed with 100ul Matrigel and a total of 200ul was injected subcutaneously. Tumor volume and body weight were measured 3X a week. Tumor volume were calculated using the formula volume = (length × (width)2)/2. Once tumors were established (~100 mm3), animals were randomized into two groups and treated with vehicle (5%DMSO, 45% PEG300 and ddH2O) or 5mg/kg T0070907 daily through intraperitoneal injection.
No inclusion or exclusion criteria parameters were used, and all animals were included in analyses. Animal wellbeing was monitored by certified veterinary staff. All mouse experiments were performed according to National Institutes of Health guidelines and received ethical approval by the University of Pennsylvania Institutional Animal Care and Use Committee.
Statistics
Statistical analyses were performed in GraphPad Prism 8 using Student’s two-tailed unpaired t-test for pairwise comparisons, one-way analysis of variance (ANOVA) for multiple comparisons, or two-way ANOVA for multiple comparisons involving two independent variables. Data are presented as mean ± SEM of at least three independent experiments. A p value less than 0.05 was considered significant. Statistical significance was defined as **** (p < 0.0001), *** (p < 0.001), ** (p < 0.01), * (p < 0.05), n.s. = not significant.
Results
Characterization of PPARγ expression in UC.
Previous reports using available sequencing data from The Cancer Genome Atlas (TCGA) demonstrated that approximately 20% of UC tumors exhibit genomic PPARG amplification and/or increased PPARG mRNA levels, while approximately 15% display genomic amplification and/or activating mutations in RXRA, the obligate heterodimer of PPARγ, in a largely mutually-exclusive pattern (16, 28). RNA-seq data from the Sanchez-Carbayo data set also showed an increased number of both PPARG and RXRA transcripts in superficial UC (Fig. 1A and B). To validate that PPARG mRNA abundance translated to increased protein expression, we performed immunohistochemistry for PPARγ on 59 independent UC patient samples from a commercially available tissue array (Fig. 1C–E). Surprisingly, our analysis revealed that nearly 37% of UC tissues stained strongly positive for nuclear PPARγ accumulation, while another 10% had more “intermediate” levels. In contrast, PPARγ levels in normal bladder epithelium was largely undetectable in these assays (Fig. 1C and D). Quantitative analysis revealed that overall PPARγ levels were significantly increased in UC samples compared to normal bladder epithelium (Fig. 1E). Interestingly, distinction between NMIBC and MIBC revealed that MIBC tissue sections have slightly higher PPARG levels than NMIBC tissue sections (Supplemental Fig. S1A).
Figure 1 – Characterization of PPARγ expression in UC.
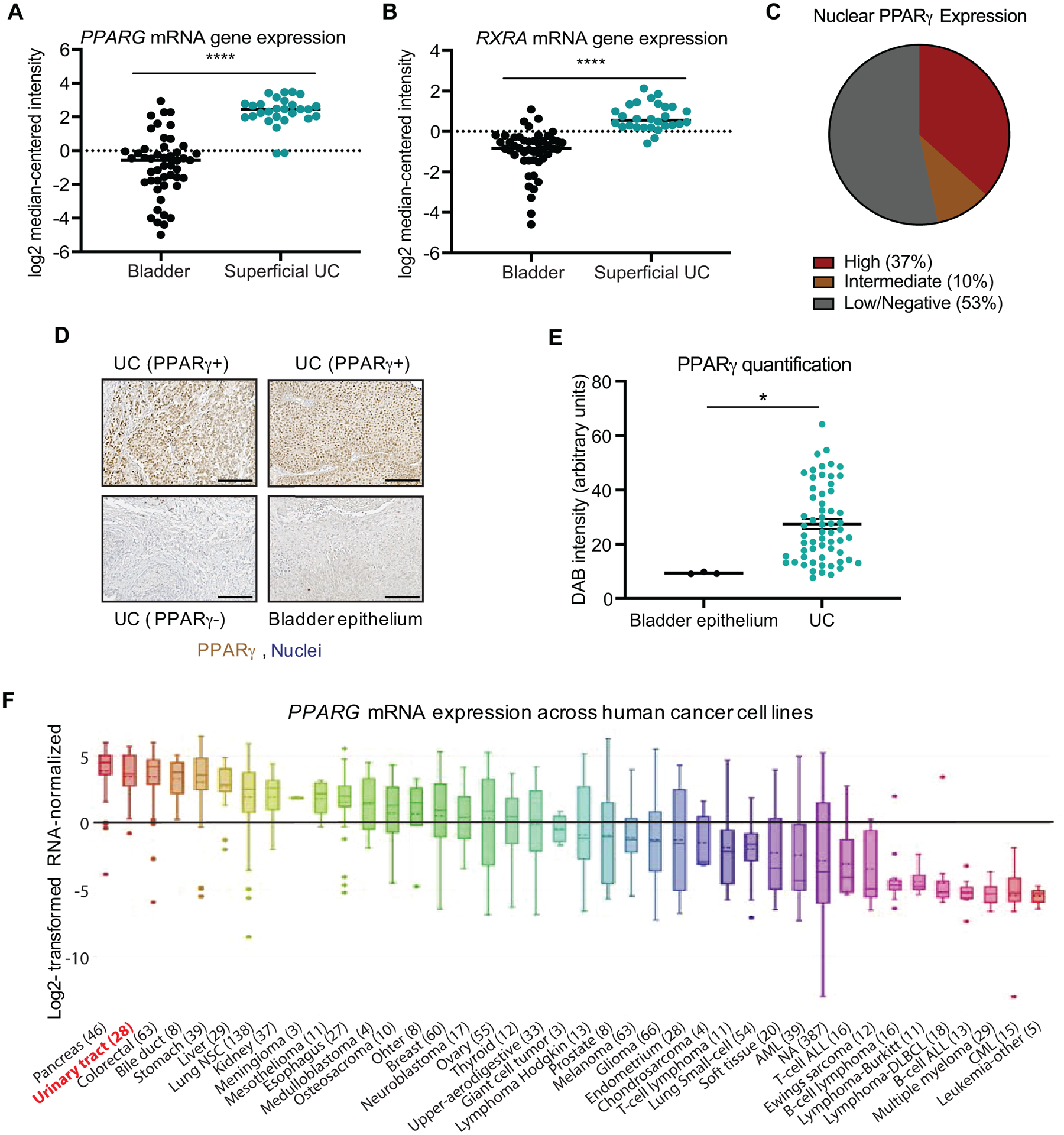
(A) PPARG mRNA expression in superficial bladder carcinoma and healthy bladder tissue from the Sanchez-Carbayo dataset. Bladder epithelium: n = 48, Superficial bladder carcinoma: n = 28, Fold change: 15.9, p = 3.72e−19 (Oncomine). (B) RXRA mRNA expression in superficial bladder carcinoma and healthy bladder tissue from the same Sanchez-Carbayo dataset. Bladder epithelium: n = 48, Superficial bladder carcinoma: n = 28, Fold change: 3.264, p = 9.05e−13 (Oncomine). (C) Subdivision of nuclear PPARγ expression in primary UC tumors in tissue array of 59 primary UC tumors and healthy bladder tissue, assessed by immunostaining. (D) High-magnification images of UC with strong PPARγ staining, low/negative PPARγ staining, and healthy bladder epithelium. Scale bar = 100 μm. (E) Quantification of PPARγ nuclear staining intensity (Diaminobenzidine [DAB] intensity) in primary UC tumors. (F) PPARG mRNA expression patterns across human cancer cell lines documented by the Cancer Cell Line Encyclopedia (CCLE). Data are presented as mean ± SEM. **** (p < 0.0001), * (p < 0.05). Unpaired student t-test. Black asterisks represent p values for comparison between healthy bladder tissue and UC.
