

Abstract
Purpose:
In preclinical studies, the LSD1 inhibitor tranylcypromine (TCP) combined with all-trans retinoic acid (ATRA) induces differentiation and impairs survival of myeloid blasts in non-APL acute myeloid leukemia (AML). We conducted a Phase I clinical trial (NCT02273102) to evaluate the safety and activity of ATRA plus TCP in patients with relapsed/refractory AML and myelodysplasia (MDS).
Experimental Design:
Seventeen patients were treated with ATRA and TCP (3 dose levels: 10 mg twice daily [BID], 20 mg BID, and 30 mg BID).
Results:
ATRA-TCP had an acceptable safety profile. The maximum tolerated dose of TCP was 20 mg BID. Best responses included 1 morphologic leukemia-free state, 1 marrow complete remission with hematologic improvement, 2 stable disease with hematologic improvement, and 2 stable disease. By intention-to-treat, the overall response rate was 23.5% and clinical benefit rate 35.3%. Gene expression profiling of patient blasts showed that responding patients had a more quiescent CD34+ cell phenotype at baseline, including decreased MYC and RARA expression, compared to non-responders that exhibited a more proliferative CD34+ phenotype, with gene expression enrichment for cell growth signaling. Upon ATRA-TCP treatment, we observed significant induction of retinoic acid (RA)-target genes in responders but not non-responders. We corroborated this in AML cell lines, showing that ATRA-TCP synergistically increased differentiation capacity and cell death by regulating the expression of key gene sets that segregate patients by their clinical response.
Conclusions:
These data indicate that LSD1 inhibition sensitizes AML cells to ATRA and may restore ATRA responsiveness in subsets of MDS and AML patients.
Keywords: acute myeloid leukemia, AML, myelodysplasia, MDS, Phase I clinical trial, LSD1 inhibitor, TCP, KDM1A, all-trans retinoic acid, ATRA, differentiation therapy, drug resistance, combinatorial therapy
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by the accumulation of myeloid blasts with blocked differentiation and abnormal proliferative capacity (1, 2). For decades, cytotoxic chemotherapy has been the standard treatment and remains largely unchanged. Chemotherapy resistance occurs in most patients leading to a 5-year overall survival (OS) rate of ~30% (2, 3). One notable exception is acute promyelocytic leukemia (APL), characterized by the PML-RARA fusion transcript, where almost all patients are cured with the combination of all-trans retinoic acid (ATRA) and arsenic trioxide (4, 5). Currently, only APL patients are treated successfully with ATRA, where induction of myeloid differentiation is regulated via activation of RARA transcription (6).
While ATRA can induce differentiation in subsets of non-APL AML in vitro (e.g., NPM1-mutated), clinically, it has no single-agent activity and minimal, if any, additive effects when given in combination with chemotherapy (7–10). Of note, a recent report demonstrated that ATRA may improve response rates and survival in untreated AML patients (older/unfit) when given in combination with the hypomethylating agent decitabine (11, 12). Little is known about the mechanisms of ATRA-resistance in non-APL AML (7–9). AML cells and primary AML blasts express low levels of RARA, which correlate with alterations in histone methylation levels rather than DNA methylation (13, 14). Methylation of H3K4 is required for ATRA-driven differentiation, and this methylation mark is lost in primary AML and cell lines due to overexpression of lysine-specific histone demethylase 1A (LSD1) (15). LSD1 acts as a transcriptional repressor through its demethylase activity on H3K4me1/me2, and genetic and pharmacological inhibition of LSD1 has been shown to re-sensitize cells to ATRA-driven differentiation (15, 16). These studies suggest that the epigenetic and transcriptional makeup of leukemia cells determines their susceptibility to ATRA.
We hypothesized that LSD1 inhibition sensitizes non-APL AML to pharmacologic doses of ATRA and would have clinical activity by inducing differentiation in myelodysplastic syndrome (MDS) and AML patients. Tranylcypromine (TCP) is an irreversible monoamine oxidase inhibitor (MAOI) in clinical use for major depressive disorder since the 1960s (17, 18). TCP has been demonstrated to inhibit LSD1 and reactivate the ATRA-mediated differentiation potential of non-APL AML cells (15, 19). Using TCP doses with established efficacy in depression and well-defined pharmacokinetic profiles, we pursued an investigator-initiated clinical trial of ATRA-TCP combination therapy in patients with relapsed/refractory (R/R) MDS and AML.
MATERIALS and METHODS
Trial Design and Endpoints
The primary endpoint of this Phase I study (NCT02273102) was to determine the maximum-tolerated dose (MTD) and safety profile of ATRA-TCP. The study employed a 3+3 dose escalation design, with dose expansion at the MTD. The MTD was defined as the dose level at which there were ≤1 dose-limiting toxicities (DLTs) out of at least 6 patients treated. A DLT was defined as any ≥grade 3 toxicity, or unacceptable grade 2 toxicity, during cycle 1 of treatment that was possibly, probably, or definitely related to ATRA or TCP. Secondary objectives included correlative studies (DNA and RNA sequencing of patient blasts) and an assessment of clinical activity using standard response criteria for MDS (International Working Group [IWG] 2006) and AML (European LeukemiaNet [ELN] 2017). The overall response rate (ORR) included complete remission (CR), CR with incomplete blood count recovery (CRi), morphologic leukemia-free state (MLFS), and partial remission (PR) for AML, and CR, marrow CR, PR, and hematologic improvement (HI) for MDS. The clinical benefit rate (CBR) included the ORR plus stable disease (SD) rate. Safety data for all patients who received any treatment on protocol are reported. Efficacy are reported both as per intention-to-treat and in response-evaluable patients (completed cycle 1 or had disease progression during cycle 1).
Treatment and Eligibility
TCP and ATRA were obtained commercially. Both drugs were administered orally. ATRA was given at 45 mg/m2 daily in divided doses without planned dose interruptions. TCP was administered at 3 escalating dose levels (10 mg twice daily [BID], 20 mg BID, and 30 mg BID), with a 3-day lead-in of TCP monotherapy in cycle 1. Cycles were 21 days in length and could repeat if the patient were experiencing clinical benefit without unacceptable toxicity. Patients had to have AML or MDS (as defined by WHO-criteria) that had failed known life-prolonging therapies (relapsed/refractory). APL was excluded. In an amendment to the protocol, patients with proliferative AML (white blood cell [WBC] count >25,000/μl) were excluded (hydroxyurea was allowed). Patients had to have an Eastern Cooperative Oncology Group (ECOG) Performance Status of 0–2. Standard inclusion and exclusion criteria for organ function in leukemia patients were followed. Precautions were employed for potential drug-drug interactions with TCP. Informed written consent was obtained from each patient or each patient’s guardian. All patients were consented on IRB-protocol 20140328, according the Declaration of Helsinki and Good Clinical Practice (GCP) guidelines.
Statistical analysis of the clinical trial
This study was an open label, Phase I, dose escalation study of TCP in combination with a fixed dose of ATRA utilizing a traditional 3+3 design. Statistical analyses were primarily descriptive and graphical in nature. No formal statistical hypothesis tests were performed. The incidence of DLT was tabulated for each dose group. To assess the relationship between toxicities and dose, the preferred term of individual toxicities was summarized by their frequency and intensity for each dose group. Safety was also be evaluated by the incidence of treatment-emergent AEs, severity and type of AEs, and by changes from baseline in the patient’s vital signs, weight and clinical laboratory results using the population evaluable for safety. AEs were tabulated according to the Medical Dictionary for Regulatory Activities (MedDRA) by system organ class, high-level terms, and preferred terms. ORR and CBR were estimated along with corresponding 95% CIs for evaluating preliminary efficacy. Median follow-up was obtained by reverse Kaplan-Meier approach. Duration on study was calculated from cycle 1 day 1 until the end-of-study visit conducted within 30 days of the patient’s last dose. Fisher’s exact test was used to examine the association between disease type and other covariates. Median, range, mean and standard deviation were used for continuous variables.
Safety precautions for TCP
Therapy with strong CYP3A4 inhibitors or CYP3A4 inducers was not permitted within 14 days prior to cycle 1 day 1. Therapy with monoamine oxidase inhibitors (MAOIs), dibenzoazepine derivatives, sympathomimetics, or selective serotonin reuptake inhibitors (SSRIs) was not permitted within 14 days prior to cycle 1 day 1. Patients were educated to follow a tyramine-free diet and avoid drug-drug interactions that might occur TCP (no events occurred).
