

Summary
Loss of the ataxia-telangiectasia mutated (ATM) kinase causes cerebellum-specific neurodegeneration in humans. We previously demonstrated that deficiency in ATM activation via oxidative stress generates insoluble protein aggregates in human cells, reminiscent of protein dysfunction in common neurodegenerative disorders. Here we show that this process is driven by poly-ADP-ribose polymerases (PARPs) and that the insoluble protein species arise from intrinsically disordered proteins associating with PAR-associated genomic sites in ATM-deficient cells. The lesions implicated in this process are single-strand DNA breaks dependent on reactive oxygen species, transcription, and R-loops. Human cells expressing Mre11 A-T-like disorder mutants also show PARP-dependent aggregation identical to ATM deficiency. Lastly, analysis of A-T patient cerebellum samples shows widespread protein aggregation as well as loss of proteins known to be critical in human spinocerebellar ataxias that is not observed in neocortex tissues. These results provide a hypothesis accounting for loss of protein integrity and cerebellum function in A-T.
Graphical Abstract
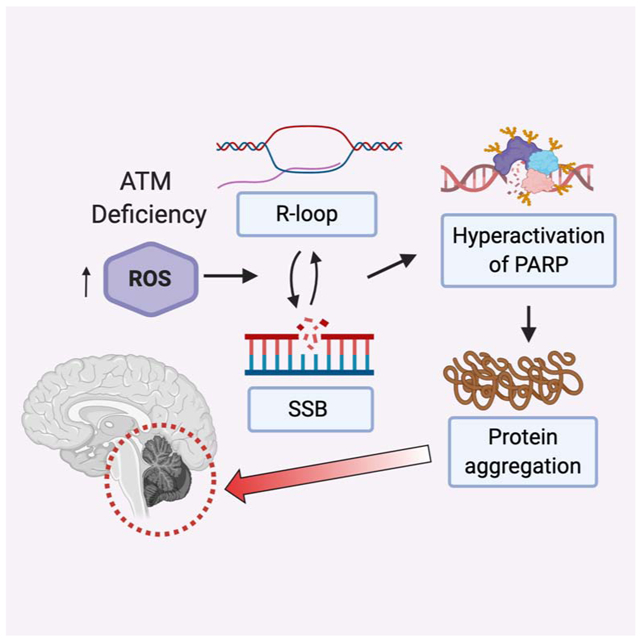
eTOC blurb:
Lee et al show that loss of protein homeostasis in ATM-deficient human cells is driven by PARP activity and transcription-associated DNA damage. poly-ADP-ribose and protein aggregates are higher in A-T patient cerebellum tissue samples, consistent with a pathological role for these species in A-T patient neurodegeneration.
Introduction
Ataxia-telangiectasia Mutated (ATM) is a master regulator of the DNA damage response in eukaryotes, responding to DNA double-strand breaks within seconds and phosphorylating several hundred targets to promote checkpoint activation, DNA repair, and many other processes related to DNA metabolism (Shiloh and Ziv, 2013). Loss of ATM in humans causes the autosomal recessive disorder ataxia-telangiectasia (A-T), a pleiotropic disease that includes a high frequency of malignancy and immunodeficiency. These features are consistent with the important role of ATM in regulation of the cell cycle and repair in response to DNA damage, particularly during immunoglobulin gene rearrangements in the immune system (Cremona and Behrens, 2014; Ghosh et al., 2018).
The primary clinical feature of A-T patients, however, is the early-onset cerebellar neurodegeneration that appears in the first few years of life and progressively worsens through early adulthood (Rothblum-Oviatt et al., 2016). The ataxia that results from this cerebellum-specific neurodegeneration in A-T patients has some similar features to that seen in other familial ataxias, including spinocerebellar ataxias (SCAs), a heterogeneous group of mostly dominantly inherited syndromes (Klockgether et al., 2019).
The source of neurotoxicity in A-T has been widely debated and is still unclear. Previous work in the field has highlighted the potential role of oxidative stress in A-T, showing that cells from A-T patients have high levels of reactive oxygen species (ROS) and signs of chronic oxidative stress, that ROS-mediated signaling in ATM-deficient cells is aberrant, and that mouse models of ATM deficiency lose stem cell populations due to insufficient antioxidant capacity (Barlow et al., 1999; Barzilai et al., 2002; Ditch and Paull, 2011; Ito et al., 2004). Several years ago we demonstrated that ATM can be directly activated by oxidative stress, independent of DNA damage, by the formation of disulfide bonds between the two monomers in an ATM dimer (Guo et al., 2010). We identified mutations that specifically block this oxidative stress pathway, one of which is associated with neurodegeneration in several A-T patients, R3047X (Chessa et al., 1992; Gilad et al., 1998; Toyoshima et al., 1998). The cerebellar degeneration observed in this subset of A-T "variants" suggests that loss of the oxidative activation pathway generates the neurodegenerative aspects of the phenotype.
Another subset of patients, those with A-T-like disorder (ATLD), are difficult to reconcile with this hypothesis, however. ATLD is caused by mutations in the gene encoding Mre11, a component of the Mre11-Rad50-Nbs1 (MRN) complex that plays a key role in activating ATM in the DNA damage response (Lee and Paull, 2005; Stewart et al., 1999; Stracker and Petrini, 2011). Rare hypomorphic mutations in Mre11 generate a clinical phenotype largely overlapping with that of A-T, including cerebellar neurodegeneration but no telangiectasia and varying cancer incidence (Regal et al., 2013; Taylor et al., 2004). ATLD mutations were originally shown to compromise ATM activation by double-strand breaks, a result generally interpreted to mean that the ataxia observed in A-T and ATLD must be due to loss of MRN activation of ATM. This is also an attractive model but is difficult to reconcile with the oxidative stress hypothesis discussed above.
To investigate ATM-dependent signaling pathways with greater specificity we previously identified alleles of ATM deficient in MRN-dependent activation by double-strand breaks, in addition to the ROS-specific separation of function alleles (Lee et al., 2018). We generated an inducible expression system for recombinant ATM in human cells combined with shRNA depletion of the endogenous protein and showed that the MRN-dependent pathway is essential for survival of DNA damage, DNA end processing, and damage-induced checkpoints and autophagy, while the oxidation-dependent pathway is essential for regulating ROS levels in human cells as well as ROS-induced autophagy and mitophagy. We also found that cells expressing ATM deficient in ROS activation, as well as A-T patient cells, exhibit widespread protein aggregation that is exacerbated by additional oxidative stress.
Here we focused on this apparent dysregulation of protein homeostasis in A-T cells because many of the more common neurodegenerative diseases in the human population are associated with aggregation of specific polypeptides in the brain, in some cases with clear causative links between the aggregation and neurotoxicity, and diverse model systems have confirmed these relationships (Bourdenx et al., 2017; Currais et al., 2017; Gidalevitz et al., 2006; Groh et al., 2017; Kikis et al., 2010; Ross and Poirier, 2004). In addition, several of the SCA ataxias, while associated with diverse genetic mutations, generate aggregation-prone mutant proteins that cause cerebellum-specific neurodegeneration similar to that observed in A-T (Buijsen et al., 2019; Klockgether et al., 2019; Seidel et al., 2012).
In the work presented here we investigate the source of the protein aggregation observed in the absence of ATM function and find that it is dependent on poly(ADP-ribose) polymerase (PARP) activation at sites of single-strand breaks in genomic DNA. The breaks are transcription-dependent and exacerbated by elevated ROS generated when ATM oxidative activation is blocked or when ATM is absent. We find that PARP-dependent aggregates also appear in ATLD cells, indicating that loss of Mre11 function and ATM deficiency have similar endpoints with respect to protein homeostasis. Finally, we show that widespread protein aggregation is also clearly apparent in A-T patient cerebellum tissue compared to control samples, and is correlated with the loss of several proteins implicated in familial SCA. These findings lead us to a model that could account for a connection between DNA damage and pathological self-assembly of intrinsically disordered proteins that occurs in A-T patients, as well as a hypothesis to explain the progression of events that drives cerebellum-specific neurodegeneration in this context.
Results
Destabilization of proteins in human cells induced by ATM deficiency
We previously demonstrated widespread protein aggregation in the absence of ATM function in several different human cell lines, including A-T patient lymphoblasts, HEK293 embryonic kidney cells, and U2OS osteosarcoma cells that was apparent in the absence of treatment and accentuated with additional oxidative stress (Lee et al., 2018). Here we used U2OS cells to identify proteins that aggregate in the absence of ATM function as we did previously, comparing changes in the total proteome (lysate) to proteins identified in the detergent-resistant fraction (aggregates)(Fig. 1). Isolation of the aggregates involves multiple rounds of intensive sonication, solubilization, and centrifugation, resulting in a final detergent-resistant aggregate fraction. Of 2,363 proteins identified by label-free mass spectrometry in the total lysate, none showed significant differences in control versus ATM-depleted cells (Fig. 1A, Table S1), both treated with a low dose of arsenite. In the aggregate fraction, however, 1584 of these proteins showed differences in ATM-depleted cells, after controlling for false discovery rate with the Benjamini-Hochberg method (Fig. 1B, Table S1). Of these, the vast majority (1,579) show higher levels of aggregation in ATM-depleted cells. Among this group of destabilized proteins is PSMB2, a subunit of the proteasome, and CK2β, a subunit of the CK2 kinase that we monitored extensively in previous work (Lee et al., 2018).
Figure 1. PARylation promotes protein aggregation in human cells lacking ATM.

(A) Human U2OS cells were grown with ATM shRNA-mediated depletion or mock treatment (3 biological replicates for each), all with arsenite (25 μM). Cell lysates were analyzed by mass spectrometry, identifying 2363 polypeptides present in all samples. Levels of each protein in control versus ATM-depleted cells are shown, with non-significant differences in grey and significant differences in red, after FDR control at 0.05. (B) Detergent-resistant aggregates were prepared from control and ATM-depleted cells as described in (A). (C) U2OS cells with control or ATM shRNA were treated with veliparib (10 μM) and arsenite (25 μM) as indicated. Lysates and detergent-resistant aggregates were analyzed by western blotting for CK2β or PSMB2. (D, E) Levels of CK2β (D) or PSMB2 (E) in aggregate fractions normalized by lysate levels were quantified in three replicates of (C); shown relative to control cells. (F) ATM-depleted U2OS cells were grown with doxycycline-induced PARP1 shRNA-mediated depletion or mock treatment as indicated, all with arsenite (25 μM). Levels of ATM, PARP1, CK2β and PSMB2 in lysates and aggregate fractions were analyzed by western blotting. (G, H) Levels of CK2β (G) or PSMB2 (H) in aggregate fractions normalized by lysate levels were quantified in three replicates of (F); shown relative to control cells. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant.
In addition to U2OS cells, we also examined protein aggregation in human brain-derived cells. We depleted endogenous ATM with shRNA in the glioblastoma-derived cell line U87-MG and found that aggregates were apparent in the absence of any exogenous stress (Fig. S1, Table S2). Mass spectrometry analysis of the total lysates did not detect any significant differences, but we identified 560 proteins showing aggregation with ATM depletion, of which all showed higher levels in the depleted cells compared to the control. As in U2OS cells (Lee et al., 2018), these aggregates were completely eliminated with anti-oxidant treatment (N-acetyl cysteine, NAC)(Fig. S1). 84% of the aggregates identified in U87-MG cells also were identified as ATM-sensitive in U2OS, indicating that, despite the differences in cell origin, there is a reproducible pattern of polypeptides destabilized with ATM depletion. Lastly, we examined the effects of ATM loss in the neuroblastoma cell line SH-SY5Y which can be induced into terminally differentiated, functional neurons in culture (Agholme et al., 2010). In these non-dividing cells, inhibition of ATM also induces aggregation of CK2β in the absence of exogenous oxidative stress (Fig. S1).
Protein aggregation observed with ATM depletion is dependent on PARP activity
Poly-ADP-ribosylation (PARylation) increases in ATM-deficient cells (Fang et al., 2016) and also is known to attract intrinsically disordered proteins (Altmeyer et al., 2015; Kai, 2016). Based on these observations, we reasoned that PARylation may be involved in the aggregation of disordered proteins in ATM-deficient cells. To test this hypothesis, we used the PARP inhibitor veliparib, specific for the dominant PARP enzymes PARP1 and PARP2 (Knezevic et al., 2016). We found that incubation of U2OS cells with veliparib reduces the CK2β and PSMB2 aggregates observed with ATM depletion and arsenite treatment (Fig. 1C, quantification in Fig. 1D, E). To validate this result, we also depleted PARP1 with shRNA and found that this reduced aggregate species substantially (Fig. 1F, quantified in 1G, H), as does deletion of PARP1 (Fig. S2). PARP2 depletion reduces aggregates as well, although to a lower extent than PARP1 depletion (Fig. S2). Taken together, this evidence suggests that protein aggregation that we have monitored in ATM-depleted cells is PARP-dependent.