Multiple UC cell lines (5637, HT1197, Cal-29, HT-1375, TCCSUP, and T24) were employed to further evaluate the role of PPARγ in this manuscript. Consistent with the patterns observed in primary patient samples, immunoblot and mRNA expression analysis showed increased but somewhat variable PPARγ expression levels (Supplemental Fig. S1B and C). Moreover, analysis of Cancer Cell Line Encyclopedia (CCLE) data confirmed our experimentally observed PPARγ expression patterns (Supplemental Fig. S1D). Across all tumor types, PPARG mRNA expression was significantly elevated in human cancer cell lines derived from the urinary tract (Figure 1F). Genome expression studies identified two distinct molecular subtypes of bladder cancer; basal and luminal, with luminal tumors representing 75% of NMIBCs and 60% of MIBCs (5). Subtyping of these cell lines using basal markers KRT5/6 and luminal marker FOXA1 indicated that our analyzed UC cell lines are comprised of luminal, basal and undetermined subtypes (Supplemental Figure S1E). Although PPARG copy number alterations are associated with luminal tumors, no clear correlation could be observed between PPARG expression levels and molecular subtypes. Overall, we concluded that PPARγ is frequently overexpressed in UC tumors compared to healthy bladder tissue, but can be detected at low, intermediate and high levels in primary patient samples.
Pharmacological inhibition of PPARγ reduces growth and migratory capacities of multiple UC cell lines in vitro.
In agreement with previously published data (14, 15), treatment of multiple human UC cell lines (5637, HT-1197, TCCSUP, and Cal29) with the PPARγ inverse agonist, T0070907, decreased clonogenic growth over the course of six to ten days, while clonogenic growth of two other cell lines (HT-1376 and T24) did not significantly change (Fig. 2A and B). We confirmed that treatment with T0070907 affected target gene expression by assessing the expression of three PPARγ regulated genes, PLIN2, SCD, and FABP4, by q-RTPCR in 5637 cells (Supplemental Fig. S2A). The cell lines displaying the strongest decrease in proliferation, 5637 and HT-1197, are notable in that they are amongst the UC cell lines with highest PPARG expression levels (see Supplemental Fig. S1D), and that they display PPARG genomic amplification and a RXRα p.S427F activating mutation (16), respectively. We next performed flow cytometric analysis of Annexin V-propidium iodide (PI) staining to determine the effect of the PPARγ inverse agonist on cell viability. Long-term treatment of six UC cell lines with T0070907 did not decrease viability of cells relative to controls (Supplemental Fig. S2B), suggesting that reduced cell number following pharmacological inhibition of PPARγ may result from cell cycle arrest rather than induction of cell death (14). To test this, we performed PI staining on 5637 cells treated with T0070907 for 5 days to analyze the percentage of cells within each phase of the cell cycle. PPARγ inverse agonist treatment significantly increased the percentage of cells in G1 phase and reduced the G2/M phase population relative to control (Fig. 2C, Supplemental Fig. S2C). Additionally, we determined that T0070907 increased the percentage of cells in G0 phase relative to control 4-fold (Fig. 2D) after seven days of treatment.
Figure 2 – Pharmacological inhibition of PPARγ reduces growth of multiple UC cell lines in vitro.
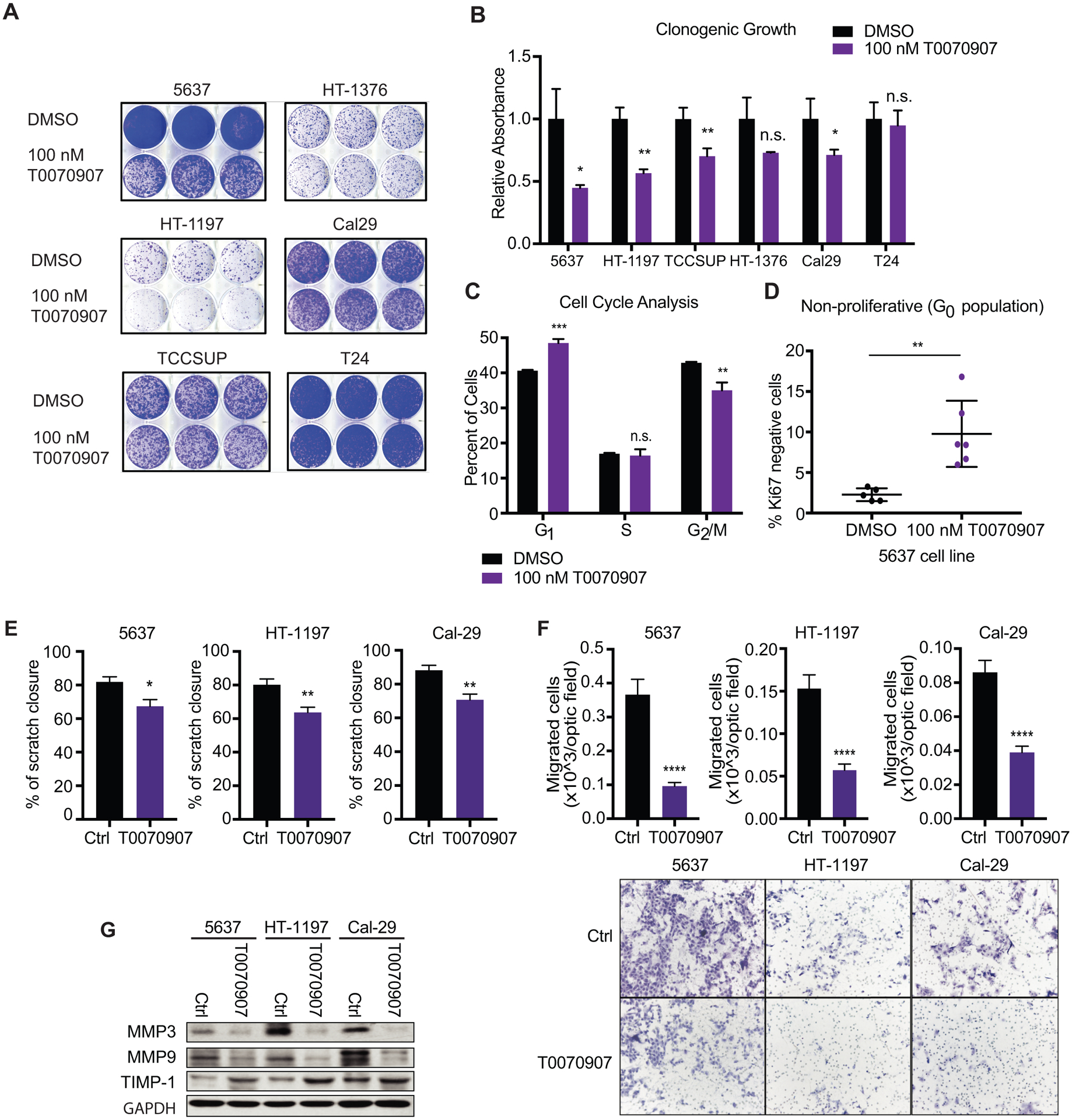
(A) Clonogenic growth of UC cell lines treated with DMSO or 100 nM T0070907 in long-term culture assayed by crystal violet staining. (B) Quantification of crystal violet staining in panel (A), depicted as relative absorbance compared to DMSO. (C) Percentage of 5637 cells in each phase of the cell cycle following DMSO or 100 nM T0070907 treatment, determined by flow cytometric analysis of propidium iodide staining. (D) Percentage of 5637 cells residing in the G0 cell cycle phase, following DMSO or 100 nM T0070907 treatment, identified as Ki67/PI double negative 5637 cells by flow cytometry. (E) Scratch wound migration assay of control and T0070907 treated cells. (F) Boyden chamber migration assay of control and T0070907 treated cells. Quantification (top), representative micrographs (bottom). (G) Western blot analysis of the migration markers Mmp3, Mmp9, and Timp1 in control and T0070907 treated cells. Data are presented as mean ± SEM. **** (p < 0.0001), *** (p < 0.001), ** (p < 0.01), * (p < 0.05), n.s. = not significant. Unpaired student t-test. Black asterisks represent p values for comparison between DMSO and 100nM T0070907 treated cells.
