

Abstract
Bacterial type IV secretion systems (T4SSs) are a functionally diverse translocation superfamily. They consist mainly of two large subfamilies: (i) conjugation systems that mediate interbacterial DNA transfer and (ii) effector translocators that deliver effector macromolecules into prokaryotic or eukaryotic cells. A few other T4SSs export DNA or proteins to the milieu, or import exogenous DNA. The T4SSs are defined by 6 or 12 conserved “core” subunits that respectively elaborate “minimized” systems in Gram-positive or -negative bacteria. However, many “expanded” T4SSs are built from “core” subunits plus numerous others that are system-specific, which presumptively broadens functional capabilities. Recently, there has been exciting progress in defining T4SS assembly pathways and architectures using a combination of fluorescence and cryoelectron microscopy. This review will highlight advances in our knowledge of structure–function relationships for model Gram-negative bacterial T4SSs, including “minimized” systems resembling the Agrobacterium tumefaciens VirB/VirD4 T4SS and “expanded” systems represented by the Helicobacter pylori Cag, Legionella pneumophila Dot/Icm, and F plasmid-encoded Tra T4SSs. Detailed studies of these model systems are generating new insights, some at atomic resolution, to long-standing questions concerning mechanisms of substrate recruitment, T4SS channel architecture, conjugative pilus assembly, and machine adaptations contributing to T4SS functional versatility.
Keywords: conjugation, cryoelectron microscopy, cryoelectron tomography, effector translocation, pilus
1 |. INTRODUCTION
Many bacterial species deploy Type IV Secretion Systems (T4SSs) to deliver DNA, protein, or other macromolecules to bacterial or eukaryotic cell targets (Li et al., 2019; Waksman, 2019). The T4SSs are composed mainly of two subfamilies, the conjugation systems and effector translocators (Cascales and Christie, 2003). Conjugation systems are of considerable medical concern for their roles in dissemination of mobile genetic elements (MGEs), often encoding resistance to heavy metals or antibiotics (Cabezon et al., 2015; Huddleston, 2014; Koraimann, 2018). The effector translocators mainly deliver proteins to eukaryotic target cells, although recent studies have also documented the interkingdom transfer of DNA, peptidoglycan, or other macromolecules (Bleves et al., 2020; Cover et al., 2020; Grohmann et al., 2018). Many effector translocator systems are integral to the virulence of Gram-negative pathogens, and there is recent evidence for their deployment by Gram-positive pathogens (Grohmann et al., 2018; Jiang et al., 2016). Conjugation systems and effector translocators typically require direct cell-to-cell contact; however, a few T4SSs function independently of target cell interactions to import exogenous DNA or export DNA or proteins to the milieu (Alvarez-Martinez and Christie, 2009; Koch et al., 2020; Stingl et al., 2010).
Recently, there has been exciting progress in defining the architectures and assembly pathways of the T4SSs. Structural studies of T4SSs began about 20 years ago with reports of X-ray structures of a few highly conserved T4SS subunits (Gomis-Ruth et al., 2001; Savvides et al., 2003; Terradot et al., 2005; Yeo et al., 2000). In the ensuing 10 years, larger (~1 megadalton; MDa) subassemblies from the R388 and pKM101 plasmid conjugation systems were solved by single-particle electron microscopy and crystallography (Chandran et al., 2009; Fronzes et al., 2009). In just the past few years, however, advances in electron microscopy have enabled visualization of larger subassemblies in isolation (Low et al., 2014; Rivera-Calzada et al., 2013) and of intact T4SSs in the native context of the bacterial cell envelope (Chetrit et al., 2018; Ghosal et al., 2017; Ghosal et al., 2019; Hu et al., 2019a; Hu et al., 2019b). These approaches, coupled with state-of-the-art fluorescence microscopy, have yielded new insights into T4SS assembly dynamics and spatial organization in intact cells (Chetrit et al., 2018; Ghosal et al., 2019; Jeong et al., 2017; Park et al., 2020). In this review, we will summarize the recent progress in structural definition of several model T4SSs functioning in Gram-negative bacteria, including the Xanthomonas spp. VirB/VirD4, Helicobacter pylori Cag, Legionella pneumophila Dot/Icm, and F plasmid-encoded Tra systems (Figure 1). At the outset, we acknowledge that a full understanding of the structural and functional diversity of T4SSs will require continued study of systems not covered in this review, such as those functioning in Gram-positive bacteria or obligate intracellular pathogens, machines that acquire substrates from the periplasm, and systems adapted for macromolecular import or export (Bhatty et al., 2013; Bleves et al., 2020; Grohmann et al., 2018).
FIGURE 1.
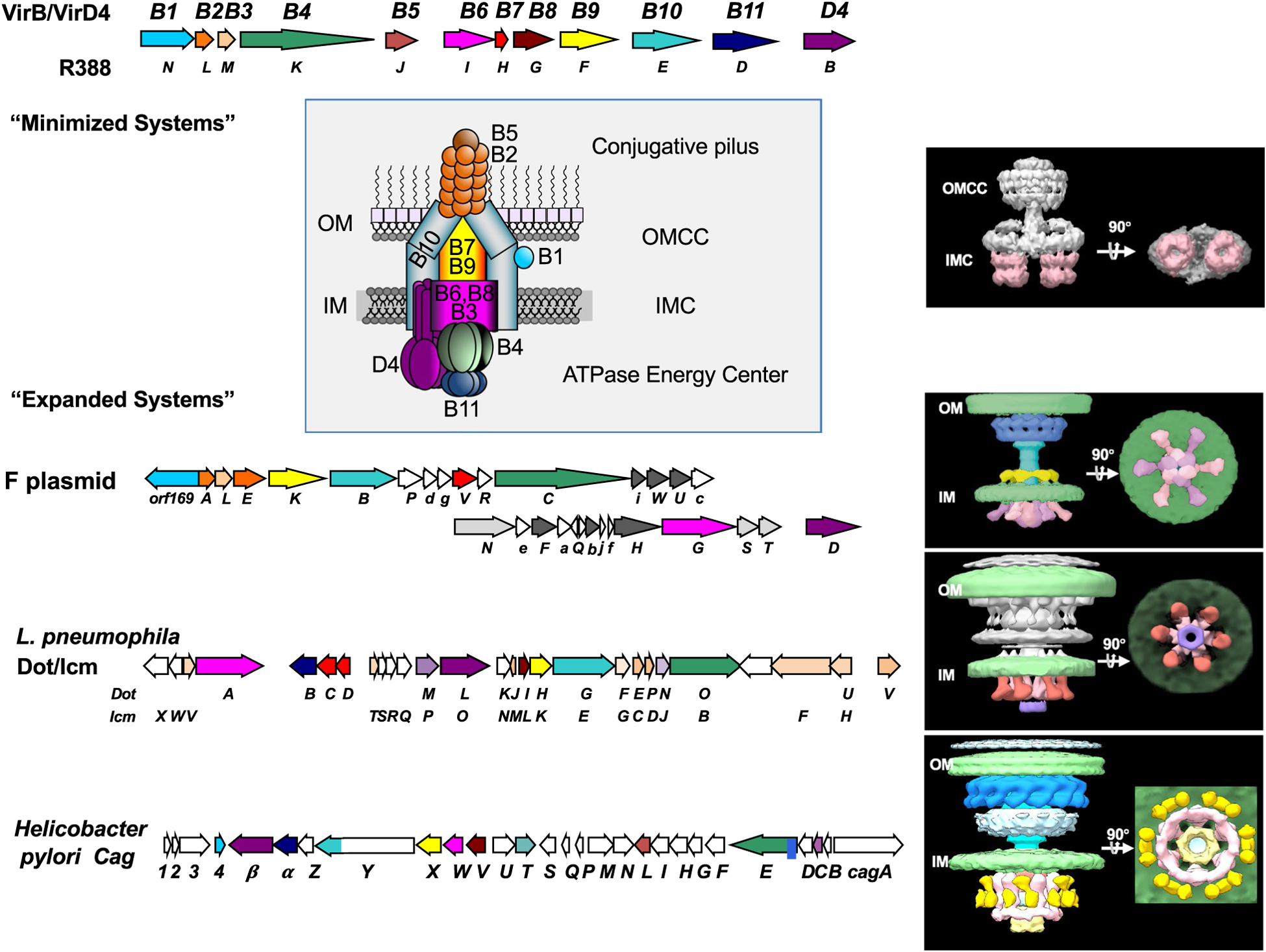
Structures of T4SSs solved to date. Upper: Operon arrangements of the A. tumefaciens VirB/VirD4 and R388 plasmid-encoded T4SSs, two representative “minimized” systems functioning in Gram-negative species. These systems are assembled from T4SS signature subunits including the 11 VirB subunits and the VirD4 receptor or type IV coupling protein (T4CP). The cartoon depicts locations of the outer membrane core complex (OMCC) and inner membrane complex (IMC) with the associated VirB/VirD4 subunits indicated. Right: Corresponding 3D reconstruction with side and bottom views at 90° angles of the R388-encoded substructure composed of the VirB3 ‒ VirB10 subunits. The substructure was visualized by single-particle negative-stain electron microscopy (EMD-2567); the two side-by-side hexameric barrels of the VirB4 ATPase are highlighted (pink shading). Lower: Operon arrangements of “expanded” systems represented by the F plasmid-encoded Tra, L. pneumophila Dot/Icm, and H. pylori Cag T4SSs. These systems are assembled from the VirB/VirD4 signature subunits (color-coded) plus system-specific subunits (no or gray shading). Right: The F plasmid-encoded Tra (F1-Channel complex, EMD-9344, EMD-9347), L. pneumophila Dot/Icm (EMD-7611, EMD-7612), and H. pylori Cag (EMD-0634, EMD-0635) structures as visualized in the bacterial cell envelope by in situ CryoET. In each of these T4SSs, the VirB4 ATPase presents as a central hexamer of dimers (pink/red-shaded). In the Dot/Icm and Cag machine, the VirB11 homologs DotB (purple-shaded) and Cagα (light yellow shading) dock at the base of the VirB4 homologs DotL and CagE, respectively. In the Cag machine, VirD4-like Cagβ contributes to peripheral densities (yellow and pink)
1.1 |. T4SS substrates and translocation signals
The T4SSs are minimally composed of a set of conserved subunits (Alvarez-Martinez and Christie, 2009; Grohmann et al., 2018). In Gram-negative bacteria, ~12 subunits are needed to build fully functional “minimized” systems (Bhatty et al., 2013). These are designated VirB1 through VirB11 and VirD4, according to a unifying nomenclature for the T4SS superfamily from the paradigmatic Agrobacterium tumefaciens VirB/VirD4 T4SS (Figure 1) (Alvarez-Martinez and Christie, 2009). Three ATPases (VirD4, VirB4, and VirB11) comprise the cytoplasmic energy center, which is situated at the base of the translocation channel. Of these ATPases, VirD4 serves as the receptor to which both DNA and protein substrates bind prior to entry into the translocation channel (Alvarez-Rodriguez et al., 2020b; Llosa and Alkorta, 2017). The channel itself consists of two large subassemblies, one spanning the inner membrane (IM) and the second situated in the periplasm and outer membrane (OM) (Christie et al., 2005; Low et al., 2014). The IM complex (IMC) consists minimally of VirB3, VirB6, VirB8, and the N-terminal region of VirB10 (Low et al., 2014). The VirB4 ATPase, which stably associates with the channel is also considered to be part of the IMC (Bleves et al., 2020; Low et al., 2014). The IMC connects via a stalk or cylinder to the outer membrane core complex (OMCC), which consists minimally of the lipoprotein VirB7, VirB9, and a C-terminal domain of VirB10 (Christie et al., 2005; Low et al., 2014).
The T4SS channel delivers substrates across the cell envelope, but it must first recruit these substrates to the cytoplasmic entrance. Recruitment of MGEs to cognate conjugation or “mating” channels is initiated by a set of processing factors termed DNA transfer and replication (Dtr) proteins, which bind the origin-of-transfer (oriT) sequence to form the relaxosome (Cabezon et al., 2015). The relaxosome, specifically the relaxase component, nicks the DNA strand destined for transfer (T-strand) and remains covalently bound to its 5′ end usually via a catalytic Tyr residue (Guzman-Herrador and Llosa, 2019). Recently, the interaction of the F plasmid-encoded TraI relaxase to its oriT target sequence was solved at atomic resolution (Ilangovan et al., 2017; Waksman, 2019). Notably, TraI harbors two translocation signals (TSs) similar in sequence to TSs carried by relaxases functioning in R388, R6112, and pKM101 plasmid transfer (Alperi et al., 2013; Ilangovan et al., 2017; Meyer, 2015; Redzej et al., 2013). These signals, designated TSA and TSB (Translocation Signals A and B), are not positioned at the N or C termini of F-encoded TraI or the related relaxases, as is characteristically the case for secreted proteins (Christie, 2019). Rather, the two TSs map to the C-proximal helicase domain, specifically, within the 2B/2B-like subdomains of an SF1A/B helicase structural fold (Ilangovan et al., 2017). These TSs likely contribute to binding of the relaxosome to cognate VirD4 receptors, also termed type IV coupling proteins (T4CPs) (Alvarez-Rodriguez et al., 2020b; Llosa and Alkorta, 2017), but details of these interactions are not yet available.
