

Abstract
Purpose:
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease characterized by an extensive fibroinflammatory stroma, which includes abundant cancer-associated fibroblast (CAF) populations. PDAC CAFs are heterogeneous, but the nature of this heterogeneity is incompletely understood. The Hedgehog (HH) pathway functions in PDAC in a paracrine manner, with ligands secreted by cancer cells signaling to stromal cells in the microenvironment. Previous reports investigating the role of HH signaling in PDAC have been contradictory, with HH signaling alternately proposed to promote or restrict tumor growth. In light of the newly discovered CAF heterogeneity, we investigated how HH pathway inhibition reprograms the PDAC microenvironment.
Experimental Design:
We used a combination of pharmacologic inhibition, gain- and loss- of-function genetic experiments, CyTOF, and single cell RNA-sequencing to study the roles of HH signaling in PDAC.
Results:
We find that HH signaling is uniquely activated in fibroblasts and differentially elevated in myofibroblastic CAFs (myCAFs) compared to inflammatory CAFs (iCAFs). SHH overexpression promotes tumor growth, while HH pathway inhibition with the Smoothened antagonist LDE225 impairs tumor growth. Further, HH pathway inhibition reduces myCAF numbers and increases iCAF numbers, which correlates with a decrease in cytotoxic T cells and an expansion in regulatory T cells, consistent with increased immune suppression.
Conclusions:
HH pathway inhibition alters fibroblast composition and immune infiltration in the pancreatic cancer microenvironment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy with a 5-year survival rate of approximately 10% [1]. The PDAC microenvironment is characterized by an extensive stroma, comprised of non-neoplastic cells and extracellular matrix (ECM) components, including cancer-associated fibroblasts (CAFs). Within the stroma, CAFs secrete growth factors, immune-modulatory ligands and ECM proteins [2–4]. The Hedgehog (HH) signaling pathway is activated in PDAC CAFs and has been associated with ECM deposition and pancreatic tumorigenesis [5–10]. In PDAC, HH signaling acts via a paracrine mechanism whereby tumor-derived HH ligands activate downstream signaling in CAFs through interactions with the canonical HH receptor, Patched (PTCH1), and the co-receptors, GAS1, CDON and BOC [11–15]. These interactions initiate a downstream signaling cascade mediated by Smoothened (SMO), which results in cellular responses driven by GLI proteins, the transcriptional effectors of the HH pathway [16]. However, reports investigating the role of HH signaling in pancreatic cancer progression have been contradictory. Initial studies indicated that HH pathway inhibition in transplantation models impairs PDAC growth [5, 6, 12, 17, 18]. Moreover, acute HH pathway inhibition in a genetically engineered mouse model (GEMM) of PDAC depleted the ECM, increased vascular perfusion, and sensitized tumors to chemotherapy, providing a modest survival advantage [19]. However, these findings failed to translate in clinical trials, which were largely unsuccessful or even detrimental to patient health, leading to early termination [20, 21]. Subsequently, genetic inactivation of the ligand Sonic Hedgehog (SHH) in the context of an oncogenic Kras-driven mouse model of pancreatic cancer led to cachexia and to poorly differentiated and highly vascularized tumors, findings that were at least in part recapitulated by prolonged pharmacologic HH pathway inhibition [22, 23]. These disparate outcomes, with HH signaling seemingly both promoting and restricting pancreatic cancer progression, indicate potentially pleiotropic roles for this pathway that have yet to be fully elucidated.
The healthy pancreas is home to distinct fibroblast populations [24, 25], which have differing potential to expand during carcinogenesis [26]. We and others recently demonstrated that also PDAC CAFs are a heterogeneous population comprised of myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs) and antigen-presenting CAFs (apCAFs) [24, 25, 27–30]. Given the different functional roles proposed for these CAF subtypes, changes in the ratio of these populations may lead to distinct outcomes in PDAC progression. Therefore, we investigated whether HH modulation differentially affects PDAC CAF subtypes. Here, we show that HH pathway activation is higher in myCAFs compared to iCAFs in both mouse and human PDAC. Our data suggest that driving higher levels of HH signaling promotes pancreatic tumor growth, whereas acute inhibition of HH signaling reduces tumor volume. The current study also defines a novel dosage-dependent role for the Indian HH ligand in pancreatic cancer. Our data collectively support the notions that HH signaling is a key pathway in the maintenance of the myCAF subtype in pancreatic cancer, and that HH pathway inhibition alters the ratio of myCAF/iCAF fibroblast populations in PDAC and shifts the inflammatory response towards a more immune-suppressive microenvironment.
Materials and Methods
Study approvals
All animal procedures and studies were conducted in compliance with the guidelines of the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan (Protocol Number PRO00007983) and at Cold Spring Harbor Laboratory.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee at which the studies were conducted and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All human organoid experiments were approved by the Institutional Review Board of Cold Spring Harbor Laboratory and conducted in accordance with recognized ethical guidelines (Declaration of Helsinki). Surgical specimens of either tumor tissue or adjacent/normal pancreas were obtained from patients referred for the Whipple procedure or distal pancreatectomy according to IRB HUM00025339. Informed written consent was obtained from each subject.
Animal experiments
Orthotopic transplantation models
C57BL/6J mice (stock number 000664) were purchased from The Jackson Laboratory. Injections of organoid cultures for the generation of orthotopically grafted organoid (OGO) tumors were conducted as previously described [29]. Typically, 1–2.5 × 105 single cells prepared from organoid cultures were re-suspended as a 45 μL suspension of 50% Matrigel in PBS and injected into the pancreas. Tumors were imaged using the Vevo 3100 Ultrasound at 2 or 3 different orientations with respect to the transducer. Tumor volumes were measured at two angles, if possible, using the Vevo LAB software program (version 2.2.0). To establish the orthotopic pancreatic cancer models with the 7940b PDAC cell line [31], 5 × 104 7940b cells (C57BL/6J strain) were injected into the pancreas of C57BL/6J or Gli1lacZ/+ heterozygous mice derived from crossing Gli1lacZ/lacZ animals maintained on a mixed genetic background (129S6/SvEvTac; C57BL/6J; Swiss Webster) with C57BL/6J animals [32]. For HH pathway inhibition, mice were subjected to once daily oral gavage for 12 days, starting 6 days after surgical implantation of tumor cells with either 20 mg/kg of LDE225 (Sonidegib, ChemieTek #NVP-LDE225) re-suspended in PEG400/5% dextrose solution (LDE-treated) or the same volume of PEG400/5% dextrose solution alone (vehicle-treated).
