

Abstract
Seizures cause retrograde amnesia, but underlying mechanisms are poorly understood. We tested whether seizure activated neuronal circuits overlap with spatial memory engram and whether seizures saturate LTP in engram cells. A seizure caused retrograde amnesia for spatial memory task. Spatial learning and a seizure caused cFos expression and synaptic plasticity overlapping set of neurons in the CA1 of the hippocampus. Recordings from learning-labeled CA1 pyramidal neurons showed potentiated synapses. Seizure-tagged neurons were also more excitable with larger rectifying excitatory postsynaptic currents than surrounding unlabeled neurons. These neurons had enlarged dendritic spines and saturated LTP. A seizure immediately after learning, reset the memory engram. Seizures cause retrograde amnesia through shared ensembles and mechanisms.
Keywords: Seizure, TRAP, T maze, EPSC, Engram, LTP
1. Introduction
Seizures cause retrograde amnesia, however underlying mechanisms are still insufficiently understood. Memory deficits in patients with temporal lobe epilepsy occur due to underlying hippocampal pathology including principal and interneuron death (Sloviter, 1987, Du et al., 1995, Bell et al., 2011) and large-scale anatomical circuit reorganizations (Houser et al., 1990, Zhang et al., 2009) including pathological hippocampal-cortical coupling (Gelinas et al., 2016). However, retrograde amnesia occurs in humans and experimental animals that do not have epilepsy and pathological changes in the hippocampus. Electroconvulsive shock-induced seizure causes a selective amnesic effect on recently acquired memories, wherein amnesic effects diminish as the interval between training and seizures increases (Duncan, 1949, Kim and Fanselow, 1992, Kopp et al., 1966; Chorover and Schiller, 1965, Squire et al.; 2001).
Newly acquired memories stabilize over time through the network, and cellular processes called consolidation and interference with these processes can lead to retrograde amnesia. Network consolidation involves interactions between the hippocampus and an array of cortical structures and neuromodulators (Dudai, 2004, Squire et al., 2000, Nader et al., 2000, McGaugh, 2000, Shimizu et al., 2000). Sharp wave ripples generated in the CA3/CA1 hippocampus facilitate consolidation and interactions between the hippocampus and cortical memory areas (Buszaki, 2015, Csicsvari and Dupret, 2014). At the cellular level, consolidation requires synaptic plasticity including long-term potentiation (LTP), long term depression (LTD) and spike-timing-dependent plasticity and de novo transcription, protein synthesis, (Bliss and Lomo, 1973; Nicoll, 2017; Scharf et al., 2002; Whitlock et al., 2006). Seizures could cause retrograde amnesia by interfering with the hippocampus –cortex interactions and by interfering with synaptic potentiation.
We investigated the potential mechanisms of seizure-induced retrograde amnesia, based on the engram hypothesis, which postulates that a specific set of neurons are active during learning, and they encode the memory trace through synaptic modification (Jocelyn and Tonegawa, 2020; Tonegawa et al., 2015). Transgenic mice using immediate-early gene promoter-driven reporter expression enable visualization of engrams at the cellular and circuit levels (Denny et al., 2014; Guenthener et al., 2013; Kitamura et al., 2017; Reijmers et al., 2007; Tanaka et al., 2014). These studies have mapped memory engrams for fear conditioning, and spatial memory, among others. Seizures activate many brain areas that are engaged in memory encoding, consolidation, and retrieval (Dabrowska et al., 2019; Kramer and Cash, 2012; Smith and Schevon, 2016; Stam, 2014). We tested seizure circuits and spatial memory engram overlap at the cellular level. We further tested whether a seizure hijacks the cellular memory mechanisms in cfos expressing neurons: potentiated synapses, and increased excitability.
2. Material and methods
2.1. Animal model:
We used TRAP mice generated by crossing mice expressing Cre-ER under cFos promoter (B6.129(Cg)-Fostm1.1(cre/ERT2)Luo/J, #021882) with mice expressing tdTomato from the Rosa locus ((B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, ME, #007909; Jackson Laboratories, Bar Harbor (Guenthner et al., 2013).
For electrophysiology recordings, we used TetTag mouse, generated by crossing transgenic mice (strain Tg (tetO-HIST1H2BJ/GFP) 47Efu/J; stock number 005104) with mice expressing tetracycline-transactivator (tTA) protein under c-fos promoter (fos-tTA, gifted by B. Wiltgen) referred to as cFos-GFP from here onwards. TetTag mice were born and raised on doxycycline containing chow (40 mg/kg) to prevent GFP expression prior to experimental manipulations. All experiments and procedures were approved by the University of Virginia Animal Care and Use Committee.
2.2. Delayed discrete trial rewarded alternation task in T maze
Pre-training, habituation, and reward familiarization:
TRAP mice (6–8 weeks, n=50) individually housed were handled 5 minutes daily for 5 days to habituate to the experimenter. Mice were food deprived to no less than 80% of their beginning body weight to increase motivation for reward. Each mouse received 2 ml of sweetened condensed milk (Nestle), provided daily in a dish for a week before the training. Mice that showed maximal response to reward feeding underwent T-maze exploration (3 trials of 5 minutes each with a 10 minute inter-trial time) followed by three reward runs in each arm with other arm blocked (see Fig. 1A).
Fig. 1. Learning curve and associated CA1 engram for a spatial memory task.
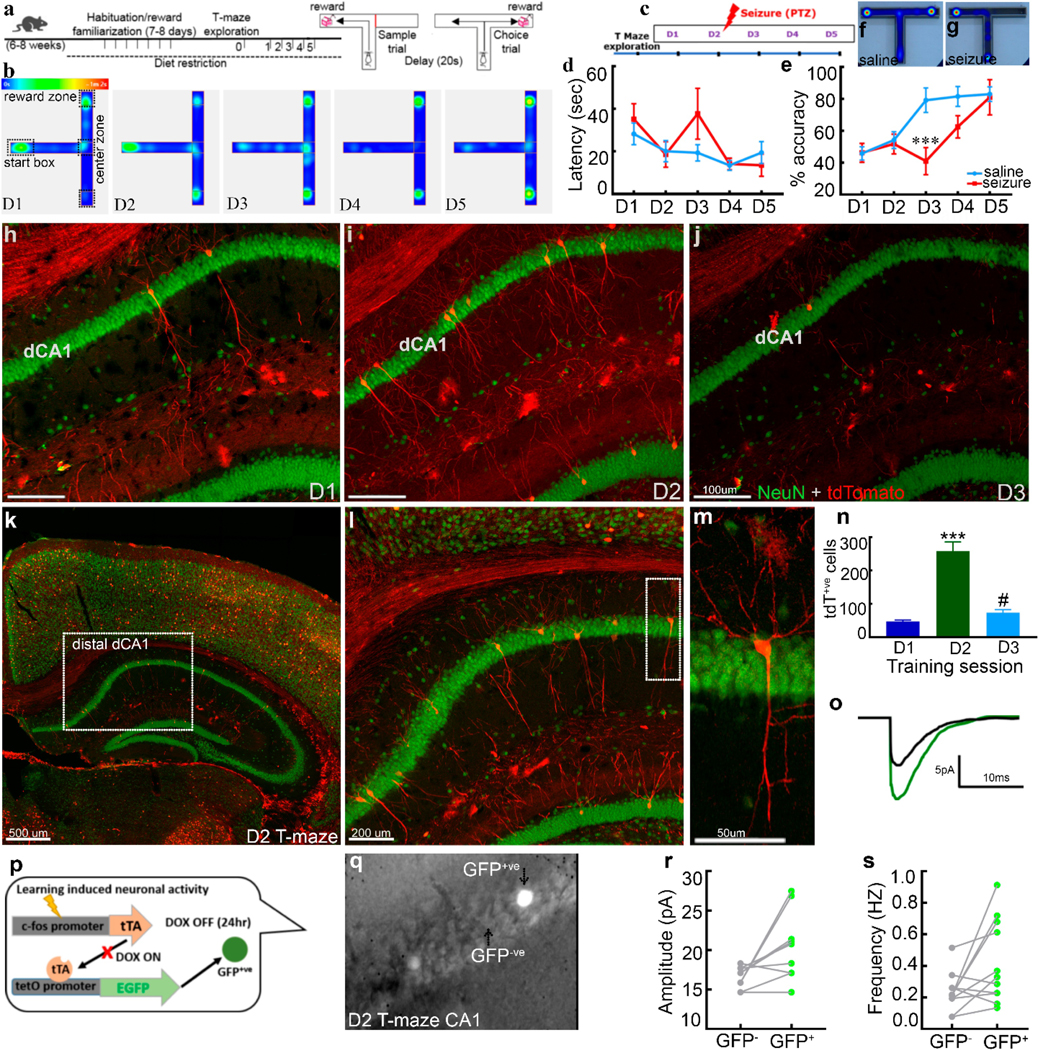
a) Schematic of experimental design for T-maze task with a graphical representation of sample and choice phases. b) Representative raw heatmap shows mouse activity on T-maze across training sessions (day1–5). Mice did not alternate and spent time spent in the start box on D1 and D2. Successive heatmaps (D3-D5) show mice alternating successfully and spending less time in the center zone. c) Schematic of the experimental plan studying the effect of seizures induced by PTZ administered 15 minutes after the second day of training. d) The latency to complete the choice trial. The latency to reach a reward arm was not different in tow groups suggesting that seizure does not cause motor deficits. e) Alternation accuracy across training sessions in saline-injected controls versus mice that had a seizure following training on the second day. The inflection inefficiency by day 3 indicates learning occurred on day 2 with stable alternation success rates maintained on days 4 and 5. A seizure immediately after training on day 2 induces retrograde amnesia. However, increased accuracy by day 4 supports the transient nature of the effect. Heatmaps show alternation success in saline (f) and alternation failure (g) in the seizure group. Representative images show tdTomato+ve neurons (training-activated, red) with NeuN (green) counterstaining in CA1 hippocampus across training sessions on days 1 to 3 from 3 different mice, day 1 (h), day 2 (i) and day 3 (j). Day 2 shows the maximum number of learning tagged CA1 neurons in distal-dorsal CA1 (distal to CA3). (k), supporting their engram-nature, (l) displays a magnified view of the dotted square.m) A magnified view of the dotted rectangle showing typical morphology and the dendritic tree of a CA1 pyramidal neuron (red, tdT labeled).n) Number of tagged CA1 pyramidal neurons across training sessions increased following second day of training. Schematic of TetTag mouse genetics (p) used for electrophysiological studies, representative averaged EPSC traces (o) from GFP+ve (training-activated, green) and neighboring GFP-ve CA1 pyramidal neuron (DIC image, bright cells on day 2, GFP+ve, (q)). Graphs show enhanced amplitude (r) and frequency (s) of EPSC’s following second day of training on T-maze (11 pairs of GFP+ve and GFP-ve CA1 neurons from 8 animals). Data are presented as mean ± SEM, n=8 for each training day (e, P<0.01), n=6 mice for each training day (n), *** represents significance with P<0.001 compared to day 1, # represents P<0.05 compared to day 2).