Patient CD34+ cell selection
Cells were isolated by Miltenyi CD34 microbead kit according to the manufacturer’s instructions. Selection was performed immediately after defrosting the cryopreserved patient bone marrow samples. Samples were thawed in 37°C and DNase-I (Sigma D4513) was added to prevent cell clumping. Cells were resuspended in 2% FBS + IMDM medium. Cell pellets were resuspended in FcR blocking reagent and CD34 microbeads; incubated for 30 minutes. Positive selection of CD34+ cells was performed on MACS MS columns (Miltenyi Biotec 130-042-201).
We defined quiescent CD34+ cells as having a dormant cellular state with a less proliferative gene expression phenotype, and proliferative CD34+ cells as having gene expression enrichment for cell growth signaling pathways.
RNA-sequencing of primary patient samples
Preparation of total RNA for sequencing on the Illumina NovaSeq 6000 NGS platform was carried out at the University of Miami’s Hussman Institute for Human Genomics (http://hihg.med.miami.edu/cgt). Briefly, 1ng of total RNA was ribo-depleted and library construction was performed using the ‘Nugen Ovation SoLo RNA-Seq (Human) (Rev. M01406 v3) with AnyDeplete Probe Standard Human/Mouse (rRNA)’ according to manufacturer’s protocol. Samples were barcoded to allow for multiplexing. Cluster generation and sequencing took place on the Illumina NovaSeq 6000 using the reagents provided in the Illumina NovaSeq 5000/6000 S1 Reagent Kit (200 cycle).
Raw FASTQ data were processed with Trimmomatic RRID:SCR_011848 (20) removing adapters and low-quality reads. Reads were aligned to the human genome (hg38) using STAR RRID:SCR_015899 (21) with default parameters. RSEM RRID:SCR_013027 (22) was used to obtain the raw counts per gene against the human transcriptome (Ensembl97). Heat maps of the Z-score row-normalized gene expression and the unsupervised hierarchical clustering were generated using clustermap (method=ward, metric=euclidean distance). We determined the baseline differential gene expression between responder and non-responder patients using DESeq2 RRID:SCR_000154 (23) at a 2-fold change cut-off (false discovery rate (FDR) < 0.05). Same protocol was applied to RNA-seq from cell lines. RNA-seq data are available at GEO under accession number GSE151594. The expressed genes were pre-ranked by the wald statistics and used for the gene set enrichment analysis (GSEA (24), RRID:SCR_003199) against “Hallmark gene sets”, and the signatures obtained from our cohort of patients (FDR<0.1) (24, 25). Additional enrichment analysis was performed by enrichr RRID:SCR_001575 (26, 27) against KEGG (28) RRID:SCR_012773, and DSigDB (29).
Cell lines and cell culture
The human AML cell lines U-937 (RRID:CVCL_0007) and SKNO-1 (RRID:CVCL_2196) (gift from Dr. Nimer’s laboratory, University of Miami, FL), were cultured in RPMI1640 medium, 10% heat-inactivated FBS (Sigma-Aldrich), 2% penicillin/streptomycin (Gibco, Fisher Scientific), and SKNO-1 supplemented with 10 ng/ml GM-CSF (Sigma-Aldrich). Cell lines were maintained in mycoplasma-free conditions and routinely tested for infection (MycoAlert, Lonza).
Analysis of myeloid differentiation
AML cell lines were treated with ATRA (Sigma), and TCP (Sigma) for 4 days. Fluorescence-activated cell sorting (FACS) analysis of CD11b and CD86 expression were performed on 5×105 cells using aphycoerythrin-conjugated human CD11b-specific (BD Pharmingen, #555388) and Alexa fluor 700 CD86 (BD Pharmingen, #561124) mouse monoclonal antibody at a 1:5 dilution on a BD LSRII FACS machine (Becton Dickinson) with CellQuest software. We performed FACS analysis on live cells, which were sorted using a LIVE/DEAD fixable green dead cell stain (ThermoFisher).
Cell cycle and apoptosis
Cell lines were plated and treated with the respective doses of ATRA, TCP, or ATRA-TCP in 96-well plates. After 4 days of treatment, cells were spun at 1500 RPM. Cells were fixed in 4% PFA following by permeabilization with 0.15% Triton-PBS and stained with DAPI. The percentage of cells in the different cell-cycle phases were determined with a BD LSRII FACS machine (Becton Dickinson) and analyzed using the FlowJo 10.6.1.
Total RNA library preparation
RNA was isolated from fresh U-937 and SKNO-1 cell pellets using Trizol reagent (Invitrogen). Total RNA was extracted using miRNeasy Micro Kit (Qiagen) following the manufacturer’s protocol. 1 μg of the RNA samples were processed with the Illumina TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold (Cat #. RS-122–2301), following the manufacturer’s instructions. Cluster generation and sequencing took place on the Illumina NovaSeq 6000 using the reagents provided in the Illumina NovaSeq 6000 S1 Reagent Kit (200 cycle) with >30M paired-end 100 base pair reads per sample.
Statistical analysis of transcriptional and in vitro data
Significant differences in mean gene expression between responders vs. non-responders for a cluster of genes was determined by the Kolmogorov–Smirnov test (p<0.05). Cell lines viability and differentiation assays from combinations of ATRA-TCP compared with ATRA alone were analyzed by one-way analysis of variance (ANOVA) (p<0.001) followed by Tukey’s post-hoc test (ATRA vs. ATRA-TCP, p<0.001). For the cell line plots, values represent the mean and the error bars denote the standard deviation (SD) of the mean from three independent experiments. The SynergyFinder web application (30) was used to calculate synergy scores and generate 2D synergy maps.
RESULTS
Patient Characteristics
Of 18 consented patients, 17 were treated on this Phase I study (1 screen failure). Patient demographics are described in Table 1 and Table S1. The median age was 73 years (range=47–82 years, Table 1). Most patients had R/R AML (11/17, 64.7%) (Table 1). All AML patients fit for induction had failed at least 2 lines of prior intensive chemotherapy, and all MDS and older/unfit AML patients had failed at least 1 line of prior therapy (including a hypomethylating agent). Of the AML patients, 81.8% (9/11) had adverse genetic risk per ELN criteria (Table 1), and 66.7% (4/6) of MDS patients were high or very-high risk by IPSS-R criteria. The median bone marrow blast percentage for all patients was 30% (range=2 to 88%). The median bone marrow blast percentage was 60% (range=28 to 88%) for AML and 6% (range=2 to 11%) for MDS. The median WBC count was 3.0 × 103/μl (range=0.8 to 39.7) for AML.
Table 1. Disease Characteristics, Best Response, and Study Duration of all 17 patients treated on study.
Patient 3 was a screen failure and not included.
| ID | Age (years) | Cancer Type | Dose Level (mg, BID) | WBC | Blast % | Risk score | Cytogenetics | Mutations | Best Response | Study Duration (months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 51 | AML | TCP 10 | 39.7 | 73% | Adverse | trisomy 8, add(12p) | FLT3-ITD | Lack of response | 2.63 |
| 2 | 69 | AML | TCP 10 | 23.7 | 81% | Adverse | Normal | TET2, ASXL1, MLL-PTD | PD | 0.43 |
| 4 | 47 | AML | TCP 10 | 4.8 | 88% | Adverse | Normal | FLT3-ITD, SF3B1, RUNX1 | Lack of response | 2.86 |
| 5 | 82 | MDS | TCP 10 | 3.0 | 4% | Intermediate | del(5q) | SETBP1, U2AF1 | SD | 2.52 |
| 6 | 62 | MDS | TCP 10 | 0.8 | 2% | High | add(14q) | None | SD w/ HI-P | 3.45 |
| 7 | 72 | MDS | TCP 20 | 1.1 | 9% | Very High | monosomy 9, del(11q), trisomy 12 | TET2, U2AF1 | Marrow CR w/ HI-P | 6.67 |
| 8 | 78 | MDS | TCP 20 | 16.4 | 8% | High | t(14;15) | BCOR, GATA2, SF3B1 | SD w/ HI-P & HI-E | 6.28 |
| 9 | 77 | AML | TCP 20 | 1.1 | 60% | Adverse | monosomy 7, trisomy 8 | U2AF1 | N/A | 0.69 |
| 10 | 79 | AML | TCP 20 | 0.8 | 29% | Adverse | Normal | TP53 | MLFS | 9.23 |
| 11 | 77 | AML | TCP 20 | 3.0 | 71% | Adverse | Multiple Complex/Monosomal [del(7q)] | TP53 | PD | 3.02 |
| 12 | 73 | AML | TCP 20 | 1.1 | 40% | Adverse | Normal | BCOR, RUNX1, TET2 | Lack of response^ | 2.53 |
| 13 | 63 | MDS | TCP 20 | 2.7 | 11% | Very High | del(7q) | RUNX1, SF3B1 | PD | 2.53 |
| 14 | 76 | AML | TCP 30 | 1.7 | 30%* | Intermediate | Normal | None | N/A | 1.61 |
| 15 | 70 | AML | TCP 30 | 5.7 | 45% | Adverse | Multiple Complex/Monosomal (monosomy 7) | TP53, EZH2 | N/A | 1.38 |
| 16 | 71 | MDS | TCP 30 | 4.7 | 2% | Low | Normal | TET2 | N/A | 1.15 |
| 17 | 78 | AML | TCP 20 | 3.6 | 28%* | Adverse | monosomy 7 | IDH2, DNMT3A, CSF3R, RUNX1 | PD | 2.76 |
| 18 | 82 | AML | TCP 20 | 0.8 | 67% | Intermediate | Normal | SETBP1, ZRSR2 | SD | 3.68 |
These patients had >30% blasts at original diagnosis.