Genetically engineered mouse models
C57BL/6J background (>20 backcrosses) KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) mice used for the LDE225 study were previously described [33]. KPC mice were monitored by abdominal palpation and ultrasound until tumors reached 6–8 mm in diameter. KPC mice were treated daily by oral gavage as described above with either vehicle or LDE225 for 2 weeks, and tumor volume was monitored by ultrasound. Tumors were imaged using the Vevo 3100 Ultrasound at 2 or 3 different orientations with respect to the transducer. Tumor volumes were measured at 2 angles, if possible, using the Vevo LAB software program (version 2.2.0). C57BL/6J background iKC-Shh mice were generated by crossing iKC (Ptf1a-CreERT; KrasLSL-G12D/+) (a gift from Chris Wright) and Shh (CLEShh) mice. For iKC-Shh mice, mutant Kras activation and/or Shh overexpression was induced by administration of 3 separate treatments of 5 mg per 30 g body weight of Tamoxifen (Sigma, T-5648) resuspended in corn oil when the mice were 8 weeks of age. In this model, these mice express mouse SHH (mRNA; GenBank #NM009170). Mice were aged for 34 weeks post Tamoxifen treatment and then pancreata were harvested for immunostaining. To localize SHH-responding cells in vivo, we used Gli1lacz/+ reporter mice [32]. Accordingly, Tamoxifen was administered and pancreata were fixed for 1 hour with 4% PFA for 1 hour on ice, frozen in Optimal Cutting Temperature Compound (OCT), and cryo-sectioned for β-Gal staining using X-GAL substrate. Sections were counterstained with nuclear Fast Red.
Single-cell RNA sequencing analysis
Datasets are from Elyada et al., [30], Peng et al., [34] and Steele et al., [35]. Dimensionality reduction was carried out in Scanpy (PMC5802054) via principal component analysis on the top 3000 highly variable genes followed by UMAP visualization using the top 30 significant components. Clustering was performed with the Louvain algorithm (https://doi.org/10.1088/1742–5468/2008/10/P10008) by iterating the resolution parameter until coarse cell types could be identified by differentially expressed canonical marker genes, which were then subjected to additional rounds of cluster refinement to annotate subtypes.
Cell lines and cell culture
Ihh WT and Ihh KO 7940b KPC cells, and Gli1lacZ/lacZ murine fibroblasts (up to passage number 15) were grown in DMEM supplemented with 10% FCS and 5% penicillin/streptomycin (Thermo Fisher, #15140163) as previously described [15, 36]. Murine PDAC T69A organoids [37] were cultured as previously described and transplanted when passage < 15 [38]. All cells were cultured at 37°C with 5% CO2. Cell line authentication was not performed. Mycoplasma testing with the MycoAlert Mycoplasma Detection Kit (LT07–318; Lonza) is performed monthly at Cold Spring Harbor Laboratory and University of Michigan, and each organoid/cell line has been tested at least once after thawing or isolation and retested prior to orthotopic transplantation experiments.
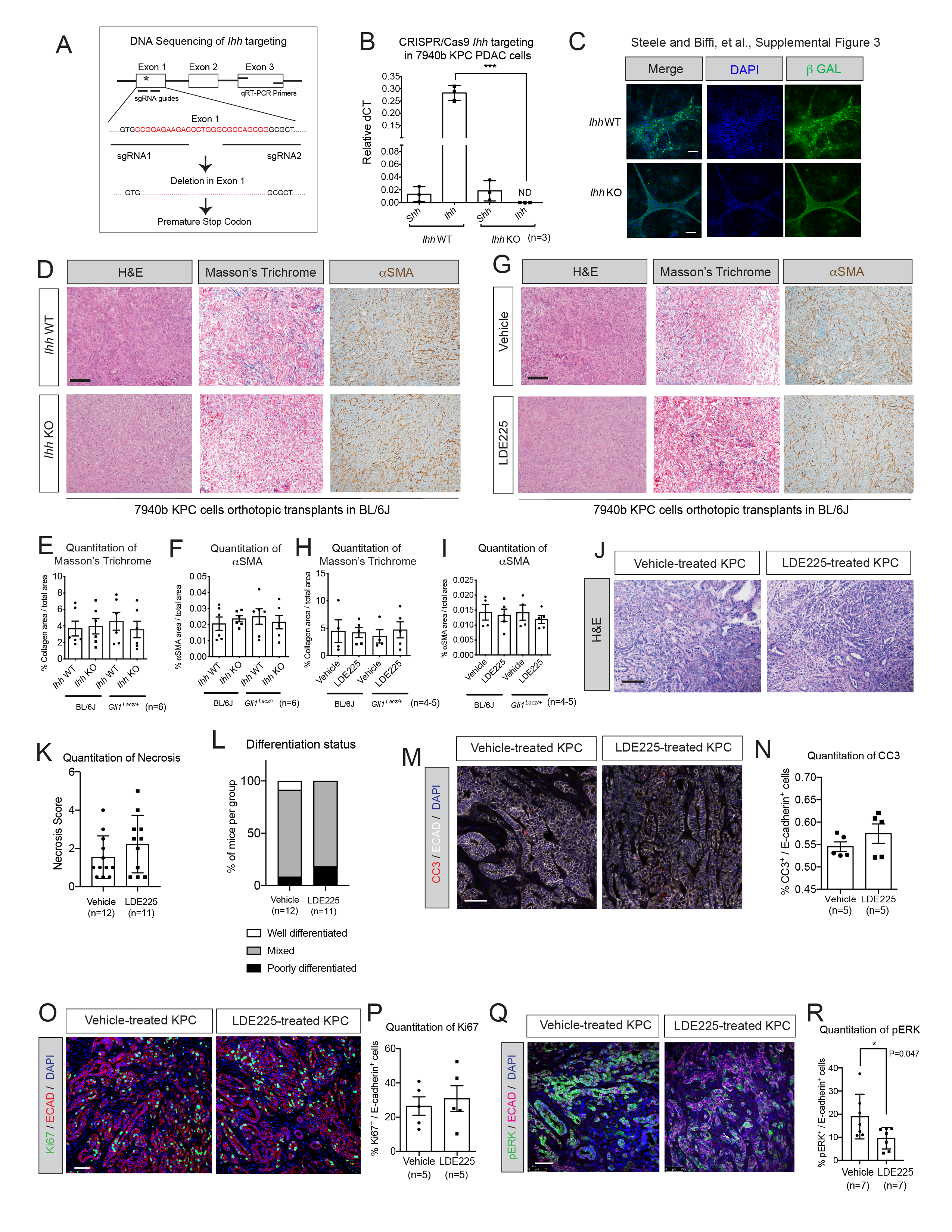
Ihh CRISPR/Cas-9 knockout and SHH ectopic expression
To knock out Ihh, single guide RNAs (sgRNAs) targeting exon 1 of the Ihh gene were selected from the GeCKO library [39] and cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector backbone (Addgene, #62988). Guide insertion was validated by Sanger sequencing. The vector with guides inserted was transfected with Lipofectamine 2000 (Thermo fisher, #11668) into 7940b KPC PDAC cells and single clones were selected by the addition of puromycin. Clones were screened by qRT-PCR with primers targeted to exon 3 of Ihh. Single clones were further validated by Sanger sequencing (data not shown). For ectopic expression of full-length SHH, Shh was cloned out from pBS mShh #CT258 (Addgene, #13999) into pMXs vector (Addgene) and expressed in T69A PDAC organoids derived from a KPC tumor [37]. Organoids were then selected with blasticidin and increased expression of SHH was confirmed by ELISA.