To quantify differences in PARylation in ATM-depleted cells, we utilized a live-cell PARylation sensor (Krastev et al., 2018)(Fig. 2) that fuses PAR-binding domains (PBZ) to split Venus proteins. In this system, simultaneous association of the PBZ-Venus(N-term) and PBZ-Venus(C-term) proteins at sites of PARylation is required for productive formation of the green fluorescent Venus protein. We expressed the PARylation sensor in U2OS cells, where it generates a nuclear punctate signal (Fig. 2A). To remove any non-stably associated Venus protein, we used detergent extraction of cells (which also removes the cytoplasm). The fluorescence yield per cell was measured by fluorescence-activated cell sorting (FACS), with approximately 10,000 cells monitored per measurement (3 biological replicates shown here). As shown in Fig. 2B, incubation of cells with arsenite alone increased the level of PAR sensor fluorescence, which increased further with concurrent ATM inhibition (AZD1390)(Durant et al., 2018). The level of PARylation observed with ATM depletion was reduced by veliparib treatment, as expected, since the majority of PAR formed in cells is generated by PARP1 and PARP2 (Gupte et al., 2017). PARylation as measured with this sensor was also reduced by NAC treatment in U2OS cells (Fig. 2C). In U87-MG cells, we also observed that the level of PARylation was reduced by both veliparib and NAC (Fig. S1).
Figure 2. Live-cell sensors for PARylation show higher levels with ATM depletion.

The PBZ-PBZ live-cell split Venus sensor, adapted from (Krastev et al., 2018), with summary of workflow and inset of fluorescence from U2OS cells with ATM inhibitor treatment. (B) FACS results from three replicates showing the mean fluorescence yield per cell from cells expressing the PBZ-PBZ PAR sensor with ATM inhibitor (ATMi, 1 μM AZD1390) and arsenite (25 μM) as indicated. At least 10,000 cells were measured in each replicate; fluorescence yield was normalized to control cells. (C) PBZ-PBZ PAR sensor results with arsenite, ATM shRNA, veliparib (10 μM), and NAC (1 mM) treatment as in (B); fluorescence yield normalized to veliparib-treated cells. (D) Diagram of PBZ-CK2β live-cell split Venus sensor: similar to the PBZ-PBZ sensor but with CK2β replacing the PBZ domain fused to VC, with inset of fluorescence signal from U2OS cells with ATM depletion. (E) FACS results from the PBZ-CK2β sensor with arsenite, ATM shRNA, veliparib, and NAC treatment as indicated; fluorescence yield normalized to veliparib-treated cells. (F) Levels of ATM in U2OS cells with ATM shRNA and expression of recombinant wild-type (WT), C2991L (CL), R2579A/R2580A (2RA), and kinase-deficient D2889A (DA) proteins by western blotting; β-actin shown for normalization. (G) FACS results from the PBZ-CK2β sensor in U2OS cells with depletion of endogenous ATM and expression of recombinant WT, CL, 2RA, or DA alleles, with arsenite as indicated. At least 10,000 cells were measured in each replicate; fluorescence yield normalized to cells expressing WT ATM. Error bars indicate standard deviation. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant.
A subset of PARylation in actively growing cells derives from nicks associated with Okazaki fragments (Hanzlikova et al., 2018). Expression of the mutant or wild-type ATM alleles does not significantly alter the cell cycle in U2OS cells (data not shown), but we also asked whether the effect of ATM loss on PAR accumulation also occurs in non-dividing cells. To do this, we incubated U2OS cells in serum-free media, allowing them to reach a non-dividing state after 7 days. Measurement of the PBZ-PBZ sensor under these conditions showed significantly lower levels of PARylation overall, but still revealed elevated levels of fluorescence with ATM inhibition similar to actively dividing cells (Fig. S3).
Based on previous work showing association of proteins with intrinsically disordered domains with PAR (Altmeyer et al., 2015; Leung, 2020), we reasoned that the oxidization-sensitive proteins forming the aggregates could be assembling at the sites of PARP1/2-mediated PARylation. We tested this hypothesis directly by modifying the PAR sensor to include a oxidation-prone protein in place of one of the PBZ domains (see Fig. 2D). We used the CK2β protein for this purpose since it has been a reliable marker for aggregation in our experiments, and fused this protein to the C-terminal domain of the split Venus fluorescent protein. Using this hybrid PBZ-CK2β sensor, we found that ATM inhibition increased the level of PAR-CK2β association, and that NAC reduces the fluorescence yield significantly (Fig. 2E).
We then tested whether the ATM separation-of-function mutants we have previously described affect the association between CK2β and PAR. In this experiment, endogenous ATM was depleted and the WT, CL, 2RA, and kinase-dead D2889A (DA) (Daniel et al., 2012) alleles of ATM were inducibly expressed (Fig. 2F). The C2991L (CL) mutant of ATM is deficient in ROS-mediated activation (Guo et al., 2010; Lee et al., 2018) while the 2RA allele is deficient in MRN-mediated activation (Lee et al., 2018). The PBZ-CK2β sensor showed significantly higher levels of fluorescence, both with and without arsenite, with expression of the CL allele compared to WT ATM (Fig. 2G). In contrast, the 2RA mutant form of ATM showed levels of PBZ-CK2β Venus fluorescence even lower than in cells with WT ATM expressed, while the kinase-deficient DA allele was similar to the CL mutant.
Transcription-dependent lesions are responsible for protein aggregation in ATM-depleted cells
The hyperactivation of PARP observed in ATM-deficient cells suggests the presence of DNA lesions. We have not observed higher levels of spontaneous double-strand breaks in the absence of ATM function (data not shown), but previous work has suggested that DNA lesions other than double-strand breaks may accumulate in the absence of ATM function (Katyal et al., 2014; Yamamoto et al., 2016). We examined levels of total single-strand DNA breaks using an alkaline comet assay in human U2OS cells depleted of endogenous ATM with concurrent expression of WT, CL, or R3047X (RX) alleles of ATM (Fig. 3A). The R3047X mutant form of ATM is deficient in ROS activation in vitro and in human cells, similar to the CL allele (Guo et al., 2010; Lee et al., 2018), and is also an A-T patient allele (Chessa et al., 1992; Gilad et al., 1998; Uhrhammer et al., 2002). We found that expression of either allele increased the levels of single-strand breaks significantly in comparison to the WT enzyme (Fig. 3B, C), in the absence of arsenite or any other exogenous stress. This pattern is consistent with the increased ROS observed with both of these alleles, measured here using the oxidation-sensitive fluorogenic probe CellROX Deep Red (Fig. 3D). We found that addition of the antioxidant N-acetyl cysteine (NAC) eliminated the increased strand breaks observed with the mutant ATM (Fig. 3C), indicating that ROS is an important factor driving the appearance of the lesions. In contrast, expression of the 2RA ATM allele, deficient only in MRN activation, complemented ATM-depleted cells similar to wild-type ATM (Fig. S4).
Figure 3. ATM-deficient cells accumulate single-strand breaks and R-loops.
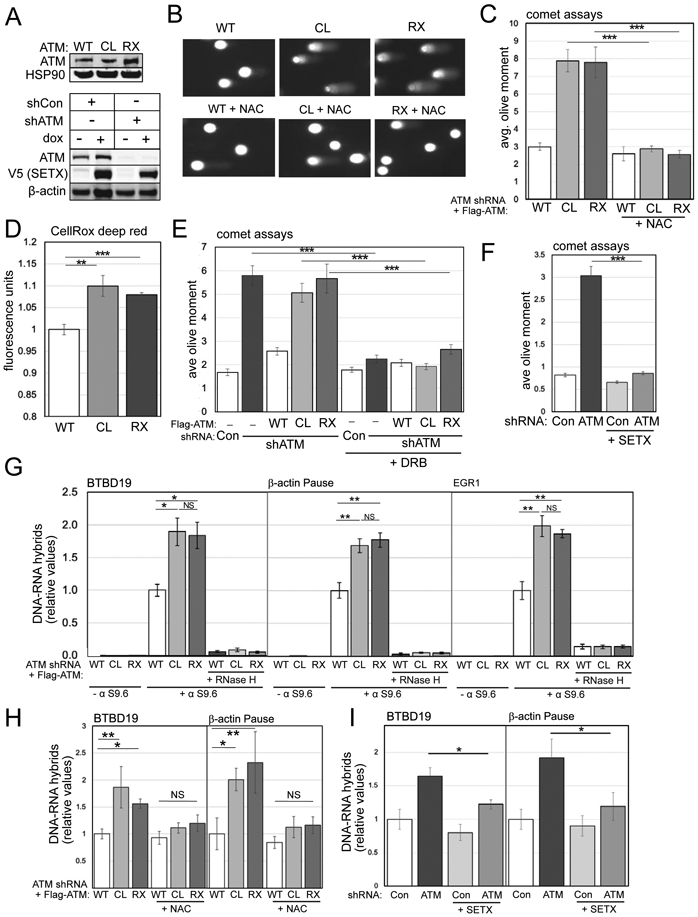
(A) Human U2OS cells with ATM shRNA depletion and wild-type (WT), C2991L (CL), or R3047X (RX) ATM expression; western blotting with anti-ATM antibody and HSP90 as loading control. Lower panel: U2OS cells with ATM shRNA depletion and inducible expression of V5-tagged Senataxin C-terminus and β-actin as loading control. (B) Alkaline comet assays were performed in U2OS cells with ATM depletion, expressing WT, CL, or RX ATM alleles with NAC treatment (1 mM) as indicated. Examples of comets are shown. (C) Quantitation of the olive moment in > 200 cells from each treatment group; error bars represent SEM. (D) Quantification of ROS levels in U2OS cells with endogenous ATM depletion and induced expression of WT, CL, or RX alleles of ATM; measured by CellROX in triplicate. (E) Alkaline comet assays as in (B,C) with 1 day DRB treatment (20 μM). (F) Alkaline comet assays as in (B,C) with ATM shRNA depletion and inducible expression of SETX. (G) DRIP-qPCR assays performed in triplicate with primers specific for the BTBD19, β-actin, or EGR1 loci in U2OS cells depleted for endogenous ATM and expressing WT, CL, or RX alleles as indicated. Immunoprecipitations were performed without S9.6 antibody or with RNaseH treatment in vitro to verify assay specificity. Levels of product were normalized to the level obtained in WT-expressing cells. Error bars indicate standard deviation. (H) DRIP-qPCR assays performed in triplicate as in (G) with addition of 1 mM NAC. (I) DRIP-qPCR assays performed in triplicate as in (G) with SETX expression indicated. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant.
Previous work has indicated that transcription may be involved in non-canonical forms of ATM activation (Bhatia et al., 2018; Britton et al., 2014; Sakasai et al., 2010; Sordet et al., 2009; Tresini et al., 2015) so we also tested the adenosine analog 5,6-dichloro-1-β-D-ribofuranosyl benzimidazole (DRB), an inhibitor of transcription elongation (Yankulov et al., 1995), and found that DRB treatment eliminates the single-strand breaks observed in the absence of ATM or with CL or RX allele expression (Fig. 3E).
We then considered the possibility that transcription-dependent single-strand breaks in ATM-deficient cells may involve R-loops, three-stranded RNA-DNA hybrid structures formed during transcription when an RNA transcript re-anneals to the template strand of the DNA (Crossley et al., 2019; Paull, 2019) since RNA-DNA hybrids have been shown to lead to strand breaks and genomic stability in many organisms (Aguilera and Gómez-González, 2017). To test for effects of R-loop removal in cells, we used inducible expression of the C-terminal catalytic domain of the RNA-DNA helicase Senataxin (SETX), an enzyme that removes RNA species from genomic DNA and also promotes transcription termination (Lavin et al., 2013). We have previously found that recombinant SETX expression reduces high levels of R-loops in CtIP-deficient human cells (Makharashvili et al., 2018). Here we found that doxycycline-induced SETX expression drastically reduced the levels of single-strand breaks in comet assays with ATM depletion (Fig. 3F and in neuronal cells in Fig. S1), consistent with the idea that R-loops are required for the DNA damage we observe in this assay.
To test this hypothesis further, we measured RNA-DNA hybrids at several genomic loci in human U2OS cells using DNA-RNA hybrid immunoprecipitation (DRIP) followed by quantitative PCR. A representative set of loci previously shown to be prone to R-loop formation (Bhatia et al., 2014; Hatchi et al., 2015; Makharashvili et al., 2018) was used for this analysis. These results showed an elevation of R-loops at the BTB19, β-actin, and EGR1 genes that was apparent with expression of the CL or RX alleles compared to expression of WT ATM (Fig. 3G). In contrast, expression of the R2579A/R2580A (2RA) allele, deficient in MRN activation (Lee et al., 2018), did not show significant differences from WT cells (Fig. S4). Appropriate controls were performed to ensure that the S9.6 antibody signal provides an accurate representation of R-loops under these conditions, including no-antibody samples and also incubation of the isolated genomic DNA with recombinant RNaseH before immunoprecipitation to test for RNA-DNA hybrid specificity. Preincubation of cells with NAC reduced the levels of R-loops observed at the BTB19 and β-actin loci (Fig. 3H), similar to the effects of NAC on single-strand breaks, indicating that ROS contributes to these lesions. Doxycycline-induced expression of SETX also reduced the levels of RNA-DNA hybrids observed in the absence of ATM, as expected (Fig. 3I).
