

Abstract
The presence of uracil in DNA calls for rapid removal facilitated by the uracil-DNA glycosylase (UNG) family of enzymes, which initiates the base excision repair (BER) pathway. In humans, uracil excision is accomplished primarily by the human UNG (hUNG) enzymes. In addition to BER, hUNG enzymes also play a key role in somatic hypermutation to generate antibody diversity. hUNG has several isoforms, with hUNG1 and hUNG2 being the two major isoforms. Both isoforms contain disordered N-terminal domains, which are responsible for a wide range of functions, with minimal direct impact on catalytic efficiency. Subcellular localization of hUNG enzymes is directed by differing N-terminal sequences, with hUNG1 dedicated to mitochondria and hUNG2 dedicated to the nucleus. An alternative isoform of hUNG1 has also been identified to localize to the nucleus in mouse and human cell models. Furthermore, hUNG2 has been observed at replication forks performing both pre- and post-replicative uracil excision to maintain genomic integrity. Replication protein A (RPA) and proliferating cell nuclear antigen (PCNA) are responsible for recruitment to replication forks via protein-protein interactions with the N-terminus of hUNG2. These interactions, along with protein degradation, are regulated by various post-translational modifications within the N-terminal tail, which are primarily cell-cycle dependent. Finally, translocation on DNA is also mediated by interactions between the N-terminus and DNA, which is enhanced under molecular crowding conditions by preventing diffusion events and compacting tail residues. This review summarizes recent research supporting the emerging roles of the N-terminal domain of hUNG.
Keywords: base excision repair, deoxyuridine, DNA damage, DNA repair, protein interaction, post-translational modification
Graphical Abstract
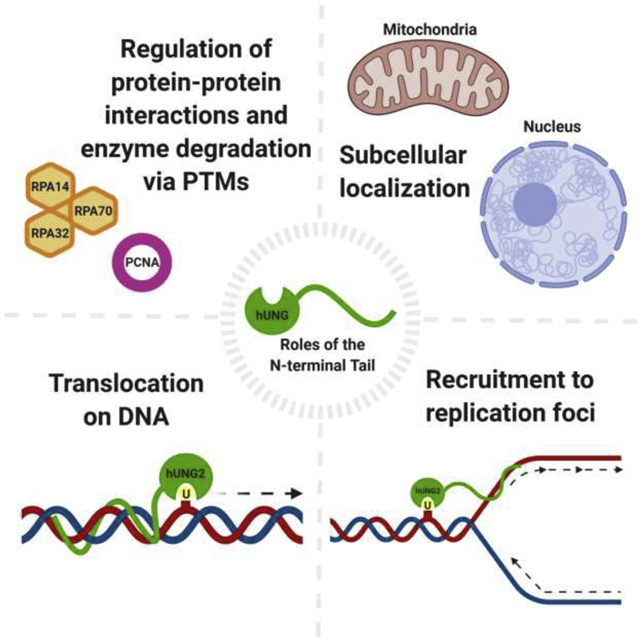
1. Introduction
Uracil is a prevalent non-canonical base in the human genome, resulting from spontaneous hydrolytic deamination of cytosine or misincorporation of deoxyuridine monophosphate (dUMP) by DNA polymerases (Lindahl 1993; Andersen et al. 2005; Chon et al. 2019). Each day, an estimated 60–500 cytosine deamination and up to 10,000 dUMP misincorporation events occur per genome (Krokan et al. 2002; Sire et al. 2008). The steady-state level of uracil in the genome is influenced by the efficiency of DNA repair, with base excision repair (BER) being the primary repair pathway and mismatch repair (MMR) being a backup. Unrepaired uracil in DNA is mutagenic, resulting in G:C to A:T transitions during DNA replication (Krokan et al. 2002). On the other hand, mutagenic processing of genomic uracil residues generated by AID/APOBEC cytidine deaminases plays an important role in adaptive and innate immunity, B-cell lymphomagenesis, and contributes to mutation signatures of many human cancers (reviewed in Krokan et al. 2014).
Critical to the processing of uracil from DNA are the human uracil-DNA glycosylase (hUNG) enzymes. hUNG enzymes are the primary UNG enzymes responsible for uracil excision in humans (Slupphaug et al. 1995; Schormann et al. 2014). hUNG searches and cleaves the uracil residue from DNA and initiates the BER pathway (Krokan et al. 2014). hUNG also plays a key role in somatic hypermutation to generate antibody diversity, supported by the association of missense mutations in the UNG gene with hyper-IgM syndrome type V, a type of immunoglobulin (Ig) deficiency (Imai et al. 2003). The catalytic domain of hUNG enzymes, which is responsible for the excision process itself, has been extensively researched and discussed (Parikh et al. 2000; Krokan et al. 2001; Visnes et al. 2009; Schormann et al. 2014). However, focused reviews of the also-important disordered N-terminal domain are absent in the literature. This article focuses on the various functions of the hUNG N-terminal domain in subcellular localization, replicative repair, protein-protein interactions, protein turnover, and translocation on DNA. For kinetic aspects of the lesion search and recognition by UNG enzymes, please refer to excellent reviews (Zharkov et al. 2010; Esadze and Stivers 2018).
2. Subcellular Localization
A single nuclear human UNG gene of 13.5 kb encodes two main isoforms, hUNG1 and hUNG2, as a result of two separate promoters and alternative splicing (Slupphaug et al. 1993; Haug et al. 1996; Nilsen et al. 1997). While the 269 amino acid catalytic domain is conserved, the two isoforms contain differing N-termini sequences, which prompt their differential subcellular localization (Fig. 1) (Nilsen et al. 1997; Otterlei et al. 1998). In the absence of binding partners, the N-terminal region is largely unstructured (Rodriguez et al. 2020). Residues 1–92 make up the N-terminus of hUNG2, which contains a nuclear localization signal comprised of positively charged residue clusters (K and R residues), rendering hUNG2 the primary uracil-DNA glycosylase enzyme in the nucleus (Slupphaug et al. 1995; Otterlei et al. 1998). N-terminal residues 1–35 of hUNG1 replace residues 1–44 of hUNG2, with the remaining 48 N-terminal residues being conserved (Nilsen et al. 1997).
Figure 1.

N-terminal amino acid sequences of hUNG1 nuclear and mitochondrial variants and hUNG2. Binding motifs are shown in yellow and labeled with their respective interacting proteins, i.e. proliferating cell nuclear antigen (PCNA) and replication protein A (RPA). Isoform-specific residues are shown under brackets. The primary mitochondrial processing peptidase (MPP) cleavage sites is denoted between residues 29 and 30, and minor MPP cleavage sites are shown between residues 75 and 76 and 77 and 78. Conserved catalytic domain residues are labeled as CD at the C-terminus. Post-translational modifications (obtained from Hornbeck et al. 2015 and Bao et al. 2020) are shown above corresponding residues with the following labels: red as acetylation, orange as ubiquitylation, and blue as phosphorylation.
