

Abstract
Mitophagy is a selective form of autophagy involving the removal of damaged mitochondria via the autophagy-lysosome pathway. PINK1-Parkin-mediated mitophagy is one of the most important mechanisms in cardiovascular disease, cerebral ischemia-reperfusion (I/R) injury, and neurodegenerative diseases. In this study we conducted an image-based screening in YFP-Parkin HeLa cells to discover new mitophagy regulators from natural xanthone compounds. We found that garciesculenxanthone B (GeB), a new xanthone compound from Garcinia esculenta, induced the formation of YFP-Parkin puncta, a well known mitophagy marker. Furthermore, treatment with GeB dose-dependently promoted the degradation of mitochondrial proteins Tom20, Tim23, and MFN1 in YFP-Parkin HeLa cells and SH-SY5Y cells. We revealed that GeB stabilized PINK1 and triggered Parkin translocation to the impaired mitochondria to induce mitophagy, and these effects were abolished by knockdown of PINK1. Finally, in vivo experiments demonstrated that GeB partially rescued ischemia-reperfusion-induced brain injury in mice. Taken together, our findings demonstrate that the natural compound GeB can promote the PINK1-Parkin-mediated mitophagy pathway, which may be implicated in protection against I/R brain injury.
Keywords: garciesculenxanthone B, mitophagy, Parkin, PINK1, ischemia-reperfusion injury
Introduction
Mitochondria are critical organelles that regulate cellular energy and cell death [1–3]. Under various detrimental conditions, such as hypoxia and nutrient depletion, mitochondria can be impaired [4, 5]. Dysfunctional mitochondria generate redundant reactive oxygen species (ROS) and promote mitochondrial depolarization [6], which is associated with Parkinson’s disease (PD), cancer, stroke, etc. [7]. The clearance of impaired mitochondria is thus essential for the maintenance of cellular homeostasis and cell survival. Selective autophagy of mitochondria, also known as mitophagy, is an important mechanism for the elimination of damaged mitochondria. During this process, damaged mitochondria are sequestered by double-membrane vesicles and subsequently delivered to lysosomes for hydrolytic degradation [8].
PTEN-induced putative kinase protein 1 (PINK1)-Parkin-mediated mitophagy plays a key role in mitochondrial quality control [9]. In healthy mitochondria, PINK1 is cleaved by PARL and degraded by the proteasome [10]. Upon mitochondrial depolarization, full-length PINK1 is stabilized on the outer mitochondrial membrane (OMM) and subsequently recruits and activates Parkin [11]. The ubiquitin E3 ligase Parkin then catalyzes the polyubiquitination of mitochondria, which mediates the autophagic elimination of damaged mitochondria [12]. Dysfunction of the PINK1/Parkin-dependent mitophagy pathway is associated with various diseases, including cerebral ischemia injury [13] and neurodegenerative diseases [14, 15]. Accumulating evidence suggests that mutations in PINK1 and Parkin are closely correlated with Parkinson’s disease [9, 16, 17]. Several studies have suggested that mitophagy plays a protective role against ischemic brain injury [18–20] and that activation of Parkin-dependent mitophagy by acidic postconditioning can protect against cerebral ischemia [21]. Notably, mitophagy-mediated mitochondrial clearance also inhibits ischemia-induced neuronal cell death [18]. Therefore, targeting the PINK1–Parkin pathway is a potential strategy for alleviating cerebral ischemia injury.
For a long time, we have focused on isolating natural xanthones from Garcinia species and investigating their bioactivity. Xanthones possess anticancer, anti-inflammatory, antioxidative, and neuroprotective activities. For example, Nujiangexathone A, which is extracted from Garcinia nujiangensis, promotes apoptosis through the ROS/JNK pathway in HeLa cancer cells [22]. Griffipavixanthone, a dimeric xanthone extracted from edible plants, inhibits esophageal tumor metastasis and proliferation by downregulating the RAF–MEK–ERK pathway [23]. Importantly, recent reports have revealed that xanthones from Garcinia mangostana exhibit activity against PD [24]. α-Mangostin, a xanthone purified from Garcinia mangostana, has been verified to inhibit the growth of several cancer cell lines and is capable of inhibiting Aβ aggregation and reducing Aβ oligomer-induced neurotoxicity [25], suggesting the potential value of xanthones in neuronal protection.
In this study, we screened natural xanthone compounds for regulators of mitophagy using YFP-Parkin HeLa cells and identified a new compound extracted from Garcinia esculenta, garciesculenxanthone B (GeB), which can induce Parkin translocation to damaged mitochondria. Our results indicated that GeB can stabilize PINK1 and induce mitophagy via the PINK1–Parkin-dependent pathway. GeB has a neuroprotective effect, as evidenced by its ability to inhibit ischemia-reperfusion injury in vivo in a middle cerebral artery occlusion (MCAO) model.
