

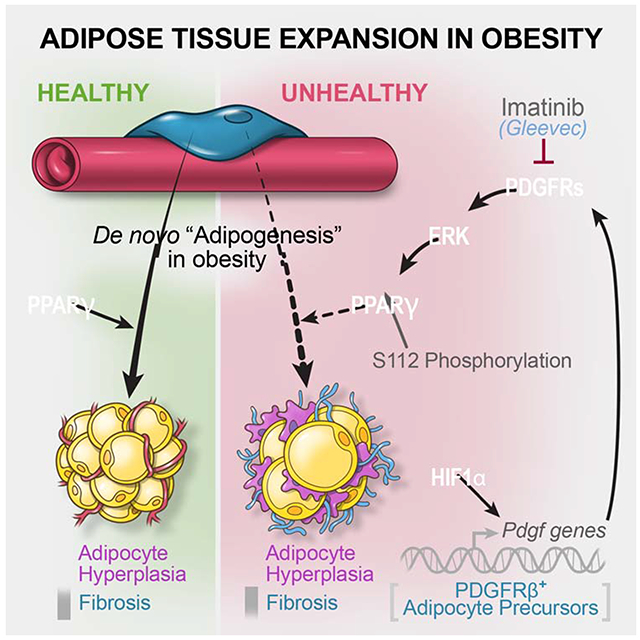
Summary
Adipose precursor cells (APCs) exhibits regional variation in response to obesity, for unclear reasons. Here, we reveal that HIFα-induced PDGFRβ signaling within murine white adipose tissue (WAT) PDGFRβ+ cells drives inhibitory serine 112 (S112) phosphorylation of PPARγ, the master regulator of adipogenesis. Levels of PPARγ S112 phosphorylation in WAT PDGFRb+ cells are depot-dependent, with levels of PPARγ phosphorylation in PDGFRβ+ cells inversely correlating with their capacity for adipogenesis upon high fat diet feeding. HIFα suppression in PDGFRβ+ progenitors promotes subcutaneous and intra-abdominal adipogenesis, healthy WAT remodeling, and improved metabolic health in obesity. These metabolic benefits are mimicked by treatment of obese mice with the PDGFR antagonist, Imatinib, which promotes adipocyte hyperplasia and glucose tolerance in a progenitor cell PPARγ-dependent manner. Our studies unveil a mechanism underlying depot-specific responses of APCs to high-fat feeding, and highlight the potential for APCs to be targeted pharmacologically to improve metabolic health in obesity.
Graphical Abstract
eTOC
How adipocyte precursor cells are regulated to control depot-specific adipose tissue expansion in obesity remains unclear. Here, Shao et al. identify a HIFα-dependent regulatory mechanism controlling adipocyte progenitor activity in mice, and demonstrate the ability of the anti-cancer drug, Imatinib, to promote metabolically beneficial adipogenesis in obesity.
Introduction
Energy-storing white adipose tissue (WAT) is exceptional in its capacity to expand in size as the demand for energy storage increases. This remarkable tissue plasticity is essential as adequate storage of lipids in WAT protects against deleterious ectopic lipid deposition in other tissues (Ghaben and Scherer, 2019). The manner by which WAT expands and remodels is a critical determinant of insulin sensitivity in obesity (Hepler and Gupta, 2017). Pathologic WAT expansion is characterized by limited expandability of metabolically favorable subcutaneous WAT and maladaptive tissue remodeling. The latter include marked adipocyte hypertrophy, chronic low-grade inflammation, and fibrosis (Hardy et al., 2011; Kloting and Bluher, 2014; Lee et al., 2010; Smith et al., 2019; Sun et al., 2014). Ensuing adipocyte dysfunction is tied to ectopic lipid deposition and insulin resistance. Healthy WAT expansion is observed in the “metabolically healthy” obese and is characterized by preferential expansion of protective subcutaneous WAT and depot expansion through adipocyte hyperplasia (Kloting et al., 2010). Adipocyte recruitment is mediated by de novo adipocyte differentiation (or “adipogenesis”) from adipocyte precursor cells (APCs). Adipocyte hyperplasia coincides with relatively lower degrees of WAT fibrosis and metabolic inflammation (Ghaben and Scherer, 2019; Vishvanath and Gupta, 2019). GWAS has pointed to genes directly involved in adipogenesis as determinants of insulin sensitivity and body fat distribution in obesity (Chu et al., 2017; Lotta et al., 2017; Lotta et al., 2018). Nevertheless, the molecular mechanisms controlling adipogenesis in obesity, and in a region-specific manner, remain unknown.
In adult mice, WAT expansion in diet-induced obesity occurs in a depot-specific manner (Gao et al., 2018; Jeffery et al., 2015; Kim et al., 2014; Vishvanath et al., 2016; Wang et al., 2013b). Gonadal WAT (gWAT) expansion in the setting of high-fat diet (HFD) induced obesity occurs through both adipocyte hypertrophy and hyperplasia. Adipocytes emerging in gWAT originate from PDGFRβ+ perivascular cells that closely resemble mural cells (Gao et al., 2018; Vishvanath et al., 2016). Importantly, the health of gWAT in obese mice is dependent on the adipogenic capacity of PDGFRβ+ cells. Inhibiting adipogenesis from PDGFRβ+ cells at the onset of HFD feeding through doxycycline (Dox)-inducible deletion of Pparg (the “master regulator” of adipogenesis) leads to pathologic WAT remodeling characterized by increased tissue fibrosis and inflammation (Shao et al., 2018). Conversely, driving adipogenesis through Dox-inducible activation of transgenic Pparg2 expression in Pdgfrb-expressing cells (Mural-PpargTG mice) results in healthy hyperplastic gWAT expansion and improvements in glucose homeostasis (Shao et al., 2018). Thus, the gWAT depot of diet-induced obese mice is a site of active de novo adipogenesis.
Interestingly, the subcutaneous inguinal WAT (iWAT) depot of HFD-fed adult male mice expands almost exclusively through adipocyte hypertrophy (Wang et al., 2013b). The frequency of iWAT adipogenesis occurring in association with HFD feeding is very low, despite the presence of multiple APC populations with a strong capacity to differentiate outside their natural environment (Merrick et al., 2019). Cell transplantations suggest that signals within the local iWAT microenvironment suppress the adipogenic potential of APCs (Jeffery et al., 2016). Notably, transgenic expression of Pparg in PDGFRβ+ cells of Mural-PpargTG mice fails to induce iWAT adipogenesis in HFD-fed mice, while readily driving adipogenesis in intra-abdominal depots (Shao et al., 2018). This suggests that such anti-adipogenic mechanisms impact PPARγ at the posttranslational level.
Here, we utilized single-cell RNA sequencing (scRNA-seq) to identify depot-dependent differences in the molecular heterogeneity of murine WAT PDGFRβ+ progenitor cells. We identified an anti-adipogenic, pro-fibrogenic, HIFα-dependent PDGFR-ERK signaling cascade that suppresses PPARγ activity in PDGFRβ+ cells through PPARγ serine 112 (S112) phosphorylation. PPARγ S112 phosphorylation is triggered by HFD within adipocyte precursors, occurring to a greater extent in APCs of iWAT than in APCs of gWAT. Inhibition of HIFα signaling in PDGFRβ+ cells attenuates PPARγ S112 phosphorylation and WAT fibrosis, while unlocking the adipogenic capacity of iWAT and gWAT adipocyte progenitors in obesity. Treatment of diet-induced obese mice with the PDGFR antagonist, Imatinib, mimics HIFα inhibition and promotes adipogenesis in both iWAT and gWAT. Our studies unveil an inhibitory mechanism underlying depot-specific responses of APCs to HFD feeding, and highlight the potential for APCs to be targeted pharmacologically as a means to improve adipose tissue health in obesity.
Results
Functional Heterogeneity of WAT PDGFRβ+ Cells is Depot-Dependent
We previously developed a genetic lineage tracing system that enables labeling and fate-mapping of PDGFRβ+ cells. This “MuralChaser” model consists of the PdgfrbrtTA transgene, TRE-Cre transgene, and the CRE-dependent Rosa26RmT/mG membrane-bound GFP reporter allele (Figure 1A) (Vishvanath et al., 2016). Our prior efforts to define APCs have focused on gWAT PDGFRβ+ cells, as these cells undergo adipogenesis in vivo in association with HFD feeding (Gupta et al., 2012; Vishvanath et al., 2016). Recently, we performed scRNA-seq analysis of genetically labeled Pdgfrb-expressing cells from gWAT of the MuralChaser model (Hepler et al., 2018). We identified molecularly and functionally distinct subpopulations of gWAT PDGFRβ+ cells. LY6C− CD9− PDGFRβ+ cells are highly adipogenic and represent APCs. LY6C+ PDGFRβ+ cells represent “Fibro-inflammatory Progenitors”, or “FIPs”. FIPs express CD9, a marker of fibrogenic cells in WAT (Marcelin et al., 2017). FIPs lack adipogenic potential, and instead are anti-adipogenic, fibrogenic, and pro-inflammatory (Hepler et al., 2018; Shan et al., 2020). Importantly, the major fibrogenic and adipogenic activity occur in distinct cell subpopulations within gWAT (Hepler et al., 2018; Marcelin et al., 2017; Spallanzani et al., 2019).
Figure 1. Single-cell RNA sequencing reveals WAT depot-dependent heterogeneity of Pdgfrb-expressing cells.
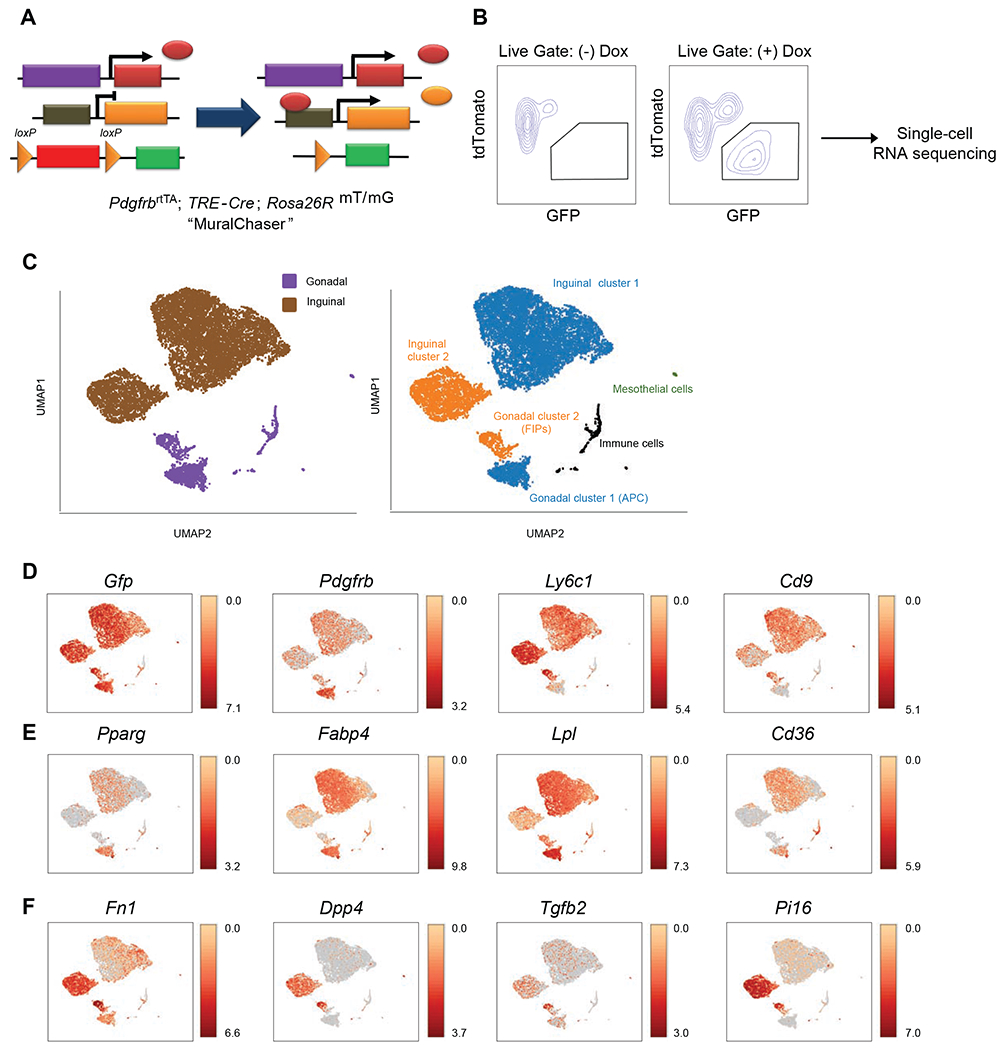
(A) MuralChaser mice: a ‘Tet-On’ system allowing for indelible labeling of Pdgfrb-expressing cells. The addition of doxycycline (Dox) leads to Cre expression and CRE-dependent activation of membrane GFP (mGFP) reporter expression.
(B) FACS strategy for the isolation of GFP+ cells from the stromal vascular fraction of inguinal WAT (iWAT) for single-cell RNA sequencing.
(C) UMAP analysis of transcriptional profiles of 8,958 iWAT GFP+ cells and 1,424 gonadal WAT (gWAT) GFP+ cells. Left: distribution of GFP+ cells by adipose depot. Right: Unique cell clusters identified in each depot.
(D) Distribution of Gfp, Pdgfrb, Ly6c1, and Cd9 expression within cell clusters shown in (C).
(E) Expression of indicated gWAT APC markers within cell clusters shown in (C).
(F) Expression of indicated gWAT FIPs markers within cell clusters shown in (C).
For panels D-F, transcript counts represent Log2 of gene expression.
Our aim in this study was to identify depot-differences in the functional heterogeneity of adipose PDGFRβ+ cells. We presumed that such depot-differences in PDGFRβ+ cell heterogeneity and functional properties underlie the differential potential to undergo adipogenesis in vivo in response to HFD feeding. To this end, we performed scRNA-seq of genetically labelled (GFP+) Pdgfrb-expressing cells isolated from the iWAT depot of 8 weeks-old MuralChaser mice (Figure 1B), and then integrated the new dataset with the previous dataset derived from gWAT mural cells. UMAP analysis identified two distinct clusters of Gfp/Pdgfrb-expressing cells within the iWAT stromal vascular fraction (Figures 1C and 1D). Both clusters were distinct from the gWAT APCs and FIPs; however, iWAT Cluster 1 was enriched in the same adipocyte-lineage transcripts that best define gWAT APCs, suggesting that they represent a committed APC population of iWAT (Figure 1E). iWAT Cluster 2 was enriched in transcripts that are enriched in FIPs (Figure 1F).