Contacts between other Dtr factors and cognate VirD4 subunits are currently better understood, in one case at atomic resolution. In the F system, the Dtr factor TraM binds the F plasmid oriT sequence and recruits the TraI relaxase to build the relaxosome (Lu et al., 2008; Wong et al., 2012). A crystal structure revealed that TraM is a member of the Ribbon–Helix–Helix (RHH) superfamily of DNA-binding proteins and that the RHH domain binds-specific sequences (sbm) within the oriT sequence (Peng et al., 2014; Wong et al., 2011). Upon binding, TraM induces a bend in oriT to allow access of the TraI relaxase to the nic site (Wong et al., 2011, 2012). TraM also has a C-terminal α-helical domain, which is involved inhomotetramerization, and it also specifically binds the C-terminal 13 residues of the VirD4-like receptor TraD (Lu et al., 2008). The TraM–TraD interaction thus forms a basis for recruitment of the cognate F plasmid to the F-encoded Tra (hereafter designated TraF) T4SS (Lu et al., 2008). Many conjugation systems of both Gram-negative and -positive species encode Dtr factors belonging to the RHH superfamily (Li and Christie, 2020; Rehman et al., 2019; Varsaki et al., 2009; Yoshida et al., 2008). Interestingly, however, deletion of the C-terminal helical domains of two such factors, TraK and PcfF, does not abolish transfer of pKM101 and pCF10, respectively, establishing that the RHH domain suffices for relaxosome assembly and at least a low level of substrate transfer (Li and Christie, 2020; Rehman et al., 2019). Taken together with other findings (see de la Cruz et al., 2010; Llosa and Alkorta, 2017), the current picture is that numerous relaxosome constituents, including the relaxase, Dtr factors, and even translocated DNA, must bind the VirD4 T4CP for specific and efficient conjugative DNA transfer.
As we await further details of relaxosome–VirD4 interactions, there has been important progress toward definition of TSs conferring recognition of protein substrates by effector translocator systems. Early on, two types of TSs were identified at the extreme C terminus, one composed of clusters of positively charged and a second of hydrophobic residues (Nagai et al., 2005; Vergunst et al., 2000, 2005). Many effectors carry such TSs, but in fact there is considerably variability among TSs recognized by different effector translocators, as illustrated by the following. First, in 2015, C. Farah and colleagues discovered a subfamily of T4SSs in Xanthomonas citri that promotes killing of neighboring bacteria through the translocation of protein toxins (Souza et al., 2015). All of these effector toxins carry C-proximal TSs consisting of a ~120 residue motif termed XVIPCD (Xanthomonas VirD4-interacting protein conserved domain) (Sgro et al., 2019). As implied by the name, effectors of the X. citri VirB/VirD4 T4SS were originally identified in screens for proteins that bind VirD4. As a result of bioinformatics screens, however, the list of candidate effectors bearing XVIPCD motifs exceeds several hundred; this list is dispersed among many species in the order Xanthomonadales and other proteobacteria (Sgro et al., 2019). The XVIPCD is characterized by a few conserved motifs in the N-terminal region and a Gln-rich C-terminal region, but the nature of the effector–VirD4 interaction is not yet specified.
Second, in Bartonella spp., effector proteins termed Beps (Bartonella effector proteins) are delivered through a VirB/VirD4 T4SS into eukaryotic cells during infection. Beps have a C-proximal TS termed the BID (Bep intracellular delivery) domain (Siamer and Dehio, 2015). BID domains are sequence-variable but adopt a conserved structural fold consisting of an extended four-helix bundle, which is intriguingly reminiscent of the C-terminal α-helical domains carried by TraM and other Dtr proteins discussed above (Stanger et al., 2017; Wagner et al., 2019). This structural fold is also implicated in VirD4 binding, yet, interestingly a given effector can have multiple BID domains distributed throughout the Bep. Furthermore, BID domains can confer effector function in the eukaryotic host cell by binding and altering protein functions (Siamer and Dehio, 2015; Stanger et al., 2017). The BID1 domain of BepA, for example, enhances proliferation of human endothelial cells by inhibiting apoptosis through binding of the catalytic subunit C2 of human adenylyl cyclase to potentiate cAMP production (Stanger et al., 2017). At least some BID domains thus have evolved dual roles as TSs for recognition by VirD4 receptors and as effectors through binding of eukaryotic cell target(s) to aid in infection.
Finally, early studies of the L. pneumophila Dot/Icm T4SS identified a number of translocated effectors that harbor C-terminal TSs composed of short polar and negatively charged amino acids (Burstein et al., 2009; Kubori et al., 2008; Nagai et al., 2005). A combination of bioinformatics and experimental approaches further identified at least 100 Dot/Icm effectors bearing clusters of glutamate residues (the E block motif) within 17 to 10 residues from the C terminus and one or more hydrophobic amino acids nearer the terminus (Huang et al., 2011). However, translocation of some effectors with the E block motif, as well as other effectors lacking this motif, can be modulated positively or negatively by adaptor proteins such as IcmS and IcmW (Burstein et al., 2016; Cambronne and Roy, 2007; Lifshitz et al., 2013). Collectively, these findings led to a proposal that Dot/Icm effectors engage with the T4SSDot/Icm in at least one of three ways, via the E block motif, through a combination of this motif and binding of adaptors, or by binding adaptors and possibly a TS other than the E block motif (Lifshitz et al., 2013). Exciting advances discussed further below have now described the structural bases underlying recruitment of potentially large numbers of effectors to the T4SSDot/Icm channel via specific contacts with one or more adaptor proteins.
1.2 |. VirD4 receptors and substrate docking
VirD4 receptors are associated with nearly all T4SSs; the few systems that lack VirD4 subunits appear to function exclusively in elaboration of antigenically variable pili or to export substrates recruited to the T4SS from the periplasm (Alvarez-Martinez and Christie, 2009; Llosa and Alkorta, 2017). R388-encoded TrwB is the structural archetype for the T4CP superfamily (Gomis-Ruth et al., 2001). On the basis of TrwB’s structure and other biochemical evidence, the VirD4 receptors are thought to assemble as homohexamers with an N-terminal transmembrane domain (NTD), a conserved cytosolic nucleotide-binding domain (NBD), and a sequence-variable all-alpha-domain (AAD) implicated in substrate binding (Gomis-Ruth and Coll, 2001; Llosa and Alkorta, 2017; Whitaker et al., 2015). Some T4CPs are considerably larger than TrwB (507 residues) due to the presence of sequence-variable extensions at one or both termini (Alvarez-Martinez and Christie, 2009; Whitaker et al., 2016). DotL associated with the L. pneumophila T4SSDot/Icm is presently the best-characterized representative of these larger receptors. DotL has a ~200-residue C-terminal domain (CTD) that binds the adaptor proteins IcmS and IcmW required for translocation of a subset of effectors through the T4SSDot/Icm (Bardill et al., 2005; Ninio et al., 2005; Sutherland et al., 2012; Vincent et al., 2012). In the last 3 years, significant advances were made in defining the roles of adaptors and DotL’s CTD to effector recruitment. First, two groups reported crystal structures of DotL’s CTD in complex with IcmS and IcmW and two other proteins, DotN and LvgA (Kwak et al., 2017; Xu et al., 2017). DotN, IcmW, IcmS, and LvgA bind successively from the N to C terminus of the CTD. In a pseudo-atomic model, the DotL holocomplex thus consists of a TrwB-like homohexamer joined to a larger bell-shaped CTD/adaptor complex, which was designated the substrate receptor or recognition module (Kwak et al., 2017). In the context of this model, most or all effector proteins are recruited to the DotL T4CP through contacts with distinct adaptor proteins.
Indeed, results of photocrosslinking assays confirmed effector contacts with an elongated hydrophobic surface of the IcmS/IcmW heterodimer (Xu et al., 2017). Furthermore, two groups have now reported crystal structures of DotL/adaptor–effector complexes (Kim et al., 2020; Meir et al., 2020). In one study, atomic structures were presented for the DotL/IcmS/IcmW/LvgA complex bound to the effectors VpdB, SetA, PieA, or SidH. Notably, a ~130 residue C-terminal fragment of VpdB is composed of 5 α-helices of which α1 forms specific and extensive contacts with a hydrophobic pocket of LvgA and α2– α5 form a four-helix bundle that weakly binds LvgA (Kim et al., 2020). C-terminal motifs of SetA, PieA, and SidH similarly bound the hydrophobic pocket of LvgA. An FxxxLxxxK motif was identified in α-helices of VpdB and SidH (but not SetA or PieA) that fitted into the hydrophobic pocket of LvgA. In a bioinformatics screen, 257 out of 2,930 proteins encoded by L. pneumophila strain Philadelphia-1 carry this motif, and of these 46 belong to the known effector repertoire of the T4SSDot/Icm (Kim et al., 2020). This FxxxLxxxK motif thus might constitute a previously unidentified TS for a large subset of Dot/Icm effectors, which mediates-specific contacts with LvgA.
In the second study, purification of the DotL/receptor complex yielded DotL bound to DotN, IcmS, IcmW, and LvgA, as well as to DotM and two previously unknown Dot proteins, DotY and DotZ (Meir et al., 2020). Modeling yielded an updated structure for the DotL homohexamer/receptor module and supplied new insights into the mechanism of recruitment of a second subset of effectors that are dependent on DotM and not IcmS, IcmW, or LvgA for translocation (Meir et al., 2018). These effectors carry the E block motif described above, and modeling of E block contacts with DotM revealed a structural basis for effector engagement with the DotL/receptor complex (Meir et al., 2020). Together, these recent findings have significantly advanced our understanding of how the T4SSDot/Icm channel recruits a large number of effectors bearing distinct TSs for translocation.
Upon binding of DNA or protein secretion substrates, how does the VirD4 receptor mediate translocation across the inner or cytoplasmic membrane? In fact, this remains one the most poorly understood aspects of type IV secretion. According to one model, VirD4 binds and directly shuttles substrates to the periplasm where they enter the VirB channel for passage to the cell exterior (Larrea et al., 2017; Meir et al., 2020). Alternatively, VirD4 recruits substrates, but then coordinates with the VirB4 and VirB11 ATPases to process and deliver them through a VirB channel that spans the entire cell envelope (Atmakuri et al., 2004; Cascales and Christie, 2004b). There is experimental support for both models, and discriminating between them is complicated by the fact that no studies have yet visualized an intact T4SS comprised of a VirD4 hexamer bound to the VirB channel. In fact, dimeric or unspecified forms VirD4 subunits were shown to bind cognate T4SSs (Hu et al., 2019b; Redzej et al., 2017), but it is not known whether these machines are competent for translocation. Indeed, there is considerable evidence that T4SSs undergo late-state assembly or structural transitions upon sensing of intracellular signals, such as substrate docking and ATP energy consumption, as well as extracellular signals such as target cell binding (see below and (Cascales et al., 2013; Cascales and Christie, 2004a; Lang et al., 2011; Li and Christie, 2020; Tato et al., 2007). Further definition of how such intra- or extracellular signals regulate channel assembly dynamics and substrate flow remain exciting areas for further exploration.
1.3 |. Structural and dynamic features of “minimized” systems
Recall that T4SSs in Gram-negative species consist of large OMCCs connected by stalks or cylinders to equally large IMCs. This current view of T4SS architecture arose from structures solved by G. Waksman and his colleagues of i) the OMCC associated with the pKM101-encoded Tra T4SS (T4SSpKM101) (Chandran et al., 2009; Fronzes et al., 2009; Rivera-Calzada et al., 2013) and ii) a nearly intact T4SS encoded by plasmid R388 (Figure 1) (Low et al., 2014). The OMCCs of both T4SSs adopt barrel-shaped structures of ~185 Å in width and height, and are composed of 14 copies each of the OM-associated VirB7 lipoprotein and VirB9 and the C-terminal region of VirB10. The barrel is composed of inner (I) and outer (O) layers, and an X-ray structure of the O-layer revealed an interior lining composed of VirB10 and an outer protective crown composed of VirB7 and VirB9 (Chandran et al., 2009). VirB10 has a two-helix bundle, originally termed the antennae projection (AP), and in the assembled OMCC the 14 APs form a helical channel that sits on top of the OMCC barrel and is presumed to span the OM. The I-layer consists of N-terminal domains of the 14 VirB9 subunits surrounding an elongated domain of VirB10, which extends to and across the IM (Jakubowski et al., 2009; Rivera-Calzada et al., 2013).
Remarkably, the IMC of the T4SSR388 nanomachine is highly asymmetric, with dimensions of 255 Å in width, 105 Å in thickness, and 134 Å in height. A prominent feature of the IMC is the presence of two side-by-side hexameric barrels of the VirB4 ATPase that extend from the IM into the cytoplasm. The IMC is composed of 12 copies of VirB3, VirB4, VirB5, VirB6, and VirB8, but 14 copies of the N-terminal region of VirB10; contacts between VirB10 and the rest of the IMC thus comprise the interface of a symmetry mismatch of possible functional importance. As noted above, an updated structure shows that two dimers of the VirD4 ATPase are integrated between the VirB4 hexamers (Redzej et al., 2017), although this likely corresponds to an incompletely assembled machine. The overall asymmetry of the T4SSR388 IMC is highly unusual among the known macromolecular translocation systems in bacteria (Christie, 2019), but at present is the only available structure for a purified IMC subassembly.