Afterward, mice were trained for discrete delayed trial rewarded alternation for five days as per Deacon and Rawlins (2006) protocol with modifications. The training paradigm involved a sample followed by a delayed-choice trial. During the sample run, a block in the one arm of T maze forced entry into the other arm, where a 100μl of condensed milk as a reward was available in the food well. Mice were gently removed from the maze after finishing the reward. The block was removed from the maze after a 20-second delay. The mice were again placed in the start arm facing the experimenter with a free choice to enter either arm. Mice received a reward for choosing the arm other than the previously visited one. The probe test duration was 120 seconds with a 3-minute inter-trial interval. Percent correct choices (alternation) and latency to sample and choice trials were recorded.
2.3. Microscopy and image analysis.
Learning-activated neuronal ensembles were tagged using transgenic TRAP mice in the CA1 region across training sessions. Separate cohorts of mice injected with 4-OHT on days 1, 2, and 3, were returned to mice room an hour after 4-OHT injection in behavior facility in order to minimize non-specific labeling during transfer or handling. A week later, mice were transcardially perfused (0.1M PBS) and fixed with 4% paraformaldehyde (PFA), harvested brains were postfixed in 4% PFA overnight, washed in PBS followed by sectioning on a Leica Vibratome (Leica VT1200, Germany). The brains were sliced on a coronal plane, and serial slices (40μm thick) through the hippocampus were carefully and sequentially mounted. Images were acquired using Nikon C2 Confocal (Nikon Instruments) at ×10 magnification (8 images per Z-stack with 5μm Z-distance between two consecutive images) using NIS-Elements software. An overlap ratio of 10% was set to stitch individual tiles of a whole slice image (typically 16 images per slice). Identical microscope settings of laser power and exposure (viz, offset, gain, overlap ratio for stitch) were used between experimental groups to prevent bias.
For unbiased manual quantification, composite images were analyzed using Imaris software 9.1.2 (Bitplane Scientific), and the experimenter was blinded to the groups until the end of the analysis. All tdTomato tagged neurons in CA1 were carefully counted across all the serial slices through hippocampus. The criteria involved counting only tdT+ve cells with well-defined cell bodies and processes in the pyramidal cell layer of CA1, whereas tdT+ve expressing processes with no evident cell body were avoided. The CA1 region in coronal slices was defined using the Allen mouse brain atlas (http://atlas.brain-map.org/atlas?atlas=1&plate=100960248#).
For automated quantification, the Z-stack images from C2 confocal were imported in Fiji ImageJ (NIH). A maximum projection file was generated for tdTomato, ARC, and NeuN or DAPI channels after subtraction of background fluorescence. For each brain region, the individual brain region was selected as the region of interest (ROI), and the rest of the image and signal was removed. For single-channel quantification of tagged neurons (tdT alone or ARC alone), a threshold was applied to individual ROI images (cleared maximum projection for CA1), particles above the threshold with size larger than 50-pixels were identified as positive and counted. All tagged neurons within a Z-stack were counted by reviewing individual frames of the stack. For the overlap analysis (tdTomato/ARC) in dual-labeling experiments, each signal was detected using red and green fluorescence channels. These same steps were carried out from the acquisition of Z-stacks in confocal to the generation of maximum projection files of the CA1 region. Next, a threshold was applied in the tdTomato channel, and particles above threshold and size larger than 50-pixels units were labeled as “positive for channel 1” by the cell-counter plugin of Fiji. Following this, similar application of a threshold to the ARC signal, and particles above the threshold, a size larger than 50-pixels were detected and labeled as “positive for channel 2”. Only the particles identified as positive for both channels (tdTomato and ARC), confirmed as cells with NeuN counterstaining were labeled as an overlap population.
Observed overlap: (tdT+ve + ARC +ve / NeuN +ve) * 100 and presented as fraction of the learning tagged CA1 neurons. Random Overlap: tdT +ve / NeuN +ve × ARC+ve / NeuN+ve * 100
2.4. Labeling neurons activated following a single seizure (PTZ)
TRAP mice (6–8 weeks) received a PTZ (40mg/kg, i.p.) injection and were observed for a seizure. Seizure activated neurons were TRAPed by injecting 4-OHT (50mg/kg) 60 minutes following a tonic-clonic seizure. Saline-injected animals served as controls. Mice were perfused a week later, and brain tissue was processed, imaged for cell counting as mentioned above.
2.5. Electrophysiology
All chemicals were obtained from Sigma (St. Louis, MO, USA) or Fisher Scientific (Hampton, NH) unless otherwise stated.
Patch-clamp recordings
Animals were deeply anesthetized using halothane and then decapitated. Brains were quickly removed and immersed in oxygenated (95% O2/5%CO2) ice-cold (0 – 4 °C) slicing buffer The slices were then transferred to an incubation chamber, where they were maintained submerged at 33ºC in artificial cerebrospinal fluid (aCSF) components of slicing buffer and aCSF were the same as previously reported from our lab (Joshi et al, 2017, 2018)
CA1 neurons activated by seizures were identified based on GFP fluorescence using a Nikon eclipse FN1 with a fluorescent source (Tokyo, Japan). During recording, slices were transferred to a recording chamber perfused with aCSF at 2–3 ml/min. Recordings were performed by using a Multiclamp 700A (Molecular Devices, Union City, CA) amplifier. The data were collected using pClamp 10.2 software (Molecular Devices), filtered at 2 kHz, and sampled at 10 kHz (Digidata 1440A; Molecular Devices); recording pipettes were fabricated from borosilicate glass (Sutter Instrument, Novato, CA) using a Flaming-Brown micropipette puller (P-1000; Sutter Instruments, Novato, CA). Recordings for EPSCs were performed in voltage-clamp mode (Joshi, et al. 2017, 2018). Current-clamp recordings were performed for measurement of cell membrane properties and for filling biocytin into the cell. Internal solution was (in mM): 135 K-gluconate, 7 KCl, 10 HEPES, 0.5 EGTA, 2.5 NaCl, 4 Mg-ATP, and 0.3 Na-GTP. For biocytin labeling, 5% (w/v) biocytin was added to the internal solution, and recording was continued for 20 to 50 minutes. Data was analyzed using Clampex 10.5 and MiniAnalysis.
2.6. Dendritic-spine analysis
In experiments for dendritic spine analyses (Fig. 5), acute hippocampal slices were prepared 30 minutes after the seizure. CA1 neurons that were tagged as having been activated in acute hippocampal slices were then identified under a fluorescence microscope. Neurons selected were filled with 5% biocytin for 20 to 50 minutes using whole cell patch-clamp techniques. After filling with biocytin, the slice was transferred to 4% PFA / PBS solution (PH 7.4) and kept at 4ºC for at least 12 hours. The fixed slices were processed with streptavidin. The slides were dried for 12 hours in the dark, then scanned using a confocal microscope (Nikon Eclipse Ti with C2 laser source, Tokyo, Japan) with 0.5 μm Z-stack steps. The spines were analyzed using imageJ and reconstruction programs.
Fig. 5: Hippocampal CA1 neurons tagged by a seizure have larger dendritic spines and are more excitable.

a) A fluorescent/DIC image showing hippocampal CA1 region in an acute hippocampal slice obtained from a c-fos/EGFP mouse after a single seizure. Tagged neurons are shown as bright ones in the image. The scale bar is 50 μm. Total 11 tagged neurons and 11 untagged surrounding neurons were tested for their membrane properties. b) A tagged CA1 neuron reconstructed through tracing using Imaris program (red). A non-tagged CA1 neuron (black) is shown for comparison. The scale bar represents to 100 μm. c) and d) Soma diameter and total dendritic length analyzed for tagged (GFP+ve) and non-tagged (GFP-ve) CA1 neurons (5 cells from 5 animals for each group, mean ± SD, P = 0.8526 for the dendritic length, P = 0.8472 for the soma diameter). e) Examples of dendritic sections obtained from a tagged and a non-tagged neurons, which were 200 to 250 μm away from the somas (s. radiatum area). Spine dimensional analysis was performed for tagged neurons and untagged neurons (5 neurons from 5 animals for each group, see F, G, H, I and J). f) Spine-head diameter for spines 200 to 250 mm away from the somas. Mean ± SD for 432 spines from 5 untagged neurons (black), and 444 spines from 5 tagged neurons (red). ***P < 0.0001. g) Spine-neck length for spines analyzed in c). P = 0.276. h) Spine number in 10 μm dendritic sections (density) for those spines analyzed in c). P = 0.931. i) A schematic illustrating classification of spines based on their morphology. j) Percentage of spines of each type (illustrated in panel i) for tagged and untagged neurons. * P = 0.0204 for mushroom shaped spines (red) and * P= 0.0431 for thin spines (blue). Long-thin spine (yellow), P = 0.4172; filopodia, black, P = 0.5861; Stubby, white, P = 0.8839. k) Action-potential (AP) threshold for tagged (red) and untagged (black) neurons (−48.23 ± 3.264 mV vs. −44.89 ± 2.891 mV, paired t-test, ** P = 0.0017). AP was evoked by current injection. AP threshold was measured from the first AP evoked during current injections. Initial point of AP upstroke phase is the threshold. l) Membrane resistance vs. rheobass for tagged (red circles) and untagged (black circles) neurons (** P = 0.007 for rheobass, P = 0.213 for Rm. Dashed lines show the mean values). m) Traces illustrate AP’s evoked at different current injections for a tagged and an untagged neurons. Current injections was performed from −100 pA to 300 pA with 20 pA increasing step, and lasted for 500 ms. To compare firing patterns between neurons, membrane potential was adjusted to −65 mV by using a small amount of current before the injections. n) Frequency-current (F-I) plot illustrating the frequency of AP’s evoked by each current injection, values are mean ± SD (n=11 per group. P < 0.0001, Two-way ANOVA). o) Instantaneous frequency vs. action potential sequence based on analysis of APs evoked by current injection, examples shown in m. The values represent mean ± SD, n=11 in each group. The lines represent two-phase exponential fit of the data (fast decay tau is 0.356, slow decay tau is 6.380).
2.7. Cell membrane properties
For testing membrane properties for tagged and untagged neurons, current injections were performed from −100 pA to 300 pA, which was increased by 20 pA steps and lasted for 500 ms. Firing patterns of neurons were compared by adjusting their membrane potential to −65 mV before the current injections.
2.8. Synaptic transmission mediated by AMPA
To test miniature excitatory postsynaptic current (mEPSC), and spontaneous excitatory postsynaptic current (sEPSC) (Fig. 6), Picrotoxin (100 μM) was added to the external solution to block inhibitory synaptic responses mediated by GABA receptors; 100 μM APV (D-2-amino-5-phosphonopentanoic acid) was added to prevent postsynaptic short-term plasticity and long-term plasticity. mEPSCs were recorded when presynaptic action-potential was blocked by 1μM Tetrodotoxin (TTX). sEPSCs were recorded when TTX was omitted. Decay kinetics were evaluated using weighted tau.
Fig. 6. AMPA synaptic transmission is enhanced in hippocampal CA1 neurons tagged by a single seizure.

