

Abstract
Cardiac action potentials are initiated by sodium ion (Na+) influx through voltage-gated Na+ channels. Na+ channel gain-of-function (GOF) can arise in inherited conditions due to mutations in the gene encoding the cardiac Na+ channel, such as Long QT syndrome type 3 (LQT3). LQT3 can be a “concealed” disease, as patients with LQT3-associated mutations can remain asymptomatic until later in life; however, arrhythmias can also arise early in life in LQT3 patients, demonstrating a complex age-associated manifestation. We and others recently demonstrated that cardiac Na+ channels preferentially localize at the intercalated disc (ID) in adult cardiac tissue, which facilitates ephaptic coupling and formation of intercellular Na+ nanodomains that regulate pro-arrhythmic early afterdepolarization (EAD) formation in tissue with Na+ channel GOF. Several properties related to ephaptic coupling vary with age, such as cell size and Na+ channel and gap junction (GJ) expression and distribution: neonatal cells have immature IDs, with Na+ channels and GJs primarily diffusively distributed, while adult myocytes have mature IDs with preferentially localized Na+ channels and GJs. Here, we perform an in silico study varying critical age-dependent parameters to investigate mechanisms underlying age-associated manifestation of Na+ channel GOF in a model of guinea pig cardiac tissue. Simulations predict that total Na+ current conductance is a critical factor in action potential duration (APD) prolongation. We find a complex cell size/ Na+ channel expression relationship: increases in cell size (without concurrent increases in Na+ channel expression) suppress EAD formation, while increases in Na+ channel expression (without concurrent increases in cell size) promotes EAD formation. Finally, simulations with neonatal and early age-associated parameters predict normal APD with minimal dependence on intercellular cleft width; however, variability in cellular properties can lead to EADs presenting in early developmental stages. In contrast, for adult-associated parameters, EAD formation is highly dependent on cleft width, consistent with a mechanism underlying the age-associated manifestation of the Na+ channel GOF.
Keywords: cardiac electrophysiology, sodium channels, computational models, long QT syndrome, intercalated disc
1. INTRODUCTION
Sodium (Na+) channel gain-of-function (GOF) is a pathological condition associated with cardiac disease, including inherited disorders, such as Long-QT syndrome type 3 (LQT3), as well as acquired diseases, such as ischemia and heart failure [1]. LQT3 is linked to mutations in the SCN5A gene that encodes the α -subunit of the cardiac voltage-gated Na+ channel (NaV1.5) [2]. Na+ channel GOF manifests as a late Na+ current, which can prolong action potential duration (APD) and generate proarrhythmic early afterdepolarizations (EADs) [3, 4]. While APD prolongation and EAD formation are reproducible phenomena in isolated myocytes with Na+ channel GOF, in intact tissue, however, this proarrhythmic response can be concealed [5, 6], requiring additional perturbations to manifest the disease phenotype [7, 8]. As such, LQT3 can be referred to as a “concealed” disease: Patients with LQT3-associated mutations often remain asymptomatic, including a normal QT interval, until later in life, with the risk of first cardiac event occurring primarily after puberty and QT prolongation becoming more prominent and increasing with age [9, 10, 11]. While LQT3 accounts for only 5–10% of all patients with long QT syndromes [9], the asymptomatic manifestation in many patients results in high lethality, with Zareba et al. reporting 20% and Schwartz et al. reporting 49% of cardiac events being lethal [12, 13]. At the same time, LQT3 can also present earlier in life, with links to sudden infant death syndrome (SIDS) [14, 15], and indeed within the same family and specific mutation, both adults and children can present as either symptomatic or asymptomatic [16, 17, 18], demonstrating complex age-associated manifestation and individual patient variability.
Recently, we and others have demonstrated that NaV1.5 channels preferentially localize at the intercalated disc (ID), the location of cell-cell junctions in cardiac tissue [19, 20, 21, 22, 23, 24]. In silico studies have predicted that Na+ current at the ID can be modulated via Na+ nanodomain signaling localized at the intercellular cleft, modulating cell-cell coupling via a mechanism known as ephaptic coupling (EpC) [19, 25, 26, 27, 28, 29, 30, 31, 32, 33]. We recently demonstrated that EpC can also be a critical modulator of EAD formation and APD prolongation in tissue comprised of myocytes with LQT3-associated Na+ channel GOF mutations [34, 35]. In this paper we consider two primary effects of EpC: Na+ ion depletion and electrical field effects in the intercellular cleft (i.e., the narrow extracellular space between electrically coupled cells at the ID). Briefly, Na+ influx in a depolarizing cell during the cardiac action potential reduces the electrical potential of the intercellular cleft. This intercellular cleft potential reduction then depolarizes the apposing cell from the extracellular, rather than the intracellular, side of the cell membrane. Additionally, Na+ influx reduces the local Na+ concentration of the intercellular cleft ([Na+]cleft), which governs the flux of the Na+ channels at the ID in both cells. Specifically, for a narrow intercellular cleft, both the electric field effects elevating the transmembrane potential () and local Na+ depletion reducing the Na+ reversal potential () at the ID collectively reduce the Na+ current driving force and thus the Na+ current. This reduction in Na+ current has been termed “self-attenuation,” and has been shown to slow conduction [19, 27, 28, 36].
We recently demonstrated that self-attenuation provides a mechanism that can also suppress APD prolongation and EAD formation in cardiac tissue with Na+ current GOF in tissue simulations and isolated guinea pig heart experiments [34, 35]. Specifically, simulations and experiments showed that expanding the intercellular cleft prolonged APD and promoted EAD formation. Further, cleft narrowing promoted localized depletion of [Na+]cleft, which reduced the late Na+ current at the ID, mitigated APD prolongation, and suppressed EADs. Notably, the “concealing” effects of the Na+ channel GOF manifestation required preferential localization of the Na+ channels at the ID, while altered gap junctional (GJ) coupling minimally altered APD prolongation [34].
Importantly, several properties related to the self-attenuation mechanism are known to change with age: Na+ channel and GJ expression are both age-dependent. Neonatal cells have immature, i.e not fully formed, IDs, and Na+ channels and GJs are primarily diffusively distributed throughout the sarcolemma. Cai et al. and Cordeiro et al. both report that pediatric cardiomyocytes produce less Na+ current than adult cardiomyocytes [37, 38], consistent with reduced Na+ current expression. Additionally, Na+ channel and GJ distribution change significantly with age. Studies found that the primary ventricular GJ protein, connexin 43 (Cx43), remains almost undetectable until 23 weeks in utero and that GJs are sporadically distributed on the sarcolemma in neonatal cardiomyocytes [39, 40, 41, 42]. Vreeker et al. noted that Cx43 gradually lateralizes at 5 months postnatal and begins to preferentially localize at the ID around 2.5 to 5 years old, with full preferential localization occurring around 7 years of age [39]. Similarly, NaV1.5 channels are primarily on the lateral membrane in neonatal cells, and begin to preferentially localize at the ID around 5 months postnatal [39]. Thus, Na+ channels are primarily localized at the ID at an earlier developmental stage than GJs [39, 43]. Additionally, cell size, which broadly influences all electrical activity by altering cell volume, surface area, ion channel expression, membrane capacitance, etc., also increases with age, as studies have shown that adult cardiomyocytes are larger than neonatal cardiomyocytes [39, 38, 44, 45].
Based on this proposed mechanism for concealment of the LQT3 phenotype, we hypothesize that the age-associated manifestation of LQT3 may be directly linked to changes in cellular and tissue properties that occur with development. In this paper, we perform a wide parameter investigation, varying age-associated parameters including Na+ channel localization at the ID (IDNa), cell size (S), Na+ channel density (ρNa), and gap junctional conductance (fgap), and measure APD in simulated guinea pig cardiac tissue with LQT3-associated Na+ channel GOF.
To our knowledge, only one study has investigated the relationship between intercellular cleft width and age in any setting; our prior work found a positive correlation between intercellular cleft width and age in atrial tissue in atrial fibrillation and control patients later in life (approximately 40–80 years old) [46]. However, no studies have investigated intercellular cleft width in ventricular tissue in either the setting of control or LQT3 tissue and the early developmental changes, i.e., neonatal to adult, relevant to the LQT3 concealment hypothesis. Thus, in our study, we investigate a range for intercellular cleft width values consistent with our prior measurements [20, 35]. In addition to cleft width, simulations predict that total Na+ current conductance is a critical factor in EAD formation. We find that for early development stages, EADs are suppressed; however, the LQT3 phenotype can be “unmasked” by multiple possible structural or cellular changes, including increases in cell size and Na+ channel density, with minimal dependence on cleft width. In contrast, for adult stages, EADs form for all conditions, unless suppressed by narrow cleft width, consistent with our prior studies in adult myocardium [34].