Several reports demonstrated a regulatory role for PPARγ signaling in cell motility and adhesion in various cancer cell types including hepatocellular carcinoma cells (HepG2) and UC cells (29–31). Accordingly, T0070907 delayed closure of scratch wound migration assays (Fig. 2E) and impaired trans-well migration and invasion (Fig. 2F, Supplemental Fig. S2D). Moreover, expression levels of MMP3 and MMP9, two matrix metalloproteases linked to tumor cell migration (32, 33) were decreased upon T0070907 treatment, while expression of the TIMP inhibitor of metalloproteases (TIMP-1) was slightly increased (Fig. 2G and Supplemental Fig. S2 E). These results suggest that pharmacological inhibition of PPARγ reduces cell cycle progression in most of the tested UC cell lines and impairs cell migration.
Genetic inhibition of PPARγ reduces growth and migratory capacities of multiple UC cell lines in vitro.
To validate the effects observed by pharmacological inhibition of PPARγ, we next depleted PPARγ protein from the panel of sensitive UC cell lines using two independent sgRNAs, and the CRISPR-Cas9 gene editing system. PPARγ depletion via CRISPR also inhibited the proliferation of these UC cell lines in vitro (Fig. 3A–C), in a similar manner to that observed through pharmacological PPARγ inhibition. In agreement, PPARG KO also resulted in a block in cell cycle progression in the 5637 cell line (Fig. 3D, Supplemental Fig. S3A), increasing the percentage of cells in G1 and lowering the G2/M phase population relative to control KO. Furthermore, we employed a shRNA-resistant PPARG cDNA to validate the effects of PPARγ depletion on cell growth in UC (Supplemental Fig. S3B and C), and confirmed functionality of the shRNA-resistant PPARG cDNA by its ability to induce adipogenesis in NIH-3T3 cells when cultured in the presence of adipogenic stimuli (Supplemental Fig. S3D). Similar to results obtained using T0070907 treatment, PPARγ depletion impaired cell migration and invasion in all UC cell lines tested, as demonstrated by scratch wound assays (Fig. 3E), Boyden chamber migration/ invasion assays (Fig. 3F, Supplemental Fig. S3E), and decreased expression of MMP3 and MMP9 while stimulating TIMP-1 expression (Fig 3G, Supplemental Fig. S3F). These data confirm the sensitivity of a subset of urinary tract-derived tumor cell lines to PPARG depletion in vitro, as predicted from Achilles heel (Broad institute) shRNA and CRISPR screens (Fig. 3H) (21, 34).
Genome-wide analysis of PPARγ-RXR binding and gene regulation in UC.
To investigate PPARγ occupancy genome-wide in UC, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) in the 5637 cell line for PPARγ, RXR, and IgG and Input controls (Fig. 4A, pre-sequencing ChIP validation shown in Supplemental Fig. S4A). Additionally, we confirmed PPARγ and RXR binding at several strong binding sites by ChIP-qPCR (Supplemental Fig. S4B–D). De novo motif analysis revealed that the majority of PPARγ-RXR binding in UC cells occurs at the direct repeat 1 (DR1) nuclear receptor motif (Fig. 4B), consistent with binding patterns in other cell types like adipocytes, macrophages (35), lung adenocarcinoma (36), and clear cell renal cell carcinoma (ccRCC) (22). A previous report demonstrated that the transcription factors FOXA1, GATA3, and PPARγ are major molecular drivers of the luminal subtype of UC (37), but did not investigate whether these proteins work coordinately based on proximal DNA binding to promote a luminal vs. basal state. Our data indicate that FOXA1, GATA3, and PPARγ likely regulate distinct genes in UC, based on the fact that neither FOXA1 nor GATA3 motifs were enriched at PPARγ-RXR binding sites (Fig. 4B). In agreement with this, the DR1 motif was not enriched under FOXA1 occupied regions in the luminal RT4 bladder cancer cell line (37). PPARγ-RXR heterodimers were predominantly located in intergenic and intronic regions of the genome at 46.08% of sites and 42.37% of sites, respectively (Fig. 4C). To elucidate the transcriptional programs regulated by PPARγ in UC, we annotated PPARγ-RXR binding sites to the nearest gene and performed gene ontology (GO) for 2581 genes: GO pathways involved in migration, adhesion, and regulation of transmembrane receptor tyrosine kinase signaling were most highly represented among this gene set (Fig. 4D).
Figure 4 – Genome-wide analysis of PPARγ-RXR binding and gene regulation in UC.