Enzyme-linked immunosorbent assays (ELISA)
For detection of SHH, we performed ELISA of media, after T69A organoid cultures were grown for 3–4 days. Media was collected, spun down and assayed using the manufacturer’s protocol (MSHH00, R&D Systems).
qRT-PCR analysis
RNA was extracted using the PureLink® Mini kit (Thermo fisher, #12183025) as described in the manufacturer’s protocol. The High-capacity cDNA Reverse Transcription Kit (Thermo fisher, #4368814) was used to reverse-transcribe RNA into cDNA. Real time PCR assays were utilized for Shh (Mm00436528_m1), Ihh (Mm_00439613_m1), and Ppia (Mm02342430_g1). PCR amplifications were done with pre-validated 20X Taqman Expression Assay probes, 2X TaqMan Universal Mastermix (Thermo fisher, #4364340) and cDNA template. Amplifications were performed in duplicate wells in a StepOne Real-Time PCR System and dCTs were normalized to Ppia housekeeping values.
Histopathological analysis and immunohistochemical stains
Tissues were fixed overnight in 10% neutral-buffered formalin, embedded in paraffin and sectioned. Standard procedures were used for IHC and IF. Briefly, tissues were de-paraffinized with xylene or Histoclear and subjected to citric acid antigen retrieval (Thermo Fisher, NC0359148) before blocking with 1% BSA in PBS for 1 hour at room temperature. Primary antibodies were added overnight at 4 °C, followed by secondary antibodies for 1 hour at room temperature. Antibodies used were: αSMA (Sigma, A2547, 1:1000 or ab5694, Abcam), Ki67 (Cell Signaling Technology, 12202, 1:100), Podoplanin (BioLegend, 127403, 1:200), Beta-galactosidase (Abcam ab9361, 1:2500), E-Cadherin (Cell Signaling Technology, 14472S, 1:50 or 610181, BD Biosciences), phospho-ERK (Cell Signaling Technology, 4370L, 1:100), CD31 (Cell Signaling Technology, 77699, 1:100), Cleaved Caspase 3 (Cell Signaling Technology, 9661, 1:200), CD8A (Cell Signaling Technology, 98941, 1:100), FOXP3 (Cell signaling Technology, 12653, 1:100). For IHC, hematoxylin was used as nuclear counterstain. For IF, Alexa Fluor secondary antibodies were used (1:500). Cell nuclei were counterstained with Prolong Gold antifade reagent (P10144, Invitrogen) or ProLong Diamond Antifade Mountant with DAPI (Invitrogen, #P36935). Images were taken using Olympus BX53F microscope, Olympus DP80 digital camera, and CellSens Standard software. Confocal microscopy images were acquired using LeicaSP5X or SP8 confocal microscopes and Leica LAS Software. X-GAL and immunofluorescence staining for β-galactosidase were done on frozen sections. For frozen sections, tissues were fixed in 4% paraformaldehyde for 1 hour at 4 °C. Tissues were washed three times in PBS and incubated for 48 hours rocking at 4 °C in 30% sucrose prior to embedding in OCT compound. Frozen sections were cut at 10 microns thickness. X-GAL staining was performed with 5 nM K3Fe(CN)6, 5 nM K4Fe(CN)6, 2 mM MgCl2, 0.01 % Na-deoxycholate, 0.02 % NP-40, and 1 mg/mL X-GAL (suspended in Dimethylformamide) made in PBS at 37 °C for 48 hours. Following this, fixed tissues were counterstained with Nuclear Fast Red for 10 minutes and subsequently mounted with Permount (VWR, #100496–550). Hematoxylin and eosin and Masson’s trichrome staining were performed according to standard protocols. Histopathological analysis of KPC tumors was performed by pancreatic pathologist in a blinded fashion on de-identified H&E slides. Tumors were graded as well, mixed or poorly differentiated adenocarcinoma. Necrosis score indicates the number of low-power field (10x objective) of necrotic area per 10 low-power field on each slide.
In situ hybridization (ISH) and dual ISH with Co-immunofluorescence
In situ hybridizations were performed with either the RNA Scope Multiplex Fluorescent Detection Kit (#323100; Advanced Cell Diagnostics), or colorimetric kits (either 322371-brown or 322360-red; Advanced Cell Diagnostics), according to the manufacturer’s protocol. Murine probes for Gli1 (311001; Advanced Cell Diagnostics) and Shh (314361; Advanced Cell Diagnostics) were used. Briefly, freshly cut paraffin embedded sections were baked for 1 hour at 60 °C prior to staining. Slides were then deparaffinized and treated with hydrogen peroxide for 10 minutes at room temperature. Target retrieval was performed in a water steamer boiling for 15 minutes and then slides were treated with the ProteasePlus reagent (Advanced Cell Diagnostics, #322381) for 30 minutes. Following this, the RNA Scope probe hybridized for 2 hours at 40 °C. The AMP5 amplification step was performed for 1 hour. For dual ISH and co-IF stains, the signal was amplified using the AMP materials provided in the ACD Multiplex Kit (320850; Advanced Cell Diagnostics). The signal was developed using 1–3 HRP channels depending on the number of probes and channel desired. Once completed, the samples were washed in PBS then blocked for 1 hour with 20% donkey serum at room temperature. Primary antibodies were incubated overnight at 4 °C (αSMA, Sigma A2547, 1:1000; Podoplanin, BioLegend 127403, 1:200; E-Cadherin, Cell Signaling Technology 14472 or 3195, 1:1000). Secondary Alexa Flour antibodies (1:500 in blocking buffer) were incubated for 1 hour at room temperature, and samples were washed 3 times in PBS. Slides were counterstained with DAPI and mounted with ProLong Gold Antifade Mountant (Thermo Fisher, P36930).