Although hUNG1 had been considered strictly mitochondrial, emerging research has shown that the unique sequence allows for dual localization of different variants of hUNG1 (Nilsen et al. 1997; Sarno et al. 2019). The N-terminus of hUNG1 residues 1–83 contains both a 29-amino acid amphiphilic helix, which acts as a mitochondrial targeting signal (MTS), and positively-charged residues of a nuclear localization signal (Bharati et al. 1998; Sarno et al. 2019). This allows for mitochondrial and nuclear variants of hUNG1, which facilitate uracil excision within both compartments. Upon entering mitochondria, hUNG1 is primarily cleaved between residues 29 and 30 by a mitochondrial processing peptidase (MPP) (Sarno et al. 2019). However, two minor forms lacking 75 and 77 N-terminal residues have also been reported (Fig. 1) (Bharati et al. 1998). Being the only known mitochondrial uracil-DNA glycosylase, hUNG1 plays an important role in retaining genomic integrity in mitochondria, though more research into the mitochondrial functions is necessary.
The identification of a novel, unprocessed isoform of hUNG1 in the nucleus of mouse and human cells has led to a better understanding of its properties and functions (Sarno et al. 2019). The variant has a higher molecular weight relative to its mitochondrial counterpart and constitutes approximately 25% of the total nuclear UNG in mouse B lymphocyte cells (Sarno et al. 2019). The nuclear UNG1 isoform assists in processing genomic uracil in mouse and human cells, though at a lower rate than hUNG2 (Sarno et al. 2019). In hUNG2-knockout cells, uracil levels increase significantly; however, the presence of the nuclear hUNG1 variant decreases the total genomic uracil levels by 5-fold compared to double knockout cells, indicating that the nuclear variant of hUNG1 plays a redundant role in removing uracil residues in the nuclear genome (Sarno et al. 2019). Comparably, the total uracil levels in mouse B lymphocyte UNG1- and UNG2-knockout cells were similar to those in wild-type cells (Sarno et al. 2019). With hUNG2 being most abundant in proliferating cells, the reduced uracil excision of hUNG1 may be the lack of the proliferating cell nuclear antigen (PCNA) binding motif for increased recruitment to replication machinery or the absence of upregulation during S-phase, as discussed in later sections. Aside from removing genomic uracil, hUNG1 also supports class switch recombination (CSR) in mouse B-cells (Sarno et al. 2019), in which hUNG2 has been considered as a major player previously (Conticello 2008). During antibody maturation in B-cells, hUNG2 cleaves AID-induced uracil to form AP sites destined for further processing in a mutagenic manner (Conticello 2008). This latter function of the nuclear hUNG1 isoform may be linked to its ability to physically interact with replication protein A (RPA) (Sarno et al. 2019), considering the presence of the RPA-binding motif in all three isoforms and the essential role of RPA in CSR (Chaudhuri et al. 2004). The lack of the PCNA interacting motif and the ability to interact with RPA have led to the proposed function of the nuclear hUNG1 isoform being dedicated to uracil in ssDNA or ssDNA-dsDNA junctions (Sarno et al. 2019), although this proposal remains to be tested explicitly. Nonetheless, the overlapping roles of hUNG1 and hUNG2 in the nucleus may prove advantageous during periods of elevated nuclear uracil.
3. hUNG at Replication Foci
hUNG2 initiates BER in replicating DNA in order to retain genomic integrity during replication. hUNG2 is most abundant in proliferating cells, and cellular assays have detected it in replisomes behind active replication forks for initiation of BER (Haug et al. 1998; Marit Otterlei, Emma Warbrick, Toril A.Nagelhus, Terje Haug, Geir Slupphaug, Mansour Akbari, Per Arne Aas, Kristin Steinsbekk, Oddmund Bakke 1999; Bjorås et al. 2017). In UNG null mice, an increased level of genomic uracil was detected in proliferating cells; however, only a slight increase in spontaneous mutation frequency was observed (Nilsen et al. 2000). In order to facilitate replicative repair, hUNG2 is several-fold upregulated in early to mid S-phase (Marit Otterlei, Emma Warbrick, Toril A.Nagelhus, Terje Haug, Geir Slupphaug, Mansour Akbari, Per Arne Aas, Kristin Steinsbekk, Oddmund Bakke 1999; Visnes et al. 2009). The high catalytic turnover of UNG enzymes also makes them suitable for uracil excision during replication, allowing them to keep up with replication machinery (Visnes et al. 2009). Indeed, hUNG2 has been shown to be a component in a multiprotein complex isolated from the nuclei of human cycling cell, with the complex containing most of the essential components of BER and is thought to be readily recruited to the lesion site during replication (Parlanti et al. 2007). hUNG2 association at replication forks is regulated and enhanced by protein-protein interactions involving the N-terminal tail. It is probable that a ternary complex forms at replication foci containing PCNA, RPA, and hUNG2, based on binding affinity assays (Weiser et al. 2017). The binding of hUNG2 to replication machinery by PCNA and RPA recruitment allows for pre- and post-replicative repair of DNA within the nucleus.
3.1. Pre-replicative Repair
hUNG2 performs pre-replicative uracil excision at replication forks to ensure the fidelity of the replicative DNA polymerases. The N-terminus of hUNG2 directly interacts with DNA at ssDNA-dsDNA junctions, and a lack of the N-terminus results in reduced uracil excision activities at junction sites (Weiser et al. 2018). The N-terminal tail is targeted to ssDNA at 5’-overhangs adjacent to the parental dsDNA, where it removes uracil near the ssDNA-dsDNA junction (Weiser et al. 2018). Furthermore, binding of hUNG2 to ssDNA-dsDNA junctions results in ~30-fold enhanced excision activity for uracil located 9-bp from the junction site and 5-fold enhancement for uracil positioned 33-bp from the junction site when compared to the catalytic domain alone (Weiser et al. 2018). This enhancement is due to the increased affinity for ssDNA at replication foci, which is 6-fold greater with the N-terminus included relative to the catalytic core alone (Weiser et al. 2018). The N-terminal tail also allows for a strong preference for ssDNA at physiological concentrations of Mg2+, which could potentially assist in its localization (Kavli et al. 2002). Additionally, in the case of a single uracil residue at ssDNA-dsDNA junctions, the position of the uracil does not affect excision activity; however, when multiple lesions are present, the N-terminus (in the presence of 5’-ssDNA overhangs) facilitates preferential targeting of uracil closest to the junction site (Weiser et al. 2018). The N-terminus, which has more conformational flexibility than that of the catalytic domain, can also orient hUNG2 bound to 5’ overhangs, which allows for excision of uracil in the 3’–5’ direction relative to the overhang (Buchinger et al. 2018; Weiser et al. 2018).