2. Methods
Full details of the computational model are provided in Supporting Material. Briefly, we simulate a 50-cell cable of guinea pig ventricular myocytes [47], incorporating a model of either a wild-type (WT) or LQT3-associated mutated (Y1795C) Na+ channel [48] as seen in Fig. 1. Na+ channel dynamics were governed by a 13-state Markov model [48] previously shown to produce a late Na+ current in the mutant channel, with channel states representing two modes of gating: a baseline background gating mode and a non-inactivating burst mode (Fig. 1C). As in our prior work [29, 34, 35] and work by others [19], we account for non-uniform Na+ subcellular localization by spatially discretizing each cell into axial membrane patches along the length of the cell and ID membrane patches at the ends of the cell. The number of axial patches varied with the size of the cell (described below), with each axial patch fixed in length (Lp = 10μm).
Figure 1:
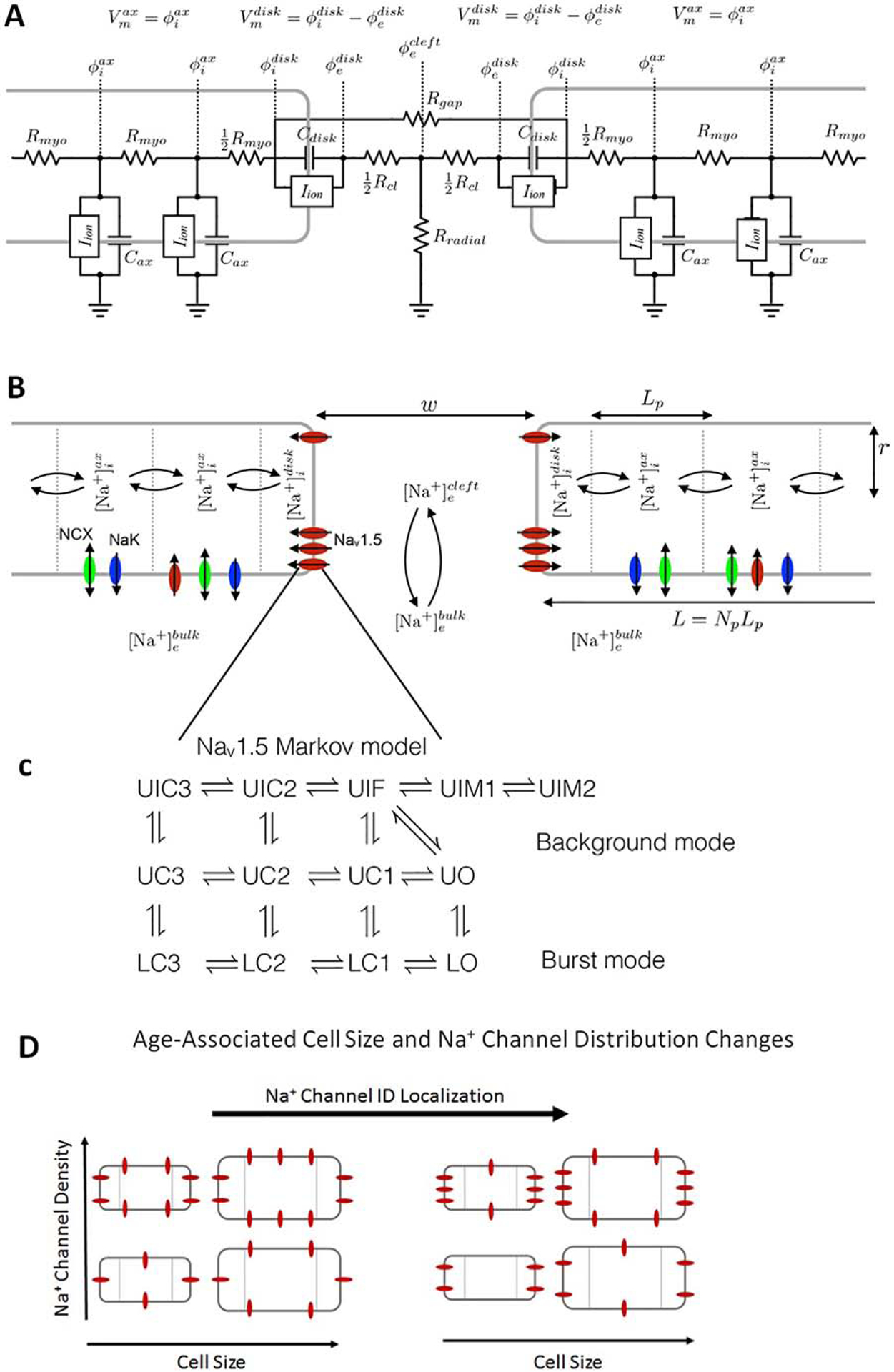
Schematic of the computational model. A: Electric circuit representation of coupled myocytes. Intracellular nodes are coupled via a myoplasmic resistance (Rmyo). End nodes are coupled via a gap junctional resistance (Rgap). Extracellular potentials at the disc and intercellular cleft ( and , respectively) are governed by a T-shaped network of two axial resistances in the intercellular cleft (Rcl) and one radial resistance (Rradial). B: Na+ concentration in diffusively coupled compartments, including intracellular Na+ in the axial and disc compartments ( and ) and extracellular Na+ in the intercellular cleft and bulk spaces ( and ). C: Markov model of the Na+ channel. Upper (U) states represent the background mode of gating and lower (L) states represent the burst mode with two open (O) conducting states. Transition rates between states depend on genotype (wild-type or mutant). D: Representation of age-associated change in model parameters, including changes in cell size (S), Na+ channel density (ρNa), and Na+ channel ID localization (IDNa).
Cells are coupled via GJs and EpC: gap junctional coupling is represented via gap junctional resistances (Rgap) coupling ID nodes of adjacent cells. EpC is represented by a T-shaped junction of a radial bulk (Rradial) and two intercellular cleft resistances (Rcl), which are inversely proportional and proportional to intercellular cleft width w, respectively. Intracellular nodes are coupled with a myoplasmic resistance (Rmyo) (Fig 1A). The nominal value for Rgap is defined as , and changes in GJ coupling are accounted for by adjusting the GJ scaling factor fgap (between 0 and 1), such that . We note that age-associated changes in GJ localization are represented as changes in GJ coupling (i.e., changes in fgap), as all GJs are located at cell ends in the one-dimensional tissue model.
We account for dynamic [Na+] in three spaces: (i) the intercellular cleft , with volume proportional to w and mediated by ID Na+ current and passive diffusion with the bulk extracellular space, (ii) the ID , mediated by and intracellular diffusion, and (iii) the axial intracellular space , mediated by axial Na+ current and intracellular diffusion. The cable was paced at one end with a specified basic cycle length (BCL). The manifestation of arrhythmias for LQT3 patients typically occurs for bradycardic conditions, i.e., slow heart rates. For most simulations, we utilize a BCL = 1000 ms or a pacing rate of 1 Hz, which is bradycardic for the guinea pig model that is utilized for this study. We also consider both faster (BCL = 500 ms) and slower (BCL = 2000 ms) pacing rates.
In addition to GJ coupling, we perform simulations in which we adjust several key age-associated properties: (1) We vary Na+ channel ID localization (IDNa) between 0.1 and 1, to account for Na+ channel redistribution, where IDNa = 1 or 100% represents all Na+ channel localized at the ID. (2) We define and vary a cell size scaling factor (S) between 0.2 and 1 to account for cell size growth. The cell geometry is assumed to be cylindrical, with radius r and length L, defined as r = Sr0 and = L = npLp, where the nominal adult radius r = 11 μm, the number of axial patches , and the maximum axial membrane patches . Note we only consider values of S such that np is a whole number, and that nominal adult cell length . Additionally, note that since S scales both cell length and radius, cell membrane surface area is scaled by S2, e.g., S = 0.4 corresponds with surface area scaled by a factor of 0.16. (3) We also vary the Na+ channel density (ρNa) between 0.2 and 1 to account for age-associated changes in Na+ channel expression. The total cellular Na+ conductance is proportional to both ρNa and total cell surface area, such that we can define a normalized total Na+ channel conductance (GNa = ρNaS2, also between 0 and 1), where the total Na+ channel conductance (in physical units) is equal to GNa, scaled by the nominal total Na+ channel conductance . Thus, for ρNa and S of 100%, total Na+ channel conductance is . (4) Finally, we vary the intercellular cleft width (w) from 10 to 50 nm, consistent with intercellular cleft width ranges measured at the perinexus in our previous work [21, 34, 35].