Our FACs sorting strategy used to isolate and separate gWAT APCs and FIPs is based on the expression of LY6C and CD9 (Hepler et al., 2018; Peics et al., 2020); however, in iWAT, Ly6c1 expression does not discriminate between Cluster 1 and Cluster 2 PDGFRβ+ cells (Figure 1D). Thus, we separated the two iWAT PDGFRβ+ clusters on the basis of DPP4 expression, which is expressed in inguinal Cluster 2 but not Cluster 1 (Figure 1F). Upon isolation (Figure S1A), we tested the ability of iWAT DPP4+ PDGFRβ+ and DPP4− PDGFRβ+ cells to undergo adipogenesis. Similar to gonadal APCs, DPP4− PDGFRβ+ cells (Cluster 1) differentiated spontaneously into adipocytes in vitro (Figure S1B) and upon transplantation into previously described lipodystrophic mice (Adiponectin-Cre; PpargloxP/loxP mice (Wang et al., 2013a)) (Figure S1C). Thus, DPP4− PDGFRβ+ cells of iWAT represent committed APCs and can undergo differentiation outside of their native environment. Given their resemblance to gWAT FIPs, we expected that iWAT DPP4+ PDGFRβ+ cells would be non-adipogenic; however, this was not the case. DPP4+ PDGFRβ+ cells differentiated to the same extent as DPP4− PDGFRβ+ cells in vitro and upon transplantation (Figures S1B and S1C). Thus, iWAT harbors two molecularly distinct subpopulations of PDGFRβ+ APCs.
As noted above, an important difference between native gWAT FIPs and APCs is the difference in expression of fibrogenic genes. Freshly isolated FIPs of chow or HFD fed animals are enriched in the expression of several genes associated with fibrosis, whereas APCs are enriched in Pparg and other genes linked to adipocyte differentiation (Figure 2A). Thus, the major fibrogenic and adipogenic activity occur in distinct cell subpopulations within gWAT of diet-induced obese mice. On the other hand, in iWAT, the expression of fibrogenic genes are co-enriched with Pparg and adipogenic genes in the committed precursor population (DPP4− cells) before and after HFD feeding (Figure 2B). Interestingly, the expression of Cd9, the aforementioned marker of fibrogenic cells, is co-enriched with Pparg in DPP4− PDGFRβ+ cells (Figure 1D). As such, regulation of the fibrogenic and adipogenic gene programs within PDGFRβ+ cells is depot-dependent.
Figure 2. Depot-dependent regulation of fibrogenic and adipogenic gene programs in WAT PDGFRβ+ cells.
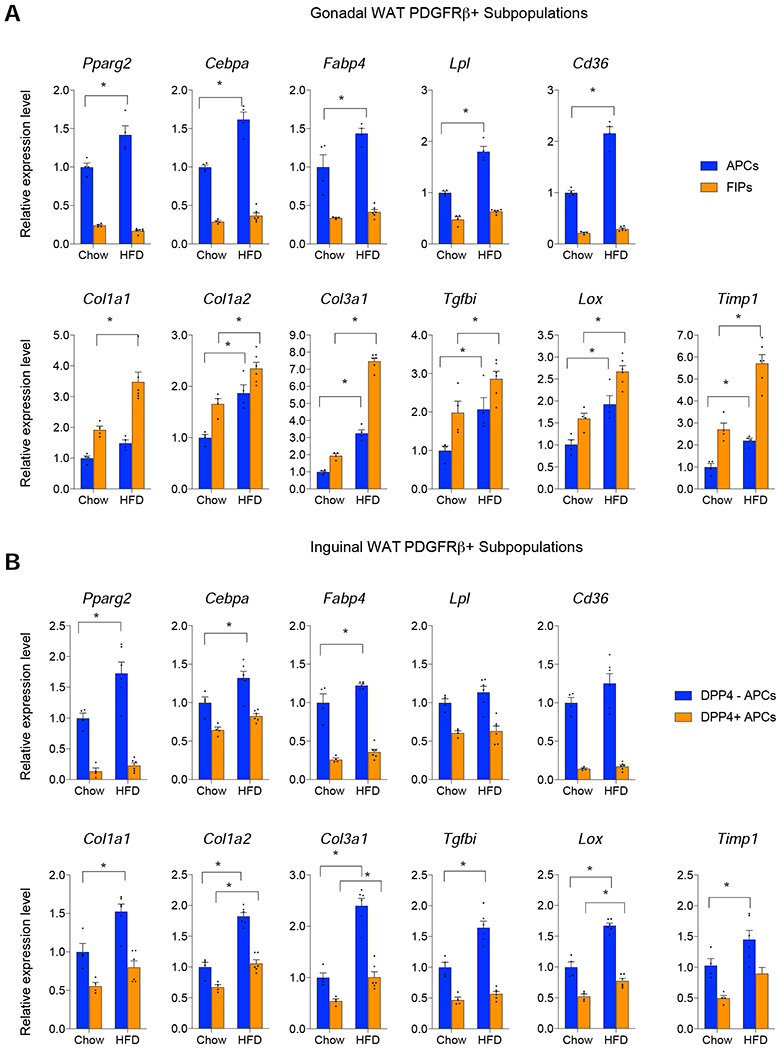
(A) mRNA levels of indicated adipogenesis (top)- and fibrosis (bottom) -related genes within freshly isolated gWAT APCs and FIPs from chow-fed mice or aged-matched animals maintained on HFD for 10 weeks.
(B) mRNA levels of indicated adipogenesis (top)- and fibrosis (bottom) -related genes within freshly isolated iWAT DPP4− PDGFRβ+ and DPP4+ PDGFRβ+ cells from chow-fed mice or aged-matched animals maintained on HFD for 10 weeks.
Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
Each sample (n) represents 10,000 freshly isolated cells from 2-3 individual depots. n=6 for HFD FIPs. n=4 for all other subpopulations analyzed.
The two clusters of PDGFRβ+ cells we identified in iWAT bear close resemblance to those populations identified in independent scRNA-seq studies. Cluster 1 (DPP4− PDGFRβ+ cells) defined here appears similar, if not identical, to the “ASC1” (adipocyte stem cell population 1) population defined by Burl et al, and contains cells representing the two committed APC populations defined by Merrick et al. (termed Group 2 and Group 3 cells by the authors) (Figure S1D–H) (Burl et al., 2018; Merrick et al., 2019). Cluster 2 (DPP4+ PDGFRβ+ cells) of the inguinal PDGFRβ+ cells appears to represent the “ASC2” population defined by Burl et al., and the Group 1 population defined by Merrick et al (Figure S1D–H). Schwalie et al. identified anti-adipogenic cells in iWAT (termed “Aregs”) that are defined by the expression of F3 (CD142) and Abcg1 (Schwalie et al., 2018). In the PDGFRβ+ subpopulations we identified, F3 expression is limited to the adipogenic DPP4− PDGFRβ+ cells in iWAT, and enriched in the committed APC population within gWAT (Figure S1I). Abcg1 was not detected within PDGFRβ+ cell populations of either depot (Figure S1I). As such, Aregs are likely to be distinct from the cells captured by the genetic reporter system utilized here, or present as small subpopulations within these cells that cannot be readily resolved.
Our functional analyses of DPP4+ and DPP4− PDGFRβ+ cells in iWAT are congruent with the recently reported findings by Merrick et al (Merrick et al., 2019). Based on transplantation studies, they propose that iWAT DPP4+ cells give rise the more committed DPP4− APC populations, forming a progenitor cell hierarchy. Our results support the idea that DPP4+ PDGFRβ+ cells of iWAT represent a more primitive APC population while DPP4− PDGFRβ+ cells of iWAT represent a more molecularly committed APC population. In fact, both DPP4+ and DPP4− APCs express the adipocyte precursor marker, Pdgfra; however, only DPP4+ PDGFRβ+ cells express Cd24a, a previously identified marker of primitive adipocyte stem cells (Figure S1J,K) (Rodeheffer et al., 2008). Merrick et al. reported that the more primitive DPP4+ APCs localize to the reticular interstitium surrounding iWAT. We assessed the localization of DPP4− and DPP4+ PDGFRβ+ cells within WAT from MuralChaser mice using indirect immunofluorescence assays. We localized iWAT DPP4+ PDGFRβ+ cells and gWAT FIPs on the basis of DPP4 and mGFP expression. We visualized DPP4− PDGFRβ+ cells on the basis of CD142 expression, whose mRNA levels (F3) are enriched in these cells. Consistent with prior studies (Gupta et al., 2012; Tang et al., 2008; Vishvanath et al., 2016), the more committed APC subpopulations in iWAT and gWAT (i.e. cells enriched in Pparg expression) are localized to the vasculature as a subset of mGFP/PDGFRβ+ cells (Figure S2). Within smaller vessels of iWAT, we also readily detect DPP4+ PDGFRβ+ cells (Figure S2). Thus, at least a subpopulation of DPP4+ stromal cells reside within the WAT vasculature. In gWAT, FIPs also reside within the vasculature (Figure S2). These molecular, functional, and histological data highlight the heterogeneity of adipose tissue PDGFRβ+ perivascular cells, and reveal that the functional heterogeneity of these cells in WAT is depot-dependent.
HIFα inhibition in PDGFRβ+ cells Attenuates WAT Fibrosis in Obesity
Gene set enrichment analysis (GSEA) revealed that gWAT FIPs are enriched in a gene signature associated with active HIF1α (hypoxia-inducible factor 1α) signaling (Hepler et al., 2018). In line with this observation, we found that HFD feeding leads to an induction of Hif1a mRNA in FIPs, but not in APCs, of gWAT (Figure S3A). Interestingly, GSEA reveals that iWAT DPP4− APCs are also enriched in this hypoxia/HIF1α gene signature (Tables S1 and S2). Thus, this gene signature is enriched in PDGFRβ+ subpopulations with greater fibrogenic gene expression. Moreover, both DPP4− and DPP4+ APCs in iWAT activate Hif1a mRNA upon HFD feeding (Figure S3A). This correlation is notable since HIF1α is a a key regulator of several aspects of WAT biology, including the adipocyte fibrogenic gene program (Jiang et al., 2011; Lee et al., 2011; Lee et al., 2014; Sun et al., 2013). As such, we hypothesized that HIF1α signaling regulates the fibrogenic gene program of adipose progenitor subpopulations.
To test this hypothesis, we first treated obese mice with PX-478, a selective chemical inhibitor of HIF1α. Seven days of treatment did not significantly impact body weight (Figure S3B); however, chemical inhibition of HIF1α impacted the frequency and gene expression profiles of PDGFRβ+ subpopulations. HIF1α inhibition led to a shift in the relative proportion of gWAT FIPs and APCs within the overall PDGFRβ+ population, with a decrease in FIPs and concomitant increase in the frequency of APCs (Figure S3C). In isolated FIPs, levels of Lox, Col1a1, and Col3a1, were lower upon HIF1α inhibition (Figure S3D). In iWAT, HIF1α inhibition led to an increase in the ratio of DPP4−to DPP4+ APCs within the total PDGFRβ+ pool (Figure S3C). Chemical HIF1α inhibition also led to reduced expression of Lox, Col1a1, and Col3a1, in these iWAT APC subpopulations (Figure S3D).
We then investigated whether HIFα functions within PDGFRβ+ cells to regulate the fibrogenic gene program of these cells. We generated a loss-of-function mouse model in which a characterized dominant negative (DN) form of HIF1α is expressed in Pdgfrb-expressing cells in a Dox-dependent manner (Figure S3E) (Chen et al., 2003; Halterman et al., 1999). This Tet-on system consists of the PdgfrbrtTA transgene and a previously reported TRE-Hif1aDN allele (herein denoted as Mural-Hif1aDN mice) (Sun et al., 2013). To validate the model, we isolated gWAT APCs from Control mice (i.e. mice carrying either PdgfrbrtTA transgene or TRE-Hif1aDN allele) and Mural-Hif1aDN mice and treated them with Dox in vitro prior to exposure to chemical HIF1α stabilizers (CoCl2 or DMOG). Treatment with either CoCl2 and DMOG blocked the spontaneous differentiation of APCs; however, this effect was lost in cells expressing HIF1αDN (Figures S3F and S3G). Thus, HIF1αDN is indeed disrupting endogenous HIFα signaling. We also administered Control and Mural-Hif1aDN mice Dox-containing chow diet (Dox-chow) for 7 days and then assessed gene expression in freshly isolated WAT PDGFRβ+ cells (Figures S3H and S3I). Following Dox exposure, mRNA of the Hif1aDN transgene was detected in PDGFRβ+ cells, but not in mature adipocytes. Importantly, acute transgene expression coincided with reduced mRNA levels of Col1a1, Col3a1, Lox, and Fn1 (Figures S3H and S3I). Collectively, these data reveal that HIFα signaling regulates the expression of genes encoding ECM components in PDGFRβ+ cells.
We asked whether HIFα inhibition in PDGFRβ+ cells influences the development of fibrosis associated with diet-induced obesity. We administered 8 weeks-old male Control and Mural-Hif1aDN mice Dox-containing high fat diet (Dox-HFD) (60% kcal from fat) for up to 10 weeks. Thus, HIFα inhibition is triggered at the onset of HFD-feeding in adult mice. Body weights and adiposity of Control and Mural-Hif1aDN mice were indistinguishable over the course of the 10-week HFD feeding period (Figures 3A–C); however, expression of HIF1αDN closely mimicked the effects of PX-478 treatment on the frequency and gene expression profile of PDGFRβ+ subpopulations in both WAT depots. The relative frequency of FIPs and DPP4+ cells in gWAT and iWAT, respectively, of Mural-Hif1aDN mice was lower than observed in corresponding depots of Control mice. On the contrary, the relative proportion of gWAT APCs and iWAT DPP4− APCs was higher in Mural-Hif1aDN mice than in Control animals (Figure 3D). HIFα inhibition led to a significant reduction in the expression of fibrosis-related genes in FIPs of gWAT and in DPP4− and DPP4+ cells of iWAT (Figure 3E). The diminished fibrogenic phenotype of PDGFRβ+ cells coincided with a 40-50% reduction in mRNA levels of Col1a1, Col3a1, Col5a1, Acta2, and Fn1, within whole WAT (Figure 3F). Moreover, picrosirius red staining revealed a strong reduction in collagen deposition within iWAT of obese Mural-Hif1aDN mice (Figure 3G). Taken together, these data reveal that HIFα signaling controls the fibrogenic phenotype of PDGFRβ+ cells, and that these cells contribute significantly to the development of WAT fibrosis associated with HFD feeding.
Figure 3. Suppression of Mural Cell HIFα Activity Attenuates Adipose Tissue Fibrosis in Obesity.