In order to increase association at replication forks for pre-replicative excision, the N-terminus of hUNG2 interacts with trimeric RPA. RPA has a high affinity for ssDNA and binds to 3’- or 5’- overhangs, subsequently recruiting hUNG2 to stalled replication forks for removal of uracil (Weiser et al. 2018; Weiser 2020). The association between hUNG2 and RPA enhances the affinity for DNA at replication foci and obviates the need for hUNG2 to diffuse to active replication forks from bulk solution (Weiser 2020). The affinity of hUNG2 for RPA-bound junction DNA is 4.5-fold greater than that of DNA substrate alone (Weiser 2020). Residues 66–88 of the N-terminus, which form an induced alpha helical conformation upon binding to RPA, predominantly interact with the winged-helix of the RPA32 subunit, which is separated from the RPA DNA binding domain by a flexible 32 amino acid linker (Mer et al. 2000; Weiser 2020). Similar to the effects of the N-terminal tail, hUNG2 bound to RPA preferentially excises uracil closer to the junction site (Weiser et al. 2018). However, when bound to RPA, hUNG2 excision is limited to the length of the flexible linker, which extends further from the junction site (Weiser 2020). Thus, excision of uracil within 30 base pairs of ssDNA-dsDNA junctions is enhanced in the presence of RPA, while excision beyond 30 base pairs is significantly decreased, with a 6-fold higher selectivity for uracil residues located at 21 bp rather than 45 bp from the junction (Weiser et al. 2018; Weiser 2020). Uracil excision kinetic assays further support the above findings and show enhanced excision activity when hUNG2 is in complex with RPA (Weiser et al. 2018; Weiser 2020). Assays containing an RPA-bound ssDNA-dsDNA junction substrate with a 5’ overhang, and a uracil located 21-bp from the junction site, showed up to a 17-fold stimulation in uracil excision compared to the tailless UNG control (Weiser 2020). With RPA, uracil excision is enhanced by ~4- to 6-fold with 5’ overhangs and up to 14-fold with 3’ overhangs (Weiser 2020). A model has been proposed where the flexible linker of RPA32 may position hUNG2 such that it can excise proximal uracils near 3′ or 5′ junction overhangs. This mechanism may be relevant for processing ssDNA behind replicative polymerases for post-replicative repair of dsDNA (Weiser et al. 2018).
RPA variants lacking the RPA32 winged-helix still interact with the N-terminus of hUNG2, suggesting the existence of multiple interacting sites between the two proteins. Experiments with different truncated forms of RPA show that hUNG2, with the binding site lying within residues 20–91, can bind another site of trimeric RPA within either its DNA binding core or flexible linkers (Weiser 2020). Enhancement of hUNG2 affinity for junction sites facilitated by the secondary interaction is 10-fold weaker than that with wild-type RPA, but still 2-fold greater than hUNG2 alone, based on binding assays (Weiser 2020). Uracil excision assays show a 3- to 4-fold increase in excision activity compared to solutions lacking RPA, even when the concentration of RPA is less than the DNA substrate (Weiser 2020). hUNG2 bound to the RPA variants can also act on DNA at further lengths from the DNA binding core than seen with wild-type RPA, with uracil excision occurring at lengths of 45-bp compared to 30-bp with the winged-helix (Weiser 2020). The role and binding location of the second RPA-binding site are unknown and require further investigation.
3.2. Post-replicative Repair
hUNG2 plays a critical role in removing incorporated uracil residues in post-replicative DNA repair. The N-terminus of hUNG2 contains a PCNA binding motif (PIP-box) consisting of residues 4–11 (Fig. 1), which assists in targeting hUNG2 to replication forks by binding to one of the three interdomain connecting loops of homotrimer PCNA (Ko and Bennett 2005; Weiser et al. 2017). This binding suggests that UNG2 is localized to newly replicated DNA for rapid removal of uracil resulting from dUMP misincorporation during replication (Buchinger et al. 2018; Sarno et al. 2019). Additionally, BER at replication forks containing PCNA switches to long-patch repair, likely as a result of PCNA interacting with the DNA polymerases involved in repair (Parlanti et al. 2007). Disruption of the PCNA binding motif greatly impairs the recruitment of hUNG2 to replication foci, though localization still occurs by means of the N-terminus or recruitment by RPA (Torseth et al. 2012). PCNA loaded on DNA substrates stimulates hUNG2 catalytic activity up to 2.6-fold, as shown in in vitro uracil excision assays (Ko and Bennett 2005). PCNA stimulation increases in the presence of replication factor C (RFC) and ATP, which are utilized in loading PCNA onto the DNA substrate (Ko and Bennett 2005). Molecular crowding agents, which may include the replication machinery, have also been shown to increase the order of PCNA-binding motif of hUNG2, which may enhance association with PCNA (Rodriguez et al. 2020). A single PCNA homotrimer has also been observed to bind to an average of two hUNG2 enzymes; therefore, it is possible that multiple hUNG2 molecules are recruited to replication forks to perform rapid post-replicative BER (Ko and Bennett 2005).
4. Post-translational Modifications of the N-terminal Domain
PTMs play an important role in regulating BER, primarily through their effects on protein-protein interactions and enzyme degradation. hUNG undergoes 30 reported PTMs, including phosphorylation, ubiquitylation, and acetylation, with many PTM sites located in the N-terminus of both isoforms (Hornbeck et al. 2015). Using two-dimensional SDS-PAGE followed by mass spectrometry analysis, a stepwise phosphorylation pattern has been identified at S23, T60, and S64 of hUNG2 in HeLa cells (Hagen et al. 2008). Phosphorylation of hUNG is facilitated by cyclin-dependent kinases and cyclins (D’Errico et al. 2017). Presumably, phosphorylation proceeds from the N-terminus to the C-terminus, although phosphorylation of S23 is not a prerequisite for subsequent modification (Hagen et al. 2008). hUNG2 expression is upregulated in late G1-phase and early S-phase, and down-regulated in late S-phase and G2-phase (Hagen et al. 2008). In early S-phase, most of the hUNG2 is unmodified or monophosphorylated (S23) (Hagen et al. 2008; Weiser et al. 2017). At the end of S-phase, phosphorylation also occurs at T60 and S64, with the amount of the di- and triphosphorylated forms dominant in late G2-phase.
With respect to the known binding motifs of hUNG2 with PCNA and RPA, S23, T60, and S64 are adjacent to but outside these motifs (Fig. 1). Nevertheless, biochemical and cellular studies have revealed the effects of phosphorylation at these residues on protein-protein interactions. For example, the phospho-mimicking variant (S23D) of hUNG2 results in a greater affinity for RPA and replicating chromatin, whereas hUNG2 variants T60D and S64D reduce the affinity for RPA (Hagen et al. 2008). On the other hand, none of the variant forms of hUNG markedly alter the binding to PCNA as compared to the wild-type protein, based upon unaffected localization to nuclei and replication foci in HeLa cells expressing these variants transiently. Protein semisynthesis, the assembly of a protein from a combination of synthetic and recombinant fragments (Thompson and Muir 2020), has been used to prepare native phosphorylated forms of hUNG2 to investigate the effects of phosphorylation. For example, within the PCNA-binding motif (residues 4–11, also known as the PIP-box motif), there are three sites for phosphorylation: T6, Y8, and S9. Phosphorylation at T6 and Y8 has been shown to inhibit PCNA binding, based on binding affinity assays (Weiser et al. 2017). Phosphorylation of Y8 within the PCNA-binding motif has a greater inhibitory effect than T6 for PCNA binding (Weiser et al. 2017), and is the most prevalent PTM within the entire enzyme (Hornbeck et al. 2015). Therefore, phosphorylation at the PIP-box motif could potentially facilitate the dissociation of hUNG2 from dsDNA (enhanced by PCNA) and its association with ssDNA (enhanced by RPA).