To further investigate the relationship between transcription-dependent DNA damage and protein homeostasis in ATM-depleted cells, we tested for the effects of DRB treatment in the protein aggregation assay as in Fig. 1. Both CK2β and PSMB2 showed significantly higher enrichment in the aggregate fraction with ATM depletion in U2OS cells, which was reduced by DRB treatment (Fig. 4A, quantification of 3 replicates in Fig. 4B). We induced SETX expression in these cells and also saw a significant reduction in the aggregate level (Fig. 4C, D), consistent with the effects of SETX on single-strand breaks and R-loops. We also monitored ROS levels in these cells and found that SETX did not reduce the elevated ROS induced by ATM depletion (Fig. 4E), thus we infer that the R-loops and transcription-induced damage in these cells are downstream of ROS.
Figure 4. Active transcription and PARylation promote protein aggregation in human cells lacking ATM.
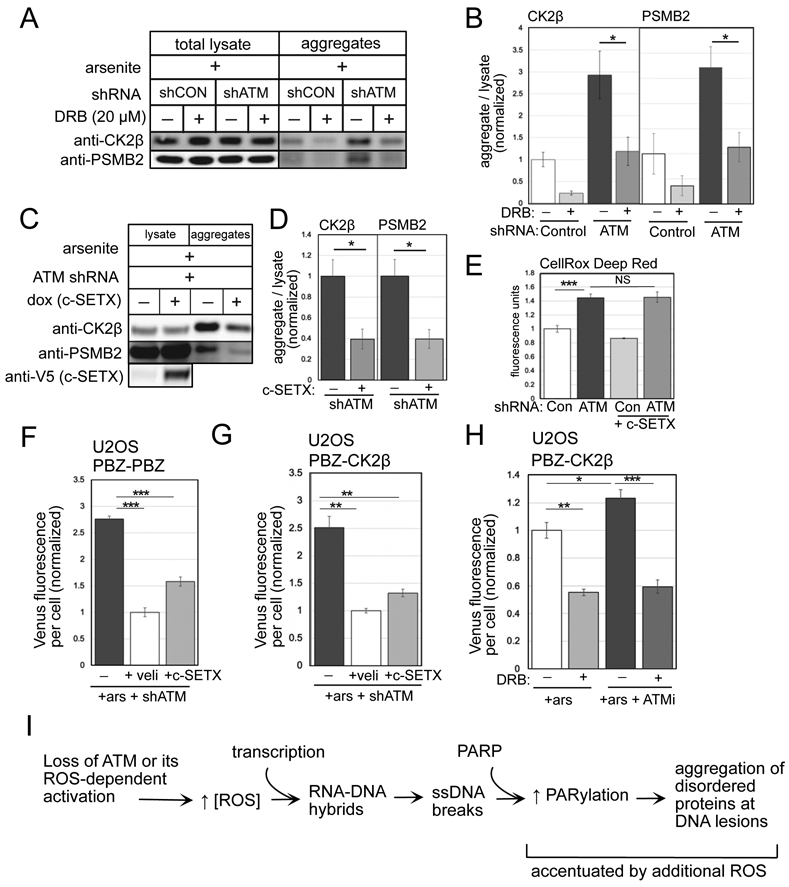
(A) Detergent-resistant aggregates were isolated in U2OS cells with shRNA-mediated depletion of ATM and arsenite (25 μM) and DRB (20 μM) added as indicated. Western blots for CK2β and PSMB2 in lysate and aggregate fractions are shown. (B) Three replicates of (A) were performed and quantified; levels of CK2β and PSMB2 in aggregate fractions normalized by lysate levels are shown relative to control cells. (C) Aggregation assays as in (A) with shRNA depletion of ATM, arsenite, and induction of SETX as indicated. (D) Three replicates of (C) were performed and quantified; levels of CK2β and PSMB2 in aggregate fractions normalized by lysate levels are shown relative to cells without SETX induction. Error (E) Quantification of ROS in U2OS cells with ATM depletion and SETX induction; measured by CellROX in triplicate. (F) Fluorescence yield from the PBZ-PBZ PAR sensor measured in triplicate as in Fig. 2B in ATM-depleted U2OS cells with arsenite and SETX expression as indicated. (G) Fluorescence yield from the PBZ-CK2β sensor measured in triplicate as in Fig. 2E in ATM-depleted U2OS cells exposed to arsenite, and treated with veliparib or induced for SETX expression. (H) Fluorescence yield from the PBZ-CK2β PAR sensor as in Fig. 2E in U2OS cells exposed to arsenite, ATM inhibitor (ATMi, 1 μM AZD1390), and DRB as indicated. Error bars indicate standard deviation. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t test; NS = not significant. (I) Diagram of events occurring in ATM-deficient or ATM oxidation activation-deficient cells. An increase in ROS, together with active transcription, generates R-loops and single-strand DNA breaks that hyperactivate PARP, leading to accumulation of disorder-prone proteins at DNA lesions.
Lastly, we measured PARylation using the live-cell sensors and found that the increase in PBZ-PBZ association caused by ATM loss was significantly reduced by SETX expression (Fig. 4F) or by RNaseH expression (Fig. S3), similar to the extent of reduction by the PARP inhibitor veliparib. The level of PBZ-CK2β association was also dramatic reduced by both SETX and by DRB (Fig. 4G, H). Thus, the elevated levels of PARylation observed in ATM-deficient cells occur in a ROS- and transcription-dependent manner under these conditions.
Taken together, this evidence shows that ATM-deficient and ATM oxidation-deficient human cells exhibit a number of pathological indicators of nuclear DNA damage that include high levels of single-strand breaks and elevated R-loops. Our evidence suggests that hyper-PARylation occurs as a result of these DNA lesions, and that direct association between oxidation-prone proteins and PAR polymer is caused by this series of events, ultimately generating the aggregation of disordered proteins at the sites of DNA lesions (Fig. 4I).
An Mre11 ATLD mutant generates single-strand breaks and protein aggregation
Our model suggests that high ROS due to loss of ATM activation by oxidation is essential for the aggregation of the proteins we have monitored and also for neurodegeneration, considering that the R3047X mutation which specifically inhibits oxidative activation (Guo et al., 2010) is the causative genetic lesion in a subset of patients with A-T (Chessa et al., 1992; Gilad et al., 1998). One complication with this hypothesis, however, is the fact that rare mutations in Mre11 also generate A-T-Like Disorder (ATLD) which includes progressive cerebellar neurodegeneration (Stewart et al., 1999; Taylor et al., 2004). If protein aggregation and neurodegeneration are in fact independent of MRN-mediated activation of ATM, then the ATLD phenotype is inconsistent with this view.
To investigate this issue, we reconstituted an ATLD mutant allele in human U2OS cells (Fig. 5A). We depleted Mre11 with shRNA and expressed either shRNA-resistant WT Mre11 or a mutant allele containing both of the mutations present in ATLD3/4 patients, R572X and N117S (Stewart et al., 1999; Taylor et al., 2004), referred to here as "ATLD". This combination of mutations results in expression of a truncated protein lacking the C-terminal 136 residues and is present at reduced levels compared to the WT allele. We confirmed that the replacement of endogenous Mre11 with the ATLD mutant compromised the activation of ATM by DNA double-strand breaks, as measured by camptothecin-induced Kap1 phosphorylation on S824 (Fig. 5A).
Figure 5. ATLD cells show elevated PARylation, single-strand DNA breaks and protein aggregation similar to ATM oxidation-deficient cells.
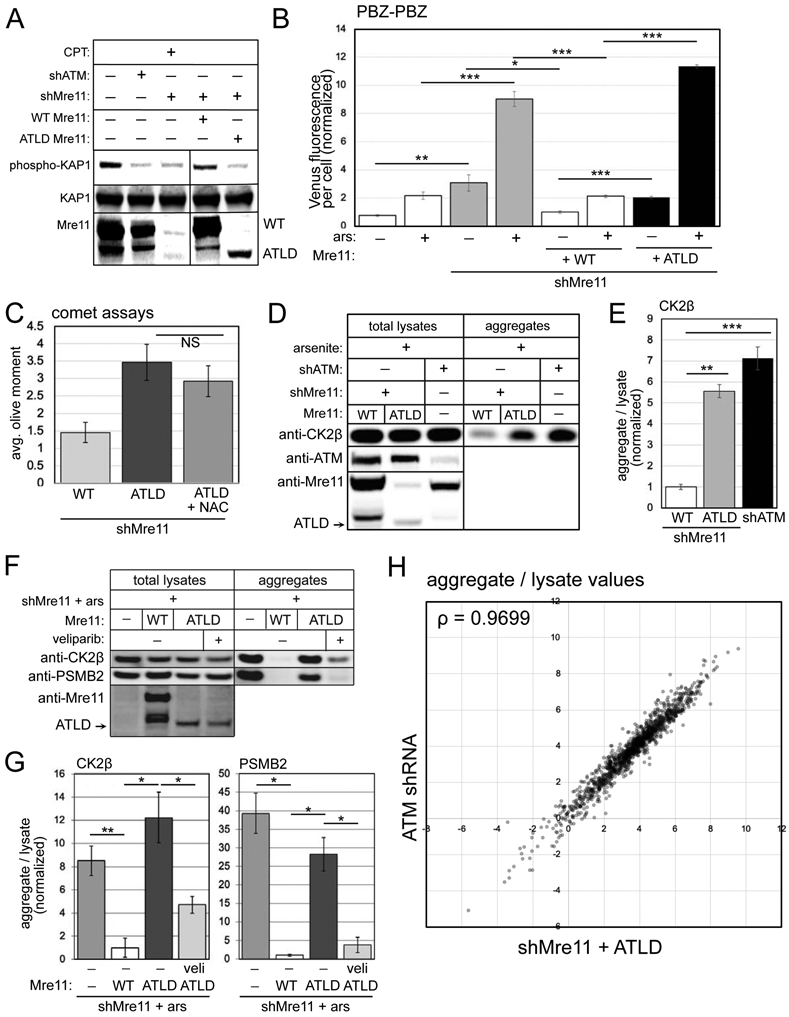
(A) shRNA depletion of Mre11 in U2OS cells with WT or ATLD3/4 expression, as indicated. Cells were exposed to camptothecin (10 μM, 1 hour) and blotted for phospho-S824 Kap1, total Kap1, and Mre11. (B) Fluorescence yield from the PBZ-PBZ PAR sensor measured in triplicate as in Fig. 2B in U2OS cells with Mre11 depletion, expression of WT or ATLD Mre11, and arsenite (25 μM) as indicated. Error bars indicate standard deviation. (C) Alkaline comet assays as in Fig. 1 with Mre11 shRNA depletion and expression of ATLD Mre11 with NAC as indicated; error bars represent SEM. (D) Aggregation assays as in Fig. 1 with Mre11 shRNA and WT or ATLD Mre11 expression compared to ATM shRNA treatment as indicated. Total lysates and aggregates analyzed by western blot for ATM, Mre11, and CK2β. (E) Three replicates of (D) were quantified; levels of CK2β and PSMB2 in aggregate fractions normalized by lysate levels are shown relative to cells expressing WT Mre11. (F) Aggregation assays as in Fig. 1 with Mre11 shRNA and either WT or ATLD Mre11 expression, veliparib added as indicated (10 μM). Lysates and aggregate fractions analyzed by western blot for Mre11, CK2β, and PSMB2. (G) Three replicates of (F) were quantified; levels of CK2β and PSMB2 in aggregate fractions normalized by lysate levels shown relative to cells expressing WT Mre11. Error bars indicate standard deviation. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant. (H) Aggregates and total lysates from 3 replicates of (D) were analyzed by mass spectrometry. Aggregate values (normalized by lysate) for each protein (mean of 3 replicates) were plotted: Mre11-depleted cells with ATLD expression (X axis) versus ATM-treated cells (Y axis); Spearman correlation coefficient = 0.9699.