a). Traces illustrating mEPSCs recorded from an untagged (black) and a tagged (red) CA1 neurons in animals which experienced PTZ-induced single seizure. These two neurons were recorded from the same acute slice. Averaged traces of mEPSCs from those two cells are also shown. b) Cumulative fraction curves illustrating distribution of mEPSC amplitudes. n = 15 cells for each group. The inserted graph shows mean of median mEPSC amplitudes andSD (paired t-test, *** P = 0.0002). c) Cumulative fraction curves illustrating distribution of mEPSC inter-event intervals. n = 15 cells for each group. The inserted graph shows the mean ± SD of mEPSC frequency (paired t – test, p = 0.5418). d) Cumulative fractions of sEPSC amplitude distributions for tagged and untagged CA1 neurons from PTZ seizure animals. Distribution of amplitudes of sEPSCs recorded from neurons activated under home cage-conditions are also plotted for comparison (blue). n= 15 each for untagged and tagged neurons from 15 seizure animals, and n= 10 tagged cells from 5 home cage-condition animals were analyzed. Inserted bar graph shows a comparison of sEPSC amplitude (means of median) for the three groups *** p<0.0001, one-way ANOVA. e) Cumulative fractions of sEPSC inter-event interval distributions for tagged and untagged CA1 neurons from PTZ seizure animals, and those neurons activated under home cage-conditions. Inserted bar graph shows a comparison of sEPSC frequency (mean ± SD) for the three groups (*** p = 0.0002, one-way ANOVA). f) Representative traces showing currents evoked at various holding potentials ranging from −45 mV to 45 mV recorded from an untagged neurons of a seizure animal. A linear I-V curve plotted from the same neuron. g) Representative traces showing currents evoked at various holding potentials from a tagged neurons of a seizure animal. Note the inwardly-rectifying currents. The I-V curve plotted from the same neuron illustrate a nonlinear I/V curve. h) Rectification index, a ratio of the current evoked at −45 mV to that at 45 mV, n= 6 in each group, *** P = 0.0007. i) Right panel shows averaged sEPSCs recorded from an untagged neuron before and after application of IEM 1460 (100 μM), a ca2+-permeable AMPA receptor blocker. Bar chart shows sEPSC amplitude for group results. n=6 neurons from 4 animals, mean ± SD, paired t-test, P = 0.0929. Pale color circles indicate mean results from individual neurons, lines linking results before and after the application of IEM1460. j) Right panel shows averaged sEPSCs recorded from a tagged neuron before and after application of IEM 1460 (100 μM), Bar chart shows sEPSC amplitude for group results. n=6 neurons from 4 animals, mean ± SD, paired t-test, *P = 0.0379. Pale color circles indicate mean results from individual neurons, lines linking results before and after the application of IEM1460
To verify GluA2-lacking AMPA receptors expressed on neurons after a seizure (Fig. 6), inwardly rectifying I/V curves were tested by using eEPSCs in response to different voltage-clamp steps. The stimulation pulse was applied to Schaffer-Collateral inputs.
The strength of stimulation was adjusted (10 to 50 μA for a 100 μs pulse) to produce a single peak EPSC with a fixed latency. Picrotoxin (100 μM) and 100 μM APV (D-2-amino-5-phosphonopentanoic acid) were added. Lidocaine (5 mM) was added to the internal solution to improve the space clamp. Membrane potentials were clamped from − 45 mV to +45 mV, 10 – 15 mV for each step. Current in response to membrane clamps was recorded, and an I/V curve was created.
2.9. LTP recording
To test LTP in hippocampal CA1 neurons (Fig. 7), LTP was evaluated by using eEPSC (Sun et al. 2018) in response to high-frequency stimulation (HFS, 100 Hz, 1s) on Schaffer-collateral axons. Stimulation was provided using a bipolar tungsten microelectrode (BAK Electronics, Mount Airy, MD) using a stimulator (Isostim A320, World Precision Instruments, Inc. Sarasota, FL USA). Learning activated neurons in the CA1 pyramidal layer from D2 trained mice were visually identified based on GFP fluorescence, patched in voltage-clamp configuration, and recorded in a holding potential of −65 mV for EPSCs.
Fig. 7. Saturated LTP in tagged neurons after a single seizure.

a). Time course of eEPSC amplitudes recorded from a tagged neuron. The amplitudes were normalized by the mean of control recordings before HFS. b). Average traces of eEPSCs for the tagged cell exhibited in A) before (a), and after (b) the application of HFS. c). Time course of eEPSC amplitude recorded from an untagged neuron. The amplitudes were normalized by the mean of control recordings before HFS. d). Average traces of eEPSCs obtained from the untagged neuron shown in panel C) before (a), and after (b) the application of HFS. e). Time course of normalized eEPSC amplitudes for tagged neurons (7 cells from 7 animals, red), and untagged neurons (6 cells from 6 animals, black), mean ± SD. f) Mean ± SD of eEPSC amplitudes after HFS for tagged and untagged neurons shown in panel E). The amplitudes were normalized by the mean of control recordings, *** P < 0.0001.
The amplitude and frequency of eEPSCs were analyzed using Clampex 10.5 and Mini Analysis. A paired t-test was used for tagged and untagged cells recorded in the same slice.
2.10. Dual labeling of neuronal ensemble activated following rewarded alternation learning and seizure in the same mouse
First, using 4-OHT injections on day 2 on T maze, we tagged the active neuronal ensemble. A week later, the same mouse received a PTZ injection, which caused a tonic-clonic seizure. Mice that had a seizure were sacrificed 60 minutes following a seizure, and ARC immunohistochemistry (1:1000, Rb polyclonal, 156003, Synaptic Systems) was used to identify seizure-induced neuronal activation. Dual labeling results were repeated and validated by cfos immunohistochemistry using an anti-cfos antibody (1:1000, Rb polyclonal, ab190289, Abcam) as well.
2.11. Statistical analysis
All statistical details with the number of animals are presented in the figure legends. All graphs show mean ± s.e.m. GraphPad Prism 8.0 was used for all statistical analysis, and for comparisons between and within the groups, two-tailed unpaired or paired Student’s t-test were used respectively. Data not modeled by a normal distribution was subjected to non- parametric Mann–Whitney U test and Wilcoxon signed-rank test for between and within-group comparisons, respectively. One-way and Two-way ANOVA and posthoc Bonferroni or Tukey’s correction were used for comparisons involving more than two groups. Statistical significance was set at * p < 0.05, ** P<0.01, ***P<.001.
3. Results
3.1. Seizure induced retrograde amnesia:
Mice were trained for five successive days to alternately enter two arms on a T-maze for a reward after they habituated to the maze and sweetened milk (Fig.1a, methods). Mice performed with similar, chance-level accuracy on days 1 and 2, but were faster on the second day. The following day mice alternated correctly more often and continued to do so on days 4 and 5. The inflection on the alternation learning curve occurred following day 2, suggested that mice were learning to alternately enter two arms on that day and consolidated and retrieved this memory on subsequent days (Fig.1b).
We tested whether a pentylene tetrazole (PTZ)-induced seizure given to mice, 15 minutes after learning (day 2), would affect their performance on the following day (Fig. 1c). Seizure-treated mice were far less accurate than saline-treated controls (Fig.1d-g). This seizure-induced memory impairment was transient, as seizure-treated animals alternated as often as saline controls on day 5 (Fig. 1e). There was a significant decrease in latency to the sample arm reward zone across training sessions from days 1 to 5 in both control and seizure group mice (Supplementary Fig. S1A). Similar latency to the choice trial across days 1 to 5 suggests that failure to alternate in these mice was not due to locomotor deficits (Fig 1d). Animals that did not have a seizure following a PTZ injection performed as accurately as saline-treated animals (Fig. S1B). A seizure following training on day 1 (Fig. S1C ) or seizure on day 5, (when the alternation accuracies had stabilized) of training did not affect alternation accuracy on the following day (Fig. S1D). Thus, mice learned to alternate on the second day of training.
3.2. Neuronal ensemble tagged while learning T-maze alternation
We tagged cfos-expressing neurons as mice learned to alternate on the T-maze. To tag learning-ensemble, we used TRAP mice and injected fast-acting 4-hydroxy tamoxifen (4-OHT) 45 minutes after the last training trial. Four cohorts, consisting of the homecage group, groups of animals studied on day 1, day 2, or day 3 of training (day 1, 2, and 3 groups respectively, see methods for details), were studied. CA1 pyramidal neurons play an essential role in encoding spatial memory, so we explored these first and in greater detail (O’Keefe and Dostrovsky, 1971, Moser and Moser, 1998, Henriksen et al., 2010). A small number of CA1 pyramidal neurons were tagged in the day 1 group as well as in the homecage group (Supplementary Fig. S2). Coronal slices through the hippocampus showed one or two tdtomato – expressing CA1 neurons from day 1 group, whereas, five to eight tagged CA1 neurons per slice in day 2 group mice (Fig.1h, i). Learning-tagged neurons in CA1 were present bi-laterally (Supplementary Fig. S2). We counted every tagged CA1 pyramidal neuron in all (60) serial sections encompassing the hippocampus in a blinded, unbiased fashion. There were three times as many labeled CA1 pyramidal neurons labeled in the day 2 group as compared to the day 1 and day 3 group (Day 1; 47.2 ± 4.33, Day 2; 258.0 ± 27.4 and Day 3; 73.4 ± 9.21, n=6; *** P<0.001, # P<0.05, Fig.1i, n, Table. 1, and Figs. S3 & S5). Labeled CA1 pyramidal neurons were more frequently present in the dorsal hippocampus than the ventral hippocampus (Figure S5, tagged CA1 neurons along the anterior-posterior axis). In CA1, there were more learning-tagged neurons in distal dorsal-CA1 (dCA1, distal to CA3) than the proximal part of CA1 (Fig1k, l, m, also see location maps in Fig. 2, Fig. S4). This activation pattern was consistent with previous studies that reported the dorsal hippocampus’s pivotal roles in learning (Fanselow and Dong, 2010). There were no tagged CA1 neurons in the most posterior slices of the hippocampus in coronal sections of the entire hippocampus (Fig. 2 a-i, k, l). This finding was consistent with previous studies, which demonstrate that CA1 pyramidal neurons encoding spatial memory are distributed heterogeneously along the septo-temporal axis (Henriksen et al., 2010, Nakazawa et al., 2016).
Table 1.
Table shows the quantification data as total tdTomato+ve CA1 pyramidal neurons (cumulative for all serial slices through hippocampal CA1) found in different experimental paradigms, sample size and detail of statistical comparsions.
| Experimental Paradigm | Total no. of tdT+ve CA1 neurons (mean ± SE) | ||
|---|---|---|---|
| T-maze Day1 | 47.20 ± 4.33 (n=6) | One-way ANOVA F=26.61 Diff. among means |
P<0.0001 |
| T-maze Day2 | 258.0 ± 27.4 (n=9) | (Yes) P<0.05 | |
| T-Maze Day 3 | 73.40 ± 9.21 (n=6) | ||
| PTZ seizure alone | 293.40 ± 22.53 (n=6) | Unpaired t-Test t=6.102 | P=0.003 |
| Saline controls | 124.60 ± 16.05 (n=6) | ||
| T-maze Day3 (D2+saline) | 31.60 ± 5.06 (n=5) | Unpaired t-Test, t=4.904 | P=0.0012 |
| T-maze Day3 (D2+ seizure) | 181.20 ± 30.09 (n=6) | ||
| Home-cage control | 41.66 ± 5.23 (n=4) | Unpaired t-test t=0.83 | P=0.45 |
| 24hr post PTZ seizure | 49.50 ± 8.50 (n=2) | ns |
Fig. 2. Location maps of learning tagged CA1 pyramidal neurons along rostrocaudal axis a-j).