Materials and methods
Cell culture and transfection
YFP-Parkin HeLa and SH-SY5Y cells were maintained in DMEM containing 10% fetal bovine serum (HyClone, Logan, UT, USA) and 0.1% penicillin/streptomycin at 37 °C in a humidified incubator with a 5% CO2 atmosphere. The cells were transfected with WT GST-Parkin and S65A GST-Parkin plasmids using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY, USA) following the manufacturer’s instructions. RNA interference was performed using RNAiMAX (Invitrogen) through a reverse transfection protocol.
Antibodies, fluorescent dyes, and other reagents
The compound GeB was isolated from Garcinia esculenta. The extraction method of GeB is shown in the Supplementary materials and methods. The 1H and 13C NMR spectra of GeB are shown in Supplementary Figs. S2 and S3. Tom20 (sc-11415, 1:3000), GFP (sc-8334, 1:1000), Ubiquitin (P4D1) (sc-8017, 1:1000) and Parkin (sc-133167, 1:1000) antibodies were purchased from Santa Cruz (Dallas, Texas, USA). Mitofusin-1 (14739, 1:1000), Mitofusin-2 (11925, 1:1000), COX IV (4850, 1:1000), GAPDH (2118, 1:1000), GST (2624, 1:1000) and PINK1 (6946, 1:1000) antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). β-Actin (A5441, 1:5000), LC3 (L7543, 1:3000) and Tubulin (T6199, 1:5000) antibodies were obtained from Sigma (St. Louis, MO, USA), and a Tim23 (611222, 1:3000) antibody was obtained from Business Development (St. Louis, MO, USA). p62 (H00008878-M01, 1:5000) was purchased from Abnova (Taipei, Taiwan, China). MitoTracker™ Red FM, anti-rabbit Alexa Fluor® 488 dye, and anti-mouse Alexa Fluor™ 594 dye were purchased from Thermo Fisher. MG132 and CCCP were purchased from Sigma (St. Louis, MO, USA).
Western blot and immunoprecipitation analysis
For Western blotting, cells were lysed in 62.5 mM Tris-HCl (pH 6.8), 20% glycerol, 2% SDS, and phosphatase inhibitor (Thermo Scientific, Waltham, MA, USA) and boiled at 100 °C for 10 min. The cell extracts were separated by SDS-PAGE and transferred onto PVDF membranes. After blocking with 5% nonfat milk, the membranes were incubated with the indicated antibodies, imaged via the enhanced chemiluminescence method and visualized with the ImageQuant LAS 4000 system (GE Healthcare, Boston, MA, USA).
For immunoprecipitation (IP) of YFP-tagged Parkin proteins, YFP-Parkin HeLa cells were lysed in IP lysis buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2.5 mM MgCl, 0.05% Triton-100, and protease inhibitors). The cell supernatants were incubated with 10 μL GFP-Trap (ChromoTek, Munich, Germany) beads overnight at 4 °C with rotation. The protein/bead complexes were washed with IP lysis buffer three times, boiled with 2× sample loading buffer and separated by SDS-PAGE for Western blotting.
Immunofluorescence staining and confocal microscopy
YFP-Parkin HeLa cells were seeded on coverslips or in chamber slides (Thermo Fisher Scientific, Waltham, MA, USA). The cells were washed with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min, blocked and permeabilized with 3% BSA (Sigma, St. Louis, MO, USA) in PBS containing 0.1% Triton X-100 for 30 min at room temperature. After washing with PBS, the cells were incubated with primary antibodies for 4 °C overnight and then incubated with secondary antibodies for 1 h at room temperature. The cells samples were observed by laser scanning confocal microscopy (OLYMPUS Corporation, Tokyo, Japan) and a FluoView Ver.1.7a Viewer.
Mitochondrial isolation
The cytoplasmic and mitochondrial fractions were isolated as described previously [26]. Briefly, cells were suspended in mitochondrial extraction buffer (200 mM mannitol, 68 mM sucrose, 50 mM Pipes-KOH (pH 7.4), 50 mM KCl, 5 mM EDTA, 2 mM MgCl2 and 1 mM dithiothreitol) containing protease inhibitors and incubated on ice for 20 min. The cells were disrupted 30–40 times with a Dounce homogenizer and then centrifuged for 10 min at 600 × g at 4 °C. The supernatant was centrifuged for 10 min at 11,000 × g at 4 °C. The pellet was the mitochondrial fraction. Twenty micrograms of protein from each sample was separated by SDS-PAGE.