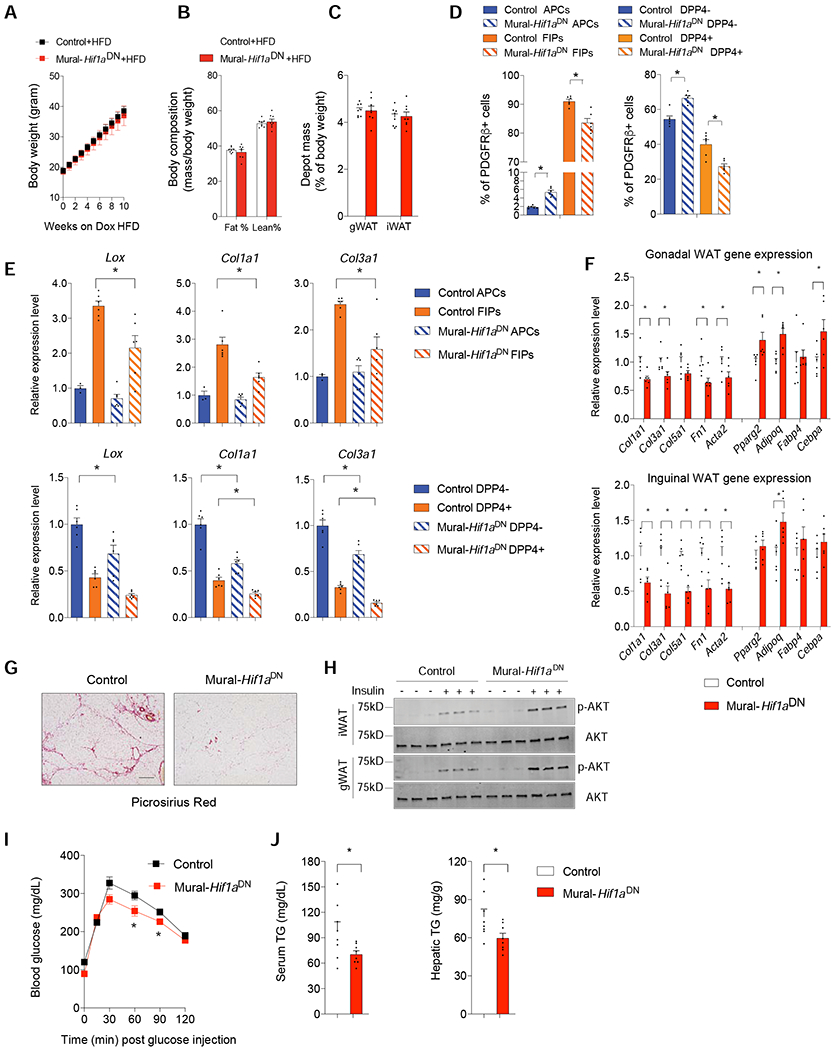
(A) Weekly body weights of Control and Mural-Hif1aDN mice following the onset of Dox-HFD feeding.
(B) Fat mass and lean mass after 10 weeks of Dox-HFD feeding. n=6 mice per genotype.
(C) Depot mass after 10 weeks of Dox-HFD feeding. n=6 mice per genotype.
(D) Relative frequency of PDGFRβ+ subpopulations within gWAT (left) and iWAT (right) after 10 weeks of Dox-HFD feeding. n= 6 mice per genotype. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
(E) mRNA levels of indicated fibrosis-related genes within gWAT APCs and FIPs (top), and iWAT DPP4− and DPP4+ PDGFRβ+ cells (bottom) after 10 weeks of Dox-HFD feeding.
Each sample (n) represents 10,000 freshly isolated cells from 2-3 individual depots. n= 3 for Control APCs. n= 6 for all other subpopulations. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
(F) mRNA levels of indicated fibrosis- and adipogenesis-related genes in whole gWAT (top) and iWAT (bottom) depots mice after 10 weeks of dox-HFD feeding. n=6 mice per genotype. Bars represent mean + s.e.m. * denotes p< 0.05 by Student’s t-test.
(G) Representative bright-field image of collagen deposition (red) in iWAT after 10 weeks of dox-HFD feeding. Scale bar denotes 200 μm.
(H) Western blot of phosphorylated AKT (p-AKT) and total AKT in iWAT and gWAT after 10 weeks of Dox-HFD feeding.
(I) Glucose tolerance tests after 10 weeks of Dox-HFD feeding. n= 8 per genotype. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
(J) Serum and hepatic triglycerides levels after 10 weeks of Dox-HFD feeding. n= 8 mice per genotype. Bars represent mean + s.e.m. * denotes p< 0.05 by Student’s t-test.
WAT fibrosis is linked to metabolic health in obesity. Consistent with this notion, insulin responsiveness, as reflected by the phosphorylation of its downstream signal transducer, AKT (pAKT), is significantly higher in both gWAT and iWAT depots of Mural-Hif1aDN mice then observed in corresponding depots from Control animals (Figure 3H). Moreover, after 10 weeks-HFD, Mural-Hif1aDN mice exhibit better glucose tolerance than Control animals, with less triglyceride accumulation in the serum and liver (Figures 3I,J). All together, these data indicate that suppression of HIFα signaling in mural cells promotes healthy adipose tissue remodeling and improved metabolic health in obesity.
HIFα inhibition in PDGFRβ+ cells Promotes iWAT and gWAT Adipogenesis
The increased frequency of PDGFRβ+ committed APCs in both WAT depots of obese Mural-Hif1aDN mice prompted us to examine whether mural cell HIFα inhibition influences the extent of de novo adipogenesis occurring in association with HFD feeding. We reconstituted the TRE-Hif1aDN transgene to the MuralChaser background; this new model (herein, “MuralChaser-Hif1aDN mice”) enables fate-mapping of mGFP labelled mural cells expressing HIF1αDN (Figure 4A). We also utilized MuralChaser-Pparg2TG mice for comparison. After 10 weeks of Dox-HFD-feeding, mGFP+ adipocytes are readily apparent within gWAT of Control mice (MuralChaser mice) (Figure 4B). Notably, the frequency of mGFP+ adipocytes in the gWAT from MuralChaser- Hif1aDN mice was significantly higher than observed in the gWAT of Control animals (Figures 4B and 4C). In fact, expression of Hif1aDN stimulated gWAT adipogenesis to a similar degree as Pparg2 overexpression. Consistent with our prior studies, mGFP+ adipocytes are only rarely observed within the iWAT depot of Control animals or MuralChaser-Pparg2TG mice (Figures 4B and 4C) (Hepler et al., 2017; Shao et al., 2018; Vishvanath and Gupta, 2019). Interestingly, mGFP+ adipocytes were readily observed in the iWAT depot of MuralChaser-Hif1aDN mice after HFD feeding (Figures 4B and 4C). Thus, HIFα inactivation in PDGFRβ+ cells permits both iWAT and gWAT adipogenesis in the setting of diet-induced obesity.
Figure 4. Suppression of mural cell HIF1α activity promotes subcutaneous and visceral adipogenesis in obesity.
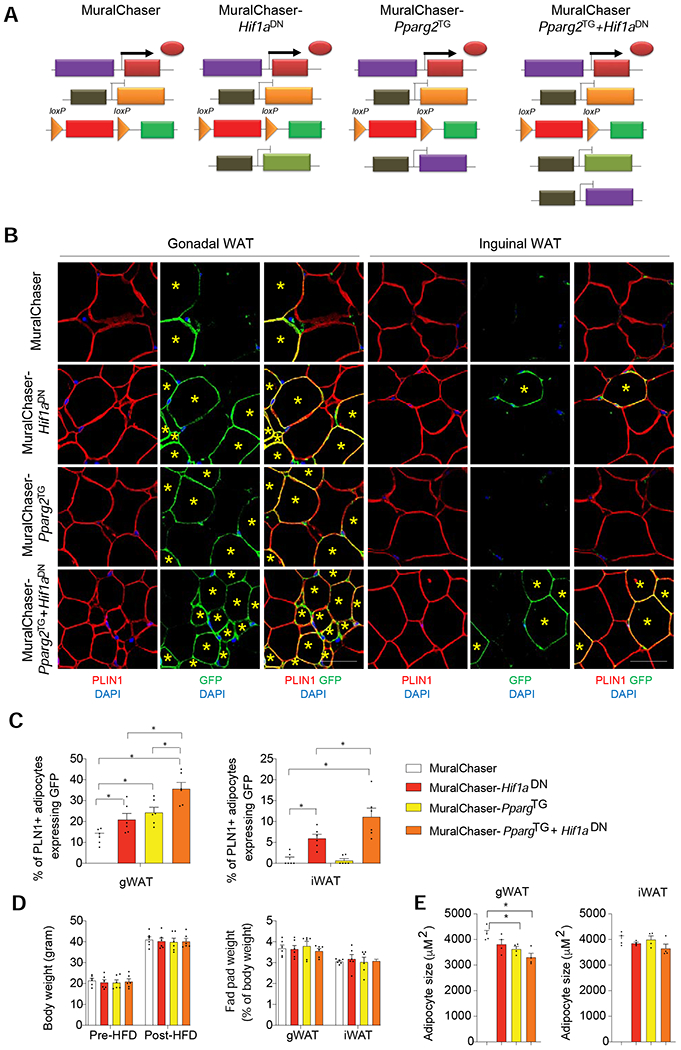
(A) Genetic models utilized in lineage tracing experiments: MuralChaser mice enable fate-mapping of PDGFRβ+ cells. MuralChaser-Hif1aDN mice enable fate-mapping of PDGFRβ+ cells expressing Hif1aDN. MuralChaser-Pparg2TG enable fate-mapping of PDGFRβ+ cells overexpressing Pparg2. MuralChaser-Pparg2TG+Hif1aDN mice enable fate-mapping of PDGFRβ+ cells co-expressing both Hif1aDN and Pparg2 transgenes.
(B) Indirect immunofluorescence images of GFP (green) and PLIN1 (red) expression in gWAT and iWAT from mice of the indicated genotypes after 10 weeks Dox-HFD feeding. New adipocytes (yellow star) emerging from PDGFRβ+ cells are marked with GFP expression. Scale bar denotes 50 μm.
(C) Percentage of PLN1+ adipocytes expressing GFP in gWAT (left) and iWAT (right) from mice of the indicated genotypes. n= 6 mice per genotype, bars represent mean + s.e.m. * denotes p< 0.05 by one-way ANOVA.
(D) (Left) Body weights of the mice utilized in the lineage tracing experiments pre- and post-HFD feeding. (Right) WAT depot mass measured after 10 weeks of dox-HFD feeding. n= 6 mice per genotype.
(E) Average adipocyte size in gWAT and iWAT from mice of the indicated genotypes after Dox-HFD feeding. n=4 per genotype. Bars represent mean + s.e.m. * denotes p< 0.05 by one-way ANOVA.
HIFα inhibition in PDGFRβ+ cells Enhances PPARγ Activity in vivo.
The ability of iWAT PDGFRβ+ cells to undergo adipogenesis when HIFα signaling is suppressed is notable given the resistance of these cells to differentiate in vivo under pro-adipogenic conditions, even when Pparg2 is overexpressed. Thus, we asked whether co-expression of HIF1αDN can enable transgenic PPARγ to induce iWAT adipogenesis. We reconstituted the TRE-Pparg2 allele into the MuralChaser-Hif1aDN background, thereby yielding a five-allele inducible model in which the fate of PDGFRβ+ cells co-expressing the Pparg2 and Hif1aDN transgenes can be tracked from the onset of HFD-feeding (herein “MuralChaser-PpargTg+Hif1aDN mice”) (Figure 4A). Average body weights were indistinguishable between any of the experimental or controls groups during Dox-HFD-feeding (Figure 4D). After 10 weeks of Dox-HFD feeding, we did not observe any significant difference in adipose tissue mass (Figure 4D); however, co-expression of HIF1αDN and PPARγ in PDGFRβ+ cells greatly enhanced the incidence of de novo adipogenesis in both gWAT and iWAT and impacted overall tissue cellularity (Figures 4B,C and E). In gWAT, co-expression of HIF1αDN and PPARβ in PDGFRβ+ cells has an additive effect on the degree of adipogenesis, with ~40% of all adipocytes representing newly formed fat cells. This degree of adipogenesis is linked to a reduction in mean adipocyte size (Figure 4E). In iWAT, co-expression of HIF1αDN and PPARγ in PDGFRβ+ cells has a synergistic effect on the degree of de novo adipogenesis, with ~10% of all adipocytes representing newly formed fat cells. Thus, inhibition of HIFα signaling unlocks PPARγ-driven iWAT adipogenesis from PDGFRβ+ in the setting of diet-induced obesity.
HIFα signaling restrains adipogenesis by promoting PPARγ S112 phosphorylation.
Prior studies revealed that HIF1α signaling inhibits 3T3-L1 adipogenesis and Pparg gene expression (Wagegg et al., 2012); however, the data above suggest that HIFα signaling also targets the activity of PPARγ to impact adipogenesis. PPARγ activity is heavily regulated by post-translational modifications (PTMs) (Brunmeir and Xu, 2018). We hypothesized that one or more PTMs of mural cell PPARγ protein can be altered by HIFα activation. We performed tandem mass spectrometry analysis of affinity-purified PPARγ from CoCl2-treated PDGFRβ+ cells. We identified two peptides carrying phosphorylated serine sites corresponding to PPARγ serine 112 (S112), with increasing signal detected after HIFα stabilization (Figure 5A). This PTM is notable, as PPARγ S112 phosphorylation inhibits PPARγ activity (Adams et al., 1997; Hu et al., 1996), and genetic disruption of this serine (S112A mutation) in mice results in a “healthy-obese” phenotype characterized by healthy inguinal WAT remodeling (Rangwala et al., 2003). We validated this finding and further investigated the effects of HIF1α activation on PPARγ S112 phosphorylation using a cellular system. We adopted a model where inguinal Pparg-deficient PDGFRβ+ cells from Dox-treated Mural-PpargKO mice are reconstituted with retroviral FLAG-tagged PPARγ2 (Figure S4A). In this system (herein, “Mural- PPARγ2 cells”), the expression of ectopic Pparg2 is stable, which allows the direct evaluation of PPARγ activity. We confirmed that CoCl2 treatment induces PPARγ S112 phosphorylation in Mural- PPARγ2 cells using site-specific PPARγ S112 antibody (Figure 5B). This was associated with ERK activation, which mediates PPARγ S112 phosphorylation (Figure 5B). S112 phosphorylation was the only known Pparγ PTM we detected by mass spectrometry under these conditions; however, ERK activation also leads to S273 (serine 273) phosphorylation of PPARγ (Banks et al., 2015). In parallel experiments, we indeed observed that CoCl2 treatment also induces PPARγ S273 phosphorylation in Mural- PPARγ2 cells (Figure 5C). To determine the relative importance of S112 and S273 phosphorylation in this context, we reconstituted Pparg-deficient PDGFRβ+ cells with either a PPARγ S112A variant or a PPARγ S273A variant and examined their ability to promote adipogenesis in the presence of HIF1α activation. Notably, PPARγ S112A, but not PPARγ S273A, is refractory to the anti-adipogenic activity of CoCl2 (Figures 5D,E). Thus, these data provide evidence that HIFα activation restrains adipogenesis by promoting PPARγ S112 phosphorylation.