Based on our results above indicating that PARP activation is a focal point that links ATM deficiency with protein aggregation, we tested the cells expressing WT or mutant Mre11 alleles for PARylation using the PBZ-PBZ sensor. This data shows that depletion of Mre11 generates higher levels of PARylation both in the presence and absence of arsenite (Fig. 5B). Expression of shRNA-resistant WT Mre11 eliminated this increase, while expression of the ATLD3/4 allele generated significantly higher PAR levels, similar to or even higher than Mre11 depletion alone. Consistent with this result, measurement of single-strand breaks using an alkaline comet assay showed that ATLD expression in the absence of endogenous Mre11 also generates higher levels of DNA damage compared to cells with WT full-length Mre11 expression (Fig. 5C). Interestingly, unlike with ATM depletion, the effect of Mre11 mutant expression on DNA breaks is not significantly reduced by antioxidant treatment (NAC)(Fig. 5C). We also do not see any change in ROS levels with depletion or mutation of Mre11 (Fig. S5).
We then measured protein aggregation using the ATLD Mre11 reconstituted cell lines, quantifying levels of CK2β lysates and in the detergent-resistant aggregate fraction. Here we observed increased aggregation of CK2β, compared to cells expressing WT Mre11, and similar to levels observed in cells with ATM depletion (Fig. 5D, E). Unlike with ATM depletion or inhibition, however, the aggregation observed with Mre11 loss was not reduced by NAC (Fig. S5). Nevertheless, ATLD-associated aggregates are reduced by veliparib (Fig. 5F, G), indicating a role for PARylation in promoting protein aggregation in the absence of normal Mre11 function, similar to what we observed with loss of ATM.
Lastly, we analyzed the lysates and detergent-resistant aggregate fractions from Mre11-depleted cells reconstituted with wild-type or ATLD Mre11, as well as ATM-depleted cells using label-free quantitative mass spectrometry. We observed a total of 2364 proteins present in all samples and calculated the abundance of each protein present in the total lysate as well as aggregate fractions, controlling the false discovery rate at 0.05 (Benjamini and Hochberg, 1995). From this analysis we determined that 1704 proteins showed significantly different aggregate values in Mre11 shRNA + ATLD expressing cells compared to WT Mre11 expressing cells (the control samples in this experiment), while 1584 targets were found to be significantly enriched in the aggregate fraction of ATM shRNA-treated cells compared to controls (Table S1). The aggregate levels (normalized by lysate amounts) of proteins showing significant differences in ATLD versus ATM shRNA cells are shown in Fig. 5H, where it is evident from visible inspection as well as the high Spearman correlation coefficient (ρ=0.9699) that the aggregation propensity of nearly all of these proteins is extremely similar in the absence of ATM function compared to the absence of Mre11 function.
From this analysis we conclude that loss of Mre11 function generates widespread protein instability extremely similar in breadth and magnitude to that observed with ATM depletion, and that expression of the ATLD allele cannot complement this deficiency. This is surprising given our previous observation that the specific ablation of MRN activation of ATM (in the 2RA mutant) does not generate protein aggregates (Lee et al., 2018), R-loops, single-strand DNA breaks, or high PARylation. The only explanation for this is that the loss of ATM activation by MRN is not functionally equivalent to the loss of the MRN complex itself.
Protein aggregation is widespread in A-T patient cerebellum tissue
Up to this point we have focused on protein aggregation in ATM-deficient cell lines in culture but here we asked if the increase in protein aggregation also occurs in human A-T patients. To test for this, we acquired fresh-frozen cerebellum tissue from 21 deceased A-T patients, ages 8 to 47 years (Table S3). For comparison we also acquired 21 control samples, with age, sex, and race matched to patients. The tissue from each individual was homogenized in dry ice prior to lysate preparation, and was sufficient to perform two replicate aggregation preparations (Fig. 6A).
Figure 6. Global proteomics analysis of A-T patient cerebellum tissues and age-matched controls shows widespread protein aggregation.

(A) Diagram of brain tissue workflow (see methods). (B) Representative western blot showing CK2β and PSMB2 levels in cerebellum lysates and aggregate fractions. (C, D) Quantification of western blotting from 42 lysates (replicates averaged) and 42 aggregate samples (normalized by lysate amounts), with all values normalized to control group. Error bars indicate standard deviation. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant. (E). Levels of 2019 proteins found in the cerebellum lysates of control and A-T patient samples by mass spectrometry. Red indicates proteins with significant differences between patient and control groups; grey indicates non-significant differences. (F) Cerebellum aggregate levels from 1512 proteins quantitated in A-T patients relative to controls, normalized by lysate levels. Red/grey indicators as in (E). (G) Neocortex aggregate levels (normalized by lysate values) for 2436 polypeptides from 5 A-T patients and age-matched controls (#1, 4, 7, 10, 11); Red/grey indicators as in (E). (H) Cerebellum aggregate levels (normalized by lysate values) for 2106 polypeptides from 5 A-T patients and age-matched controls (#1, 4, 7, 10, 11). Red/grey indicators as in (E). Significance was determined by Benjamini-Hochberg with FDR 0.05 for the analysis of 21 pairs (E,F) and 0.1 for the analysis of 5 pairs (G,H).
The lysate and aggregate fractions from each of the 42 individuals (with technical replicates for each) were analyzed first by western blotting, as done previously with cell line-derived samples. An example of a portion of this dataset is shown in Fig. 6B, blotted for CK2β and PSMB2. The results show generally similar levels of the proteins in lysates, but enrichment of these factors in the detergent-resistant aggregate fractions. Analysis performed for all of the samples show a statistically significant increase (2 to 2.5-fold) in CK2β and PSMB2 aggregates when comparing all 21 A-T patients as a group to the control individuals, while no differences were observed in lysate levels (Fig. 6C, D).
We then analyzed all samples by quantitative mass spectrometry which identified a total of 4350 proteins, using a minimum of 2 unique peptides identified with high confidence as a threshold. Replicates of the lysate and aggregate fractions were analyzed, with a high level of similarity between replicates (average Spearman rho correlation coefficients for normalized aggregate values were 0.945 and 0.898, respectively, for controls and patients). 2019 proteins were identified in all lysate samples from all individuals, while 2577 proteins were identified in all aggregate samples. From this analysis we identified 1512 proteins for which we obtained a complete set of quantitative values for all lysate and all aggregate samples in all individuals in both replicates, which was used to calculate normalized aggregate levels for each protein (aggregate/lysate ratio)(Table S4).
The total lysates from patients and control individuals show relatively few statistically significant differences (5 proteins among the 2019 proteins with complete data for all individuals, Fig. 6E, Table S4). Nevertheless, it is notable that the proteins in this group include IP3R1 and CA8, and that INPP5A and CBLN1 are also strongly reduced in the patient lysate samples (Fig. S6). Mutations in the gene encoding IP3R1 cause familial spinocerebellar ataxia, and IP3R1, CA8, INPP5A, and CBLN1 are all important for inositol-regulated calcium signaling (Dudding et al., 2004; Huang et al., 2012; Klar et al., 2017; van de Leemput et al., 2007; Yang et al., 2015).
In contrast to the lysates, the number of proteins with significantly different levels of aggregates in A-T patient cerebellum samples compared to controls was very high (1184 of 1512 total)(Fig. 6F, Table S4). The vast majority of these (1183 of 1184) exhibited higher aggregate levels in A-T patients compared to controls, reminiscent of the results we observed in human cell lines (Fig. 1).
To determine if this aggregation propensity is specific to the cerebellum, we also obtained prefrontal neocortex tissue (Brodmann areas 9 and 10) from a subset (5) A-T patients and from their respective controls. Analysis of these samples, also with technical replicates, showed no differences between A-T patients and the control tissues in either the lysates or the aggregate fractions (Fig. 6G, Table S4). Thus the marked tendency toward protein destabilization is only seen in A-T patient cerebellum, the site of neurodegeneration. Analysis of the cerebellum aggregates specifically from the subset of 5 A-T patients analyzed for neocortex are shown for comparison in Fig. 6H.
We previously showed that aggregation-prone proteins observed in the absence of ATM oxidative activation in cultured human cells exhibit higher predicted disorder scores when measured by the TANGO algorithm for aggregation propensity (Fernandez-Escamilla et al., 2004; Lee et al., 2018). The aggregation scores for the proteins found to be more enriched in A-T cerebellum aggregate samples were also analyzed in this manner and were found to have even higher scores (average of 2792.5 compared to the average for the total proteome, 1910.4) than the aggregates previously identified.
Analysis of the aggregate-prone proteins identified in the cerebellum samples shows enrichment for metabolic enzymes (top 10% of hits analyzed for gene ontology term enrichment shown in Fig. S7). Other enriched categories include NADH regeneration and sodium/potassium transport proteins, oxidoreductases, and protein chaperones. Lastly, it is notable that proteins associated with other forms of neurodegeneration are found enriched in the aggregate list, including Ataxin-10, a protein required for the survival of cerebellar neurons and mutated in Spinocerebellar ataxia type 10 (Teive et al., 2011), and alpha-synuclein, well-known for its aggregation propensity in Parkinson's Disease (De Mattos et al., 2020).
Comparison of the aggregation-prone polypeptides in U2OS cells (including proteins found to be aggregated both ATM-depleted as well as ATLD cells), U87-MG cells, and in A-T patient samples identifies a core set of 189 aggregation-prone proteins (Table S4). This comparison indicates similar global aggregation propensity in cultured cells and in patient cerebellum samples, particularly when considering the fact that the overlap in total proteome among this diverse set of cell lines and brain tissue includes only 705 polypeptides.
Aggregate and PARylation levels distinguish A-T patients from controls
We also used the normalized aggregate levels for all of the individuals to perform unsupervised hierarchical clustering (Fig. S7). Consistent with the strong bias for aggregation observed in patients as a group (Fig. 6F), this analysis separates most of the patients from the controls. However, it is clear that there is variation among the patients, and a group of 5 patients (A-T 3, 6, 17, 19, and 21) cluster separately from the rest. Included in this subset are the two youngest patients (A-T 3 and 21), both 8 years old at the time of sampling.
A subset of the patient cerebellum tissues were also available as formalin-fixed samples. Several of these were examined using immunohistochemistry (IHC) for overall organization of the cerebellum, as well as for levels of poly-ADP-ribose using an anti-PAR antibody (Fig. 7A). Analysis of patient samples showed that all contained identifiable molecular, granular, and Purkinje cell layers, thus the patient tissue is not grossly dissimilar to that of the controls with respect to cerebellum organization (A-T 6 shown here as an example). Blinded scoring of 5 patient tissue samples and 5 controls revealed a significant increase in overall PAR staining intensity in the patient group as compared to the controls (Fig. 7B).
Figure 7. Elevated PAR is present in A-T patient cerebellum tissue.

(A) Examples of cerebellum tissue (formalin-fixed) from an A-T patient (#6) and control, analyzed by immunohistochemistry with an antibody directed against poly-ADP-ribose, HRP-conjugated secondary antibody, and 3,3'Diaminobenzidine with methyl green counterstain at two resolutions as indicated. (B) Subjective scoring of the level of PAR staining of cells in the granular layer of 5 A-T patient cerebellum samples (5, 6, 12, 18, 21) compared to age-matched controls was performed using a 0 to 3 scale of blinded samples by 3 individuals. Error bars indicate standard deviation. *, **, ***, and **** indicate p<0.05, 0.005, and 0.0005 by Student two-tailed t-test; NS = not significant. (C) Examples of A-T patient formalin-fixed cerebellum tissues stained with antibodies directed against PAR, CALB1 (Purkinje cells), GFAP (astrocytes), and TMEM119 (microglia) as indicated from A-T patients #5, 12, and 1, respectively, using fluorescence-labeled secondary antibodies and DAPI. White arrows indicate Purkinje cells in the CALB1/PAR-stained images.
From the IHC analysis, we observed PAR staining in the nuclei of cells in both the granule and molecular layers, as well as in Purkinje cells. To confirm this, we also performed similar experiments with antibodies directed to specific cell populations in the cerebellum using fluorescence detection (Fig. 7C). This analysis confirms the presence of PAR in A-T Purkinje neurons (CALB1) and microglia (TMEM119), but we did not observe colocalization with GFAP (generally considered to be a marker for astrocytes). PAR is also present in these cell types in control tissues, however, so the cell type specificity of PAR here does not distinguish the patient from control samples.
Discussion
Widespread protein aggregation is a characteristic of A-T patient cerebellum tissue
All of the prevalent neurodegenerative disorders in humans are associated with a loss of protein homeostasis that ultimately results in aggregation of specific proteins in disease-specific patterns in the brain (Bourdenx et al., 2017). A-T has not previously been considered in this way, but here we present evidence showing that this cerebellum-specific neurodegenerative disorder also exhibits widespread protein aggregation, affecting more than 1000 polypeptides in human patient cerebellum tissue. Proteins found in these aggregates range from metabolic enzymes to signaling components that are, on average, much higher in predicted intrinsic disorder compared to the total proteome. In addition, we observed this aggregation propensity in the cerebellum but not in the pre-frontal cortex, thus it is correlated with the known site of neurodegeneration in A-T patients.