Quantitative Image Analysis
Quantitative analysis was performed in at least 3 random non-overlapping fields (20X magnification) in each sample using ImageJ, Fiji version 2.0.0-rc-69/1.52p or Halo (Indica Labs, Corrales, NM) software to measure the percentage of positive area. At least three representative samples per group were analyzed in a blinded manner. For Gli1 ISH staining in Fig. 2M, Pannoramic SCAN scanner (Perkin Elmer) images were quantified using the Halo software algorithms to identify tissue architecture and analyze red puncta staining area across the total tissue area. For all quantitation, blood vessels, necrotic areas and connective tissues were excluded. For quantitation of DAPI, color channels of each captured image were split and converted to binary. Cells were counted automatically (Analyze → Analyze Particles) and results were normalized to the total number of cells in the image. To quantify particles (such as Gli1 puncta) within a specific stain, captured images were first converted to binary. Particles were selected using a binary mask of the channel containing the desired stain (Edit → Selection → Create Mask) and counted automatically (Analyze → Analyze Particles). Results were then normalized to the total number of cells positive for the given marker. Fluorescence imaging of tissues was done with a Leica TCS SP8 laser scanning confocal (Leica Microsystems, Wetzlar, Germany), controlled by the LAS AF 3.3.10134 software. Immunofluorescence images were quantified using the population analysis module in Volocity (Improvision, Lexington, MA). Bright field images of tissues were obtained with an Axio Imager.A2 (ZEISS). Stained sections were scanned with Aperio ScanScope CS and analyzed using the ImageScope Positive Pixel Count algorithm. To quantify Masson’s trichrome stain, hue values for blue and pink were measured using an average hue value of 0.6 and a hue width of 0.854. Percent collagen area was then determined by calculating percentage of blue pixels relative to the entire stained area. To quantify αSMA and PDPN IHC stain, the percentage of strong positive pixels was calculated relative to the entire section with the ImageScope software.
Figure 2.

HH pathway inhibition impairs PDAC growth. (A) Schematic of orthotopic transplants of 7940b KPC PDAC cells with Ihh wild-type (WT) or knockout (KO) in C57BL/6J (BL/6J) or Gli1lacZ/+ mice. (B) Tumor weights at day 18 post-transplantation of the experiment from (A). Results show mean ± SEM of 6 biological replicates. ** P < 0.01, one-way ANOVA. (C) Representative RNA ISH images of Gli1 in Ihh WT or KO tumors in BL/6J mice (left panels), and of X-GAL stain and co-IF of αSMA (magenta), beta-galactosidase (β−GAL) (green) and DAPI (blue) in Ihh WT or KO tumors in Gli1lacZ/+ mice (middle and right panels). Inserts, magnifications. Scale bars, 50 μm (left panels), 100 μm (middle panels), 25 μm (right panels). (D) Quantitation of Gli1 RNA ISH in Ihh WT or KO tumors in BL/6J mice. Results show mean ± SEM on 6 biological replicates. *** P < 0.001, unpaired Student t test. (E) Quantitation of X-GAL stain in Ihh WT or KO tumors in Gli1lacZ/+ mice. Results show mean ± SEM on 6 biological replicates. ** P < 0.01, unpaired Student t test. (F) Schematic of orthotopic transplants of 7940b KPC PDAC cells into BL/6J or Gli1lacZ/+ mice, followed by a 12-day treatment with 20 mg/kg smoothened (SMO) inhibitor LDE225 or vehicle by daily oral gavage. (G) Tumor weights at day 18 post-transplantation of the experiment from (F). Results show mean ± SEM of 4–6 biological replicates. *** P < 0.001, one-way ANOVA. (H) Representative RNA ISH images of Gli1 in vehicle- or LDE225- treated tumors in BL/6J mice (left panels), and of X-GAL stain and co-IF of αSMA (red), βGal (green) and DAPI (blue) in vehicle- or LDE225- treated tumors in Gli1lacZ/+ mice (middle and right panels). Inserts, magnifications. Scale bars, 50 μm (left panels), 100 μm (middle panels), 25 μm (right panels). (I) Quantitation of Gli1 RNA ISH in LDE225- or vehicle- treated tumors in BL/6J mice. Results show mean ± SEM on 4 biological replicates. *** P < 0.001, unpaired Student t test. (J) Quantitation of X-GAL stain in LDE225- or vehicle- treated tumors in Gli1lacZ/+ mice. Results show mean ± SEM on 4 biological replicates. *** P < 0.001, unpaired Student t test. (K) Schematic of 2-week treatment of tumor-bearing KPC mice with 20 mg/kg LDE225 or vehicle by daily oral gavage. U/S, ultrasound. (L) Tumor volume as measured by U/S of vehicle- (n=9) and LDE225- (n=8) treated KPC tumors from (K). Results show mean ± SEM. * P < 0.05, unpaired Student t test. (M) Representative RNA ISH of Gli1 in 2-week vehicle- (n=11) and LDE225- (n=10) treated KPC tumors. Scale bar, 100 μm. (N) Quantitation of Gli1 stain in vehicle- (n=11) and LDE225- (n=10) treated KPC tumors. Results show mean ± SEM. *** P < 0.001, unpaired Student t test.
Flow cytometry and FACS Sorting
Tumors were processed as previously described [29]. For flow cytometric analysis of myCAF, iCAF and apCAF populations, cells were stained for 30 minutes with anti-mouse CD31-PE/Cy7 (102418, Biolegend), CD45-PerCP/Cy5.5 (103132, Biolegend), CD326 (Ep-CAM)-Alexa Fluor 488 (118210, BioLegend), PDPN-APC/Cy7 (127418; BioLegend), CD140a (PDGFRα)-PE (135905, BioLegend), Ly6C-APC (128015, Biolegend), I-A/I-E (MHC-II)-BV785 (107645, Biolegend), and for 15 minutes with DAPI. Flow plots were generated with the Flowjo software. For sorting of fibroblasts from KPC tumors, cells were stained for 30 minutes with CD45-Alexa Fluor 488 (103122; BioLegend), CD326 (EpCAM)-Alexa Fluor 647 (118212; BioLegend), CD324 (E-cadherin)-Alexa Fluor 647 (147307; BioLegend), PDPN-APC/Cy7 (127418; BioLegend), and for 15 minutes with DAPI. DAPI- CD45- EpCAM- ECAD- PDPN+ cells were sorted on the FACSAria cell sorter (BD) and processed for bulk RNA-seq.
RNA sequencing and data analysis
Total RNA samples were isolated using Trizol, according to manufacturer’s instructions. RNA concentration and quality were determined and ribosomal depleted mRNA libraries were prepared by the University of Michigan Advanced Genomics Core. Libraries were sequenced using 150 paired end NovaSeq 6000 (Illumina). Data are available at the NCBI Gene Expression Omnibus database under the accession number GSE156867. RNA-seq libraries were mapped to GENCODE GRCm38 primary assembly using STAR (v2.7.3), and gene expression estimations were performed using RSEM (v1.2.29). DESeq2 (v1.22.2) was used for identifying differentially expressed genes with default parameters. Genes with adjusted p-values below 0.05 were selected as significant genes. Pathway enrichment analysis was conducted using GSEA (v4.0.3) program and canonical pathway collections (cp_v7.0_all) available in MSigDB. To create signature gene sets, top 200 up- and down- regulated differential expression genes in iCAFs vs myCAFs were selected from the study by Öhlund et al., [27], and defined as myCAF and iCAF gene signatures, respectively. Fisher’s exact test available in R was used to perform association test between differential expression genes and myCAF/iCAF signatures. All plots were produced using R. RNA-seq data of human pancreatic normal and tumor organoids were from Tiriac et al. [40].