Transient MCAO mouse model and drug treatment
Adult male C57BL/6 mice weighing 22–25 g were maintained at room temperature (20–25 °C). All mice were randomly divided into sham-operated, 20 mg/kg, 40 mg/kg GeB after MCAO-operated, or 40 mg/kg GeB for per-treated 2 h groups. A transient MCAO mouse model was established as described previously [18]. Briefly, the mice were anesthetized with 3% chloral hydrate. Under a microscope, the left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed. A nylon monofilament was inserted into the ICA to occlude the origin of the MCA. After 1 h of occlusion, the monofilament was removed to allow reperfusion. Transient MCAO model mice were administered 20 or 40 mg/kg GeB by intraperitoneal injection during reperfusion.
To measure the cerebral infarct volume, mouse brains were collected and cut into 2-mm slices. Then, slices were stained with 2,3,5-triphenyltetrazolium chloride (TTC) (LJ0508B1009J, BIO BASICINC). The infarcted areas were analyzed by Photoshop software. The percentage of infarct volume = infarct volume/total contralateral hemispheric volume × 100%.
TUNEL staining assay
The TUNEL assay is a method for identifying and quantifying apoptotic cells. Briefly, tissue slides were permeabilized in proteinase K solution for 5–15 min at 37 °C. TdT enzyme and fluorescein-12-dUTP were then added to the tissue slides and incubated for 1–3 h at 37 °C. Following incubation, stopping buffer was added to the tissue section for 5 min, and then the slides were washed with PBS. Finally, TUNEL-positive sections were visualized using fluorescence microscopy.
Statistical analysis
All data are presented as the mean ± SD. Statistical analysis of more than two groups was performed by one-way analysis of variance followed by the Student–Newman–Keuls test. P < 0.05 and P < 0.01 were considered statistically significant.
Results
Identification of natural xanthones that are novel mitophagy regulators using image-based screening
To study the mechanism of mitophagy and identify small molecules that can regulate mitophagy, we treated HeLa cells stably expressing YFP-Parkin with a series of xanthones from Garcinia species and examined the formation of puncta of YFP-Parkin, which is a well-known mitophagy marker (Supplementary Fig. S1). Interestingly, the results showed that GeB (Fig. 1a and Supplementary Figs. S2, S3), a new compound from Garcinia esculenta, dramatically induced the formation of YFP-Parkin puncta in a concentration-dependent manner (Fig. 1b, c). Parkin translocates to damaged mitochondria and then targets OMM proteins for polyubiquitination and degradation upon mitochondrial damage [27, 28]. Based on this, we investigated whether GeB promotes the degradation of these OMM proteins. As expected, the levels of these mitochondrial proteins, including Tom20 and MFN1, dramatically decreased in a time- and dose-dependent manner in YFP-Parkin HeLa and SH-SY5Y cells after treatment with GeB, with GeB having similar effects as CCCP (Fig. 1d–k). Moreover, GeB reduced the expression of the inner mitochondrial protein Tim23 (Fig. 1d–k). Collectively, these results suggest that GeB promotes Parkin translocation to induce mitophagy.
Fig. 1. Identification of natural xanthone compounds that are regulators of mitophagy.

a Chemical structure of GeB. b, c GeB promotes the translocation of Parkin. YFP-Parkin HeLa cells were treated with GeB (20 μM) for 4 h. Cells were fixed and analyzed by confocal microscopy. Scale bar, 10 μm. CCCP was used as a positive control. The graph shows the number of cells with puncta (mean ± S.D.); n = 3, *P < 0.05; **P < 0.01. d–g GeB promotes the degradation of mitochondrial proteins. YFP-Parkin HeLa cells were treated with GeB for the indicated times and doses and analyzed by Western blotting. CCCP was used as a positive control to induce mitophagy. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05. h–k SH-SY5Y cells were treated with GeB or CCCP for the indicated times and doses and analyzed by Western blotting. CCCP was used as a positive control to induce mitophagy. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05.
GeB promotes Parkin recruitment to mitochondria
To address whether GeB promotes Parkin recruitment to mitochondria, we treated YFP-Parkin HeLa cells with GeB. We found that GeB greatly promoted the formation of puncta of Parkin, which was extensively colocalized with Tom20, an OMM protein (Fig. 2a, b). Recent studies have shown that the autophagic cargo adaptor p62 binds to ubiquitylated mitochondria and then promotes the clearance of damaged mitochondria via mitophagy [29]. We next investigated whether GeB promotes the recruitment of p62 to mitochondria. Figure 2c, d showed that treatment with GeB significantly enhanced the colocalization of Parkin with p62, suggesting that the autophagy receptor p62 is recruited to damaged mitochondria to drive mitophagy. Consistently, p62 was enriched in the mitochondrial fraction in SH-SY5Y cells following GeB treatment and that this change was accompanied by increased LC3-II levels (Fig. 2e, f). Taken together, these data suggest that GeB promoted Parkin-dependent mitophagy following the recruitment of the autophagy receptor p62.
Fig. 2. GeB promotes Parkin-dependent mitophagy.

