

Summary
Integrin activation controls cell adhesion, migration, invasion, and extracellular matrix remodeling. RIAM (RAP1-GTP-interacting adaptor molecule) is recruited by activated RAP1 to the plasma membrane (PM) to mediate integrin activation via an inside-out signaling pathway. This process requires the association of the pleckstrin homology (PH) domain of RIAM with the membrane PIP2. We identify a conserved inter-molecular interface that masks the PIP2 binding site in the PH domains of RIAM. Our data indicate that phosphorylation of RIAM by Src family kinases disrupts this PH-mediated interface, unmasks the membrane PIP2-binding site, and promotes integrin activation. We further demonstrate that this process requires phosphorylation of Tyr267 and Tyr427 in the RIAM PH domain by Src. Our data reveal an unorthodox regulatory mechanism of small GTPase effector proteins by phosphorylation-dependent PM association of the PH domain, and provide new insights into the link between Src kinases and integrin signaling.
Keywords: RIAM; RAP1; lamellipodin; Integrin Signaling; Crystal Structure; PIP2 Binding; Src Kinase; LCK; FYN; Phosphorylation, PH Domain
Graphical Abstract
eTOC Blurb
CHO et al. discovered that phosphorylation of RIAM by Src disrupts a common interaction that mediates RIAM oligomerization and promotes RIAM translocation to the plasma membrane. These findings elucidate a new regulatory mechanism for RIAM-mediated integrin activation.
Introduction
Adaptive immunity is an advanced defense mechanism in vertebrates that responses to specific pathogen via immunological memory (Vestweber, 2015). The adaptive immune system is mainly composed of lymphocytes, particularly T cells and B cells. Adhesion of lymphocytes to target cells or tissues is a critical phase for adaptive immune responses (Mor et al., 2007). Integrins, a group of cell adhesion molecules, are heterodimeric receptors that mediate the attachment of cells to neighboring cells or extracellular matrix. In particular, several integrin species abundant in lymphocytes, such as αLβ2 and α4β1 (also known as LFA-1 and VLA-4, respectively), play crucial roles in lymphocyte adhesion (Berlin-Rufenach et al., 1999; Mor et al., 2007; Schmits et al., 1996). RIAM (RAP1-interacting adaptor molecule), a RAP1 effector protein, mediates integrin activation via a RAP1-RIAM-talin-integrin signaling cascade, known as the inside-out integrin signaling pathway (Lagarrigue et al., 2016; Lee et al., 2009). RIAM recruits talin, a cytoskeletal protein, to the plasma membrane (PM) and promotes a direct interaction of talin with the cytoplasmic moiety of integrin β subunit (Klapholz and Brown, 2017; Lee et al., 2009). This interaction leads to conformational changes of extracellular ligand-binding domains of integrins from an inactive state to an active state (Ginsberg et al., 2005). Integrins of various species serve specialized functions in different tissues. Recent studies have shown that integrins in platelets and T lymphocytes regulate hemostasis and T cell mediated immune responses (Barczyk et al., 2010). Hence, the integrin signaling pathway components have become appealing targets for clinical intervention, particularly in autoimmune diseases and other immune disorders (Ley et al., 2016; Park and Jeen, 2018). In this study, we reveal new insight into the connection between integrin signaling and nonreceptor tyrosine kinase mediated by small GTPase effector proteins.
RIAM is abundantly expressed in leukocytes and is required for activation of β1 integrins and β2 integrins via the inside-out pathway upon RAP1 activation (Klapproth et al., 2015; Su et al., 2015). Lymphocytes in RIAM-null mice exhibit diminished integrin-mediated adhesion, reduced trafficking, and impaired humoral immunity (Klapproth et al., 2015; Su et al., 2015). Indeed, recent studies at the molecular level have demonstrated that RIAM is able to drive cell protrusion by forming a molecular complex called “sticky finger” with talin and integrin at the tips of growing actin filament during cell migration (Lagarrigue et al., 2015). RIAM is a typical adaptor molecule that interacts with various signaling partners, many of which are involved in integrin signaling, through specific binding sites (Lafuente et al., 2004). An N-terminal TBS (talin-binding site) in RIAM is responsible for talin association (Lee et al., 2009). An inhibitory segment (IN) succeeding TBS masks the RAP1-binding site on the Ras Associating (RA) domain, resulting in an autoinhibitory configuration of RIAM (Chang et al., 2019). The PM translocation of RIAM is dependent on a signature structural module consisting of an RA and a pleckstrin homology (PH) domains, known as RA-PH, that interacts with activated RAP1 and phosphoinositol 4,5-bisphosphate (PIP2) respectively (Wynne et al., 2012; Zhang et al., 2014). RIAM also regulates cytoskeletal dynamics by interacting with actin cytoskeleton regulator proteins including Ena/VASP proteins and profilin via its poly-proline motifs (PP) near the C-terminal end (Holt and Daly, 2005; Krause et al., 2003; Lafuente et al., 2004) (Fig. 1A).
Figure 1. Identifying a conserved intermolecular interface in RIAM.

A. Schematic representation of the domain organization of RIAM. Full length mouse RIAM (FL) possesses 670 residues. Talin-binding site (TBS) is colored in green; inhibitory (IN) segment is in orange; Coiled coil (CC) segment is in red; poly-proline (PP) segment is in black; the Ras-associating domain (RA) is in yellow; and the Pleckstrin-Homology domain (PH) is in cyan. The length of each structural region is proportional to the number of their amino acids. A scale bar corresponding to 100 amino acids is shown in gray. B. A conserved asymmetrical interface observed in the previously reported RIAM structures (3TCA: the original CC-RA-PHa; 4KVG: RIAM RA-PH in complex with RAP1-GTP; 6E31: RIAM in an autoinhibited configuration). One molecule was labeled as RA and PH, and the other molecule was labeled as RA’ and PH’. C. Left: superposition of the RIAM RA-PH core domain structure (wheat/pale cyan) and the new RIAM CC-RA-PHb structure (yellow/cyan). For each structure, two molecules (A and B) the form the asymmetrical interface are included in the superposition to demonstrate the identical interface (in dashed box). Right: conserved side-chain interactions that mediate the asymmetrical interface. Salt-bridge interactions are illustrated by yellow dash line; residues making Van der Waals contacts are indicated by surface representation. phosphoinositide-binding residues (K327, K328, K331, R332, and R333) are also indicated in stick representation. Ala435 is shown in the right panel to indicate its close contact with the neighboring molecule.
The function of RIAM in mediating integrin activation is tightly regulated. RIAM adopts an autoinhibitory configuration that can be relieved when phosphorylated by focal adhesion kinase (FAK) on the Tyr45 residue of the IN segment (Chang et al., 2019). Two Src family kinases, lymphocyte-specific protein tyrosine kinase (LCK) and Fyn are essential for T cell development and activation (Laird and Hayes, 2010; Lovatt et al., 2006; Palacios and Weiss, 2004), and have also been shown to phosphorylate RIAM in T cells (Patsoukis et al., 2009). Interestingly, RIAM possesses 20 tyrosines in total, 15 of which are located in the PH domain and the adjacent RA-PH linker, and are predicted to be substrates for Src family kinases by Group-based Prediction System (GPS), a computational prediction algorithm of cognate protein kinase (Zhou et al., 2004). Nonetheless, the regulatory role of RIAM’s function in mediating integrin activity through phosphorylation in the PH domain by Src kinases remains unknown. In this study, we identified a biologically relevant, intermolecular interaction between two RIAM PH domains that is regulated by Src phosphorylation. Strikingly, this interaction masks the membrane phosphoinositide-binding site in the PH domain. Disruption of this interaction promotes the PM translocation of RIAM, and leads to increased integrin activity. Our data indicate that phosphorylation of the PH domain by Src kinases unmasks of the phosphoinositide-binding site in the PH domain and RAP1-induced PM translocation of RIAM, and the inside-out integrin signaling pathway is activated as a result.