Recently, a structure of an OMCC from another “minimized” T4SS encoded by Xanthomonas citri T4SS (T4SSXan) was reported (Figure 2) (Sgro et al., 2018). Remarkably, the T4SSXan functions not in DNA transfer or delivery of effectors to eukaryotic cells but to translocate protein toxins to neighboring bacteria for niche establishment. Most VirB and VirD4 subunits of the T4SSXan closely resemble their counterparts in the A. tumefaciens or pKM101 systems. A prominent exception is the VirB7 lipoprotein, which is considerably larger (~14–20 kDa) than other VirB7 subunits (~4.5 kDa) due to the presence of a C-terminal N0 domain. Interestingly, N0 domains are prominent features of secretions associated with types II and III secretion systems, as well as the DotD lipoprotein associated with the T4SSDot/Icm (Nakano et al., 2010; Souza et al., 2011). In the X. citri OMCC, the N0 domains splay out from the central core of the OMCC, giving rise to an overall “flying saucer” shape, as opposed to the barrel shapes of the OMCCs from the R388, pKM101 and A. tumefaciens VirB/VirD4 systems (Figure 2) (Fronzes et al., 2009; Gordon et al., 2017; Low et al., 2014; Sgro et al., 2018). Other than the N0 extensions, the overall architecture of the OMCC from T4SSXan superimposes very well over that from the T4SSpKM101, and possesses characteristic features such as AP helical projections that likely form the OM channel. VirB10 also lines the interior and VirB9 forms the exterior of the O-layer, and VirB10 extends to and across the IM. Importantly, the entire Xanthomonas OMCC was solved at high resolution (3.28 Å), giving rise to the first atomic structure of the I-layer. Notably, this structure revealed the presence of linker regions between the domains of both VirB9 and VirB10, which allow for considerable flexibility and even the possibility of independent movement of the O- and I-layers.
FIGURE 2.
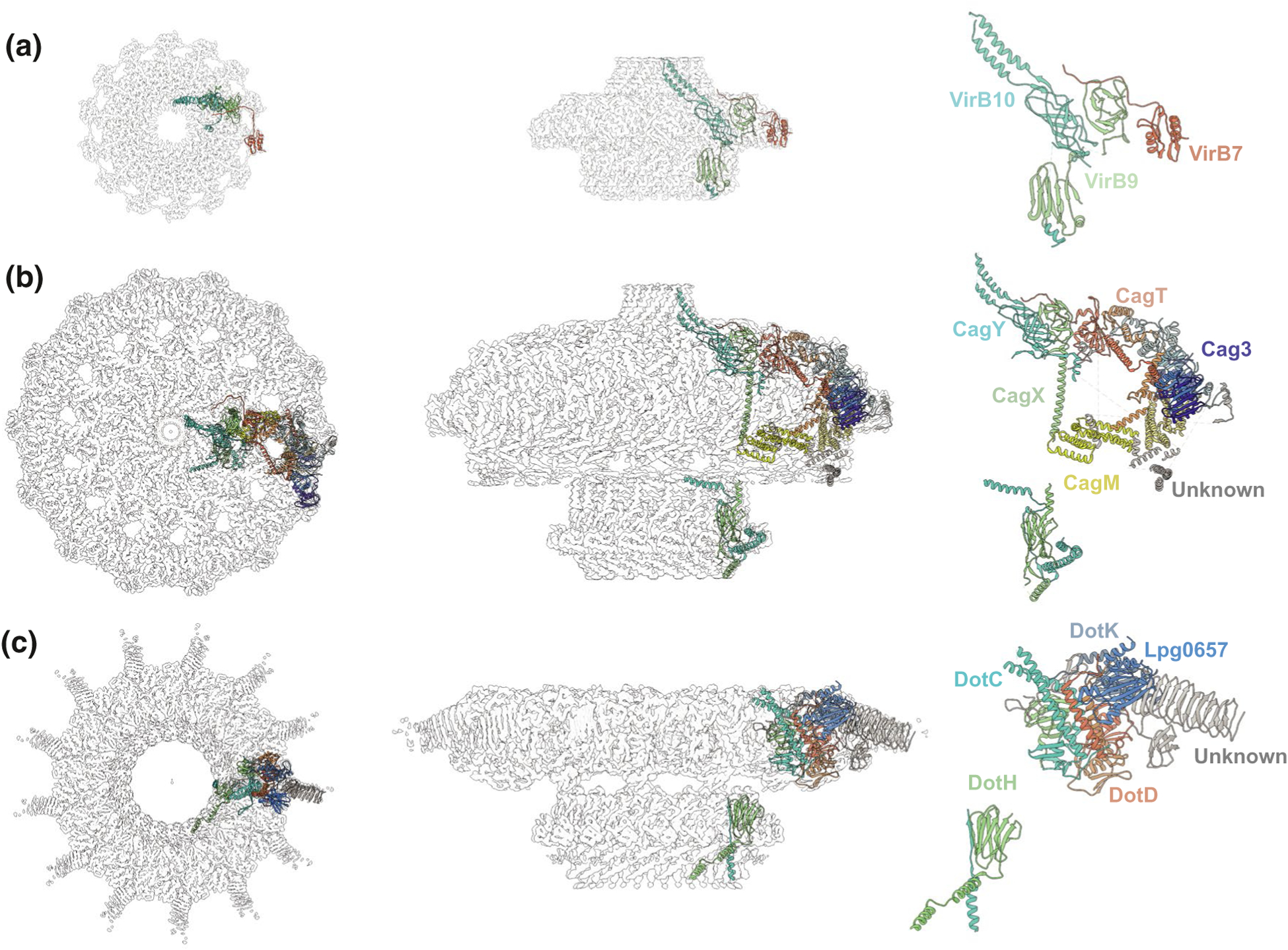
High-resolution CryoEM structures from different T4SS outer membrane complexes. (a) The X. citri CryoEM density map (EMDB-0089) resolved at 3.28 Å with 14-fold symmetry; the map is fitted with an asymmetric unit from PDB model 6gyb. (b) The H. pylori CryoEM density maps of the OMC and PR subdomains (OMC:EMD-22081 and PR:20021) resolved at 3.4 Å with 14-fold symmetry and 17-fold symmetry, respectively; the maps are fitted with asymmetric units from PDB models 6×6j and 6×6s. C) The L. pneumophila OMC disk (EMD-22068) resolved at 3.5 Å with 13-fold symmetry and the PR (EMD-22069) resolved at 3.7 Å with 18-fold symmetry, the maps are fitted with asymmetric units from PDB models 6×62 and 6×64. Locations of confirmed subunits are shown, with similar color-coding for the VirB7, VirB9, and VirB10 homologs or orthologs in each system
The importance of an intrinsic flexibility in OMCCs is underscored by early findings that the A. tumefaciens VirB/VirD4 T4SS is activated by sensing and transduction of signals that accompany substrate binding and ATP energy consumption by the VirD4 and VirB11 ATPases. In turn, the distal portion of VirB10 undergoes a conformational change required for substrate passage (Banta et al., 2011; Cascales et al., 2013; Cascales and Christie, 2004a). Recently, the notion that OMCCs, and specifically the VirB10 components, undergo dynamic conformational changes has gained further support from extended molecular dynamics simulations of the C-terminal domains of native VirB10 from A. tumefaciens and a mutant variant (G272R), which is locked in the energy-activated state (Banta et al., 2011; Darbari et al., 2020). Most interestingly, the ATP-insensitive open state conferred by the G272R mutation exhibits a more rigid conformation compared to the WT complex, consistent with a reduced conformational flexibility that might impact channel gating in response to perception of intracellular signals (Darbari et al., 2020). Comparisons of OMCC architectures from quiescent versus energy-locked T4SSs will yield a better understanding of the structural consequences of signal activation. However, a full accounting of T4SSs in their open states remains a formidable challenge, as contacts between T4SSs and target cell receptors also have been proposed to stimulate late-stage channel assembly or gating as a prerequisite for substrate transfer (Kwok et al., 2007; Lu and Frost, 2005).
1.4 |. Architecture , assembly,and spatial organization of “expanded” T4SSs
Remarkable progress has been made in structural definition of three “expanded” T4SSs, the H. pylori Cag, L. pneumophila Dot/Icm, and F plasmid-encoded T4SSs by a combination of single-particle CryoEM, in situ CryoET, and fluorescence microscopy approaches (Figures 1 and 2).
1.4.1 |. The H. pylori Cag T4SS (T4SSCag)
The T4SSCag is deployed by H. pylori strains to attach and translocate substrates into human gastric epithelial cells during infection (Cover et al., 2020). These substrates include not only the CagA “oncoprotein,” but also chromosomal DNA (Varga et al., 2016), peptidoglycan (Viala et al., 2004) and D-glycero-b-D-manno-heptose 1,7 bisphosphate (HBP), which is an LPS metabolite (Gall et al., 2017; Stein et al., 2017; Zimmermann et al., 2017). Translocation of CagA induces various changes in eukaryotic target cells, including but not limited to cytoskeletal alterations and changes in cell polarity and adhesion, disruption of cell–cell junctions and increased cell motility and cell migration (Backert and Tegtmeyer, 2017; Hatakeyama, 2014). Independently of CagA translocation and, curiously, VirD4-like Cagβ, the T4SSCag stimulates interleukin-8 (IL-8) induction (Fischer et al., 2001), and also activates the Toll-like receptor 9 (TLR9) through DNA translocation (Cover et al., 2020; Lin et al., 2020; Varga et al., 2016). T4SSCag-mediated translocation of HBP was implicated in activation of the NF-kB pathway, and HBP was thus designated as a new pathogen-associated molecular pattern or PAMP (Zimmermann et al., 2017). In a recent update, however, H. pylori was shown to produce HBP at levels too low to account for NF-kB activation. Instead, a derivative of HBP, ADP-glycero-β-D-manno-heptose (ADP heptose), was identified as the key PAMP responsible for H. pylori-induced NF-kB activation in human epithelial cells (Pfannkuch et al., 2019).
The H. pylori T4SSCag is unique among the known effector translocator systems in its capacity to elaborate extracellular pilus structures when cocultivated with human epithelial cells. The T4SSCag thus resembles the Gram-negative bacterial conjugation machines and the A. tumefaciens VirB/VirD4 T4SS, which produce conjugative pili to promote attachment (Johnson et al., 2014; Rohde et al., 2003; Shaffer et al., 2011). Some similarities between the Cag and conjugative pili have been noted; for example, CagL and its homolog, VirB5, both localize on the respective pilus and are implicated in target cell binding (Aly and Baron, 2007; Kwok et al., 2007). Curiously, however, homologs or orthologs of VirB subunits required for production of conjugative pili are dispensable for Cag pili, such as CagC and CagY, which are orthologs of the VirB2 pilin and VirB10 subunits, respectively (Johnson et al., 2014; Shaffer et al., 2011; Skoog et al., 2018). H. pylori also has been reported to elaborate large sheathed tube structures whose relationship to Cag pili is not yet defined (Chang et al., 2018; Rohde et al., 2003; Tanaka et al., 2003). Finally, the CagA secretion substrate has been localized to the tips of Cag pili (Jimenez-Soto et al., 2009; Kwok et al., 2007). Among the conjugation systems, by contrast, there is no direct evidence for association of DNA or protein substrates within or attached to conjugative pili (Cascales and Christie, 2004b). Thus, at this time, the genetic requirements, nature of association with the T4SSCag, and functional roles of Cag pili and sheathed tubes are still under study.
The Cag OMCC was amenable to extraction from H. pylori for high-resolution analysis (Figure 2) (Frick-Cheng et al., 2016). Strikingly, purification of the Cag OMCC utilized an epitope tag to the chaperone CagF, which is thought to stably associate with the cytoplasmic portion (IMC) of the Cag machine. Yet, the purified complex lacks CagF and the IMC, and instead consists of the OMCC built from VirB10-like CagY, VirB9-like CagX, VirB7-like CagT, and two Cag-specific subunits, Cag3 and CagM. The OMCC is a ring-shaped structure with dimensions of ~400 Å in width and ~250 Å in height, and thus is considerably larger than OMCCs of minimized systems (Figures 1 and 2). Production of CagY, CagX, and CagM are required for detection of the OMCC, whereas OMCCs assembled in the absence of CagT and Cag3 are narrower, suggesting that CagT and Cag3 form the periphery (Frick-Cheng et al., 2016).
Recent reports have defined the 3D structure of the OMCC at a resolution as high as 3.4 Å (Figure 2) (Chung et al., 2019; Sheedlo et al., 2020). The OMCC has three distinct structural features, an outer membrane cap (designated as the OMC) consisting of an outer layer (O-layer) and inner layer (I-layer), a periplasmic ring (PR), and a stalk (Chang et al., 2018; Chung et al., 2019; Hu et al., 2019). Most strikingly, there is an apparent symmetry mismatch between the OMC (14-fold symmetry) and the PR (17-fold symmetry). The initial sub-4Å structure established that central portions of the OMC are composed of the three VirB orthologs, CagT, CagX, and CagY, but left unanswered the composition of the PR or locations of Cag3 and CagM (Chung et al., 2019). The latest ~3.4 Å structure reveals several remarkable features (Sheedlo et al., 2020). First, positions of all five components (CagX, CagY, CagM, CagT, and Cag3) within the OMCC and stoichiometries (1:1:2:2:5) were defined. The OMCC thus consists of 14 copies each of CagX and CagY, 28 of CagM, and CagT and 70 of Cag3, which explains its large size. The high-resolution structure also defines the nature of contacts among all five subunits within and between asymmetric units. Most importantly, the structure highlights the fact that both CagX and CagY form parts of the OMC and PR and, consequently, bridge the symmetry mismatch. Portions of CagX and CagY within the PR also resemble those comprising the I-layer of the X. citri, leading to the proposal that the T4SSCag PR and the I-layers of “minimized” systems are analogous structures (Sheedlo et al., 2020). If so, it is remarkable that the two highly conserved core subunits of T4SSs, VirB9, and VirB10, span two portions of OMCCs that in the “minimized” systems have equivalent 14-fold symmetries but in the T4SSCag have 14- and 17-fold symmetries.