Statistics
GraphPad Prism software was used for graphical representation of data. Statistical analysis was performed using Two-tailed Student’s t test, and p<0.05 was considered statistically significant. One-way ANOVA analysis was performed to determine if there was any significance within the dataset. This was followed by post hoc tests using Tukey’s difference in means method with a p<0.05 considered statistically significant.
Cytometry by Time-of-Flight (CyTOF)
Tumors were digested as previously described [29]. Up to 1×106 cells were stained with Cell-ID Cisplatin (1.67 μM) for 5 minutes at room temperature. Briefly, after quenching Cisplatin reaction with 5X volume of MaxPar® Cell Staining Buffer, cells were centrifuged at 300 × g for 5 minutes. Then cells were stained with cell surface antibody cocktail (See Table S1 for catalogue numbers and dilutions) in 100 μl volume of MaxPar® Cell Staining Buffer for 30 minutes at room temperature. After being washed twice in 1 ml MaxPar® Cell Staining Buffer cells were fixed in 1.6% freshly made formaldehyde solution for 10 minutes at room temperature. Cells were washed twice with 2 ml MaxPar® Cell Staining Buffer, then were re-suspended in 1 ml cell intercalation solution (125 nM Cell-ID Intercalator-Ir in Maxpar Fix and Perm Buffer) and shipped to the Flow Cytometry core at the University of Rochester Medical Center where sample preparation was finalized and CyTOF2 Mass Cytometer analyses were performed.
CyTOF data analysis
Raw FCS files were analyzed using the Premium CytoBank Software (cytobank.org). Data were checked for quality of staining and normalized by the use of internal bead standards. Live singlet cells were identified using a combination of Ir191 DNA Intercalator and Pt195 Cisplatin stain intensity. Manual gating was performed.
Results
HH pathway activation is higher in myCAFs compared to iCAFs in PDAC
Pre-neoplastic pancreatic Intraepithelial Neoplasia (PanIN) lesions and invasive PDAC are associated with increased expression of HH ligands, which are low or undetectable in the normal pancreas [5, 6]. To evaluate the expression of HH pathway components in different cellular compartments of the pancreas, we analyzed single-cell RNA-sequencing data (scRNA-seq) from human adjacent normal or normal pancreas (n=3) and PDAC (n=16) samples (Fig. 1A) [35]. Consistent with previous studies [5, 6, 13], the expression of the Sonic hedgehog (SHH) and Indian hedgehog (IHH) ligands was restricted to epithelial cells and increased in PDAC compared to adjacent normal/normal tissues, whereas the ligand Desert hedgehog (DHH) was not detected in epithelial cells, but was expressed at low levels in endothelial cells (Fig. 1B). Similarly, analysis of bulk RNA sequencing (RNA-seq) of human adjacent normal/normal pancreas (n=4) and PDAC patient-derived (n=178) tissues from The Cancer Genome Atlas (TCGA) dataset indicated a significant increase in SHH and IHH in PDAC samples compared to controls, whereas DHH levels did not significantly change (Fig. S1A). SHH and IHH transcripts were also significantly increased in PDAC patient-derived organoids (n=63) compared to organoids isolated from normal human pancreata (n=14) (Fig. S1B–S1C) [40]. On the contrary, expression of HH transcriptional targets, including GLI1, PTCH1, PTCH2, and HHIP, was enriched in PDAC fibroblasts, along with pathway components, such as SMO [41], GAS1, CDON, and BOC [11–13, 15, 42, 43] (Fig. 1B). Finally, analysis of two previously published scRNA-seq datasets of human PDAC and normal pancreata [30, 34] confirmed that SHH and IHH were confined to the epithelial compartment and higher in PDAC compared to normal tissues, whereas HH target gene expression was largely restricted to fibroblasts, and generally higher in PDAC compared to non-malignant samples (Fig. S1D–S1G).
Figure 1.

HH pathway activation is higher in myCAFs compared to iCAFs in PDAC. (A) Uniform Manifold Approximation and Projection (UMAP) visualization of cell populations from single-cell RNA sequencing (scRNA-seq) of human adjacent normal/normal pancreas (n=3) and PDAC (n=16) tissues. The normal samples (n=3) were isolated from patients undergoing surgery for duodenal adenoma, ampullary carcinoma or PDAC, where an uninvolved portion of the pancreas was included in the resection. Different cell type clusters are color-coded. Data are from Steele et al. [35]. (B) Dot blot visualization of HH pathway gene expression level (color intensity) and frequency (size of dot) in different cell populations of human adjacent normal/normal pancreas (blue, n=3) and PDAC (red, n=16) samples from (A). Boxes highlight HH ligands (SHH, IHH, DHH) and HH targets (GLI1, PTCH1, PTCH2, HHIP). (C) UMAP visualization of scRNA-seq of the fibroblast clusters in pooled human adjacent normal pancreas (n=2) and PDAC (n=6) samples. Different CAF subtype clusters are color-coded. Data are from Elyada et al. [30]. (D) UMAP visualization of myCAF (ACTA2 coding for αSMA, TAGLN) and iCAF (IL6, CLEC3B) marker expression in the fibroblast clusters in human PDAC samples from (C). (E) UMAP visualization of HH target (GLI1, PTCH1) expression in the fibroblast clusters in human PDAC samples from (C). (F) Heatmaps of normalized expression of HH targets (GLI1, PTCH1, PTCH2, HHIP) and of HH receptor (SMO) and co-receptors (GAS1, CDON, BOC) in each fibroblast cluster in human PDAC samples from (C). Colors indicate log-scale gene counts. (G) Heatmap of scaled expression of HH ligands (Shh, Ihh, Dhh), HH targets (Gli1, Ptch1, Ptch2, Hhip), HH receptor (Smo) and co-receptors (Gas1, Cdon, Boc) in different cell populations of pancreatic tumors of the KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) mouse model of PDAC (n=4). Data are scaled such that the cluster with the lowest average expression = 0 and the highest = 1 for each gene. Data are from Elyada et al. [30]. (H) Representative RNA in situ hybridization (ISH) of Gli1 (white) and co-immunofluorescence (co-IF) of podoplanin (PDPN, green) in a KPC tumor. Counterstain, DAPI (blue). Scale bar, 20 μm. (I) Quantitation of Gli1 stain in PDPN+ (CAFs) and PDPN- (non-CAFs) cells in KPC tumors. Results show mean ± standard error of the mean (SEM) of 7 biological replicates. *** P < 0.001, unpaired Student t test. (J) UMAP visualization of scRNA-seq of the fibroblast clusters in KPC tumors (n=4) from Elyada et al. [30]. Different CAF subtype clusters are color-coded. (K) Heatmaps of normalized expression of HH targets (Gli1, Ptch1, Ptch2, Hhip), and of HH receptor (Smo) and co-receptors (Gas1, Cdon, Boc) in each fibroblast cluster from (J). Colors indicate log-scale gene counts. (L) Representative RNA ISH of Gli1 (white) and co-IF of PDPN (green) and alpha smooth muscle actin (αSMA, red) in a KPC tumor. Counterstain, DAPI (blue). Scale bar, 20 μm. The arrowhead points at a PDPN+αSMA+ cell with lower Gli1 expression; solid arrow points at a PDPN+αSMA+ cell with higher Gli1 expression. (M) Quantitation of Gli1 stain in αSMA+ PDPN+ (myCAFs) and αSMA- PDPN+ (non-myCAFs) cells in KPC tumors. Results show mean ± SEM of 7 biological replicates. ** P < 0.01, unpaired Student t test.