a, b GeB promotes Parkin translocation to mitochondria. YFP-Parkin HeLa cells were treated with GeB (20 μM) for 4 h. Immunostaining for Tom20 (red) was used to identify mitochondria. Cells were analyzed by confocal microscopy. Scale bar, 10 μm. n = 3, **P < 0.01. c, d GeB induces the colocalization of Parkin with p62. YFP-Parkin HeLa cells were treated with GeB (20 μM) for 4 h. Parkin and endogenous p62 were analyzed by confocal microscopy. Colocalization was quantified by Pearson’s correlation coefficient (PCC); n = 3, **P < 0.01. e, f GeB induces the accumulation of p62 in mitochondria. Mitochondrial fractions and whole-cell lysates were prepared from SH-SY5Y cells treated with GeB (20 μM) for different periods of time as indicated and then analyzed by Western blotting. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05, **P < 0.01.
GeB promotes Parkin ubiquitination
Parkin is the main E3 ubiquitin ligase responsible for mediating ubiquitination on the OMM in the course of mitophagy [28]. At the same time, ubiquitination of Parkin itself plays a critical role in mediating mitophagy [30]. It is thus of interest to know whether GeB-induced mitophagy depends on Parkin ubiquitination. First, we conducted an immunofluorescence assay. As shown in Fig. 3a, b, YFP-Parkin puncta were markedly colocalized with ubiquitin in the presence of GeB. Second, the ubiquitination level of Parkin was elevated following GeB treatment (Fig. 3c). Finally, we tested whether the degradation of OMM proteins induced by GeB relies on the E3 ligase activity of Parkin. To this end, we treated YFP-Parkin HeLa cells with GeB or CCCP in the presence or absence of MG132, a proteasome inhibitor. Consistent with a previous study [30], treatment with MG132 restored the protein levels of Tom20 and MFN1 in YFP-Parkin HeLa and SH-SY5Y cells treated with GeB or CCCP (Fig. 3d–g), suggesting that GeB promotes Parkin-mediated ubiquitination and proteasomal degradation of OMM proteins.
Fig. 3. GeB increases Parkin ubiquitination.

a, b GeB promotes Parkin and ubiquitin colocalization. YFP-Parkin HeLa cells were treated with GeB (20 μM) for the indicated times. Ubiquitin immunostaining was performed, and cells were analyzed by confocal microscopy. Colocalization was quantified by PCC; n = 3, **P < 0.01. c YFP-Parkin HeLa cells were treated with GeB (20 μM) or CCCP (20 μM) for 4 h and analyzed using immunoprecipitation followed by Western blotting. CCCP was used as a positive control. d–g YFP-Parkin HeLa or SH-SY5Y cells were cotreated with GeB (20 μM) or CCCP (20 μM) and MG132 for 8 h and analyzed by Western blotting. CCCP was used as a positive control. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05. h–j HeLa cells were transfected with GST-Parkin WT or GST-Parkin S65A after treatment with GeB or CCCP and examined by immunostaining and Western blotting. CCCP was used as a positive control. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05.
Phosphorylation of Parkin at S65 within the UBL domain stimulates Parkin recruitment to the mitochondria and degradation [31, 32]. We thus wanted to know whether GeB-induced mitophagy relies on such phosphorylation. We transfected GST-Parkin WT or GST-Parkin S65A mutant plasmids into HeLa cells to examine the translocation of Parkin. Immunostaining results showed that GeB or CCCP treatment induced the formation of WT Parkin puncta, while the S65A mutant failed to form such puncta (Fig. 3h), which is consistent with previous reports [33, 34]. Phosphorylated and ubiquitinated Parkin is prone to proteasomal degradation [32, 35]. Consistently, we observed that GeB promoted the degradation of WT Parkin, but not the Parkin S65A mutant (Fig. 3i, j). Thus, our results indicate that GeB promoted mitophagy by regulating the E3 ligase activity of Parkin.
GeB regulates Parkin translocation and activation by stabilizing PINK1
To further understand how GeB affects Parkin translocation to damaged mitochondria, we examined the protein level of PINK1, a serine/threonine protein kinase that is stabilized and accumulates on the OMM and then triggers Parkin translocation to impaired mitochondria [36]. Consistently, treatment with GeB induced the accumulation of PINK1 in a time-dependent manner (Fig. 4a). Furthermore, we prepared mitochondrial and cytosolic fractions to detect the distribution of PINK1 following treatment with GeB. Immunoblotting analysis suggested that GeB promoted PINK1 accumulation in the mitochondrial fraction in YFP-Parkin HeLa cells (Fig. 4b). Similar results were found in SH-SY5Y cells (Supplementary Fig. S4a, b), confirming that GeB can stabilize PINK1.
Fig. 4. GeB-induced Parkin translocation is PINK1-dependent.