The effect of phosphorylation on the catalytic activity of hUNG2 has been implicated from enzyme kinetic analysis using phospho-mimicking variants (Table 1) (Hagen et al. 2008; Weiser et al. 2017). Compared to wild-type hUNG2, three variant forms (S23D, S23D/T60D, and S23D/T60D/S64D) showed elevated kcat and Km values, with varying extents for different proteins (Hagen et al. 2008). The catalytic efficiency (kcat/Km) stays unaltered overall, with the exception of the S23D/T60D/S64D variant exhibiting an approximately 30% increase relative to wild-type hUNG2. Similarly, the K78Q variant, an acetylation mimic of hUNG2, shows no change in the enzymatic activities relative to wild-type hUNG2 (Bao et al. 2020). An S12C variant and proteins with phosphorylated residue pT6 (UV-induced phosphorylation of T6 has been observed in vivo) or pY8 also show catalytic activity comparable to the wild-type enzyme (Lu et al. 2004; Weiser et al. 2017). Therefore, PTMs of the N-terminal domain have minimal effects on the uracil excision kinetics of hUNG2, although the modifications may affect the glycosylase activity via protein-protein interactions (Weiser 2020).
Table 1.
Enzyme kinetic parameters of wild-type and variant hUNG2. Data are from (Weiser et al. 2017) and (Hagen et al. 2008).
| Enzyme | Substrate | kcat (min−1) | Km (μM) | kcat/Km (min−1 μM−1) | Relative efficiency (kcat/Km)variant/(kcat/Km)wt |
|---|---|---|---|---|---|
| wild-type | dsDNA | 449 | 1 | 449 | |
| S23D | 870 | 1.8 | 483 | 1.07 | |
| S23D/T60D | 641 | 1.4 | 458 | 1.02 | |
| S23D/T60D/S 64D |
726 | 1.2 | 605 | 1.35 | |
| wild-type | ssDNA | 663 | 0.05 | 13300 | |
| S23D | 1981 | 0.14 | 14200 | 1.07 | |
| S23D/T60D | 1141 | 0.09 | 12700 | 0.96 | |
| S23D/T60D/S 64D |
1360 | 0.11 | 12400 | 0.93 | |
| wild-type | dsDNA | 636 ± 68 | 0.7 ± 0.2 | 900 ± 280 | |
| S12C | 546 ± 36 | 0.6 ± 0.1 | 900 ± 160 | 1.0 ± 0.36 | |
| pT6 | 585 ± 74 | 0.7 ± 0.3 | 800 ± 370 | 0.9 ± 0.50 | |
| pY8 | 687 ± 58 | 0.8 ± 0.2 | 900 ± 230 | 1.0 ± 0.40 |
hUNG2 turnover is dependent on the PTMs in the N-terminal tail, with phosphorylation and acetylation signaling for ubiquitylation and subsequent protein degradation. In late G2-phase, the level of the triphosphorylated form (S23D/T60D/S64D) of hUNG2 dominates over the mono- and di-phosphorylated forms (Hagen et al. 2008). Site-directed mutagenesis experiments support the role of the sequence encompassing T60 and S64 in hUNG2 as a phosphodegron (Hagen et al. 2008). Roscovitine, a protein kinase inhibitor, prevents hUNG2 degradation, further implicating the role of phosphorylation in protein degradation (Fischer et al. 2004). Following the formation of the triphosphorylated form (the phosphodegron), another form has a higher molecular weight, suggesting that ubiquitylation follows phosphorylation in the late G2-phase (Hagen et al. 2008). Supporting this notion, assays involving proteasome degradative activity inhibitors identify a ubiquitylated form of hUNG2 (Fischer et al. 2004). Therefore, the triphosphorylated form (S23/T60/S64) of hUNG2 is thought to direct ubiquitylation and proteolytic degradation (Hagen et al. 2008). PhosphoSitePlus reports four ubiquitylation sites in the N-terminus, with K78 being the most common site of ubiquitylation within the entire enzyme (Hornbeck et al. 2015). Recently, acetylation of hUNG2 (primarily at K78) has been shown to facilitate its degradation via interactions with UHRF1 (Ubiquitin Like With PHD And Ring Finger Domains 1), a type E3 ubiquitin ligase (Bao et al. 2020). Supporting the role of E3 ubiquitin ligases in hUNG2 degradation is the observation of scaffolding proteins cullin 4A (CUL4A) and cullin 1 (CUL1) during human immunodeficiency virus type 1 (HIV-1) Vpr-induced degradation of hUNG2 (Schröfelbauer et al. 2005; Schröfelbauer et al. 2007). ROS-induced deacetylation of hUNG2 by class I or II histone deacetylases (HDACs) then prevents interactions between UHRF1 and the hUNG2 N-terminus, and confers cancer cell resistance to H2O2-induced cytotoxicity (Bao et al. 2020). Therefore, it has been proposed that HDAC inhibitors can be used to induce a synergistic killing effect during cancer treatment by promoting hUNG2 degradation resulting from PTMs of the N-terminus (Bao et al. 2020). Exploring novel approaches involving modulating the level and activity of hUNG2 in cancer treatment is a promising area of research.
5. N-terminal Domain of hUNG Facilitates Translocation on DNA
Similar to other DNA glycosylases, hUNG2 searches for uracil by two methods of facilitated diffusion: DNA sliding and hopping (Fig. 3). Sliding, also referred to as associative translocation, involves hUNG2 binding to DNA and ‘sliding’ along the major groove to scan for lesions (Schormann et al. 2014; Rodriguez et al. 2017). While sliding, hUNG2 is transiently bound to DNA as it diffuses past the ion cloud (Schonhoft et al. 2013). Hopping, also referred to as dissociative translocation, involves hUNG2 dissociating from the DNA strand and reassociating at another site (Schormann et al. 2014; Rodriguez et al. 2017). Hopping allows hUNG2 to bypass protein-bound regions of DNA, which would otherwise disrupt translocation when sliding (Esadze and Stivers 2018). In order to search efficiently for lesions, hUNG2 utilizes both modes of diffusion.
Figure 3.