This patient had blast reduction and neutrophil recovery.
Dose-limiting Toxicity and Maximum Tolerated Dose
There were 3 DLTs on study: none at dose level 1 (TCP 10 mg BID), 1/6 patients at dose level 2 (TCP 20 mg BID), and 2/3 patients at dose level 3 (TCP 30 mg BID). The first DLT was persistent grade 2 dizziness at TCP 20 mg BID, requiring the patient (#9) to be removed from the study in week 2. The second DLT, at TCP 30 mg BID, was grade 2 asthenia and bilateral leg weakness, starting after the first dose of TCP and worsening until the patient (#14) was removed from study in week 2. The third DLT, at TCP 30 mg BID, was grade 2 nausea and vomiting after every TCP dose despite pre-medications. This patient (#16) was removed from study on day 4. No DLTs required hospitalization. All DLTs were determined to be related to TCP and resolved rapidly upon drug discontinuation. At dose level 2, there were 1/6 DLTs, so we proceeded to dose level 3, where 2/3 patients had DLTs. Therefore, dose level 2 (20 mg TCP BID) was declared the MTD. We expanded dose level 2 with 3 additional patients (9 total patients treated at this dose) with no further DLTs (Table 1).
Safety
Treatment-emergent adverse events (TEAEs), regardless of causality, are summarized in Table S2. The most common TEAEs (all grades, ≥20% of patients) were fatigue (6/17, 35%), creatinine increased (5/17, 29%), dizziness (5/17, 29%), dry mouth (5/17, 29%), headache (5/17, 29%), rash (5/17, 29%), dry skin (4/17, 24%), febrile neutropenia (4/17, 24%), pneumonia (4/17, 24%), and urinary frequency (4/17, 24%) (Table S2, Figure S1). The most common grade 3/4 TEAEs (≥15%) included febrile neutropenia (4/17, 24%) and pneumonia (4/17, 24%) (Table S3). Other notable grade 3–5 TEAEs (≥10%) included anemia (2/17, 12%), sepsis (2/17, 12%), neutropenia (2/17, 12%), and thrombocytopenia (2/17, 12%) (Table S3). One patient [#2] expired during cycle 1 due to sepsis and disease progression. Two grade 3–5 AEs were attributed to study treatment by investigator assessment (1 case of thrombocytopenia and 1 of anemia, both grade 3 and possibly related) (Table S4). Five patients did not complete cycle 1 due to DLT (3 patients), sepsis (1 patient), and sepsis with disease progression (1 patient) (Figure 1).
Figure 1. Clinical course of the patients and disease type distributions.
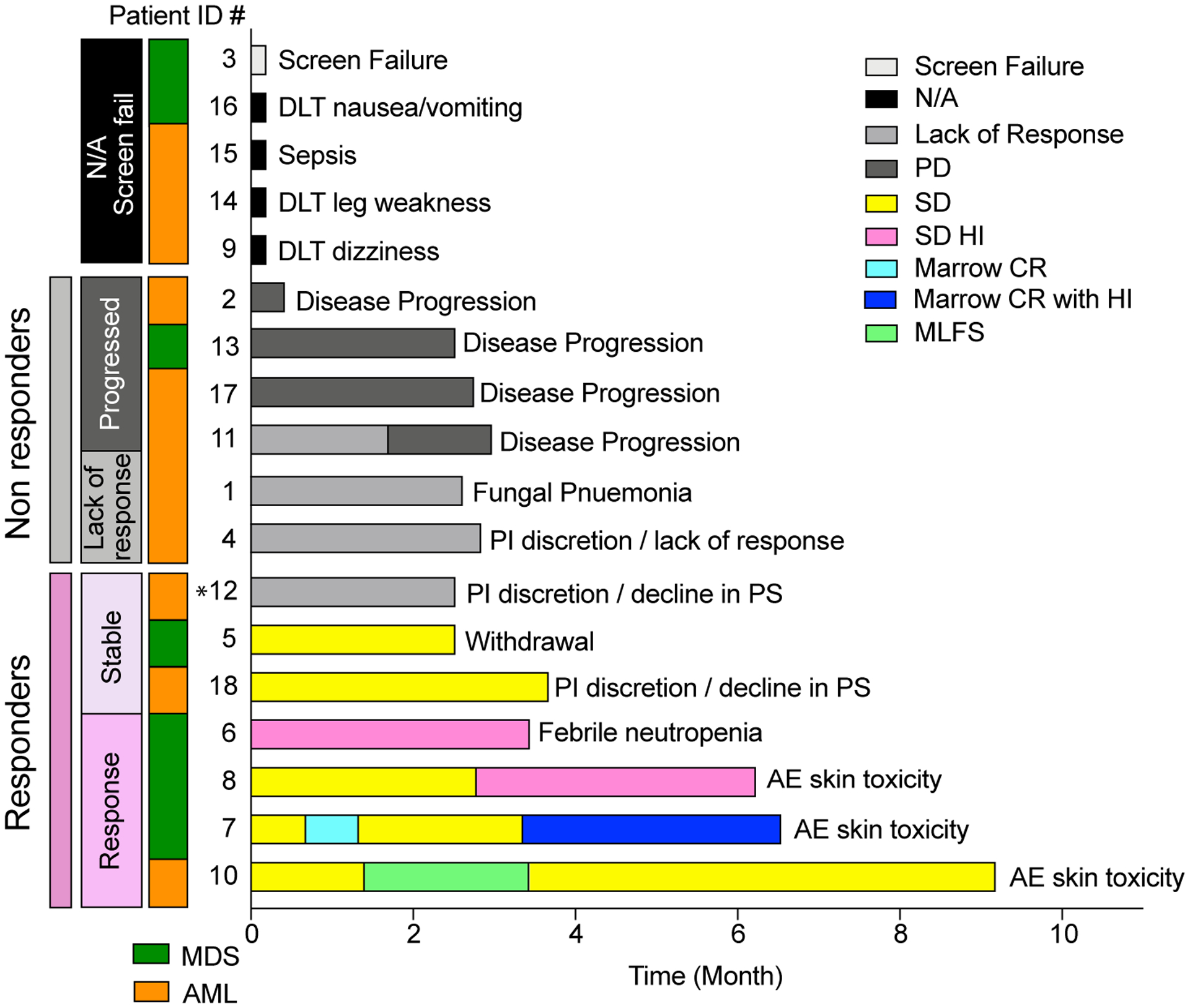
A) Swimmer’s plot where horizontal bars represent the duration on study for a cohort of 18 patients (17 treated and 1 screen-failure [Patient #3]). Bars are color-coded by the patient’s clinical working group response (Screen failure and N/A, non-evaluable response; PD, progressive disease; SD, stable disease; SD HI, stable disease with hematologic improvement; Marrow CR, marrow complete remission; Marrow CR with HI, marrow complete remission with hematologic improvement; MLFS, morphologic leukemia-free state). The reason for study discontinuation is listed at the end of each bar. Among the response evaluable patients, patients are grouped as responders (response/SD) by the pink vertical bar, and non-responders (lack or response/PD) by the grey vertical bar. An additional vertical bar color-codes the diagnosed disease type (MDS in green, AML in orange). *Patient #12 had neutrophil recovery and blast reduction but did not qualify for a clinical response. This patient’s RNA-seq signature clustered with the other responders.