Complementing these findings, the intact T4SSCag was recently visualized in the H. pylori cell envelope by in situ CryoET, albeit at a lower resolution (Figure 1) (Chang et al., 2018; Hu et al., 2019b). The T4SSCag machine is readily visualized as ring-shaped structures at several locations around the H. pylori cell. The outer portion corresponding to the OMCC resembles that solved by CryoEM in its 14-fold symmetrical ring-like features. Interestingly, the radial spokes of the OMCC, presumably corresponding to the 22-β-strand density of unknown composition in the CryoEM structure (Figure 2), exhibit a change in chirality. At the proximal face of the I-layer facing the IM, the spokes have an overall counterclockwise rotation, whereas near the OM they have a clockwise rotation. Furthermore, near the IM the spokes terminate in two distinct knobs, whereas near the OM they terminate in one knob. These findings might reflect a dynamic movement of the spokes, as well as the presence of additional subunits at the spoke termini that are dissociated during purification of the OMCC. As also shown by single-particle CryoEM, analyses of cag mutant machines confirmed that assembly of the OMCC in situ requires the Cag subunits CagX, CagY, and CagM, and also that CagT and Cag3 associate peripherally with the OMCC. Finally, at the OMCC–OM junction, the OM is pinched inward, indicating that the OMCC spans and causes local distortion of the inner and outer leaf-lets of the OM (Hu et al., 2019b).
In situ CryoET also enabled visualization of other portions of the T4SSCag not yet detected by single-particle CryoEM, including a central periplasmic cylinder and surrounding collar and a large IMC (Figure 1) (Hu et al., 2019b). The cylinder has a central channel through which CagA and other substrates are likely translocated. The IMC was visualized initially at a resolution sufficient to show the presence of four tiers of densities extending into the cytoplasm that were interpreted as side-by-side hexamers of the VirB4-like subunit CagE, reminiscent of the R388 structure (Figure 1) (Chang et al., 2018). However, a higher resolution structure clearly established that the IMC consists of three concentric rings of ~ 12, ~22, and ~ 36 nm in diameter, denoted as the I-, M-, and O-rings, respectively (Figure 1)(Hu et al., 2019b). Analyses of mutant machines lacking each one of the three ATPases, VirB4-like CagE, VirB11-like Cagα, and VirD4-like Cagβ, supplied evidence for specific contributions of each ATPase to the IMC architecture. First, CagE contributes to both the I- and M-rings by assembling as a hexamer of dimers that contribute to parts of the I- and M-rings. Second, VirB11-like Cagα is stably associated with the cytoplasmic face of the I-ring through direct contacts with CagE. Third, VirD4-like Cagβ is required for detection of densities contributing to the O-ring as well as cytoplasmic extensions of the I- and M-rings. It is unlikely that Cagβ accounts for all of the densities that are missing in a Δcagβ mutant, suggesting that the IMC is composed of additional subunits that are dependent on Cagβ for a stable association While further studies are needed to define contributions of Cagβ and unspecified subunits to the IMC, it is noteworthy that this is the first visualized ATPase energy center for any T4SS in which all three ATPases clearly make structural contributions. Overall, the results support an ordered assembly pathway for the T4SSCag, whereby the OMCC assembles first, and then, nucleates assembly of the integral membrane portion of the IMC. The ATP energy center assembles at the cytoplasmic base of the IMC through successive recruitment of CagE, Cagα, and Cagβ. Finally, one or more unspecified components are recruited through contacts with Cagβ and likely one or both of the other ATPases (Hu et al., 2019b).
1.4.2 |. The L. pneumophila Dot/Icm T4SS
L. pneumophila is an environmental parasite of amebae that coincidentally infects human hosts typically from inhalation of water droplets (Sherwood and Roy, 2016). The T4SSDot/Icm is used by L. pneumophila to colonize human alveolar macrophages, which triggers a severe pneumonia called Legionnaire’s disease (Sherwood and Roy, 2016). To evade killing by the host, L. pneumophila convert phagosomes into a protective compartment termed the Legionella-containing vacuole (LCV), whose formation requires the T4SSDot/Icm. As noted above, the T4SSDot/Icm translocates over 300 protein effectors during infection, many of which have been shown to target host cellular pathway controlling membrane transport processes (Isaac and Isberg, 2014; Sherwood and Roy, 2016). The T4SSDot/Icm also has retained a functional vestige of its ancestral conjugation machine in its capacity to transfer the mobilizable IncQ plasmid RSF1010 to recipient bacteria (Vogel et al., 1998).
The T4SSDot/Icm was first visualized as a ring-shaped complex on L. pneumophila cells by transmission electron microscopy (Kubori et al., 2014). The OMCC subcomplex was purified by ultracentrifugation and gel filtration (Kubori and Nagai, 2019). It is composed of five subunits, of which DotD, DotH, and DotG are the functional counterparts of VirB7, VirB9, and VirB10, and DotF and DotC are system-specific (Kubori and Nagai, 2019; Vincent et al., 2006). Interestingly, although deletion of VirB10 abolishes assembly of the OMCCs of “minimized” systems, deletion of DotG does not abolish ring formation by the other OMCC components. Rather, the central density is missing, supporting the notion that DotG forms part of the central channel (Kubori et al., 2014). Very recently, the structure of the OMCC was solved at a global resolution of ~4.6 Å without imposed symmetry (Durie et al., 2020). The OMCC approximates that of the Cag system in width (~400 Å) but is more compressed in height (~165 Å) (Figure 2). Also reminiscent of the Cag system, the OMCC is divisible into an outer ring-like OMC and central PR. The OMC has two prominent features, a central dome and a disk with 13 spokes that extend outward. The dome could not be resolved but its position on top of the disk and its size (~100 Å wide, ~50 Å in height) with a narrow opening of ~40 Å suggests it is equivalent to the α-helical channels built from VirB10-like APs. With symmetry imposed, the OMC and PR were solved at 3.5 Å and 3.7 Å resolution, respectively. The OMC and PR exhibit symmetry mismatch, but in this case the OMC has 13-fold symmetry and the PR has 18-fold symmetry. Positions of DotC, DotD, and DotH, as well as two subunits not previously identified, DotK and Lpg0657, were successfully mapped within the OMC and shown to exist in stoichiometries of 1:2:1:1:1, respectively. DotK and Lpg0657 have folds resembling peptidoglycan-binding domains, but no peptidoglycan was observed in the binding clefts of either protein. Despite the high resolution of this structure, several densities could not be definitively assigned, including a polypeptide sitting on top of the OMC that might link the OMC with the OM. Thirteen copies of a second, 22-strand β-helix extend outward to form the radial spokes (Figure 2). Initially, these were thought to correspond to a β-helix structure formed by a central repeat region of DotG, but a ΔdotG mutant machine retains these spokes as well as the OMC disk and instead lacks the dome and the PR. Thus, as shown for minimized systems and, more equivalently, the OMCC from the Cag system, VirB10-like DotG forms the central region of the OMCC as well as the dome presumptively attached to or spanning the OM. Another notable feature that recapitulates findings for the T4SSCag OMCC is that the VirB9-like DotH forms part of the OMC and PR, intriguingly bridging the symmetry mismatch between these two large subdomains (Kubori et al., 2014).
The Dot/Icm system is currently one of the most intensively studied T4SSs. Besides the high-resolution structure of the OMCC, recent studies with complementary live-cell imaging and in situ CryoET have identified a couple of important features. First, the channel preferentially assembles at the poles of L. pneumophila cells and, in fact, polar localization is essential for function of this T4SS during infection (Jeong et al., 2017; Ghosal et al., 2019). Cells in the exponential phase of growth have clusters of machines at the lateral mid-cell region, which can account for polar localization once cells divide and the septum becomes the new cell poles. However, even cells growing in stationary phase have lateral machines (Ghosal et al., 2019; Jeong et al., 2017; Park et al., 2020). The lateral machines might correspond to assembly intermediates, but it was recently shown that both polar and lateral machines consist of heterogeneous populations of assembly intermediates and presumptively intact machines (Park et al., 2020). Thus, the biological role of lateral machines in stationary phase cells, if any, remains to be determined. Second, analyses of mutant machines defined an assembly pathway for the T4SSDot/Icm, which resembles out-to-in assembly pathways proposed in early studies for the A. tumefaciens T4SSVirB/VirD4 and more recently for the H. pylori T4SSCag (Christie et al., 2005; Ghosal et al., 2019; Hu et al., 2019b; Park et al., 2020). A distinctive feature of the T4SSDot/Icm assembly pathway, however, is that two proteins, DotU and IcmF, are responsible for polar targeting of the T4SSDot/Icm (Ghosal et al., 20191). These proteins are not found associated with other T4SSs, but rather are homologs of two components (TssL, TssM) of type VI secretion systems (T6SSs). TssL and TssM do not promote polar targeting of T6SSs, yet, do function as nucleators of the T6SS in conjunction with TssJ (Durand et al., 2015). The DotU/IcmF complex is postulated to recruit the lipoprotein DotC, DotD, and DotH, which comprise the OMC (Durie et al., 2020; Ghosal et al., 2019). This ring-shaped complex then recruits the bitopic IM proteins DotG and DotF to build the PR and periplasmic densities including the central cylinder and surrounding collar. Once the OMCC/periplasmic substructures are formed, the IMC is assembled by recruitment of one or more unspecified IM subunits and finally the DotO and DotB ATPases (Chetrit et al., 2018; Ghosal et al., 2019; Park et al., 2020).
The architecture and dynamic activities of the IMC also have been further illuminated through in situ CryoET and fluorescence microscopy. In the first in situ CryoET structure, the OMCC presented as a “Wi-Fi” like structure of 400 Å in diameter consisting of the DotC, DotD, DotF, DotG, and DotH subunits (Ghosal et al., 2017). Initially, densities projecting into cytoplasm were interpreted as side-by-side hexamers of VirB4-like DotO, but higher resolution structures established that DotO adopts the same hexamer of dimers arrangement as visualized for VirB4-like CagE (Figure 1) (Chetrit et al., 2018; Hu et al., 2019b; Park et al., 2020). In end-on view, DotO presents as two concentric rings of approximately the same diameters as the I- and M-rings formed by CagE (Chetrit et al., 2018). A hexameric ring at the base of the I-ring was postulated to correspond to VirB11-like DotB, which was confirmed by density tracing of a GFP tag appended to DotB. The overall dimensions of DotB visualized in situ also fit well with the recently solved X-ray structure of the DotB homohexamer (Prevost and Waksman, 2018). Remarkably, DotB-GFP was shown to dynamically cycle off and on the Dot/Icm IMC by live-cell imaging. A mutant protein (E191K) that binds but does not hydrolyze ATP stably associates with the IMC, indicating that the ATP-bound state of DotB docks stably on the IMC (Chetrit et al., 2018). Very recently, a focused refinement of cytoplasmic complexes also supplied evidence for DotB-induced conformational changes in the IMC, which is interpreted as an opening in the channel in response to ATP hydrolysis by DotB (Park et al., 2020). Thus, ATP energy consumption by VirD4/VirB-like ATPases has now been shown to induce conformational changes in both the proximal (IMC) and distal (OMCC) portions of the T4SS channel (Cascales and Christie, 2004a; Park et al., 2020).
1.4.3 |. The F plasmid Tra T4SS
The F plasmid-encoded T4SSF was the first described bacterial conjugation system (Lederberg and Tatum, 1946). There has been a resurgence of interest in F plasmids largely due to their prominent in dissemination of genes encoding antibiotic and heavy metal resistance, virulence factors, and fitness traits among clinically important Enterobacterial species (Koraimann, 2018). The T4SSF also is an ideal model system for investigating fundamental mechanistic and structural properties of these nanomachines, because it is highly amenable to genetic manipulation and is presently the only T4SS known to produce pili that dynamically extend and retract (Clarke et al., 2008).
Recently, our views of how F pili assemble at the cell surface and how they mediate intercellular contacts have been completely reshaped through in situ CryoET studies of the F-encoded T4SS and structural analyses of F pili (Costa et al., 2016; Hu et al., 2019a; Zheng et al., 2020). Analyses of F-encoded structures in the E. coli cell envelope identified several distinct structures, including one interpreted as the translocation channel through which the F plasmid transfer intermediate passes to recipient cells (Figure 1). Like the other characterized T4SSs, this structure (named the F1-CH complex) is composed of OMCC and IMC subassemblies joined by a thin periplasmic density. The OMCC adopts a “flying-saucer” structure of 250 Å in width and 115 Å in height, which in top view has 13 knobbed-spokes joined to a central ring of 130 Å in diameter. Both in overall architecture and fold symmetry, the OMCC of the F1-CH more closely resembles that associated with the T4SSDot/Icm than those of the “minimized” systems or the T4SSCag (Figure 1). The OMCC surrounds a cylindrical density with a central channel that extends to and through the IMC. The cytoplasmic portion of the IMC closely resembles those of the Dot/Icm and Cag machines, insofar as VirB4-like TraC is configured as a hexamer of dimers surrounding the central channel. F plasmids lack VirB11 homologs and VirD4-like TraD was not visible, suggesting that TraD associates asymmetrically or transiently with the channel (Hu et al., 2019a).