Figure 5. HIF1α activation induces inhibitory PPARγ S112 phosphorylation in WAT PDGFRβ+ cells.
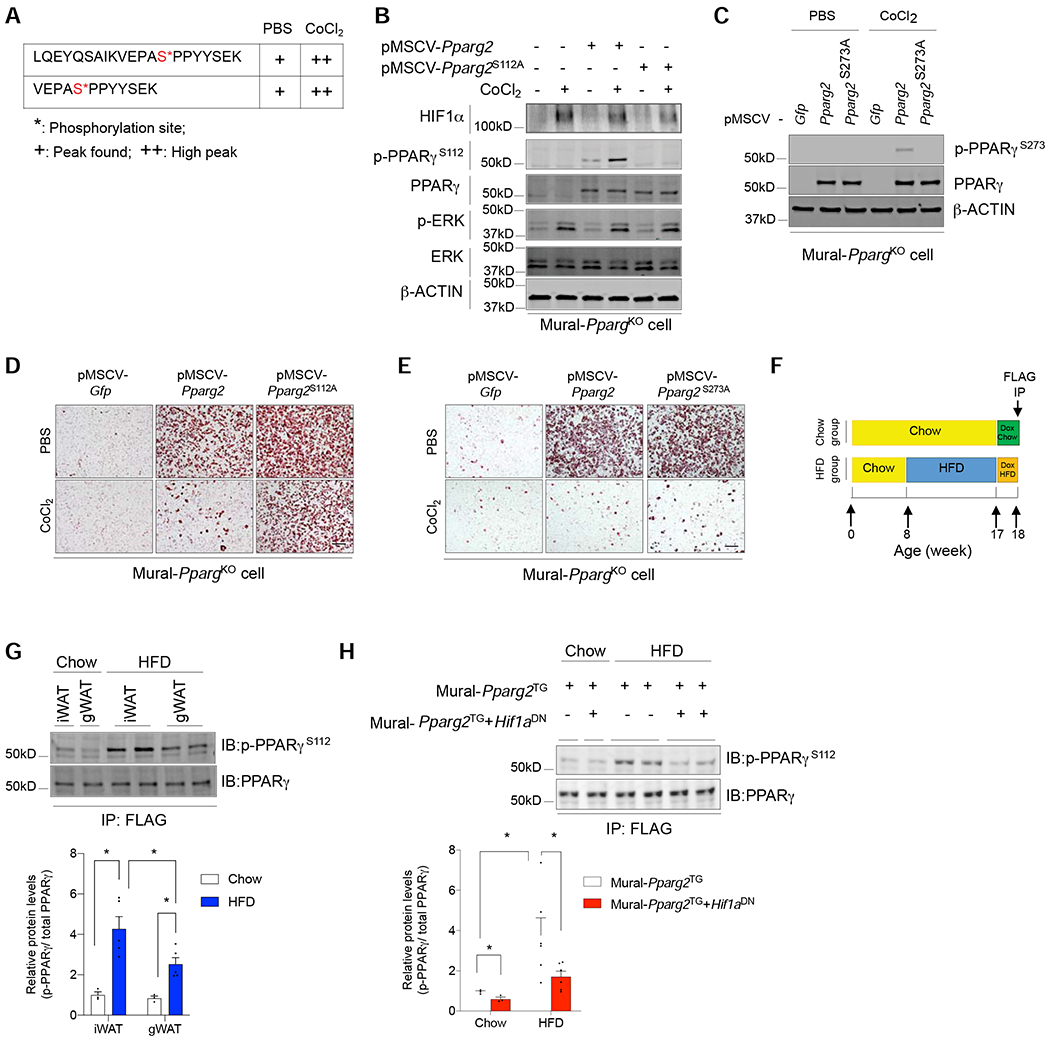
(A) Mass spectrometry analysis of affinity purified FLAG-PPARγ isolated from iWAT PDGFRβ+ cells identifies two peptides carrying phosphorylated serine sites corresponding to PPARγ serine 112 (S112), with increasing signal (+) detected after the addition of CoCl2 for 24 hours.
(B) Western blot analysis of levels of indicated proteins in Pparg-deficient iWAT PDGFRβ+ cells transduced with retrovirus expressing either GFP, PPARγ2, or the PPARγ2 S112A (serine 112 to alanine) variant. Cells were treated with 50 μM CoCl2 or vehicle for 24 hours prior to lysis.
(C) Western blot analysis of levels of indicated proteins in Pparg-deficient iWAT PDGFRβ+ cells transduced with retrovirus expressing either GFP, PPARγ2, or the PPARγ2 S273A (serine 273 to alanine) variant. Cells were treated with 50 μM CoCl2 or vehicle for 24 hours prior to lysis.
(D) Oil-red O staining of adipocytes differentiated from CoCl2-treated Pparg-deficient iWAT PDGFRβ+ cells transduced with virus expressing GFP, PPARγ2, or the PPARγ2 S112A variant.
(E) Oil-red O staining of adipocytes differentiated from CoCl2-treated Pparg-deficient iWAT PDGFRβ+ cells transduced with virus expressing GFP, PPARγ2, or the PPARγ2 S273A variant. For D and E, scale bar denotes 200 μm.
(F) Assay of phosphorylated PPARγ in mural cells in vivo: 8 weeks-old Mural-PpargTG mice and Mural-Pparg2TG+Hif1aDN mice were maintained on chow or HFD for 10 weeks. Doxycycline was added during the last 7 days to induce expression of FLAG-tagged PPARγ protein in mural cells of Mural-PpargTG mice or FLAG-tagged PPARγ protein and HIF1αDN in Mural-Pparg2TG+Hif1aDN mice. Levels of PPARγ S112 phosphorylation were analyzed by western blot of affinity purified FLAG-tagged PPARγ protein (FLAG IP).
(G) Western blot (top) and quantification (bottom) of phosphorylated PPARγ within iWAT and gWAT mural cells of chow and HFD-fed mice. n= 3 for chow samples, n=5 for HFD samples. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
(H) Western blot (top) and quantification (bottom) of phosphorylated PPARγ within iWAT mural cells of chow and HFD-fed Mural-PpargTG mice and Mural-Pparg2TG+Hif1aDN mice. n= 3 for chow samples, n=6 for HFD samples. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
PPARγ S112 phosphorylation is triggered by HFD feeding in PDGFRβ+ cells.
We asked whether PPARγ S112 phosphorylation occurs within mural cells in vivo, and is physiologically regulated. We took advantage of the FLAG-epitope encoded on transgenic Pparg2 in Mural-PpargTG animals. Mural-PpargTG mice and Mural-PpargTg+Hif1aDN mice maintained on chow or HFD for 9 weeks were administered Dox for 7 days. Following 7 days of transgene expression, we assayed PPARγ S112 phosphorylation by western blot of immunoprecipitated FLAG-tagged PPARγ protein (anti-FLAG antibody) from freshly isolated WAT depots (Figure 5F). This allowed us to assess PPARγ S112 phosphorylation specifically in PDGFRβ+ cells of WAT. We observed that levels of phosphorylated PPARγ in PDGFRβ+ cells were higher in iWAT than in gWAT in chow-fed mice (Figure 5G). In obese mice, levels of PPARγ S112 phosphorylation in PDGFRβ+ cells of both depots are higher than observed in chow-fed mice; however, the induction in mural cell PPARγ S112 phosphorylation is more robust in iWAT (Figure 5G). Thus, levels of PPARγ S112 phosphorylation in PDGFRβ+ cells inversely correlate with their ability to undergo adipogenesis in association with HFD feeding. Importantly, inhibition of mural cell HIFα signaling strongly reduces HFD-driven PPARγ S112 phosphorylation in mural cells in vivo (Figure 5H). Thus, PPARγ S112 phosphorylation in mural cells is dependent on HIFα signaling.
HIF1α drives PPARγ S112 phosphorylation through autocrine/paracrine signaling.
HIF1α drives the expression of numerous secreted factors (Pugh and Ratcliffe, 2003). In principle, many could activate ERK signaling and thus PPARγ phosphorylation. We performed co-culture experiments to determine if HIFα activation in PDGFRβ+ cells drives the production of secreted molecules that can induce PPARγ S112 phosphorylation in an autocrine/paracrine manner (Figure S4B). Indeed, we observed that co-culture of CoCl2 or DMOG-treated PDGFRβ+ cells with untreated Mural-PPARγ2 cells (cells with stable FLAG- PPARγ2) leads to ERK phosphorylation/activation, and PPARγ S112 phosphorylation in Mural-PPARγ2 cells (Figures S4C,D). Moreover, Mural-PPARγ2 cells co-cultured with CoCl2 or DMOG-treated PDGFRβ+ cells lack adipogenic potential (Figures S4E,F). Importantly, these effects on PPARγ phosphorylation and adipogenesis were abolished when HIF1αDN is expressed in CoCl2 or DMOG-treated cells, highlighting the dependence of these effects on HIFα signaling (Figure S4C–F). In principle, HIF1αDN can inhibit the activity of both HIF1α and HIF2α therefore, we performed complementary co-culture experiments in which either or both Hif1a and Epas1 (encoding HIF2α) are inactivated in PDGFRβ+ cells treated with DMOG (Figure S5A). CRISPR-Cas9 mediated inactivation of Hif1a, but not Epas1, mimics the effects of HIF1αDN expression, and blocks the inhibitory effects of DMOG on PPARγ phosphorylation and adipogenesis (Figures S5B,C). Collectively, these data provide evidence that the activation of HIF1γ in mural cells drives PPARγ S112 phosphorylation and the inhibition of adipogenesis through an autocrine/paracrine signaling mechanism.
PDGFR inhibition attenuates HIFα-mediated suppression of PPARγ and adipogenesis.
We next performed proteomics analysis of cell culture supernatants from cultures of HIFα-activated PDGFRβ+ cells. Many well-known downstream targets of HIF1α signaling were identified in the media (Table S3); however, both PDGF-C and PDGF-D were amongst the factors most robustly induced by HIF1α stabilization (Figure S6A). PDGFs have been implicated in the regulation of adipogenesis, and activate signal transduction cascades that converge on ERK. Moreover, constitutive activation of PDGFR signaling drives WAT fibrosis and pathologic WAT remodeling in obesity (Iwayama et al., 2015; Marcelin et al., 2017; Sun et al., 2017). We confirmed that DMOG treatment of iWAT PDGFRβ+ cells leads to increased mRNA levels of Pdgfc and Pdgfd in a HIF1α-dependent manner (Figures S6B,C). Moreover, PDGF-CC and PDGF-DD treatment induced ERK phosphorylation, PPARγ S112 phosphorylation, and inhibited PPARγ-driven adipogenesis in Mural-PPARγ2 cells (Figures S6D,E). Pparg-deficient PDGFRβ+ cells reconstituted with a PPARγ S112A variant were largely resistant to the inhibitory effects of PDGF-CC and PDGF-DD, which indicates that PDGFR signaling blocks PPARγ activity by promoting S112 phosphorylation. We tested the contribution of this signaling pathway to HIFα-driven phosphorylation of PPARγ in PDGFRβ+ cells through the addition of PDGFR antagonists to our co-culture system (Figure 6A). Indeed, co-culture of DMOG-treated PDGFRβ+ cells with untreated Mural-PPARγ2 cells no longer induced PDGFR phosphorylation, ERK phosphorylation, and PPARγ S112 phosphorylation in Mural-PPARγ2 cells when the PDGFR antagonist, Imatinib, was present (Figure 6B). Moreover, the inhibitory effects of HIFα activation on PPARγ-driven adipogenesis are largely abolished in the presence of Imatinib (10μM) (Figure 6C). Similar results were observed in co-cultures treated with Axinitib (100nM) or Toceranib (100nM), other known PDGFR inhibitors (Figures 6D,E). Together, these data point to PDGFR signaling as an important link between HIFα activation and the inhibition of mural cell PPARγ and adipogenesis.
Figure 6. HIF1α-dependent autocrine/paracrine PDGFR signaling drives PPARγ S112 phosphorylation and inhibition of adipogenesis in PDGFRβ+ mural cells.
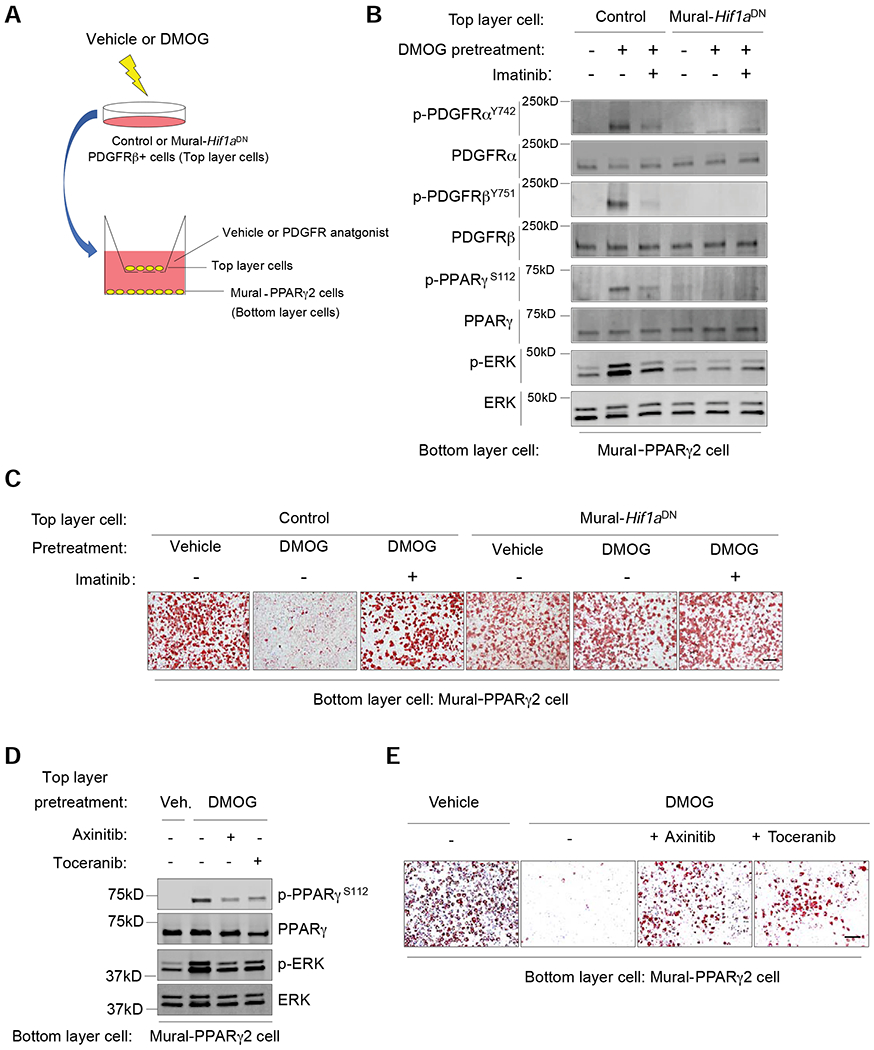
(A) Experimental design: iWAT PDGFRβ+ cells from Control or Mural-Hif1aDN mice were treated with vehicle or 50 μM DMOG (48 hours). Washed cells were transferred to transwell membranes (Top layer cells) overlaying stable Mural-PPARγ2 cells (without DMOG) (Bottom later cells). Co-cultures were maintained in serum free media (+/− PDGFR antagonist) for 6 hours before bottom layer cells were harvested for western blot analysis. Parallel co-cultures were established to assay for adipogenesis of Mural-PPARγ2 cells, with PDGFR antagonist or vehicle added to the adipogenic induction media for the first 48 hours (See Material and Methods for additional details).