a GeB elevated PINK1 expression. YFP-Parkin HeLa cells were treated with GeB (20 μM) for the indicated time period. The protein level of PINK1 was quantified by ImageJ software; n = 3, *P < 0.05. b GeB stabilized PINK1 in the mitochondria. Mitochondrial fractions and whole-cell lysates were prepared from YFP-Parkin HeLa cells treated with GeB (20 μM) for different periods of time as indicated. The protein level of PINK1 was analyzed by Western blotting and quantified by ImageJ software; n = 3, *P < 0.05. c The promotion of Parkin translocation by GeB is PINK1-dependent. YFP-Parkin HeLa cells were transfected with the indicated siRNAs for 48 h, treated with GeB (20 μM) or CCCP (20 μM), and analyzed by immunostaining and Western blotting. The graph shows the number of cells with puncta (mean ± S.D.); n = 3, **P < 0.01. d–g YFP-Parkin HeLa and SH-SY5Y cells were transfected with the indicated siRNAs for 48 h, treated with GeB (20 μM) or CCCP (20 μM), and analyzed by immunostaining and Western blotting. The Western blot bands were quantified by ImageJ software; n = 3, *P < 0.05. h YFP-Parkin HeLa cells were transfected with the indicated siRNAs for 48 h and then treated with GeB (20 μM) or CCCP (20 μM) for 4 h. The cell lysates were analyzed using immunoprecipitation followed by Western blotting. CCCP was used as a positive control; n = 3.
To further determine whether the functions of GeB in mitophagy are dependent on PINK1, we knocked down PINK1 in YFP-Parkin HeLa cells and observed that the effect of GeB on Parkin translocation was abolished (Fig. 4c). Furthermore, to determine whether knocking down PINK1 delays GeB-induced mitophagy, we treated YFP-Parkin HeLa cells with GeB. Compared with control cells, PINK1 knockdown cells showed delayed mitophagy, as evidenced by the stabilization of Parkin, OMM proteins such as Tom20, and IMM proteins such as Tim23 (Fig. 4d, e). Similar results were observed in SH-SY5Y cells (Fig. 4f, g). It has been reported that PINK1 first phosphorylates Parkin on the OMM and that the binding of phospho-Parkin to phospho-ubiquitin fully activates Parkin activity [37]. We next determined whether GeB affects Parkin activity in a PINK1-dependent manner by detecting Parkin ubiquitination. To this end, we immunoprecipitated Parkin from YFP-Parkin HeLa cells treated with GeB and detected the ubiquitination level of Parkin. As shown in Fig. 4h, GeB treatment effectively induced Parkin ubiquitination, which was efficiently blocked by knockdown of PINK1. Taken together, these results demonstrate that GeB stabilized PINK1 on the OMM and thereby induced Parkin translocation and activation to promote mitophagy.
GeB attenuates brain ischemia/reperfusion (I/R) injury in vivo
Several studies have suggested that Parkin-dependent mitophagy is involved in cerebral I/R-induced injury [38, 39]. Our above results indicate that GeB promoted PINK1/Parkin-mediated mitophagy. Therefore, we studied its potential protective effects in the transient MCAO model, which is a well-established model for studying ischemia/reperfusion in vivo [40]. GeB was injected i.p. at the indicated concentrations 2 h before or 1 h after the operation (Fig. 5a).
Fig. 5. GeB attenuates brain ischemia-reperfusion (I/R) injury in vivo.

a The animal model of ischemia-reperfusion (I/R) injury. GeB was injected at the indicated concentrations 2 h before or 1 h after the operation (n = 5 mice/group). b, c Mouse brain slices were stained with TTC, and the infarcted areas were analyzed by Photoshop software (n = 5 mice/group); *P < 0.05. d, e The protein levels of Tom20 and Parkin were analyzed by Western blotting (n = 5 mice/group); *P < 0.05. f, g TUNEL staining of mouse brains (n = 5 mice/group); *P < 0.05.
Infarct volume is one of the most common indexes used to assess the extent of ischemic brain injury following focal cerebral ischemia [40]. Therefore, 24 h after the designated treatments, the brain infarct volume was measured by the TTC assay. As shown in Fig. 5b, the high-dose GeB (40 mg/kg) group exhibited a significantly reduced infarct volume compared with that of the MCAO group. Interestingly, pretreatment with GeB was more effective in reducing the infarct volume than treatment at 1 h after reperfusion (Fig. 5b, c).
To further investigate whether GeB activates mitophagy in ischemic brain injury, we measured the protein levels of Tom20 in vivo. Western blot analysis showed that treatment with GeB before or after I/R significantly reduced Tom20 levels, suggesting that GeB increased mitophagy in vivo (Fig. 5d, e). In addition, cerebral I/R injury led to an increase in TUNEL fluorescence staining, indicating that apoptosis was increased in the affected regions of the brain. After GeB pretreatment, however, this TUNEL staining intensity was significantly decreased (Fig. 5f, g), suggesting that GeB can exert protective effects against brain I/R injury in mice by preventing apoptosis. Overall, these results indicate that GeB attenuated brain I/R injury via mitophagy and antiapoptotic mechanisms.