Remarkably, three other F-encoded structures were bound at the cell surface by F pili (Hu et al., 2019a). One such structure, termed the F2-Channel/Pilus or F2-CH/P, resembles F1-CH complex but with an attached pilus. These structures are nonabundant (~5% of visualized F-encoded structures), and their most remarkable feature is that the F pilus sits on top of the structure and clearly does not extend across the OM. This observation, coupled with analyses of mutant machines, firmly establish that the mature F pilus assembles at the OM and not on an IM platform as previously envisioned. This distinguishes the F pilus from type IV pili (T4P; phylogenetically unrelated to conjugative pili) and the needle complexes of Type III secretion machines (T3SSs), which both nucleate on an IM platform and extend across the OM (see (Hu et al., 2019a). A second, a very abundant structure (~60% of visualized F-encoded structures) presents as a thin (~12 nm) stalk that spans the periplasm and a wider “mushroom cap” near the OM, to which the F pilus is attached at the outer surface of the OM. These F3-Stalk/Pilus or F3-ST/P structures lack a central channel or IMC densities corresponding to the VirB4-like TraC ATPase. Finally, a fourth structure, designated the F4-Outer membrane/Pilus or F4-OM/P complex, is again nonabundant (~5% of visualized F-encoded structures) and consists of the F pilus attached to a small density at the OM without any associated periplasmic density.
Taken together, these findings establish for the first time that F pili are docked onto alternative basal platforms around the cell surface (Hu et al., 2019a). In view of the genetic requirements for F pilus biogenesis, a working model posits that the F1-CH structure functions as a translocation channel when donor cells are in the “mating” mode, for example, in direct contact with target cells. However, in the absence of target cell contact, the F1-CH transition to a pilus assembly platform, giving rise to F2-CH/P structures. The F2-CH/P structure dynamically builds pili through a process involving extraction of TraA pilin subunits from an IM pool, assembly of a thin protofilament that extends through the central channel to the cell surface, and nucleation of the mature pilus as a helical fiber at the OM. Due to its highly dynamic nature, the F2-CH/P structure is difficult to capture in structural snapshots generated by CryoET, hence, its nonabundance. Once F pili polymerize, they can either retract through a reversal of the assembly process, or be deposited around the cell surface on alternative basal platforms conforming to the visualized F3 and F4 complexes. Because these complexes lack densities known to correspond to components that are essential for F pilus assembly/retraction dynamics, for example, TraC, the associated pili are presumed to be static structures. How these alternative platforms arise and what biological functions the associated F pili serve remain intriguing questions for future study (Hu et al., 2019a).
1.4.4 |. Structure–function studies of F pili
F pili are filaments that extend into the bacterial extracellular space to mediate attachment to target cells. Upon attachment, F pili retract to draw the two cells together, allowing for establishment of direct cell–cell contacts and, ultimately, an SDS- and shear-resistant “mating junction” (Arutyunov and Frost, 2013). Previous studies established that F pili are composed of thousands of TraA (pilin) molecules polymerized as a helical assembly (Paranchych and Frost, 1988; Wang et al., 2009). However, recent structures of two members of the F pilus family (encoded by the pED208 and pOX38 plasmids) solved by single-particle CryoEM revealed not only the expected electron density of the TraA pilin, but also a neighboring density attributed to a phospholipid from the PG (phosphatidylglycerol) family (Figure 3a) (Costa et al., 2016). Similarly, a recent CryoEM structure of an F pilus encoded by K. pneumoniae pKpQIL (Zheng et al., 2020) and a CryoET structure of the F pilus bound with MS2 phage, also shows the presence of both molecular species in the structure (Meng et al., 2019; Zheng et al., 2020). Thus, all F pilus structures solved to date confirm the peculiar arrangement of copious amounts of stoichiometric 1:1 pilin–phospholipid complexes as the building block.
FIGURE 3.
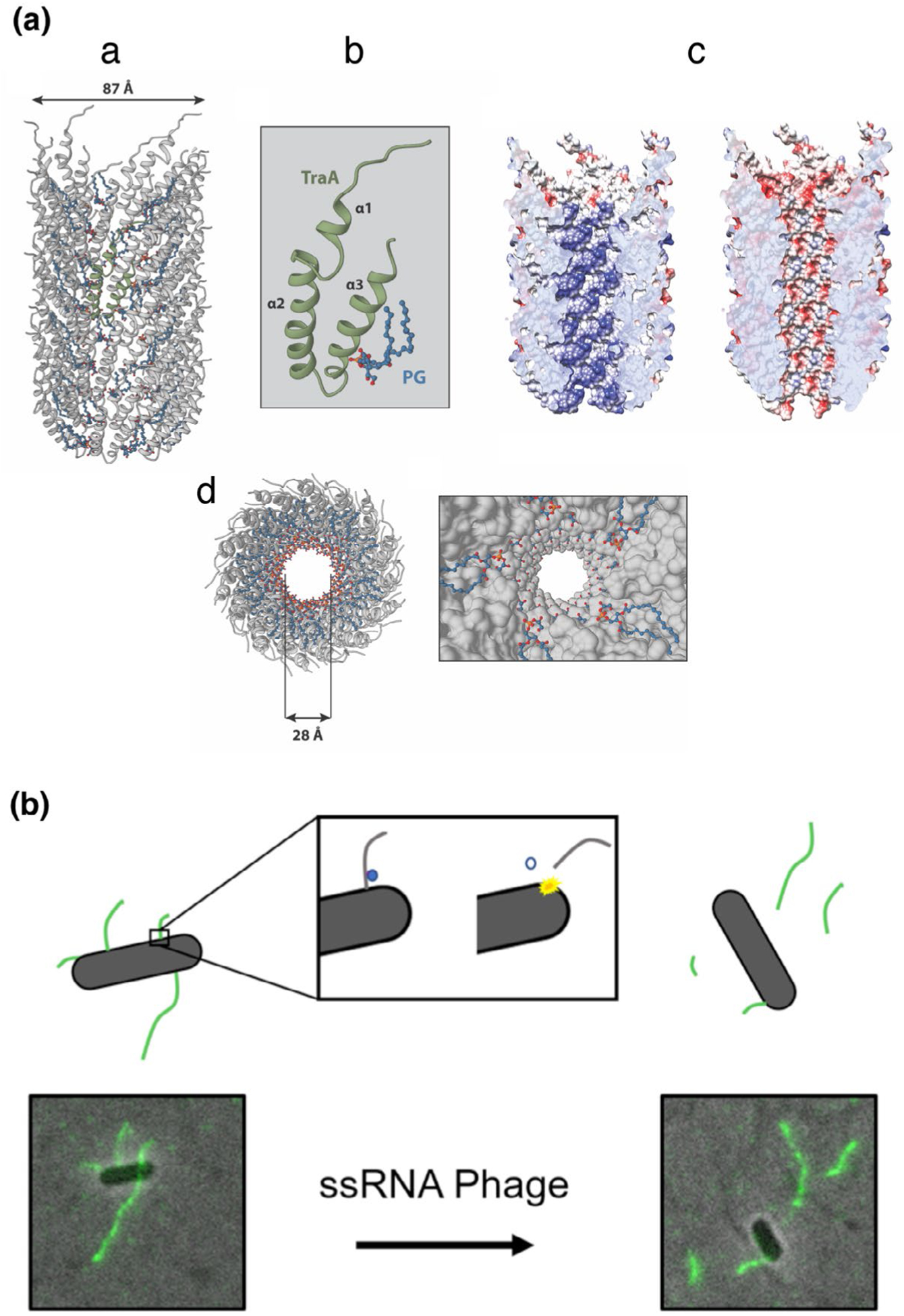
F pilus structure and dynamics. A) Architecture of the FpED208 pilus. (a) Representation of the overall molecular model from the FpED208 pilus structure (PDB:5LEG) derived from its cryoelectron density map (EMDB:4042). (b) Molecular model of the F pilus subunit formed by a complex of TraA and PG (phosphatidylglycerol). (c) The electrostatic potential in the pilus lumen in the absence (left) or presence (right) of PG. The blue color represents an electron positive surface, whereas red color represents electron negative. d) Inset view of the pilus lumen showing the PG head group array pointing to the central channel. (b) ssRNA phage penetration causes F pilus detachment. sfGFP fused to the MS2 Coat protein allows for visualization of F pili by proxy of phage binding. Prior to phage infection, F pili are observed protruding from the cell surface. However, once phage is added under infectious conditions, F pili rapidly detach from the cell surface in a time frame matching the MS2 RNA entry period, suggesting that viral RNA penetration triggers detachment of the F pilus
The pED208-encoded F pilus has been solved at the highest resolution (3.6 Å) among the available structures (Costa et al., 2016). The structure shows a filament with an internal lumen diameter of 28 Å and an external diameter of 87 Å (Figure 3a). The structure can be described as TraA-phosphatidylglycerol (PG) pentameric complexes stacked on top of each other related by an axial rise of 12.2 Å and with a rotation angle of 28-degrees or, alternatively, a five-start helical filament. Minor variations in terms of axial rise, rotation angle and start helix were observed in the CryoEM structures of F pili elaborated by the F plasmid and pKpQIL (Costa et al., 2016; Zheng et al., 2020). The high-resolution map obtained from pED208-encoded F pili yielded an atomic structure of TraA (Figure 3a). Each TraA subunit is composed of three α-helices (α1, α2, α3), where the loop between α2 and α3 projects to the lumen of the pilus and the N- and C-termini are exposed at the pilus surface where they are accessible for phage attachment (Zheng et al., 2020). The overall atomic structures of all individual TraA proteins are likely very similar given the very high degree of amino acid sequence identity between pilin subunits of different F systems.
In the vicinity of each TraA protein density, an unconnected electron density consistent with a phospholipid head group (pointing to the pilus lumen) and two acyl chains (embedded in the pilus wall) was resolved (Figure 3a) (Costa et al., 2016). Mass spectrometry experiments identified the two PG species present in the pilus as phosphatidylglycerol 32:1 and phosphatidylglycerol 34:1. The negatively charged head group from the PG phospholipids, reverses the electrostatic potential from the lumen surface from positive to negative. In addition to the electrostatic remodeling properties produced by the PG head group, the flexible acyl chains embedded in the pilus wall could likely contribute for the unique F pilus bio-mechanical properties such as bending, extension, and retraction (Clarke et al., 2008).
Our knowledge of F pilus structure and function has been further enriched by studies of F pili and their phages (Figure 3b). Many canonical ssRNA coliphages (e.g., MS2, R17, Qβ, etc.) utilize F pili to facilitate host recognition and delivery of viral RNA into the host cytoplasm during infection. Infection begins with phage adsorption to host pili. Recent CryoEM studies determined the molecular and structural details describing MS2 adsorption to the F pilus (Meng et al., 2019). Adsorption is mediated by the MS2 maturation protein (Mat), specifically, the β-region of Mat binds the N- and C- termini of four pilin subunits through a series of electrostatic interactions between residues at the Mat-pilin interface. This interaction can be abolished by eliminating charged residues at the interface (Manchak et al., 2002). Additionally, the orientation of F pilin monomers in assembled pili was revealed, where the N- and C- termini of each pilin are angled toward the cell envelope (Meng et al., 2019). This orientation may be favorable to promote pilus extension or retraction through the T4SS channel.
After adsorption, the ssRNA phage penetrates the cell envelope to deliver its genomic RNA into the cytoplasm. This penetration step is arguably the least understood aspect of the infection process. Since the F pili are naturally involved in conjugation, a process requiring intimate contact between donor and recipient cells brought together through pilus retraction, it seems intuitive that the pilus bound phage is brought to the cell surface through pilus retraction. As shown by use of dye-labeled phage R17 (Clarke et al., 2008), F pili undergo cycles of extension and retraction presumably as a means of surveying the area around the cells. Pilus retraction is typically completed within 5 min after initiation at a rate of 16 nm/sec, whereas extension occurs more rapidly at a rate of 40 nm/sec. Additionally, pili tend to supercoil when their distal ends contact a surface, suggesting that pili rotate during extension and retraction.
Direct evidence that F pili bound by phage are capable of retracting prompted further investigations of F pilus behavior in the presence of ssRNA phages. Recently, fluorescently labeled MS2 phage was used to detect and measure the length of F pili after ssRNA phage infection (Figure 3b) (Harb et al., 2020). These studies established that F pili detach from host cells during infection by ssRNA phages. The duration of pilus detachment coincided with the entry period of MS2, suggesting that pili detach during penetration of viral RNA. Additionally, the length of detached pili increased with higher multiplicities of infection, supporting the notion that pilus detachment, and likely viral RNA penetration, is facilitated through a process involving pilus retraction. In the context of the recent CryoET structure of the F2-CH/P complex (Hu et al., 2019a), it is envisioned that as the pilus-bound phage are drawn to the T4SS channel through pilus retraction, a torsional stress imposed at the channel–pilus junction causes breakage of the mature F pilus from the protofilament and release of F pili into the milieu (Figure 3). Interestingly, this process of phage-induced pilus release might represent a novel form of superinfection exclusion, insofar as it prevents subsequent binding of phages in the vicinity to cell-bound pilus receptors. These studies further established that MS2 phage retained the ability to bind F pili produced by mutant machines lacking the TraD T4CP, but the phage genomic RNA does not enter the cytoplasm of the ΔtraD mutant cells. TraD thus appears to serve as a gatekeeper of the F-encoded channel from both directions, in its capacity to regulate F plasmid transfer out of cells during conjugation and phage uptake during infection (Harb et al., 2020). Finally, the authors note that phage infection is also known to transiently inhibit cells from producing F pili, which represents another form of superinfection exclusion. Given the selective advantage of superinfection exclusion mechanisms, it is reasonable to propose that other phages that use pili as receptors might deploy similar strategies of pilus release and pilus assembly inhibition to prevent further infection.