Results
Structures of RIAM reveal a PH domain-mediated intermolecular interaction.
We have previously determined three crystal structures that contain the RA-PH module of RIAM. The first structure (PDB: 3TCA) possesses RA-PH and the proceeding CC segment, which is disordered in the crystal (Wynne et al., 2012). The RIAM:RAP1 complex structure (PDB:4KVG) includes the RA-PH domain and the GTPase domain of RAP1 (Zhang et al., 2014). The recent RIAM autoinhibitory structure (PDB: 6E31) contains RA-PH, CC segment, and the inhibitory “IN” segment (Chang et al., 2019). Among the three structures, we observed two distinct intermolecular interfaces. A pseudo-symmetrical interface of a 590-Å2 buried area was found in the structures of CC-RA-PH and RA-PH:RAP1 complex. This interface involves extensive side-chain contacts centered at residues His389 and Tyr398 of the PH domain, and is thus named the “H-Y” interface (Fig. S1). Interestingly, we also identified another interface with an asymmetrical, smaller buried area of 410-Å2. This PH-mediated, asymmetrical interface is observed in all reported RIAM RA-PH structures including CC-RA-PH, RA-PH:RAP1 complex, and the autoinhibited RIAM (Fig. 1B).
To further validate the intermolecular interfaces, we determined the structure of the core RA-PH domains (residues 179-437) of RIAM. This 1.9-Å structure reveals an RA-PH structural module virtually identical to all previously reported RA-PH module structures, indicating that the RA-PH module is structurally rigid (Fig. 1C). This structure of the RA-PH core domains does not afford the pseudo-symmetrical “H-Y” interface. In contrast, the asymmetrical, PH-mediated interface remains intact (Fig. 1C). Moreover, we also determined a structure of RIAM CC-RA-PH in a new crystal form with a space group of P21212. The new crystal form of CC-RA-PH (referred as CC-RA-PHb,) also possesses the same asymmetrical interface as seen in the crystal structure of RA-PH core domains, but not the “H-Y” interface that was seen in the previous CC-RA-PH structure (referred as CC-RA-PHa, PDB code: 3TCA, space group P21). Thus, the asymmetrical interface is conserved in all five structures that contain the RA-PH module of RIAM (Fig. 1C). This asymmetrical interface involves the C-terminal helical extension from one PH domain and the β1-β2 loop/β2 region of the other PH domain. It is noteworthy that the β1-β2 loop and the β2 strand of the PH domain is the membrane phosphoinositide-binding site, which possesses multiple positively-charged residues (Lys327, Lys328, Lys331, Arg332, and Arg333) (Wynne et al., 2012). The interface is further stabilized by two salt-bridge interactions (Glu299A:Lys378B and Arg432A:Glu405B) and a polar interaction (Arg436A:Tyr321B) (Fig. 1C). Since binding to PIP2 through this site is essential for the translocation of RIAM to the PM (Wynne et al., 2012), we hypothesize that this intermolecular interaction suppresses RIAM-mediated integrin signaling that requires the PM translocation of RIAM.
Disruption of the PH-mediated asymmetrical interface in RIAM leads to PM translocation and increased integrin activity.
To test whether the RAP1-induced PM translocation of RIAM is suppressed by the asymmetrical interface observed in all RA-PH structures, we substituted Ala435 in the C-terminal helical extension of the PH domain with a bulky and stiff residue, which, based on the structural analysis, is expected to disrupt the asymmetrical interface (Fig. 1C). We also generated a mutation H389A/Y398A to diminish the other putative “H-Y” interaction (Fig. S1). These mutations were expressed in cells to assess their impact on the PM translocation of RIAM and RIAM-medicated integrin activation.
The PM translocation of RIAM requires simultaneous interaction of the RA and PH domains with RAP1 and membrane phosphoinositide. To test if the phosphoinositide-binding site of RIAM is masked by the PH-mediated interface as seen in the crystal structures, we transfected Jurkat T cells with constitutively active RAP1G12V and RIAM (wild type and mutants), and examined the RAP1-dependent PM translocation of RIAM. Wild type RIAM remained in cytosol when co-expressed with RAP1G12V in unstimulated Jurkat T cells. RIAM-H389A/Y398A exhibited little effect on the PM translocation of RIAM. In contrast, RIAM-A435Y exhibited strong PM translocation in a RAP1-dependent manner (Fig. 2A and 2B). These results suggest that the phosphoinositide-binding site of RIAM is masked by the PH-mediated interface, and the interface mutation A435Y disrupts the interaction of C-terminal helix and PIP2-binding site, promoting the RAP1-mediated PM translocation of RIAM.
Figure 2. Mutation disrupting the interface enhances RIAM function.

A. Left: Jurkat T cells co-transfected with HA-tagged RAP1G12V and GFP-tagged RIAM. Cellular distribution of RIAM and RAP1 was plotted after densitometric analysis. Portions defined for the plasma membrane distribution are indicated by the shadowed areas in the plots. Right: Jurkat T cells transfected with GFP-RIAM alone. B. The PM distribution of RIAM (wild type and various mutations) is shown in the scatter plot. Statistical significances were calculated using a two-tailed t-test for unpaired samples. Additional cells used for analyzing PM distribution are shown in Fig. S2. C. αIIbβ3 over-expressed stable CHO cells (CHO-A5) were transfected with GFP or assorted GFP-RIAM constructs (WT, H389A/Y398A, and A435Y). The cells were incubated with PAC1 antibody (indicator of the integrin activation) and Alexa 647 to quantify GFP-tagged protein expression and integrin αIIbβ3 activity. Data were analyzed on a LSR flow cytometer using 30,000 cells per measurement.
We then examined the effect of the interface-disrupting mutation on RIAM-mediated integrin activation. A CHO cell model that stably expresses αIIbβ3 integrin (CHO-A5) has been widely used to demonstrate RIAM-mediated integrin activation (Chang et al., 2014; Lagarrigue et al., 2015; Lee et al., 2009; Yang et al., 2014; Zhang et al., 2016). We transfected CHO-A5 cells with RIAM (wild type or mutations) and examined the integrin αIIbβ3 activity. CHO-A5 cells expressing RIAM-A435Y exhibited a significant increase in integrin activity compared with those of wild type RIAM and RIAM-H389A/Y398A (Fig. 2C). Because integrin activation can be promoted by the PM translocation of RIAM, this result is consistent with the observation that RIAM-A435Y enhances RIAM translocation to the PM. Together these data indicate that the PH-mediated interface inhibits the PM translocation of RIAM, and mutation-induced dissociation of this inhibitory interface unmasks the phosphoinositide-binding site in the PH domain, leading to RIAM translocation to the PM and increased integrin activity.