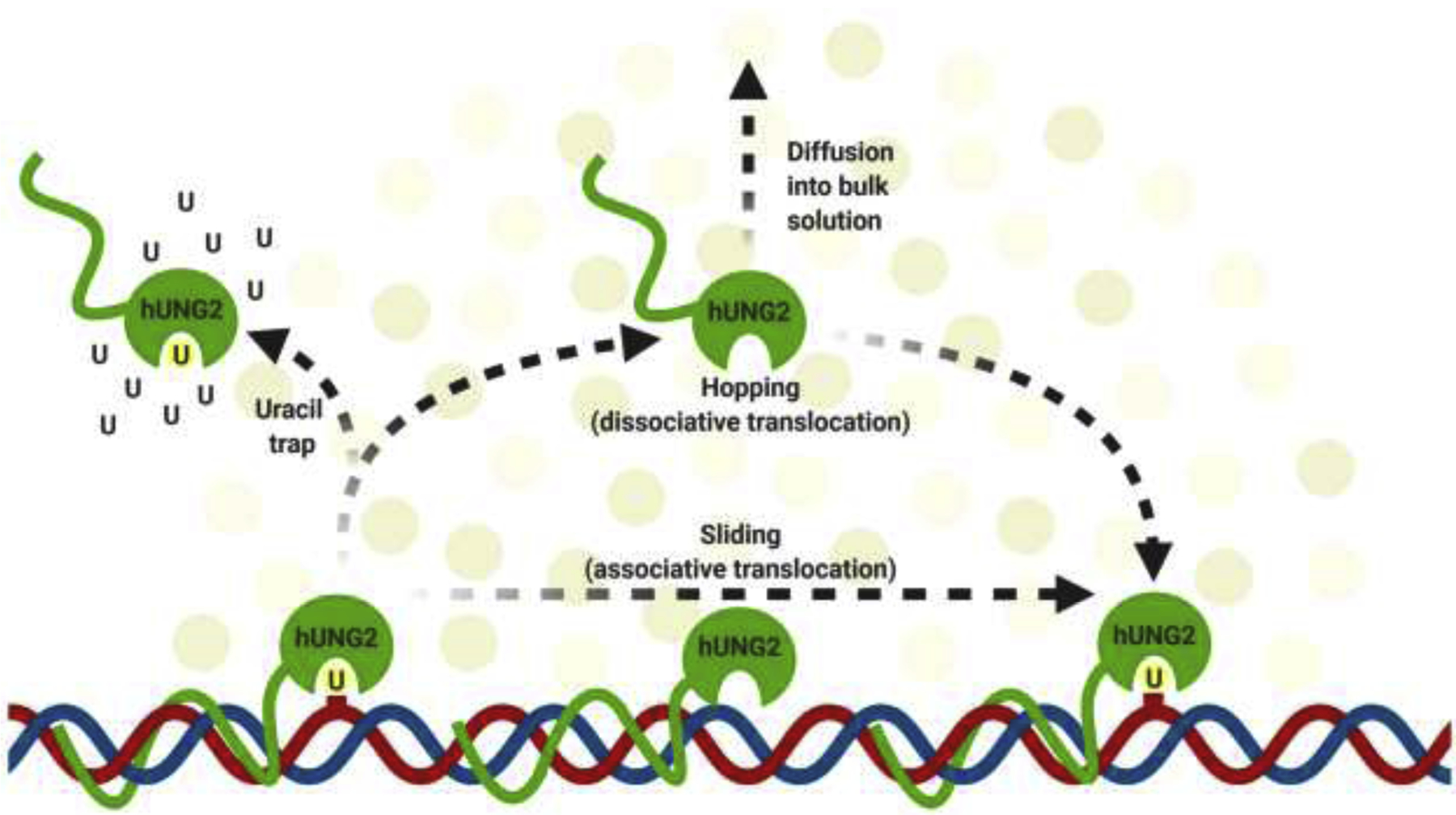
Graphical representation of hUNG2 translocation on DNA by hopping (the dissociative pathway) and sliding (the associative pathway). Uracil trap assays are represented by an abundance of uracil (U), which prevents hUNG2 from translocating via hopping, essentially deactivating the enzyme and leaving only translocation by sliding. For details, see Schonhoft and Stivers 2012.
The N-terminal tail plays an important role in enhancing the translocation of hUNG2 on DNA, especially under molecular crowding conditions. Contact between DNA and the N-terminal tail, which increases under molecular crowding conditions, allows for efficient sliding by preventing diffusion into the bulk solution. Wild-type hUNG2 sliding is 3-fold greater than that of the catalytic core alone, as evidenced in translocation assays (Rodriguez et al. 2017). The addition of the crowding agent polyethylene glycol 8000 (PEG8K) increases the sliding range and site transfer probability of hUNG2 even further (Rodriguez et al. 2017). The transfer probability is defined as the fraction of hUNG2 molecules that excise one uracil site followed by translocation and excision of the other uracil in the same DNA molecule without diffusing into the bulk solution (Schonhoft and Stivers 2012). Uracil traps are utilized to determine the rate of sliding from the total rate of translocation (Fig. 3) (Schonhoft and Stivers 2012). In such an assay, data show that under conditions lacking molecular crowding, hUNG2 sliding is abolished, indicating that hopping dominates under such conditions (Rodriguez et al. 2017). However, with PEG8K and uracil traps in solution, the translocation efficiency occurring at 20-bp spacing is about 68% of that in the absence of the trap (Rodriguez et al. 2017). The overall probabilities of sliding at the 20-bp spacing are 40% for wild-type hUNG2 and only 10% for the catalytic domain, supporting the role of the N-terminus of hUNG2 in facilitating sliding (Rodriguez et al. 2017). Similar effects of hUNG2 have also been demonstrated in human cells (Esadze et al. 2017). In human cells transfected with fluorescent DNA, hUNG2 translocates (both sliding and hopping) across 10-bp with a probability of 100% (Esadze et al. 2017). The likelihood decreases to 54% with 40-bp and 10% with 80-bp spacing, both higher than those observed in the in vitro assays (Esadze et al. 2017). At 40-bp spacing, there is a probability of ~8% that sliding will occur and 46% that hopping will occur (Esadze et al. 2017). Even with crowding conditions in human cells, the major method of DNA translocation is hopping, particularly in cases of larger spacing between uracil (Esadze et al. 2017). Therefore, sliding is primarily observed with short lengths of DNA (≤10 base pairs), the range of a helical turn, but this still allows for scanning of more than a single base pair (Porecha and Stivers 2008). In addition, the hopping mechanism of DNA translocation also shows a 35% increase for hUNG2 enzymes containing the N-terminal tail, and an additional 13% increase in the presence of crowding agents, for a total of 48% increased lesion search by hopping (Dey and Bhattacherjee 2019). The amplified hopping is a result of molecular crowding preventing hUNG2 from fully diffusing into the bulk solution when transferring to another site on the DNA strand (Cravens et al. 2015; Esadze and Stivers 2018). By means of the N-terminal tail and molecular crowding, hUNG2 translocation on DNA is significantly increased compared to the catalytic domain alone.
Mechanistically, the crowding agents surround hUNG2 and confine it to the DNA ion cloud, reducing diffusion events (Esadze and Stivers 2018). Such effects are not unique to hUNG and have also been demonstrated with human 8-oxoguanine DNA glycosylase (hOGG1) (Esadze and Stivers 2018). The 92-amino acid N-terminal domain of hUNG2 interacts with ssDNA and dsDNA weakly and nonspecifically only in the presence of PEG8K, with Kd values being 23 uM and 10 uM, respectively (Rodriguez et al. 2020). Presumably, volume exclusion collapses the N-terminus in order to favor a more compact enzyme-substrate complex, which enhances association with DNA and overshadows negative effects of salt ions in the nucleus (Esadze et al. 2017; Rodriguez et al. 2017; Rodriguez et al. 2020). Such properties support the role of the N-terminus in modulating the DNA-binding activity of hUNG. NMR chemical shifts show the primary points of conformational changes within the N-terminal tail lie between residues S67 and A68 and within residues 84–89 at the C-terminus of the helix, though the remainder of the N-terminus is required for full DNA binding affinity (Rodriguez et al. 2020). The junction of the extended coil region lies between S67 and A68 and the RPA-binding helix, allowing for conformational flexibility (Rodriguez et al. 2020). S67 is also a site for serine phosphorylation, which may negatively impact conformational changes needed for DNA binding (Rodriguez et al. 2020). Furthermore, molecular dynamic simulations suggest the role of positively-charged residues 5–19 and 73–88, particularly R76, K78, R84, and R88 of the RPA-binding motif, in facilitating nonspecific and electrostatic interactions with DNA (Dey and Bhattacherjee 2019; Rodriguez et al. 2020). Overall, the effects associated with the N-terminus under crowding conditions (PEG8K) result in a 5-fold higher kcat and a 4-fold lower Km, as measured by steady-state analysis (Rodriguez et al. 2017). The N-terminal tail, protein conformational plasticity, and crowding agents act synergistically to facilitate the diffusion of hUNG in a crowded environment.