In other neurological diseases where mutations in specific disorder-prone factors predispose individuals to neurodegeneration, the presence of the mutant aggregating protein clearly plays a causal role in tissue dysfunction (Currais et al., 2017; Soto and Pritzkow, 2018). The progression toward disease endpoints is also influenced by the levels of protein dysfunction, natural aging, as well as levels of oxidative stress and DNA damage (Madabhushi et al., 2014; Maynard et al., 2015). In A-T patient cerebellum tissues, protein aggregation is clearly elevated when comparing patients as a group to control individuals. Hierarchical clustering of patients and controls by aggregate values groups most of the patients together, but five patients (including the two youngest) show a different pattern of modest aggregation levels (Fig. S7). It is possible that these subtle patterns represent earlier stages of the disease, with relatively few aggregating proteins present. If so, these individuals illustrate patterns early in the degenerative process and could yield further insights into the initial molecular events that drive tissue degeneration.
A-T cerebellum contains lower levels of proteins associated with dominant cerebellar ataxia syndromes
The total proteome quantifications we performed on the patient and control lysate samples only identified a few differences compared to the aggregate comparisons. However, among this small group are proteins that are very important in other forms of cerebellar ataxia. For instance, the intracellular calcium channel IP3R1 (expressed from the ITPR1 gene) is 7-fold lower in A-T patients relative to the control distribution (Fig. S6). ITPR1 deletions in humans cause a dominant form of spinocerebellar ataxia, SCA15/16 (Dudding et al., 2004; van de Leemput et al., 2007), and missense mutations in these genes are linked to congenital nonprogressive ataxia known as SCA29 (Huang et al., 2012; Klar et al., 2017). IP3R1 is regulated by levels of inositol 1, 4, 5-triphosphate, a signaling molecule hydrolyzed by the INPP5A phosphatase. Deletion of the INPP5A gene causes cerebellar degeneration in mice (Yang et al., 2015), emphasizing the important role for this signaling pathway in Purkinje cells. We also found that INPP5A levels are low in A-T patients (2.8-fold, Fig. S6). Lastly, carbonic anhydrase 8 (CA8), a binding partner of IP3R1, is also significantly decreased in A-T patient cerebellum tissue. Missense mutations in ITPR1 that abolish IP3R1 binding to CA8 are pathological mutations in SCA29 (Ando et al., 2018). Mutant ataxin-2 and ataxin-3 proteins responsible for the more common SCA2 and SCA3 disorders, respectively, have also been shown to interact abnormally with IP3R1 (Kasumu and Bezprozvanny, 2012). Taken together, the observations in our dataset strongly suggest the possibility of deficient inositol phosphate-regulated calcium signaling in A-T patients. It is notable in this regard that the early A-T literature documented "aberrant sensing" of calcium by A-T cells in culture, reduced duration of calcium currents in A-T neuronal cells, as well as deficiencies in phosphoinositol metabolism (Famulski and Paterson, 1999; Famulski et al., 2003; Khanna et al., 1997; Yorek et al., 1999), but the molecular basis of these observations was not resolved.
PARP activity promotes protein aggregation in the absence of ATM oxidative activation
Our results show that poly-ADP-ribosylation by PARP1 and 2 is important for the aggregation of proteins observed in the absence of ATM function in cultured cells. PARP has previously been shown to be hyperactivated in ATM-deficient cells (Fang et al., 2016), although the depletion of NAD+ caused by PARP overactivation was implicated as the critical factor in that study. We have not observed any effects of NAD+ replenishment on ROS levels, R-loops, or DNA damage (using nicotinamide riboside as an NAD+ donor), thus we conclude that it is the hyper-PARylation itself and not the depletion of NAD+ that generates the protein aggregates in ATM-deficient cells.
PARylation at sites of DNA damage has already been shown to attract intrinsically disordered proteins (Altmeyer et al., 2015; Chen et al., 2018; Hong et al., 2013; Mastrocola et al., 2013; Rulten et al., 2014; Singatulina et al., 2019). In a few specific cases, phosphorylation of these disordered proteins by ATM has been shown to disperse them from PARylation sites (Chen et al., 2018; Gardiner et al., 2008), suggesting that ATM function is important for reversal of PAR-induced insoluble protein domains. We propose that loss of ATM combined with excessive PARylation and disordered protein accumulation leads ultimately to the irreversible loss of protein solubility that we observe in the tissues. In agreement with this idea, there is a growing literature surrounding PAR-mediated protein condensates, the association of disordered proteins with these condensates, and their appearance in pathological situations (Leung, 2020). PARP1 hyperactivity promotes cerebellar ataxia in mice in the absence of XRCC1 for instance (Hoch et al., 2017), also a situation in which excess single-strand breaks generate PARP overactivation. PARP also promotes the neurotoxicity of several RNA-binding proteins in a Drosophila model of ALS (Duan et al., 2019), and has been shown to accelerate alpha-synuclein-dependent neurodegeneration in Parkinson's disease (Kam et al., 2018). All of this evidence is consistent with a role for PAR in generating stable, toxic protein aggregates. PARP activity does facilitate single-strand break repair, however (Gupte et al., 2017), and deletion of ATM and PARP1 simultaneously in the mouse generates synthetic lethality (Murcia et al., 2001). It is possible that PARP activity is required for the rapid cell divisions during early mammalian embryogenesis in the context of ATM-deficiency, but then becomes toxic in later stages of growth by promoting the assembly of protein aggregates.
Transcription-associated lesions in ATM-deficient cells generate single-strand breaks, PARP hyperactivation, and protein insolubility
Our results strongly suggest a role for ATM in preventing global single-strand breaks, with important components of this phenotype contributed by excess ROS as well as active transcription. The canonical view of ATM activation via DNA damage requires DNA double-strand breaks, an activation pathway we have previously demonstrated in vitro with purified components and many groups have confirmed in mammalian cells (Lee and Paull, 2005; Paull, 2015; Shiloh, 2003). Others have suggested more recently that ATM can also be activated by single-strand breaks in DNA where it acts to delay S phase entry (Khoronenkova and Dianov, 2015), and that base excision repair (BER) efficiency is decreased upon loss of ATM protein, coincident with the accumulation of oxidized protein and increases in nuclear proteasome activity (Poletto et al., 2017). Changes in BER-mediated repair of single-strand breaks in the absence of ATM may contribute to the spontaneous breaks we have observed.
In addition, ATM activation can be induced by Top1 poisons such as camptothecin, which was reported to generate ATM activation in a transcription-dependent manner (Sakasai et al., 2010; Sordet et al., 2009), although double-strand breaks were invoked in this scenario as the activating lesion. Based on this evidence as well as observations of Top1 and Top2 conjugates accumulating in the absence of ATM function (Alagoz et al., 2013; Katyal et al., 2014; Yamamoto et al., 2016) or Mre11 (Hoa et al., 2016), we overexpressed Tdp1 or Tdp2 in ATM-depleted cells since these enzymes directly promote resolution of topoisomerase conjugates (Pommier et al., 2014). However, this did not alleviate the protein aggregation we observed (data not shown). We note that Top2a was slightly elevated in aggregate fractions from ATLD-expressing U2OS cells compared to WT Mre11-expressing cells but was unchanged in ATM-depleted cells (Table S1). Top1 levels were not statistically different in either case, nor was it found to be elevated in aggregate fractions from A-T patients (Table S4). Lastly, camptothecin treatment does not induce aggregation in the absence of ATM function (data not shown), despite the fact that Top1 conjugates are strongly induced under these conditions (Pommier, 2006). We conclude that Top1 conjugates are not likely to be the critical lesion that induces the aggregation.
The fact that elevated ROS is required for hyperactivation of PAR, accumulation of single-strand breaks, and aggregation in the absence of ATM oxidative activation suggests an alternative model where ROS-induced transcriptional stalling generates R-loops and, ultimately, single-strand breaks (Fig. 4I). This has been suggested in other contexts where active transcription in the presence of oxygen stress was shown to generate R-loops that trigger transcription-coupled DNA repair (Teng et al., 2018). Our data showing that SETX overexpression alleviates not only the R-loops but also single-strand breaks, hyper-PARylation, and protein aggregation suggests that RNA-DNA hybrids are a critical intermediate in this pathway. Consistent with this model, mutations in SETX are linked to neurodegenerative disorders AOA2 (ataxia with oculomotor apraxia type 2) and amyotrophic lateral sclerosis type 4 (ALS4), and expansions in C9ORF72 that are causative for ALS are also known to generate pathological R-loops (Wang et al., 2015).
It is not clear how loss of ATM leads to R-loop accumulation. ATM phosphorylation of splicing-related targets was shown to alter irradiation-induced spliceosome dynamics (Tresini et al., 2015), which could be important considering the close relationship between splicing efficiency and R-loop formation (Santos-Pereira and Aguilera, 2015). R-loops were not found to be present at higher levels in ATM-deficient mice (Yeo et al., 2014), although the mouse models of A-T do not show overt cerebellar degeneration or ataxia (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996).
ATLD is not functionally equivalent to loss of ATM activation by MRN
The association between the MRN complex and ATM was initiated by the discovery of MRE11 mutations in patients diagnosed with A-T, identified as "A-T-like Disorder" (ATLD) patients (Stewart et al., 1999). Subsequent work showed that MRN is required for ATM activation by double-strand breaks in mammalian cells and in purified systems (Lee and Paull, 2004, 2005; Uziel et al., 2003), solidifying the model that the clinical features observed in ATLD are attributable to a loss of ATM activation via MRN. It is certainly the case that MRN plays a critical role in facilitating ATM activation at double-strand breaks; however, a series of recent observations suggest that the neurodegeneration in ATLD patients may not be related to this specific function.
First, three cerebellar ataxia patients were identified in France with previously unreported biallelic mutations in the MRE11 gene, diagnosed with ATLD (Fiévet et al., 2019). Unlike other ATLD patients, cells from these individuals showed no defects in ATM activation despite relatively low levels of MRN complex. This data indicates that the ATLD phenotype does not necessarily include a global loss of ATM-mediated DNA damage responses.
Second, we identified a separation-of-function allele of ATM (2RA) that is specifically deficient in ATM activation via the MRN complex and DNA double-strand breaks in vitro with purified components as well as in human cells (Lee et al., 2018). Expression of the 2RA allele of ATM in place of the WT allele did not generate high ROS or protein aggregation, even in the presence of additional oxidative stress, while alleles that block the oxidative pathway show high levels of aggregates. Considering the correlation between protein aggregates and A-T status shown in this study, the results with the 2RA allele suggests that loss of MRN-stimulated ATM activity is not the critical event leading to loss of protein homeostasis observed in ATM-deficient cells.
Third, the analysis we performed in this study suggests that loss of the MRN complex, accentuated by expression of ATLD alleles (a combination of ATLD3/4 R572X and N117S used in this work), generates many of the same functional consequences as loss of ATM, including high single-strand breaks, hyper-activation of PARP, and protein aggregation. A detailed comparison of the levels of each misfolded polypeptide shows remarkable convergence between ATLD and loss of ATM, analogous to the neurological similarities between ATLD and A-T patients. Combined with the additional observation that the ROS activation-specific allele of ATM (R3047X) also causes neurodegeneration (Chessa et al., 1992; Gilad et al., 1998), this suggests that ATLD shares a defect with ATM-deficient cells and cells lacking ATM oxidative activation.
Interestingly, cells lacking Mre11 (either with or without ATLD allele expression) show the same loss of protein homeostasis observed with ATM-deficient cells, but ROS does not appear to play a critical role. For instance, the antioxidant NAC does not significantly reduce the levels of single-strand DNA breaks in ATLD cells, nor does it block protein aggregation. ROS levels are also not elevated in Mre11-depleted cells, in contrast to ATM-depleted cells. We do not know the origin of single-strand DNA breaks in cells lacking MRN, but this will be an important area of future study.
Limitations
Overall, our investigation of human cerebellum tissue as well as cell lines is consistent with the proposal that loss of protein homeostasis is a marked feature of both ATM and Mre11 deficiency in human cells. Results in tissue culture clearly show that the activities of PARP enzymes are important for this phenomenon, stimulated by single-strand breaks that are created in a transcription-dependent manner. Our investigation of protein aggregates in A-T patient and control brain tissue is consistent with this hypothesis; however, a retrospective analysis cannot prove causality. More detailed analysis of PARylation targets and dynamics will help us to understand the trajectory of events from initial lesion to the ultimate loss of cerebellum function that occurs in this untreatable disease but ultimately an animal model or manipulable system is critical for demonstrating these relationships.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tanya Paull, (tpaull@utexas.edu).
Materials Availability
Recombinant DNA and cell lines generated in this study are available on request without restriction.