3. Results
3.1. Mechanisms of EAD formation in mutant cardiac tissue
We simulate cardiac tissue comprised of myocytes with mutant (Y1795C) Na+ channels and modulate cell size (S), Na+ channel density (ρNa), Na+ channel localization at the ID (IDNa), and intercellular cleft width (w). Motivated by our previous findings that intercellular cleft width is a key regulator of APD changes [34, 35] and our hypothesis that cell size is a critical factor in age-dependent regulation, we first investigate the mechanism for APD modulation via intercellular cleft width (w) and cell size (S). We consider cases of narrow and wide intercellular cleft spacing, with preferential localization of Na+ channels at the ID (IDNa = 90%) and high whole cell Na+ channel density (ρNa = 100%). In Fig. 2, the time series for transmembrane voltage (Vm), Na+ current at the ID , Na+ channel open probability at the ID , Na+ concentration in the intercellular cleft ([Na+]cleft), and the reversal potential are shown for a bradycardic pacing rate (BCL of 1000 ms). For simulated mutant tissue with narrow intercellular cleft width (Fig. 2A, left, w = 10 nm), we find that APD is comparable to wild-type tissue (black line). That is, no EADs form regardless of cell size. For all sizes, is increased due to the mutant channel kinetics, resulting in a small amplitude late Na+ current (Fig. 2B, C, left). However, as we previously demonstrated [34, 35], the initial Na+ influx during the action potential upstroke drives a transient local depletion of [Na+]cleft. This depletion results in an associated decrease in , reducing the Na+ current driving force and in fact producing a brief outward Na+ current (due to transiently). Subsequently, the late Na+ current drives a secondary depletion in [Na+]cleft, further reducing the and driving force. This secondary depletion is a negative feedback that suppresses EAD formation, which serves as a protective mechanism against arrhythmia triggers.
Figure 2:
Early after-depolarizations (EADs) depend on cleft width and cell size. (A) Transmembrane voltage (Vm), (B) Na+ current at the ID , (C) Na+ channel open probability at the ID , (D) cleft Na+ concentration ([Na+]cleft), and (D) the Na+ reversal potential at the ID are shown in mutant tissue for narrow (left) and wide (right) intercellular cleft widths (w). Traces for wild-type (WT) are shown for 100% cell size for comparison (black lines). For clarity, traces are shown for cell 25 out of 50 in the one-dimensional chain of cells. Parameters: BCL = 1000 ms, IDNa = 50%, ρNa = 100%, fgap = 100%.
For an expanded cleft and thus larger intercellular cleft volume, [Na+]cleft is depleted to a lesser extent, maintaining an elevated and larger driving force (Fig. 2B–E, right), compared to tissue with narrow clefts. For larger cells (S = 80 and 100%, green and magenta lines), the late Na+ current drives EAD formation; however, for smaller cells (S = 60%), no EADs form (Fig. 2A, right, red line) and APD is comparable to wild-type tissue (black line). Thus, we find that for a wide cleft width, the interactions between cell size and EAD formation are complex and involve two opposing influences: a larger cell has a larger Na+ current conductance (due to larger membrane surface area) and thus larger late Na+ current (Fig. 2B, right), which tends to promote EADs. However, the larger current leads to greater [Na+]cleft depletion (Fig. 2E, right), which tends to suppress EADs. In this setting of a wide cleft, the cell size influence on total Na+ current conductance is the more critical factor, and thus EADs are promoted for large cell size and suppressed for small cell size.
3.2. Cell size, Na+ channel density and distribution, and intercellular cleft width govern action potential duration in mutant cardiac tissue
We next more broadly investigate the interdependence between cell size (S), Na+ channel density (ρNa), Na+ channel ID localization (IDNa), and intercellular cleft width (w) in mutant cardiac tissue on APD and EAD formation (Fig. 3). We investigate a wide range of parameters to assess the influence of their respective age-associated changes on APD. For high preferential Na+ channel localization at the ID (IDNa = 90%, Fig. 3, right column) and narrow cleft width, no EADs form (as in Fig. 2, left), such that APD is negligibly dependent on cell size S and Na+ channel density ρNa, with APD values comparable to wild-type tissue (black lines). For tissue with expanded clefts (w = 40 nm, bottom row), APD increases as both S and ρNa increase. Thus, these simulations predict that for conditions associated with adult tissue (dashed black box), i.e., preferential Na+ channel ID localization, high Na+ channel density, and large cell size, EAD formation requires cleft expansion. In contrast, EADs are suppressed due to cleft narrowing, low Na+ channel density, or small cell size, such that APD is comparable to wild-type tissue.
Figure 3:

Action potential duration (APD) prolongation due to increased cell size and Na+ channel density in mutant tissue. APD is shown as a function of cell size (S) for different values of Na+ channel density (ρNa) for 10% (left column), 50% (middle column), and 90% (right column) Na+ channel ID localization (IDNa) for intercellular cleft widths of w = 10 nm (top row), w = 20 nm (middle row), and w = 40 nm (bottom row). APD for wild-type (WT) is shown for 100% ρNa for comparison (black lines). Parameters: BCL = 1000 ms, fgap = 100%. Parameter regimes associated with neonatal (gray boxes) and adult (black boxes) tissue are highlighted.
In the setting of more uniform Na+ channel distribution (Fig. 3, left and middle columns), we find similar trends: APD prolongs with increased cell size and Na+ channel density. Additionally, consistent with our prior findings [34, 35], more uniform Na+ channel distribution (i.e., reduced IDNa) further promotes EAD formation, due to less [Na+]cleft depletion (similar to the mechanism described above). EADs are similarly suppressed by low Na+ channel density and small cell size, however in a manner that is weakly dependent on cleft width. Importantly, simulations predict that for conditions associated with neonatal tissue (dashed gray box), i.e., more uniform Na+ channel localization, low Na+ channel density, and small cell size, EADs are typically suppressed; however, an increase in either cell size or Na+ channel density promotes EADs, independent of cleft width.
APD is shown as a function of cleft width in Fig. S1, which further illustrates that cleft width-dependence is mitigated as ID localization decreases. Simulations in wild-type tissue for a wide range of conditions illustrate minimal dependence on all of the investigated properties, with APD slightly increasing with increasing size (Fig. S2). This is consistent with APD in mutant tissue depending specifically on regulation of the Na+ channel GOF.
3.3. Age-associated changes in conduction velocity in mutant cardiac tissue
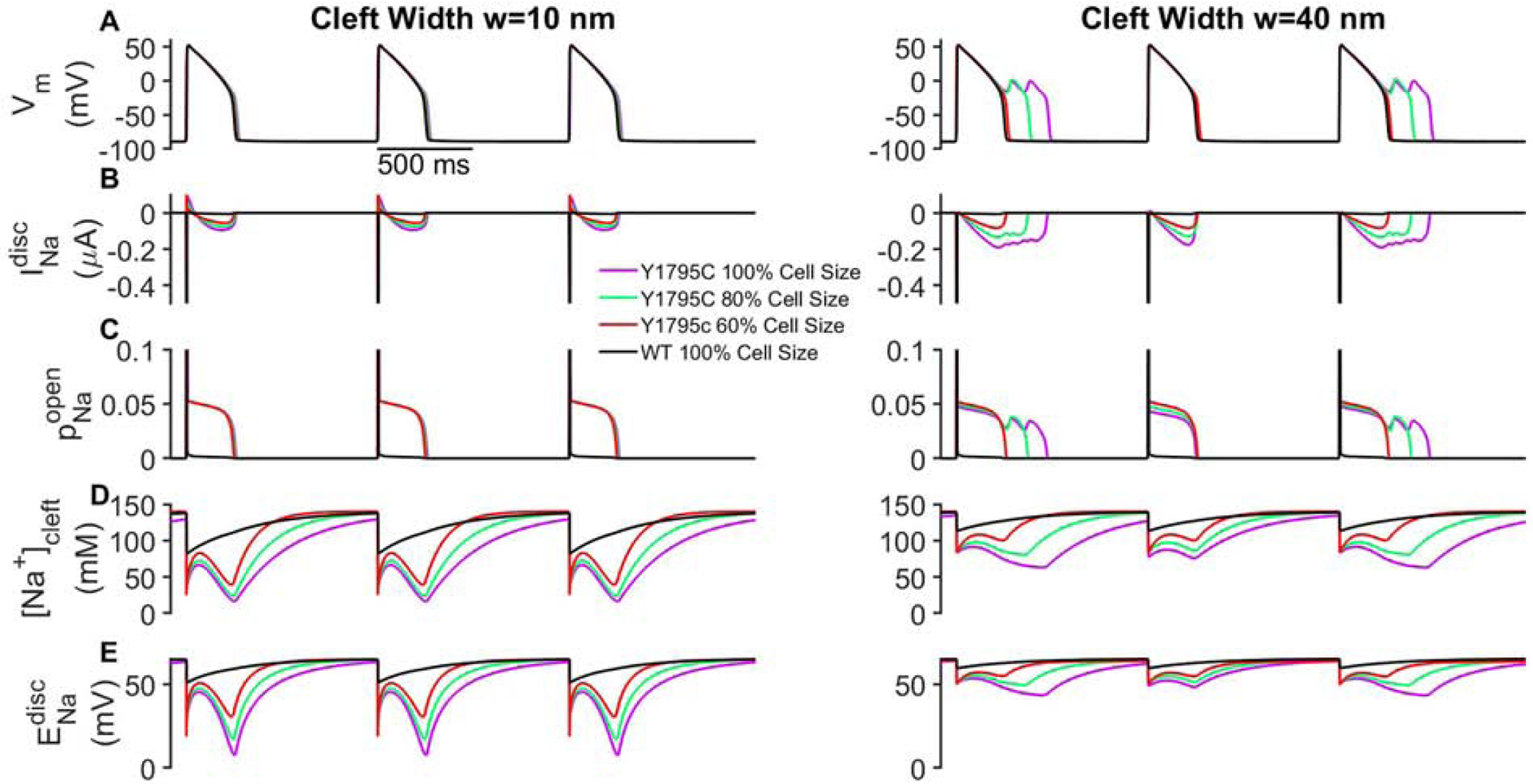
As the above studies reveal the complex age-associated regulation of EAD formation by subcellular and cellular level properties, in particular properties governing Na+ channels, we next investigate how these age-associated changes also impact CV in mutant cardiac tissue, considering different levels of GJ coupling (fgap), which also varies with maturation. We plot CV as a function of S, for different levels of ρNa and IDNa, for weak GJ coupling, associated with early development (fgap = 4%, Fig. 4A), and strong GJ coupling, associated with adult myocytes (fgap = 100%, Fig. 4B). As expected, CV is faster for strong GJ coupling, compared with weak coupling. Across all conditions, increasing ρNa increases CV, i.e., greater Na+ channel density promotes faster conduction. Interestingly, increasing cell size has opposing effects, depending on GJ coupling: for weak coupling, increasing S decreases CV, while for strong coupling, increasing S increases CV. The regulation by Na+ channel localization (IDNa) is relatively small, with high Na+ ID localization (IDNa = 90%) generally increasing CV for weak coupling and decreasing CV for strong coupling, consistent with prior studies of EpC [19, 29].