Phosphorylation of RIAM by Src kinase promotes the PM translocation of RIAM and RIAM-mediated integrin activation
Intermolecular interactions are commonly seen in proteins that form oligomeric assembly. We have shown previously that the IN segment of RIAM interacts with the RA domain and inhibits RA-PH:RAP1 interaction in trans, and phosphorylation of the IN segment disrupts IN:RA interaction (Chang et al., 2019), suggesting an inhibitory oligomeric assembly of RIAM. To assess the oligomeric assembly of RIAM and examine the impact of phosphorylation, we co-expressed HA-tagged RIAM and GFP-tagged RIAM in HEK293 cells, then treated the cell lysate with ATP to promote phosphorylation. Indeed, untreated RIAM exhibited an oligomeric assembly, which is diminished upon phosphorylation (Fig. 3A). Thus, the oligomeric assembly of RIAM further supports the biological relevance of the PH-mediated intermolecular interaction that blocks the PIP2-binding site.
Figure 3. RIAM phosphorylation by Src Promotes the PM translocation of RIAM and integrin activation.

A. Co-expression of HA-tagged RIAM and GFP-tagged RIAM in HEK293 cells. The cell lysate was treated with or without ATP, then subject to co-immunoprecipitation using anti-HA antibody, and immunoblotted with anti-GFP, anti-HA, and anti-pTyr antibodies. B. GST-RA-PH was treated with Jurkat T cell lysates (unstimulated or stimulated by anti-CD3), then pulled down by glutathione beads to examine for tyrosine phosphorylation. Lane 1: unstimulated; lane 2: stimulated; lane 3: stimulated and with 400 nM FAK inhibitor (PF431396); and lane 4: stimulated and with 400 nM Src kinase inhibitor (RK-24466). C. Purified RIAM-NT or RIAM RA-PH treated with LCK were examined for tyrosine phosphorylation. D. Jurkat T cells were starved and treated with RK-24466 and/or anti-CD3 antibodies. Cells were sub-fractionated and loaded for western blotting of cytosol and plasma membrane. A representative result of a triplicate experiment is shown. E. The PM distribution of RIAM indicated in 3D is shown in the bar graph. F. Jurkat T cells were starved and treated with RK-24466 and stimulated with or without anti-CD3 antibodies. Adhesion assay was performed and adherence was determined by plate reader (595 nm) with crystal violet. G. CHO cells were treated Src inhibitor for 30 min and adhesion assay was performed and adherence was determined by plate reader (595 nm) with crystal violet. E, F, and G: Data shown are mean ± SD, n = 3, also seen in Supplemental Table 1. Statistical significances were calculated using a two-tailed t-test for unpaired samples. *: p < 0.03; **: p < 0.005; #: p = 0.06 (NS).
Both FAK and Src kinases have been shown to phosphorylate RIAM. To identify the kinase responsible for unmasking the PM binding site in the PH domain, we treated purified RIAM RA-PH with activated T cell lysate in the presence of kinase inhibitors of FAK or Src. Indeed, RIAM RA-PH was strongly phosphorylated upon T cell activation, and the phosphorylation can be suppressed by a Src kinase-specific inhibitor (RK-24466), but not by a FAK kinase-specific inhibitor (PF431396) (Fig. 3B). We then demonstrated that LCK, the most abundant Src family kinase in T lymphocytes (Lagarrigue et al., 2015; Su et al., 2015), is able to phosphorylate the RA-PH structural module. In contrast, no tyrosine phosphorylation by LCK was observed in the N-terminal segment of RIAM (RIAM-NT, residues 1-93) (Fig. 3C). We performed in vitro kinase assays using RIAM-NT with LCK and Fyn kinases, and confirmed that no phosphorylated tyrosine in RIAM-NT was detected by mass spectrometry (data not shown). We also confirmed that Fyn kinase is able to phosphorylate RA-PH (Fig. S3A). These data indicate that Src kinases, but not FAK kinase, phosphorylate the RA-PH module.
We then examined the effect of Src kinase inhibitor on the PM translocation of RIAM. Jurkat T cells were treated with RK-24466, then stimulated by anti-CD3 antibodies. Cells were then lysed, fractionated, and analyzed for subcellular distribution of RIAM, RAP1, and LCK. Consistent with previous reports (Ley et al., 1994; Wynne et al., 2012), endogenous RAP1 and LCK were found predominately in the membrane fraction (Fig. 3D). Although the PM translocation of RIAM is strongly promoted upon T cell stimulation in the absence of RK-24466, it is markedly suppressed when the cells are subsequently treated by RK-24466 (Fig. 3E). Together, these results indicate that LCK phosphorylates the RA-PH module of RIAM and promotes RIAM PM translocation.
RIAM mediates the inside-out integrin signaling pathway by linking talin to the PM-anchored RAP1 (Lee et al., 2009). We next examined the effect of Src kinase inhibitor on RIAM-mediated integrin activation. Because RIAM is abundantly expressed in T cells, we first assessed T cell adhesion to ICAM-1 via LFA-1 (αLβ2 integrin). RK-24466 inhibited T cell adhesion to ICAM-1 in a dose-dependent manner (Fig. 3F). We then examined the effect of Src kinases on β1 integrin activation. We assessed the effect of Src inhibitor on β1 integrin activity in CHO cells which express high level of endogenous β1 integrin (Fig. S3B) (De Franceschi et al., 2015; Weitzman et al., 1995). CHO cells were transfected with HA-RIAM and assessed for β1 integrin-mediated cell adhesion to fibronectin upon RK-24466 treatment. Indeed, RK-24466 also suppressed the attachment of CHO cells to fibronectin-coated plates in a dose-dependent manner (Fig. 3G). These data demonstrated that the RIAM-mediated integrin activation can be promoted by Src kinases through phosphorylation of RIAM.
Src regulates RIAM’s function in integrin signaling by phosphorylating Tyr267 and Tyr427
To identify the phosphorylation sites in the RA-PH module that are essential for the PM translocation of RIAM, we treated purified RIAM RA-PH protein with two representative Src kinases, LCK and Fyn, respectively, and analyzed the phosphorylation sites by mass spectrometry. We identified eight tyrosine phosphorylation sites by LCK and six tyrosine phosphorylation sites by Fyn kinase (Fig. S3C). Among these sites, four sites are found in both samples. Interestingly, these four sites include Tyr398, which mediates the crystallographic “H-Y” interface, and its mutation had showed no significant impact on RIAM translocation and function (Fig. 2). Tyr277 is located in the highly flexible part of the RA-PH linker and is immediately adjacent to the disordered sequence of residues 278-292 (Fig. 4A). Tyr277 only makes moderate contact within the RA-PH linker. No direct or indirect contact with the C-terminal helix or the PIP2 bindings site was observed in all crystal forms. Therefore, it is highly unlikely the phosphorylation (or inhibiting the phosphorylation) of Tyr277 would affect the PM translocation of RIAM. In contrast, Tyr427 resides on the C-terminal helix that blocks the PIP2 binding site, and Tyr267 makes direct contact with the C-terminal helix. We therefore examined the effect of nonphosphorylatable mutations of Tyr267 and Tyr427 on RIAM translocation and function.
Figure 4. Roles of Tyr267 and Tyr427 of RIAM in regulating the PM translocation of RIAM and integrin activation.