(B) Western blot of indicated proteins in Mural-PPARγ2 cells following culture with the indicated cells/treatments.
(C) Oil Red-O staining 7 days after inducing adipogenesis of Mural-PPARγ2 cells exposed to the indicated cells/treatments. Scale bar denotes 200 μm.
(D) Western blot of indicated proteins in Mural-PPARγ2 cells following culture with the indicated cells/treatments.
(E) Oil Red-O staining 7 days after inducing adipogenesis of Mural-PPARγ2 cells exposed to the indicated cells/treatments. Scale bar denotes 200 μm.
Imatinib Treatment Triggers iWAT and gWAT Adipogenesis in Obese Mice.
The ability of Imatinib to block HIFα-mediated inhibition of PPARγ and adipogenesis is notable. Imatinib (GLEEVEC) has potent anti-diabetic effects in obese mice and in patients with both chronic myeloid leukemia and type 2 diabetes (Choi et al., 2016; Gomez-Samano et al., 2018). We examined the potential for Imatinib to modulate mural cell PPARγ S112 phosphorylation and adipogenesis in diet-induced obese mice. 8 weeks-old mice were placed on HFD for 10 weeks prior to the administration of Imatinib for 4 weeks (weeks 11-14 of HFD) (Figure 7A). Imatinib administration did not alter body weight gain or WAT mass in obese mice; however, Imatinib treatment lead to a notable improvement in systemic glucose tolerance (Figures S7B–D). This was associated with reduced mRNA levels of fibrogenic and inflammation-related genes in iWAT and gWAT, along with an increase in the expression of Ucp1 and Cidea in iWAT (Figures S7E–S7H). Imatinib treatment led to an increase in the frequency of DPP4− committed APCs and a concomitant decrease in the frequency of the DPP4+ population in iWAT (Figures 7A,B). A significant increase of APCs in gWAT was also observed (Figure 7C). Importantly, levels of adipose mural cell PPARγ S112 phosphorylation were lower in mice receiving Imatinib treatment (Figures 7D–F). We performed parallel lineage tracing experiments using the MuralChaser mice. Specifically during weeks 11-14 of HFD feeding, very few GFP+ adipocytes (~2-3% of adipocytes in gWAT, <<1% in iWAT) emerge from PDGFRβ+ cells (Figures 7H–I). Remarkably, treatment with Imatinib induces adipogenesis from PDGFRβ+ cells in both the iWAT and gWAT depots during this same period (~10% of adipocytes in gWAT, ~7% in iWAT) (Figures 7H–I). In gWAT, this degree of adipogenesis was associated with a decrease in mean adipocyte size (Figures 7J,K). As such, Imatinib’s effect on PDGFRβ+ subpopulations and WAT remodeling mimic the effects of mural cell HIFα suppression.
Figure 7: Imatinib treatment reduces mural cell PPARγ S112 phosphorylation and triggers iWAT and gWAT adipogenesis in obese mice.
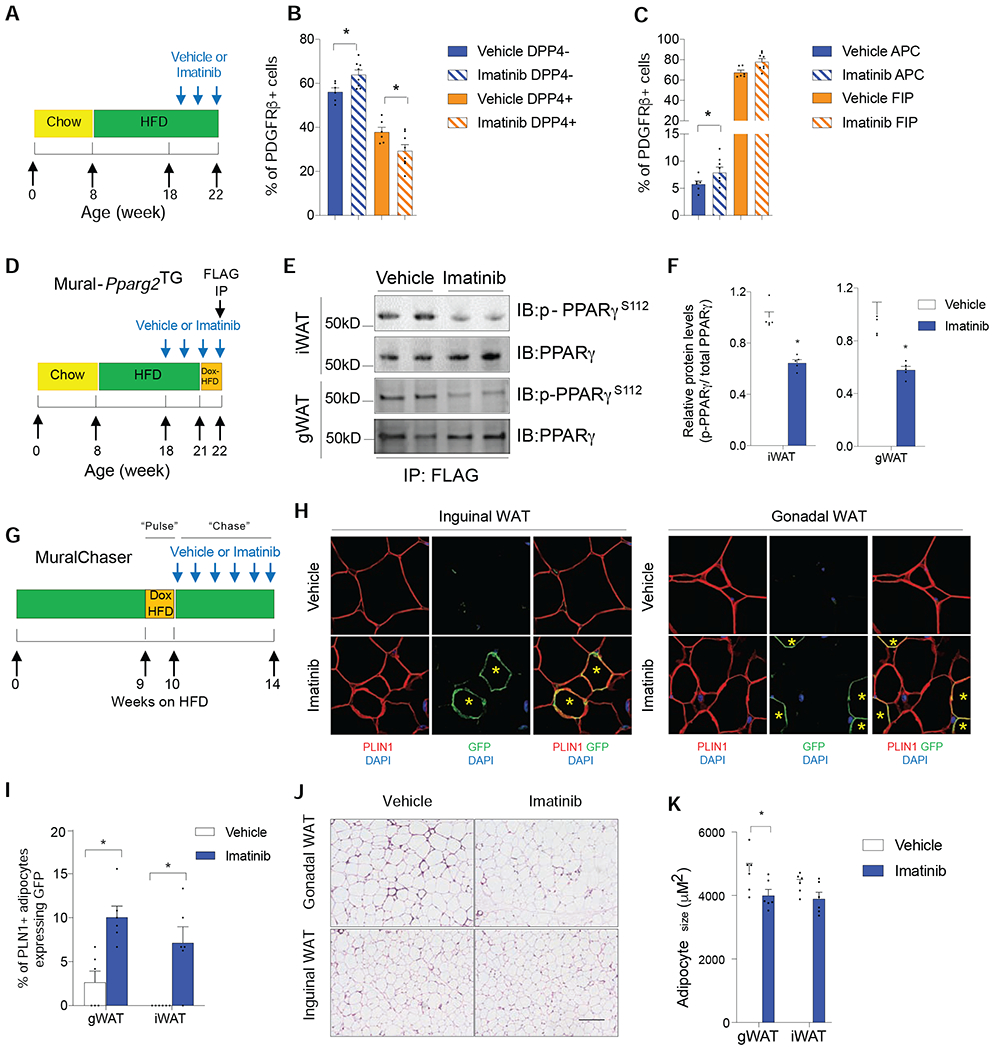
(A) 8 weeks-old C57BL/6 wildtype mice were fed a a HFD for 14 weeks. During the last 4 weeks, vehicle or Imatinib was administered four times weekly.
(B) Relative frequency of gWAT APCs and FIPs following treatment with Imatinib.
(C) Relative frequency of iWAT DPP4− APCs and DPP4+ APCs following treatment with Imatinib.
For panels B and C, n= 6. Bars represent mean + s.e.m. * denotes p< 0.05 by two-way ANOVA.
(D) Assay of PPARγ S112 phosphorylation in obese mice treated with Imatinib: 8 weeks-old Mural-Pparg2TG were fed a a HFD for 14 weeks. During the last 4 weeks, vehicle or Imatinib was administered four times weekly. During the last week of Imatinib treatment, mice were switched to Dox-HFD to induce the expression of FLAG-tagged Pparg2 transgene in PDGFRβ+ mural cells. FLAG-tagged PPARγ protein (anti-FLAG antibody) was then affinity purified from whole WAT depots for western blot analysis.
(E) Western blot of mural cell PPARγ S112 phosphorylation in WAT of obese Mural-Pparg2TG mice treated with vehicle or Imatinib.
(F) Quantification of mural cell PPARγ S112 phosphorylation in iWAT and gWAT of obese Mural-Pparg2TG mice treated with vehicle or Imatinib. n= 6 per group. Bars represent mean + s.e.m. * denotes p< 0.05 by Student’s t-test.
(G) Pulse-chase lineage tracing: Following 9 weeks of HFD-feeding, MuralChaser mice were switched to Dox-containing HFD for 1 week to induce labelling of existing PDGFRβ+ cells (“Pulse”). Mice were then switched back to HFD (without Dox) for an additional 4 weeks (“Chase”), during which vehicle or Imatinib was administered four times weekly.
(H) Indirect immunofluorescence images of GFP (green) and PLIN1 (red) expression in WAT of obese MuralChaser mice treated with either vehicle or Imatinib. Scale bar denotes 50 μm.
(I) Frequency of of GFP+ PLN1+ adipocytes in WAT of MuralChaser mice following Imatinib treatment. n= 6. Bars represent mean + s.e.m. * denotes p< 0.05 by Student’s t-test.
(J) H&E staining of WAT sections from mice treated with vehicle or Imatinib. Scale bar denotes 200 μm.
(K) Adipocyte size following treatment with vehicle or Imatinib. n=6 per group. Bars represent mean + s.e.m. * denotes p< 0.05 by Student’s t-test.
We tested whether progenitor cell Pparg is required for the anti-diabetic effects of Imatinib by inducing deletion of mural cell Pparg in obese mice at the onset of Imatinib treatment. 8 weeks-old Mural-PpargKO mice were first placed on HFD for 10 weeks. Then animals were switched to Dox-HFD and treated with vehicle or Imatinib for 4 additional weeks (Figure S7A). Thus, Pparg is inactivated in PDGFRβ+ cells at the onset of Imatinib treatment. Pparg deficiency itself during this period of HFD feeding did not impact body weight, adiposity, glucose tolerance, or fibrogenic and inflammatory gene expression in WAT (Figures S7B–F). However, loss of mural cell Pparg rendered Imatinib largely ineffective. In Mural-PpargKO mice, Imatinib treatment did not improve glucose tolerance, reduce WAT fibrogenic and pro-inflammatory gene expression, or induce thermogenic gene expression (Figures S7D–H). These data indicate that the anti-diabetic effects of Imatinib in mice depend on the actions of PPARγ in PDGFRβ+ progenitor cells.
Discussion
In recent years, considerable progress has been made in defining the heterogeneity of adipocyte progenitor cells. Nevertheless, the molecular mechanisms controlling de novo adipogenesis in obesity, and in a region-specific manner, have remained unknown. Here, we propose a model in which mural cell HIF1α signaling drives the production of secreted factors that act in an autocrine/paracrine manner to suppress PPARγ activity and limit de novo adipogenesis in obesity.
HIFα signaling inhibits mural cell PPARγ activity, in large part, through activation of signaling events leading to PPARγ S112 phosphorylation. S112 phosphorylation inhibits PPARγ transcriptional activity and its ability to promote adipogenesis (Hu et al., 1996). HIFα activation also leads to S273 phosphorylation of PPARγ in PDGFRβ+ cells; however, S273 phosphorylation does not impact adipogenesis per se (Choi et al., 2010; Choi et al., 2011; Hall et al., 2020; Wang et al., 2016). Rangwala et al. previously reported that homozygous PpargS112A knockin mice exhibit an “healthy obese” phenotype in which the animals are relatively protected from insulin resistance (Rangwala et al., 2003). iWAT depots of PpargS112A mice contain smaller and more numerous adipocytes, suggestive of enhanced adipogenesis. More recently, Olefsky and colleagues highlight the importance of PPARγ S112 phosphorylation in mature adipocytes (El Ouarrat et al., 2019). Our data here reveal the additional importance of PPARγ S112 phosphorylation in APCs in vivo. Levels of PPARγ S112 phosphorylation in mural cells are induced by HFD-feeding in a HIFα-signaling dependent manner. PPARγ S112 phosphorylation naturally occurs to a greater extent in the iWAT than gWAT, suggesting that this PTM is at least one key determinant of depot-specific responses of APCs to HFD-feeding.
Our data reveal that the molecular and functional heterogeneity of WAT progenitors is depot-dependent. In adult gWAT, we identified one highly adipogenic subpopulation of PDGFRβ+ cells (APCs), as well as a distinct population (FIPs) that lack adipogenic potential and exert a pro-inflammatory and fibrogenic phenotype. Our results from this depot are congruent with the earlier work from Marcelin et al. highlighting the separation between fibrogenic and adipogenic signaling within gWAT stromal cell subpopulations (Marcelin et al., 2017). Here, we identify two subpopulations of PDGFRβ+ cells in iWAT that harbor adipogenic potential. In alignment with Merrick et al, DPP4+ PDGFRβ+ cells likely represent a less committed adipocyte progenitor cell subpopulation, whereas DPP4− PDGFRβ+ cells are further along the differentiation pathway. Importantly, our analysis here identifies an important difference between committed DPP4− APCs of iWAT and committed APCs of gWAT. In iWAT, the fibrogenic and adipogenic gene programs appear co-enriched in the same subpopulation of cells. These data highlight the importance of treating each anatomically distinct WAT depot as a distinct entity, with each tissue containing its own functionally distinct adipocyte progenitor populations.
HIF1α functions in mature adipocytes, in part, to regulate fibrogenic and pro-inflammatory gene expression (Jiang et al., 2011; Lee et al., 2011; Lee et al., 2014; Sun et al., 2013). Our work here reveals an important role for HIFα signaling in controlling the fibrogenic vs. adipogenic gene programs of WAT progenitor cells. Inhibition of mural cell HIFα signaling has a significant impact on collagen deposition in iWAT of HFD-fed mice. These data suggest that both progenitor cells and adipocytes contribute to adipose tissue collagen deposition in a HIFα-signaling dependent manner. Whether the fibrogenic responses of mural cells and adipocytes in WAT are coordinated through inter-cellular communication in this context remains unclear.