Figure 4:
Conduction velocity (CV) dependence on cell size and Na+ channel density in mutant tissue. CV is shown as a function of cell size (S) for different values of Na+ channel density (ρNa) for 10% (left column), 50% (middle column), and 90% (right column) Na+ channel ID localization (IDNa) for (A) weak (fgap = 4%) and (B) strong (fgap = 100%) gap junctional (GJ) coupling. CV for wild-type (WT) is shown for 100% ρNa for comparison (black lines). Parameters: BCL = 1000 ms, cleft width w = 40 nm.
Importantly, we note that for all conditions, CV in wild-type tissue (black lines) is comparable to the mutant tissue with comparable Na+ channel density, suggesting that the mutation has minimal influence on CV. Thus, while age-associated changes in subcellular and cellular properties can influence conduction, the primary influence of developmental regulation of the Na+ channel GOF occurs via changes in APD, as might be expected in the setting of a LQT3-associated mutation.
3.4. Total cell Na+ channel conductance governs EAD formation in mutant cardiac tissue
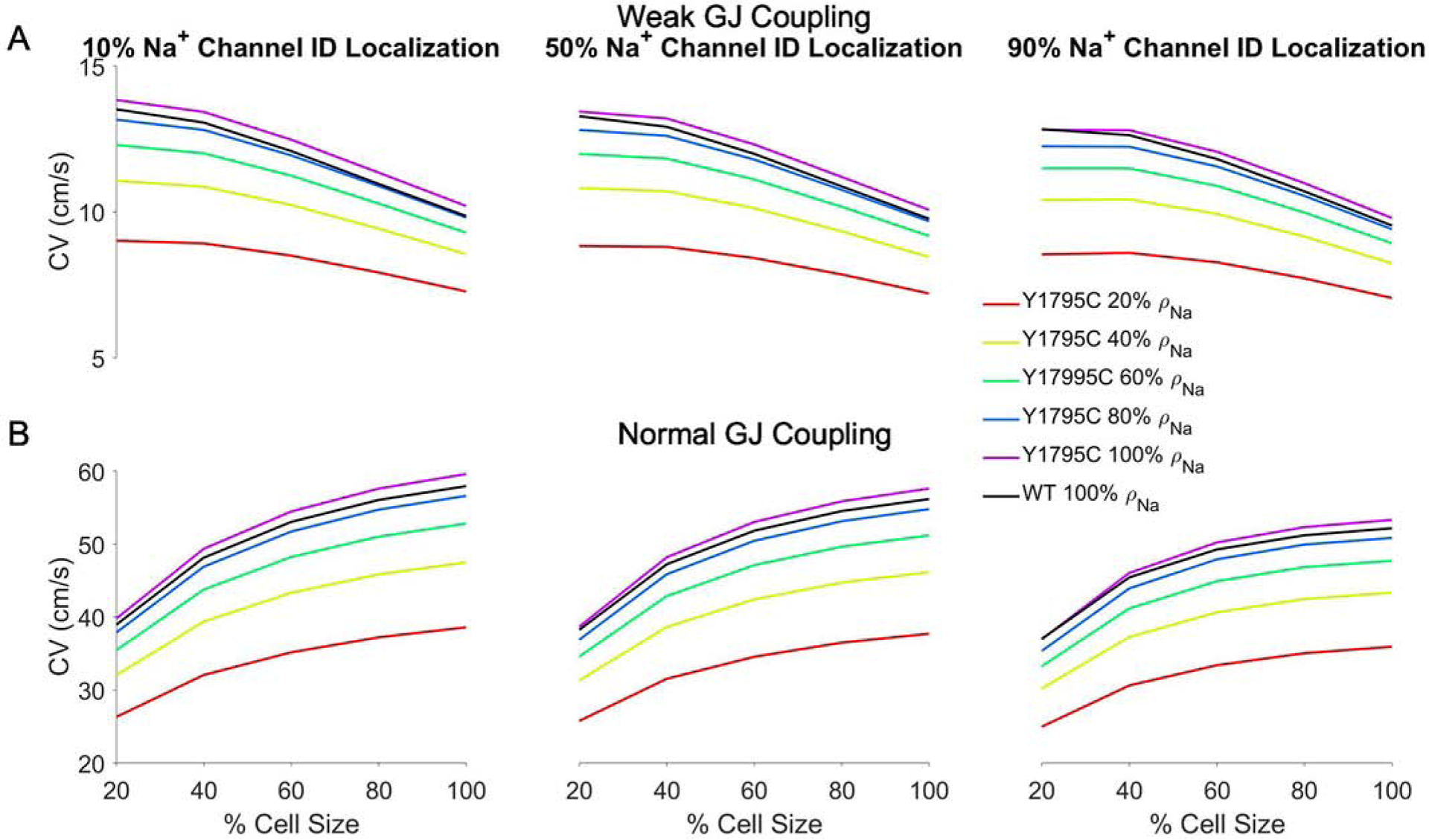
We next further investigate the properties governing the age-associated changes in APD. The previous results demonstrate that EAD formation in mutant tissue can be driven by large cell size or high Na+ channel density, both of which correspond with increases in total cell Na+ current conductance GNa, i.e., more total Na+ channels. Therefore, we next generate a scatter plot of APD as a function of the normalized GNa (Fig. 5A), where GNa = 1 corresponds with adult levels of both ρNa and S, for different values of Na+ ID localization IDNa and cleft width w. We find that for smaller values of GNa, APD gradually increases as GNa increases, with minimal dependence on either IDNa or w. However, above a threshold level of GNa, APD dramatically increases as a result of EAD formation.
Figure 5:
Action potential duration (APD) generally prolongs with increased total cell Na+ conductance in mutant tissue. A: APD is shown for specific values of normalized total cell Na+ conductance (GNa) (determined by varying cell size and Na+ channel density), for different Na+ channel ID localization (IDNa) values and for narrow (w = 10 nm), moderate (w = 20 nm), and wide (w = 40 nm) cleft widths. Different symbols denote cell size. B: The total cell Na+ conductance threshold for EADs is shown as a function of IDNa for varying cleft widths. Parameters: BCL = 1000 ms, fgap = 100%.
In Fig. 5B, we plot the GNa threshold value for EADs, defined as , as a function of IDNa and w, and we find that generally increases as IDNa increases. Thus, as Na+ channels are more preferentially localized at the ID, a larger whole cell Na+ current conductance is necessary to promote EADs. This is consistent with the finding that the age-associated redistribution of Na+ channels to the ID is protective (i.e., suppresses EADs), in the context of the concurrent age-associated increase in total Na+ current conductance (due to both increases in cell size and channel density).
Interestingly, for a given Na+ ID localization, the threshold total conductance tends to decrease as cleft width increases, consistent with wider cleft width (associated with less [Na+]cleft depletion) requiring fewer Na+ channel to promote EADs. However, the dependence on cleft width is not a strictly decreasing relationship for low Na+ ID localization (Fig. S3), a condition in which cleft width dependence is relatively weaker. This complex relationship arises due to increased GNa both increasing late Na+ current and enhancing cleft depletion.