A. Four tyrosine phosphorylation sites (Tyr267, Tyr277, Tyr 398, and Tyr 427) in RIAM RA-PH (yellow and cyan) by LCK and Fyn kinases are indicated by sticks in green. A neighboring RA-PH molecule is shown in dark yellow (RA domain) and light blue (PH domain) with the PIP2 binding site highlighted in purple. B. Jurkat T cells were co-transfected with HA-tagged RAP1G12V and GFP-tagged RIAM. Cellular distribution of RIAM and RAP1 along the indicated bars was plotted after densitometric analysis. C. The PM distribution of RIAM (wild type and various mutations) is shown in the scatter plot. D. HEK293 cells co-transfected with HA-tagged RAP1G12V and GFP-tagged RIAM were sub-fractionated. Fractions of plasma membrane and cytosol were analyzed by Western blot. E. The PM distribution of RIAM indicated in 4D is shown in the bar graph. Data shown are mean ± SD, n = 3, also seen in Supplemental Table 1. F. CHO-A5 cells transfected with GFP-tagged RIAM were analyzed for αIIbβ3 integrin activity by FACS using PAC-1 antibody. Statistical significances were calculated using a two-tailed t-test for unpaired samples. *: p < 0.05.
We first examined the RAP1-dependent PM translocation of RIAM (wild type and mutants). Jurkat T cells were transfected with constitutively active RAP1-G12V bearing two nonphosphorylatable mutations (Y267F or Y427F), and three phosphomimetic mutations (Y267E, Y427E, or Y267E/Y427E (YY/EE)). Both Y267F and Y427F significantly suppressed the RAP1-induced PM translocation of RIAM, and Y267E, Y427E, and YY/EE enhanced PM translocation (Fig. 4B, C). To confirm that the nonphosphorylatable mutations suppress the PM translocation of RIAM, we transfected HEK293 cells with GFP-RIAM and HA-RAP1-G12V and examined subcellular distribution of RIAM (Fig. 4D, E). Constitutively active RAP1-G12V mainly distributes on the PM. Although RIAM are observed in both PM and cytosol fractions, the PM fractions of Y267F and Y427F are significantly reduced, and that of A435Y is strongly enhanced. The effect of the YY/EE mutation is moderate compared with that of the wild type RIAM, which is conceivable because a flexible glutamate residue can only recapitulate the effect of a bulky phosphotyrosine to a limited extent, and a small population of wild type RIAM is expected to be phosphorylated in HEK293 cells without starvation.
We then assessed the effect of the nonphosphorylatable mutations on RIAM’s function in mediating integrin activation. CHO-A5 cells transfected with GFP-RIAM (wild type or mutations) were subject to FACS analysis for integrin αIIbβ3 activity. While the cells expressing wild type RIAM exhibited an increased integrin activity, this increase is significantly diminished in the cells expressing the Y267F or the Y427F mutations. Moreover, the CHO-A5 cells expressing A435Y or Y267E/Y427E exhibited increased integrin compared with those of wild type RIAM. Together, these results indicate that phosphorylation of RIAM by Src kinases at Tyr267 and Tyr427 is required for promoting the PM translocation of RIAM and the subsequent RIAM-mediated integrin activation.
Discussion
Adaptor proteins play important roles in cell signaling that modulates cell function and behavior. Partner binding avidity and accessibility to the partner binding site of adaptor proteins are tightly linked to their oligomerization states and associated intermolecular interactions. In particular, another RA-PH-containing protein, Grb10, enhances its inhibitory function towards insulin receptor kinase via a PH-mediated intermolecular interaction (Depetris et al., 2009). The biologically relevant intermolecular interaction of proteins can often be revealed in the crystal structures. However, Crystallization of the protein molecules also inevitably generates biologically irrelevant protein-protein interfaces due to crystal packing. Xu et al. developed a structural bioinformatics tool known as ProtCID to help identify the biologically relevant interfaces by searching for common interfaces from other reported structures with similar structural elements (Xu and Dunbrack, 2011). When we performed interface search for the original RIAM CC-RA-PHa structure (PDB code: 3TCA) using ProtCID, both the asymmetric interface and the pseudo-symmetric “H-Y” interface were identified. Interestingly, although the “H-Y” interface affords a larger buried area, ProtCID ranked the asymmetric interface at the top position for biological relevance. Indeed, in addition to the aforementioned RIAM structures, ProtCID also identified a conserved interface in the crystal structure of Lpd CC-RA-PH segment (PDB code: 4GMV), in which the phosphoinositide-binding site of one Lpd PH domain is masked by the C-terminal helical extension of a neighboring PH domain (Fig. S4A). Moreover, in the crystal structure of Lpd RA-PH (PDB code: 4GN1), we also identified an interface in which the phosphoinositide-binding site is masked by the helical extension of a neighboring PH domain in a similar manner (Fig. S4B). This observation suggests that Lpd may also adopt oligomeric assemblies that suppress its interaction with the PM. Nevertheless, the regulatory mechanism for the oligomerization of Lpd on its functions such as lamellipodia formation requires further investigation.
In T cells, RIAM has been shown to regulate the intracellular localization and activation of PLC-γ1. PLC-γ1 constitutively interacts with RIAM, and knock-down of RIAM in T cells leads to changes of intracellular localization of PLC-γ1 from the PM to the cytoplasm (Patsoukis et al., 2009). Although it has been shown that both RIAM and PLC-γ1 are substrates of LCK upon T cells activation (Patsoukis et al., 2009), it was unclear how phosphorylation of RIAM by LCK triggers the PM translocation of PLC-γ1. Our data confirm that RIAM is phosphorylated by LCK upon T cell activation, and reveal that phosphorylation of RIAM by LCK leads to the PM translocation of RIAM and β2 integrin activation. Thus, our study provides a rational mechanism for the RIAM-dependent PM translocation of PLC-γ1: phosphorylation of RIAM by LCK unmasks the phosphoinositide-binding site in the PH domain, allowing RIAM to transport the bound PLC-γ1 to the PM. Moreover, our data indicated that phosphorylation of RIAM by Src promotes β1 integrin-mediated cell adhesion in CHO cells (Fig. 3G), and β3 integrin activation in CHO-A5 cells (Fig. 4F). Despite the recently reported alternative RIAM-independent pathways, these results underscore the close involvement of RIAM in integrin signaling.
Although Tyr267 appears to be buried inside of the molecule, the PH domain in the RA-PH module affords unique features that would allow the exposure of Tyr267 for phosphorylation. Here we showed the superposition of the RIAM PH domain with two canonical PH domains of SHARPIN (PDB code: 4EMO) and IRS-1 (PDB code: 1QQG) (Fig. S5) (Dhe-Paganon et al., 1999; Stieglitz et al., 2012). The C-terminal helical extension in RIAM is not conserved in the canonical PH domains, and is distal from the hydrophobic core of the PH domain. Moreover, the segment where Tyr267 resides is not seen in the conserved PH domains. This segment, connecting the PH domain and the RA domain, is also quite dynamic, as a part of the segment (residues 278-292) is disordered in all crystal forms. Moreover, this analysis is supported by the mass spectrometry of RIAM treated by either Fyn or Lck, that confirmed the phosphorylation of Tyr267 with high abundance in both samples. Therefore, the flexibility of the C-terminal helical extension and dynamic environment for the Tyr267 would allow the exposure of Tyr267 for phosphorylation.