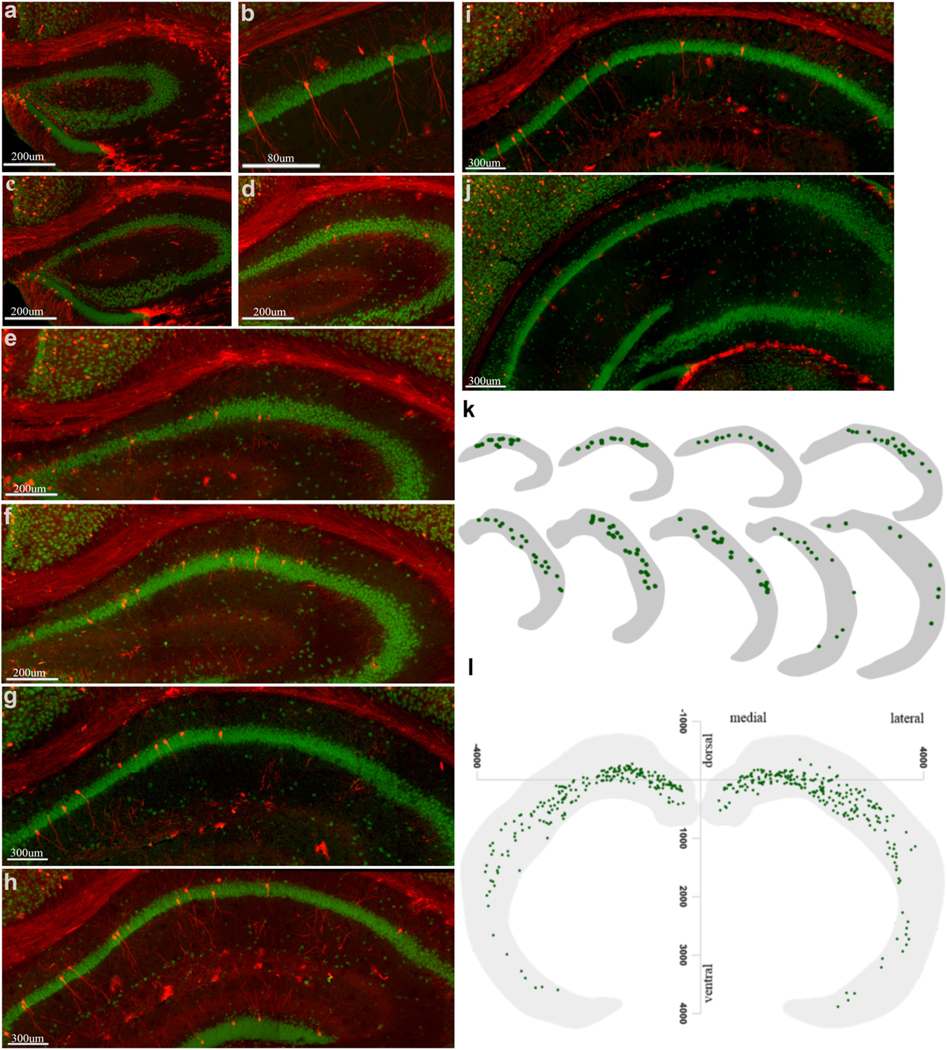
Representative images show learning-activated tagged CA1 pyramidal neurons (tdTomato) with NeuN (green) on the second day of training across the anterior dorsal-posterior ventral axis. There were more active neurons in dorsal CA1 (dCA1). b) Morphology of typical learning-activated dorsal CA1 pyramidal neurons. k) The location of tagged CA1 pyramidal neurons from 5 hippocampal slices was mapped in a section. Sections were arranged along the rostrocaudal axis. Note the greater density of labeled neurons in the anterior-dorsal region than in larger ventral slices. l) A composite location map of all learning activated CA1 pyramidal neurons, which shows anterior-posterior and medial-lateral gradients in the distribution.
Since the hippocampus is engaged in the retrieval of recent memories (Riesel et al., 2003, Frankland and Bontempi, 2005; Tanaka et al., 2014), we expected a similar population of tagged pyramidal neurons in CA1 in the day 3 group as observed in day 2 group. Surprisingly, this was not the case. There were fewer labeled CA1 pyramidal neurons in CA1 in the day 3 group compared to the day 2 group (Fig. 1j, n, Table. 1, Fig. S5). The retrieval of memory on the third day may have occurred by activation of a small number of tagged CA1 pyramidal neurons, as previously reported by Reijmers et al., 2007.
3.3. Synaptic potentiation of tagged neurons suggested participation in learning
Several lines of evidence indicate that labeled neurons in the day 2 group participate in memory formation. There was an inflection in the learning curve following the second training day, seizure following learning that day diminished performance on the following day (Fig.1e). Finally, more tagged CA1 pyramidal neurons were present in dorsal CA1 in the day 2 group than the previous day (Fig.1n). There is learning-induced synaptic potentiation in engram cells (Poo et al. 2016; Ryan et al. 2015); therefore, we determined whether excitatory synaptic currents in tagged CA1 pyramidal neurons in the day 2 group were also potentiated. TRAP mice were not suitable for electrophysiological studies of tagged neurons following learning as it takes several days for the fluorescent protein to accumulate sufficiently for visualization in a thick slice. To overcome this, we used TetTag mice (Fig. 1p, see methods for details), which allowed immediate visualization of tagged neurons with EGFP. We confirmed that the rewarded alternation learning curve for TetTag mice was similar to TRAP and c57 Bl6 mice, with an inflection following the second day of training (Fig. S6). CA1 pyramidal neurons were tagged (GFP+ve) following training on day 2 in these TetTag mice (DIC image, bright cells, Fig. 1q), as in TRAP mice (Fig.1m).
We investigated AMPA-mediated synaptic transmission in tagged and surrounding untagged neurons after learning T-maze on day 2. The amplitude of AMPA receptor-mediated spontaneous excitatory postsynaptic currents (sEPSCs) recorded from tagged neurons was larger than those obtained from neighboring untagged neurons (Fig.1o). We confirmed this finding using 11 pairs of neurons (one labeled and one untagged neuron) from 8 animals (Fig.1r). The sEPSCs recorded from tagged neurons were more frequent than in untagged neurons (Fig.1s). There were no differences in sEPSC rise and decay time between tagged and untagged neurons.
3.4. Memory and seizure-activated neuronal circuits overlap
We tested whether seizures activate neuronal circuits similar to the memory engram. A seizure labeled more CA1 pyramidal neurons than a saline injection (Fig.3, Table. 1). There were many seizure-tagged neurons in dorsal CA1, and there were far fewer tagged cells in the posterior-ventral regions (Fig. 3a-g).
Fig. 3. Seizure-tagged CA1 neuronal ensemble map is similar to memory map:
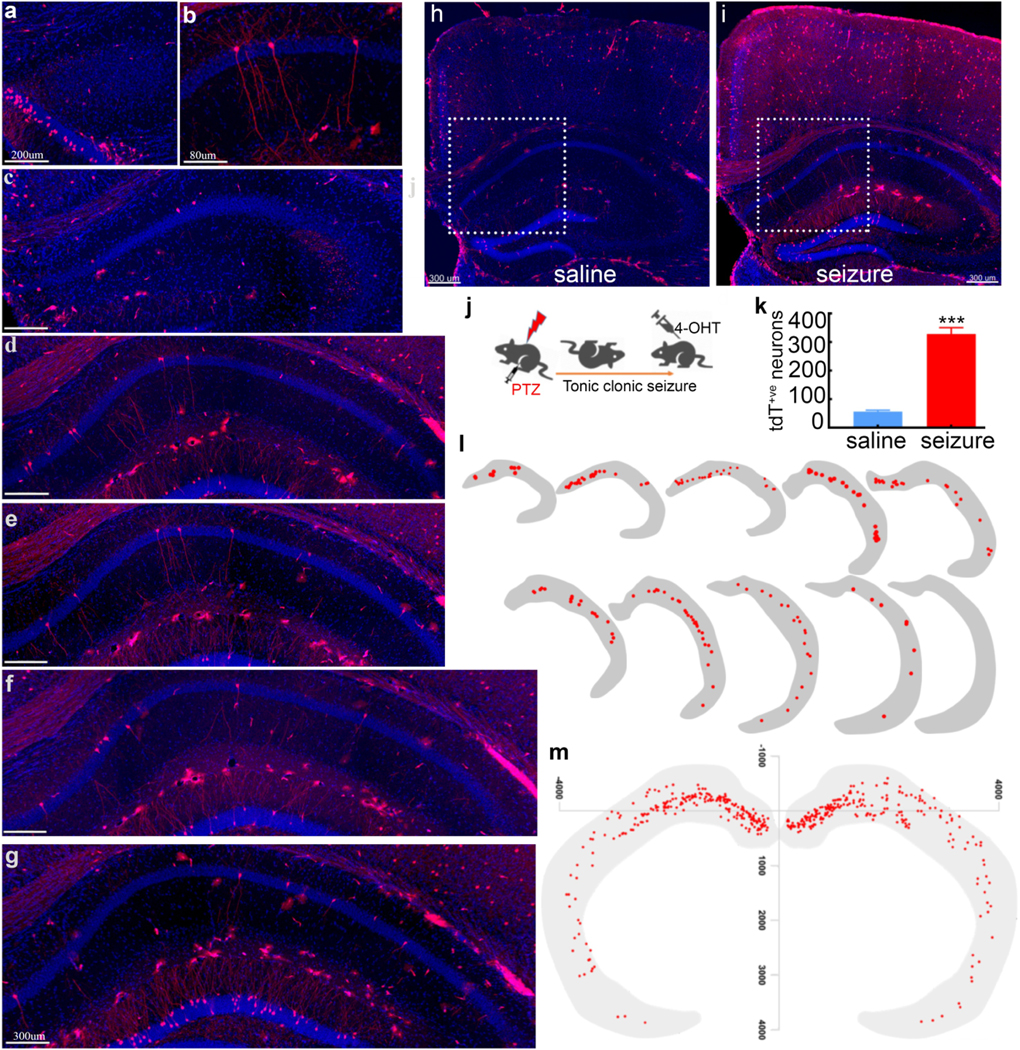
a-i) Representative images show tagged CA1 pyramidal neurons (red) activated following a seizure across the anterior-posterior axis, with DAPI (blue) counterstaining. Anterior dorsal CA1 had more tagged neurons than the ventral part. h) There were fewer tdT+ve neurons in representative images of the hippocampus from saline-injected mice, compared to seizure treated mice. There were one to two tagged CA1 pyramidal neurons (dotted square) per 40μm hippocampal slice. (i), with 5–7 tagged pyramidal neurons in distal dCA1 (dotted square). j) The experimental design for tagging neurons by a seizure. k) Seizure tagged more CA1 pyramidal neurons than saline injection (n=6, ***p<0.001). l) Unilateral location maps (10) representing 50 (40μm) hippocampal slices show the distribution of seizure-tagged pyramidal CA1 neurons rostrocaudal axis. m) Combined location map of seizure-tagged CA1 ensemble showing their distribution across the rostrocaudal and medial-lateral axis.