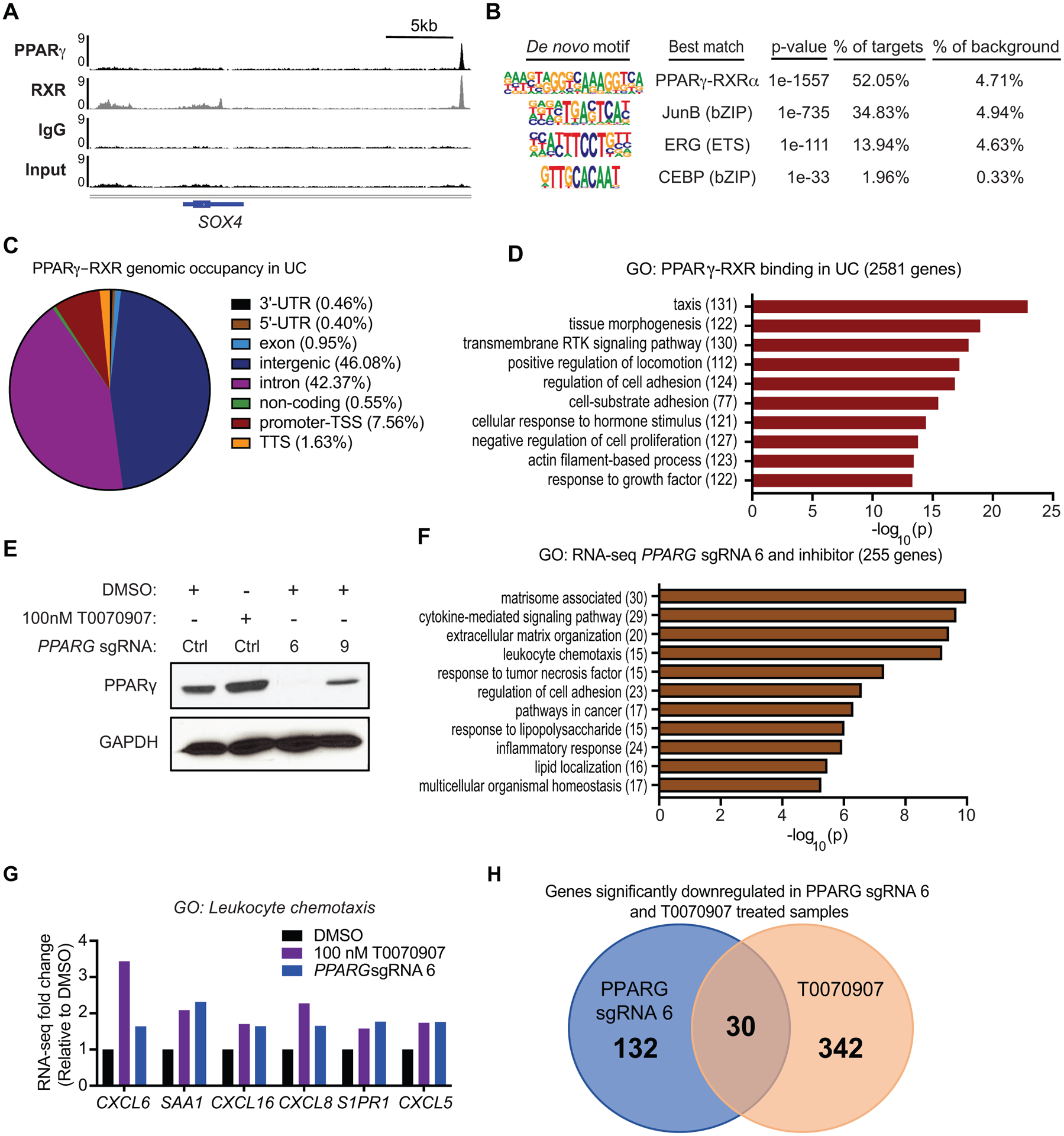
(A) ChIP-seq tracks of PPARγ, RXR, IgG and Input, showing PPARγ-RXR occupancy ~22 kb downstream of SOX4. (B) De novo motif analysis of PPARγ-RXR binding sites in UC. (C) PPARγ-RXR genomic occupancy in UC, defined by HOMER annotate peaks. For bioinformatics analyses displayed in panels C and D, sites were defined by the following criteria: peak score ≥ 10 (≥ 1 read per million) and RXR peak called with strict overlap. (D) Gene ontology of PPARγ-RXR binding sites in UC, annotated to the nearest gene. (E) RNA-seq experimental design and Western blot for PPARγ expression following 100 nM T0070907, PPARG sgRNA 6 or PPARG sgRNA 9. GAPDH was used as loading control. (F) Gene ontology of significantly altered genes by both 100 nM T0070907 treatment and knockout by PPARG sgRNA 6. (G) RNA-seq gene expression changes for select GO category: leukocyte chemotaxis genes following 100 nM T0070907 or PPARG sgRNA 6, relative to DMSO. (H) Venn Diagram showing significantly down regulated genes in both PPARG sgRNA 6 and T0070907 treated 5637 samples as assessed by RNA-seq experiments.
To determine the effects of PPARγ inhibition on gene expression, we performed RNA-seq in the 5637 cell line with four experimental conditions involving either inverse-agonist treatment or gene knockout by two PPARG sgRNA. As the knockout of PPARG was incomplete at the protein level for sgRNA 9 (Fig. 4E), we proceeded to narrow our targets of interest based on overlap between inhibitor treatment and PPARG KO by sgRNA 6. Significantly altered genes by both PPARG CRISPR KO and inhibitor were enriched in pathways related to inflammatory responses (Fig. 4F), including leukocyte chemotaxis, response to tumor necrosis factor, and response to lipopolysaccharide. Interestingly, six chemokines whose expression increased following PPARγ inhibition (Fig. 4G) did not have PPARγ-RXR binding sites within 200 kb of the TSS (data not shown), suggesting that genomic binding of another transcription factor such as NF-κB mediates the inflammatory response. Of note, transrepression of NF-κB targets by PPARγ has been previously suggested (17) to explain the effects of PPARγ signaling in UC, yet a protein complex whereby PPARγ is tethered to chromatin-bound NF-κB is incompatible with the absence of PPARγ occupancy at inflammatory gene loci.
While PPARG sgRNA 6 generated a list of 132 genes significantly downregulated at least 1.5-fold (padj < 0.05), T0070907 treatment generated a list of 342 genes significantly downregulated. We therefore predict that PPARγ targets of interest in regulating UC proliferation fall into a list of 30 protein-coding genes by both inhibitor treatment and PPARG KO by sgRNA 6 (Fig. 4H, Table 1). Of the 30 genes, 7 had a significant positive association with PPARG expression in RNA-seq data from 126 primary bladder urothelial carcinoma samples (2, 18).