Data Availability
The gel and microscopy images generated during this study are available at Mendeley Data [6wxfjgyk5k].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines, cell culture and recombinant protein induction
Human osteosarcoma U2OS cells (wild-type and ΔPARP1), human U87-MG glioblastoma cells, and human SH-SY5Y neuroblastoma cells were used. U2OS T-Rex FLP-in cells containing plasmids with ATM and MRE11 genes were cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) containing 15 μg/ml Blasticidin (A1113903, Life Technology), 100 units/ml penicillin-streptomycin (15140-122, Life Technology), and 200 μg/ml Hygromycin (400052-50ml, Life Technology). Depletion of endogenous ATM was performed by incubating cells with lentivirus containing shRNA cassettes overnight and selecting with media containing 1 μg/ml puromycin (Invitrogen) for 5-7 days. To induce genes introduced through the FLP-in system, doxycycline (1 μg/ml; Thermo Fisher, #BP-2653-5) was added to the medium as final concentration 3 days before treatment with arsenite (25 mM; Sigma, #S7400-500G). U2OS with PARP1 deletion were a generous gift from Nicholas Lakin (Ronson et al., 2018). Lentivirus was prepared in HEK-293T cells as previously described (Lee et al., 2018). U87-MG glioblastoma cells were obtained from Vishwanath Iyer and were grown under the same conditions as U2OS cells. SH-SY5Y cells were obtained from the Bioresource Collection and Resource Center in Taiwan and were grown in DMEM with 10% FBS for maintenance (adherent cells only). The cells were induced to differentiate with a 3 day treatment with 1% FBS media and 10 μM retinoic acid followed by 4 days with zero FBS, retinoic acid, and 50 ng/ml BDNF. In these cells, C-term-V5 SETX (a.a. 1851 to 2677) was expressed from a bacmam (pTP4483) under the control of a tet-inducible promoter.
Human tissues
Frozen cerebellum and cortex tissues were obtained for 21 A-T patients and 21 age/gender-matched controls (total 14 female, 28 male, average age 28 yrs) (see Table S3). Formalin-fixed tissues for 5 A-T patients and age/gender matched controls as well as frozen tissues were obtained. All tissues were obtained from the NIH NeuroBiobank and are de-identified.
METHOD DETAILS
Gene expression constructs
Recombinant ATM expression in U2OS cells was performed with pcDNA5-FRT/TO-intron derived vectors containing shRNA-resistant ATM alleles, including WT (pTP3540), C2991L (pTP3362), R2579A/R2580A (pTP4011), D2889A (pTP4012), and R3047X (pTP3552). Depletion of endogenous ATM was performed by incubating cells with lentivirus containing shRNA toward ATM (sc-29761-SH, Santa Cruz Biotechnology). The C-terminus of Senataxin was expressed from a pcDNA5-FRT-TO derivative containing C-term-V5 SETX, a.a. 1851 to 2677 (pTP3531)(Makharashvili et al., 2018). RNaseH was expressed from a pcDNA5-FRT-TO derivative containing E. coli rnhA fused to mCherry, pTP3492. The original rnhA plasmid was a gift from Patrick Calsou (Addgene #60365)(Britton et al., 2014). The PBZ-PBZ sensor was derived from pBiFC-PBZ-VC and pBiFC-PBZ-VN (Addgene #110648 and 110646, respectively) but combined into one bacmam plasmid, pTP4623, using the backbone of pAceBac1 (Bieniossek et al., 2008). The original pBiFC PBZ constructs were gifts from Chris Lord (Krastev et al., 2018). The PBZ domain fused to VC in the PBZ-PBZ plasmid pTP4623 was also replaced with human CK2β, creating the PBZ-CK2β sensor in pTP4658. These plasmids were used to generate bacmids using standard methods with the bac-to-bac system (Life Technologies), and virus was produced in Sf21 insect cells. Depletion of endogenous ATM was performed by lentivirus-encoded shRNA as described previously (Lee et al., 2018). Depletion of endogenous Mre11 was performed with lentivirus expressing shRNA (5′-ACAGGAGAAGAGAUCAACUUUGUUAAUAUUCAUAGCAAAGUUGAUCUCUUCUCCUGU-3′) from the H1 promoter using the backbone from lenticrispr v2 (Addgene #52961), a gift from Feng Zhang (Sanjana et al., 2014), with Cas9 deleted, pTP4359. WT shRNA-resistant Mre11 with a C-terminal Flag tag was expressed from a pcDNA5-FRT-TO derivative, pTP4099. A mutant version of this plasmid containing Mre11 N117S and R572X mutations was generated using quikchange mutagenesis (Agilent) to create the ATLD expression plasmid pTP4225. Parp1 and Parp2 were depleted with shRNA expressed from a pRSITEP (Cellecta) backbone (PARP1: 5′-GCCUCCGCUCCUGAACAAUGCGUUAAUAUUCAUAGCGCAUUGUUCAGGAGCGGAGGC-3′ ; PARP2: 5′-UCAGUGUAAUGAACUACUAGAGCUAUGAAUAUUAACUCUAGUAGUUCAUUACACUGA-3′), pTP4960 and pTP4961 respectively. Cloning details and sequence files are available upon request.
Split venus PBZ sensors
U2OS cells were plated in T75 flasks until cells reached 70% confluency. 5 ml baculovirus second amplification supernatant containing either PARylation sensor or PBZ/CK2ß sensor bacmam was added to each flask with 5ml of DMEM containing 10% FBS. A day after infection, cells were plated in multiple wells of a 6-well plate at approximately X% confluence. Cells were treated with either 25μM arsenite, 1μM AZD1390, 1mM NAC, 1μg/ml Doxocycline or 10μM Veliparib as indicated in the figure legends. The following day, cells were harvested, washed with cold PBS containing 0.9mM Ca2+, 0.5mM Mg2+, and resuspended in Triton X-100 extraction buffer (0.5% Triton X-100, 20 mM Hepes-KOH (pH 7.9), 50 mM NaCl, 3 mM MgCl2, 300 mM Sucrose) for 2 minutes. Extracted samples were recovered by centrifugation at 1200Xg at 4°C for 3 minutes, then resuspended in 500 uL cold PBS and kept on ice until FACS analysis (no more than 2 hours). Flow Cytometry: Extracted cells were analyzed with a BD LSRII Fortessa Flow cytometer using fluorescence detection parameters for Alexa Fluor® 488 (excitation 488nm; emission 519nm) for detection of split venus. Extracted cells were gated according to forward scatter and side scatter in order to remove any partial or non-extracted cells. For all experiments at least 10,000 events measured. Average and standard deviation were generated with the median of fluorescence value from three biological replicates. The absolute levels of laser output can vary so all results are shown as fold differences in comparison to either untreated cells or cells with veliparib exposure.
Comet assay
U2OS cells were grown in DMEM (10% FBS) media in the presence of doxycycline (1 μg/ml) in 6-well plates for 3 days and treated with NAC (1 mM) as indicated in the figure legends for 18 hours before harvesting. Alkaline comet assays were performed using OxiSelect Comet assay Kit (Cellbiolabs, #STA-350) following the manufacturer’s protocol. Samples were observed under a Zeiss Axiovert 200M fluorescence microscope and analyzed with Fiji imageJ program. SH-SY5Y cells were analyzed similarly after 7 days differentiation as described above with doxycycline (1 μg/ml) to induce shRNA targeted to ATM.
ROS measurements
Cells treated with CellRox deep red reagent (Thermo Fisher, #C10422) at a final concentration of 5 mM for 30 min at 37°C. Cells were harvested with trypsin, washed with PBS, and then transferred to 5 ml of polystyrene round bottom tubes (VWR, #60818-496) in 1 ml of PBS for analysis by flow cytometry according to the manufacturer’s protocol.
DNA-RNA immunoprecipitation (DRIP) assay
Cells were harvested with trypsin, resuspended in 5 ml of PBS supplemented with 0.5% SDS and 1 mg/ul Proteinase K (Gold biotechnology, #P-480-500) as a final concentration, and digested at 37°C overnight. Genomic DNA was isolated and performed DRIP assay as previously described (Makharashvili et al., 2018).
Aggregation assay
To prepare cell lysates, the pellets were resuspended in lysis buffer (20 mM Na-phosphate pH 6.8, 10 mM DTT, 1mM EDTA, 0.1% Tween 20, 1 mM PMSF, and EDTA-free protease inhibitor mini tablets (Thermo Fisher, #A32955)) and rotated at 4°C for 30 min. Cells were sonicated in a 4°C water bath-based sonicator (Bioruptor: 8 times at level 4.5 and 50% duty cycle) and centrifuged for 20 min at 200g at 4°C. The concentration of protein in the supernatant was calculated using the Bradford reagent (Thermo Fisher, #23236) and adjusted to a same concentration for all samples. Protein aggregates were pelleted at 16,000g for 20 min at 4°C. After removing supernatants, protein aggregates were washed 2 times with NP-40 buffer (20 mM Na-phosphate pH 6.8, 2% NP-40, 1mM PMSF, and EDTA-free protease inhibitor mini tablets), sonicated 6 times at level 4.5 and 50% duty cycle, and centrifuged at 16,000g for 20 min at 4°C. Aggregated proteins were washed in wash buffer (20 mM Na-phosphate pH 6.8, 1mM PMSF, and EDTA-free protease inhibitor mini tablets), sonicated 4 times at level 3 and 50% duty cycle, and boiled in 2X SDS sample buffer. Resuspended aggregated proteins were analyzed by western blotting with antibodies directed against CK2β (Abcam ab76025) and PSMB2 (Abcam ab166628), or processed for mass spectrometry as described below.
Brain tissue homogenization and aggregate preparation
Fresh-frozen cerebellum tissue (vermis, approximately 0.5 g) or prefrontal cortex tissue (Brodmann areas 9 or 10) was obtained for 21 A-T patients ranging in age from 8 to 50 years as well as age/gender-matched controls. Tissue samples were pulverized with 15 ml powdered dry ice for 2 min in a commercial blender on the highest setting. The tissue and dry ice mixtures were brushed into disposable weigh dishes then poured into 50 ml conical tubes and placed on ice prior to cell lysis. All steps were performed in a 4°C cold room with materials kept chilled on dry ice. Powdered brain tissues were incubated with lysis buffer (see Aggregation assay, above) for 20 min on ice, sonicated 5 times at level 4.5 and 50% duty cycle, and centrifuged for 5 min at 200g. Supernatants were processed using the standard aggregation assay by starting with sonication 8 times at level 4.5 and 50% duty cycle.
Filter aided sample preparation
Frozen tissue lysates and pellets were kept in SDS loading buffer prior to experiment. Sample preparation was performed as previously described (Lee et al., 2018) with the following changes. 8μg of protein for lysates or whole samples for pellets in SDS loading buffer were boiled for 5 min. before dilution to 800μL of UA (8M Urea, 0.1M Tris-HCl pH8.8) total volume. After filter aided sample preparation, label free quantification LC-MS/MS was performed by the proteomics facility in the University of Texas at Austin following previously described procedures (Ryu et al., 2020). The raw data was processed by Proteome Discoverer 2.2.
Statistics testing and analysis
Results from Proteome Discoverer 2.2 (Thermo Fisher) were further refined by removing common known contaminates and any proteins identified with less than two unique polypeptides. Then refined data was normalized by western blot results of corresponding samples using CK2β amount in order to correct for variation in sample loading. Pellet over lysate values were generated by dividing pellet values with corresponding lysate values then taking log (base 2) of the results. Proteins with missing data were dropped for the analysis. For U2OS cells, P-values comparing control to shATM or shMre11 were generated using Welch’s T-test. Two technical replicates from brain samples were averaged before the Welch’s T test comparing control patients to A-T patients. Benjamini-Hochberg procedure was used in order to control for multiple hypothesis testing using either 0.05 FDR (except for 5 patient comparisons which used 0.1 FDR as noted in the legends). Heatmap and hierarchical clustering were generated using R studio version 1.2.5001. with heatmap.2 function under gplots package in conjugation with colorRampPalette function in RColorBrewer package. The hierarchical clustering was performed using default hclust method with “euclidean” distance calculation and “complete” clustering.
Immunohistochemistry
Formalin-fixed cerebellar tissue blocks were dehydrated, paraffinized, and cut into 4 μm thick sections using standard methods. Tissue sections were mounted on charged glass slides, deparaffinized with xylene then rehydrated with graded alcohols. Antigen retrieval was performed by incubation with Proteinase K for 20 min at 37°C.