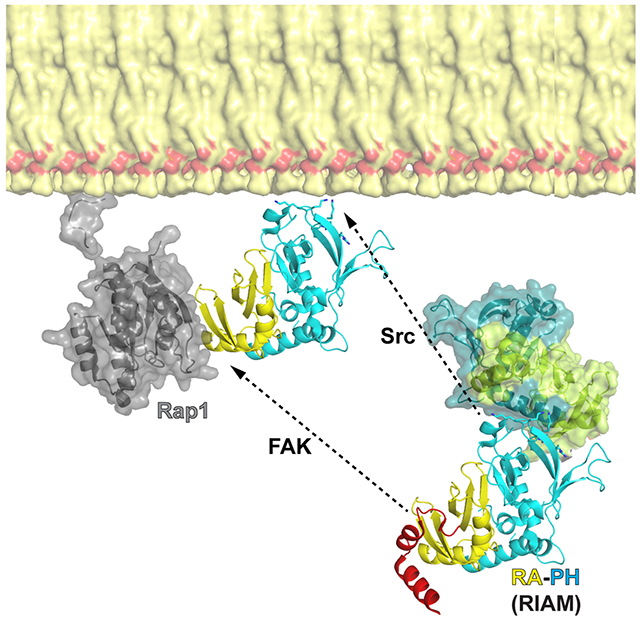
The PM translocation of RIAM requires both RA and PH domains as they function as a proximity detector for GTP-bound RAP1 and PIP2. Interestingly, the PH domain appears to be even more essential than the RA domain for this event. Indeed, ectopically expressed tandem PH domains spontaneously locate on the PM, whereas the tandem RA domains remain in the cytosol even with the presence of constitutively activated RAP1 (Wynne et al., 2012). An autoinhibitory configuration of RIAM was recently unveiled, in which the RAP1-binding site in the RA domain is masked by an N-terminal IN segment (Chang et al., 2019). Phosphorylation of the IN segment by FAK disrupts this configuration, exposing the RA domain for RAP1 association. Here we identify a new inhibitory interface that suppresses the association of the PH domain with the PM. Our data also indicate that both phosphorylation of the IN segment by FAK kinase and phosphorylation of the PH domain by Src kinase are essential for RIAM’s function in mediating integrin activation. Thus, our results yield a comprehensive understanding of the RIAM-mediated integrin signaling pathway. In summary, the signaling functions of RA and PH domains in RIAM are tightly regulated, as the binding sites to their partners on the PM, RAP1 and PIP2, respectively, are both masked, resulting in an autoinhibitory assembly of RIAM. Upon activation of the inside-out signaling pathway, phosphorylation of the IN segment by FAK kinase unmasks the RAP1-binding site in the RA domain and phosphorylation of the RA-PH module by Src kinases unmasks the phosphoinositide-binding site in the PH domain, leading to the PM translocation of RIAM. Activated RIAM recruits and activates talin, which in turn coordinates with kindlin to bind to integrin β chain, resulting in integrin activation and clustering (Lee et al., 2009; Sun et al., 2019; Yang et al., 2014; Ye et al., 2014) (Fig. 5).
Figure 5. A model of RIAM activation by FAK and Src kinases.

Dormant RIAM adopts an autoinhibited configuration with intermolecular interactions mediated by the PH domain and the N-terminal region. This autoinhibited configuration is released upon phosphorylation by FAK and Src kinases. FAK kinase phosphorylates the IN segment, unmasking the RAP1-binding site in the RA domain. Src kinase phosphorylates of the PH domain, unmasking the phosphoinositide-binding site in the PH domain. The two activating mechanisms of RA-PH by phosphorylation cooperatively activate RIAM, promoting the PM translocation of RIAM by interacting with RAP1 and PIP2.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jinhua Wu (Jinhua.wu@fccc.edu)
Materials Availability
All plasmids generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The accession numbers for the data reported in this paper are PDB: 6OLU (RA-PH) and 6O6H (CC-RA-PHb).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RIAM RA-PH and CC-RA-PH were expressed in BL21(DE3) in LB media. Protein expression was induced by supplement of 0.2 mM IPTG at 16°C for overnight.
Cell lines: HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM). Jurkat T cells were cultured in Roswell Park Memorial Institute medium (RPMI). CHO cells and CHO-stably expresses αIIbβ3 integrin (CHO-A5) were cultured in Ham’s F-12. All media were enriched with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin, and the cells were maintained in a 5% CO2 incubator at 37°C. HEK293T cells, Jurkat T cells, and CHO cells were obtained from ATCC, Manassas, VA, USA. And CHO-A5 cells were generated as reported in JC Loftus et al., 1990, Science 249;4971:915-918.
METHOD DETAILS
Plasmid construction, protein purification, and size exclusion chromatography
Various mouse RIAM constructs (gene name: amyloid beta (A4) precursor protein-binding, family B, member 1 interacting protein; gene id: 54519; and uniprot identifier: B1AYC9) containing the RA-PH domains were generated for expression in E. coli, and His6-tagged protein purification was carried out as previously described (Wynne et al., 2012; Zhang et al., 2014). Purified RIAM proteins were concentrated to 10 mg/ml. Crystals of RA-PH alone were grown in 0.2 M sodium citrate, and 20% (w/v) PEG 3350. Crystals of the CC-RA-PHb form were grown in 0.1 M sodium malonate, pH 5.8, and 10% (w/v) PEG 3350. The crystals were briefly soaked in the crystallization solution with 30% ethylene glycol then flash-frozen in liquid nitrogen prior to the diffraction experiments. Site directed mutagenesis (following the QuikChange site-directed mutagenesis instruction manual). Primers; Y398A-F: cag aag gaa tct caa gct atc aag tat ctc tgc, Y398A-R: gca gag ata ctt gat agc ttg aga ttc ctt ctg, H389A-F: tgc ttt gtt tta aag gct cct caa att cag aag, H389A-R: ctt ctg aat ttg agg agc ctt taa aac aaa gca, A435Y-F: tat cag cgg gct gtg tat aga gct gga ctt gcc, A435Y-R: ggc aag tcc agc tct ata cac agc ccg ctg ata, Y267E-F: caa agt act gtt ttt gga gaa aga gga aag aga ggc tgt gtt taa aaa ccc, Y267E-R: ggg ttt tta aac aca gcc tct ctt tcc tct ttc tcc aaa aac agt act ttg, Y267F-F: aag tac tgt ttt tgg aga aag agg aaa gat ttg ctg tgt tta aaa ac, Y267F-R: gtt ttt aaa cac agc aaa tct ttc ctc ttt ctc caa aaa cag tac tt, Y427E-F: caa gta tgg gaa gac tct cga gga taa cta tca gcg ggc tg, Y427E-R: cag ccc gct gat agt tat cct cga gag tct tcc cat act tg, Y427F-F: gcc aag tat ggg aag act ctc ttt gat aac tat cag c, Y427F-R: gct gat agt tat caa aga gag tct tcc cat act tgg c.