Table 1 –
protein-coding genes significantly downregulated by both inhibitor treatment and PPARG KO by sgRNA 6
| Ensembl Stable ID | Gene name | CTT_v_CTD log2FoldChange | CTT_v_CTD padj | CR6_v_CTD log2FoldChange | CR6_v_CTD padj | CR9_v_CTD log2FoldChange | CR9_v_CTD padj |
|---|---|---|---|---|---|---|---|
| ENSG00000099194 | SCD | −1.84836 | 4.75E-32 | −0.967383 | 1.31E-08 | −0.752986 | 3.78E-05 |
| ENSG00000164687 | FABP5 | −0.99932 | 4.64E-22 | −0.643733 | 6.05E-09 | −0.495873 | 2.96E-05 |
| ENSG00000112769 | LAMA4 | −1.61257 | 4.18E-35 | −1.02366 | 5.47E-14 | −0.627017 | 3.51E-05 |
| ENSG00000152661 | GJA1 | −1.07888 | 2.00E-35 | −0.964049 | 5.57E-28 | −0.699964 | 1.50E-14 |
| ENSG00000147872 | PLIN2 | −1.81531 | 1.33E-71 | −0.719112 | 1.49E-11 | −0.346714 | 0.008121 |
| ENSG00000175445 | LPL | −0.689581 | 0.000155974 | −0.642038 | 0.00112915 | −0.507711 | 0.0223404 |
| ENSG00000145819 | ARHGAP26 | −0.974373 | 2.95E-06 | −0.699078 | 0.00319829 | −0.338475 | 0.346469 |
| ENSG00000139668 | WDFY2 | −1.10349 | 0.000942173 | −0.882333 | 0.0209325 | −0.44463 | 0.450809 |
| ENSG00000166582 | CENPV | −1.09435 | 3.02E-13 | −0.723569 | 9.47E-06 | −0.471268 | 0.0148911 |
| ENSG00000272410 | AC022384.1 | −0.733766 | 0.029615 | −1.26105 | 5.31E-05 | −0.406835 | 0.459134 |
| ENSG00000164690 | SHH | −3.85968 | 8.48E-10 | −1.70939 | 0.0220824 | −1.21403 | 0.199725 |
| ENSG00000186205 | MARC1 | −0.872443 | 2.55E-05 | −0.67909 | 0.00324381 | −0.579206 | 0.0234548 |
| ENSG00000270885 | RASL10B | −0.980824 | 0.00119155 | −1.01761 | 0.00131968 | −0.406437 | 0.436146 |
| ENSG00000196636 | SDHAF3 | −0.81483 | 0.00725144 | −0.748726 | 0.0259025 | −0.16698 | 0.825209 |
| ENSG00000162769 | FLVCR1 | −0.598071 | 0.0041765 | −0.606917 | 0.00618048 | −0.15439 | 0.747874 |
| ENSG00000088280 | ASAP3 | −0.752019 | 0.0467213 | −0.905955 | 0.0207149 | −0.362481 | 0.589076 |
| ENSG00000187772 | LIN28B | −1.18362 | 4.01E-12 | −0.698968 | 0.000145048 | −0.354951 | 0.155858 |
| ENSG00000107614 | TRDMT1 | −0.802833 | 0.0411313 | −0.930975 | 0.0236378 | −0.00304135 | 0.997887 |
| ENSG00000185306 | C12orf56 | −0.98203 | 0.000131536 | −0.688257 | 0.0220165 | −0.673972 | 0.0349877 |
| ENSG00000056277 | ZNF280C | −0.661477 | 0.0299821 | −0.726138 | 0.0259025 | −0.370599 | 0.45078 |
| ENSG00000126562 | WNK4 | −1.33002 | 2.29E-08 | −1.05148 | 1.72E-05 | −0.563283 | 0.0770588 |
| ENSG00000107295 | SH3GL2 | −1.52589 | 4.99E-06 | −1.25267 | 0.000556914 | −0.989043 | 0.0159139 |
| ENSG00000176532 | PRR15 | −1.768 | 4.23E-11 | −0.967951 | 0.000823952 | −0.848442 | 0.00536503 |
| ENSG00000137819 | PAQR5 | −0.896192 | 0.0303572 | −0.992695 | 0.0215294 | −0.798751 | 0.115044 |
| ENSG00000007944 | MYLIP | −0.669156 | 0.0307392 | −0.925459 | 0.00324381 | −0.374242 | 0.449459 |
| ENSG00000123576 | ESX1 | −2.43788 | 7.88E-15 | −0.878463 | 0.00763728 | −0.740077 | 0.0428575 |
| ENSG00000198691 | ABCA4 | −2.34148 | 0.0153891 | −2.67712 | 0.0112151 | −5.37503 | 1.70E-07 |
| ENSG00000100433 | KCNK10 | −3.23619 | 0.00525737 | −3.05194 | 0.00622587 | −3.73567 | 0.000766383 |
| ENSG00000257446 | ZNF878 | −1.10965 | 0.0307597 | −1.31489 | 0.0164442 | −0.776613 | 0.279924 |
| ENSG00000068976 | PYGM | −2.27423 | 0.000138108 | −2.30913 | 0.00023289 | −2.65603 | 3.46E-05 |
Denotes significant positive association with PPARG mRNA expression in primary samples
CTD - Control gRNA, DMSO
CTT - Control gRNA, 100 nM T0070907
CR6 - PPARG sgRNA 6
CR9 - PPARG sgRNA 9
PPARG directly regulates SHH expression to ensure proliferation and migration
PPARγ-RXR heterodimers bind several locations up and downstream of sonic hedgehog (SHH), one of the most downregulated genes among all groups in our data set and a regulator of embryonic development and maintenance of tissue polarity (38, 39), within a 200 kb window (Fig. 5A and B). Of note, SHH-expressing cells have been previously shown to give rise to MIBC in a carcinogen-induced mouse model (40), but its connection to PPARγ signaling has not yet been described.
SHH exhibited a strong positive correlation with PPARG expression in primary patient samples according to TCGA data (Fig. 5C). Furthermore, silencing of PPARG reduced SHH expression in three UC cell lines (Fig. 5D and E) and treatment of 5637 cells with the PPARγ agonist rosiglitazone and the inverse agonist T0070907, significantly increased and decreased SHH gene expression, respectively (Supplemental Fig. S5A and B). Interestingly, SHH ablation using two SHH shRNA, reducing SHH expression by ~80% (Supplemental Fig. S5C and D), decreased EdU incorporation in UC cells and cell growth over the course of six days (Fig. 5F, Supplemental Fig. S5E), thus phenocopying the proliferation defect observed upon PPARγ depletion. Previous studies have reported SHH to be involved in invasion and metastasis (41, 42). Accordingly, we observed decreased scratch wound closure and reduced transmembrane migration upon SHH inhibition (Fig. 5G and H). In addition, SHH inhibition impaired UC cell invasion capacities (Supplemental Fig. S5F).
To demonstrate the downstream effect of SHH on PPARγ dependent cellular functions, we overexpressed SHH in PPARG KO cells (Fig. 5I). SHH overexpression upon PPARG inhibition resulted in a partial rescue in cell proliferation (Fig. 5J, supplemental Fig. S5G) and migration/invasion (Fig. 5K and L, Supplemental Fig. S5H), demonstrating that the SHH functions downstream of PPARγ signaling and that PPARγ dependent cell activities in UC cells are at least in part dependent on PPARγ mediated SHH expression.
PPARγ is required to maintain in vivo bladder cancer xenograft growth.
To assess the function of PPARγ in UC tumor growth in vivo, we implanted 5637 control and PPARG sgRNA 6 cells subcutaneously into opposing flanks of 6–7week-old BALB/c nude Mice. We noticed a marked decrease in the growth of PPARG KO tumors relative to controls over a course of 28 days (Fig. 6A and B) with no reports of body weight loss (Fig. 6C). Importantly, expression analyzes of tumor lysates showed that PPARγ depletion was sustained during the 28 days of tumor growth (Fig. 6D–F). In agreement with our in vitro data, PPARG KO tumors showed lower SHH expression levels than control tumors (Fig. 6D, G, H), suggesting that PPARγ is required for UC proliferation and SHH expression in vivo. Additionally, pharmacological PPARγ inhibition as result of daily intraperitoneal T0070907 injections also impaired UC tumor growth (Fig. 6I). No weight loss was reported during the treatment period (Fig. 6J) and tumor weight measured at day 28 post injection showed smaller tumors in T0070907 treated mice than control mice (Fig. 6K). As expected, T0070907 treatment reduced SHH expression levels in UC xenograft tumors (Fig. 6L–N). These data confirm our findings observed in in vitro experiments and demonstrate the importance of PPARγ signaling in SHH expression and UC tumor growth.