Tissue hypoxia is an obvious candidate regulator of HIF1α signaling in adipose progenitors; changes in WAT oxygen tension may occur in obesity as enlarged cells outstrip their vascular supply. Alternatively, hypermetabolic cells can experience a state of “pseudo-hypoxia” triggered by cellular oxygen consumption (Lee et al., 2014; Seo et al., 2019). In this context, HIF1α stabilization may be regulated by cellular metabolites. Wang et al. reported that metabolism of adipocyte-derived beta-hydroxybutyrate in APCs impacts HIF1α activity to facilitate cold-induced beige adipocyte differentiation (Wang et al., 2019). HIF1α activity in white adipocyte progenitors might also be regulated by cellular metabolites accumulating in association with HFD-feeding. In fact, the accompanying study by Joffin et al. describe the differential metabolic activities of PDGFRβ+ cell subpopulations in adipose tissue, and illustrate the sensitivity of the adipogenic program to alterations in mitochondrial activity and glycolysis. HIF1α is well-known driver of glycolysis and regulator of cellular metabolism (Denko, 2008). As such, altered cellular metabolism may act as an upstream or downstream mediator of HIF1α activity in adipose progenitors.
Our work implicates PDGFR signaling as a link between HIF1α activation and the inhibition of PPARγ activity. PDGFR signaling has been linked to fibrosis in many tissues, including WAT (Iwayama et al., 2015; Marcelin et al., 2017; Sun et al., 2017). Notably, treatment of obese mice with Imatinib triggers de novo adipogenesis in both iWAT and gWAT, alongside improvements in glucose tolerance. The anti-diabetic effects of Imatinib may be mediated through a number of known mechanisms (AlAsfoor et al., 2018; Choi et al., 2016; Hagerkvist et al., 2007; King et al., 2016); however, our data here suggest that the activation of mural cell PPARγ through the suppression of S112 phosphorylation represents a contributor to Imatinib-driven improvements in glucose homeostasis. These observations are notable given the observed anti-diabetic effects of GLEEVEC treatment in patients with both chronic myeloid leukemia and type 2 diabetes (Gomez-Samano et al., 2018). Whether GLEEVEC itself would be safe and effective as an anti-diabetic therapy in the broader population of obese individuals is unclear. Nevertheless, our data provide proof of concept that such anti-adipogenic regulatory mechanisms within APCs can be targeted pharmacologically as a means to stimulate adipogenesis and promote healthy WAT remodeling. Exploiting depot-specific differences in adipocyte precursor behavior may inform strategies to alter body fat distribution, improve adipose tissue health, and uncouple obesity from metabolic disease.
Limitations of Study
A limitation to our study is the inability to directly quantify endogenous HIF1α or phosphorylated PPARγ levels within progenitor subpopulations. Detection of HIF1α in cells isolated by FACS is challenging given the well-known instability of the protein and the lengthy cell isolation process. The notion that HIFα signaling is active in these cell populations is supported by regulation of Hif1a mRNA, known HIF1α target-genes, and ultimately loss of function analysis. Moreover, a caveat to our mouse model is that HIF1αDN can, at least in principle, inhibit the activity of both HIF1α and HIF2α. HIF1α and HIF2α have both overlapping and distinct functional roles in cells (Higgins et al., 2008). CRE-mediated inactivation of loxP-flanked Hif1a alleles in PDGFRβ+ cells can be achieved; however, a drawback to this approach is that Hif1a inactivation will carry forth into mature adipocytes derived from these cells. This would confound the analysis given the established role of HIF1α in adipocytes. Nevertheless, our in vitro experiments indicate that HIF1α, but not HIF2α, is mediating the effects of chemical HIFα stabilization on PPARγ phosphorylation and adipogenesis. Importantly, treatment of obese mice with the HIF1α-selective inhibitor, PX-478, mimics the effects of HIF1αDN expression, providing further evidence of the involvement of HIF1α in regulating the function of PDGFRβ+ progenitors.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rana K. Gupta (Rana.Gupta@utsouthwestern.edu).
Material Availability
Unique materials and reagents generated in this study are available upon request from the Lead Contact with a completed Material Transfer Agreement.
Data and Code Availability
The single-cell RNA sequencing datasets are available at GEO Accession viewer (https://www.ncbi.nlm.nih.gov/geo) under the accession number GSE144090; the mass spectrometry datasets are available at MassIVE (https://massive.ucsd.edu) under the accession number MSV000084788.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
PpargloxP/loxP (B6.129-Ppargtm2Rev/J; JAX 004584), Rosa26RmT/mG (B6 129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; JAX 007676) and TRE-Cre (B6.Cg-Tg(tetO-cre)1Jaw/J; JAX 006234) strains were obtained from Jackson laboratory. PdgfrbrTA transgenic mice (C57BL/6-Tg(Pdgfrb-rtTA)58Gpta/J; JAX 028570), TRE-Pparg2, and TRE-Hif1aDN have been described previously (Shao et al., 2018; Sun et al., 2013). All animals used in this study were male and on a pure C57BL/6 background. Mice were maintained with a 12-hour light/dark cycle and free access to food and water.
All animal experiments were performed according to procedures approved by the UTSW Institutional Animal Care and Use Committee.
Isolation of adipose stromal vascular fraction and flow cytometric assays
The stromal vascular fraction (SVF) of white adipose tissue was isolated as previously described (Hepler et al., 2018). In brief, minced white fat depots were incubated for 1 hour in digestion buffer (1×Hank’s Balanced Salted Solution, 1.5% bovine serum albumin and 1 mg/ml Collagenase D [Roche, #11088882001]) at 37°C within a shaking water bath. The digested mixture was then filtered through a 100 μm cell strainer and then a 40 μm cell strainer. The red blood cells in SVFs were lysed by short incubation in 1 ml 1×RBC lysis buffer (eBioscience, #00-4300-54) and the SVF cells were then resuspended in blocking buffer (2% FBS/PBS containing anti-mouse CD16/CD32 Fc 758 Block 1:200). For FACS isolation, primary antibodies were added to the cells in blocking buffer for 15 min while incubating at 4°C. The cells were then washed once and resuspended in 2% FBS/PBS before sorting. FACS was performed using a BD Biosciences FACS Aria cytometer at the Flow Cytometry Core Facility at UT Southwestern.
The primary antibodies and the working concentrations are as following: CD45-PerCP/Cyanine5.5 1:400 (Biolegend, clone 30-F11, #103132), CD31-PerCP/Cyanine 5.5 1:400 (Biolegend, clone 390, #102420), PDGFRβ-PE 1:75 (Biolegend, clone APB5, 765 #136006), LY6C-APC 1:400 (Biolegend, clone HK1.4, #128016), CD9-FITC 1:400 (Biolegend, clone MZ3, #124808). DPP4-BV421 1:300 (BD, clone H194-112, #740021). All the flow cytometry datasets were analyzed and graphed using FlowJo V10.6.1.
Cell culture and cellular assays
Freshly isolated inguinal WAT PDGFRβ+ cells were cultured in DMEM/F12 plus 10% FBS, Pen/Strep, and gentamicin (growth media). For in vitro differentiation, PDGFRβ+ cells were cultured in 10% CO2 at 37°C until confluency. Confluent cultures were stimulated with adipogenic induction media (growth media supplemented with 5 μg ml−1 insulin, 1 μM dexamethasone, and 0.5 mM isobutylmethyxanthine) for 48 hours. Subsequently, cells were maintained in growth media supplemented with 5 μg ml−1 insulin (maintenance media) until harvest. For co-culture experiments, inguinal WAT PDGFRβ+ cells (~1×105 cells per well) isolated from mice of the indicated genetic backgrounds were first cultured in 8 μM polycarbonate filter upper chambers of transwell plates (Corning, #3428) and treated with 50 μM DMOG or 50 μM CoCl2 as described in the text. Following the treatments, cells were washed with fresh media for 3 times and transferred into new transwell plate lower chambers containing confluent untreated inguinal PDGFRβ+ cells which had been cultured in DMEM/F12 supplemented with 1% FBS, Pen/strep, and gentamicin. Cells were harvested 6 hours after co-culture for signaling analysis by immunoblotting. For co-cultured differentiation assays, culture media was replaced by adipogenic induction media immediately after upper chamber transfer and the top and bottom layers of cells were co-cultured for 48 hours. The upper chambers were removed after 48 hours co-culture, and cells in the lower chambers were kept in maintenance media until harvest for oil-red O staining. For the preparation of conditioned media samples, inguinal WAT PDGFRβ+ cells from wildtype C57BL/6 male mice were cultured in growth media till confluency and treated with vehicle or DMOG (50μM) for 24 hours in serum free growth media. Conditioned media were harvested and centrifuged at 600g for 5 min to remove cell debris. Supernatants were transferred and concentrated by Amicon Ultra 3K filters (Sigma, #Z740200). The concentrated supernatants were sequentially added with 4x volumes of methanol, 1x volume of chloroform, and 3x volumes for protein precipitation. After centrifugation at 14,000g for 5 min, the top aqueous layer was carefully removed. 4x volume of methanol was added to the remaining flake of protein and bottom chloroform layer. After thorough mixing, the precipitated protein was pelleted by centrifugation at 14,000g for 5 min. After brief drying, the protein pellets were dissolved in loading buffer and run into SDS-PAGE gels prior to the submission to the UTSW Proteomics Core for proteomics analysis.
METHOD DETAILS
Rodent diets and drug treatments
Mice were maintained on a standard rodent chow diet or chow diet containing 600 mg/kg doxycycline (DOX) (Bio-Serv, S4107). For high-fat diet studies, mice were fed a standard high-fat diet (60 kcal% fat, Research Diets, D12492i) or doxycycline-containing high fat diet (600 mg/kg dox, 60% kcal% fat, Bio-Serv, S5867) as indicated. For Imatinib administration, mice were i.p. injected four times weekly with saline or 25 mg/kg of Imatinib (Cayman Chemical) for 4 weeks.
Body composition analysis
Body fat mass and lean mass were measured in conscious mice using the Bruker Minispec mq10 NMR (UTSW Metabolic Phenotyping Core).
Glucose tolerance test
For glucose tolerance tests (GTT), mice were injected i.p. with glucose (Sigma) at the dosage of 1g per kg body weight after an overnight fast. Blood was collected by venous bleeding from the tail vein at 0, 15, 30, 60, 90, and 120, minutes post injection. Glucose concentrations were measured using Bayer Contour glucometers.
Serum and liver measurements
Serum levels of triglycerides were determined using a triglyceride determination kit (Sigma, triglyceride reagent T2449 and free glycerol reagent F6428). For liver triglyceride measurements, ~50 mg of liver tissue was homogenized in PBS and mixed sufficiently with 1.6 ml of CHCL3-CH3OH (2:1, v/v). After centrifugation at 3,000 r.p.m. for 10 min at room temperature, the lower organic phase was transferred and air-dried completely in a chemical hood. Samples were re-suspended using 1% Triton X-100 in absolute ethanol and triglycerides were measured using the serum triglyceride determination kit (Sigma, triglyceride reagent T2449 and free glycerol reagent F6428).
Retroviral production and infection
Retrovirus was packaged in phoenix cells as previously described (Shao et al., 2016). Briefly, phoenix packaging cells were co-transfected with 10 μg of the pMSCV overexpression plasmids (pMSCV-Gfp, or pMSCV-Pparg2) and 5 μg gag-pol and 5 μg VSV plasmids using Lipofectamine LTX (ThermoFisher Scientific, #15338100). Media containing viral particles were harvested 48 hours after transfection and centrifugated at 600g for 5 min. Supernatants were collected and used for following experiments. For retroviral infection, Mural-PpargKO mice were fed on dox-containing (600mg/kg) chow diet for 7 days. Inguinal PDGFRβ+ cells isolated from Mural-PpargKO mice after Dox exposure were infected with diluted virus-containing supernatants (1:1 ratio) in DMEM/F12 media containing 8 μg/ml polybrene (Sigma, #TR-1003) for 24 hours. Following infection, cells were maintained in fresh media for > 48 hours and then used for experiments.
CRISPR/Cas9 gene targeting in cells
Lentiviral CRISPR plasmids targeting Hif1a and Epas1 were constructed by cloning gRNAs (Hif1a #1: 5’-GCTAACAGATGACGGCGACA-3’; Hif1a #2: 5’-CGTCCTCCCCCGGCTTGTTA-3’; Epas1 #1: 5’-AGAAATCCCGTGATGCCGCG-3’; Epas1 #2: 5’-CATGAAGAAGTCACGCTCGG-3’) into the LentiCRISPR V2 (Addgene #52961) plasmid backbone. For lentivirus production, 10 μg lentiviral CRISPR plasmids were transfected using Lipofectamine LTX (Invitrogen) into Phoenix packaging cells along with 5 μg psPAX2 (Addgene #12260) and 5 μg pMD2.G (Addgene # 12259). Viral supernatants were harvested 48 hours after transfection. For viral infection, viral supernatants were added to isolated PDGFRβ+ cells for 24 hours in the presence of 8 μg/ml polybrene (Sigma). After switching into virus-free media, cells were cultured for another 24 hours before being used for the following experiments as described.
Oil red O staining
Differentiated cells were fixed in 10% formalin for 10 min at room temperature. Following fixation, the cells were washed with deionized water twice and incubated in 60% isopropanol for 5 min. Cells were completely air dried at room temperature before Oil red O working solution (2 g l−1 Oil red O in 60% isopropanol) was applied. After incubation at room temperature for 10 min, the Oil red O solution was removed and the cells were washed with deionized water for 4 times before the images were acquired for analysis.
Single-cell RNA-sequencing and analysis
Six-week-old male MuralChaser mice were fed doxycycline-containing chow diet for 9 days, followed by standard chow diet for 5 days. Following the 5-day washout period, inguinal WAT was isolated and digested as described above. tdTomato- mGFP+ cells were collected by FACS. Single cell library preparation was performed using the 10X Genomics Single Cell 3’ v2 according to the manufacturer’s instructions.
After FACS isolation of gonadal WAT tdTomato-mGFP+ cells from MuralChaser mice, 10,000 cells were partitioned into droplets containing a barcoded bead, a single cell, and reverse transcription enzyme mix using the GemCode instrument. This was followed by cDNA amplification, fragmentation, end repair and A-tailing, adaptor ligation, and index PCR. Cleanup and size selection were performed using Dynabeads MyOne Silane beads (Thermo Fisher Scientific) and SPRIselect Reagent beads (Beckman Coulter). Libraries were sequenced on an Illumina NextSeq 500 High Output (400M) by the UT Southwestern McDermott Center Next Generation Sequencing Core. 75 paired-end reads were obtained using one flow cell with the following length input: 26 bp Read 1, 66 bp Read 2, 0 bp Index 1, and 0 bp Index 2.