Clinical Efficacy
In the 17 patients treated on study, 4 patients (3 MDS, 1 AML) had an objective working group response (ORR 23.5% [95% CI=6.8 to 49.9]): 1 MLFS, 1 marrow CR with platelet HI, 1 SD with erythroid HI (including transfusion independence) and platelet HI, and 1 SD with platelet HI (Figure 1). The patient with marrow CR/platelet HI also achieved a cytogenetic CR, with resolution of the abnormal clone (complex cytogenetics). Two other patients (1 MDS, 1 AML) had SD for a CBR of 6/17 patients or 35.3% [95% CI=14.2 to 61.7]. In the 13 response-evaluable patients (completed cycle 1 or had disease progression in cycle 1), the ORR was 30.8% [95% CI=10.4 to 61.1%] and CBR 46.2% [95% CI=20.4 to 73.9]. In the 7 patients on study who had MDS or low blast AML, the ORR was 57.1% (4/7 patients) and the CBR was 71.4% (5/7 patients). Hydroxyurea was used in 4 AML patients to control the WBC, none of whom responded or had SD.
Of the 6 patients with an objective response or SD, the median duration of response/SD was 5.1 months (range=2.5 to 9.2 months). The 4 responders were on study for 3.5, 6.3, 6.7, and 9.2 months, respectively. The 3 responders on study for >6 months all discontinued treatment for skin toxicity related to ATRA (not treatment failure or loss of response) (Figure 1). The patient with MLFS (#10) continued to receive ATRA-TCP off study using an intermittent dosing schedule of ATRA for another 5.8 months. The 2 patients with SD discontinued treatment for declining performance status (#18) and withdrawal of consent for personal reasons (#5). Patient #12 (AML) had neutrophil recovery (0.62 to 14.75 × 103/μl) and stability of bone marrow (40% to 28%) and peripheral (14% to 9%) blasts but did not qualify for a response/SD by ELN criteria (Figure 1).
The median duration on study for all patients was 2.8 months (range=0.4–9.2 months), and the median OS was 5.0 months (95% CI=3.2 to 21.6). The median OS was 15.3 months (95% CI=8.9 to 21.6) for the 6 patients with a response or clinical benefit. For the 7 patients with lack of response or PD, the median OS was 3.9 months (95% CI=0.4 to 5.0).
Although numerically more MDS patients responded (Figure 1), when examining the intent-to-treat population (17 patients), having MDS vs. AML was not associated with an objective response (p= 0.19) or clinical benefit (p=0.11) (Table S1). There was a trend towards lower baseline blast percentage and response or SD (p=0.063). Notably, the 1 AML patient who had an objective response (MLFS followed by prolonged SD [#10]) had low blast AML with a TP53 mutation and myelodysplasia-related changes and had failed prior azacitidine therapy (Table 1).
Gene expression signature of patient response to ATRA-TCP
We performed RNA-sequencing on CD34+ cells isolated from the bone marrow (BM) aspirates of patients at baseline (6 AML and 2 MDS). Using an unsupervised hierarchical clustering we segregate patients into responders/non-responders at baseline (Figure 2A, (RPKM/row>=50). While clusters 1–3 represent patient-specific gene signatures, we identified a subset of genes (cluster 4) that were consistently lower expressed in responders vs. non-responders (Figure 2A–B). Of the 3 responders, 2 had AML and 1 MDS, and of the 5 non-responders, 4 had AML and 1 MDS. Bone marrow and peripheral blood blast percentages, blood counts, and best response in patients at baseline and after treatment have been summarized in Table S5.
Figure 2. The transcriptomes of CD34+ cells from AML and MDS patients at baseline correlate with response to ATRA-TCP treatment.
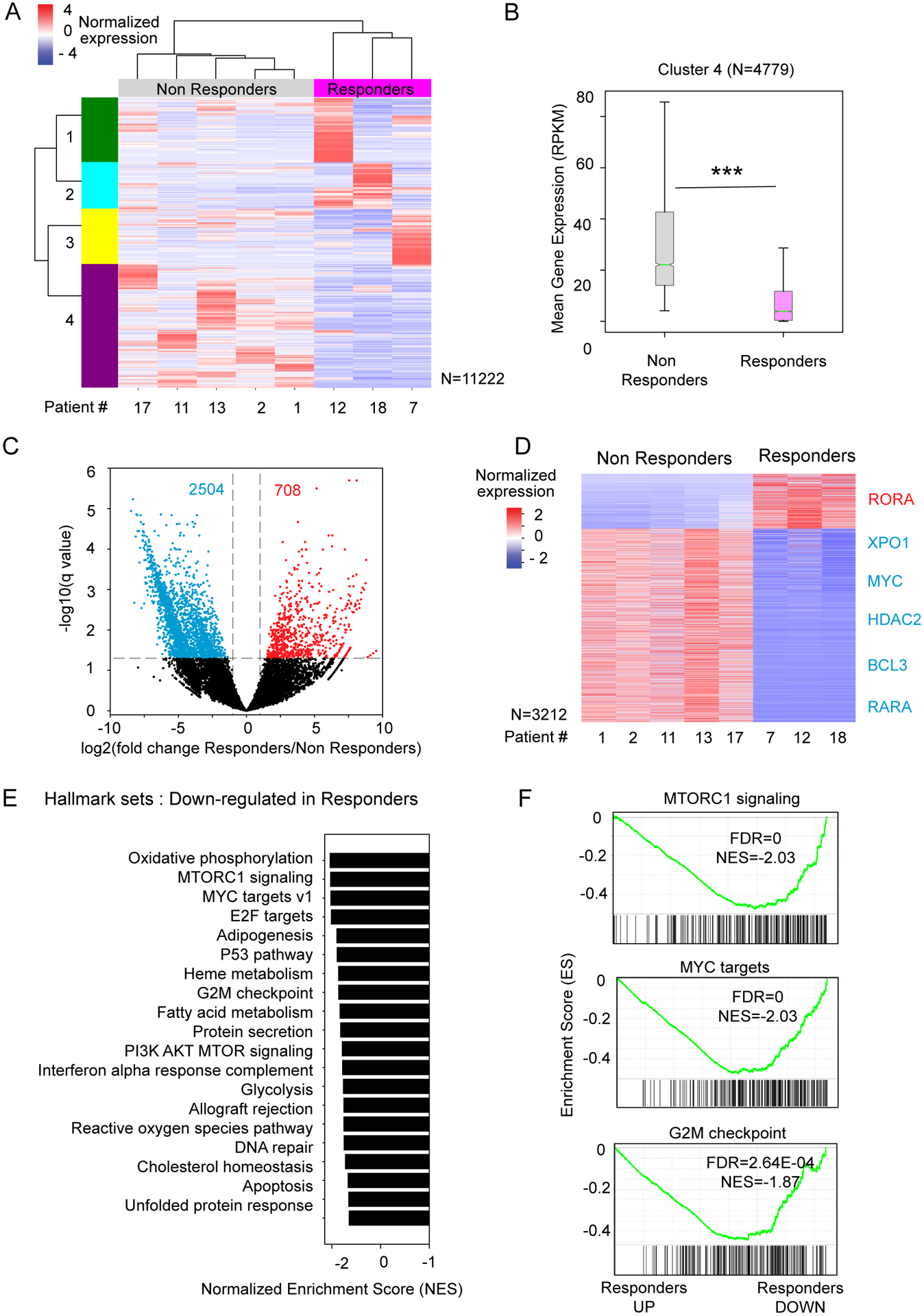
A) Heat map of normalized gene expression from RNA-seq of 8 patient samples before treatment (Z-score of the RPKM, per row). An unsupervised hierarchical clustering was applied to rows (all expressed genes with sum(expression/row>=50 RPKM)) and columns (patient samples). B) Boxplot distributions of the mean gene expression (RPKM) for cluster 4 showing consistent lower gene expression in responders vs. non-responders before treatment (Kolmogorov–Smirnov test, p<0.0001). C) Volcano plot showing the differential expressed genes between responders (n=3) and non-responders (n=5) before treatment (2-fold change cut-off, and FDR<0.05, blue=down-regulated genes, red=up-regulated genes). D) Heat map of normalized gene expression of the differential expressed genes from panel C, sorted by fold change. E) Bar plot of the normalized enrichment score (NES) of the Top20 negatively-enriched “Hallmark genes sets” by GSEA. F) GSEA plots of the top-ranked negatively-enriched gene sets from panel E. The enrichment score (ES) is indicated by a green line. Below, each vertical line of the barcode represents a gene. Genes are ranked by their differential expression between responders and non-responders at baseline (from left=up-regulated to right=down-regulated in responders compared to non-responders).