The F pilus is the best-characterized member of the conjugative pili, but is unique among these pili in its demonstrated capacity to dynamically extend and retract. Other characterized conjugative pili naturally slough from cells, which has complicated efforts to study assembly dynamics. Indeed, such pili are thought to promote donor–target cell contacts through release and establishment of hydrophobic interactions that mediate nonspecific pilus clumping and cell aggregation. A common, and, perhaps, only function of conjugative pili thus might be to serve as attachment organelles for adherence of donors to surfaces and for biofilm development, rather than contributing directly to translocation of DNA or other substrates to target cells. At least two lines of evidence support this notion. First, “uncoupling” mutations have been isolated that block detectable pilus production without affecting substrate transfer (Jakubowski et al., 2005, 2009). Second, conjugation machines functioning in Gram-positive species do not elaborate pili, yet, can function highly efficiently in DNA transfer (Bhatty et al., 2013; Grohmann et al., 2017). Nevertheless, there is some evidence for transfer of the F plasmid from donor to recipient cells at a distance, which is proposed to be mediated by the extended F pilus (Babic et al., 2008). The latest structures of F pilus basal platforms, for example, the F2-CH complex, and the discovery of a phospholipid lining of the F pilus lumen, allow for this possibility at least as a secondary and low-efficiency mode of transfer. Clearly, further work is needed to “visualize” real-time transfer events through state-of-the-art fluorescence approaches or by capturing CryoET snapshots of donor-recipient pairs in the act of DNA transfer.
2 |. SUMMARY AND FUTURE DIRECTIONS
In this review, we have updated the reader on three remarkable advances in structural definition of model T4SSs obtained by single-particle CryoEM. First, the DotL/adaptor receptor module, solved in the absence and presence of docked effectors, offer exciting new insights into early stage substrate docking reactions generally as well as specific mechanisms for regulating the flow of hundreds of effectors through the T4SSDot/Icm. (Kim et al., 2020; Kwak et al., 2017; Meir et al., 2020; Xu et al., 2017). Second, structural resolution of F pili revealed TraA pilin–phospholipid as the fundamental building block, which raises intriguing new questions concerning the pilus biogenesis pathway, the possible functional significance of the PG-lined lumen, and the generality of these findings to other conjugative pili. Third, structural definition of the intrinsically stable OMCCs from the X. citri VirB/VirD4, H. pylori Cag and L. pneumophila Dot/Icm systems revealed that only the latter “expanded” systems display a striking symmetry mismatch between the outer and inner layers (Chung et al., 2019; Costa et al., 2016; Durie et al., 2020; Sgro et al., 2018; Sheedlo et al., 2020; Zheng et al., 2020). This symmetry mismatch is highly curious, especially in view of the fact that “minimized” systems are fully capable of translocating DNA or proteins to other bacteria as well as to eukaryotic cell targets, as best illustrated with the paradigmatic A. tumefaciens VirB/VirD4 T4SS. It should be noted that the “minimized” systems in fact exhibit symmetry mismatch, but between their OMCCs and IMCs. At this time, we can only speculate that symmetry mismatches between or within large subassemblies of T4SS nanomachines affords greater conformational flexibility. For example, symmetry mismatch between the IMC and OMCC may be necessary for conversion of intra- and extracellular signals into structural transitions associated with channel gating. Asymmetry within the OMCCs of the “expanded” Cag and Dot/Icm systems might afford more profound structural transitions necessary for recruitment of nonprotein substrates, spatiotemporal control in delivery of many hundreds of effectors, or elaboration of pili or large sheathed structures upon binding of eukaryotic cell receptors.
We also summarized exciting new insights gained through visualization of “expanded” T4SSs by in situ CryoET (Chetrit et al., 2018; Ghosal et al., 2017; Ghosal et al., 2019; Hu et al., 2019a #4672; Hu et al., 2019b; Jeong et al., 2017; Park et al., 2020). The studies have enabled comparisons of the OMCC structures solved in isolation and in the native environment of the cell envelope, and also defined for the first time structures of central periplasmic channels and IMCs from machines that have been refractory to purification. Live-cell imaging has further advanced our knowledge of T4SS machine assembly dynamics, as nicely illustrated by discoveries that the T4SSDot/Icm localizes and functions at cell poles, and the VirB11-like DotB ATPase associates dynamically and induces structural changes in the channel through rounds of ATP hydrolysis. With this new information in hand, it is clear that the T4SS field is well-poised for further basic studies aimed at deciphering the mechanistic details of substrate trafficking and exploring T4SS structural and biological diversity.
Continued structure–function studies of T4SSs also have important translational applications. One area of active investigation is in the design of small-molecule inhibitors of T4SSs with the broad goals of suppressing dissemination of antibiotic resistance or mitigating virulence of clinically important pathogens (reviewed in Alvarez-Rodriguez et al., 2020a; Boudaher and Shaffer, 2019). In some cases, inhibitors are being identified through high throughput screens for molecules blocking a T4SS-mediated process, for example, conjugation (Cabezon et al., 2017; Casu et al., 2017; Shaffer et al., 2016). In others, specific requirements for machine assembly or function are targeted for inhibition, such as VirB8 dimerization (Casu et al., 2016; Paschos et al., 2011; Smith, 2012 #3395), or catalytic activities of the VirD4, VirB4, or VirB11 ATPases (Arya et al., 2019; Garcia-Cazorla et al., 2018; Ripoll-Rozada et al., 2016). This latter approach, in particular, will benefit through structural definition of intersubunit contacts critical to T4SS integrity, as these are prime targets for small-molecule disruption of machine assembly. As we learn more of the infection processes of male-specific phages that parasitize T4SSs to gain entry into cells (Harb et al., 2020; Meng et al., 2019), novel strategies may arise for deployment of phages to kill cells harboring T4SSs or selectively inactivate these systems. Finally, there is developing interest in manipulation of T4SSs for therapeutic applications through delivery of toxic DNA or protein substrates to kill-specific target cells of interest. The T4SSs are excellent candidates as programed delivery systems as they are the only bacterial secretion system known to translocate DNA and proteins to both bacterial and human cell targets (Guzman-Herrador et al., 2017).
In summary, if the recent structural advances are any indication, the future of basic and applied studies of T4SSs shines brighter than ever.
ACKNOWLEDGMENTS
We thank the entire T4SS community for valuable contributions to this field, and we apologize for omissions of published work on the many other exciting aspects of type IV secretion not covered here due to page limitations. We thank members of the Zeng, Hu, and Christie labs for helpful comments and critiques. This work was supported by NSF Grant 1902392, NIH Grant R21 AI156846, and Texas A & M University X-Grant 290386 to L.Z, NIH Grant R21 AI 142378 to B.H. and P.J.C., McGovern Medical School start-up funds and Welch Foundation Grant AU-1953-20180324 to B.H., Wellcome Trust 215164/Z/18/Z grant to T.R.D.C., and NIH Grant R35 GM131892 to PJC. None of the authors declare a conflict of interest. Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Alperi A, Larrea D, Fernandez-Gonzalez E, Dehio C, Zechner EL and Llosa M (2013) A translocation motif in relaxase TrwC specifically affects recruitment by its conjugative type IV secretion system. Journal of Bacteriology, 195, 4999–5006. 10.1128/JB.00367-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Martinez CE and Christie PJ (2009) Biological diversity of prokaryotic type IV secretion systems. Microbiology and Molecular Biology Reviews, 73, 775–808. 10.1128/MMBR.00023-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Rodriguez I, Arana L, Ugarte-Uribe B, Gomez-Rubio E, Martin-Santamaria S, Garbisu C et al. (2020a) Type IV coupling proteins as potential targets to control the dissemination of antibiotic resistance. Frontiers in Molecular Biosciences, 7, 201. 10.3389/fmolb.2020.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Rodriguez I, Ugarte-Uribe B, de la Arada I, Arrondo JLR, Garbisu C and Alkorta I (2020b) Conjugative coupling proteins and the role of their domains in conjugation, secondary structure and in vivo subcellular location. Frontiers in Molecular Biosciences, 7, 185. 10.3389/fmolb.2020.00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly KA and Baron C (2007) The VirB5 protein localizes to the T-pilus tips in Agrobacterium tumefaciens. Microbiology, 153, 3766–3775. 10.1099/mic.0.2007/010462-0 [DOI] [PubMed] [Google Scholar]
- Arutyunov D and Frost LS (2013) F conjugation: back to the beginning. Plasmid, 70, 18–32. 10.1016/j.plasmid.2013.03.010 [DOI] [PubMed] [Google Scholar]
- Arya T, Oudouhou F, Casu B, Bessette B, Sygusch J and Baron C (2019) Fragment-based screening identifies inhibitors of ATPase activity and of hexamer formation of Cagalpha from the Helicobacter pylori type IV secretion system. Scientific Reports, 9, 6474. 10.1038/s41598-019-42876-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmakuri K, Cascales E and Christie PJ (2004) Energetic components VirD4, VirB11 and VirB4 mediate early DNA transfer reactions required for bacterial type IV secretion. Molecular Microbiology, 54, 1199–1211. 10.1111/j.1365-2958.2004.04345.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic A, Lindner AB, Vulic M, Stewart EJ and Radman M (2008) Direct visualization of horizontal gene transfer. Science, 319, 1533–1536. 10.1126/science.1153498 [DOI] [PubMed] [Google Scholar]
- Backert S and Tegtmeyer N (2017) Type IV secretion and signal transduction of Helicobacter pylori CagA through interactions with host cell receptors. Toxins (Basel), 9, 115. 10.3390/toxins9040115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta LM, Kerr JE, Cascales E, Giuliano ME, Bailey ME, McKay C et al. (2011) An Agrobacterium VirB10 mutation conferring a type IV secretion system gating defect. Journal of Bacteriology, 193, 2566–2574. 10.1128/JB.00038-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardill JP, Miller JL and Vogel JP (2005) IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Molecular Microbiology, 56, 90–103. 10.1111/j.1365-2958.2005.04539.x [DOI] [PubMed] [Google Scholar]
- Bhatty M, Laverde Gomez JA & Christie PJ (2013) The expanding bacterial type IV secretion lexicon. Research in Microbiology, 164, 620–639. 10.1016/j.resmic.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleves S, Galan JE and Llosa M (2020) Bacterial injection machines: evolutionary diverse but functionally convergent. Cellular Microbiology, 22, e13157. 10.1111/cmi.13157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaher E and Shaffer CL (2019) Inhibiting bacterial secretion systems in the fight against antibiotic resistance. Med Chem Comm, 10, 682–692. 10.1039/C9MD00076C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein D, Amaro F, Zusman T, Lifshitz Z, Cohen O, Gilbert JA et al. (2016) Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nature Genetics, 48, 167–175. 10.1038/ng.3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein D, Zusman T, Degtyar E, Viner R, Segal G & Pupko T (2009) Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathogens, 5, e1000508. 10.1371/journal.ppat.1000508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezon E, de la Cruz F and Arechaga I (2017) Conjugation inhibitors and their potential use to prevent dissemination of antibiotic resistance genes in bacteria. Frontiers in Microbiology, 8, 2329. 10.3389/fmicb.2017.02329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezon E, Ripoll-Rozada J, Pena A, de la Cruz F and and Arechaga I (2015) Towards an integrated model of bacterial conjugation. FEMS Microbiology Reviews, 39, 81–95. [DOI] [PubMed] [Google Scholar]