Closer examination of the results in Fig. 5 illustrate that, while APD generally increases with GNa, this is not strictly a monotonic relationship. Noting that increases in GNa can arise from increases in either cell size S or Na+ channel density ρNa, we next investigated the APD-GNa relationship for GNa values near , separately for each value of IDNa and for an expanded cleft width (w = 40 nm) that promotes EADs (Fig. 6). We find that there are typically several pairs of points with nearly identical values for GNa, but different APD measurements; we consider two such pairs, points labeled as A1 and A2, and B1 and B2. We first consider A1 and A2, which correspond with (S, ρNa) values of (80%, 100%) and (100%, 60%), respectively. For small IDNa, we find that APD for point A1 is consistently longer, compared with point A2. Similarly, for B1 and B2, which correspond with (S, ρNa) values of (60%, 100%) and (80%, 60%), respectively, APD for point B1 is consistently longer, compared with point B2. Thus, for comparable levels of total Na+ current conductance, a smaller cell size and higher Na+ channel density are associated with longer APD and enhanced EAD formation. Interestingly, as IDNa increases, EAD formation is suppressed (as described above), such that the APD measurements for the corresponding pairs tend to converge to similar values. Importantly, this demonstrates that variations in cell size and Na+ channel density are most critical in EAD formation for small IDNa, which occurs during early developmental stages.
Figure 6:

Action potential duration (APD) in mutant tissue near the EAD transition regime. APD is shown for specific values of normalized total cell Na+ conductance (GNa), with key points are labeled for each Na+ ID localization value (IDNa). Corresponding points (A1 and A2) represent APD measurements for similar GNa values, with different cell size (S) and Na+ channel density (ρNa), with a similar relation for GNa values for points B1 and B2. Parameters: BCL = 1000 ms, cleft width w = 40 nm, fgap = 100%. Different symbols denote cell size (same legend as in Fig. 5).
3.5. Rate-dependent properties of action potential duration prolongation in mutant cardiac tissue
We next investigate the rate-dependence of the age-associated changes in mutant tissue and the manifestation of the LQT3 phenotype. Our prior analysis considered a BCL of 1000 ms, which as noted above is a bradycardic rate for guinea pigs. In Fig. 7, we plot APD as of a function of cell size, and Na+ channel density and localization, for both a faster pacing rate (A, shorter BCL of 500 ms) and slower pacing rate (B, longer BCL of 2000 ms) and an expanded cleft width (w = 40 nm). For a faster pacing rate (Fig. 7A), increasing cell size and Na+ channel density both prolong APD, as above, but with minimal dependence on Na+ channel localization. Importantly, for all conditions, APD is only marginally longer compared with wild-type tissue (black lines) and no EADs form for all conditions.
Figure 7:

Action potential duration (APD) prolongation is suppressed at shorter cycle lengths and promoted at longer cycle lengths in mutant tissue. APD is shown as a function of cell size (S) for different values of Na+ channel density (ρNa) for 10% (left column), 50% (middle column), and 90% (right column) Na+ channel ID localization (IDNa). APD for wild-type (WT) is shown for 100% ρNa for comparison (black lines). Parameters: (A) BCL = 500 and (B) 2000 ms, cleft width w = 40 nm, fgap = 100%.
For a slower pacing rate (Fig. 7B), we find the same trends as for BCL of 1000 ms, i.e., increasing cell size and Na+ channel density both prolong APD while increasing Na+ channel ID localization shortens APD. Further, for the more bradycardic pacing rate, APD is prolonged relative to values for BCL of 1000 ms, such that EADs form for a wider range of conditions. Thus, we find that faster pacing rate suppresses EADs, while slower pacing rate promotes EADs across all age-associated conditions, in agreement with our previous findings in adult myocytes [34]. In Supporting Material, we demonstrate that narrow cleft width similarly suppresses EADs for the slower pacing rate (Fig. S4), as described above, while APD negligibly depends on cleft width for the faster pacing rate (Fig. S5).
Thus, collectively these results demonstrate the following age-associated changes in mutant cardiac tissue: 1. For all conditions, both Na+ channel redistribution to the ID and narrowing of the intercellular cleft suppress EAD formation. 2. Cell size increases without proportional increases in Na+ channel expression (thus reducing Na+ channel density) are similarly protective and suppress EADs. 3. In contrast, Na+ channel expression increases without proportional increases in cell size (thus increasing Na+ channel density) promote EADs. 4. Increases in total Na+ channel conductance (due to either increased cell size or channel density) prolong APD, with a larger conductance required to promote EADs as Na+ channel ID localization increases. 5. Faster pacing rate suppresses EAD formation and slower pacing rate promotes EAD formation for all age-associated conditions.
3.6. Developmental changes in action potential duration and EAD formation in LQT3
The above results illustrate a complex relationship between APD in cardiac tissue with a LQT3-associated Na+ channel GOF mutation, and how EAD formation depends on a wide range of properties for age-associated expression and tissue structural changes. As discussed in the Introduction, neonatal tissue properties are associated with small cell size and low Na+ channel expression and ID localization, while adult tissue properties are associated with large cell size and high Na+ channel expression and ID localization. During both development and disease progression, we hypothesize that these key properties are variable in time and also exhibit variability between individual patients. For a final analysis, we illustrate ranges of APD values for an age-associated progression, considering neonatal, intermediate developmental stages, and adult tissue with a LQT3 mutation (Fig. 8). For each stage, to account for individual variability and parameter uncertainty, we consider a range of values for each of the key parameter values (S, ρNa, and IDNa), varying these parameters over a 20% range, and plot the minimum and maximum APD values over all of the parameter conditions for that stage. Additionally, due to a lack of evidence of the precise order of developmental changes, we consider different possible parameter conditions for the intermediate stages. Finally, we illustrate the changes in minimum, maximum, and mean APD values during the developmental progression for different values of cleft width. The figure legend (bottom) illustrates the developmental changes in cell size and Na+ channel density and localization, and parameter ranges for each developmental stage are listed in Table S2.
Figure 8:
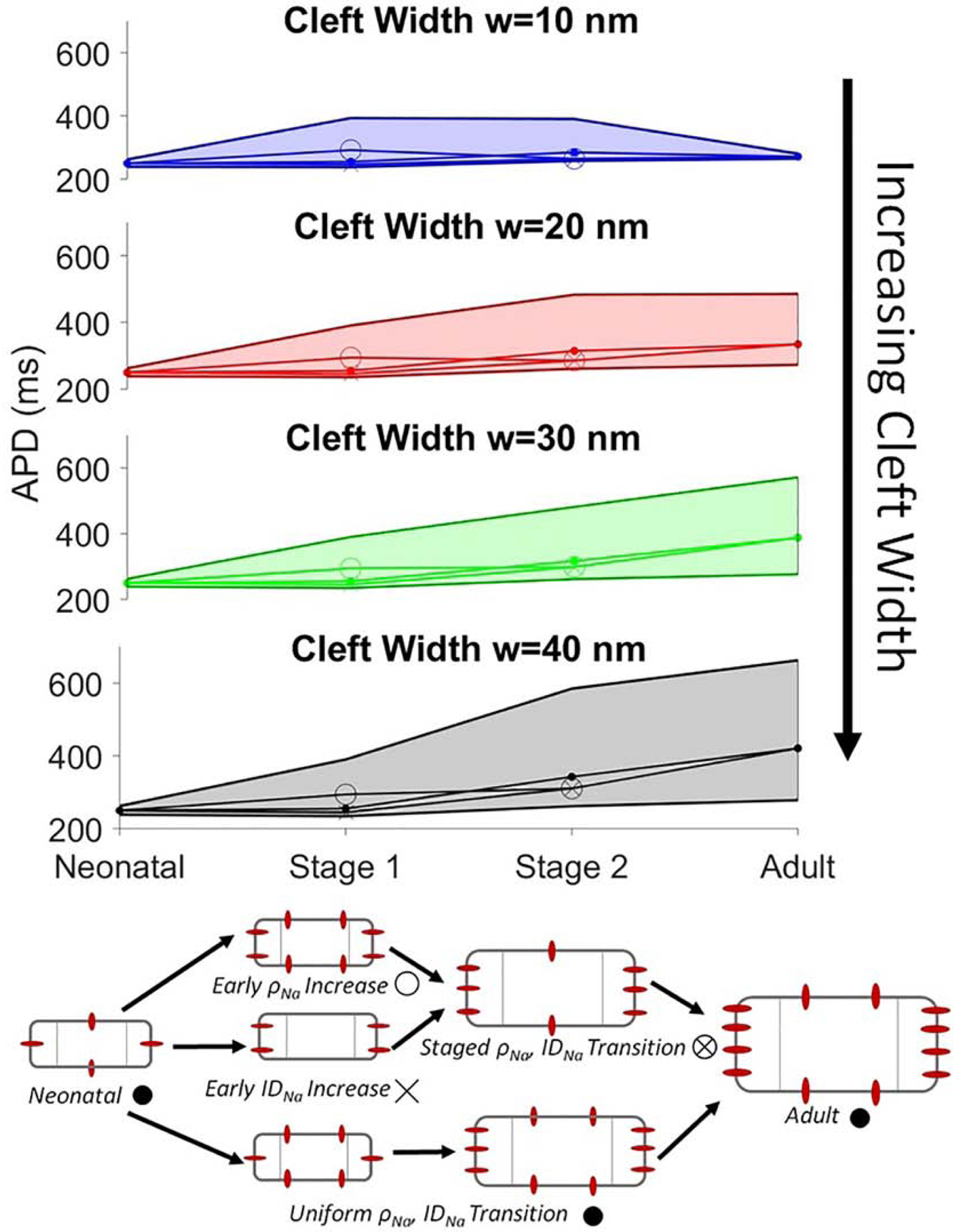
Age-associated manifestation of action potential duration (APD) prolongation. The range of APD values (see text for details) are shown as functions of age-dependent progression, for different cleft widths w. Parameter ranges for each stage are shown in Table S2. Markers for each stage are shown in the illustration (bottom), with the value shown for each stage representing the average APD value over all conditions within the specified parameter ranges. BCL = 1000 ms.