X-ray crystallography
X-ray diffraction data were collected using the X4C beamline at the National Synchrotron Light Source, Brookhaven, NY for RIAM RA-PH alone, and the F1 beamline at Cornell High Energy Synchrotron Source (CHESS), Ithaca, NY for RIAM CC-RA-PHb. Molecular replacement with the RIAM RA-PH domains (PDB code 3TCA) as the initial search model was carried out for structure determination. The atomic models were built and refined using Coot (Emsley and Cowtan, 2004) and REFMAC (Murshudov et al., 1997). Data collection and refinement statistics are listed in Table 1. Structural figures were generated using the PyMOL program suite (http://pymol.sourceforge.net). The atomic coordinates and structure factors have been submitted to Protein Data Bank with accession number 6OLU for RA-PH alone, and 6O6H for CC-RA-PHb.
Table 1.
Data collection and refinement statistics
| RIAM | CC-RA- PHb (6O6H) |
RA-PH (6OLU) |
|---|---|---|
| Data Collection | ||
| Space group | P21212 | P212121 |
| a, b, c (Å) | 84.146, 92.981, 44.100 | 45.315, 80.624, 82.714 |
| α, β, γ(°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 50.00-2.50 | 50.00-1.90 |
| Completeness (%) | 100.0 (100.0) | 98.0 (92.4) |
| Rsym (%) | 10.4 (65.9) | 7.2 (41.5) |
| I / σ(I) | 20.3 (2.0) | 16.0 (2.0) |
| Unique reflections | 12656 | 24074 |
| Redundancy | 6.2 (6.3) | 3.2 (2.6) |
| Refinement | ||
| Resolution (Å) | 50.00-2.50 | 50.00-1.90 |
| Rwork/Rfree (%) | 22.9/28.1 (26.4/28.9) | 21.2/25.6 (25.7/27.3) |
| RMSD bonds (Å) | 0.009 | 0.008 |
| RMSD angle (°) | 1.359 | 1.239 |
| Protein atoms | 2094 | 2026 |
| Solvent atoms | 38 | 293 |
| Average b-factors (Å2) | 57.0 | 23.0 |
| Favored/Allowed regions | 98%/100% | 100%/100% |
Parentheses denoted highest resolution bin, 2.57-2.50 Å for CC-RA-PHb; 1.95-1.90 Å for RA-PH.
Transient transfection, co-immunoprecipitation, and Western blot analysis
The cells were split in 6-mm dishes 1 day prior to transfection, and the subconfluent cells were co-transfected with 1 μg of RIAM constructs, together with additional DNA when necessary, using the Lipofectamine® 2000 (Thermo Fisher Scientific). Control cells were transfected with reagents without DNA or with empty vectors. Twenty-four hours later, the transfected cells were rinsed twice with phosphate-buffered saline and lysed in a lysis buffer containing 50 mM Tris 8.0, 120 mM NaCl, 0.5% NP-40 with and protease/ phosphatase inhibitor cocktails. For RIAM phosphorylation, HEK293 cells overexpressing HA-RIAM and GFP-RIAM were lysed and treated 0.5 mM ATP. Co-IP was performed using Surebeads™ magnetic beads (Bio-Rad, Hercules, CA, USA). Samples were eluted in Laemmli sample buffer by heating the samples to 70°C for 10 min, and then separated on a 10-15% SDS-polyacrylamide gel. The proteins were detected by reacting with an enhanced chemiluminescence reagent (Thermo Fisher Scientific, Waltham, MA USA). The resulting image was recorded using a luminescent image analyzer (FluorChem E, ProteinSimple, San Jose, CA USA). The antibodies used in this study are: anti-GFP and anti-HA (Sigma), anti-pY (Cell signaling), anti-LCK and anti-GAPDH (Santa Cruz Biotech), anti-RIAM and anti-RAP1 (Abcam), and anti-integrin β1 (Proteintech).
In vitro Phosphorylation of RIAM and GST pull-down
Jurkat T cells were starved in RPMI serum free medium with 0.5% FBS for 2 h and incubated with DMSO or 400 nM of FAK inhibitor PF431396 or Src inhibitor RK24466 (Cayman Chemical, Ann Arbor, MI USA) at 37°C for 1 h, and then stimulated by 2.5 μg/ml anti-CD3 antibodies (Abcam, Cambridge, MA USA) for 10 min. The cells were lysed in 200 ul of lysis buffer (1% Triton X-100, 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.25 mM ATP) supplemented with protease/ phosphatase inhibitor cocktails. Clarified lysate was incubated with 10 μg of GST-RA-PH for 1 h at 4 °C with gentle shaking. GST beads were then added to mixture of lysate and GST-RA-PH and gently agitated for 1 h at 4 °C. Samples were eluted in Laemmli sample buffer and boiled to 95°C for 10 min. For In vitro kinase assays, purified RIAM-NT or RA-PH were incubated with 100 nM FYN (Carna Biosciences) or 50 nM LCK (Carna Biosciences) in the reaction buffer (5 mM ATP, 20 mM MgCl2, 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 50 μM Na3VO4 at room temperature. Reactions were stopped by adding EDTA to the samples to a final concentration of 50 mM. Samples were separated by 10-15% SDS-PAGE gels.
Subcellular fractionation
Jurkat T cells were pretreated with 10 μM of Src inhibitor and stimulated by anti-CD3 antibodies. Cells were harvested in fractionation buffer (20 mM Tris-HCl pH 7.4, 50 mM NaCl, 2 mM MgCl2, 5 mM KCl with protease/phosphatase inhibitor) and incubated on ice for 10 min. Swollen cells were disrupted by shearing through 27-gauge needles. Lysates were centrifuged at 2,000 rpm for 10 min to pellet nuclei and intact cells. The supernatant was further centrifuged at 13,200 rpm for 30 min to precipitate the membrane- and cytoskeleton-containing fraction. The pellet was washed and then solubilized in fractionation buffer containing 1% NP-40 on ice. Each sample was then mixed with Laemmli sample buffer and boiled at 95 °C for 10 min. Samples were run on SDS-PAGE gels and expression of RIAM in cytosol and plasma membrane fractions were analyzed by western blot assay. RAP1 and GAPDH were used as membrane and cytosolic markers, respectively.
Mass Spectrometry
Gel bands of interest were digested from SDS-PAGE. Proteolytic peptides were resuspended in 0.1% formic acid and separated with a Thermo RSLC Ultimate 3000 on a Thermo Easy-Spray C18 PepMap 75 μm x 50 cm C-18 2 μm column with a 70-min gradient of 4-25% acetonitrile with 0.1% formic acid at 300 nL/min and 50°C. Eluted peptides were analyzed by a Thermo Q Exactive plus mass spectrometer. The AGC for MS1 was set to 3x106 with a max injection time of 120 ms, and the AGC for MS2 ions was set to 1x105 with a max injection time of 150 ms and the dynamic exclusion was set to 90 s. Protein identification was performed by searching MS/MS data against the swiss-prot human protein database downloaded on July 26, 2017 using sequest HT built in Proteome Discoverer 2.2. The search was set up for full tryptic peptides with a maximum of two missed cleavage sites. Acetylation of protein N-terminus, phosphorylation of serine, threonine, tyrosine and oxidized methionine were included as variable modifications and carbamidomethylation of cysteine was set as fixed modification. The precursor mass tolerance threshold was set 10 ppm for and maximum fragment mass error was 0.02 Da. The significance threshold of the ion score was calculated based on a false discovery rate of < 1%.