- Cambronne ED and Roy CR (2007) The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathogens, 3, e188. 10.1371/journal.ppat.0030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E, Atmakuri K, Sarkar MK and Christie PJ (2013) DNA substrate-induced activation of the Agrobacterium VirB/VirD4 type IV secretion system. Journal of Bacteriology, 195, 2691–2704. 10.1128/JB.00114-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E and Christie PJ (2003) The versatile bacterial type IV secretion systems. Nature Reviews Microbiology, 1, 137–150. 10.1038/nrmicro753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E and Christie PJ (2004a) Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proceedings of the National Academy of Sciences USA, 101, 17228–17233. 10.1073/pnas.0405843101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E and Christie PJ (2004b) Definition of a bacterial type IV secretion pathway for a DNA substrate. Science, 304, 1170–1173. 10.1126/science.1095211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu B, Arya T, Bessette B and Baron C (2017) Fragment-based screening identifies novel targets for inhibitors of conjugative transfer of antimicrobial resistance by plasmid pKM101. Scientific Reports, 7, 14907. 10.1038/s41598-017-14953-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu B, Smart J, Hancock MA, Smith M, Sygusch J and Baron C (2016) Structural analysis and inhibition of TraE from the pKM101 type IV secretion system. Journal of Biological Chemistry, 291, 23817–23829. 10.1074/jbc.M116.753327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J and Waksman G (2009) Structure of the outer membrane complex of a type IV secretion system. Nature, 462, 1011–1015. 10.1038/nature08588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, Shaffer CL, Rettberg LA, Ghosal D and Jensen GJ (2018) In vivo structures of the Helicobacter pylori Cag type IV secretion system. Cell Reports, 23, 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetrit D, Hu B, Christie PJ, Roy CR and Liu J (2018) A unique cytoplasmic ATPase complex defines the Legionella pneumophila type IV secretion channel. Nature Microbiology, 3, 678–686. 10.1038/s41564-018-0165-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ (2019) The rich tapestry of bacterial protein translocation systems. Protein Journal, 38, 389–408. 10.1007/s10930-019-09862-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S and Cascales E (2005) Biogenesis, architecture, and function of bacterial type IV secretion systems. Annual Review of Microbiology, 59, 451–485. 10.1146/annurev.micro.58.030603.123630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JM, Sheedlo MJ, Campbell AM, Sawhney N, Frick-Cheng AE, Lacy DB et al. (2019) Structure of the Helicobacter pylori Cag type IV secretion system. Elife, 8, e47644. 10.7554/eLife.47644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M, Maddera L, Harris RL and Silverman PM (2008) F-pili dynamics by live-cell imaging. Proceedings of the National Academy of Sciences USA, 105, 17978–17981. 10.1073/pnas.0806786105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa TR, Ilangovan A, Ukleja M, Redzej A, Santini JM, Smith TK et al. (2016) Structure of the bacterial sex F pilus reveals an assembly of a stoichiometric protein-phospholipid complex. Cell, 166(1436–1444), e1410. 10.1016/j.cell.2016.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover TL, Lacy DB and Ohi MD (2020) The Helicobacter pylori Cag type IV secretion system. Trends in Microbiology, 28, 682–695. 10.1016/j.tim.2020.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbari VC, Ciccone J, Patel JS, Islam B, Agarwal PK and Haider S (2020) Electrostatic switching controls channel dynamics of the sensor protein VirB10 in A. tumefaciens type IV secretion system. ACS Omega, 5, 3271–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz F, Frost LS, Meyer RJ and Zechner EL (2010) Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiology Reviews, 34, 18–40. 10.1111/j.1574-6976.2009.00195.x [DOI] [PubMed] [Google Scholar]
- Durand E, Nguyen VS, Zoued A, Logger L, Pehau-Arnaudet G, Aschtgen MS et al. (2015) Biogenesis and structure of a type VI secretion membrane core complex. Nature, 523, 555–560. 10.1038/nature14667 [DOI] [PubMed] [Google Scholar]
- Durie CL, Sheedlo MJ, Chung JM, Byrne BG, Su M, Knight T et al. (2020) Structural analysis of the Legionella pneumophila Dot/Icm type IV secretion system core complex. Elife, 9, e59530. 10.7554/eLife.59530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Püls J, Buhrdorf R, Gebert B, Odenbreit S and Haas R (2001) Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Molecular Microbiology, 42, 1337–1348. 10.1046/j.1365-2958.2001.02714.x [DOI] [PubMed] [Google Scholar]
- Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD and Cover TL (2016) Molecular and structural analysis of the Helicobacter pylori Cag type IV secretion system core complex. Mbio, 7, e02001–e02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV and Waksman G (2009) Structure of a type IV secretion system core complex. Science, 323, 266–268. 10.1126/science.1166101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Gaudet RG, Gray-Owen SD & Salama NR (2017) TIFA signaling in gastric epithelial cells initiates the Cag type 4 secretion system-dependent innate immune response to Helicobacter pylori infection. Mbio, 8, e01168–e1217. 10.1128/mBio.01168-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cazorla Y, Getino M, Sanabria-Rios DJ, Carballeira NM, de la Cruz F, Arechaga I et al. (2018) Conjugation inhibitors compete with palmitic acid for binding to the conjugative traffic ATPase TrwD, providing a mechanism to inhibit bacterial conjugation. Journal of Biological Chemistry, 293, 16923–16930. 10.1074/jbc.RA118.004716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal D, Chang YW, Jeong KC, Vogel JP and Jensen GJ (2017) In situ structure of the Legionella Dot/Icm type IV secretion system by electron cryotomography. EMBO Reports, 18, 726–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal D, Jeong KC, Chang YW, Gyore J, Teng L, Gardner A et al. (2019) Molecular architecture, polar targeting and biogenesis of the Legionella Dot/Icm T4SS. Nat Microbiol, 4, 1173–1182. 10.1038/s41564-019-0427-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Ruth FX and Coll M (2001) Structure of TrwB, a gatekeeper in bacterial conjugation. International Journal of Biochemistry and Cell Biology, 33, 839–843. 10.1016/S1357-2725(01)00060-7 [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth FX, Moncalian G, Perez-Luque R, Gonzalez A, Cabezon E, de la Cruz F et al. (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1- ATPase. Nature, 409, 637–641. 10.1038/35054586 [DOI] [PubMed] [Google Scholar]
- Gordon JE, Costa TRD, Patel RS, Gonzalez-Rivera C, Sarkar MK, Orlova EV et al. (2017) Use of chimeric type IV secretion systems to define contributions of outer membrane subassemblies for contact-dependent translocation. Molecular Microbiology, 105, 273–293. 10.1111/mmi.13700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann E, Christie PJ, Waksman G and Backert S (2018) Type IV secretion in Gram-negative and Gram-positive bacteria. Molecular Microbiology, 107, 455–471. 10.1111/mmi.13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann E, Keller W and Muth G (2017) Mechanisms of conjugative transfer and type IV secretion-mediated effector transport in Gram-positive bacteria. Current Topics in Microbiology and Immunology, 413, 115–141. [DOI] [PubMed] [Google Scholar]
- Guzman-Herrador DL & Llosa M (2019) The secret life of conjugative relaxases. Plasmid, 104, 102415. 10.1016/j.plasmid.2019.102415 [DOI] [PubMed] [Google Scholar]
- Guzman-Herrador DL, Steiner S, Alperi A, Gonzalez-Prieto C, Roy CR and Llosa M (2017) DNA delivery and genomic integration into mammalian target cells through type IV A and B secretion systems of human pathogens. Frontiers in Microbiology, 8, 1503. 10.3389/fmicb.2017.01503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb L, Chamakura K, Khara P, Christie PJ, Young R and Zeng L (2020) (2020) ssRNA phage penetration triggers detachment of the F-pilus. Proceedings of the National Academy of Sciences USA, 117, 25751–25758. 10.1073/pnas.2011901117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M (2014) Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host and Microbe, 15, 306–316. 10.1016/j.chom.2014.02.008 [DOI] [PubMed] [Google Scholar]
- Hu B, Khara P & Christie PJ (2019a) Structural bases for F plasmid conjugation and F pilus biogenesis in Escherichia coli. Proceedings of the National Academy of Sciences USA, 116, 14222–14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Khara P, Song L, Lin AS, Frick-Cheng AE, Harvey ML et al. (2019b) In situ molecular architecture of the Helicobacter pylori Cag type IV secretion system. Mbio, 10, e00849–19. 10.1128/mBio.00849-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston JR (2014) Horizontal gene transfer in the human gastrointestinal tract: potential spread of antibiotic resistance genes. Infection and Drug Resistance, 7, 167–176. 10.2147/IDR.S48820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilangovan A, Kay CWM, Roier S, El Mkami H, Salvadori E, Zechner EL et al. (2017) Cryo-EM structure of a relaxase reveals the molecular basis of DNA unwinding during bacterial conjugation. Cell, 169(708–721), e712. 10.1016/j.cell.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac DT and Isberg R (2014) Master manipulators: an update on Legionella pneumophila Icm/Dot translocated substrates and their host targets. Future Microbiology, 9, 343–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski SJ, Cascales E, Krishnamoorthy V and Christie PJ (2005) Agrobacterium tumefaciens VirB9, an outer-membrane-associated component of a type IV secretion system, regulates substrate selection and T-pilus biogenesis. Journal of Bacteriology, 187, 3486–3495. 10.1128/JB.187.10.3486-3495.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski SJ, Kerr JE, Garza I, Krishnamoorthy V, Bayliss R, Waksman G et al. (2009) Agrobacterium VirB10 domain requirements for type IV secretion and T pilus biogenesis. Molecular Microbiology, 71, 779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong KC, Ghosal D, Chang YW, Jensen GJ and Vogel JP (2017) Polar delivery of Legionella type IV secretion system substrates is essential for virulence. Proceedings of the National Academy of Sciences USA, 114, 8077–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Yang Y, Zhou J, Zhu L, Gu Y, Zhang X et al. (2016) Roles of the putative type IV-like secretion system key component VirD4 and PrsA in pathogenesis of Streptococcus suis type 2. Frontiers in Cellular and Infection Microbiology, 6, 172. 10.3389/fcimb.2016.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U et al. (2009) Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathogens, 5, e1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Gaddy JA, Voss BJ, Hennig EE and Cover TL (2014) Genes required for assembly of pili associated with the Helicobacter pylori Cag type IV secretion system. Infection and Immunity, 82, 3457–3470. 10.1128/IAI.01640-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kubori T, Yamazaki K, Kwak MJ, Park SY, Nagai H et al. (2020) Structural basis for effector protein recognition by the Dot/Icm Type IVB coupling protein complex. Nature Communications, 11, 2623. 10.1038/s41467-020-16397-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch B, Callaghan MM, Tellechea-Luzardo J, Seeger AY, Dillard JP and Krasnogor N (2020) Protein interactions within and between two F-type type IV secretion systems. Molecular Microbiology, 114, 823–838. 10.1111/mmi.14582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koraimann G (2018) Spread and persistence of virulence and antibiotic resistance genes: A ride on the F plasmid conjugation module. EcoSal plus, 8, 10.1128/ecosalplus.ESP-0003-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T, Hyakutake A and Nagai H (2008) Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Molecular Microbiology, 67, 1307–1319. 10.1111/j.1365-2958.2008.06124.x [DOI] [PubMed] [Google Scholar]
- Kubori T, Koike M, Bui XT, Higaki S, Aizawa S and Nagai H (2014) Native structure of a type IV secretion system core complex essential for Legionella pathogenesis. Proceedings of the National Academy of Sciences USA, 111, 11804–11809. 10.1073/pnas.1404506111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubori T and Nagai H (2019) Isolation of the Dot/Icm type IV secretion system core complex from Legionella pneumophila. Methods in Molecular Biology, 1921, 241–247. [DOI] [PubMed] [Google Scholar]
- Kwak MJ, Kim JD, Kim H, Kim C, Bowman JW, Kim S et al. (2017) Architecture of the type IV coupling protein complex of Legionella pneumophila. Nature Microbiology, 2, 17114. 10.1038/nmicrobiol.2017.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S et al. (2007) Helicobacter exploits integrin for type IV secretion and kinase activation. Nature, 449, 862–866. 10.1038/nature06187 [DOI] [PubMed] [Google Scholar]
- Lang S, Kirchberger PC, Gruber CJ, Redzej A, Raffl S, Zellnig G et al. (2011) An activation domain of plasmid R1 TraI protein delineates stages of gene transfer initiation. Molecular Microbiology, 82, 1071–1085. 10.1111/j.1365-2958.2011.07872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrea D, de Paz HD, Matilla I, Guzman-Herrador DL, Lasso G, de la Cruz F et al. (2017) Substrate translocation involves specific lysine residues of the central channel of the conjugative coupling protein TrwB. Molecular Genetics and Genomics, 292, 1037–1049. 10.1007/s00438-017-1331-3 [DOI] [PubMed] [Google Scholar]
- Lederberg J and Tatum EL (1946) Gene recombination in Escherichia coli. Nature, 158, 558. 10.1038/158558a0 [DOI] [PubMed] [Google Scholar]
- Li YG and Christie PJ (2020) The TraK accessory factor activates substrate transfer through the pKM101 type IV secretion system independently of its role in relaxosome assembly. Molecular Microbiology, 114, 214–229. 10.1111/mmi.14507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YG, Hu B and Christie PJ (2019) Biological and structural diversity of type IV secretion systems. Microbiology Spectrum, 7. 10.1128/microbiolspec.PSIB-0012-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA et al. (2013) Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proceedings of the National Academy of Sciences USA, 110, E707–E715. 10.1073/pnas.1215278110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AS, Dooyema SDR, Frick-Cheng AE, Harvey ML, Suarez G, Loh JT et al. (2020) Bacterial energetic requirements for Helicobacter pylori Cag type IV secretion system-dependent alterations in gastric epithelial cells. Infection and Immunity, 88, e00790–19. 10.1128/IAI.00790-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa M and Alkorta I (2017) Coupling proteins in type IV secretion. Current Topics in Microbiology and Immunology, 413, 143–168. [DOI] [PubMed] [Google Scholar]
- Low HH, Gubellini F, Rivera-Calzada A, Braun N, Connery S, Dujeancourt A et al. (2014) Structure of a type IV secretion system. Nature, 508, 550–553. 10.1038/nature13081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J & Frost LS (2005) Mutations in the C-terminal region of TraM provide evidence for in vivo TraM-TraD interactions during F-plasmid conjugation. Journal of Bacteriology, 187, 4767–4773. 10.1128/JB.187.14.4767-4773.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Wong JJ, Edwards RA, Manchak J, Frost LS and Glover JN (2008) Structural basis of specific TraD-TraM recognition during F plasmid-mediated bacterial conjugation. Molecular Microbiology, 70, 89–99. 10.1111/j.1365-2958.2008.06391.x [DOI] [PubMed] [Google Scholar]
- Manchak J, Anthony KG and Frost LS (2002) Mutational analysis of F-pilin reveals domains for pilus assembly, phage infection and DNA transfer. Molecular Microbiology, 43, 195–205. 10.1046/j.1365-2958.2002.02731.x [DOI] [PubMed] [Google Scholar]