Cell Ranger software (v3.1.0) was used to perform demultiplexing, aligning reads, filtering, clustering, and gene expression analyses, using default parameters. The reference used was 10x Genomics mm10 (version 3.0.0) with GFP included, as per the mkref instructions. Previously published single cell sequencing data from gonadal WAT (GSE111588) (Hepler et al., 2018) was reanalyzed as above and combined with the inguinal WAT sample using 10x Genomics Cell Ranger aggr command. The resulting aggregate resolved 10,382 cells (1,424 from gonadal WAT, and 8,958 from inguinal WAT) with a median UMI count of 4,828 per cell, a mean reads per cell of 25,835, and a median genes per cell of 1,620.
The raw sequencing data from the inguinal WAT sample was deposited to Gene Expression Omnibus.
Gene Expression Analysis
Total RNA from tissue or cells was extracted and purified using the TRIzol reagent (Invitrogen) and the RNeasy Mini Kit (Qiagen). Total RNA from FACS-sorted cells was extracted using RNAqueous micro RNA isolation kit (Thermo Fisher Scientific). cDNA was synthesized with M-MLV reverse transcriptase (Invitrogen) and random hexamer primers (Invitrogen). Relative expression of mRNAs was determined by quantitative PCR using SYBR Green PCR system (Applied Biosystems) and values were normalized to levels of Rps18 using the ΔΔ-Ct method. All qPCR primer sequences are listed in Table S4.
Histological analysis and indirect immunofluorescence
Dissected tissues were fixed in 4% paraformaldehyde overnight. Paraffin embedding, sectioning, and picrosirius red staining, were performed at the Molecular Pathology Core Facility at UTSW. For indirect immunofluorescence assays, the following antibodies and concentrations were used: guinea pig anti-perilipin 1:1500 (Fitzgerald 20R-PP004); chicken anti-GFP 1:700 (Abcam, ab13970); rabbit anti-CD31 prediluted (Abcam, ab 28365); hamster anti-CD31 1:500 (Millipore, clone 2H8, MAB1398Z); goat anti-DPP4 1:500 (R&D Systems, AF954); rabbit anti-CD142 1:500 (Sino Biological, 50413-R001); goat anti-chicken Alexa 488 1:200 (Invitrogen, A-11039); goat anti-guinea pig Alexa 647 1:200 (Invitrogen, A-21450); donkey anti-rabbit Alexa 594 1:200 (Invitrogen, A-21207); donkey anti-chicken Alexa 488 1:200 (Jackson ImmunoResearch, 703-545-155); donkey anti-goat Alexa 647 1:200 (Invitrogen, A-21447), goat anti-hamster Alexa 594 1:200 (Invitrogen, A-21113); and goat anti-rabbit Alexa 647 (Invitrogen, A-32795). Dewaxed and rehydrated slides were placed in chambers containing 1x R-Buffer A pH 6.0 solution and antigen retrieval was performed using Antigen Retriever 2100 (Electron Microscopy Sciences) for 2 hours. Following one PBS wash for 5 minutes, Fx Signal Enhancer (Invitrogen) was added to the slides for 30 minutes at room temperature. Slides were then blocked for 30 minutes in PBS containing 10% normal goat serum at room temperature. Primary antibodies were diluted in PBS containing 10% normal goat serum and added to paraffin sections overnight at 4°C. Following overnight incubation, slides were washed in PBS and incubated with secondary antibodies diluted in PBS containing 10% normal goat serum for 2 hours at room temperature. Washed slides were mounted with Prolong Anti-Fade mounting medium containing DAPI (Invitrogen) before images were acquired for analysis. Bright-field and fluorescent images were acquired using Keyence BZ-X710 microscope. Confocal images were captured using Zeiss LSM880 Airyscan system at the Live Cell Imaging Core at UTSW.
Protein affinity purification
For the detection of PPARγ phosphorylation in cultured inguinal WAT PDGFRβ+ cells, isolated inguinal WAT PDGFRβ+ cells were infected with pMSCV-Pparg2 retroviral vectors. 48 hours after infection, cells were treated with PBS or CoCl2 (50μM) for 24 hours. Following the treatment, cells were lysed by Pierce IP lysis buffer (ThermoFisher Scientific, #87787) supplemented with 1% Protease Inhibitor Cocktail (Sigma, #P8340), and Phosphatase Inhibitor Cocktails (Sigma, #P5726 and #P0044). Cell lysates were then incubated with anti-PPARγ antibodies (1:100 dilution, Cell Signaling Technology #2443) at 4°C for overnight. Following the primary antibody incubation, the tissue protein lysates were mixed with Protein G Sepharose 4 Fast Flow (GE Healthcare Biosciences, #17-0618-01) for 2 hours at 4°C prior to three sequential washes with Pierce IP lysis buffer. PPARγ protein was eluted by Pierce IgG Elution Buffer (Thermo Fisher Scientific, #21004) and resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) prior to the submission to the UTSW Proteomics Core for proteomics analysis.
For measurements of PPARγ phosphorylation in PDGFRβ+ cells in vivo, Mural-PpargTG mice were fed with dox-containing (600mg/kg) chow or HFD diets for another 7 days following the treatments as described in the text, prior to the harvest of inguinal and gonadal WAT. ~1 g of adipose tissues were pooled and homogenized in Pierce IP lysis buffer (ThermoFisher Scientific, #87787) supplemented with 1% Protease Inhibitor Cocktail (Sigma, #P8340), and Phosphatase Inhibitor Cocktails (Sigma, #P5726 and #P0044). Following the brief sonication using Bioruptor 300 and centrifugation at 4°C, tissue protein lysates were harvested and incubated with anti-FLAG antibodies (1:250 dilution, Sigma #F1804) at 4°C for overnight. Following the primary antibody incubation, the tissue protein lysates was mixed with Protein G Sepharose 4 Fast Flow (GE Healthcare Bio-sciences, #17-0618-01) for 2 hour at 4°C to capture immune complexes. After three washes with Pierce IP lysis buffer, protein samples were eluted by boiling in 2×SDS loading buffer and subjected to SDS-PAGE and immunoblotting.
Immunoblotting and antibodies
Protein extracts from cells or tissues were prepared by homogenization in RIPA lysis buffer (Santa Cruz) supplemented with Protease Inhibitor Cocktail (Sigma, #P8340), and Phosphatase Inhibitor Cocktails (Sigma, #P5726 and #P0044). Protein extracts were separated by SDS-PAGE electrophoresis and transferred onto PVDF membrane (Millipore, #IPFL00010). After incubation with the indicated primary antibodies at 4°C overnight, the blots were incubated with IR Dye- coupled secondary antibodies (Ll-COR) and visualized by the LI-COR Odyssey infrared imaging system.
The primary antibodies and the working concentrations are as following:
Phospho-AKT (Ser473): 1:1000 dilution; Cell Signaling Technology, #9271
AKT: 1:1000 dilution, Cell Signaling Technology, #2920
β-ACTIN: 1:10000 dilution, Sigma, #A1978
HIF1α: 1:1000 dilution, Cell Signaling Technology, #36169
Phospho-PPARγ (Ser112): 1:1000 dilution, Millipore, #04-816
PPARγ: 1:1000 dilution, Cell Signaling Technology, #2443
Phospho-ERK1/2 (Thr202/Tyr204): 1:1000 dilution, Cell Signaling Technology, #9101
ERK1/2: 1:1000 dilution, Cell Signaling Technology, #4695
Phospho-PDGFRα (Tyr742): 1:1000 dilution, Abcam, #ab5452
PDGFRα: 1:1000 dilution, Cell Signaling Technology, #3174
Phospho-PDGFRβ (Tyr751): 1:1000 dilution, Cell Signaling Technology, #4549
PDGFRβ: 1:1000 dilution, Cell Signaling Technology, #3169
β–TUBULIN: 1:1000 dilution, Cell Signaling Technology, #2128
Proteomics analysis
Protein gel fragments was reduced and alkylated with DTT (20 mM) and iodoacetamide (27.5 mM). A 0.1 μg/μL solution of trypsin in 50 mM triethylammonium bicarbonate (TEAB) was added to completely cover the gel. Following incubation on mice, 50 μL of 50 mM TEAB was added and the gel pieces were digested overnight (Pierce). Following solid-phase extraction cleanup with an Oasis MCX μelution plate (Waters), the resulting peptides were reconstituted in 10 uL of 2% (v/v) acetonitrile (ACN) and 0.1% trifluoroacetic acid in water. 5 uL of this solution was injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography systems (Dionex). Samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo), and eluted with a gradient from 1-28% buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 2.2 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2-7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification and oxidation of Met was set as a variable modification. Phosphorylation of Ser, Thr, and Tyr was also set as a variable modification for phosphorylation samples and the ptmRS node was used for phosphorylation site location. The false-discovery rate (FDR) cutoff was 1% for all peptides.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were expressed as the mean + SEM. We used GraphPad Prism 7.0 (GraphPad Software, Inc., La Jolla, CA, USA) to perform the statistical analyses. For comparisons between two independent groups, a Student’s t-test was used and p<0.05 was considered statistically significant. For in vitro studies, we estimated the approximate effect size based on independent preliminary studies. Studies designed to characterize an in vitro difference in gene expression were estimated to have a slightly larger effect size of 30% with assumed 15% standard deviation of group means. To detect this difference at a power of 80% and an alpha of 0.05, we predicted we would need 4 independent replicates per group. We estimated this effect size based on independent preliminary studies. Statistical information, including p values, samples sizes, and repetitions, for all datasets are provided in Table S5.
Supplementary Material
Table S1: Related to Figure 1. GSEA Results: Gene sets enriched in inguinal WAT DPP4− PDGFRβ+ cells
Table S2: Related to Figure 1. GSEA Results: Gene sets enriched in inguinal WAT DPP4+ PDGFRβ+ cells
Table S3: Related to Figure 6. Mass spectrometry results: Proteins secreted from PDGFRβ+cells following DMOG exposure.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-phospho-AKT (Ser473) antibody | Cell Signaling Technology | Cat# 9271; RRID:AB_329825 |
| Anti-AKT antibody | Cell Signaling Technology | Cat# 2920; RRID:AB_1147620 |
| Anti-HIF1α antibody | Cell Signaling Technology | Cat# 36169 RRID: AB_2799095 |
| Anti-phospho-PPARγ (Ser112) antibody | Millipore | Cat# 04-816 RRID: AB_10563102 |
| Anti-PPARγ antibody | Cell Signaling Technology | Cat# 2443 RRID: AB_823598 |
| Anti-phospho-ERK1/2 (Thr202/Tyr204) antibody | Cell Signaling Technology | Cat# 9101 RRID: AB_331646 |
| Anti-ERK1/2 antibody | Cell Signaling Technology | Cat# 4695 RRID: AB_390779 |
| Anti-β-ACTIN antibody | Sigma | Cat# A1978 RRID: AB_476692 |
| Anti-phospho-PPARγ (Ser273) antibody | Banks et al., 2015 | N/A |
| Anti-phospho-PDGFRα (Tyr742) antibody | Abcam | Cat# ab5452 RRID: AB_304899 |
| Anti-PDGFRα antibody | Cell Signaling Technology | Cat# 3174 RRID: AB_2162345 |
| Anti-phospho-PDGFRβ (Tyr751) antibody | Cell Signaling Technology | Cat# 4549 RRID: AB_1147704 |
| Anti-PDGFRβ antibody | Cell Signaling Technology | Cat# 3169 RRID: AB_2162497 |
| Anti-HIF2α antibody | Novus | Cat# NB100-122 RRID: AB_10002593 |
| Anti-FLAG antibody | Sigma | Cat# F1804 RRID: AB_262044 |
| Anti-β-TUBULIN antibody | Cell Signaling Technology | Cat# 2128 RRID: AB_823664 |
| Anti-PLIN1 antibody | Fitzgerald Industries International | Cat# 20R-PP004 RRID: AB_1288416 |
| Anti-GFP antibody | Abcam | Cat# ab13970 RRID: AB_300798 |
| Anti-CD31 antibody | Abcam | Cat# ab28365 RRID: AB_726365 |
| Anti-CD31 antibody | Millipore | Cat# MAB1398Z RRID: AB_94207 |
| Anti-DPP4 antibody | R&D Systems | Cat# AF954 RRID: AB_355739 |
| Anti-CD142 antibody | Sino Biological | Cat# 50413-R001 RRID: AB_2860267 |
| Alexa Fluor 488 goat anti-chicken secondary antibody | Invitrogen | Cat# A-11039 RRID: AB_142924 |
| Alexa Fluor 647 goat anti-guinea pig secondary antibody | Invitrogen | Cat# A-21450 RRID: AB_141882 |
| Alexa Fluor 594 donkey anti-rabbit secondary antibody | Invitrogen | Cat# A-21207 RRID: AB_141637 |
| Alexa Fluor 488 donkey anti-chicken secondary antibody | Jackson ImmunoResearch Labs | Cat# 703-545-155 RRID: AB_2340375 |
| Alexa Fluor 647 donkey anti-goat secondary antibody | Invitrogen | Cat# A-21447 RRID: AB_141844 |
| Alexa Fluor 594 goat anti- guinea pig secondary antibody | Invitrogen | Cat# A-21113 RRID: AB_2535762 |
| Alexa Fluor 647 donkey anti- rabbit secondary antibody | Invitrogen | Cat# A-32795 RRID: AB_2762835 |
| PerCP/Cyanine5.5 anti-mouse CD45 antibody | Biolegend | Cat# 103132 RRID: AB_893340 |
| PerCP/Cyanine5.5 anti-mouse CD31 antibody | Biolegend | Cat# 102420 RRID: AB_10613644 |
| PE anti-mouse CD140b (PDGFRβ) antibody | Biolegend | Cat# 136006 RRID: AB_1953271 |
| APC anti-mouse Ly6C antibody | Biolegend | Cat# 128016 RRID: AB_1732076 |
| FITC anti-mouse CD9 antibody | Biolegend | Cat# 124808 RRID: AB_1279321 |
| BV421 anti-mouse CD26 (DPP4) antibody | BD Biosciences | Cat# 740021 RRID: AB_2739793 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imatinib (mesylate) | Cayman Chemical | Cat# 13139 |
| Glucose | Sigma | Cat# G7021 |
| Collagenase D | Roche | Cat# 11088882001 |
| RBC lysis buffer | eBioscience | Cat# 00-4300-54 |
| Lipofectamine LTX | ThermoFisher Scientific | Cat# 15338100 |
| Polybrene | Sigma | Cat# TR-1003 |
| DMEM/F12 GlutaMAX | Life Technologies | Cat# 10565042 |
| DMEM | Cellgro | Cat# 10-014-CV |
| Insulin | Sigma | Cat# I6634 |
| IBMX | Sigma | Cat# I7018 |
| Dexamethasone | Sigma | Cat# D4902 |
| Cobalt chloride | Sigma | Cat# 15862 |
| DMOG | Cayman Chemical | Cat# 71210 |
| Oil Red O | Sigma | Cat# O0625 |
| Dynabeads MyOne Silane beads | ThermoFisher Scientific | Cat# 37002D |
| SPRIselect Reagent beads | Beckman Coulter | Cat# B23317 |
| TRIzol | Invitrogen | Cat# 15596018 |
| Power SYBR Green | Life Technologies | Cat# 4368708 |
| Iamge-iT Fx Signal Enhancer | Invitrogen | Cat# I36933 |
| Prolong Anti-Fade mounting reagent with DAPI | Life Technologies | Cat# P36931 |
| Pierce IP lysis buffer | ThermoFisher Scientific | Cat# 87787 |
| Protease Inhibitor Cocktail | Sigma | Cat# P8340 |
| Phosphatase Inhibitor Cocktail | Sigma | Cat# P5726&P0044 |
| Protein G Sepharose 4 Fast Flow beads | GE Healthcare Bio-sciences | Cat# 17-0618-01 |
| Pierce IgG elution buffer | ThermoFisher Scientific | Cat# 21004 |
| RIPA Lysis Buffer System | Santa Cruz Biotechnology | Cat# sc-24948A |
| PVDF membrane | Millipore | Cat# IPFL00010 |
| Critical Commercial Assays | ||
| Triglyceride determination kit | Sigma | Cat# T2449&F6428 |
| RNeasy Mini Kit | Qiagen | Cat# 5805 |
| RNAqueous micro RNA isolation kit | ThermoFisher Scientific | Cat# AM1931 |
| M-MLV Reverse Transcriptase | Invitrogen | Cat# 28025-013 |
| Deposited Data | ||
| Single cell RNA-seq dataset | This paper | GEO Accession viewer: GSE144090 |
| Mass spectrometry dataset | This paper | MassIVE: MSV000084788 |
| Single cell RNA-seq dataset | Hepler et al., 2018 | GEO Accession viewer: GSE111588 |
| Experimental Models: Cell Lines | ||
| Phoenix cell | ATCC | Cat# CRL-3213 RRID: CVCL_H716 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129-Ppargtm2Rev/J | The Jackson Laboratory | JAX 004584 |
| Mouse: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX 007676 |
| Mouse: B6.Cg-Tg(tetO-cre)1Jaw/J | The Jackson Laboratory | JAX 006234 |
| Mouse: C57BL/6-Tg(Pdgfrb-rtTA)58Gpta/J | The Jackson Laboratory | JAX 028570 |
| Mouse: TRE-Pparg2 | Shao et al., 2018 | N/A |
| Mouse: TRE-Hif1aDN | Sun et al., 2013 | N/A |
| Oligonucleotides | ||
| A full list of qPCR primers, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| pMSCV-GFP | Shao et al., 2016 | N/A |
| pMSCV-PPARγ2 | Shao et al., 2016 | N/A |
| pMSCV-PPARγ2 S112A | This paper | N/A |
| pMSCV-PPARγ2 S273A | This paper | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| LentiCRISPR V2 | Addgene | Cat# 52961 |
| pUMVC | Addgene | Cat# 8449 |
| pCMV-VSV-G | Addgene | Cat# 8454 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc | https://www.graphpad.com |
| FlowJo | FlowJo, LLC | https://www.flowjo.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Cell Ranger | 10x Genomics | https://github.com/10XGenomics/cellranger |
| Proteome Discoverer | ThermoFisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30810#/OPTON-30810 |
| Other | ||
| Doxycycline chow diet | Bio-Serv | Cat# S4107 |
| High fat diet | Research Diets | Cat# D12492i |
| Doxycycline high fat diet | Bio-Serv | Cat# S5867 |
Highlights.