HRP imaging:
After rinsing in TBST, sections were incubated overnight at 4°C with primary antibody for PAR (Abcam PN: ab14459, 1:100 in IHC World diluent PN: IW1000). Endogenous peroxidase activity was quenched with 3% H2O2 in water for 15 min before incubation with poly-HRP secondary antibody (Thermo Fisher PN: 32230, 1:400 in TBS-1% BSA) for 1 hr at room temperature. Sections were stained using BD DAB substrate kit (PN: 550880), counterstained with methyl green, dehydrated in ethanol, cleared in xylene and mounted with DPX mountant (Millipore Sigma). A negative control for non-specific secondary antibody binding was generated following the above procedure omitting primary antibody addition. Nuclear staining with DAB was observed in slides incubated with PAR antibody while only methyl green counterstain was present in negative control. Five images of each A-T patient or control tissue at 40X magnification were scored by 3 individuals, with patient/control identity blinded. The scoring was performed as previously described (Emre et al., 2020), with a scale of 0 (not detected), 0.5 (very low), 1 (low), 1.5 (low to moderate), 2 (moderate), 2.5 (moderate to strong), and 3 (strong), analyzing only cells in the granular layer.
Fluorescence imaging:
Tissue sections were deparaffinized as above before performing antigen retrieval in 10mM sodium citrate, 0.05% Tween 20, pH 6.0 for 15 min in a pressure cooker. Sections were incubated overnight at 4°C with primary antibodies for PAR (Abcam ab14459, 1:100) and either Calbindin D28K (Abclonal A0802, 1:100), GFAP (Abclonal A0237, 1:100), or TMEM119 (Atlas Antibodies HPA051870, 1:200), followed by a 1 hr room temperature incubation with Goat anti-Mouse Alexa Fluor 647 (Invitrogen A32728, 1:500) and Goat anti-Rabbit Alexa Fluor 488 (Invitrogen A32732, 1: 500). A negative control for non-specific secondary antibody binding was generated following the above procedure omitting primary antibody addition. All slides were counterstained with DAPI to visualize nuclei. Multispectral laser scanning confocal microscopy was performed using a Zeiss LSM880 and the 20X (0.8 NA) Plan/Apo objective with a pinhole aperture of 1-1.5 AU. Sequential scanning was used to capture DAPI, 488 and 647 channels and tile scans with a 10% overlap were stitched using ZenBlue to create seamless mosaics. Patient and normal samples were imaged in parallel using the same instrument parameters (e.g., transmission, gain, offset).
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blot analysis: Fluorescence-based signals were quantified using a Licor Odyssey system after subtraction of background. At least 3 biological replicates were used for quantification and the Student t-test was used for statistical analysis, as noted in the figure legends. Standard deviation and standard error were used as indicated in the figure legends.
Mass spectrometry:
Label-free quantification of peaks was performed with Proteome Discoverer 2.2 (Thermo Fisher) as discussed above. Protein identifications with 2 or more unique peptides with used for quantification. Welch's t-test was used for p value calculation and the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) was used to control the false discovery rate at 0.05 (all comparisons in the study, including the A-T cerebellum analysis) or 0.1 (comparisons of 5 patients and 5 controls). Excel and R Studio were used for the analysis of the mass spectrometry data. Zen software was used for fluorescence-based IHC analysis and Fiji was used for DAB analysis.
Supplementary Material
Supplementary Table 1: Quantitation of polypeptides in U2OS cells with ATM shRNA depletion or Mre11 shRNA depletion with either wild-type Mre11 or ATLD expression by mass spectrometry. Lysates and aggregate fractions shown separately. Related to Figure 1.
Supplementary Table 2: Quantitation of polypeptides in U87-MG cells with ATM shRNA depletion, ATM shRNA depletion plus NAC treatment, or control cells, by mass spectrometry. Lysates and aggregate fractions shown separately. Related to Figure 1 and Figure S1.
Supplementary Table 4: Quantitation of polypeptides in human cerebellum and cortex samples by mass spectrometry. Lysates and aggregate fractions shown separately. Aggregated proteins found in U2OS, U87-MG, and cerebellum. Related to Figures 6 and 7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| poly-ADP-ribose | Abcam | ab14459 |
| Calbindin D28K | ABclonal | A0802 |
| GFAP | ABclonal | A0237 |
| TMEM119 | Atlas Antibodies | HPA051870 |
| Goat anti-Mouse Alexa Fluor 647 | Invitrogen | A32728 |
| Goat anti-Rabbit Alexa Fluor 488 | Invitrogen | A32732 |
| Goat anti-Rabbit Alexa Fluor 680 | Thermo Scientific | A-21076 |
| Goat anti-Mouse IRDye 800CW | Li-cor Biosciences | 926-32210 |
| PARP2 | Genetex | GTX34116 |
| PARP1 | Genetex | GTX75098 |
| CK2β | Abcam | ab76025 |
| PSMB2 | Abcam | ab166628 |
| ATM | Santa Cruz | sc-135663 |
| Mre11 | Genetex | GTX118741 |
| SETX | Santa Cruz | sc-100319 |
| Biological Samples | ||
| human brain tissue, see Table S3 | NeuroBioBank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATM inhibitor AZD1390 | Selleckchem | S8680 |
| veliparib | Fisher Scientific | S100410 |
| retinoic acid | Cayman Chemical Company | 11017 |
| BDNF | Fisher Scientific | CYT20710UG |
| sodium arsenite | Sigma | S7400-100G |
| Critical Commercial Assays | ||
| comet assay | Cellbiolabs | OxiSelect Comet assay Kit, #STA-350 |
| ROS indicator | Thermo Fisher | CellRox deep red reagent, #C10422 |
| DAB substrate kit | BD | 550880 |
| Deposited Data | ||
| gel and IHC original images | Mendeley Data | doi: 10.17632/6wxfjgyk5k.1 |
| Experimental Models: Cell Lines | ||
| U2OS osteosarcoma cells | Blerta Xhelmace | U2OS |
| U87-MG glioblastoma cells | Vishy Iyer | U87-MG |
| SH-SY5Y neuroblastoma cells | Bioresource Collection and Resource Center (Taiwan) | BCRC 960450 |
| U2OS with PARP1 deletion | Nicholas Lakin | (Ronson et al., 2018) |
| Oligonucleotides | ||
| ATM shRNA expression (lentivirus) | Santa Cruz Biotechnology | sc-29761-SH |
| Mre11 shRNA expression (lentivirus); (5′-ACAGGAGAAGAGAUCAACUUUGUUAAUAUUCAUAGCAAAGUUGAUCUCUUCUCCUGU-3′) in lenticrispr v2 | this work | N/A |
| Parp1 shRNA expression (lentivirus); (5'-GCCUCCGCUCCUGAACAAUGCGUUAAUAUUCAUAGCGCAUUGUUCAGGAGCGGAGGC-3') in pRSITEP | this work | N/A |
| Parp2 shRNA expression (lentivirus); (5'-UCAGUGUAAUGAACUACUAGAGCUAUGAAUAUUAACUCUAGUAGUUCAUUACACUGA-3') in pRSITEP | this work | N/A |
| Recombinant DNA | ||
| WT ATM shRNA-resistant expression construct based on pcDNA5 | (Lee et al., 2018) | N/A |
| C2991L ATM shRNA-resistant expression construct based on pcDNA5 | (Lee et al., 2018) | N/A |
| R2579A/2580A ATM shRNA-resistant expression construct based on pcDNA5 | (Lee et al., 2018) | N/A |
| D2889A kinase-deficient ATM shRNA-resistant expression constructs based on pcDNA5 | (Lee et al., 2018) | N/A |
| R3047X ATM shRNA-resistant expression constructs based on pcDNA5 | (Lee et al., 2018) | N/A |
| c-term SETX expression in pcDNA5 | (Makharashvili et al., 2018) | N/A |
| E. coli RNAseH expression in pcDNA5 | (Makharashvili et al., 2018) | N/A |
| PBZ-PBZ sensor in pAceBac1 bacmam | this work | N/A |
| PBZ-CK2β sensor in pAceBac1 bacmam | this work | N/A |
| Flag-hMre11, shRNA-resistant, in pcDNA5 | this work | N/A |
| Flag-hMre11 with N117S and R572X mutations in pcDNA5 | this work | N/A |
| Software and Algorithms | ||
| Proteome Discoverer 2.2 | Thermo Fisher | N/A |
| R Studio | rstudio.com | N/A |
| ZenBlue | Zeiss | N/A |
| Fiji | imagej.net | N/A |
Highlights:
PARP activity drives protein aggregation in ATM-deficient human cells
reactive oxygen species and R-loops contribute to the aggregation propensity
Loss of Mre11 is similar to ATM deficiency
Protein aggregates are abundant in A-T patient cerebellum tissue but not in cortex
Acknowledgments
We thank members of the Paull laboratory for helpful discussion and help with blinded scoring, as well as Domenico Delia for critical suggestions, to Vishwanath Iyer for the glioblastoma cell line, to Nicholas Lakin for PARP1 deletion cells, and to Rajashree Deshpande, Oshadi Wimalarathne, and Colleen Jeter for important reagents and imaging contributions. The graphical abstract was created in BioRender. We are indebted to the families of the A-T patients and control individuals who contributed the autopsy material for research use, as well as the Neurobiobank for providing us with the frozen and fixed tissue samples. We acknowledge the Howard Hughes Medical Institute for funding this project as well as Cancer Prevention and Research Institute grant RP170628. Laser scanning confocal microscopy was performed at the MD Anderson Cancer Center Science Park Flow Cytometry and Cell Imaging Core with funding support provided by the CPRIT core facility grant RP170628.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agholme L, Lindström T, Kågedal K, Marcusson J, and Hallbeck M (2010). An In Vitro Model for Neuroscience: Differentiation of SH-SY5Y Cells into Cells with Morphological and Biochemical Characteristics of Mature Neurons. J. Alzheimers Dis 20, 1069–1082. [DOI] [PubMed] [Google Scholar]
- Aguilera A, and Gómez-González B (2017). DNA–RNA hybrids: the risks of DNA breakage during transcription. Nat. Struct. Mol. Biol 24, 439–443. [DOI] [PubMed] [Google Scholar]
- Alagoz M, Chiang SC, Sharma A, and El-Khamisy SF (2013). ATM deficiency results in accumulation of DNA-topoisomerase I covalent intermediates in neural cells. PLoS One 8, e58239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grøfte M, Rask M-BD, Streicher W, Jungmichel S, Nielsen ML, et al. (2015). Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun 6, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H, Hirose M, and Mikoshiba K (2018). Aberrant IP 3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc. Natl. Acad. Sci 115, 12259–12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–171. [DOI] [PubMed] [Google Scholar]
- Barlow C, Dennery PA, Shigenaga MK, Smith MA, Morrow JD, Roberts LJ, Wynshaw-Boris A, and Levine RL (1999). Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc Natl Acad Sci U A 96, 9915–9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai A, Rotman G, and Shiloh Y (2002). ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair Amst 1, 3–25. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. [Google Scholar]
- Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365. [DOI] [PubMed] [Google Scholar]
- Bhatia V, Valdés-Sánchez L, Rodriguez-Martinez D, and Bhattacharya SS (2018). Formation of 53BP1 foci and ATM activation under oxidative stress is facilitated by RNA:DNA hybrids and loss of ATM-53BP1 expression promotes photoreceptor cell survival in mice. F1000Research 7, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniossek C, Richmond TJ, and Berger I (2008). MultiBac: multigene baculovirus-based eukaryotic protein complex production. Curr. Protoc. Protein Sci Chapter 5, Unit 5.20. [DOI] [PubMed] [Google Scholar]
- Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, and Tsarbopoulos A (2017). Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog. Neurobiol 155, 171–193. [DOI] [PubMed] [Google Scholar]
- Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, Frit P, and Calsou P (2014). DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 42, 9047–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijsen RAM, Toonen LJA, Gardiner SL, and van Roon-Mom WMC (2019). Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 16, 263–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-K, Lin W-L, Chen Z, and Liu H (2018). PARP-1–dependent recruitment of cold-inducible RNA-binding protein promotes double-strand break repair and genome stability. Proc. Natl. Acad. Sci 115, E1759–E1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessa L, Petrinelli P, Antonelli A, Fiorilli M, Elli R, Marcucci L, Federico A, and Gandini E (1992). Heterogeneity in ataxia-telangiectasia: classical phenotype associated with intermediate cellular radiosensitivity. Am J Med Genet 42, 741–746. [DOI] [PubMed] [Google Scholar]
- Cremona CA, and Behrens A (2014). ATM signalling and cancer. Oncogene 33, 3351–3360. [DOI] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, and Cimprich KA (2019). R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 73, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A, Fischer W, Maher P, and Schubert D (2017). Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 31, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Pellegrini M, Lee BS, Guo Z, Filsuf D, Belkina NV, You Z, Paull TT, Sleckman BP, Feigenbaum L, et al. (2012). Loss of ATM kinase activity leads to embryonic lethality in mice. J Cell Biol 198, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mattos EP, Wentink A, Nussbaum-Krammer C, Hansen C, Bergink S, Melki R, and Kampinga HH (2020). Protein Quality Control Pathways at the Crossroad of Synucleinopathies. J. Park. Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditch S, and Paull TT (2011). The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 37, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, Ma Z, Qian B, Deng X, Zhang K, et al. (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 29, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudding TE, Friend K, Schofield PW, Lee S, Wilkinson IA, and Richards RI (2004). Autosomal dominant congenital non-progressive ataxia overlaps with the SCA15 locus. Neurology 63, 2288–2292. [DOI] [PubMed] [Google Scholar]
- Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, Fok JHL, et al. (2018). The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv 4, eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, and Leder P (1996). Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U A 93, 13084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre C, Hjorth E, Bharani K, Carroll S, Granholm A, and Schultzberg M (2020). Receptors for pro-resolving mediators are increased in Alzheimer’s disease brain. Brain Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski KS, and Paterson MC (1999). Defective regulation of Ca2+/calmodulin-dependent protein kinase II in gamma-irradiated ataxia telangiectasia fibroblasts. FEBS Lett. 453, 183–186. [DOI] [PubMed] [Google Scholar]