We next investigated whether HH signaling was differentially active in distinct human PDAC CAF subtypes. Analysis of fibroblasts from our previously published scRNA-seq dataset of human PDAC [30] revealed an enrichment of HH target genes (GLI1, PTCH1, PTCH2 and HHIP) in myCAFs compared to iCAFs (Fig. 1C–1F), whereas core HH pathway components (SMO, GAS1, CDON, BOC) were expressed in both compartments (Fig. 1F). These observations were confirmed in a second scRNA-seq dataset of human PDAC and normal pancreata [34] (Fig. S1H–S1K). To determine whether these observations were recapitulated in mouse models of PDAC, we analyzed our previously published scRNA-seq dataset of autochthonous pancreatic tumors from the KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) mouse model of PDAC [30, 33]. Similar to human PDAC, murine KPC tumors express HH ligands predominantly in epithelial cells, while fibroblasts are the main HH-responsive cell type, based on target gene expression (Fig. 1G). RNA in situ hybridization (ISH) of Gli1, a target gene and transcriptional mediator of the HH pathway [32], combined with co-immunofluorescence (co-IF) of the pan-fibroblast marker podoplanin (PDPN) confirmed that Gli1 expression was restricted to PDPN+ cells in KPC tumors (Fig. 1H–1I). Finally, similar to what we observed in human PDAC samples, scRNA-seq analysis of CAFs from KPC tumors demonstrated enrichment of HH target gene expression in myCAFs compared to other CAF subtypes (Fig. 1J–1K; S1L). Accordingly, RNA ISH of Gli1 with co-IF of PDPN and the myCAF marker alpha smooth muscle actin (αSMA) showed higher Gli1 expression in myCAFs (αSMA+ PDPN+) compared to non-myofibroblastic CAFs (αSMA- PDPN+) in KPC tumors (Fig. 1L–1M). Notably, within the myCAF population, we detected variable Gli1 levels (Fig. 1L), consistent with the heterogeneity observed in our scRNA-seq analysis of human PDAC fibroblasts (Fig. 1E).
Together, these data suggest that although all CAF subtypes are capable of responding to tumor-derived SHH and IHH ligands based on SMO, GAS1, CDON and BOC expression, HH signaling is upregulated in myCAFs in murine and human PDAC.
HH pathway inhibition impairs PDAC growth
To gain insight into the functional role of HH signaling, we examined the consequence of HH pathway hyperactivation on PDAC progression. SHH was ectopically expressed in KPC tumor-derived PDAC organoids [37], which we then used to generate an orthotopically grafted organoid (OGO) mouse model of PDAC [38] (Fig. S2A–S2B). Consistent with previous evidence [7], ectopic SHH expression in the epithelial compartment resulted in significantly increased tumor volume compared to controls (Fig. S2C). Increased epithelial Shh expression was validated by RNA ISH of Shh with co-IF of the epithelial marker E-cadherin (ECAD) and the fibroblast marker PDPN (Fig. S2D–S2E). Moreover, increased HH pathway activation in fibroblasts was confirmed by RNA ISH of Gli1 with co-IF of αSMA and PDPN (Fig. S2F–S2G). Although SHH expression has been previously linked to formation of fibrotic stroma [7–9, 12], in this transplantation model we did not observe a significant change in desmoplasia, as measured by Masson’s Trichrome and αSMA stains (Fig. S2H–S2J). Instead, SHH overexpression led to a significant increase in phospho-ERK (p-ERK) and a decrease in cleaved caspase 3 (CC3) levels in epithelial cells (Fig. S2H; S2K–S2L), consistent with the change in tumor growth.
To further evaluate the role of HH signaling in the early stages of PDAC development, we activated the epithelial expression of Shh in the iKC (Ptf1a-CreERT; KrasLSL-G12D/+) tamoxifen-inducible GEMM of PDAC [44] (Fig. S2M). In iKC;Shh mice, mutant Kras activation and SHH overexpression are simultaneously induced in acinar cells by Cre recombination upon Tamoxifen administration. To validate the increase in HH activation, Ptf1aCreERT;Shh mice were crossed to Gli1lacZ/+ animals, which read out HH pathway activity through the measurement of β-galactosidase activity [32]. The resulting Ptf1aCreERT;Gli1lacZ/+;Shh mice were then treated with Tamoxifen at 8 weeks old and aged for 34 weeks (Fig. S2N). Compared to control Ptf1aCreERT;Gli1lacz/+ pancreata, Ptf1aCreERT;Gli1lacZ/+;Shh pancreata had increased numbers of X-GAL+ cells (Fig. S2O), confirming that SHH overexpression in acinar cells results in increased HH activation. Notably, iKC;Shh mice exhibited an increase in pre-neoplastic PanIN lesions, compared to Tamoxifen-treated iKC mice, wild-type (WT) and Ptf1aCreERT;Shh controls (Fig. S2P), consistent with the notion that HH pathway activation promotes PDAC formation [7–9, 12]. Further, consistent with the more advanced lesions, pancreata from iKC;Shh mice displayed more abundant phospho-ERK compared to Tamoxifen-treated iKC, WT and Ptf1aCreERT;Shh controls (Fig. S2P). SHH overexpression in iKC;Shh mice also led to increased desmoplasia, as evaluated by Masson’s Trichrome and αSMA stains (Fig. S2P), indicating that HH pathway activation prior to tumor formation in this GEMM has a greater effect on stroma remodeling compared to transplantation models of murine PDAC cells (Fig. S2H–S2J).