6. Conclusions and Future Perspectives
The N-terminal tail of hUNG enzymes directs and assists in their enzymatic functions within the cell. By means of signal sequences within the domain, hUNG enzymes are located to the nucleus and mitochondria, though overlapping N-terminal sequences result in dual localization of hUNG1 (Sarno et al. 2019). In order to prevent mutagenic processing, hUNG2 initiates BER at replication foci in pre- and post-replicative processes (Weiser et al. 2018; Weiser 2020). hUNG2 and nuclear hUNG1 also function in mutagenic processing of uracil residues in Ig diversification in activated B-cells (Sarno et al. 2019). Localization at replication forks is enhanced by association with RPA and PCNA, with these interactions being modulated by certain types of PTMs within their respective binding motifs (Ko and Bennett 2005; Hagen et al. 2008; Weiser et al. 2017; Weiser 2020). Aside from protein-protein interactions, N-terminal PTMs also regulate protein turnover, with phosphorylation, acetylation, and ubiquitylation likely involved in its degradation (Hagen et al. 2008; Bao et al. 2020). Additionally, the N-terminal tail enhances association with DNA and translocation across it in the search for uracil (Esadze et al. 2017; Rodriguez et al. 2017). This effect increases further in the presence of molecular crowding agents such as those present in the nucleus, which compact the N-terminal tail and decrease diffusion into the bulk solution (Rodriguez et al. 2020). Overall, investigating the N-terminus provides mechanistic insights into the complex DNA repair processes initiated by hUNG enzymes and its modes of regulation.
Many of the known PTMs of hUNG enzymes lie within the N-terminal tail. For hUNG2, it carries 19 out of the 29 reported PTMs; in hUNG1, it carries 9 out of the 19 reported PTMs (Bao et al. 2020). As discussed, these PTMs have roles in regulating the functions and degradation of hUNG enzymes. Modulating N-terminal PTMs to increase the degradation of hUNG2 may be a novel strategy for cancer therapy. Specifically, HDACs counter ROS treatment by deacetylating hUNG2 and preventing protein degradation following UHRF1 ubiquitylation, which ultimately increases cancer cell survival (Bao et al. 2020). Treating cancer cells with therapeutics that include HDAC inhibitors may be utilized to allow deacetylation of hUNG2, increase its turnover, and prevent repair of ROS-induced uracil incorporation within DNA (Bao et al. 2020). N-terminal phosphorylation events may also represent a step in the degradative process for further investigation (Hagen et al. 2008). Additionally, PTMs may modulate DNA substrate specificity of hUNG enzymes via protein-protein interactions. Interactions with PCNA and RPA can be modulated by PTMs within their respective N-terminal binding motifs (Torseth et al. 2012; Weiser et al. 2017). For example, phosphorylation of T6 and Y8 have been shown to exert inhibitory effects on the binding of hUNG2 to PCNA, and an Arg88Cys variant of hUNG2 also shows impaired RPA binding (Torseth et al. 2012; Weiser et al. 2017). Importantly, PCNA slides along dsDNA while RPA binds to ssDNA, recruiting hUNG enzymes to their respective substrates (Weiser et al. 2017). By modulating these protein-protein interactions through PTMs, the substrate specificity and thus, the roles of hUNG enzymes (replicative repair, antibody maturation, etc.) might be altered. Therefore, it is important to understand the relative abundance of different PTMs, their dynamics, and their biological effects in each hUNG isoform. Use of modern quantitative proteomic approaches is likely expected to answer these questions.
The discovery of the nuclear hUNG1 raised additional questions regarding this isoform. It is clear that hUNG1 can compensate for the loss of hUNG2 in uracil removal and CSR in mammalian cells (Sarno et al. 2019); however, whether nuclear hUNG1 functions in a specific biological context remains to be explored. For example, it has been proposed that the nuclear hUNG1 can be specialized in uracil cleavage in ssDNA-dsDNA junctions by interacting with RPA (Sarno et al. 2019). Although the presence of hUNG2 in BER complexes was reported, whether nuclear hUNG1 is present in such complexes has not yet been tested.
The division of labor among the different uracil-DNA glycosylases remains an intriguing question. While other UNGs exist in humans, including SMUG1, TDG, and MBD4, their lower catalytic rates or different substrate specificities make them inadequate to fulfill the functions of hUNG enzymes (Kavli et al. 2005; Doseth et al. 2012). For instance, SMUG1, which is the second major uracil-DNA glycosylase in mammalian cells, has been shown to participate in uracil excision, but does not fully compensate for the loss of UNG enzymes (Kavli et al. 2005; Kemmerich et al. 2012; Dingler et al. 2014; Alsøe et al. 2017). In UNG-deficient mice (UNG−/−), SMUG1 is the major contributor to the residual genomic uracil excision, significantly decreasing uracil levels compared to double knockout cells (UNG−/−SMUG1−/−), though a modest increase in uracil levels is still observed in multiple tissue types (Alsøe et al. 2017). Furthermore, uracil excision activity is abolished on single-stranded uracil substrates in mouse UNG-knockout cells, even in the presence of SMUG1 (Alsøe et al. 2017). This is consistent with studies on UNG-deficient mice displaying inhibited CSR and SHM, which occurs on ssDNA substrates, resulting in reduced lifespans and substantially increased development of B-cell lymphomas (Nilsen et al. 2003; Kemmerich et al. 2012; Alsøe et al. 2017). Human patients exhibiting hUNG deficiency or mutations also show severely impaired CSR and altered SHM, with SMUG1 and other uracil-DNA glycosylases again falling short (Imai et al. 2003; Kavli et al. 2005). Such defects of hUNG cause hyper-IgM syndrome (HIGM), which is an immunodeficiency disorder triggering a susceptibility to bacterial infections and lymphoid hyperplasia, among other symptoms (Imai et al. 2003; Kavli et al. 2005). With ssDNA being ~100-fold more prone to deamination, along with its importance in antibody maturation following AID-induced deamination, abolishing uracil excision from ssDNA is problematic (Kavli et al. 2007). However, the nuclear variant of hUNG1 is able to conduct CSR and SHM with the same efficiency as hUNG2, suggesting its presence as a potential alternative (Sarno et al. 2019). The conserved N-terminal RPA-binding motif also allows for increased association with ssDNA substrates (Sarno et al. 2019). Sharing multiple nuclear functions, substrate specificity, catalytic rates, and the N-terminal RPA binding motif makes the nuclear variant of hUNG1 an ideal backup enzyme to hUNG2. Additional research is required to fully elucidate the respective contribution of uracil DNA glycosylases within the nucleus.
Figure 2.