For the neonatal stage, the entire APD range is within a normal range, i.e., near values for wild-type tissue, with no EADs and with minimal dependence on cleft width. For intermediate developmental stages, simulations predict that the average APD values are also near wild-type values; however the range of APD values is much larger, with the maximum APD values associated with significant APD prolongation and EAD formation. For the earlier intermediate stage (Stage 1), we find minimal dependence on cleft width, while for the later stage (Stage 2), we find that cleft width narrowing mitigates APD prolongation; however, APD values are still observed over a wide range, with maximum values associated with EADs. For the adult stage, as discussed above, EADs are suppressed by cleft narrowing, and we further observe that cleft narrowing reduces the range of APD values. Thus, we find that while early developmental stages are nominally associated with normal APD values and no EADs, variability in cellular properties can lead to a wide range of APD values and promote EAD formation. In contrast, for adult-associated parameters, EAD formation is only suppressed by cleft width narrowing. We note that, consistent with Fig. 3, the maximum APD values for each stage are associated with the largest cell size and Na+ channel density parameters for that stage.
4. Discussion
In this study, we demonstrate that in guinea pig cardiac tissue model with a Na+ channel GOF, age-associated increases of total Na+ current conductance are a critical factor governing APD prolongation and EAD formation, in addition to previously identified regulation by intercellular cleft width and Na+ channel localization [34, 35]. However, regulation by total Na+ current conductance is complex and depends on both cell size and Na+ channel density, such that simulations predict that increases in total Na+ current conductance due to increased Na+ channel density are more likely to promote APD prolongation, compared with increases due to cell size. We also find that cell size is a key mediator of CV that can influence CV in differing manners depending on GJ coupling. Our study utilizes simulations to predict and extrapolate findings made in a guinea pig computational model to human patients with LQT3-association mutations. Consistent with clinical studies demonstrating an age-associated manifestation of the LQT3 phenotype [5, 11], our simulations predict on average normal APD for parameters associated with neonatal and early development stages, primarily due to small cell size and lower levels of Na+ channel expression, comparable to a normal QT interval in asymptomatic patients with LQT3-associated mutations. Critically, for small cell size and low Na+ channel density associated with earlier developmental stages, APD is more weakly dependent on intercellular cleft width and Na+ channel ID localization, the key determinants of EAD formation at adult stages (Fig. 3), which suggests an age-dependent protective mechanism while these subcellular and tissue properties are in flux due to normal development. In particular, the low preferential Na+ channel ID localization associated with early development does not promote EADs, due to low overall total Na+ channel conductance, while this subcellular Na+ channel distribution would be pro-arrhythmic in adult myocardium. However, for these early developmental stage, variability in cellular properties, in particular increases in cell size and Na+ channel density, can lead to APD prolongation and EAD formation, consistent with early life manifestation of arrhythmias observed in some LQT3 patients.
In contrast, simulations with adult-associated parameters predict APD prolongation, relative to earlier developmental stages, consistent with significant QT interval prolongation found in adult LQT3 patients [10, 49, 50, 51, 11], and further that APD strongly depends on intercellular cleft width, with expanded cleft width prolonging APD and promoting EAD formation. We and others have previously shown that CV depends prominently on intercellular cleft width for strong GJ coupling (i.e., conditions associated with adult myocardium) [19, 34, 29, 26]. Interestingly, Beaufort-Krol et al. found in LQT3 patients that, while QT prolongation with age occurred as early as immediately after birth, conduction defects occurred later in life [10]; these clinical findings are consistent with a common underlying defect, specifically intercellular cleft expansion, being responsible for both conduction slowing and EAD formation, both critical arrhythmia triggers that would promote symptom manifestation in LQT3 patients later in life. While to our knowledge, no study has investigated intercellular cleft width in symptomatic versus asymptomatic LQT3 patients, these findings are consistent with an age-associated cleft expansion-mediated manifestation of disease.
Symptomatic LQT3 patients are commonly treated with Na+ channel blocking drugs, such as mexiletine, a class 1B anti-arrhythmic structurally related to lidocaine [52], in addition to β-blockers. However, channel-drug interactions are complex, in addition to potential off-target effects. For example, the efficacy of Na+ channel block using mexiletine has been shown to depend on the specific LQT3 mutation [53], due to different mutant-specific channel kinetics that in turn alter drug binding. This suggests that drug therapies targeting ion channels may require patient-specific considerations. Our study suggests that therapies that modulate structural components of the cardiac tissue, specifically the intercellular cleft width, may prove to be an effective alternative therapeutic target, in particular considering our prior work that suggests that the mechanism underlying EAD suppression is relevant in multiple LQT3-associated mutations [34]. While such an approach would potentially mitigate patient-specific challenges, additional work is needed to investigate the mechanism in a wider range of mutations. We recently showed that osmotic agents can alter the cleft width in regions adjacent to GJs, and specifically that albumin narrows the cleft [35], which suggests that modulation of cardiac fluid balance may also be a potential therapy; further work is needed to develop such interventions to assess therapeutic efficacy. However, our findings also suggest that age-specific considerations may be relevant with such a therapy. Simulations predict that these agents may be an effective therapy in adults, while cleft width modulation would not be effective for early developmental stages, as EAD formation is generally independent of cleft width at these stages.
As discussed above, LQT3 has a complex presentation in which symptoms may be concealed until later in life in some individuals but can also present with fatal arrhythmias during infancy in other individuals. In families with the specific mutation investigated in this study (Y1795C), both adults and children frequently exhibited prolonged corrected QT interval; however, there are examples of symptomatic (specifically, arrhythmia events) and asymptomatic adults and also symptomatic and asymptomatic children [16, 17, 18, 54], highlighting that the manifestation of arrhythmia triggering events is complex and patient-specific. In Fig. 8, we account for developmental variability in individual cellular properties, specifically cell size and Na+ channel localization and density, and predict that EADs can form for parameter conditions associated with both earlier and later in life. For individual LQT3 patients, variability in expression of other ion channels contributing to repolarization will also influence age-related symptoms and arrhythmia triggers. Further, while we previously demonstrated that the mechanism of the concealed LQT3 phenotype are applicable to other LQT3-associated Na+ channel mutations (specifically I1768V) [34], both variability in channel kinetics for a specific mutation and differences between different mutations also likely contribute to patient variability in age-associated disease manifestation. Indeed, as EAD suppression depends on a negative feedback mechanism in which the late Na+ current promotes cleft Na+ depletion and thus is highly dependent on the specific dynamics of the late current activation, it may be the case that this mechanism is only plausible in a subset of LQT3-associated Na+ channel mutations. This investigation is a focus of on-going and future work.
Finally, we acknowledge limitations of our study. While our modeling approach incorporates significant subcellular details in the representation of cardiac tissue, this representation is still a simplification of the complex cardiac tissue structure. Specifically, the geometry of individual cells and the ID is heterogeneous, whereas our model assumes a simplified cylindrical cell geometry and uniform intercellular cleft. Further, cardiac tissue is a three-dimensional structure, such that a one-dimensional tissue representation cannot fully represent all aspects of developmental changes, such as GJ localization as noted above. However, our model formulation does represent key subcellular and structural details, such as subcellular ion channel distribution and intercellular cleft space dynamics, which we find are critical in EAD formation in tissue with LQT3-associated mutations. Additionally, heterogeneity in tissue properties exists at multiple spatial scales, such as transmural gradients and left and right ventricle differences. Interestingly, there is evidence that suggests that EAD-mediated arrhythmia triggers can originate in the Purkinje network in experimental models of LQT3 [55, 56], for which the structure is closer to the one-dimensional tissue simulated in this study. However, the ID within the Purkinje network [57], and the extent to which similar Na+ nanodomain regulation of EAD formation occurs, requires further investigation.