Visual examination suggested that CA1 neurons tagged by a seizure were at similar positions as memory labeled neurons. We created a detailed map of activated neurons in CA1 following learning (day 2) and after a single seizure across the rostrocaudal axis to test for similarities in distribution. We recorded all tagged CA1 pyramidal neurons’ positions on a horizontal plane across all serial coronal slices and generated location maps (Figs. 2 a-l & 3 a-m). We merged maps from the two experiences and found that the learning and seizure activated neuronal ensembles were quite close to each other, raising the possibility of overlap between the two (Fig.4 g).
Fig. 4. Memory and seizure-activated CA1 pyramidal neuronal ensembles overlap:
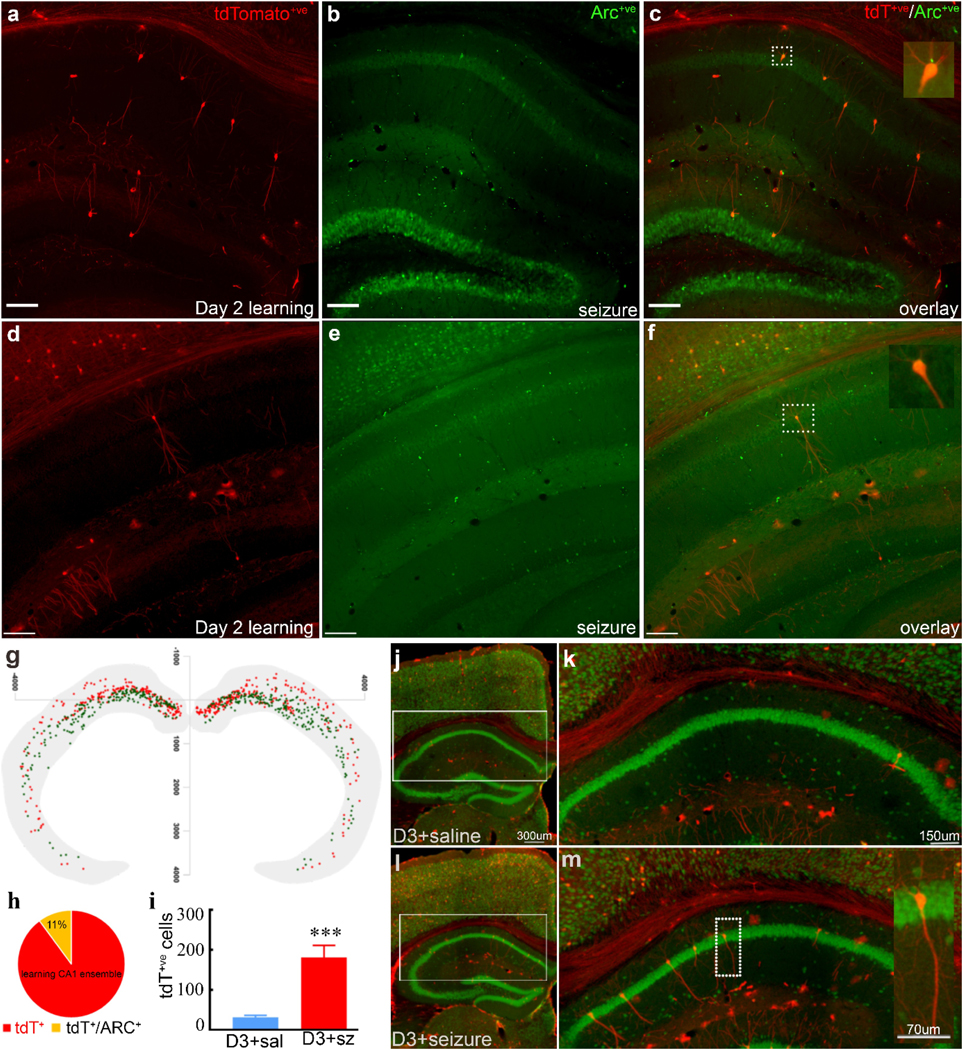
We detected the memory ensemble by TRAP method and seizure activated neurons by ARC immunohistochemistry in the same animal and analyzed them for overlap. a) Learning activated CA1 pyramidal neurons (red), and seizure-tagged neurons (ARC+ve, green, b) in the anterior-dorsal region, and c) the overlap of ensembles. Inset shows a magnified view of a dual labeled CA1 pyramidal neuron (dotted square). Images of CA1 pyramidal neurons tagged by learning d), seizure e) and merged f) from the posterior ventral part of the hippocampus. The inset is a magnified view of the neuron activated by learning and seizure (dotted square, yellow). g) A composite location map of CA1 from memory ensemble (green dots) and a seizure activated neurons (red dots) also shows proximity overlap in the distal dorsal part of CA1. h) A pie chart shows that 11 percent of learning-tagged tdT+ve CA1 neurons colocalized for ARC (seizure-tagged). i) More tagged CA1 pyramidal neurons on day 3 in mice that had experienced a seizure a day before, as compared to saline controls. CA1 pyramidal neurons in the hippocampus TRAPed on the 3rd day of training after mice received either saline (j, k) or tonic-clonic seizure (l, m) on day 2. Magnified images (k, m) from the dotted rectangle area of dorsal CA1 show significantly more tagged CA1 pyramidal neurons in the seizure group than saline. The inset image shows an enlarged view of a typical pyramidal neuron. (n=6 / group,*** P<0.001).
We simultaneously labeled for learning and seizure activated neurons to determine the precise overlap between learning and seizure-activated neuronal ensemble. In TRAP mice, we tagged neurons after training on the second day of training (Fig. 4 a,d). A week later, we delivered a seizure and detected seizure-activated neurons using ARC-immunohistochemistry (Fig. 4 b,e). ARC immunolabel and tdTomato colocalized in neurons in the CA1 region of the hippocampus (Fig.4 c, f, Fig. S7). tdTomato and ARC positive neurons in the CA1 region were counted within a frame were expressed as a percent fraction of all the NeuN labeled neurons for calculating chance overlap. Chance levels for double labeling of tdTomato and ARC were less than 0.06 ((total number of tdT+ve / total number of NeuN +ve) * (total number of ARC+ve / total number of NeuN+ve)*100 %). There was a significant overlap between memory and seizure activated ensemble in the CA1, 11% of tdTomato labeled CA1 pyramidal neurons co-labeled for ARC (Fig. 4 h). This fraction was more than the probability of random colocalization (see methods for details). As a negative control, we tested colocalization in mice injected with 4-OHT in their homecage and later received a single seizure, (Supplementary Fig. S7 H-K). Only a smaller fraction (2.5%) of tdTomato+ve homecage activated CA1 pyramidal neurons colocalized forseizure activated ARC in this group (Supplementary Fig. S8).
3.5. Seizure resets the memory circuit
We found that a seizure following learning on the second day induced memory impairment, which improved on subsequent days (Fig. 1e). Furthermore, we saw peak tagging of neurons in the day 2 group (Fig. 1k, l, n, Table. 1). Finally, there was an overlap in seizure and learning activated neurons (Fig. 4 a-h, Supplementary Fig. S7). We tested whether a seizure following the second day would shift peak activation to the third day of training when these animals learned to alternate on the T-maze. Hence, we randomized TRAP mice after training on the second day of training to receiving a seizure or saline and TRAPed neurons after 3rd day of training (Fig. 4 j-i).
In the saline-treated group, there were few tagged CA1 pyramidal neurons (Fig. 4j, k, Table. 1), whereas in the seizure–treated group (Fig. 4 l, m), there were far more tagged neurons as compared to saline controls (Fig. 4 I, Table. 1). These labeled CA1 pyramidal neurons were present bilaterally, and the density was more in anterior-dorsal than posterior-ventral regions. Following the 3rd day of training, seizure-treated mice exhibited a day 2-like tagged CA1 pyramidal neuron population, consistent with the learning curve for these mice. A seizure after learning on the second day had reset the hippocampal trace, which was engaged in the usual manner the following day. Tagged CA1 pyramidal cells on day 3 could represent residual cfos activation after a seizure the day before. We injected 2 mice with 4-OHT 24 hours after a seizure, and the number of tagged neurons was similar to homecage controls (Fig. S9, Table. 1).
3.6. Seizure engages memory mechanisms
We tested whether neurons tagged following seizure engaged memory mechanisms in CA1 pyramidal neurons. First, we compared tagged and untagged neuron morphology (Fig. 5 a, b). Forty-eight hours before inducing a seizure, we removed doxycycline from the diet of TetTag mice and prepared hippocampal slices 30 minutes following a seizure. We filled tagged, and surrounding untagged neurons with biocytin visualized using a glass micropipette. Biocytin-filled neurons were visualized using fluorescent streptavidin and imaged using a confocal microscope (Fig. 5a).
Although dendritic lengths and soma diameters were similar in seizure-tagged and untagged neurons (Fig. 5b-d), there were differences in the density of distal dendritic spines. Dendritic spines are enlarged in engram cells (Ryan et al., 2015), and following LTP (Nicoll, 2017). Therefore, we observed whether there was a similar enhancement of spines in seizure-tagged CA1 pyramidal neurons. We analyzed mesial dendritic spines, 100 μm or closer to the soma, and distal spines, 200 to 250 μm from the soma, within the stratum radiatum (5e). There were no differences in mesial spine head diameter, neck length, or density between tagged and untagged neurons (Fig. S10). The distal spine head diameter was 14% larger in tagged neurons than untagged neurons, with no differences in neck length and density (Fig. 5 f-h). We also analyzed types of distal spines (Fig. 5i) based on characteristics described previously by Risher et al., (2014). Mushroom spines were more numerous, while there were fewer thin-type spines in tagged neurons. Of the distal spines in tagged neurons, 31.3% were mushroom-shaped, where the diameter of the spine head is twice the length of its neck (red area in the pie chart Fig. 5j), in contrast to 14.8% in the untagged neurons (P = 0.0204). Thin spines were more abundant in untagged neurons (blue area, 46.9%) compared to tagged neurons (34.8%, P = 0.043). Seizure-labeled neurons had enlarged dendritic spines.
3.7. Active and passive membrane properties
Experiential learning can enhance neuronal excitability (Ryan et al., 2015), so we tested whether seizure-tagged CA1 pyramidal neurons were more excitable than surrounding untagged cells. We analyzed the resting membrane potential, membrane resistance, and membrane time constant of tagged and surrounding untagged CA1 neurons in hippocampal slices from mice that had experienced a seizure (Fig. 5 k-m). Passive membrane properties, including resting membrane potential, membrane time constant, and membrane resistance, were similar between these two cell types (Fig. S11). Tagged and untagged neuron action potential amplitude and width were similar in (Fig. S11 D & E). However, the action potential threshold was closer to the resting membrane potential compared to surrounding untagged cells (Fig. 5k). We confirmed this by analyzing the relationship between membrane resistance and rheobase current. The membrane resistance of tagged and untagged cells was similar, whereas the rheobase current was lower in tagged cells (Fig. 5l). We then compared the frequency of spikes in response to intracellular depolarizations (Fig. 5m), and it was larger in tagged neurons compared to untagged neurons (F-I plot, Fig. 5n). Spike-frequency adaptation was less in tagged neurons compared to untagged neurons (Fig. 5o). In summary, labeled neurons were more excitable.