- Meir A, Chetrit D, Liu L, Roy CR and Waksman G (2018) Legionella DotM structure reveals a role in effector recruiting to the Type 4B secretion system. Nature Communications, 9, 507. 10.1038/s41467-017-02578-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Mace K, Lukoyanova N, Chetrit D, Hospenthal MK, Redzej A et al. (2020) Mechanism of effector capture and delivery by the type IV secretion system from Legionella pneumophila. Nature Communications, 11, 2864. 10.1038/s41467-020-16681-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng R, Jiang M, Cui Z, Chang JY, Yang K, Jakana J et al. (2019) Structural basis for the adsorption of a single-stranded RNA bacteriophage. Nature Communications, 10, 3130. 10.1038/s41467-019-11126-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R (2015) Mapping type IV secretion signals on the primase encoded by the broad-host-range plasmid R1162 (RSF1010). Journal of Bacteriology, 197, 3245–3254. 10.1128/JB.00443-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA and Roy CR (2005) A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proceedings of the National Academy of Sciences USA, 102, 826–831. 10.1073/pnas.0406239101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano N, Kubori T, Kinoshita M, Imada K and Nagai H (2010) Crystal structure of Legionella DotD: insights into the relationship between type IVB and type II/III secretion systems. PLoS Pathogens, 6, e1001129. 10.1371/journal.ppat.1001129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio S, Zuckman-Cholon DM, Cambronne ED, Roy CR (2004) The Legionella IcmS-IcmW protein complex is important for Dot/Icmmediated protein translocation. Molecular Microbiology, 55, 912–926. 10.1111/j.1365-2958.2004.04435.x [DOI] [PubMed] [Google Scholar]
- Paranchych W and Frost LS (1988) The physiology and biochemistry of pili. Adv Microb Phys, 29, 53–114. [DOI] [PubMed] [Google Scholar]
- Park D, Chetrit D, Hu B, Roy CR & Liu J (2020) Analysis of Dot/Icm type IVB secretion system subassemblies by cryoelectron tomography reveals conformational changes induced by DotB Binding. mBio, 11. 10.1128/mBio.03328-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM et al. (2011) An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infection and Immunity, 79, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Lu J, Wong JJW, Edwards RA, Frost LS and Mark Glover JN (2014) Mechanistic basis of plasmid-specific DNA binding of the F plasmid regulatory protein, TraM. Journal of Molecular Biology, 426, 3783–3795. 10.1016/j.jmb.2014.09.018 [DOI] [PubMed] [Google Scholar]
- Huang L, Boyd D, Amyot WM, Hempstead AD, Luo Z-Q, O’Connor TJ, (2011) The E Block motif is associated with Legionella pneumophila translocated substrates. Cellular Microbiology, 13, 227–245. 10.1111/j.1462-5822.2010.01531.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannkuch L, Hurwitz R, Traulsen J, Sigulla J, Poeschke M, Matzner L et al. (2019) ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. The FASEB Journal, 33, 9087–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevost MS and Waksman G (2018) X-ray crystal structures of the type IVb secretion system DotB ATPases. Protein Science, 27, 1464–1475. 10.1002/pro.3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redzej A, Ilangovan A, Lang S, Gruber CJ, Topf M, Zangger K et al. (2013) Structure of a translocation signal domain mediating conjugative transfer by type IV secretion systems. Molecular Microbiology, 89, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redzej A, Ukleja M, Connery S, Trokter M, Felisberto-Rodrigues C, Cryar A et al. (2017) Structure of a VirD4 coupling protein bound to a VirB type IV secretion machinery. EMBO Journal, 36, 3080–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S, Li YG, Schmitt A, Lassinantti L, Christie PJ and Berntsson RP (2019) Enterococcal PcfF is a ribbon-helix-helix protein that recruits the relaxase PcfG through binding and bending of the oriT sequence. Frontiers in Microbiology, 10, 958. 10.3389/fmicb.2019.00958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll-Rozada J, Garcia-Cazorla Y, Getino M, Machon C, Sanabria-Rios D, de la Cruz F et al. (2016) Type IV traffic ATPase TrwD as molecular target to inhibit bacterial conjugation. Molecular Microbiology, 100, 912–921. 10.1111/mmi.13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Calzada A, Fronzes R, Savva CG, Chandran V, Lian PW, Laeremans T et al. (2013) Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO Journal, 32, 1195–1204. 10.1038/emboj.2013.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde M, Puls J, Buhrdorf R, Fischer W and Haas R (2003) A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Molecular Microbiology, 49, 219–234. 10.1046/j.1365-2958.2003.03549.x [DOI] [PubMed] [Google Scholar]
- Savvides SN, Yeo HJ, Beck MR, Blaesing F, Lurz R, Lanka E et al. (2003) VirB11 ATPases are dynamic hexameric assemblies: new insights into bacterial type IV secretion. EMBO Journal, 22, 1969–1980. 10.1093/emboj/cdg223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgro GG, Costa TR, Cenens W, Souza DP, Cassago A, Oliverira LD et al. (2018) CryoEM structure of the core complex of a bacterial killing type IV secretion system. Nature Microbiology, 3, 1429–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgro GG, Oka GU, Souza DP, Cenens W, Bayer-Santos E, Matsuyama BY et al. (2019) Bacteria-killing type IV secretion systems. Frontiers in Microbiology, 10, 1078. 10.3389/fmicb.2019.01078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer CL, Gaddy JA, Loh JT, Johnson EM, Hill S, Hennig EE et al. (2011) Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathogens, 7, e1002237. 10.1371/journal.ppat.1002237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer CL, Good JA, Kumar S, Krishnan KS, Gaddy JA, Loh JT et al. (2016) Peptidomimetic small molecules disrupt type IV secretion system activity in diverse bacterial pathogens. Mbio, 7, e00221–16. 10.1128/mBio.00221-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedlo MJ, Chung JM, Sawhney N, Durie CL, Cover TL, Ohi MD et al. (2020) Cryo-EM reveals species-specific components within the Helicobacter pylori Cag type IV secretion system core complex. Elife, 9, e59495. 10.7554/eLife.59495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RK and Roy CR (2016) Autophagy evasion and endoplasmic reticulum subversion: the yin and yang of Legionella intracellular infection. Annual Review of Microbiology, 70, 413–433. [DOI] [PubMed] [Google Scholar]
- Siamer S and Dehio C (2015) New insights into the role of Bartonella effector proteins in pathogenesis. Current Opinion in Microbiology, 23, 80–85. 10.1016/j.mib.2014.11.007 [DOI] [PubMed] [Google Scholar]
- Skoog EC, Morikis VA, Martin ME, Foster GA, Cai LP, Hansen LM et al. (2018) CagY-dependent regulation of type IV secretion in Helicobacter pylori is associated with alterations in integrin binding. Mbio, 9, e00717–18. 10.1128/mBio.00717-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Coinçon M, Paschos A, Jolicoeur B, Lavallée P, Sygusch J, Baron C (2012) Identification of the Binding Site of Brucella VirB8 Interaction Inhibitors. Chemistry & Biology, 19, 1041–1048. 10.1016/j.chembiol.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Souza DP, Andrade MO, Alvarez-Martinez CE, Arantes GM, Farah CS and Salinas RK (2011) A component of the Xanthomonadaceae type IV secretion system combines a VirB7 motif with a N0 domain found in outer membrane transport proteins. PLoS Pathogens, 7, e1002031. 10.1371/journal.ppat.1002031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DP, Oka GU, Alvarez-Martinez CE, Bisson-Filho AW, Dunger G, Hobeika L et al. (2015) Bacterial killing via a type IV secretion system. Nature Communications, 6, 6453. 10.1038/ncomms7453 [DOI] [PubMed] [Google Scholar]
- Stanger FV, de Beer TAP, Dranow DM, Schirmer T, Phan I & Dehio C (2017) The BID Domain of type IV secretion substrates forms a conserved four-helix bundle topped with a hook. Structure, 25, 203–211. 10.1016/j.str.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SC, Faber E, Bats SH, Murillo T, Speidel Y, Coombs N et al. (2017) Helicobacter pylori modulates host cell responses by Cag T4SS-dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathogens, 13, e1006514. 10.1371/journal.ppat.1006514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl K, Muller S, Scheidgen-Kleyboldt G, Clausen M and Maier B (2010) Composite system mediates two-step DNA uptake into Helicobacter pylori. Proceedings of the National Academy of Sciences USA, 107, 1184–1189. 10.1073/pnas.0909955107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland MC, Nguyen TL, Tseng V and Vogel JP (2012) The Legionella IcmSW complex directly interacts with DotL to mediate translocation of adaptor-dependent substrates. PLoS Pathogens, 8, e1002910. 10.1371/journal.ppat.1002910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Suzuki T, Mimuro H and Sasakawa C (2003) Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cellular Microbiology, 5, 395–404. [DOI] [PubMed] [Google Scholar]
- Tato I, Matilla I, Arechaga I, Zunzunegui S, de la Cruz F and Cabezon E (2007) The ATPase activity of the DNA transporter TrwB is modulated by protein TrwA: implications for a common assembly mechanism of DNA translocating motors. Journal of Biological Chemistry, 282, 25569–25576. 10.1074/jbc.M703464200 [DOI] [PubMed] [Google Scholar]
- Terradot L, Bayliss R, Oomen C, Leonard GA, Baron C and Waksman G (2005) Structures of two core subunits of the bacterial type IV secretion system, VirB8 from Brucella suis and ComB10 from Helicobacter pylori. Proceedings of the National Academy of Sciences USA, 102, 4956–4961. 10.1073/pnas.0408927102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga MG, Shaffer CL, Sierra JC, Suarez G, Piazuelo MB, Whitaker ME et al. (2016) Pathogenic Helicobacter pylori strains translocate DNA and activate TLR9 via the cancer-associated cag type IV secretion system. Oncogene, 35, 6262–6269. 10.1038/onc.2016.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsaki A, Moncalian G, Garcillan-Barcia Mdel P, Drainas C and de la Cruz F (2009) Analysis of ColE1 MbeC unveils an extended ribbon-helix-helix family of nicking accessory proteins. Journal of Bacteriology, 191, 1446–1455. 10.1128/JB.01342-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergunst AC, Schrammeijer B, den Dulk-Ras A, de Vlaam CM, Regensburg-Tuink TJ and Hooykaas PJ (2000) VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science, 290, 979–982. 10.1126/science.290.5493.979 [DOI] [PubMed] [Google Scholar]
- Vergunst AC, van Lier MC, den Dulk-Ras A, Grosse Stuve TA, Ouwehand A and Hooykaas PJ (2005) Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proceedings of the National Academy of Sciences USA, 102, 832–837. 10.1073/pnas.0406241102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunology, 5, 1166–1174. 10.1038/ni1131 [DOI] [PubMed] [Google Scholar]
- Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL and Vogel JP (2006) Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Molecular Microbiology, 62, 1278–1291. 10.1111/j.1365-2958.2006.05446.x [DOI] [PubMed] [Google Scholar]
- Vincent CD, Friedman JR, Jeong KC, Sutherland MC and Vogel JP (2012) Identification of the DotL coupling protein subcomplex of the Legionella Dot/Icm type IV secretion system. Molecular Microbiology, 85, 378–391. 10.1111/j.1365-2958.2012.08118.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong SK and Isberg RR (1998) Conjugative transfer by the virulence system of Legionella pneumophila. Science, 279, 873–876. 10.1126/science.279.5352.873 [DOI] [PubMed] [Google Scholar]
- Wagner A, Tittes C and Dehio C (2019) Versatility of the BID domain: Conserved function as type-IV-secretion-signal and secondarily evolved effector functions within Bartonella-infected host cells. Frontiers in Microbiology, 10, 921. 10.3389/fmicb.2019.00921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman G (2019) From conjugation to T4S systems in Gram-negative bacteria: a mechanistic biology perspective. EMBO Reports, 20, e47012. 10.15252/embr.201847012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YA, Yu X, Silverman PM, Harris RL and Egelman EH (2009) The structure of F-pili. Journal of Molecular Biology, 385, 22–29. 10.1016/j.jmb.2008.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker N, Berry TM, Rosenthal N, Gordon JE, Gonzalez-Rivera C, Sheehan KB et al. (2016) Chimeric coupling proteins mediate transfer of heterologous type IV effectors through the Escherichia coli pKM101-encoded conjugation machine. Journal of Bacteriology, 198, 2701–2718. 10.1128/JB.00378-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker N, Chen Y, Jakubowski SJ, Sarkar MK, Li F and Christie PJ (2015) The all-alpha domains of coupling proteins from the Agrobacterium tumefaciens VirB/VirD4 and Enterococcus faecalis pCF10-encoded type IV secretion systems confer specificity to binding of cognate DNA substrates. Journal of Bacteriology, 197, 2335–2349. 10.1128/JB.00189-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJ, Lu J, Edwards RA, Frost LS and Glover JN (2011) Structural basis of cooperative DNA recognition by the plasmid conjugation factor, TraM. Nucl Acids Res, 39, 6775–6788. 10.1093/nar/gkr296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJ, Lu J and Glover JN (2012) Relaxosome function and conjugation regulation in F-like plasmids - a structural biology perspective. Molecular Microbiology, 85, 602–617. 10.1111/j.1365-2958.2012.08131.x [DOI] [PubMed] [Google Scholar]
- Xu J, Xu D, Wan M, Yin L, Wang X, Wu L et al. (2017) Structural insights into the roles of the IcmS-IcmW complex in the type IVb secretion system of Legionella pneumophila. Proceedings of the National Academy of Sciences U S A, 114, 13543–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo HJ, Savvides SN, Herr AB, Lanka E and Waksman G (2000) Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Molecular Cell, 6, 1461–1472. 10.1016/S1097-2765(00)00142-8 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Furuya N, Lin YJ, Guntert P, Komano T and Kainosho M (2008) Structural basis of the role of the NikA ribbon-helix-helix domain in initiating bacterial conjugation. Journal of Molecular Biology, 384, 690–701. 10.1016/j.jmb.2008.09.067 [DOI] [PubMed] [Google Scholar]
- Zheng W, Pena A, Low WW, Wong JLC, Frankel G and Egelman EH (2020) Cryoelectron-microscopic structure of the pKpQIL conjugative pili from carbapenem-resistant Klebsiella pneumoniae. Structure, 28, 1321–1328.e2. 10.1016/j.str.2020.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Pfannkuch L, Al-Zeer MA, Bartfeld S, Koch M, Liu J et al. (2017) ALPK1- and TIFA-dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Reports, 20, 2384–2395. 10.1016/j.celrep.2017.08.039 [DOI] [PubMed] [Google Scholar]