PPARγ S112 phosphorylation underlies depot-differences in preadipocyte activity
Mural cell HIFα drives PPARγ phosphorylation and suppression of adipogenesis
Inhibition of mural cell HIFα promotes adipogenesis and limits fibrosis in obesity
Anti-diabetic effects of Imatinib dependent on mural cell PPARγ and adipogenesis
ACKNOWLEDGMENTS
The authors thank P. Scherer for providing the TRE-Hif1aDN transgenic mice and C. Kusminski and N. Joffin for critical reading of the manuscript. The authors thank C. Lee, the UTSW Animal Resource Center, Metabolic Phenotyping Core, Pathology Core, Live Cell Imaging Core, Flow Cytometry Core, McDermott Sequencing Center, and Proteomics Core, for excellent assistance with experiments performed here, along with A. Banks (Boston, MA) for useful and discussion and critical reagents. This study and/or personnel were supported in part by NIDDK F31DK113696 to C.H., ADA 1-17-IBS-181 and NIDDK awards R01 DK104789, RC2 DK118620, and R01 DK119163 to R.K.G., AHA awards 16POST26420136 and 19CDA34670007 to M.S., and NIDDK R01 DK115477 to D.W.S. Y.W. is supported by the Grant-in-aid for Scientific Research (B) 18H02425 from the Japan Society for the Promotion of Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that they have no competing financial interests.
References
- Adams M, Reginato MJ, Shao D, Lazar MA, and Chatterjee VK (1997). Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272, 5128–5132. [DOI] [PubMed] [Google Scholar]
- AlAsfoor S, Rohm TV, Bosch AJT, Dervos T, Calabrese D, Matter MS, Weber A, and Cavelti-Weder C (2018). Imatinib reduces non-alcoholic fatty liver disease in obese mice by targeting inflammatory and lipogenic pathways in macrophages and liver. Sci Rep 8, 15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, and Spiegelman BM (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, and Xu F (2018). Functional Regulation of PPARs through Post-Translational Modifications. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burl RB, Ramseyer VD, Rondini EA, Pique-Regi R, Lee YH, and Granneman JG (2018). Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab 28, 300–309 e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhao S, Nakada K, Kuge Y, Tamaki N, Okada F, Wang J, Shindo M, Higashino F, Takeda K, et al. (2003). Dominant-negative hypoxia-inducible factor-1 alpha reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am J Pathol 162, 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, et al. (2011). Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Kim ES, Jung JE, Marciano DP, Jo A, Koo JY, Choi SY, Yang YR, Jang HJ, Kim EK, et al. (2016). PPARgamma Antagonist Gleevec Improves Insulin Sensitivity and Promotes the Browning of White Adipose Tissue. Diabetes 65, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AY, Deng X, Fisher VA, Drong A, Zhang Y, Feitosa MF, Liu CT, Weeks O, Choh AC, Duan Q, et al. (2017). Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat Genet 49, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8, 705–713. [DOI] [PubMed] [Google Scholar]
- El Ouarrat D, Isaac R, Lee YS, Oh DY, Wollam J, Lackey D, Riopel M, Bandyopadhyay G, Seo JB, Sampath-Kumar R, et al. (2019). TAZ Is a Negative Regulator of PPARgamma Activity in Adipocytes and TAZ Deletion Improves Insulin Sensitivity and Glucose Tolerance. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Daquinag AC, Su F, Snyder B, and Kolonin MG (2018). PDGFRalpha/PDGFRbeta signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaben AL, and Scherer PE (2019). Adipogenesis and metabolic health. Nat Rev Mol Cell Biol 20, 242–258. [DOI] [PubMed] [Google Scholar]
- Gomez-Samano MA, Baquerizo-Burgos JE, Coronel MFC, Wong-Campoverde BD, Villanueva-Martinez F, Molina-Botello D, Avila-Rojo JA, Palacios-Baez L, Cuevas-Ramos D, Gomez-Perez FJ, et al. (2018). Effect of imatinib on plasma glucose concentration in subjects with chronic myeloid leukemia and gastrointestinal stromal tumor. BMC Endocr Disord 18, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, Frontini A, Bhowmick DC, Ye L, Cinti S, et al. (2012). Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab 15, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerkvist R, Sandler S, Mokhtari D, and Welsh N (2007). Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J 21, 618–628. [DOI] [PubMed] [Google Scholar]
- Hall JA, Ramachandran D, Roh HC, DiSpirito JR, Belchior T, Zushin PH, Palmer C, Hong S, Mina AI, Liu B, et al. (2020). Obesity-Linked PPARgamma S273 Phosphorylation Promotes Insulin Resistance through Growth Differentiation Factor 3. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman MW, Miller CC, and Federoff HJ (1999). Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci 19, 6818–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy OT, Perugini RA, Nicoloro SM, Gallagher-Dorval K, Puri V, Straubhaar J, and Czech MP (2011). Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis 7, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, and Gupta RK (2017). The expanding problem of adipose depot remodeling and postnatal adipocyte progenitor recruitment. Mol Cell Endocrinol 445, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, Shan B, Zhang Q, Henry GH, Shao M, Vishvanath L, Ghaben AL, Mobley AB, Strand D, Hon GC, et al. (2018). Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, Shao M, Xia JY, Ghaben AL, Pearson MJ, Vishvanath L, Sharma AX, Morley TS, Holland WL, and Gupta RK (2017). Directing visceral white adipocyte precursors to a thermogenic adipocyte fate improves insulin sensitivity in obese mice. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Iwano M, and Haase VH (2008). Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 7, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, and Olson LE (2015). PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 29, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, and Rodeheffer MS (2015). Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol 17, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra-Pena R, Church CD, Colman L, Berry R, and Rodeheffer MS (2016). The Adipose Tissue Microenvironment Regulates Depot-Specific Adipogenesis in Obesity. Cell Metab 24, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, and Gonzalez FJ (2011). Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, and Steinhauser ML (2014). Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab 20, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJ, Griffiths LA, Persaud SJ, Jones PM, Howell SL, and Welsh N (2016). Imatinib prevents beta cell death in vitro but does not improve islet transplantation outcome. Ups J Med Sci 121, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloting N, and Bluher M (2014). Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord 15, 277–287. [DOI] [PubMed] [Google Scholar]
- Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, Stumvoll M, and Bluher M (2010). Insulin-sensitive obesity. Am J Physiol Endocrinol Metab 299, E506–515. [DOI] [PubMed] [Google Scholar]
- Lee KY, Gesta S, Boucher J, Wang XL, and Kahn CR (2011). The differential role of Hif1beta/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab 14, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Wu Y, and Fried SK (2010). Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care 13, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al. (2014). Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, et al. (2017). Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta LA, Wittemans LBL, Zuber V, Stewart ID, Sharp SJ, Luan J, Day FR, Li C, Bowker N, Cai L, et al. (2018). Association of Genetic Variants Related to Gluteofemoral vs Abdominal Fat Distribution With Type 2 Diabetes, Coronary Disease, and Cardiovascular Risk Factors. JAMA 320, 2553–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, et al. (2017). A PDGFRalpha-Mediated Switch toward CD9(high) Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab 25, 673–685. [DOI] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P (2019). Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peics J, Vishvanath L, Zhang Q, Shan B, Pedersen TA, and Gupta RK (2020). Isolation of Adipogenic and Fibro-Inflammatory Stromal Cell Subpopulations from Murine Intra-Abdominal Adipose Depots. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Pugh CW, and Ratcliffe PJ (2003). Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9, 677–684. [DOI] [PubMed] [Google Scholar]
- Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, and Lazar MA (2003). Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev Cell 5, 657–663. [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, and Friedman JM (2008). Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–249. [DOI] [PubMed] [Google Scholar]
- Schwalie PC, Dong H, Zachara M, Russeil J, Alpern D, Akchiche N, Caprara C, Sun W, Schlaudraff KU, Soldati G, et al. (2018). A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559, 103–108. [DOI] [PubMed] [Google Scholar]
- Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK, Andreyev AY, Murphy AN, Beeman SC, Smith GI, Klein S, et al. (2019). Knockdown of Ant2 Reduces Adipocyte Hypoxia And Improves Insulin Resistance in Obesity. Nat Metab 1, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Shao M, Zhang Q, Hepler C, Paschoal VA, Barnes SD, Vishvanath L, An YA, Jia L, Malladi VS, et al. (2020). Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity. Nat Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Ishibashi J, Kusminski CM, Wang QA, Hepler C, Vishvanath L, MacPherson KA, Spurgin SB, Sun K, Holland WL, et al. (2016). Zfp423 Maintains White Adipocyte Identity through Suppression of the Beige Cell Thermogenic Gene Program. Cell Metab 23, 1167–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Vishvanath L, Busbuso NC, Hepler C, Shan B, Sharma AX, Chen S, Yu X, An YA, Zhu Y, et al. (2018). De novo adipocyte differentiation from Pdgfrbeta(+) preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat Commun 9, 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GI, Mittendorfer B, and Klein S (2019). Metabolically healthy obesity: facts and fantasies. J Clin Invest 129, 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spallanzani RG, Zemmour D, Xiao T, Jayewickreme T, Li C, Bryce PJ, Benoist C, and Mathis D (2019). Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Berry WL, and Olson LE (2017). PDGFRalpha controls the balance of stromal and adipogenic cells during adipose tissue organogenesis. Development 144, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, and Scherer PE (2013). Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol Cell Biol 33, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Park J, Gupta OT, Holland WL, Auerbach P, Zhang N, Goncalves Marangoni R, Nicoloro SM, Czech MP, Varga J, et al. (2014). Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun 5, 3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, and Graff JM (2008). White fat progenitor cells reside in the adipose vasculature. Science 322, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, and Gupta RK (2019). Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J Clin Invest 129, 4022–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, and Gupta RK (2016). Pdgfrbeta+ Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell Metab 23, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagegg M, Gaber T, Lohanatha FL, Hahne M, Strehl C, Fangradt M, Tran CL, Schonbeck K, Hoff P, Ode A, et al. (2012). Hypoxia promotes osteogenesis but suppresses adipogenesis of human mesenchymal stromal cells in a hypoxia-inducible factor-1 dependent manner. PLoS One 7, e46483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Mullican SE, DiSpirito JR, Peed LC, and Lazar MA (2013a). Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc Natl Acad Sci U S A 110, 18656–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu L, Lin JZ, Aprahamian TR, and Farmer SR (2016). Browning of White Adipose Tissue with Roscovitine Induces a Distinct Population of UCP1(+) Adipocytes. Cell Metab 24, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, and Scherer PE (2013b). Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19, 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ishibashi J, Trefely S, Shao M, Cowan AJ, Sakers A, Lim HW, O’Connor S, Doan MT, Cohen P, et al. (2019). A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab 30, 174–189 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Related to Figure 1. GSEA Results: Gene sets enriched in inguinal WAT DPP4− PDGFRβ+ cells
Table S2: Related to Figure 1. GSEA Results: Gene sets enriched in inguinal WAT DPP4+ PDGFRβ+ cells
Table S3: Related to Figure 6. Mass spectrometry results: Proteins secreted from PDGFRβ+cells following DMOG exposure.
Data Availability Statement
The single-cell RNA sequencing datasets are available at GEO Accession viewer (https://www.ncbi.nlm.nih.gov/geo) under the accession number GSE144090; the mass spectrometry datasets are available at MassIVE (https://massive.ucsd.edu) under the accession number MSV000084788.