We next investigated the effects of HH ligand depletion on PDAC progression. Whereas the effect of SHH deletion in PDAC has been previously investigated [22, 23], the role of IHH has not been addressed, despite the observation that both ligands are expressed in both human and mouse pancreatic cancer. We therefore deleted Ihh using CRISPR/Cas9 in a KPC PDAC cell line that expressed low levels of Shh [31] (Fig. S3A–S3B). Ihh WT PDAC cells induced HH pathway activation in co-cultured Gli1lacZ/lacZ fibroblasts [15], as evidenced by staining for β-galactosidase (β-GAL), whereas Ihh knock-out (KO) cells failed to do so (Fig. S3C). Orthotopic transplantation of Ihh KO PDAC cells in C57BL/6J (BL/6J) mice showed no difference in tumor growth compared to control WT cells (Fig. 2A–2B; Fig. S3D–S3F), although HH pathway inhibition was confirmed by RNA ISH of Gli1 (Fig. 2C–2D). We reasoned that Ihh KO PDAC cells transplanted in BL/6J mice potentially retained low-level HH activity. As we have previously identified a dosage-dependent role of HH signaling in pancreatic cancer progression [15], we evaluated the impact of Ihh deletion on PDAC progression in Gli1lacz/+ mice that lack one copy of Gli1. These mice have systemically lower HH signaling levels and display defects in HH-responsive adult homeostatic and repair processes [45–47]. Notably, transplantation of Ihh KO PDAC cells into the pancreas of Gli1lacz/+ mice resulted in significantly smaller tumors compared to control WT cells (Fig. 2A–2B; Fig. S3D–S3F). HH pathway inhibition was confirmed by X-GAL staining and co-IF for β-GAL and αSMA (Fig. 2C; 2E). We next utilized a selective SMO antagonist to pharmacologically inhibit HH signaling in both BL/6J and Gli1lacz/+ mice orthotopically transplanted with PDAC cells. Once a palpable mass was detected, mice were treated once daily by oral gavage with the SMO inhibitor LDE225 [48] for 12 days (Fig. 2F). LDE225 is an effective inhibitor of the HH pathway, acting downstream of all HH ligands by targeting Smoothened, and is in active clinical use [48–50]. Similar to what was observed with Ihh deletion, pharmacologic HH pathway inhibition resulted in smaller tumors only in Gli1lacz/+ mice (Fig. 2G; Fig. S3G–S3I), which lack one copy of Gli1 and thus have lower levels of HH signaling at baseline before tumor cells are transplanted. RNA ISH of Gli1, X-GAL staining and β-GAL/αSMA co-IF confirmed HH pathway inhibition in both models (Fig. 2H–2J). Overall, these data are consistent with previous reports in which Gli1 null animals exhibited decreased progression of pancreatic preneoplastic lesions, and the loss of one copy of Gli1 had an impact on pancreatic tissue homeostasis [45, 51].
As the accelerated tumor growth kinetics of these transplantation models of PDAC could prevent the observation of more subtle changes in tumor volume upon HH pathway inhibition, we investigated the effect of using LDE225 in the KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) mouse model. KPC mice were enrolled in either the control or treatment arm of the study once tumors were detected by ultrasound. Interestingly, a 2-week treatment with LDE225 led to an impairment of tumor growth in the KPC model (Fig. 2K–2L; S3J). Ablation of HH signaling was confirmed by RNA ISH of Gli1 (Fig. 2M–2N). Although no changes in tumor necrosis, differentiation, epithelial apoptosis and epithelial proliferation were observed (Fig. S3K–S3P), we observed a moderate, but significant reduction in p-ERK levels in epithelial cells upon HH pathway inhibition (Fig. S3Q–S3R).
Taken together, these data indicate that, consistent with prior studies, HH signaling generally promotes PDAC progression, that both SHH and IHH contribute to pancreatic cancer growth, and that the degree of contribution of HH signaling to tumor growth is dependent on the level of HH pathway activity.
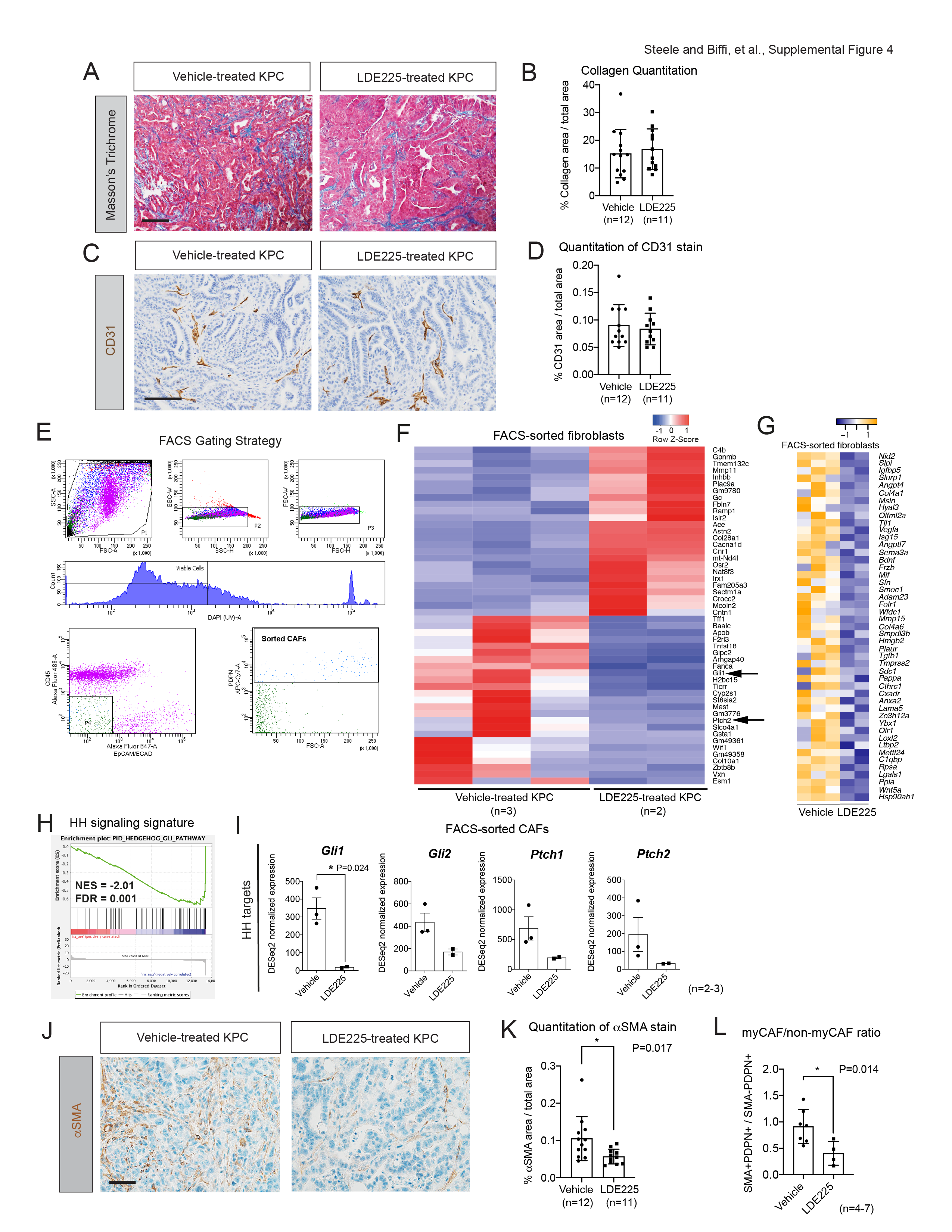
HH pathway inhibition alters the fibroblast compartment in PDAC
In contrast to previous work that inhibited HH signaling [19, 22, 23], 2-week LDE225 treatment did not alter the amount of collagen deposition, as measured by Masson’s Trichrome stain (Fig. S4A–S4B), or vasculature, as evaluated by immunohistochemistry (IHC) for CD31 (Fig. S4C–S4D), in KPC tumors. However, 2-week HH pathway inhibition resulted in a reduction in PDPN+ cells (Fig. 3A–3B). To further investigate this, we isolated PDPN+ DAPI- CD45- EpCAM- CAFs from 2-week vehicle- and LDE225- treated KPC tumors by fluorescence-activated cell sorting (FACS) and performed bulk RNA-seq (Fig. 3C; Fig. S4E). Gene set enrichment analysis (GSEA) and differential expression of HH targets confirmed significant downregulation of HH signaling in CAFs isolated from LDE225-treated tumors compared to vehicle-treated controls (Fig. S4F–S4H). We then assessed what secreted factors were downregulated by LDE225 treatment in the RNA-seq dataset. We found that growth factors, including Wnt5a, Tgfb1 and Sema3a, and angiogenic factors, such as Angpt4 and Vegfa, were downregulated in fibroblasts isolated from LDE225-treated KPC tumors