Comparing all transcriptionally active genes from responders and non-responders, we identified 3212 differentially expressed genes (708 up-regulated genes and 2,504 down-regulated genes in responders, Figure 2C). Several genes, such as MYC (31), XPO1 (32), RARA, BCL3 (33), and HDAC2 (34) showed lower expression in responders vs. non-responders, whereas RORA was more expressed in responders (Figure 2D, Figure S2).
By GSEA, we found gene signatures related to growth signaling, metabolism, and DNA repair to be negatively-enriched in responders at baseline, indicating that their CD34+ cells were more quiescent and less proliferative (i.e., genes down-regulated in responders vs. non-responders, Figure 2E). Non-responders exhibited a transcriptional signature at baseline enriched for cell growth signaling pathways, such as mTOR signaling, MYC-regulated genes, and G2M checkpoint genes, which is associated with a more proliferative phenotype (Figure 2E–F). In summary, our results suggest that the patient responders to ATRA-TCP therapy have less proliferative CD34+ leukemic blast cells at baseline compared to the non-responders.
LSD1 inhibitor sensitizes AML cell lines to ATRA
Unlike APL and APL-like cells, the majority of non-APL AML sub-types are known to be less responsive to the differentiation or cell killing effects of ATRA (10, 35, 36). Previously it has been shown that different LSD1 inhibitors in combination with ATRA decrease cell viability with substantial synergy in a number of AML cell lines (37).
To validate that TCP specifically is an effective LSD1 inhibitor, we treated 6 AML cell lines with ATRA and TCP: U-937, SKNO-1, MOLM-13, THP-1, SET-2 and MV4–11, cultured for 4 days (Figure 3 and Figure S3). Treatment of U-937 cells with ATRA alone induced cell death, a block in the cell cycle and myeloid differentiation, evidence by increased surface expression of CD11b and CD86, and these effects were enhanced by the co-treatment of ATRA with TCP (Figure 3A–C, Figure S3A). For SKNO-1, we also observed that despite a relative insensitivity to ATRA alone, combination treatment significantly reduced cell viability and increased cell death, despite no observable changes in CD11b or CD86 surface expression (Figure 3D–F, Figure S3B). Following combination of ATRA-TCP, U-937 showed additive effect with zip score 6.251 (Figure 3G) and a synergist effect was observed in SKNO-1 cells with zip score 17.267.
Figure 3. The LSD1 inhibitor TCP sensitizes SKNO-1 cells to ATRA treatment.
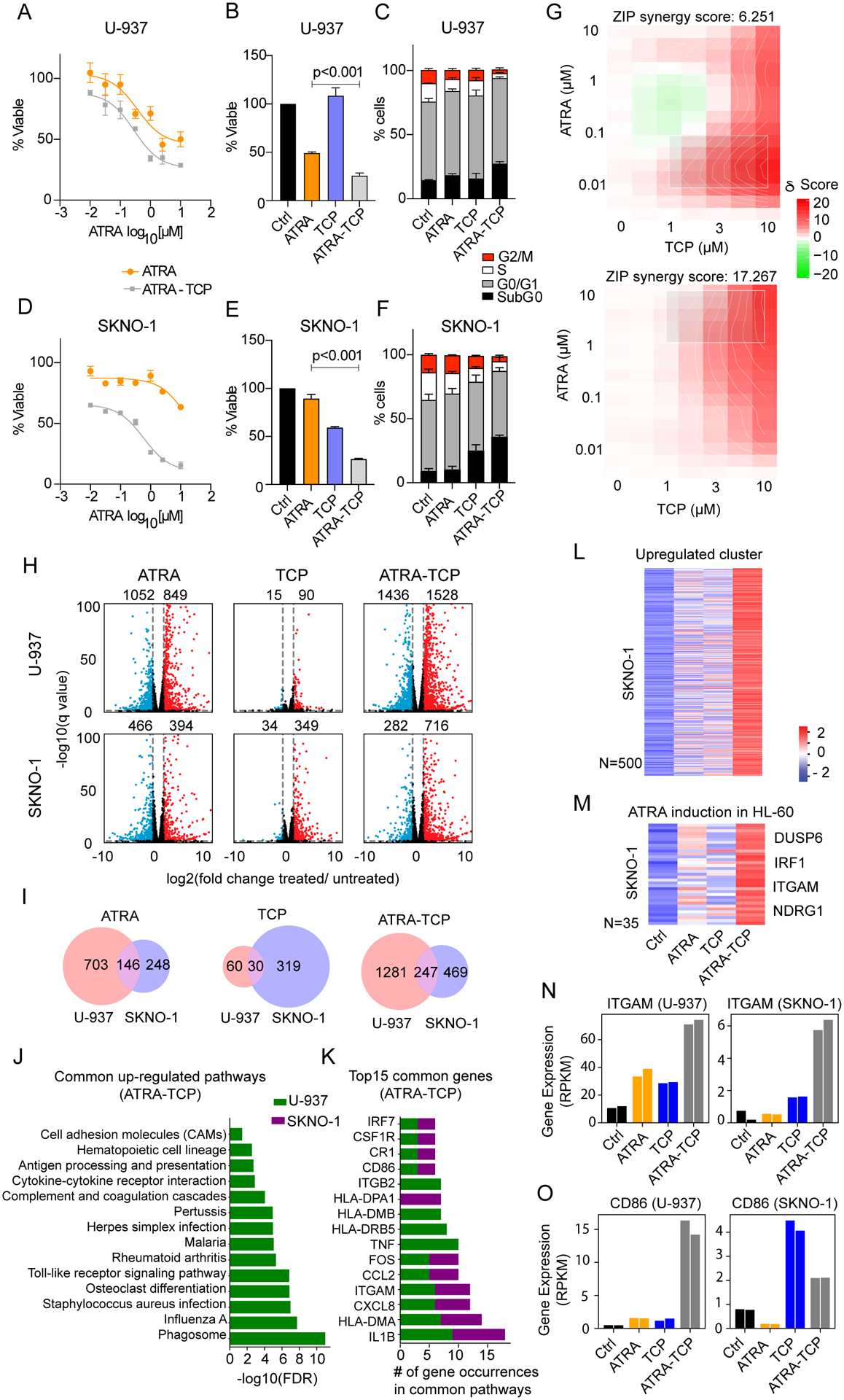
A) LSD1 inhibitor TCP (10 μM) and ATRA dose response plot. AML cell lines were treated with either ATRA (1 μM), TCP (10 μM), or ATRA-TCP (1 μM-10 μM) for 4 days and measured for B) cell viability and C) cell-cycle percentages in U-937, and D-F) SKNO-1 cells. Data were analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s post-hoc test (ATRA vs. ATRA-TCP). Data represent the mean and the error bars represent the standard deviation of the mean for three independent experiments. G) Synergy maps for U-937 (top) and SKNO-1 cells (bottom). The 2D synergy matrix was generated with SynergyFinder 2.0 using ZIP model. H) Volcano plots showing differential expressed genes in U-937 (top) and SKNO-1 (bottom) upon treatment compared to the untreated control cells (treatments from left to right: ATRA, TCP or ATRA-TCP) (2 fold-change cutoff, and FDR<0.05, blue=down-regulated genes, red=up-regulated genes). I) Venn diagrams of the significantly up-regulated genes in U-937 and SKNO-1 from panel H (treatments from left to right: ATRA, TCP, or ATRA-TCP). J) Bar plot of the 14 significantly up-regulated pathways upon ATRA-TCP treatment in U-937 that are also up-regulated in SKNO-1 upon ATRA-TCP. K) Top-ranked genes from the pathways in panel I, sorted by the cumulative count in both cell lines. L) Heat map of the Z-score normalized gene expression in SKNO-1 for a subset of genes obtained by unsupervised clustering of the up-regulated genes upon any of the treatments in the ATRA-resistant cell line (SKNO-1). M) Heat map of the Z-score normalized gene expression in SKNO-1 for a subset of genes from panel K with positive-enrichment in the ATRA-induction signature reported for HL-60 cells (15) showing ATRA-TCP enhances the expression of known ATRA-responsive genes. Heat maps are color-coded from blue (lower expression compared to the row’s average), to white (no change), to red (higher expression compared to the row’s average). N-O) Normalized mRNA expression of the differentiation markers ITGAM (CD11b) and CD86, respectively, for untreated control cells, ATRA, TCP, or ATRA-TCP treatment in U-937 (left), and in SKNO-1 (right). Same-color bars represent two independent experiments.