Confocal microscopy, flow cytometry, and cell adhesion assay
For confocal microscopy imaging, Jurkat cells were plated in 12-well plate on the cover glass and incubated until they became 60% confluent. The cells were transfected with RIAM and RAP1G12V, and after 24 h, washed the cells and fixed with methanol for 10 min and permeated with acetone for 1min. After blocking with 2% BSA for 1 h, the cells were incubated with an anti-HA antibody (1:500) for 2 h, followed by incubation with Alexa 647(goat anti-mouse IgG, 1:200) and DAPI for 15 min. Mounted with prolong gold antifade reagent (Invitrogen, Carlsbad, CA, USA) and the cells were observed with a confocal microscope (Leica SP8 advanced). 25 cells from three independent experiments representing the majority of the cellular patterns for each sample were selected and ranked based on the membrane distribution of RIAM. In each sample, six cells with rankings of 1, 5, 9, 13, 17, 21 were selected rankings in each sample were used to calculate the profile plots. One representative cell of each sample is shown. The remaining five cells for Figure 2A are shown in Figure S2. To calculate the profile plot, the RIAM distribution of fluorescence intensities were measured using Fiji upon drawing the line cross the cells. The percentage of the fluorescence intensity within the 10% distance from both sides for the six selected cells was calculated for means to make the scatter plots shown in Figure 2B and 4C. The statistical analysis was carried out with two tailed unpaired t-test.
For flow cytometry, CHO-A5 cells were transfected with RIAM and incubated for 24 h. Then, the cells were washed twice with phosphate-buffered saline, harvested in trypsin-EDTA, and spun at 2,000 rpm for 5 min. The cells were incubated in Tyrode’s buffer (136.9 mM NaCl, 10 mM HEPES, 5.5 mM Glucose, 11.9 mM NaHCO3, 2.7 mM KCl, 0.5 mM CaCl2, 1.5 mM MgCl2, 0.4 mM NaH2PO4, pH 7.4) containing PAC1 antibody for 1 h at 30 °C and then stained with Alexa 647 (goat anti-mouse IgM) for 30 min. The cells were quantified with an LSR flow cytometer using 30,000 cells per measurement.
For cell adhesion analyses, 5x106 Jurkat T cells or 4x105 CHO cells were spread into ICAM-1 (10 μg/ml) and fibronectin (FN) (20 μg/ml) coated 24-well plates respectively and blocked with PBS containing 0.5% BSA. For Jurkat T cells, cells were starved by 0.5% FBS for 2 h before anti-CD3 antibodies (2.5 μg/ml) was used for stimulation. Src inhibitor was used to pretreat cells for 1 h with warm 0.5% BSA in PBS containing 2 mM MgCl2 and 1 mM CaCl2 (adhesion buffer). Unbound cells were removed by washing the plate with warm adhesion buffer and stained with crystal violet for 10 min, and then measured by plate-reader (570 nm). For CHO cells, Src inhibitor pretreated cells were placed to the FN-coated plate for 30 min and unbound cells were removed and stained with crystal violet. PMA was used for positive control and unstimulated cells or uncoated plates were used for negative control.
QUANTIFICATION AND STATISTICAL ANALYSIS
To quantify the PM distribution of RIAM (wild type and various mutations) which is shown in the Figure 2A, scatter plot was shown mean ± SD, n = 6 using Prism. Statistical significances were calculated using a two-tailed t-test for unpaired samples using excel. *: p < 0.005: p = 0.17 (NS) (Figure 2B). To show the bar graph for the PM distribution of RIAM indicated in 3D and for β2 and β1 integrin adhesion, data shown are mean ± SD, n = 3, also seen in Supplemental Table 1. Statistical significances were calculated using a two-tailed t-test for unpaired samples using excel. *: p < 0.03; **: p < 0.005; #: p = 0.06 (NS) (Figure 3E, F, and G). To quantify the PM distribution of RIAM (wild type and various mutations) and integrin activation, the scatter plot was shown with mean ± SD, n = 6 (Figure 4C) and with mean ± SD, n = 4 (Figure 4F) using Prism, and the bar graph was shown the PM distribution of RIAM indicated in 4D with mean ± SD, n = 3 (Figure 4E) using excel. Statistical significances were calculated using a two-tailed t-test for unpaired samples using excel. *: p < 0.05.
ADDITIONAL RESOURCES.
Not applicable
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GFP (N-terminal) | Sigma-Aldrich | Cat# G1544 |
| Mouse monoclonal anti-HA (HA-7) | Sigma-Aldrich | Cat# H9658 |
| Mouse anti-p-Tyr-100 | Cell Signaling | Cat# 9411S |
| Mouse monoclonal anti-LCK (3A5) | Santa Cruz Biotech | Cat# sc-433 |
| Mouse monoclonal anti-GAPDH (G-9) | Santa Cruz Biotech | Cat# sc-365062 |
| Rabbit monoclonal anti-RIAM | Abcam | Cat# ab92537 |
| Rabbit monoclonal anti-RAP1(RAP1A+RAP1B) | Abcam | Cat# ab75871 |
| Mouse monoclonal anti-Integrin β1 | Proteintech | Cat# 66315-1 |
| Rabbit polyclonal anti-CD3 antibody | Abcam | Cat# ab5690 |
| Mouse anti-PAC1 | BD Biosciences | Cat# 340535 |
| Goat polyclonal anti-Mouse IgM, Alexa 647 | Invitrogen | Cat# A21238 |
| Goat polyclonal anti-Mouse IgG1, Alexa 647 | Invitrogen | Cat# A21240 |
| Bacterial and Virus Strains | ||
| Bacteria: E.coli DH5α | Thermo Scientific | Cat# EC0112 |
| Bacteria: E.coli BL21(DE3) | NEB | Cat# C2527I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine® 2000 | Invitrogen | Cat# 11668019 |
| FAK inhibitor PF431396 | Cayman Chemical | Cat# 17665 |
| Src inhibitor RK24466 | Cayman Chemical | Cat# 15135 |
| ICAM-1 | Bio-techne | Cat# 720-IC-200 |
| Fibronectin | Sigma-Aldrich | Cat# F0895 |
| PMA (phorbol 12-myristate 13-acetate) | Abcam | Cat# ab120297 |
| Full-length human GST-FYN (Isoform a) | Carna Biosciences | Cat# 08-168 |
| Full-length human GST-LCK | Carna Biosciences | Cat# 08-170 |
| Critical Commercial Assays | ||
| Surebeads™ protein G magnetic beads | Bio-Rad | Cat# 161-4023 |
| Deposited Data | ||
| RIAM RA-PH core structure | This study | PDB: 6OLU |
| RIAM CC-RA-PHb | This study | PDB: 6O6H |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | CRL-11268 |
| Human: Jurkat T cells | ATCC | TIB-152 |
| Hamster: CHO cells | ATCC | CLR-12023 |
| Oligonucleotides | ||
| Primers for mutagenesis, see Method Details | This paper | N/A |
| Software and Algorithms | ||
| Fiji | Schindelin and Frise, E. et al. (2012) | https://imagej.net/Fiji |
| FlowJo | Version: 9.9.6 | https://www.flowjo.com/solutions/flowjo/downloads/previous-versions |
| GraphPad Prism | Prism 8.4.2 | https://www.graphpad.com/scientific-software/prism/ |
| Pymol | Schrödinger | https://pymol.org/ |
| coot | Emsley and Cowtan (2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| REFMAC | Murshudov et al. (1997) | http://www.ccp4.ac.uk/html/refmac5.html |
Highlights.