Discussion
Damaged mitochondria contribute to the production of ROS and a decrease in the mitochondrial membrane potential, which is closely associated with Parkinson’s disease, cancer, stroke, and other diseases [3]. The clearance of damaged mitochondria via mitophagy is an important mechanism for maintaining cellular homeostasis [41]. Mitophagy has been studied intensively during the past decade. However, few compounds involved in mitophagy regulation have been identified [42]. In this study, we identified a novel compound, GeB, which can stabilize PINK1 on the OMM and then recruit and activate Parkin, thereby promoting mitophagy. Importantly, GeB possesses protective effects against brain I/R injury.
Recently, increasing attention has been focused on the discovery of natural compounds extracted from traditional medicinal herbs that can protect against cerebral ischemia. Various compounds that have been identified to protect against ischemic brain injury have been shown to be associated with their function in mitophagy upregulation. For example, esculetin is a natural compound that alleviates cognitive impairments in transient cerebral ischemia and reperfusion via upregulation of mitophagy [43]. Tunicamycin and thapsigargin protect against transient ischemic brain injury through Parkin-dependent mitophagy [44]. Such protective effects of tunicamycin and thapsigargin are largely attenuated by treatment with the mitophagy inhibitor mdivi-1 [45], indicating the importance of mitophagy in protection against ischemic brain injury. Consistent with these findings, the results of our study also showed that GeB can effectively induce mitophagy (Fig. 1). In this study, we showed that GeB treatment strikingly increases the PINK1 level, leading to Parkin recruitment and activation and subsequent mitophagy induction. To date, it has been reported that the PINK1 protein level can be regulated via the following pathways: (a) NRF2- or FOXO3a-dependent transcriptional upregulation of PINK1 as an antioxidative stress response [46, 47] and (b) PINK1 import via the Tim23 complex and its proteolytic destabilization by PARL [10]. It would be interesting to test whether GeB regulates PINK1 levels through these pathways. In addition, unbiased proteomics analysis with biotin-labeled GeB is necessary to identify the potential targets of GeB that contribute to PINK1 stabilization in future studies.
Under the conditions of I/R, the restoration of blood flow can cause damage to ischemic tissue through ROS accumulation and cell death with inflammatory responses [48]. Dysfunctional mitochondria resulted from I/R not only generate toxic ROS but also trigger the endogenous apoptosis pathway, which is associated with I/R injury [49]. Emerging evidence suggests that mitophagy can remove damaged mitochondria and play a vital role in protection against cerebral ischemia [18, 21, 38]. The PINK1/Parkin pathway is one of the most important pathways in mitophagy and is involved in the removal of dysfunctional mitochondria during cerebral I/R injury [44, 50]. Several studies have indicated that the activated NLRP3 inflammasome and its related cytokines significantly contribute to cerebral I/R injury [48, 51], while mitophagy has been shown to inhibit the NLRP3 inflammasome and hence relieve cerebral I/R injury [52]. Conversely, inhibition of mitophagy promotes cerebral I/R injury [53]. These studies are consistent with our findings, which suggest that mitophagy is a protective mechanism against I/R injury. However, the exact mechanism(s) by which mitophagy attenuates I/R injury are not fully understood, and further investigation is needed in the future.
In summary, we identified a new xanthone compound, GeB, which can effectively activate mitophagy in a Parkin-PINK1-dependent manner and inhibit apoptosis, which contributes to protection against ischemia-reperfusion injury (Fig. 6). The results of our study thus provide valuable information on the potential application of xanthones in future drug developments against ischemia reperfusion-induced brain injury.
Fig. 6. Schematic representation of the protective effects of GeB against ischemia-reperfusion injury.