Consistent with previous studies (13), ATRA-TCP induced CD11b and/or CD86 upregulation and differentiation in the MLL-AF9+ cell lines MOLM-13 (Figure S3C–F), THP-1 (Figure S3G–J), and SET-2 (Figure S3K–N), whereas in the MLL-AF4+ cell line MV4–11, CD11b and CD86 remained unchanged despite enhanced cell killing with ATRA-TCP (Figure S3O–R). In all 6 AML cell lines tested, ATRA in combination with TCP decreased cell viability and enhanced cell death more than ATRA or TCP alone (Figure 3A–F, Figure S3C–R).
Up-regulation of differentiation gene signatures in AML cell lines upon ATRA-TCP treatment
To determine the transcriptional effect of ATRA-TCP, we performed RNA-sequencing in U-937 and SKNO-1 cell lines. In U-937, 1901 genes were differentially expressed upon ATRA treatment, with 849 up-regulated and 1052 down-regulated genes, respectively, compared to the vehicle control (Figure 3H). TCP alone resulted in minor transcriptomic changes, affecting only 105 genes (90 up-regulated and 15 down-regulated) whereas the combination of ATRA-TCP induced the greatest expression change (1528 up-regulated and 1436 down-regulated genes compared to control, Figure 3H). In SKNO-1, ATRA altered the expression of fewer genes compared to U-937, with 860 genes becoming differentially regulated compared to vehicle control (394 up-regulated, and 466 down-regulated genes). TCP alone induced greater transcriptional changes in SKNO-1 (349 up-regulated and 34 down-regulated genes) compared to U-937, and ATRA-TCP led to 716 up-regulated and 282 down-regulated genes, respectively compared to control (Figure 3H). In both cell lines, the combination of ATRA-TCP almost doubled the number of genes up-regulated when treated with ATRA alone (Figure 3H).
Given the known repressive effect of LSD1 on gene expression, the increased number of up-regulated genes upon ATRA-TCP treatment suggests that de-repressing chromatin, via LSD1 inhibition, sensitizes resistant cells to ATRA. We therefore compared the overlap between the genes up-regulated in SKNO-1 and U-937 upon treatment. We found an overlap of 146 genes that became up-regulated upon ATRA treatment, 30 genes upon TCP treatment, and 247 genes with combined ATRA-TCP (Figure 3I).
We performed a gene set enrichment analysis (enrichr) against a KEGG database on the up-regulated and down-regulated genes in each treatment group from Figure 3G. Interestingly, we found 55 up-regulated pathways were significantly enriched in U-937 and/or SKNO-1 upon ATRA, TCP, and/or ATRA-TCP (Table S6). Of the top most enriched pathways upon ATRA-TCP treatment, we observed the up-regulation of genes involved in inflammation (rheumatoid arthritis), immune responses (infection pathways), differentiation (osteoclast differentiation), Toll-like receptor signaling, cell adhesion, and cytokine signaling (Figure 3J, Table S6). Genes associated with apoptosis were also up-regulated upon ATRA-TCP in U-937, or by TCP alone in SKNO-1 (Table S6). The most common genes in the top-ranked enriched pathways from Figure 3J included IL1B, HLA-DMA, CXCL8, ITGAM (encoding for CD11b), CCL2, and FOS (Figure 3K). These data support the hypothesis that TCP enhances ATRA sensitivity by acting as a de-repressor of ATRA-induced pathway genes in ATRA-resistant AML cell lines.
We also performed an unsupervised hierarchical clustering of genes up-regulated in the ATRA-resistant cell line SKNO-1 upon ATRA, TCP, or ATRA-TCP. A cluster of 500 genes showed consistent induction when comparing ATRA-TCP with ATRA alone, suggesting that these genes could be co-regulated by both retinoic acid (RA) receptors and LSD1 (Figure 3L). Additionally, we confirmed that LSD1 inhibition enhances the transcription of ATRA-responsive genes previously reported for another AML cell line (HL-60, APL-like) (29), validating the de-repression of the RA pathway in the ATRA-resistant SKNO-1 cell line (Figure 3M). Examples of ATRA-responsive genes in this gene set, include DUSP6, IRF1, ITGAM, and NDRG1 (Figure 3M, Figure S3S–T). Importantly, the genes of the differentiation markers CD11b (ITGAM) and CD86 were both found to be more expressed upon treatment of both SKNO-1 and U-937 with ATRA-TCP (Figure 3N–O).
Interestingly, genes such as XPO1, MYC and HDAC2, which were lower expressed in patient responders vs. non-responders (Figure S2), were further down-regulated in U-937 after either ATRA or ATRA-TCP compared to control cells. However, for SKNO-1, the changes in expression of these genes were not significant (Figure S3V–X). In summary, we validated that TCP sensitizes AML cells to ATRA, leading to the upregulation of a greater number of known ATRA-responsive genes, even in non-APL-like cell lines.
Gene expression induction of RA-responsive genes in patient responders and AML cell lines upon ATRA-TCP treatment
Using available ChIP-seq data (38), we found 314 genes among the 3212 differentially expressed genes from patients in Figure 2D, with RARA occupancy and at least 1 RARE motif (Figure 4A). Of those, there was a subset of 86 RA-responsive genes that become up-regulated in the cell lines upon at least one of the treatments (Figure 4A). For most of these genes there was a strong induction with ATRA-TCP that cannot be explained by ATRA alone (Figure 4B). In fact, in AML cell lines these 86 genes showed significantly higher expression when treated with ATRA-TCP than with each treatment alone (Figure 4C).
Figure 4. ATRA-TCP induces the expression of a RA-responsive gene signature in patient responders.
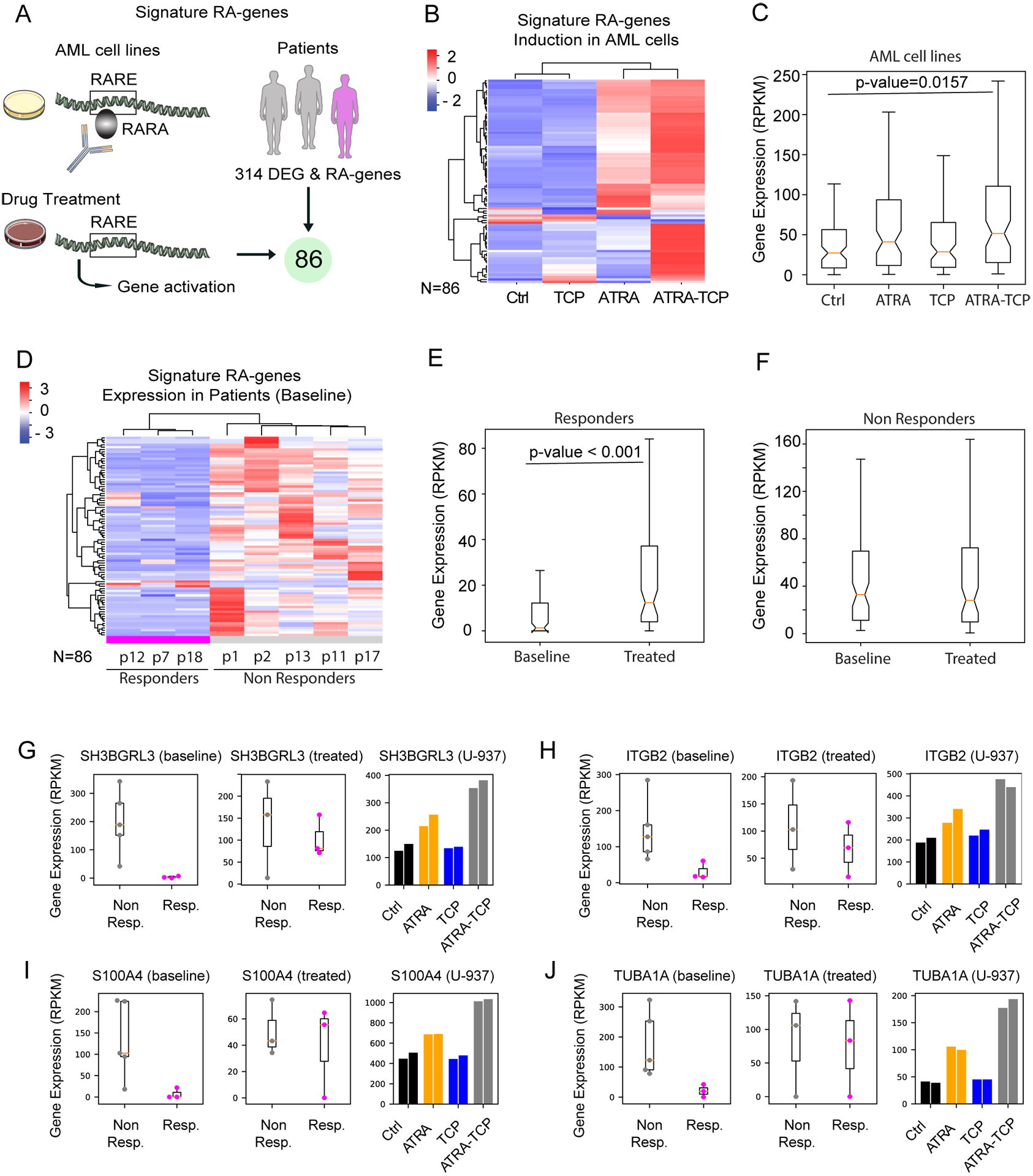
A) Signature of RA-responsive genes: genes with RARA occupancy and RARE motif from (38) that are up-regulated upon any of the treatments in the AML cell lines (U-937 and/or SKNO-1), that further overlapped with the gene signature of response from patients at baseline (N=3212 genes). Cartoons were downloaded from https://smart.servier.com. B) Heat map of mean normalized gene expression in AML cell lines for control and treatments (Z-score of the RPKM, per row). Unsupervised hierarchical clustering was applied to rows and columns. C) Boxplot distributions of the mean gene expression (RPKM) showing significant gene induction of RA-genes after ATRA-TCP treatment (Kolmogorov–Smirnov test). D) Heat map of mean normalized gene expression in patients before treatment (Z-score of the RPKM, per row). Unsupervised hierarchical clustering was applied to rows and columns. E-F) Boxplot distributions of the mean gene expression (RPKM) showing significant gene induction of RA-genes after ATRA-TCP treatment in responders but not in non-responders (Kolmogorov–Smirnov test).See Figure S4 for related heat map. G-J) Examples of RA-responsive genes: Gene expression plots for patients (before/after treatment), and for U-937 cells (from left to right: untreated control cells, ATRA, TCP, or ATRA-TCP treatment. Same-color bars represent two independent experiments).