We also utilize the same mutant Na+ channel model parameters for all conditions, thus not specifically accounting for potential age-associated changes in channel kinetics, such as differences in steady-state Na+ channel availability that have been identified in neonatal and adult rat ventricular myocytes [58, 59]. However, to our knowledge, no studies have identified if or to what extent similar developmental changes in kinetics occur for LQT3-associated mutant Na+ channels. In our study, we find that faster pacing rates suppress EADs for all age-associated conditions; however these simulations do not account for modulation by β-adrenergic signaling, which enhances late Na+ current but also other currents contributing to repolarization, including calcium-mediated effects via the sodium-calcium exchanger [60, 61], and has been shown to shorten APD in a LQT3 model [7]. However, these influences are likely more significant in arrhythmias triggered by exercise and arousal, which are more common in other long QT syndrome genotypes and rare for LQT3 [62, 13].
Finally, as noted above, individual expression levels of all ion channels vary across individual patients, and these individual differences likely contribute to the different manifestation of symptoms observed within populations of LQT3 patients, in addition to Na+ channel, GJ, and tissue structural properties discussed in this study. While population-based simulation studies, previously demonstrated in single cell simulations [63, 64], are not feasible due to high computational cost associated with this tissue model formulation, the wide ranging age-associated parameter study across multiple values for cell size, Na+ channel density and distribution, GJ coupling, and intercellular cleft width (comprised of 16,034 simulations) illustrate key relationships between these subcellular, cellular, and tissue properties and mechanisms underlying conduction and repolarization defects in the settings of Na+ channel GOF. Overall, while these limitations may account for quantitative differences, importantly our simulations predict qualitative trends similar to clinical and experimental findings, demonstrating their utility in assessing mechanisms underlying age progression and disease manifestation.
Supplementary Material
Highlights:
Long QT syndrome type 3 can present early in life or be concealed to adulthood.
Age-dependent properties regulate LQT3-associated Na+ channel gain-of-function.
Cell size and Na+ channel density increases promote arrhythmias in neonatal tissue.
Intercellular cleft width increases promote arrhythmias in adult tissue.
Cellular property variability can lead to arrhythmias for all developmental stages.
Acknowledgements
This study was supported by funding from the National Institutes of Health, grant numbers R01HL138003 (SHW, SP) and R01HL102298 (SP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- [1].Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC, Cardiac late na+ current: proarrhythmic effects, roles in long qt syndromes, and pathological relationship to camkii and oxidative stress, Heart Rhythm 12 (2015) 440–448. [DOI] [PubMed] [Google Scholar]
- [2].Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT, Scn5a mutations associated with an inherited cardiac arrhythmia, long qt syndrome, Cell 80 (1995) 805–811. [DOI] [PubMed] [Google Scholar]
- [3].Burashnikov A, Antzelevitch C, Acceleration-induced action potential prolongation and early afterdepolarizations, Journal of cardiovascular electrophysiology 9 (1998) 934–948. [DOI] [PubMed] [Google Scholar]
- [4].Clancy CE, Rudy Y, Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia, Nature 400 (1999) 566–569. [DOI] [PubMed] [Google Scholar]
- [5].Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, et al. , Risk for life-threatening cardiac events in patients with genotype-confirmed long-qt syndrome and normal-range corrected qt intervals, Journal of the American College of Cardiology 57 (2011) 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barsheshet A, Dotsenko O, Goldenberg I, Genotype-specific risk stratification and management of patients with long qt syndrome, Annals of Noninvasive Electrocardiology 18 (2013) 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shimizu W, Antzelevitch C, Differential effects of beta-adrenergic agonists and antagonists in lqt1, lqt2 and lqt3 models of the long qt syndrome, Journal of the American College of Cardiology 35 (2000) 778–786. [DOI] [PubMed] [Google Scholar]
- [8].de Lange E, Xie Y, Qu Z, Synchronization of early afterdepolarizations and arrhythmogenesis in heterogeneous cardiac tissue models, Biophysical journal 103 (2012) 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wilde AA, Moss AJ, Kaufman ES, Shimizu W, Peterson DR, Benhorin J, Lopes C, Towbin JA, Spazzolini C, Crotti L, et al. , Clinical aspects of type 3 long-qt syndrome: an international multicenter study, Circulation 134 (2016) 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beaufort-Krol GC, van den Berg MP, Wilde AA, van Tintelen JP, Viersma JW, Bezzina CR, Bink-Boelkens MTE, Developmental aspects of long qt syndrome type 3 and brugada syndrome on the basis of a single scn5amutation in childhood, Journal of the American College of Cardiology 46 (2005) 331–337. [DOI] [PubMed] [Google Scholar]
- [11].Kutyifa V, Daimee UA, McNitt S, Polonsky B, Lowenstein C, Cutter K, Lopes C, Zareba W, Moss AJ, Clinical aspects of the three major genetic forms of long qt syndrome (lqt 1, lqt 2, lqt 3), Annals of Noninvasive Electrocardiology 23 (2018) e12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, G. S, Benhorin J, Locati EH, Towbin JA, Keating MT, et al. , Influence of the genotype on the clinical course of the long-qt syndrome, New England Journal of Medicine 339 (1998) 960–965. [DOI] [PubMed] [Google Scholar]
- [13].Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, et al. , Genotype-phenotype correlation in the long-qt syndrome: gene-specific triggers for life-threatening arrhythmias, Circulation 103 (2001) 89–95. [DOI] [PubMed] [Google Scholar]
- [14].Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL Jr, et al. , Prevalence of long-qt syndrome gene variants in sudden infant death syndrome, Circulation 115 (2007) 361–367. [DOI] [PubMed] [Google Scholar]
- [15].Huang H, Millat G, Rodriguez-Lafrasse C, Rousson R, Kugener B, Chevalier P, Chahine M, Biophysical characterization of a new scn5a mutation s1333y in a sids infant linked to long qt syndrome, FEBS letters 583 (2009) 890–896. [DOI] [PubMed] [Google Scholar]
- [16].Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS, Inherited brugada and long qt-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes, Journal of Biological Chemistry 276 (2001) 30623–30630. [DOI] [PubMed] [Google Scholar]
- [17].Vecchietti S, Grandi E, Severi S, Rivolta I, Napolitano C, Priori SG, Cavalcanti S, In silico assessment of y1795c and y1795h scn5a mutations: implication for inherited arrhythmogenic syndromes, American Journal of Physiology-Heart and Circulatory Physiology 292 (2007) H56–H65. [DOI] [PubMed] [Google Scholar]
- [18].Benito B, Brugada R, Perich RM, Lizotte E, Cinca J, Mont L, Berruezo A, Tolosana JM, Freixa X, Brugada P, et al. , A mutation in the sodium channel is responsible for the association of long qt syndrome and familial atrial fibrillation, Heart Rhythm 5 (2008) 1434–1440. [DOI] [PubMed] [Google Scholar]
- [19].Kucera JP, Rohr S, Rudy Y, Localization of sodium channels in intercalated disks modulates cardiac conduction, Circulation research 91 (2002) 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S, Sodium channels in the cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study, Pflügers Archiv-European Journal of Physiology 467 (2015) 2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Veeraraghavan R, Gourdie RG, Stochastic optical reconstruction microscopy–based relative localization analysis (storm-rla) for quantitative nanoscale assessment of spatial protein organization, Molecular biology of the cell 27 (2016) 3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, Keegan S, Lin X, Arcos T, Korchev YE, Gorelik J, Fenyö D, Rothenberg E, et al. , Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc, Nature communications 7 (2016) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Liang F-X, Li Z, Pfenniger A, Lübkemeier I, Keegan S, Fenyö D, et al. , Super-resolution imaging reveals that loss of the c-terminus of connexin43 limits microtubule plus-end capture and nav1. 5 localization at the intercalated disc, Cardiovascular research 104 (2014) 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rhett JM, Ongstad EL, Jourdan J, Gourdie RG, Cx43 associates with na v 1.5 in the cardiomyocyte perinexus, The Journal of membrane biology 245 (2012) 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin J, Keener JP, Modeling electrical activity of myocardial cells incorporating the effects of ephaptic coupling, Proceedings of the National Academy of Sciences 107 (2010) 20935–20940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mori Y, Fishman GI, Peskin CS, Ephaptic conduction in a cardiac strand model with 3d electrodiffusion, Proceedings of the National Academy of Sciences 105 (2008) 6463–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sperelakis N, An electric field mechanism for transmission of excitation between myocardial cells, 2002. [DOI] [PubMed]