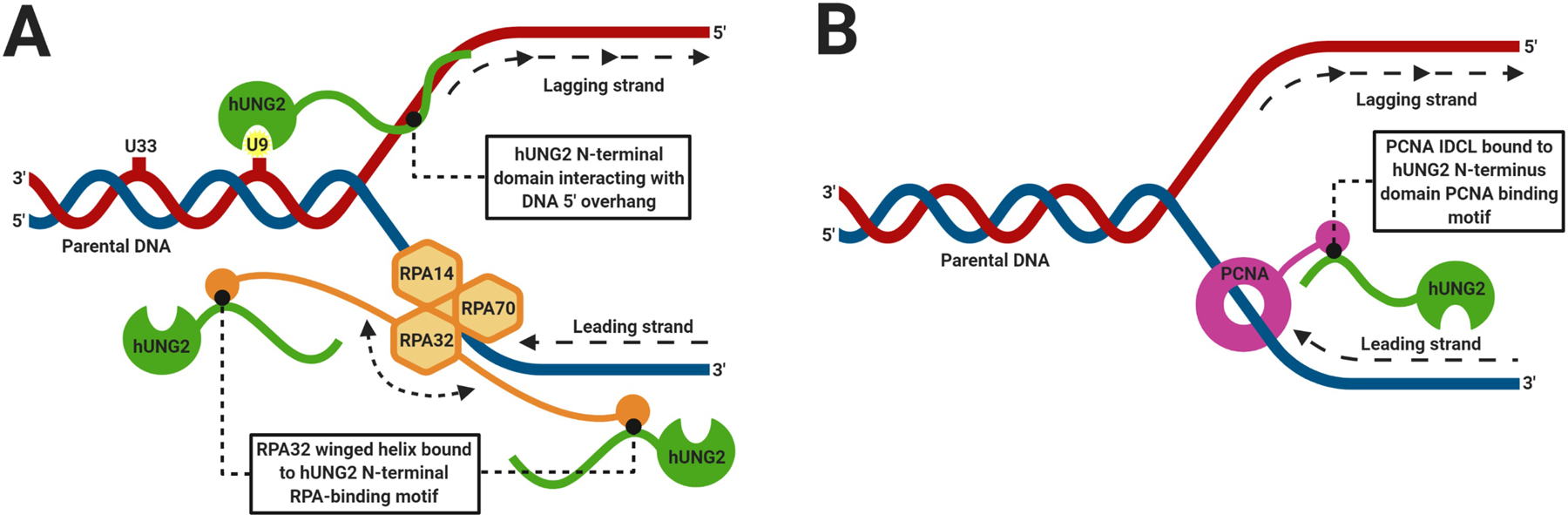
Graphical representations of pre- and post-replicative repair at replication forks. (A) hUNG2 is depicted interacting with the 5’ overhang at ssDNA-dsDNA junction sites, where it initiates pre-replicative BER of parental DNA with specificity for uracil closer to the junction site. The winged helix of RPA32 is shown bound to the RPA-binding motif of hUNG2, where it initiates either pre-replicative BER of parental DNA or ssDNA, or it initiates post-replicative BER following replicative polymerases. (B) The PCNA interdomain connecting loop (IDCL) is shown bound to the PCNA-binding motif (PIP-box) of hUNG2, where it initiates rapid post-replicative BER following replicative polymerases. Other proteins involved in replicative machinery are not shown.
Acknowledgments
We thank Dr. Laurie Kaguni and Dr. James Stivers for critical reading the manuscript.
Funding
We acknowledge funding support from the National Institutes of Health (NIH) Grant R35 GM128854 (to L.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare that there are no conflicts of interest
Disclosure statement
The authors report no declarations of interest.
References
- Alsøe L, Sarno A, Carracedo S, Domanska D, Dingler F, Lirussi L, Sengupta T, Tekin NB, Jobert L, Alexandrov LB, et al. 2017. Uracil Accumulation and Mutagenesis Dominated by Cytosine Deamination in CpG Dinucleotides in Mice Lacking UNG and SMUG. Sci Rep. 7(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen S, Heine T, Sneve R, König I, Krokan HE, Epe B, Nilsen H. 2005. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 26(3):547–555. [DOI] [PubMed] [Google Scholar]
- Bao Y, Tong L, Song B, Liu G, Zhu Q, Lu X, Zhang J, Lu Y, Wen H, Tian Y, et al. 2020. Free Radical Biology and Medicine UNG2 deacetylation confers cancer cell resistance to hydrogen peroxide- induced cytotoxicity. Free Radic Biol Med. 160(1066):403–417. 10.1016/j.freeradbiomed.2020.06.010 [DOI] [PubMed] [Google Scholar]
- Bharati S, Krokan HE, Kristiansen L, Otterlei M, Slupphaug G. 1998. Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res. 26(21):4953–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorås K, Sousa MML, Sharma A, Fonseca DM, S Gaard CK, Bjorås M, Otterlei M. 2017. Monitoring of the spatial and temporal dynamics of BER/SSBR pathway proteins, including MYH, UNG2, MPG, NTH1 and NEIL1–3, during DNA replication. Nucleic Acids Res. 45(14):8291–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchinger E, Wiik S, Kusnierczyk A, Rabe R, Aas PA, Kavli B, Slupphaug G, Aachmann FL. 2018. Backbone 1H, 13C and 15N chemical shift assignment of full-length human uracil DNA glycosylase UNG2. Biomol NMR Assign. 12(1):15–22. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. 2004. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 430(7003):992–998. [DOI] [PubMed] [Google Scholar]
- Chon J, Field MS, Stover PJ. 2019. Deoxyuracil in DNA and disease: Genomic signal or managed situation? DNA Repair (Amst). 77:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG. 2008. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravens SL, Schonhoft JD, Rowland MM, Rodriguez AA, Anderson BG, Stivers JT. 2015. Molecular crowding enhances facilitated diffusion of two human DNA glycosylases. Nucleic Acids Res. 43(8):4087–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Errico M, Parlanti E, Pascucci B, Fortini P, Baccarini S, Simonelli V, Dogliotti E. 2017. Single nucleotide polymorphisms in DNA glycosylases: From function to disease. Free Radic Biol Med. 107(December 2016):278–291. [DOI] [PubMed] [Google Scholar]
- Dey P, Bhattacherjee A. 2019. Mechanism of Facilitated Diffusion of DNA Repair Proteins in Crowded Environment: Case Study with Human Uracil DNA Glycosylase. J Phys Chem. 123:10354–10364. [DOI] [PubMed] [Google Scholar]
- Dingler FA, Kemmerich K, Neuberger MS, Rada C. 2014. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur J Immunol. 44(7):1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doseth B, Ekre C, Slupphaug G, Krokan HE, Kavli B. 2012. Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair (Amst). 11(6):587–593. 10.1016/j.dnarep.2012.03.003 [DOI] [PubMed] [Google Scholar]
- Esadze A, Rodriguez G, Weiser BP, Cole PA, Stivers JT. 2017. Measurement of nanoscale DNA translocation by uracil DNA glycosylase in human cells. Nucleic Acids Res. 45(21):12413–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esadze A, Stivers JT. 2018. Facilitated Diffusion Mechanisms in DNA Base Excision Repair and Transcriptional Activation. Chem Rev. 118(23):11298–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JA, Muller-Weeks S, Caradonna S. 2004. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair (Amst). 3(5):505–513. [DOI] [PubMed] [Google Scholar]
- Hagen L, Kavli B, Sousa MML, Torseth K, Liabakk NB, Sundheim O, Peňa-Diaz J, Otterlei M, Hørning O, Jensen ON, et al. 2008. Cell cycle-specific UNG2 phosphorylations regulate protein turnover, activity and association with RPA. EMBO J. 27(1):51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug T, Skorpen F, Aas PA, Malm V, Skjelbred C, Krokan HE. 1998. Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res. 26(6):1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug T, Skorpen F, KvalØy K, Eftedal I, Lund H, Krokan HE. 1996. Human uracil-DNA glycosylase gene: Sequence organization methylation pattern mapping to chromosome 12q23-q24.1. Genomics. 36(3):408–416. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. 2015. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43(D1):D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 4(10):1023–1028. [DOI] [PubMed] [Google Scholar]
- Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, Durandy A, Krokan HE, Slupphaug G. 2005. B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. J Exp Med. 201(12):2011–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Otterlei M, Slupphaug G, Krokan HE. 2007. Uracil in DNA-General mutagen, but normal intermediate in acquired immunity. DNA Repair (Amst). 6(4):505–516. [DOI] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. 2002. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 277(42):39926–39936. [DOI] [PubMed] [Google Scholar]