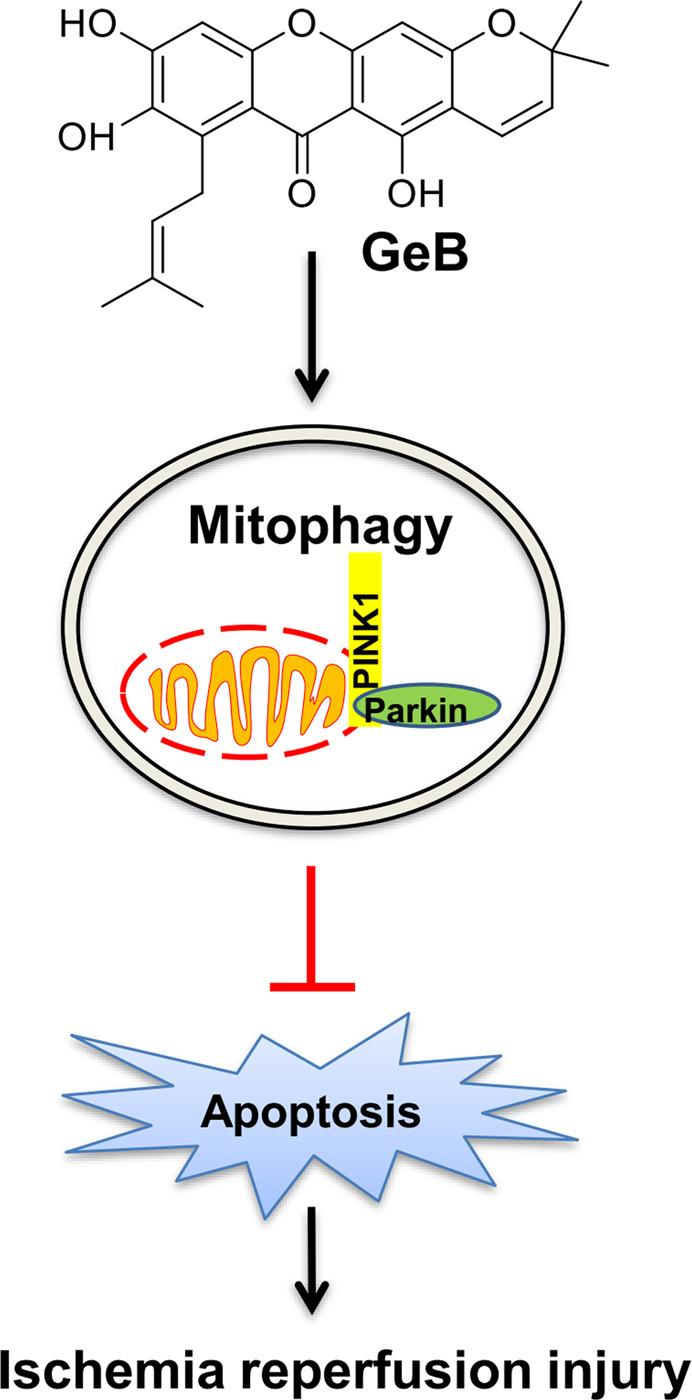
GeB induces PINK1/Parkin-dependent mitophagy, which contributes to the suppression of apoptosis caused by ischemia-reperfusion-induced stress. This is believed to be the mechanism by which GeB protects against ischemia reperfusion injury.
Supplementary information
Acknowledgements
We are grateful to Richard J. Youle for providing the YFP-Parkin HeLa cells and plasmids. This work was supported by the National Natural Science Foundation of China (Nos. 81773951 and 81303188), the National Major Scientific and Technological Special Project for “Significant New Drugs Development” in 2018 (No. 2019ZX09301140), Key-Area Research and Development Program of Guangdong Province (2020B1111110003), the Three-Year Development Plan Project for Traditional Chinese Medicine (ZY(2018–2020)-CCCX-2001-02) to HXX and research grants from NMRC (NMRC/CIRG/1430/2015) to HMS.
Author contributions
MW and GL performed the experiments and wrote the original draft; ZQZ, JY, and QX analyzed the data; YZL, HMS, and HXX designed the research and edited the paper; HZ and DZ extracted the compounds; LMW, HST, and HZ reviewed and edited the paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Man Wu, Guang Lu, Yuan-zhi Lao
Contributor Information
Han-ming Shen, Email: phsshm@nus.edu.sg.
Hong-xi Xu, Email: xuhongxi88@gmail.com.
Supplementary information
The online version of this article (10.1038/s41401-020-0480-9) contains supplementary material, which is available to authorized users.