All crystal structures of RIAM RA-PH possess a common interface.
The PIP2 binding site of the PH domain is masked by the interface.
Phosphorylation of RIAM by Src family kinases promotes RIAM translocation to the PM.
Phosphorylation of Y267 and Y427 is essential for RIAM-mediated integrin activation.
Acknowledgements:
We thank Roland L. Dunbrack for help on ProtCID analyses. We thank the beamline staff of X4C at NSLS and F1 at MacCHESS for help on data collection. This work was supported by NIH Grants GM119560 (to J.W.), CA211670 (to J.S.D.), American Cancer Society Grant RSG-15-167-01-DMC (to J.W.), and a Cancer Center Support Grant Supported Pilot Projects Award 5P30CA006927-51 (to J.W.). E.A.C. was supported by Grant 5T32-CA-009035-42 to FCCC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES:
- Barczyk M, Carracedo S, and Gullberg D (2010). Integrins. Cell Tissue Res 339, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A, and Hogg N (1999). Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J Exp Med 189, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Su W, Cho EA, Zhang H, Huang Q, Philips MR, and Wu J (2019). Molecular basis for autoinhibition of RIAM regulated by FAK in integrin activation. Proc Natl Acad Sci U S A 116, 3524–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Zhang H, Franco-Barraza J, Brennan ML, Patel T, Cukierman E, and Wu J (2014). Structural and mechanistic insights into the recruitment of talin by RIAM in integrin signaling. Structure 22, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi N, Peuhu E, Parsons M, Rissanen S, Vattulainen I, Salmi M, Ivaska J, and Pouwels J (2015). Mutually Exclusive Roles of SHARPIN in Integrin Inactivation and NF-kappaB Signaling. PLoS One 10, e0143423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depetris RS, Wu J, and Hubbard SR (2009). Structural and functional studies of the Ras-associating and pleckstrin-homology domains of Grb10 and Grb14. Nat Struct Mol Biol 16, 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhe-Paganon S, Ottinger EA, Nolte RT, Eck MJ, and Shoelson SE (1999). Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc Natl Acad Sci U S A 96, 8378–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, and Shattil SJ (2005). Integrin regulation. Curr Opin Cell Biol 17, 509–516. [DOI] [PubMed] [Google Scholar]
- Holt LJ, and Daly RJ (2005). Adapter protein connections: the MRL and Grb7 protein families. Growth Factors 23, 193–201. [DOI] [PubMed] [Google Scholar]
- Klapholz B, and Brown NH (2017). Talin - the master of integrin adhesions. J Cell Sci 130, 2435–2446. [DOI] [PubMed] [Google Scholar]
- Klapproth S, Sperandio M, Pinheiro EM, Prunster M, Soehnlein O, Gertler FB, Fassler R, and Moser M (2015). Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood 126, 2704–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, and Gertler FB (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol 19, 541–564. [DOI] [PubMed] [Google Scholar]
- Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, Constantine E, Springer TA, Gertler FB, and Boussiotis VA (2004). RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell 7, 585–595. [DOI] [PubMed] [Google Scholar]
- Lagarrigue F, Kim C, and Ginsberg MH (2016). The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 128, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarrigue F, Vikas Anekal P, Lee HS, Bachir AI, Ablack JN, Horwitz AF, and Ginsberg MH (2015). A RIAM/lamellipodin-talin-integrin complex forms the tip of sticky fingers that guide cell migration. Nat Commun 6, 8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird RM, and Hayes SM (2010). Roles of the Src tyrosine kinases Lck and Fyn in regulating gammadeltaTCR signal strength. PLoS One 5, e8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, and Ginsberg MH (2009). RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem 284, 5119–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Rivera-Nieves J, Sandborn WJ, and Shattil S (2016). Integrin-based therapeutics: biological basis, clinical use and new drugs. Nature reviews. Drug discovery 15, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley SC, Marsh M, Bebbington CR, Proudfoot K, and Jordan P (1994). Distinct intracellular localization of Lck and Fyn protein tyrosine kinases in human T lymphocytes. J Cell Biol 125, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt M, Filby A, Parravicini V, Werlen G, Palmer E, and Zamoyska R (2006). Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol Cell Biol 26, 8655–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Dustin ML, and Philips MR (2007). Small GTPases and LFA-1 reciprocally modulate adhesion and signaling. Immunol Rev 218, 114–125. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Palacios EH, and Weiss A (2004). Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000. [DOI] [PubMed] [Google Scholar]
- Park SC, and Jeen YT (2018). Anti-integrin therapy for inflammatory bowel disease. World J Gastroenterol 24, 1868–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Lafuente EM, Meraner P, Kim J, Dombkowski D, Li L, and Boussiotis VA (2009). RIAM regulates the cytoskeletal distribution and activation of PLC-gamma1 in T cells. Sci Signal 2, ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, Wakeham A, Shahinian A, van der Heiden A, Bachmann MF, et al. (1996). LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med 183, 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz B, Haire LF, Dikic I, and Rittinger K (2012). Structural analysis of SHARPIN, a subunit of a large multi-protein E3 ubiquitin ligase, reveals a novel dimerization function for the pleckstrin homology superfold. J Biol Chem 287, 20823–20829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, Berger J, Paul DS, Bergmeier W, Gertler FB, et al. (2015). Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood 126, 2695–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Costell M, and Fässler R (2019). Integrin activation by talin, kindlin and mechanical forces. Nature Cell Biology 21, 25–31. [DOI] [PubMed] [Google Scholar]
- Vestweber D (2015). How leukocytes cross the vascular endothelium. Nat Rev Immunol 15, 692–704. [DOI] [PubMed] [Google Scholar]
- Weitzman JB, Chen A, and Hemler ME (1995). Investigation of the role of beta 1 integrins in cell-cell adhesion. J Cell Sci 108 (Pt 11), 3635–3644. [DOI] [PubMed] [Google Scholar]
- Wynne JP, Wu J, Su W, Mor A, Patsoukis N, Boussiotis VA, Hubbard SR, and Philips MR (2012). Rap1-interacting adapter molecule (RIAM) associates with the plasma membrane via a proximity detector. J Cell Biol 199, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, and Dunbrack RL Jr. (2011). The protein common interface database (ProtCID)--a comprehensive database of interactions of homologous proteins in multiple crystal forms. Nucleic Acids Res 39, D761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhu L, Zhang H, Hirbawi J, Fukuda K, Dwivedi P, Liu J, Byzova T, Plow EF, Wu J et al. (2014). Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat Commun 5, 5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Snider AK, and Ginsberg MH (2014). Talin and kindlin: the one-two punch in integrin activation. Front Med 8, 6–16. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chang YC, Brennan ML, and Wu J (2014). The structure of Rap1 in complex with RIAM reveals specificity determinants and recruitment mechanism. J Mol Cell Biol 6, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chang YC, Huang Q, Brennan ML, and Wu J (2016). Structural and Functional Analysis of a Talin Triple-Domain Module Suggests an Alternative Talin Autoinhibitory Configuration. Structure 24, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FF, Xue Y, Chen GL, and Yao X (2004). GPS: a novel group-based phosphorylation predicting and scoring method. Biochem Biophys Res Commun 325, 1443–1448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the data reported in this paper are PDB: 6OLU (RA-PH) and 6O6H (CC-RA-PHb).