- Kemmerich K, Dingler FA, Rada C, Neuberger MS. 2012. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil-and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/− mice. Nucleic Acids Res. 40(13):6016–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko R, Bennett SE. 2005. Physical and functional interaction of human nuclear uracil-DNA glycosylase with proliferating cell nuclear antigen. DNA Repair (Amst). 4(12):1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Drabløs F, Slupphaug G. 2002. Uracil in DNA - Occurrence, consequences and repair. Oncogene. 21(58 REV. ISS. 8):8935–8948. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Otterlei M, Nilsen H, Kavli B, Skorpen F, Andersen S, Skjelbred C, Akbari M, Aas PA, Slupphaug G. 2001. Properties and functions of human uracil-DNA glycosylase from the UNG gene. Prog Nucleic Acid Res Mol Biol. 68:365–386. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Sætrom P, Aas PA, Pettersen HS, Kavli B, Slupphaug G. 2014. Error-free versus mutagenic processing of genomic uracil-Relevance to cancer. DNA Repair (Amst). 19:38–47. 10.1016/j.dnarep.2014.03.028 [DOI] [PubMed] [Google Scholar]
- Lindahl T 1993. Instability and decay of the primary structure of DNA. Nature. 362(6422):709–715. [DOI] [PubMed] [Google Scholar]
- Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. 2004. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 15(4):621–634. [DOI] [PubMed] [Google Scholar]
- Otterlei Marit, Warbrick Emma, Nagelhus Toril A., Haug Terje, Slupphaug Geir, Akbari Mansour, Aas Per Arne, Steinsbekk Kristin, Bakke Oddmund and HEK. 1999. Post-replicative base excision repair in replication foci. EMBO J. 18(13):3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. 2000. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 103(3):449–456. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. 1997. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 25(4):750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. 2000. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 5(6):1059–1065. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, Barnes DE. 2003. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. 22(35):5381–5386. [DOI] [PubMed] [Google Scholar]
- Otterlei M, Haug T, Nagelhus TA, Slupphaug G, Lindmo T, Krokan HE. 1998. Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 26(20):4611–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Putnam CD, Tainer JA. 2000. Lessons learned from structural results on uracil-DNA glycosylase. Mutat Res - DNA Repair. 460(3–4):183–199. [DOI] [PubMed] [Google Scholar]
- Parlanti E, Locatelli G, Maga G, Dogliotti E. 2007. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 35(5):1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porecha RH, Stivers JT. 2008. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc Natl Acad Sci U S A. 105(31):10791–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G, Esadze A, Weiser BP, Schonhoft JD, Cole PA, Stivers JT. 2017. Disordered N-Terminal Domain of Human Uracil DNA Glycosylase (hUNG2) Enhances DNA Translocation. ACS Chem Biol. 12(9):2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G, Orris B, Majumdar A, Bhat S, Stivers JT. 2020. Macromolecular crowding induces compaction and DNA binding in the disordered N-terminal domain of hUNG2. DNA Repair (Amst). 86(November 2019):102764. 10.1016/j.dnarep.2019.102764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno A, Lundbæk M, Liabakk NB, Arne Aas P, Mjelle R, Hagen L, Sousa MML, Krokan HE, Kavli B. 2019. Uracil-DNA glycosylase UNG1 isoform variant supports class switch recombination and repairs nuclear genomic uracil. Nucleic Acids Res. 47(9):4569–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoft JD, Kosowicz JG, Stivers JT. 2013. DNA Translocation by Human Uracil DNA Glycosylase: Role of DNA Phosphate Charge. Biochemistry. 52:2526–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoft JD, Stivers JT. 2012. Timing facilitated site transfer of an enzyme on DNA. Nat Chem Biol. 8(2):205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormann N, Ricciardi R, Chattopadhyay D. 2014. Uracil-DNA glycosylases - Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 23(12):1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, Hakata Y, Landau NR. 2007. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc Natl Acad Sci U S A. 104(10):4130–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, Yu Q, Zeitlin SG, Landau NR. 2005. Human Immunodeficiency Virus Type 1 Vpr Induces the Degradation of the UNG and SMUG Uracil-DNA Glycosylases. J Virol. 79(17):10978–10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sire J, Quérat G, Esnault C, Priet S. 2008. Uracil within DNA: An actor of antiviral immunity. Retrovirology. 5(45). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G, Eftedal I, Kavli B, Bharati S, Hellet NM, Haug T, Krokan HE, Levine DW. 1995. Properties of a Recombinant Human Uracil-DNA Glycosylase from the UNG Gene and Evidence that UNG Encodes the Major Uracil-DNA Glycosylase. Biochemistry. 34(1):128–138. [DOI] [PubMed] [Google Scholar]
- Slupphaug G, hugo Markussen F, Olosen LC, Aasland R, Aarsæther N, Bakke O, Krokan HE, Helland DE. 1993. Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 21(11):2579–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE, Muir TW. 2020. Chemoenzymatic Semisynthesis of Proteins. Chem Rev. 120(6):3051–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torseth K, Doseth B, Hagen L, Olaisen C, Liabakk NB, Græsmann H, Durandy A, Otterlei M, Krokan HE, Kavli B, Slupphaug G. 2012. The UNG2 Arg88Cys variant abrogates RPA-mediated recruitment of UNG2 to single-stranded DNA. DNA Repair (Amst). 11(6):559–569. 10.1016/j.dnarep.2012.03.006 [DOI] [PubMed] [Google Scholar]
- Visnes T, Doseth B, Pettersen HS, Hagen L, Sousa MML, Akbari M, Otterlei M, Kavli B, Slupphaug G, Krokan HE. 2009. Uracil in DNA and its processing by different DNA glycosylases. Philos Trans R Soc B Biol Sci. 364(1517):563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser BP. 2020. Analysis of uracil DNA glycosylase (UNG2) stimulation by replication protein A (RPA) at ssDNA-dsDNA junctions. Biochim Biophys Acta - Proteins Proteomics. 1868(3):140347. 10.1016/j.bbapap.2019.140347 [DOI] [PubMed] [Google Scholar]
- Weiser BP, Rodriguez G, Cole PA, Stivers JT. 2018. N-terminal domain of human uracil DNA glycosylase (hUNG2) promotes targeting to uracil sites adjacent to ssDNA-dsDNA junctions. Nucleic Acids Res. 46(14):7169–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser BP, Stivers JT, Cole PA. 2017. Investigation of N-Terminal Phospho-Regulation of Uracil DNA Glycosylase Using Protein Semisynthesis. Biophys J. 113(2):393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharkov DO, Mechetin GV., Nevinsky GA. 2010. Uracil-DNA glycosylase: Structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat Res - Fundam Mol Mech Mutagen. 685(1–2):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]