Figure 6 – PPARγ inhibition suppresses tumor growth in vivo in xenograft models of UC.
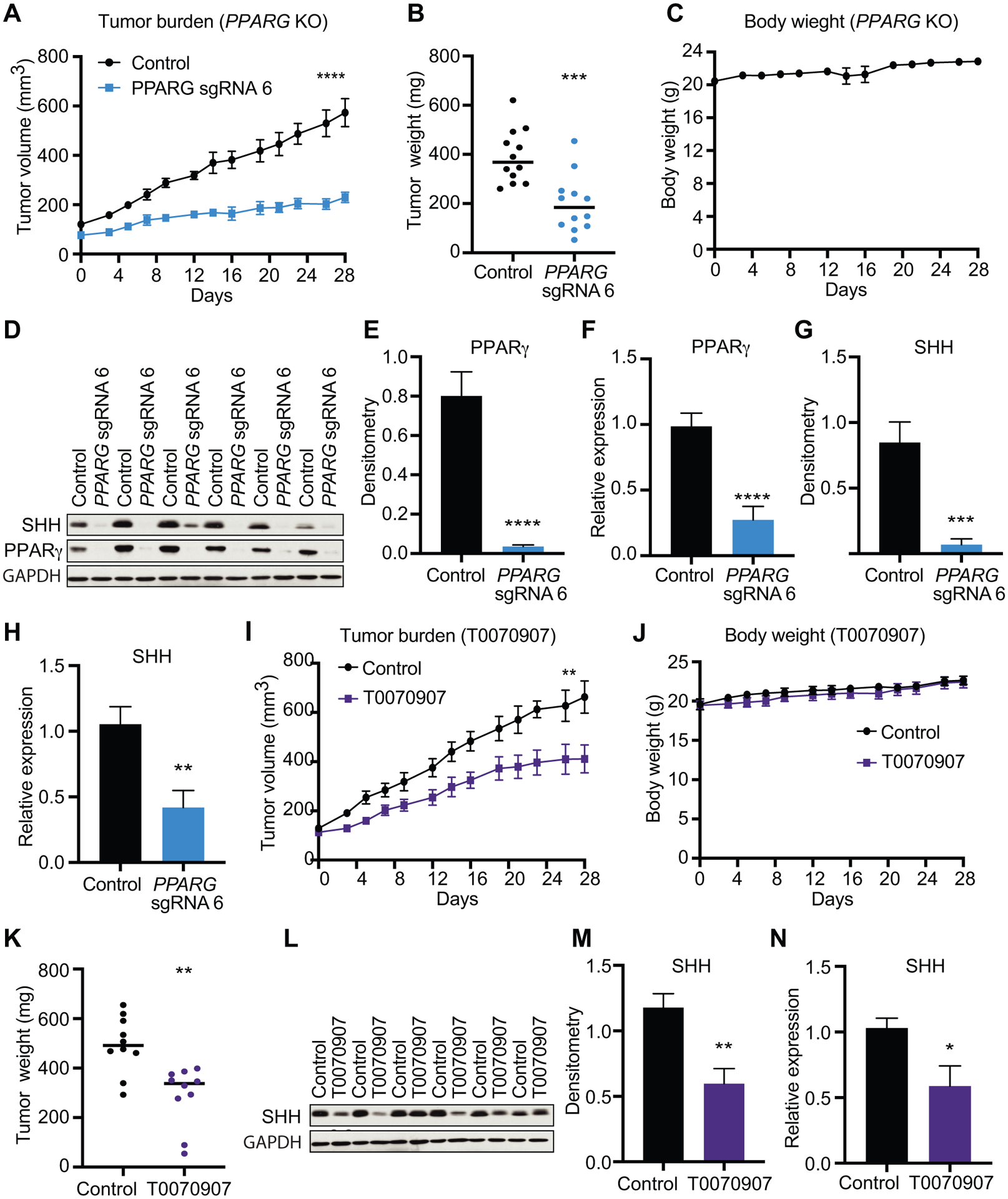
(A) Tumor volume measurements for 5637 control and PPARG sgRNA 6 subcutaneous xenografts at indicated timepoints. (B) Tumor weights of 5637 control and PPARG sgRNA 6 subcutaneous xenografts at day 28 post-injection. (C) Body weight measurements of mice baring 5637 control and PPARG sgRNA 6 subcutaneous xenografts. (D) Western blot analysis for SHH and PPARγ of control and PPARG sgRNA 6 xenografts at day 28 post-injection. GAPDH is used as loading control. (E) Densitometric analysis of PPARγ of control and PPARG sgRNA 6 xenografts at day 28 post-injection. (F) mRNA expression levels of PPARG of control and PPARG sgRNA 6 xenografts at day 28 post-injection. (G) Densitometric analysis of SHH of control and PPARG sgRNA 6 xenografts at day 28 post-injection. (H) mRNA expression levels of SHH of control and PPARG sgRNA 6 xenografts at day 28 post-injection. (I) Tumor volume measurements for control and T0070907 treated 5637 subcutaneous xenografts at indicated timepoints. (J) Body weight measurements of mice baring control or T0070907 treated 5637 subcutaneous xenografts at indicated timepoints. (K) Tumor weights of control and T0070907 treated 5637 subcutaneous xenografts at day 28 post-injection. (L) Western blot analysis for SHH of control and T0070907 treated at day 28 post-injection. GAPDH is used as loading control. (M) Densitometric analysis of SHH of control and T0070907 treated at day 28 post-injection. (N) mRNA expression levels of SHH of control and T0070907 treated at day 28 post-injection. Data are presented as mean ± SEM. **** (p < 0.0001), *** (p < 0.001), ** (p < 0.01), * (p < 0.05). Student’s two-tailed unpaired t-test for pairwise comparisons, or two-way ANOVA for multiple comparisons involving two independent variables. Asterisks represent p values for comparison between control and used experimental conditions.
Discussion
Active PPARγ-RXR signaling has been implicated as a molecular signature of luminal MIBC, yet the cell-autonomous mechanisms by which PPARγ contributes to UC growth are incompletely understood. In this study, we confirmed that multiple UC cell lines exhibit reduced growth upon treatment with PPARγ inverse agonist T0070907 in vitro as previously reported (15), and demonstrated that pharmacological PPARγ inhibition results in reduced cell cycle progression rather than induction of apoptosis. Moreover, T0070907 treatment impaired UC cell migration and invasion. The block in the G1 phase of the cell cycle and the migration/invasion defect observed following treatment with inverse agonist were also replicated by PPARG genetic knockout via CRISPR-Cas9. Interestingly, patient MIBC tissue sections showed higher levels of PPARγ than NMIBC tissue sections. agreeing with our findings that UC cell migration and invasion is PPARγ dependent, which suggests that the invasive nature of MIBC tumors might be PPARγ dependent. To further elucidate the effects of genetic and pharmacological inhibition of PPARγ signaling in UC, we performed ChIP- and RNA-seq to identify commonly deregulated pathways. Our findings corroborate recently published data showing an inverse correlation between PPARγ activity and immune infiltration in UC (17), and also reveal novel PPARγ-RXR transcriptional targets which may be responsible for promoting growth in UC.