- [28].Hichri E, Abriel H, Kucera JP, Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc, The Journal of physiology 596 (2018) 563–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weinberg S, Ephaptic coupling rescues conduction failure in weakly coupled cardiac tissue with voltage-gated gap junctions, Chaos: An Interdisciplinary Journal of Nonlinear Science 27 (2017) 093908. [DOI] [PubMed] [Google Scholar]
- [30].Jæger KH, Edwards AG, McCulloch A, Tveito A, Properties of cardiac conduction in a cell-based computational model, PLoS computational biology 15 (2019) e1007042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tveito A, Jæger KH, Kuchta M, Mardal K-A, Rognes ME, A cell-based framework for numerical modeling of electrical conduction in cardiac tissue, Frontiers in Physics 5 (2017) 48. [Google Scholar]
- [32].Wei N, Mori Y, Tolkacheva EG, The dual effect of ephaptic coupling on cardiac conduction with heterogeneous expression of connexin 43, Journal of theoretical biology 397 (2016) 103–114. [DOI] [PubMed] [Google Scholar]
- [33].Wei N, Tolkacheva EG, Interplay between ephaptic coupling and complex geometry of border zone during acute myocardial ischemia: Effect on arrhythmogeneity, Chaos: An Interdisciplinary Journal of Nonlinear Science 30 (2020) 033111. [DOI] [PubMed] [Google Scholar]
- [34].Greer-Short A, George SA, Poelzing S, Weinberg SH, Revealing the concealed nature of long-qt type 3 syndrome, Circulation: Arrhythmia and Electrophysiology 10 (2017) e004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nowak MB, Greer-Short A, Wan X, Wu X, Deschênes I, Weinberg SH, Poelzing S, Intercellular sodium regulates repolarization in cardiac tissue with sodium channel gain-of-function, Biophysical Journal (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].George SA, Bonakdar M, Zeitz M, Davalos RV, Smyth JW, Poelzing S, Extracellular sodium dependence of the conduction velocity-calcium relationship: evidence of ephaptic self-attenuation, American Journal of Physiology-Heart and Circulatory Physiology 310 (2016) H1129–H1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cai B, Mu X, Gong D, Jiang S, Li J, Meng Q, Bai Y, Liu Y, Wang X, Tan X, et al. , Difference of sodium currents between pediatric and adult human atrial myocytes: evidence for developmental changes of sodium channels, International journal of biological sciences 7 (2011) 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cordeiro JM, Panama BK, Goodrow R, Zygmunt AC, White C, Treat JA, Zeina T, Nesterenko VV, Di Diego JM, Burashnikov A, et al. , Developmental changes in expression and biophysics of ion channels in the canine ventricle, Journal of molecular and cellular cardiology 64 (2013) 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vreeker A, Van Stuijvenberg L, Hund TJ, Mohler PJ, Nikkels PG, Van Veen TA, Assembly of the cardiac intercalated disk during pre-and postnatal development of the human heart, PloS one 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hirschy A, Schatzmann F, Ehler E, Perriard J-C, Establishment of cardiac cytoarchitecture in the developing mouse heart, Developmental biology 289 (2006) 430–441. [DOI] [PubMed] [Google Scholar]
- [41].Swift LM, Burke M, Guerrelli D, Reilly M, Ramadan M, McCullough D, Prudencio T, Mulvany C, Chaluvadi A, Jaimes R, et al. , Age-dependent changes in electrophysiology and calcium handling: implications for pediatric cardiac research, American Journal of Physiology-Heart and Circulatory Physiology 318 (2020) H354–H365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH, Green CR, Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium., Circulation 90 (1994) 713–725. [DOI] [PubMed] [Google Scholar]
- [43].Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA, Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development, Physiological genomics 28 (2007) 273–283. [DOI] [PubMed] [Google Scholar]
- [44].Spach MS, Heidlage JF, Dolber PC, Barr RC, Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth, Circulation research 86 (2000) 302–311. [DOI] [PubMed] [Google Scholar]
- [45].Kato Y, Masumiya H, Agata N, Tanaka H, Shigenobu K, Developmental changes in action potential and membrane currents in fetal, neonatal and adult guinea-pig ventricular myocytes, Journal of molecular and cellular cardiology 28 (1996) 1515–1522. [DOI] [PubMed] [Google Scholar]
- [46].Raisch TB, Yanoff MS, Larsen TR, Farooqui MA, King DR, Veeraraghavan R, Gourdie RG, Baker JW, Arnold WS, AlMahameed ST, et al. , Intercalated disk extracellular nanodomain expansion in patients with atrial fibrillation, Frontiers in physiology 9 (2018) 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Livshitz LM, Rudy Y, Regulation of ca2+ and electrical alternans in cardiac myocytes: role of camkii and repolarizing currents, American Journal of Physiology-Heart and Circulatory Physiology 292 (2007) H2854–H2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Clancy CE, Tateyama M, Kass RS, et al. , Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long qt-3 syndrome, The Journal of clinical investigation 110 (2002) 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, et al. , Risk stratification in the long-qt syndrome, New England Journal of Medicine 348 (2003) 1866–1874. [DOI] [PubMed] [Google Scholar]
- [50].Van den Berg MP, Wilde AA, Viersma JW, Brouwer J, Haaksma J, Van Der Hout AH, Stolte-Dijkstra I, Bezzina CR, Van Langen IM, Beaufort-Krol GC, et al. , Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long qt syndrome type 3 and brugada syndrome, Journal of cardiovascular electrophysiology 12 (2001) 630–636. [DOI] [PubMed] [Google Scholar]
- [51].Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen L-SK, et al. , Long qt syndrome patients with mutations of the scn5a and herg genes have differential responses to na+ channel blockade and to increases in heart rate: implications for gene-specific therapy, Circulation 92 (1995) 3381–3386. [DOI] [PubMed] [Google Scholar]
- [52].Shimizu W, Antzelevitch C, Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in lqt2 and lqt3 models of the long-qt syndrome, Circulation 96 (1997) 2038–2047. [DOI] [PubMed] [Google Scholar]
- [53].Zhu W, Mazzanti A, Voelker TL, Hou P, Moreno JD, Angsutararux P, Naegle KM, Priori SG, Silva JR, Predicting patient response to the antiarrhythmic mexiletine based on genetic variation: personalized medicine for long qt syndrome, Circulation research 124 (2019) 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kilinc OU, Tuzcu V, Successful elimination of significant arrhythmia burden with flecainide in an adolescent with long qt syndrome type 3, Congenital Heart Disease 7 (2012) E42–E45. [DOI] [PubMed] [Google Scholar]
- [55].Iyer V, Roman-Campos D, Sampson KJ, Kang G, Fishman GI, Kass RS, Purkinje cells as sources of arrhythmias in long qt syndrome type 3, Scientific reports 5 (2015) 13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Caref EB, Boutjdir M, Himel HD, El-Sherif N, Role of subendocardial purkinje network in triggering torsade de pointes arrhythmia in experimental long qt syndrome, Europace 10 (2008) 1218–1223. [DOI] [PubMed] [Google Scholar]
- [57].Shimada T, Kawazato H, Yasuda A, Ono N, Sueda K, Cytoarchitecture and intercalated disks of the working myocardium and the conduction system in the mammalian heart, The Anatomical Record Part A: Discoveries in Molecular, Cellular, and Evolutionary Biology: An Official Publication of the American Association of Anatomists 280 (2004) 940–951. [DOI] [PubMed] [Google Scholar]
- [58].Xu Y-Q, Pickoff AS, Clarkson CW, Evidence for developmental changes in sodium channel inactivation gating and sodium channel block by phenytoin in rat cardiac myocytes., Circulation research 69 (1991) 644–656. [DOI] [PubMed] [Google Scholar]
- [59].Wu C-C, Su M-J, Chi J-F, Wu M-H, Lee Y-T, Comparison of aging and hypercholesterolemic effects on the sodium inward currents in cardiac myocytes, Life sciences 61 (1997) 1539–1551. [DOI] [PubMed] [Google Scholar]
- [60].Hegyi B, Bányász T, Izu LT, Belardinelli L, Bers DM, Chen-Izu Y, β-adrenergic regulation of late na+ current during cardiac action potential is mediated by both pka and camkii, Journal of molecular and cellular cardiology 123 (2018) 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hegyi B, Morotti S, Liu C, Ginsburg KS, Bossuyt J, Belardinelli L, Izu LT, Chen-Izu Y, Bányász T, Grandi E, et al. , Enhanced depolarization drive in failing rabbit ventricular myocytes: Calcium-dependent and β-adrenergic effects on late sodium, l-type calcium, and sodium-calcium exchange currents, Circulation: Arrhythmia and Electrophysiology 12 (2019) e007061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ali RHH, Zareba W, Moss AJ, Schwartz PJ, Benhorin J, Vincent GM, Locati EH, Priori S, Napolitano C, Towbin JA, et al. , Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with the long qt syndrome, The American journal of cardiology 85 (2000) 457–461. [DOI] [PubMed] [Google Scholar]
- [63].Sarkar AX, Christini DJ, Sobie EA, Exploiting mathematical models to illuminate electrophysiological variability between individuals, The Journal of physiology 590 (2012) 2555–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ, Rodriguez B, Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology, Proceedings of the National Academy of Sciences 110 (2013) E2098–E2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.