References
- 1.Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426–31. doi: 10.1016/j.bbrc.2016.11.088. [DOI] [PubMed] [Google Scholar]
- 2.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Magalhaes J, Venditti P, Adhihetty PJ, Ramsey JJ, Ascensao A. Mitochondria in health and disease. Oxid Med Cell Longev. 2014;2014:814042. doi: 10.1155/2014/814042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–80. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 5.Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–15. doi: 10.1016/j.redox.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts RF, Tang MY, Fon EA, Durcan TM. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int J Biochem Cell Biol. 2016;79:427–36. doi: 10.1016/j.biocel.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 7.Jomova K, Valko M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharmacol Des. 2011;17:3460–73. doi: 10.2174/138161211798072463. [DOI] [PubMed] [Google Scholar]
- 8.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–42. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–80. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 12.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Y, Zhang X, Zheng Y, Chen Z. Regulation of mitophagy in ischemic brain injury. Neurosci Bull. 2015;31:395–406. doi: 10.1007/s12264-015-1544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Lievens JC. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015;6:e1617. doi: 10.1038/cddis.2014.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. 2015;24:2938–51. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R, et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet. 2005;13:1086–93. doi: 10.1038/sj.ejhg.5201455. [DOI] [PubMed] [Google Scholar]
- 17.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–33. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 19.Tang YC, Tian HX, Yi T, Chen HB. The critical roles of mitophagy in cerebral ischemia. Protein Cell. 2016;7:699–713. doi: 10.1007/s13238-016-0307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Q, Luo CL, Tao LY. Dynamin-related protein 1 (Drp1) mediating mitophagy contributes to the pathophysiology of nervous system diseases and brain injury. Histol Histopathol. 2017;32:551–9. doi: 10.14670/HH-11-841. [DOI] [PubMed] [Google Scholar]
- 21.Shen Z, Zheng Y, Wu J, Chen Y, Wu X, Zhou Y, et al. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy. 2017;13:473–85. doi: 10.1080/15548627.2016.1274596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Feng J, Kong S, Wu M, Xi Z, Zhang B, et al. Nujiangexathone A, a novel compound from Garcinia nujiangensis, suppresses cervical cancer growth by targeting hnRNPK. Cancer Lett. 2016;380:447–56. doi: 10.1016/j.canlet.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Ding Z, Lao Y, Zhang H, Fu W, Zhu L, Tan H, et al. Griffipavixanthone, a dimeric xanthone extracted from edible plants, inhibits tumor metastasis and proliferation via downregulation of the RAF pathway in esophageal cancer. Oncotarget. 2016;7:1826–37. doi: 10.18632/oncotarget.6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SN, Li Q, Jing MH, Alba E, Yang XH, Sabate R, et al. Natural xanthones from Garcinia mangostana with multifunctional activities for the therapy of Alzheimer’s disease. Neurochem Res. 2016;41:1806–17. doi: 10.1007/s11064-016-1896-y. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Xia Z, Xu JR, Wang YX, Hou LN, Qiu Y, et al. α-Mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates β-amyloid oligomers-induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology. 2012;62:871–81. doi: 10.1016/j.neuropharm.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Cho YL, Tang Y, Wang J, Park JE, Wu Y, et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res. 2018;28:787–802. doi: 10.1038/s41422-018-0056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26:733–44. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210–22. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 29.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 30.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–37. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, et al. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A. 2015;112:6637–42. doi: 10.1073/pnas.1506593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–39. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–60. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–6. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 37.Kazlauskaite A, Martinez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015;16:939–54. doi: 10.15252/embr.201540352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X, Li X, Liu Y, Yuan N, Li C, Kang Z, et al. Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway. Brain Res. 2018;1698:89–98. doi: 10.1016/j.brainres.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 39.Feng J, Chen X, Guan B, Li C, Qiu J, Shen J. Inhibition of peroxynitrite-induced mitophagy activation attenuates cerebral ischemia-reperfusion injury. Mol Neurobiol. 2018;55:6369–86. doi: 10.1007/s12035-017-0859-x. [DOI] [PubMed] [Google Scholar]
- 40.Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445–54. doi: 10.2147/DDDT.S56071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ivankovic D, Chau KY, Schapira H, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136:388–402. doi: 10.1111/jnc.13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol. 2017;13:136–46. doi: 10.1038/nchembio.2287. [DOI] [PubMed] [Google Scholar]
- 43.Xu B, Zhu L, Chu J, Ma Z, Fu Q, Wei W, et al. Esculetin improves cognitive impairments induced by transient cerebral ischemia and reperfusion in mice via regulation of mitochondrial fragmentation and mitophagy. Behav Brain Res. 2019;372:112007. doi: 10.1016/j.bbr.2019.112007. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801–13. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M. NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS One. 2015;10:e0142438. doi: 10.1371/journal.pone.0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci U S A. 2009;106:5153–8. doi: 10.1073/pnas.0901104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016;2016:2183026. doi: 10.1155/2016/2183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West AP. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology. 2017;391:54–63. doi: 10.1016/j.tox.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 50.Lan R, Wu JT, Wu T, Ma YZ, Wang BQ, Zheng HZ, et al. Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the PINK1/Parkin/p62 signaling pathway. Brain Res Bull. 2018;142:63–77. doi: 10.1016/j.brainresbull.2018.06.018. [DOI] [PubMed] [Google Scholar]
- 51.Gong Z, Pan J, Shen Q, Li M, Peng Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation. 2018;15:242. doi: 10.1186/s12974-018-1282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Q, Li Z, Meng C, Wu J, Zhao Y, Zhao J. Parkin-dependent mitophagy is required for the inhibition of ATF4 on NLRP3 inflammasome activation in cerebral ischemia-reperfusion injury in rats. Cells. 2019;8:897. doi: 10.3390/cells8080897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z, Yu J. NR4A1 promotes cerebral ischemia reperfusion injury by repressing mfn2-mediated mitophagy and inactivating the MAPK-ERK-CREB signaling pathway. Neurochem Res. 2018;43:1963–77. doi: 10.1007/s11064-018-2618-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.