Interestingly, lipid and glucose metabolism-related pathways were not among the most highly enriched pathways in our ChIP-seq dataset, suggesting that the PPARγ-RXR cistrome is distinct in UC relative to other cell types including adipocytes and ccRCC (22, 43). However, analyses of downregulated genes comparing control to either PPARγ inhibitor-treated or PPARG sgRNA 6-treated cells suggest that some “canonical” targets are affected in UC, as evidenced by repression of several targets within the KEGG hsa03320: PPAR signaling pathway (FABP4, PLIN2, LPL, SCD) by both manipulations (Supplemental Fig. S5I). The absence of widespread alterations in gene expression involved in lipid and glucose metabolism pathways may be explained by the lack of C/EBP transcription factor co-occupancy with PPARγ in this cistrome. We hypothesize that the targets most important for UC proliferation are related to extracellular matrix organization (matrisome associated), adhesion, and cytokine-mediated signaling pathways (Fig. 4D and F). Future studies performing ChIP-seq in primary urothelial carcinoma tissue would be beneficial to validate the genomic occupancy patterns observed in the 5637 cell line, and to further correlate genomic binding with PPARγ-responsive genes in vivo.
Based on our ChIP- and RNA-seq data, we propose SHH as a novel target gene of PPARγ in UC. Interactions between PPARγ and SHH have not been fully investigated. Several studies demonstrated that SHH signaling leads to PPARγ phosphorylation at Ser112 in adipocytes, and subsequent protein degradation (41, 44), but to our knowledge, no reports are available showing a regulatory role for PPARγ in SHH expression. Briefly, SHH is a signaling effector that binds extracellularly to the transmembrane receptor Patched (PTCH). The PTCH-SHH interaction inactivates the 7-pass transmembrane receptor Smoothened, which allows for the transcription factor GLI1 to translocate to the nucleus and regulate genes involved in proliferation and stem cell self-renewal (39). SHH expression has a weak, yet statistically significant positive association with PPARG expression in primary patient samples according to TCGA data, and treatment of the 5637 cell line with the PPARγ agonist rosiglitazone, increased expression of this gene 4-fold relative to control. These gain- and loss-of-function experiments suggest for the first time that SHH is a direct target gene of PPARγ in in vitro cell culture models of UC (Table 1). The varied levels of natural endogenous ligands of PPARγ in vitro and in vivo certainly affect gene expression, and is an important consideration when trying to identify targets likely to be most relevant to the human disease. Functionally, SHH, a gene known to be essential for growth and proliferation of cells during embryonic development in several tissue types such as neural tube, lungs, skin, hair, and teeth, has been implicated in normal bladder development and in the maintenance of bladder cancer stemness (45, 46). Moreover, our data demonstrate that genetic inhibition of SHH reduces cell proliferation to levels observed in PPARG silenced UC cells. Genetic inhibition of SHH phenocopied the effect of PPARγ inhibition, resulting in impaired UC cell proliferation and migration/invasion. Importantly. SHH overexpression following PPARγ inhibition only partially rescued the PPARγ dependent cell activity defects, suggesting that although SHH seems to be a major factor involved in PPARγ dependent cell activities, additional factors likely contribute. SHH positivity is also associated with the luminal-papillary subclass of MIBC, the same as PPARγ (2). The role of the SHH pathway in UC is controversial, with reports indicating both pro- (47) and anti-tumorigenic (48) roles in UC initiation and progression. Therefore, the relationship between PPARγ signaling, SHH expression, and UC proliferation and migration remains to be further clarified.
As stated above, it is unlikely that a single target gene would underlie PPARγ dependence, other PPARγ targets need to be investigated to elucidate the complete PPARγ dependence in UC. Interestingly, in addition to SHH, our Chip-seq revealed Proline-rich 15 (PRR15) to be another putative PPARγ target with two PPARγ-RXR binding sites within a 200 kb window, which is also significantly downregulated following PPARγ inhibition in all three experimental conditions (data not shown). Although several observations link PRR15 to cell cycle progression and tumorigenesis (49, 50), its role and expression patterns in UC have not been previously described, making the involvement of PRR15 in the PPARγ mediated UC tumorigenesis an interesting factor for future projects.
Finally, we demonstrated that in vivo PPARγ inhibition in UC xenografts impaired tumorigenesis, suggesting that inhibition of PPARγ could be a potential therapeutic strategy for treatment of PPARγ dependent bladder cancer.
In conclusion, our study supports a cell-autonomous, pro-oncogenic role for PPARγ signaling in luminal bladder urothelial carcinoma and provides a candidate list of target genes responsible for these phenotypes from the intersection of in vitro genome-wide approaches and in vivo primary patient sequencing data.
Supplementary Material
Implications:
Genome wide analysis of DNA binding sites for oncogenic factor PPARγ revealed SHH as a novel downstream target involved in UC progression, providing important insight into the tumorigenic nature and molecular mechanism of PPARγ signaling in UC.
Acknowledgements:
The authors thank John Tobias for bioinformatics assistance on the ChIP-seq and RNA-seq datasets, and the Simon lab for helpful discussions. This work was supported by P32HD083185 and F31CA206381 (D.J.S.1,2), Belgian American Education Fellowship (R.M.), R35CA220483 (N.S.), R01 DK098542 (D.J.S.3), and R35CA220483 and NCIPO1CA104838 (M.C.S.).
Footnotes
Conflict of interest disclosure statement: The authors declare no potential conflicts of interest.