The RA-responsive gene signature was lower expressed in patient responders compared to non-responders at baseline. Remarkably, upon ATRA-TCP treatment, there was a significant induction of the signature genes in patient responders, but not in the non-responders (Figure 4E–F). Figure 4 G–J shows the transcriptional behavior of notable genes from the RA-responsive signature that were upregulated in cell lines upon ATRA-TCP treatment as well as in patient responders vs. non-responders, where induction of these genes correlated with clinical response. Genes included in the RA-responsive signature were enriched for SPI1 transcription factor targets (Figure 4G–I, Table S7), in line with prior studies (38). We found enrichment of interleukin-2 signaling pathway genes (Figure 4H–I, Table S7), genes regulating apoptosis (Figure 4I–J, Table S7), as well as genes regulating neutrophil mediated immune response, macrophage/dendritic cells differentiation, and RUNX1 transcription factor targets (Table S8). These findings suggest that lower basal expression of RA-responsive genes sensitize patients to ATRA-based differentiation therapy when combined with LSD1 inhibition.
Up-regulation of TCP-driven gene signature in treated patients
To evaluate whether TCP reached the bone marrow of patients, we first defined a TCP-driven gene induction signature (N=244) including genes up-regulated by TCP alone and ATRA-TCP, but not up-regulated by ATRA alone, in any of the AML cell lines (SKNO-1 and/or U-937) (Figure S5A). Treated patients show gene induction of this TCP-driven signature (Figure S5B), which is broader for responders. Indeed, we observed that patient responders show a significant increase in the expression of these TCP-driven genes upon treatment (Figure S5C–D). For examples of gene induction see Figure S5E–G. Overall, these results suggest that TCP reached and had effect in the bone marrow.
DISCUSSION
In a Phase I clinical study, we demonstrated ATRA-TCP to be safe (MTD 20 mg BID) and observed clinical responses in R/R MDS and AML patients. All DLTs were related to TCP and consistent with known effects of MAOIs (39). Disease-related AEs occurred at an expected rate and drug-related AEs were low-grade and manageable. Notable AEs included rash, dry mouth and dry skin related to continuous ATRA exposure, and an intermittent dosing schedule should be considered in future studies.
We demonstrated clinical activity for this drug combination that would not be expected from ATRA alone (10), with 4/13 response-evaluable patients having blast clearance and/or HI. Some responses were durable (Figure 1), with the 3 best responders discontinuing study for ATRA-related skin toxicity. Patients with a hypoproliferative phenotype (MDS/low blast AML) appeared more likely to respond (4 out of 7; ORR=57.1% [95% CI=18.4 to 90.1]). Patients with more proliferative AML did poorly, and if these patients are further studied clinically with ATRA-LSD1 inhibitor, a third, cytoreductive or pro-apoptotic agent should be employed. Patient responders appeared to be enriched for TET2 and splicing factor mutations and did not have proliferative mutations such as FLT3-ITD (Table 1), although this did not reach statistical significance. Whether there is a biological reason related to the mechanism of action of LSD1 accounting for these differences in response, or it is reflecting the proliferative index of disease and response kinetics to epigenetic therapy, is an important question to answer in future studies.
Using a whole-transcriptome sequencing approach we demonstrated a unique baseline gene expression signature in patients that correlated with clinical responsiveness to ATRA-TCP. We acknowledge that these preliminary results include a limited number of RNA-seq samples (N=8). Low RARA expression has previously been proposed as the reason why non-APL AML patients are resistant to ATRA therapy (13, 14); however, we found that non-responders had higher levels of RARA expression compared to responders (Figure S2D) in agreement with previous reports (40) that ATRA alone fails to show efficacy in RARA-high models. Non-responders were enriched for genes in the mTOR signaling pathway, which may confer resistance to ATRA-TCP and supports the rationale for combining LSD1 and mTOR inhibitors in AML (41). Non-responders also expressed higher levels of XPO1, which codes for an exportin that regulates the cytoplasmic localization of nuclear receptors like RARs, preventing their transcriptional activity, and is associated with poor outcomes in AML (32, 42). This may explain why patients with higher levels of RARA at baseline did not respond to ATRA-TCP therapy. We also showed that basal HDAC2 expression is higher in non-responders compared to responders. HDAC2 is often overexpressed or mutated in AML, and plays a critical role in transcriptional regulation and cell cycle progression (34). Other reports have shown that the effects of LSD1 inhibition are noticeably enhanced in combination with HDAC inhibitors (15, 43).
The preliminary activity we observed is supportive of further studies targeting LSD1, and other irreversible LSD1 inhibitors have been tested in combination with ATRA in R/R AML. GSK2879552 (GlaxoSmithKline) is a potent, selective inhibitor of LSD1 that showed encouraging anti-proliferative activity (44), but the Phase I study was stopped due to safety concerns (NCT02177812). Preclinically, IMG7289 (Imago Biosciences) showed promising therapeutic efficacy coinciding with the induction of myeloid differentiation in MLL-rearranged AML models (45). Results from the clinical study of IMG7289 with ATRA (NCT02842827) have not yet been reported. Wass and colleagues reported results from a different clinical trial testing TCP and ATRA in R/R AML, with comparable safety and efficacy (ORR=20%) to our study. They demonstrated intracellular uptake of TCP and increased H3K4me1 and me2 levels in some ATRA-TCP treated patients, supporting on-target activity (46). In our study, the transcriptome results suggest that TCP likely reached the bone marrow of treated patients as they showed induction of the expression of TCP-driven genes.
In summary, ATRA-TCP is a feasible and safe oral regimen for patients with R/R MDS and AML. Our study was the first to include MDS, and intriguingly, significant clinical activity was observed in these patients. We identified a gene expression signature delineating responder from non-responder at baseline, with non-responders having a more proliferative phenotype and increased RARA expression (independent of MDS vs. AML diagnosis). We observed transcriptional changes in potential biomarkers of response upon ATRA-TCP treatment of both AML cell lines and primary patient samples. Additionally, we demonstrated that adding the LSD1 inhibitor TCP to ATRA enhances the expression of ATRA-responsive genes, leading to in vitro differentiation and cell death. These findings support that AML and MDS patients with decreased basal expression of RA-responsive genes are re-sensitized to ATRA when combined with LSD1 inhibition, and this should be validated in subsequent studies. A triplet combination of ATRA, LSD1 inhibitor, and azacitidine is planned in frontline higher risk MDS.
Supplementary Material
KEY POINTS.