3.8. Larger EPSCs recorded from tagged neurons
We evaluated whether excitatory transmission on seizure- tagged CA1 neurons was enhanced. We investigated AMPA-mediated synaptic transmission after induction of a single seizure (Fig. 6). The amplitude of AMPA receptor-mediated miniature excitatory postsynaptic currents (mEPSCs) recorded from a tagged neuron was larger than neighboring untagged neurons (15 pairs of tagged and untagged neurons recorded from 15 animals; Fig. 6a-c). The frequencies of mEPSCs and their rise times and decay times recorded were similar (Fig. S12).
We further confirmed the enhancement of EPSC amplitude by studying action potential-dependent spontaneous EPSCs (sEPSCs). sEPSCs recorded from tagged neurons were larger and more frequent than surrounding untagged neurons (Fig. 6d-h). Other characteristics of sEPSCs were not different (Fig. 6 i,j). To verify that larger EPSCs in tagged neurons were because of seizure and not due to fluorescent protein expression (tagging), we studied labeled neurons in naive mice 48 hours after taking them off the doxycycline diet. The amplitude and frequency of sEPSCs recorded from tagged neurons (tagged following homecage activity) in these naïve mice were similar to those recorded from untagged neurons in mice with a seizure (results in blue color in Fig. 6d, e). Thus, there was a specific enhancement of AMPA-mediated transmission in neurons activated by a seizure.
3.9. Inwardly rectifying I/V curves in tagged neurons
The larger EPSCs recorded from seizure-activated tagged neurons could be due to the insertion of a GluA1 subunit, which occurs after LTP (Shimshek et al., 2017). The AMPARs expressed on the CA1 neurons are primarily heterodimers consisting of GluA1 and GluA2 subunits, where insertion of a GluA1 subunit will alter the proportion of GluA1/GluA2 subunits, making them increasingly Ca2+ permeable. The presence of a GluA2-lacking subunit in the AMPARs was probed by determining the current-voltage relationship to look for an inwardly rectifying current (Bowie and Mayer, 1995). We evoked EPSCs (eEPSCs) by stimulating Schaffer-collateral inputs in CA1 neurons voltage-clamped to voltages ranging from −45 mV to +45 mV. (Fig. 6). In a tagged neuron, eEPSCs at positive holding potentials were smaller than those produced at equivalent negative holding potentials (Fig. 6b), which resulted in an inwardly rectifying I/V curve (Fig. 6d). In contrast, eEPSCs recorded from the surrounding untagged neurons (Fig. 6a) demonstrated a linear I/V relationship (Fig. 6f, g). The rectification index, the ratio of current amplitude recorded at −45 mV to that recorded at +45 mV, was greater in tagged neurons compared to untagged neurons (Fig. 6h).
We evaluated IEM 1460 sensitivity of AMPAR-mediated sEPSCs in tagged and untagged neurons because GluA2-lacking AMPARs are sensitive to it. After a 5-minute baseline recording, we applied the drug for 10 minutes. In tagged neurons, IEM 1460 reduced the amplitude of sEPSCs (Fig. 6i, six cells from 4 animals, paired t-test, P = 0.0379) but did not affect the sEPSCs recorded from untagged neurons (Fig. 6j). These findings indicate increased expression of GluA2 subunit-lacking AMPARs in CA1 neurons tagged by a single seizure.
3.10. HFS failed to potentiate the seizure-tagged CA1 neurons
We hypothesized that long-term synaptic potentiation (LTP) would be saturated in tagged CA1 neurons because seizure had already engaged the cellular mechanism of LTP. We stimulated Schaffer-collateral inputs to CA1 neurons to consistently evoke a single eEPSC. After recording baseline eEPSCs from a tagged neuron for 5 minutes, we administered a high-frequency stimulus train (100 Hz, 1 second), and continued recording for 40 minutes (Fig. 7a). HFS caused transient post-tetanic augmentation of the eEPSC amplitude, which then dropped to baseline (Fig. 7a, b). In contrast, in the untagged neurons, the enhancement of eEPSC amplitude persisted for 40 minutes following high-frequency stimulation (Fig. 7c, d). There were similar eEPSC amplitudes in both tagged and untagged cells after high-frequency stimulation (Fig. 7e). In contrast, the labeled neurons had larger eEPSC amplitudes in response to threshold stimuli (Fig. 7e). We tested seven tagged neurons from 7 animals and six untagged neurons from 6 animals, and following high-frequency stimulus, the normalized eEPSC amplitude in labeled neurons was 67% less than that in untagged neurons (P < 0.0001, Fig. 7e, 7f).
4. Discussion
We propose that seizures cause retrograde amnesia by engaging neuronal circuits and cellular mechanisms involved in memory formation and consolidation. A generalized tonic-clonic seizure after learning caused retrograde amnesia for a hippocampus-dependant memory task. Memory formation and a seizure caused cfos expression in specific neurons (engram cells) in the hippocampus. Neurons labeled during memory encoding and after a seizure physically proximate and overlapped more than that expected by chance in the CA1 hippocampus. A seizure potentiates excitatory synapses on CA1 neurons and causes LTP saturation. A concert of pathological overlap at the cellular and neuronal ensemble levels between memory and seizure, along with altered physiological state, explains retrograde amnesia following a seizure.
We studied the mechanisms of seizure-induced retrograde amnesia at the cellular and circuit levels from the perspective of engram theory. Seminal studies by Duncan (1949) and others (McGaugh, 2000) showed that seizures cause retrograde amnesia. Seizure-induced retrograde amnesia occurs in epilepsy (Butler and Zeman, 2008, Shuman et al., 2020). Seizures administered as electroconvulsive therapy (ECT) to treat medically refractory major depression also cause retrograde amnesia (Brus O et al., 2017; Meeter et al., 2011; O’ Connor et al., 2008; Squire et al., 1976).
A hypothetical physical means of storing new memory, engram refers to enduring bio-physical and biochemical modifications elicited by learning in a population of neurons called engram cells (Asok et al., 2019; Frankland et al., 2019; Jocelyn and Tonegawa, 2020; Poo et al., 2016). Recent advances in tagging specific neurons activated during an experience have enabled researchers to identify associated neurons and microcircuits. Within a brain region, a population of such engram cells involved in a particular computation constitutes an engram cell ensemble. The synchronous activity of cells following learning within a specific time window results in the formation of neuronal memory ensemble. We leveraged TRAP and TetTag transgenic mice to investigate the nature of retrograde amnesia from the perspective of the engram theory. CA1 pyramidal neurons propagate seizures and help form memories; thus, they are a likely site for the intersection between seizure and memory engrams. These neurons fire actively during temporal lobe seizures, which induce excitotoxicity and loss of these neurons, commonly referred to as mesial temporal sclerosis (Blumcke et al., 2007, Fujita et al., 2014, Goldstein and Abrahams, 2013). PTZ blocks GABA-A receptors, and resulting disinhibition can initiate recurrent synchronous bursting of a population of CA1 neurons, their input cells CA3 pyramidal neurons (Wong et al., 1986). The blockage of GABAergic inhibition compromises the gating function of dentate granule cells, allowing passage of seizures into the hippocampus (Dengler and Coulter, 2016; K-Magnuson, 2017).
CA1 pyramidal neurons are necessary for acquisition, consolidation, and retrieval of spatial memory, and we observed activation of cfos in a subset of these neurons following T-maze learning (Nakazawa et al., 2004; Reisel et al., 2003; Tanaka et al., 2014). In addition to this role in memory, CA1 place cell firing generates a spatial cognitive map (O’Keefe and Nadel, 1978, O’Keefe, 1999). It is likely that tagged CA1 neurons observed in our study are memory engram cells and not place cells, for several reasons. Place cells have characteristic stable place fields, whereas there were more tagged neurons on the second day of training compared to the first or the third day. A study by Henriksen et al., (2010) found that distal CA1 encodes contextual rather than place information.
Furthermore, synapses on tagged CA1 pyramidal neurons were potentiated, which is a property of engram cells, not place cells. Previous animal studies have also reported such synaptic potentiation following fear learning in the hippocampus (Whitlock et al., 2006) and amygdala (McKernan and Shinnick-Gallagher, 1997, Rumpel et al., 2005). We found a higher concentration of tagged neurons in the distal dorsal CA1, and lesioning this hippocampus area leads to spatial memory deficits. Yoon et al (2008) demonstrated that targeted infusions of muscimol in the dorsal hippocampus of rats before the delayed alternation task impaired reference memory as well as choice alternation accuracy.
Finally, Tanaka et al., (2017) demonstrated that tagged cfos-expressing CA1 neurons following spatial learning map experience rather than space. Memory index theory posits that CA1 pyramidal neurons serve as index cells that provide efficient access to memories stored in the neocortex.
We found that seizure- tagged CA1 pyramidal neurons were more excitable and had potentiated synapses, similar to engram cells. Intrinsic excitability can determine the recruitment of a neuron in a representative engram (Pignateli et al., 2019). Also, overexpressing CREB in specific neurons helps recruit them in a fear engram (Park et al., 2020). Seizure tagged neurons in our study were similar to the burst firing CA1 pyramidal neurons reported by Graves et al. 2016. Three parameters were initial firing frequency, post-spike depolarization (ADP), and threshold. However, we did not find differences in other parameters, which could be due to a smaller sample in our study and differences between rat and mouse CA1 pyramidal neurons. A subset of CA1 pyramidal neurons are likely more excitable (Graves et al. 2016), and memory or seizures recruited into the engram network. Several other studies suggest that increasing excitability in a small, random population of neurons preferentially recruits them into the engram (Brightwell et al. 2007; Sekeres et al. 2010, 2012, Park et al. 2016, Josselyn 2010, Rogerson et al. 2014, Frankland & Josselyn 2015, Kim et al. 2016). Park et al. (2019) also showed that increasing CREB function in a small but random population of principal neurons in the lateral amygdala minutes before training preferentially allocated them to the underlying memory engram.
We found synaptic potentiation in tagged CA1 neurons following learning and interestingly, also, after a single seizure. LTP saturation in seizure-tagged may render them unfit for being recruited in a memory trace. Notably, previous studies have associated LTP saturation with impaired spatial learning (Barnes et al., 1994, Nicoll, 2017). Here, we report for the first time that a single seizure-induced enlargement of spines on the distal, but not proximal, apical dendrites of activated-CA1 pyramidal neurons. Distal apical dendrites of CA1 pyramidal neurons receive inputs from the entorhinal cortex. The coordination of the entorhinal cortex with CA1 pyramidal neurons plays a role in associative learning (Colgin et al., 2015; Li et al., 2017). Entorhinal cortex and CA1 pyramidal neurons are likely fire together during a seizure, which potentiates synapses.