References:
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2020. CA Cancer J Clin, 2020. 70(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. , Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell, 2017. 171(3): p. 540–556. e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, et al. , Bladder cancer. Nature reviews Disease primers, 2017. 3: p. 17022. [DOI] [PubMed] [Google Scholar]
- 4.Choi W, Czerniak B, Ochoa A, Su X, Siefker-Radtke A, Dinney C, et al. , Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nature Reviews Urology, 2014. 11(7): p. 400. [DOI] [PubMed] [Google Scholar]
- 5.Rochel N, Krucker C, Coutos-Thévenot L, Osz J, Zhang R, Guyon E, et al. , Recurrent activating mutations of PPARγ associated with luminal bladder tumors. Nature communications, 2019. 10(1): p. 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. , PPARγ signaling and metabolism: the good, the bad and the future. Nature medicine, 2013. 19(5): p. 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna NJ and O’Malley BW, Combinatorial control of gene expression by nuclear receptors and coregulators. Cell, 2002. 108(4): p. 465–474. [DOI] [PubMed] [Google Scholar]
- 8.Tontonoz P, Hu E, and Spiegelman BM, Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell, 1994. 79(7): p. 1147–1156. [DOI] [PubMed] [Google Scholar]
- 9.Shapira SN and Seale P, Transcriptional Control of Brown and Beige Fat Development and Function. Obesity, 2019. 27(1): p. 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis JD, Ferrara A, Peng T, Hedderson M, Bilker WB, Quesenberry CP, et al. , Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes care, 2011. 34(4): p. 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann A, Weill A, Ricordeau P, Fagot J, Alla F, and Allemand H, Pioglitazone and risk of bladder cancer among diabetic patients in France: a population-based cohort study. Diabetologia, 2012. 55(7): p. 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuccori M, Filion KB, Yin H, Oriana HY, Platt RW, and Azoulay L, Pioglitazone use and risk of bladder cancer: population based cohort study. Bmj, 2016. 352: p. i1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lubet RA, Fischer SM, Steele VE, Juliana MM, Desmond R, and Grubbs CJ, Rosiglitazone, a PPAR gamma agonist: Potent promoter of hydroxybutyl (butyl) nitrosamine‐induced urinary bladder cancers. International Journal of Cancer, 2008. 123(10): p. 2254–2259. [DOI] [PubMed] [Google Scholar]
- 14.Biton A, Bernard-Pierrot I, Lou Y, Krucker C, Chapeaublanc E, Rubio-Pérez C, et al. , Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell reports, 2014. 9(4): p. 1235–1245. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein JT, Berger AC, Shih J, Duke FF, Furst L, Kwiatkowski DJ, et al. , Genomic activation of PPARG reveals a candidate therapeutic axis in bladder cancer. Cancer research, 2017: p. canres. 1701.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halstead AM, Kapadia CD, Bolzenius J, Chu CE, Schriefer A, Wartman LD, et al. , Bladder-cancer-associated mutations in RXRA activate peroxisome proliferator-activated receptors to drive urothelial proliferation. eLife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korpal M, Puyang X, Wu ZJ, Seiler R, Furman C, Oo HZ, et al. , Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nature communications, 2017. 8(1): p. 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Network CGAR, Comprehensive molecular characterization of urothelial bladder carcinoma. Nature, 2014. 507(7492): p. 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal, 2013. 6(269): p. pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. 2012, AACR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. , Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nature genetics, 2017. 49(12): p. 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez DJ, Steger DJ, Skuli N, Bansal A, and Simon MC, PPARγ is dispensable for clear cell renal cell carcinoma progression. Molecular metabolism, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013. 29(1): p. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell, 2010. 38(4): p. 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nature methods, 2017. 14(4): p. 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soneson C, Love MI, and Robinson MD, Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, 2014. 15(12): p. 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rochel N, Krucker C, Coutos-Thevenot L, Osz J, Zhang R, Guyon E, et al. , Recurrent activating mutations of PPARgamma associated with luminal bladder tumors. Nat Commun, 2019. 10(1): p. 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HJ, Su Y, Yin PH, Lee HC, and Chi CW, PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer Res, 2009. 29(12): p. 5057–63. [PubMed] [Google Scholar]
- 30.Cheng S, Qian K, Wang Y, Wang G, Liu X, Xiao Y, et al. , PPARgamma inhibition regulates the cell cycle, proliferation and motility of bladder cancer cells. J Cell Mol Med, 2019. 23(5): p. 3724–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang DR, Lin SJ, Ding XF, Miyamoto H, Messing E, Li LQ, et al. , Higher expression of peroxisome proliferator-activated receptor gamma or its activation by agonist thiazolidinedione-rosiglitazone promotes bladder cancer cell migration and invasion. Urology, 2013. 81(5): p. 1109 e1–6. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JF, Wang P, Yan YJ, Li Y, Guan MW, Yu JJ, et al. , IL33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2-NF-kappaB pathway. Oncol Rep, 2017. 38(4): p. 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merchant N, Nagaraju GP, Rajitha B, Lammata S, Jella KK, Buchwald ZS, et al. , Matrix metalloproteinases: their functional role in lung cancer. Carcinogenesis, 2017. 38(8): p. 766–780. [DOI] [PubMed] [Google Scholar]
- 34.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, et al. , Cell-specific determinants of peroxisome proliferator-activated receptor γ function in adipocytes and macrophages. Molecular and cellular biology, 2010. 30(9): p. 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srivastava N, Kollipara RK, Singh DK, Sudderth J, Hu Z, Nguyen H, et al. , Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell metabolism, 2014. 20(4): p. 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warrick JI, Walter V, Yamashita H, Chung E, Shuman L, Amponsa VO, et al. , FOXA1, GATA3 and PPARɣ Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Scientific reports, 2016. 6: p. 38531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villavicencio EH, Walterhouse DO, and Iannaccone PM, The sonic hedgehog–patched–gli pathway in human development and disease. The American Journal of Human Genetics, 2000. 67(5): p. 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rimkus T, Carpenter R, Qasem S, Chan M, and Lo H-W, Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers, 2016. 8(2): p. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin K, Lim A, Odegaard JI, Honeycutt JD, Kawano S, Hsieh MH, et al. , Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nature cell biology, 2014. 16(5): p. 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao Q, Liu J, Xiao L, and Wang N, Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARgamma stability. J Biol Chem, 2019. 294(9): p. 3284–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J-S, Huang X.-h., Wang Q, Huang J-Q, Zhang L.-j., Chen X-L, et al. , Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis, 2013. 34(1): p. 10–19. [DOI] [PubMed] [Google Scholar]
- 43.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, et al. , PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes & development, 2008. 22(21): p. 2941–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh JM, Gao X, McKay J, McKay R, Salo Z, and Graff JM, Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab, 2006. 3(1): p. 25–34. [DOI] [PubMed] [Google Scholar]
- 45.Rupika Sunidhi C, Jeyaprakash MR, and Rajeshkumar R, Sonic Hedgehog gene as a potential target for the early prophylactic detection of cancer. Med Hypotheses, 2019. 137: p. 109534. [DOI] [PubMed] [Google Scholar]
- 46.Syed IS, Pedram A, and Farhat WA, Role of Sonic Hedgehog (Shh) signaling in bladder cancer stemness and tumorigenesis. Current urology reports, 2016. 17(2): p. 11. [DOI] [PubMed] [Google Scholar]
- 47.Islam S, Mokhtari R, Noman A, Uddin M, Rahman M, Azadi M, et al. , Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial‐to‐mesenchymal transition (EMT) in bladder cancer. Molecular carcinogenesis, 2016. 55(5): p. 537–551. [DOI] [PubMed] [Google Scholar]
- 48.Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, et al. , Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer cell, 2014. 26(4): p. 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gates KC, Goetzmann LN, Cantlon JD, Jeckel KM, and Anthony RV, Effect of proline rich 15-deficiency on trophoblast viability and survival. PloS one, 2017. 12(4): p. e0174976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meunier D, Patra K, Smits R, Hägebarth A, Lüttges A, Jaussi R, et al. , Expression analysis of proline rich 15 (Prr15) in mouse and human gastrointestinal tumors. Molecular carcinogenesis, 2011. 50(1): p. 8–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.