- Famulski KS, Al-Hijailan RS, Dobler K, Pienkowska M, Al-Mohanna F, and Paterson MC (2003). Aberrant sensing of extracellular Ca2+ by cultured ataxia telangiectasia fibroblasts. Oncogene 22, 471–475. [DOI] [PubMed] [Google Scholar]
- Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, et al. (2016). NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 24, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, and Serrano L (2004). Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22, 1302–1306. [DOI] [PubMed] [Google Scholar]
- Fiévet A, Bellanger D, Valence S, Mobuchon L, Afenjar A, Giuliano F, Dubois d’Enghien C, Parfait B, Pedespan J, Auger N, et al. (2019). Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect. Hum. Mutat 40, 1690–1699. [DOI] [PubMed] [Google Scholar]
- Gardiner M, Toth R, Vandermoere F, Morrice NA, and Rouse J (2008). Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J 415, 297–307. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Das D, and Franco S (2018). The Role for the DSB Response Pathway in Regulating Chromosome Translocations. Adv. Exp. Med. Biol 1044, 65–87. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, and Morimoto RI (2006). Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474. [DOI] [PubMed] [Google Scholar]
- Gilad S, Chessa L, Khosravi R, Russell P, Galanty Y, Piane M, Gatti RA, Jorgensen TJ, Shiloh Y, and Bar-Shira A (1998). Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet 62, 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh N, Bühler A, Huang C, Li KW, van Nierop P, Smit AB, Fändrich M, Baumann F, and David DC (2017). Age-Dependent Protein Aggregation Initiates Amyloid-β Aggregation. Front. Aging Neurosci 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, and Paull TT (2010). ATM activation by oxidative stress. Science 330, 517–521. [DOI] [PubMed] [Google Scholar]
- Gupte R, Liu Z, and Kraus WL (2017). PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, and Caldecott KW (2018). The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 71, 319–331.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 57, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsusui K, et al. (2016). Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 64, 580–592. [DOI] [PubMed] [Google Scholar]
- Hoch NC, Hanzlikova H, Rulten SL, Tétreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, et al. (2017). XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Ma J, Dai S, Xu T, Li H, and Yasui A (2013). The Role of hnRPUL1 Involved in DNA Damage Response Is Related to PARP1. PLoS ONE 8, e60208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Chardon JW, Carter MT, Friend KL, Dudding TE, Schwartzentruber J, Zou R, Schofield PW, Douglas S, Bulman DE, et al. (2012). Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis 7, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002. [DOI] [PubMed] [Google Scholar]
- Kai M (2016). Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci 2016, 17, 310. Int. J. Mol. Sci. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam T-I, Mao X, Park H, Chou S-C, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, et al. (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumu A, and Bezprozvanny I (2012). Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum Lond. Engl 11, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, Zhao J, Russell HR, Petrini JHJ, Nitiss JL, et al. (2014). Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci 17, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Yan J, Watters D, Hobson K, Beamish H, Spring K, Shiloh Y, Gatti RA, and Lavin MF (1997). Defective signaling through the B cell antigen receptor in Epstein-Barr virus-transformed ataxia-telangiectasia cells. J. Biol. Chem 272, 9489–9495. [DOI] [PubMed] [Google Scholar]
- Khoronenkova SV, and Dianov GL (2015). ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. U. S. A 112, 3997–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikis EA, Gidalevitz T, and Morimoto RI (2010). Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol 694, 138–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar J, Ali Z, Farooq M, Khan K, Wikström J, Iqbal M, Zulfiqar S, Faryal S, Baig SM, and Dahl N (2017). A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur. J. Hum. Genet 25, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T, Mariotti C, and Paulson HL (2019). Spinocerebellar ataxia. Nat. Rev. Dis. Primer 5, 24. [DOI] [PubMed] [Google Scholar]
- Knezevic CE, Wright G, Rix LLR, Kim W, Kuenzi BM, Luo Y, Watters JM, Koomen JM, Haura EB, Monteiro AN, et al. (2016). Proteome-wide Profiling of Clinical PARP Inhibitors Reveals Compound-Specific Secondary Targets. Cell Chem. Biol 23, 1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krastev DB, Pettitt SJ, Campbell J, Song F, Tanos BE, Stoynov SS, Ashworth A, and Lord CJ (2018). Coupling bimolecular PARylation biosensors with genetic screens to identify PARylation targets. Nat. Commun 9, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF, Yeo AJ, and Becherel OJ (2013). Senataxin protects the genome: Implications for neurodegeneration and other abnormalities. Rare Dis. Austin Tex 1, e25230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, and Paull TT (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96. [DOI] [PubMed] [Google Scholar]
- Lee J-H, and Paull TT (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Mand MR, Kao C-H, Zhou Y, Ryu SW, Richards AL, Coon JJ, and Paull TT (2018). ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci. Signal 11, eaan5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung H-C, Lin X, et al. (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 3, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKL (2020). Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol. 30, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, and Tsai L-H (2014). DNA Damage and Its Links to Neurodegeneration. Neuron 83, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makharashvili N, Arora S, Yin Y, Fu Q, Wen X, Lee J-H, Kao C-H, Leung JW, Miller KM, and Paull TT (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. ELife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, and Tibbetts RS (2013). The RNA-binding Protein Fused in Sarcoma (FUS) Functions Downstream of Poly(ADP-ribose) Polymerase (PARP) in Response to DNA Damage. J. Biol. Chem 288, 24731–24741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, and Bohr VA (2015). DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med 5, a025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murcia J.M. -d., Mark M, Wendling O, Wynshaw-Boris A, and de Murcia G (2001). Early Embryonic Lethality in PARP-1 Atm Double-Mutant Mice Suggests a Functional Synergy in Cell Proliferation during Development. Mol. Cell. Biol 21, 1828–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT (2015). Mechanisms of ATM Activation. Annu Rev Biochem 84, 711–738. [DOI] [PubMed] [Google Scholar]
- Paull TT (2019). RNA-DNA hybrids and the convergence with DNA repair. Crit. Rev. Biochem. Mol. Biol 54, 371–384. [DOI] [PubMed] [Google Scholar]
- Poletto M, Yang D, Fletcher SC, Vendrell I, Fischer R, Legrand AJ, and Dianov GL (2017). Modulation of proteostasis counteracts oxidative stress and affects DNA base excision repair capacity in ATM-deficient cells. Nucleic Acids Res. 45, 10042–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y (2006). Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Huang SN, Gao R, Das BB, Murai J, and Marchand C (2014). Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 19, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regal JA, Festerling TA, Buis JM, and Ferguson DO (2013). Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet 22, 5146–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, Petermann E, and Lakin ND (2018). PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun 9, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, and Poirier MA (2004). Protein aggregation and neurodegenerative disease. Nat. Med 10 Suppl, S10–17. [DOI] [PubMed] [Google Scholar]
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, and Lederman HM (2016). Ataxia telangiectasia: a review. Orphanet J. Rare Dis 11, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DAQ, Gómez-Herreros F, Hafezparast M, and Caldecott KW (2014). PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 42, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SW, Stewart R, Pectol DC, Ender NA, Wimalarathne O, Lee J-H, Zanini CP, Harvey A, Huibregtse JM, Mueller P, et al. (2020). Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. PLOS Biol. 18, e3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakasai R, Teraoka H, Takagi M, and Tibbetts RS (2010). Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison. J. Biol. Chem 285, 15201–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, and Aguilera A (2015). R loops: new modulators of genome dynamics and function. Nat. Rev. Genet 16, 583–597. [DOI] [PubMed] [Google Scholar]
- Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf H-W, and Rüb U (2012). Brain pathology of spinocerebellar ataxias. Acta Neuropathol. (Berl.) 124, 1–21. [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003). ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3, 155–68. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, and Ziv Y (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14, 197–210. [PubMed] [Google Scholar]
- Singatulina AS, Hamon L, Sukhanova MV, Desforges B, Joshi V, Bouhss A, Lavrik OI, and Pastré D (2019). PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Rep. 27, 1809–1821.e5. [DOI] [PubMed] [Google Scholar]
- Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, et al. (2009). Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 10, 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, and Pritzkow S (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci 21, 1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, and Taylor AM (1999). The DNA double-strand break repair gene hMre11 is mutated in individuals with an Ataxia-Telangiectasia-like disorder. Cell 99, 577–587. [DOI] [PubMed] [Google Scholar]
- Stracker TH, and Petrini JH (2011). The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Groom A, and Byrd PJ (2004). Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair Amst 3, 1219–1225. [DOI] [PubMed] [Google Scholar]
- Teive HAG, Munhoz RP, Arruda WO, Raskin S, Werneck LC, and Ashizawa T (2011). Spinocerebellar ataxia type 10 - A review. Parkinsonism Relat. Disord. 17, 655–661. [DOI] [PubMed] [Google Scholar]
- Teng Y, Yadav T, Duan M, Tan J, Xiang Y, Gao B, Xu J, Liang Z, Liu Y, Nakajima S, et al. (2018). ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun 9, 4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M, Hara T, Zhang H, Yamamoto T, Akaboshi S, Nanba E, Ohno K, Hori N, Sato K, and Takeshita K (1998). Ataxia-telangiectasia without immunodeficiency: novel point mutations within and adjacent to the phosphatidylinositol 3-kinase-like domain. Am J Med Genet 75, 141–144. [PubMed] [Google Scholar]
- Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JA, van Ij.W.F., Grosveld FG, Medema RH, Hoeijmakers JH, et al. (2015). The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrhammer N, Bay J-O, Perlman S, and Gatti RA (2002). Ataxia-Telangiectasia and variants. [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, and Shiloh Y (2003). Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22, 5612–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Haeusler AR, and Simko EA (2015). Emerging role of RNA•DNA hybrids in C9orf72 -linked neurodegeneration. Cell Cycle 14, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, and Baltimore D (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10, 2411–22. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Wang J, Sprinzen L, Xu J, Haddock CJ, Li C, Lee BJ, Loredan DG, Jiang W, Vindigni A, et al. (2016). Kinase-dead ATM protein is highly oncogenic and can be preferentially targeted by Topo-isomerase I inhibitors. Elife Camb. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AW, Sachs AJ, and Nystuen AM (2015). Deletion of Inpp5a causes ataxia and cerebellar degeneration in mice. Neurogenetics 16, 277–285. [DOI] [PubMed] [Google Scholar]
- Yankulov K, Yamashita K, Roy R, Egly J-M, and Bentley DL (1995). The Transcriptional Elongation Inhibitor 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole Inhibits Transcription Factor IIH-associated Protein Kinase. J. Biol. Chem 270, 23922–23925. [DOI] [PubMed] [Google Scholar]
- Yeo AJ, Becherel OJ, Luff JE, Cullen JK, Wongsurawat T, Jenjaroenpun P, Jenjaroenpoon P, Kuznetsov VA, McKinnon PJ, and Lavin MF (2014). R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PloS One 9, e90219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorek MA, Dunlap JA, Manzo-Fontes A, Bianchi R, Berry GT, and Eichberg J (1999). Abnormal myo-inositol and phospholipid metabolism in cultured fibroblasts from patients with ataxia telangiectasia. Biochim. Biophys. Acta 1437, 287–300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Quantitation of polypeptides in U2OS cells with ATM shRNA depletion or Mre11 shRNA depletion with either wild-type Mre11 or ATLD expression by mass spectrometry. Lysates and aggregate fractions shown separately. Related to Figure 1.
Supplementary Table 2: Quantitation of polypeptides in U87-MG cells with ATM shRNA depletion, ATM shRNA depletion plus NAC treatment, or control cells, by mass spectrometry. Lysates and aggregate fractions shown separately. Related to Figure 1 and Figure S1.
Supplementary Table 4: Quantitation of polypeptides in human cerebellum and cortex samples by mass spectrometry. Lysates and aggregate fractions shown separately. Aggregated proteins found in U2OS, U87-MG, and cerebellum. Related to Figures 6 and 7.
Data Availability Statement
The gel and microscopy images generated during this study are available at Mendeley Data [6wxfjgyk5k].