Many previous studies have suggested that seizures and memory share mechanisms. William Gowers (1881) made a seminal observation regarding seizures that “the tendency of the disease is to self-perpetuate.” Careful studies in patients with temporal lobe epilepsy and in animal models suggest that individual seizures leave a memory trace that renders the subject more susceptible to the next seizure (McNamara et al., 1980, McNamara, 1988). Several signaling pathways are involved in memory and seizure-induced plasticity, including Ca2+ entry, BDNF, cAMP, MAP kinase ERK, and CREB (Flavell and Greenberg, 2008, Asok et al., 2020).
Transient retrograde amnesia reported here is distinct from fixed inter-ictal memory dysfunction observed in persons with temporal lobe epilepsy and animal models of the disease (Baker and Zeman, 2017; Bui et al., 2018; Coras et al., 2014; Muller et al., 2009; Reyes et al., 2018;). Mechanistic studies have primarily focused on hippocampal pathology, including principal and inter-neuron death and pathological and anatomical circuit reorganizations (Bell et al., 2011; Santini and Scott, 2015). These pathological changes result in the reduced synchrony of interneuron firing and diminished stability and reliability of place fields (Holmes, 2015; Liu et al., 2003; Santini and Holmes, 2008; Shuman et al., 2019).
A seizure following learning T-maze alternation shifted the learning curve to the right and reset the CA1 engram, which was likely due to overlap and proximity of neuronal ensembles activated by seizures and learning. The overlap amongst the learning-tagged and seizure tagged neurons in CA1 was more than expected by chance. The original engram studies (Reijmers et al., 2007) found that only 12% of all neurons tagged during the fear learning paradigm were active during retrieval. Seizure – activated cells may have interfered with the memory network through potentiated synapses and saturated LTP. This overlap during the critical consolidation window may impair neuronal ensemble stabilization, which likely depends on synaptic potentiation.Thus seizures could erode the recently formed memories by hijacking the memory mechanisms.
Many of these seizure-activated neurons were close to engram cells in the CA1 region and expressed ARC. ARC is reported to be released from neurons in capsid like form and moves across neighboring neurons (Pastuzyn et al., 2018). Finally, some studies have shown that immediate early gene ARC induces synaptic depotentiation (Husi et al., 2000, Okuno et al., 2012;). A seizure may have interfered with memory at sites outside the hippocampus, and we are currently investigating this possibility.
Hijacking of the cellular mechanisms may cause the failure of sharp-wave ripple mediated hippocampal-cortical interaction, thereby perturbing systems consolidation (Buzsaki, 2015; Wang et al, 2006). In patients with epilepsy and animal models of temporal lobe epilepsy, epileptiform discharges interfere with sharp-wave ripples and impair memory consolidation (Buzsaki, 2015, Gelinas et al., 2016). CA1 ensembles are activated during high-frequency oscillations, and seizure induces pathological high-frequency oscillations (pHFO) in the network. A recent study by Ewell et al., 2020, showed disruption of hippocampal place code in epileptic animals is associated with co-generation of pHFO and ripple oscillations in the CA1 network. This finding that pathological and physiological ripple events could occur in the same seizure-genic network supports the failure of systems consolidation. Convincing evidence supports the crucial role of NMDA receptors and its downstream signaling molecules like Ca2+/CAMKII in early phase LTP and memory encoding and consolidation (Lisman and Zhabotinsky, 2001; Malinov et al., 1989; Mayford et al., 1995). Since we showed seizure-induced plasticity changes, on the one hand, this supports the engram nature of seizure and on other possible interference points for memory consolidation. However, memory impairments may not necessarily stem from interference in the hippocampus alone, as memory has a brain-wide engram. Future studies will dissect the circuits at the level of the engram complex.
Supplementary Material
Highlights:
A single seizure caused transient retrograde amnesia
Learning and seizure activated CA1 hippocampal neuronal ensembles overlapped.
Seizure potentiated CA1 neuron synapses such that further LTP did not occur
Seizures hijack memory mechanisms to interfere with storage and retrieval.
Acknowledgments:
We thank Dr. Brian J. Wiltgen and Dr. Mark Mayford for sharing TetTag transgenic mice, Dr. Suchitra Joshi, and Dr. John Williamson for discussing experimental design and results. Funding: This work was supported by the UVA Brain Institute and NIH (NINDS) grants R01 NS 040337 and R01 NS 044370 to J.K.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Du F, Eid T, Lothman EW, Kohler C, Schwarcz R. 1995. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J. Neurosci 15.10, 6301–6313. 10.1523/jneurosci.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell B, Lin JJ, Seidenberg M, Hermann B, 2011. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat. Rev. Neurol 7.3, 154–164. 10.1038/nrneurol.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sloviter RS,1994. On the relationship between neuropathology and pathophysiology in the epileptic hippocampus of humans and experimental animals. Hippocampus. 4.3, 250–253. 10.1002/hipo.450040304 [DOI] [PubMed] [Google Scholar]
- 4.Houser CR, 1990. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain res. 535.2, 195–204. 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Lu G, Zhong Y, Tan Q, Yang Z, Liao W, Liu Y, 2009. Impaired attention network in temporal lobe epilepsy: a resting FMRI study. Neurosci. Lett 458.3, 97–101. 10.1016/j.neulet.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 6.Gelinas JN, Khodagholy D, Thesen T, Devinsky O, Buzsáki G, 2016. Interictal epileptiform discharges induce hippocampal–cortical coupling in temporal lobe epilepsy. Nat. med 22.6, 641–648. 10.1038/nm.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chorover SL, Schiller PH, 1965. Short-term retrograde amnesia in rats. J. comp. Physiol. Psychol 59.1, 73. 10.1037/h0021606 [DOI] [PubMed] [Google Scholar]
- 8.Duncan CP,1949. The retroactive effect of electroshock on learning. J. comp. Physiol. Psychol 42, 32. 10.1037/h0058173. [DOI] [PubMed] [Google Scholar]
- 9.Kim JJ, Fanselow MS, 1992. Modality-specific retrograde amnesia of fear. Science. 256, 675–677. 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 10.Kopp R, Bohdanecky Z, Jarvik ME, 1966. Long temporal gradient of retrograde amnesia for a well-discriminated stimulus. Science. 153, 1547–1549. 10.1126/science.153.3743.1547. [DOI] [PubMed] [Google Scholar]
- 11.Squire LR, Clark RE, Knowlton BJ, 2001. Retrograde amnesia. Hippocampus. 11, 50–55. 10.1002/1098-1063(2001)11:1<50::aid-hipo1019>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 12.Dudai Y, Eisenberg M, 2004. Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron. 44.1, 93–100. 10.1016/j.neuron.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 13.McGaugh JL, 2000. Memory--a century of consolidation. Science. 287, 248–251. 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 14.Nader K, Schafe GE, Le Doux JE, 2000. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 406, 722–726. 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu E, Tang YP, Rampon C, Tsien JZ, 2000. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 290, 1170–1174. 10.1126/science.290.5494.1170. [DOI] [PubMed] [Google Scholar]
- 16.Squire LR, Genzel L, Wixted JT, Morris RG, 2015. Memory consolidation. Cold Spring Harb. Perspect. Biol 7.8, a021766. 10.1101/cshperspect.a021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buzsáki G, 2015. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus. 25.10, 1073–1188. 10.1002/hipo.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csicsvari J, Dupret D, 2014. Sharp wave/ripple network oscillations and learning-associated hippocampal maps. Philos. Trans. R. Soc. Lond., Biol. Sci 369, 20120528. 10.1098/rstb.2012.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bliss TV, Lomo T, 1973. Long-lasting Potentiation of Synaptic Transmission in the Dentate Area of the Anaesthetized Rabbit Following Stimulation of the Perforant Path. J. Physiol 232, 331–356. 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicoll RA, 2017. A brief history of long-term potentiation. Neuron. 93.2, 281290. 10.1016/j.neuron.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T, 2002. Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J. Neurophysiol 87, 2770–2777. 10.1152/jn.2002.87.6.2770. [DOI] [PubMed] [Google Scholar]
- 22.Whitlock JR, Heynen AJ, Shuler MG, Bear MF, 2006. Learning induces long-term potentiation in the hippocampus. Science. 313, 1093–1097. 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 23.Josselyn SA, Tonegawa S, 2020. Memory engrams: Recalling the past and imagining the future. Science. 367. 10.1126/science.aaw4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tonegawa S, Liu X, Ramirez S, Redondo R, 2015. Memory engram cells have come of age. Neuron. 87, 918–931. [DOI] [PubMed] [Google Scholar]
- 25.Denny CA, Lebois E, Ramirez S, 2017. From engrams to pathologies of the brain. Front. Neural Circuits. 11, 23. 10.3389/fncir.2017.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L, 2013. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron. 78, 773–784. 10.1016/j.neuron.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura T, Ogawa SK, Roy DS, Okuyama T, Morrissey MD, Smith LM, Redondo RL, Tonegawa S, 2017. Engrams and circuits crucial for systems consolidation of a memory. Science. 356, 73–78. 10.1126/science.aam6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reijmers LG, Perkins BL, Matsuo N, Mayford M, 2007. Localization of a stable neural correlate of associative memory. Science. 317, 1230–1233. 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka KZ, Pevzner A, Hamidi AB, Nakazawa Y, Graham J, Wiltgen BJ, 2014. Cortical representations are reinstated by the hippocampus during memory retrieval. Neuron. 84, 347–354. 10.1016/j.neuron.2014.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Dabrowska N, Joshi S, Williamson J, Lewczuk E, Lu Y, Oberoi S, Brodovskaya A, Kapur J, 2019. Parallel pathways of seizure generalization. Brain.142, 2336–2351. 10.1093/brain/awz170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer MA, Cash SS, 2012. Epilepsy as a disorder of cortical network organization. The Neuroscientist. 18, 360–372. 10.1177/1073858411422754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith EH, Schevon CA, 2016. Toward a mechanistic understanding of epileptic networks. Curr. Neurol. Neurosci. Rep 16, 97. 10.1007/s11910-016-0701-2. [DOI] [PubMed] [Google Scholar]
- 33.Stam CJ, 2014. Modern network science of neurological disorders. Nat. Rev. Neurosci 15, 683–695. 10.1177/1073858409334423. [DOI] [PubMed] [Google Scholar]