ATRA-TCP combination therapy has clinical activity and an acceptable safety profile in relapsed/refractory AML and MDS patients.
Prior to treatment, non-responder patients had more proliferative CD34+ leukemic blasts compared to patients that responded to ATRA-TCP.
TRANSLATIONAL RELEVANCE.
Differentiation therapy with all-trans retinoic acid (ATRA) transformed the management of acute promyelocytic leukemia (APL), but has minimal clinical activity in AML. We sought to de-repress the retinoic acid (RA) pathway and re-sensitize AML cells to the effects of ATRA by combining it with the LSD1 inhibitor tranylcypromine. We conducted a phase I clinical trial and demonstrated this combination to be safe and clinically active, particularly in MDS and less proliferative AML patients. We found that patient responders, whether MDS or AML and across risk categories, had a more quiescent gene expression profile and decreased basal RARA expression. These gene signatures inverted upon ATRA-TCP treatment of responsive patients and ATRA-resistant AML cell lines. ATRA-LSD1 inhibitor therapy is being further studied in combination with azacitidine and could establish a new treatment paradigm of differentiation therapy in MDS and AML patients with low baseline RARA expression.
ACKNOWLEDGMENTS
We gratefully acknowledge the help provided by Dr. Maria “Ken” Figueroa and Dr. Stephen D. Nimer for their constructive comments. We would like to thank Dr. Felipe Beckedorff for his assistance on the preliminary analysis of the patient samples, and his insightful advice. We thank our laboratory members and the Bioinformatics core at Sylvester Comprehensive Cancer Center for support and helpful discussions. We also thank the Oncogenomics core facility for performing the high-throughput sequencing. This work was supported by grants from The Leukemia & Lymphoma Society, Specialized Center of Research Program (Nimer PI), Project 1 (Shiekhattar/Morey/Watts); NCI 5R21CA202488-02 (Swords/Watts PI); NIH/NCI, The Sylvester Cancer Center Support Grant, 1P30CA240139-01 (Nimer PI); and the Sylvester Comprehensive Cancer Center.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(26):6285–95. [DOI] [PubMed] [Google Scholar]
- 2.Lowenberg B. Acute myeloid leukemia: the challenge of capturing disease variety. Hematology Am Soc Hematol Educ Program. 2008:1–11. [DOI] [PubMed] [Google Scholar]
- 3.Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010. [DOI] [PubMed] [Google Scholar]
- 4.Huang ME, Ye YC, Chen SR, Zhao JC, Gu LJ, Cai JR, et al. All-trans retinoic acid with or without low dose cytosine arabinoside in acute promyelocytic leukemia. Report of 6 cases. Chin Med J (Engl). 1987;100(12):949–53. [PubMed] [Google Scholar]
- 5.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–72. [PubMed] [Google Scholar]
- 6.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113(16):3655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlenk RF, Lubbert M, Benner A, Lamparter A, Krauter J, Herr W, et al. All-trans retinoic acid as adjunct to intensive treatment in younger adult patients with acute myeloid leukemia: results of the randomized AMLSG 07–04 study. Ann Hematol. 2016;95(12):1931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martelli MP, Gionfriddo I, Mezzasoma F, Milano F, Pierangeli S, Mulas F, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125(22):3455–65. [DOI] [PubMed] [Google Scholar]
- 9.El Hajj H, Dassouki Z, Berthier C, Raffoux E, Ades L, Legrand O, et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125(22):3447–54. [DOI] [PubMed] [Google Scholar]
- 10.Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109(6):1114–24. [DOI] [PubMed] [Google Scholar]
- 11.Lübbert M, Rüter BH, Claus R, Schmoor C, Schmid M, Germing U, et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica. 2012;97(3):393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lübbert M, Grishina O, Schmoor C, Schlenk RF, Jost E, Crysandt M, et al. Valproate and Retinoic Acid in Combination With Decitabine in Elderly Nonfit Patients With Acute Myeloid Leukemia: Results of a Multicenter, Randomized, 2 × 2, Phase II Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2020;38(3):257–70. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto K, Imamura T, Yano M, Yoshida H, Fujiki A, Hirashima Y, et al. Sensitivity of MLL-rearranged AML cells to all-trans retinoic acid is associated with the level of H3K4me2 in the RARalpha promoter region. Blood cancer journal. 2014;4:e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasow A, Barrett A, Petrie K, Gupta R, Boix-Chornet M, Zhou DC, et al. DNA methylation-independent loss of RARA gene expression in acute myeloid leukemia. Blood. 2008;111(4):2374–7. [DOI] [PubMed] [Google Scholar]
- 15.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19(6):857–64. [DOI] [PubMed] [Google Scholar]
- 17.Kruse W. Trifluoperazine and Tranylcypromine in chronic refractory schizophrenics. Am J Psychiatry. 1960;117:548–9. [DOI] [PubMed] [Google Scholar]
- 18.Barsa JA, Saunders JC. Tranylcypromine in the treatment of chronic schizophrenics. Am J Psychiatry. 1962;118:933–4. [DOI] [PubMed] [Google Scholar]
- 19.Ravasio R, Ceccacci E, Nicosia L, Hosseini A, Rossi PL, Barozzi I, et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid–induced differentiation. Science Advances. 2020;6(15):eaax2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73. [DOI] [PubMed] [Google Scholar]
- 26.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research. 2016;44(W1):W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo M, Shin J, Kim J, Ryall KA, Lee K, Lee S, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic acids research. 2020;48(W1):W488–w93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delgado MD, Albajar M, Gomez-Casares MT, Batlle A, Leon J. MYC oncogene in myeloid neoplasias. Clin Transl Oncol. 2013;15(2):87–94. [DOI] [PubMed] [Google Scholar]
- 32.Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121(20):4166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niu Y, Yang X, Chen Y, Zhang L, Jin X, Tang Y, et al. BCL3 Expression Is a Potential Prognostic and Predictive Biomarker in Acute Myeloid Leukemia of FAB Subtype M2. Pathology oncology research : POR. 2019;25(2):541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conte M, Dell’Aversana C, Benedetti R, Petraglia F, Carissimo A, Petrizzi VB, et al. HDAC2 deregulation in tumorigenesis is causally connected to repression of immune modulation and defense escape. Oncotarget. 2015;6(2):886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milligan DW, Wheatley K, Littlewood T, Craig JI, Burnett AK. Fludarabine and cytosine are less effective than standard ADE chemotherapy in high-risk acute myeloid leukemia, and addition of G-CSF and ATRA are not beneficial: results of the MRC AML-HR randomized trial. Blood. 2006;107(12):4614–22. [DOI] [PubMed] [Google Scholar]
- 36.Howell AL, Stukel TA, Bloomfield CD, Davey FR, Ball ED. Induction of differentiation in blast cells and leukemia colony-forming cells from patients with acute myeloid leukemia. Blood. 1990;75(3):721–9. [PubMed] [Google Scholar]
- 37.McGrath JP, Williamson KE, Balasubramanian S, Odate S, Arora S, Hatton C, et al. Pharmacological Inhibition of the Histone Lysine Demethylase KDM1A Suppresses the Growth of Multiple Acute Myeloid Leukemia Subtypes. Cancer Res. 2016;76(7):1975–88. [DOI] [PubMed] [Google Scholar]
- 38.Wang K, Wang P, Shi J, Zhu X, He M, Jia X, et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer cell. 2010;17(2):186–97. [DOI] [PubMed] [Google Scholar]
- 39.Safferman AZ, Masiar SJ. Central nervous system toxicity after abrupt monoamine oxidase inhibitor switch: a case report. Ann Pharmacother. 1992;26(3):337–8. [DOI] [PubMed] [Google Scholar]
- 40.McKeown MR, Corces MR, Eaton ML, Fiore C, Lee E, Lopez JT, et al. Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARα Dependency Targetable by SY-1425, a Potent and Selective RARα Agonist. Cancer discovery. 2017;7(10):1136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deb G, Wingelhofer B, Amaral FMR, Maiques-Diaz A, Chadwick JA, Spencer GJ, et al. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia. 2020;34(5):1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. Embo j. 1999;18(6):1660–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28(11):2155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer cell. 2015;28(1):57–69. [DOI] [PubMed] [Google Scholar]
- 45.Cusan M, Cai SF, Mohammad HP, Krivtsov A, Chramiec A, Loizou E, et al. LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPalpha-dependent enhancers in AML. Blood. 2018;131(15):1730–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wass M, Göllner S, Besenbeck B, Schlenk RF, Mundmann P, Göthert JR, et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.