- 34.O’Keefe J, Dostrovsky J, 1971. The hippocampus as a spatial map: Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 34, 171–175. 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- 35.Moser MB, Moser EI, 1998. Functional differentiation in the hippocampus. Hippocampus. 8, 608–619. 10.1002/(SICI)1098-1063(1998)8:6<608::AID-HIPO3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Henriksen EJ, Colgin LL, Barnes CA, Witter MP, Moser MB, Moser EI, 2010. Spatial representation along the proximodistal axis of CA1. Neuron. 68, 127–137. 10.1016/j.neuron.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fanselow MS, Dong HW, 2010. Are the dorsal and ventral hippocampus functionally distinct structures?. Neuron. 65, 7–19. 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakazawa Y, Pevzner A, Tanaka KZ, Wiltgen BJ, 2016. Memory retrieval along the proximodistal axis of CA1. Hippocampus. 26, 1140–1148. 10.1007/7854_2016_462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JNP, 2002. Spatial memory dissociations in mice lacking GluR1. Nature Neurosci. 5, 868–873. 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- 40.Frankland PW, Bontempi B, 2005. The organization of recent and remote memories. Nat. Rev. Neurosci 6(2), 119–130. 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- 41.Poo MM, Pignatelli M, Ryan TJ, Tonegawa S, Bonhoeffer T, Martin KC, Rudenko A, Tsai LH, Tsien RW, Fishell G, Mullins C, 2016. What is memory? The present state of the engram. BMC Biol.14, 40. 10.1186/s12915-016-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S, 2015. Engram cells retain memory under retrograde amnesia. Science. 348, 1007–1013. 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Risher WC, Ustunkaya T, Alvarado JS, Eroglu C, 2014. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PloS one. 9, e107591. 10.1371/journal.pone.0107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimshek DR, Bus T, Schupp B, Jensen V, Marx V, Layer LE, Kohr G, Sprengel R, 2017. Different forms of AMPA receptor mediated LTP and their correlation to the spatial working memory formation. Front. Mol. Neurosci 10, 214. 10.3389/fnmol.2017.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowie D, Mayer ML 1995. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 15, 453–462. 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 46.Butler CR, Zeman AZ, 2008. Recent insights into the impairment of memory in epilepsy: transient epileptic amnesia, accelerated long-term forgetting and remote memory impairment. Brain. 131, 2243–2263. 10.1093/brain/awn127. [DOI] [PubMed] [Google Scholar]
- 47.Shuman T, Aharoni D, Cai DJ, Lee CR, Chavlis S, Page-Harley L, Vetere LM, Feng Y, Yang CY, Mollinedo-Gajate I, Chen L, 2020. Breakdown of spatial coding and interneuron synchronization in epileptic mice. Nat. Neurosci 1–10. 10.1101/358580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brus O, Cao Y, Gustafsson E, Hultén M, Landen M, Lundberg J, Nordanskog P, Nordenskjöld A, 2017. Self-assessed remission rates after electroconvulsive therapy of depressive disorders. Eur. Psych 45, 154–60. 10.1016/j.eurpsy.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 49.Meeter M, Murre JM, Janssen SM, Birkenhager T, van den Broek WW, 2011. Retrograde amnesia after electroconvulsive therapy: a temporary effect?. J. Affective Dis 132, 216–222. 10.1016/j.jad.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 50.O’Connor M, Lebowitz BK, Ly J, Panizzon MS, Elkin-Frankston S, Dey S, Bloomingdale K, Thall M, Pearlman C, 2008. A dissociation between anterograde and retrograde amnesia after treatment with electroconvulsive therapy: a naturalistic investigation. The Journal of ECT. 24,146–51. 10.1097/yct.0b013e318158792f. [DOI] [PubMed] [Google Scholar]
- 51.Squire LR, Slater PC, Chace PM, 1976. Reactivation of recent or remote memory before electroconvulsive therapy does not produce retrograde amnesia. Behav. Bio 18, 335–343. 10.1016/S0091-6773(76)92295-1. [DOI] [PubMed] [Google Scholar]
- 52.Asok A, Leroy F, Rayman JB, Kandel ER, 2019. Molecular mechanisms of the memory trace. Trends Neurosci. 42, 14–22. 10.1016/j.tins.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frankland PW, Josselyn SA, Köhler S, 2019. The neurobiological foundation of memory retrieval. Nat. Neurosci 22, 1576–1585. 10.1038/s41593-019-0493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blümcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C, Merschhemke M, Meencke HJ, Lehmann T, von Deimling A, Scheiwe C, 2007. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropath.113, 235–44. 10.1007/s00401-006-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujita S, Toyoda I, Thamattoor AK, & Buckmaster PS, 2014. Preictal activity of subicular, CA1, and dentate gyrus principal neurons in the dorsal hippocampus before spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci 34, 16671–16687. 10.1523/jneurosci.0584-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldstein LH, Abrahams S, 2013. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol. 12, 368–380. 10.1016/S1474-4422(13)70026-7. [DOI] [PubMed] [Google Scholar]
- 57.Wong PTH, Teo WL, 1986. The effect of phenytoin on glutamate and GABA transport. Neurochem. Res 11, 1379–1382. 10.1007/BF00966131. [DOI] [PubMed] [Google Scholar]
- 58.Dengler CG, Coulter DA, 2016. Normal and epilepsy-associated pathologic function of the dentate gyrus. In Progress in brain research (Vol. 226, pp. 155–178). Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krook-Magnuson E, 2017. The gate and the source? The dentate gyrus takes central stage in temporal lobe epilepsy. Epilepsy Curr. 17, 48–49. doi: 10.5698/1535-7511-17.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S, 2004. NMDA receptors, place cells and hippocampal spatial memory. Nat. Rev. Neurosci 5, 361–372. 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- 61.O’keefe J, Nadel L, 1978. The hippocampus as a cognitive map. Oxford: Clarendon Press. [Google Scholar]
- 62.O’Keefe J, 1999. Do hippocampal pyramidal cells signal non-spatial as well as spatial information?. Hippocampus. 9, 352–364. 10.1002/(SICI)1098-1063(1999)9:4<352::AID-HIPO3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 63.McKernan MG, Shinnick-Gallagher P, 1997. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 390, 607–611. 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 64.Rumpel S, LeDoux J, Zador A, Malinow R, 2005. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 308, 83–88. 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- 65.Yoon T, Okada J, Jung MW, Kim JJ, 2008. Prefrontal cortex and hippocampus subserve different components of working memory in rats. Learn. Mem 15, 97–105. http://www.learnmem.org/cgi/doi/10.1101/lm.850808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pignatelli M, Ryan TJ, Roy DS, Lovett C, Smith LM, Muralidhar S, Tonegawa S, 2019. Engram cell excitability state determines the efficacy of memory retrieval. Neuron. 101, 274–284. 10.1016/j.neuron.2018.11.029. [DOI] [PubMed] [Google Scholar]
- 67.Park A, Jacob AD, Walters BJ, Park S, Rashid AJ, Jung JH, Lau J, Woolley GA, Frankland PW, Josselyn SA, 2020. A time-dependent role for the transcription factor CREB in neuronal allocation to an engram underlying a fear memory revealed using a novel in vivo optogenetic tool to modulate CREB function. Neuropsychopharmacol. 45, 916–24. 10.1038/s41386-019-0588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barnes CA, Jung MW, McNaughton BL, Korol DL, Andreasson K, Worley PF, 1994. LTP saturation and spatial learning disruption: effects of task variables and saturation levels. J. Neurosci 14, 5793–5806. 10.1523/JNEUROSCI.14-10-05793.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colgin LL, 2015. Do slow and fast gamma rhythms correspond to distinct functional states in the hippocampal network?. Brain Res. 1621, 309–315. 10.1016/j.brainres.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Xu J, Liu Y, Zhu J, Liu N, Zeng W, Li X, 2017. A distinct entorhinal cortex to hippocampal CA1 direct circuit for olfactory associative learning. Nat. Neurosci 20, 559–570. 10.1038/nn.4517. [DOI] [PubMed] [Google Scholar]
- 71.Gowers WR, 1881. Epilepsy and other chronic convulsive disorders: their causes, symptoms, and treatment. Churchill. [Google Scholar]
- 72.McNamara JO, Byrne MC, Dasheiff RM, Fitz JG, 1980. The kindling model of epilepsy: a review. Prog. Neurobiol 15, 139–159. 10.1016/0301-0082(80)90006-4. [DOI] [PubMed] [Google Scholar]
- 73.McNamara JO, 1988. Pursuit of the mechanisms of kindling. Trends Neurosci. 11, 33–36. 10.1016/0166-2236(88)90047-1. [DOI] [PubMed] [Google Scholar]
- 74.Flavell SW, Greenberg ME, 2008. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci 31, 563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baker J, Zeman A, 2017. Accelerated long-term forgetting in epilepsy—and beyond. In Cognitive neuroscience of memory consolidation. Springer, Cham. (401–417). [Google Scholar]
- 76.Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, Maroso M, Soltesz I, 2018. Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science. 359, 787–790. 10.1126/science.aan4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coras R, Pauli E, Li J, Schwarz M, Rössler K, Buchfelder M, Blumcke I, 2014. Differential influence of hippocampal subfields to memory formation: insights from patients with temporal lobe epilepsy. Brain. 137, 1945–1957. 10.1093/brain/awu100. [DOI] [PubMed] [Google Scholar]
- 78.Müller CJ, Gröticke I, Bankstahl M, Löscher W, 2009. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Exp. Neurol 219, 284–297. 10.1016/j.expneurol.2009.05.035 [DOI] [PubMed] [Google Scholar]
- 79.Reyes A, Holden HM, Chang YH, Uttarwar VS, Sheppard DP, DeFord NE, DeJesus SY, Kansal L, Gilbert PE, McDonald CR, 2018. Impaired spatial pattern separation performance in temporal lobe epilepsy is associated with visuospatial memory deficits and hippocampal volume loss. Neuropsychologia. 11, 209–15. 10.1016/j.neuropsychologia.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bell B, Lin JJ, Seidenberg M, Hermann B, 2011. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat. Rev. Neurol 7, 154–164. 10.1038/nrneurol.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lenck-Santini PP, Scott RC, 2015. Mechanisms responsible for cognitive impairment in epilepsy. Cold Spring Harb. Perspect. med 5, a022772. 10.1101/cshperspect.a022772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holmes GL, 2015. Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Dis. 17, 101–116. 10.1684/epd.2015.0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu RS, Lemieux L, Bell GS, Hammers A, Sisodiya SM, Bartlett PA, Shorvon SD, Sander JW, Duncan JS, 2003. Progressive neocortical damage in epilepsy. Ann. Neurol 53, 312–324. 10.1002/ana.10463. [DOI] [PubMed] [Google Scholar]
- 84.Lenck-Santini PP, Holmes GL, 2008. Altered phase precession and compression of temporal sequences by place cells in epileptic rats. J. Neurosci 28, 5053–5062. 10.1523/jneurosci.5024-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okuno H, Akashi K, Ishii Y, Yagishita-Kyo N, Suzuki K, Nonaka M, Kawashima., Fujii H, Takemoto-Kimura S, Abe M, Natsume R, 2012. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3. 1 with CaMKIIβ. Cell. 149. 886–898. 10.1016/j.cell.2012.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang H, Hu Y, Tsien JZ, 2006. Molecular and systems mechanisms of memory consolidation and storage. Prog. Neurobiol 79, 123–135. 10.1016/j.pneurobio.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 87.Ewell LA, Fischer KB, Leibold C, Leutgeb S, Leutgeb JK, 2019. The impact of pathological high-frequency oscillations on hippocampal network activity in rats with chronic epilepsy. Elife. 8, e42148. 10.7554/eLife.42148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lisman JE, & Zhabotinsky AM, 2001. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 31, 191–201. 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 89.Malinow R, Schulman H, Tsien RW, 1989. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 245, 862–866. 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 90.Mayford M, Wang J, Kandel ER, O’Dell TJ, 1995. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 81, 891–904. 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.