compared to controls (Fig. S4I). Additionally, genes encoding ECM proteins, including Col4a1, Col4a6, and Mmp15, were also downregulated upon LDE225 treatment (Fig. S4I). Thus, we detected HH inhibition-dependent ECM-related gene expression alterations and additional changes in fibroblast-secreted factors that could provide an explanation for the reduction in tumor growth. Moreover, in accordance with the observed decrease in fibroblasts, the cell proliferation gene signature and proliferation markers were significantly reduced in CAFs isolated from LDE225-treated tumors compared to vehicle-treated controls (Fig. 3D–3E). Given that HH pathway activity is higher in myCAFs compared to other CAF subtypes in KPC tumors (Fig. 1K–1M), we investigated the consequences of LDE225 treatment on myCAF and non-myCAF populations. IHC of αSMA and co-IF detection of αSMA and PDPN to identify myCAFs (PDPN+ αSMA+) and non-myCAFs (PDPN+ αSMA-) not only revealed a decrease in αSMA+ myofibroblasts (Fig. 3F–3G; S4J–S4K), consistent with prior studies [19, 22, 23], but also showed an increase in non-myCAFs (Fig. 3F; 3H) in LDE225-treated tumors compared to vehicle-treated controls. Overall, these fibroblast changes resulted in a significant decrease in the myCAF/non-myCAF ratio (Fig. S4L).
Figure 3.

Two-week HH pathway inhibition alters the fibroblast compartment in PDAC. (A) Representative immunohistochemistry (IHC) of PDPN in vehicle- (n=10) and LDE225- (n=11) treated KPC tumors. Scale bar, 50 μm. (B) Quantitation of PDPN stain in vehicle- (n=10) and LDE225- (n=11) treated KPC tumors. Results show mean ± SEM. ** P < 0.01, unpaired Student t test. (C) Schematic of fluorescence-activated cell sorting (FACS) strategy for bulk RNA-seq of fibroblasts from vehicle- and LDE225- treated KPC tumors. (D) Gene set enrichment analysis (GSEA) of cell cycle signature in FACS-sorted CAFs from LDE225-treated KPC tumors (n=2) compared to FACS-sorted CAFs from vehicle-treated controls (n=3). (E) RNA-seq expression of proliferation markers (Mki67, Ccnb2 and Cks2) in FACS-sorted CAFs from vehicle- (n=3) and LDE225- (n=2) treated KPC tumors. Results show mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, unpaired Student t test. (F) Co-IF of αSMA (red) and PDPN (green) in vehicle- (n=7) and LDE225- (n=4) treated KPC tumors. Counterstain, DAPI. Scale bar, 20 μm. (G) Quantitation of myofibroblastic CAFs (αSMA+ PDPN+ DAPI+) in vehicle- (n=7) and LDE225- (n=4) treated KPC tumors. Results show mean ± SEM. P = 0.05, unpaired Student t test. (H) Quantitation of non-myofibroblastic CAFs (αSMA- PDPN+ DAPI+) in vehicle- (n=7) and LDE225- (n=4) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student t test.
Our findings indicate that fibroblast proliferation is affected upon LDE225 treatment, which may partly explain the reduction in myofibroblasts seen in HH inhibitor-treated tumors. Overall, our data corroborate a role for the HH pathway inhibitor LDE225 in targeting the fibroblast compartment and preferentially depleting myofibroblasts over non-myofibroblastic CAFs in PDAC.
HH pathway inhibition alters the ratio of myCAFs and iCAFs in PDAC
We next used a previously established flow cytometry approach [29, 30] to further evaluate myCAFs (PDPN+ MHCII- LY6C-), iCAFs (PDPN+ MHCII- LY6C+), and apCAFs (PDPN+ LY6C- MHCII+) in 2-week vehicle- and LDE225- treated KPC tumors (Fig. 4A). HH pathway inhibition resulted in a reduction in myCAF numbers and a significant expansion of iCAFs (Fig. 4B–4E), leading to an increase in the iCAF/myCAF ratio (Fig. 4F).
Figure 4.

Two-week HH pathway inhibition alters the ratio of myCAFs and iCAFs in PDAC. (A) Schematic of flow cytometry strategy for myCAFs and iCAFs from 2-week vehicle- and LDE225- treated KPC tumors. (B) Representative flow plots showing the gating strategy for the analysis of DAPI- CD45- CD31- EpCAM- PDPN+ CAFs in 2-week vehicle- (n=7) and LDE225- (n=6) treated KPC tumors. (C) Flow cytometry analysis of myCAFs (calculated from DAPI+ singlets) in vehicle- (n=7) and LDE225- (n=6) treated KPC tumors. Results show mean ± SEM. Unpaired Student t test. (D) Flow cytometry analysis of iCAFs (calculated from DAPI+ singlets) in vehicle- (n=7) and LDE225- (n=6) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student t test. (E) Proportions of myCAF, iCAF and apCAF subtypes from the PDPN+ gate in vehicle- (n=7, top) and LDE225- (n=6, bottom) treated KPC tumors, as measured by flow cytometry analysis. Results show average % of biological replicates. (F) Flow cytometric analysis of the iCAF/myCAF ratio from the PDPN+ gate in vehicle- (n=7) and LDE225- (n=6) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student t test. (G) GSEA of the myCAF gene signature in FACS-sorted CAFs from 2-week LDE225-treated KPC tumors (n=2) compared to FACS-sorted CAFs from vehicle-treated controls (n=3). The myCAF gene signature was defined from the study by Öhlund et al. [27]. (H) RNA-seq expression of myCAF markers (Acta2, Thy1, Tagln, Tgfb1) in FACS-sorted CAFs from vehicle- (n=3) and LDE225- (n=2) treated KPC tumors. Results show mean ± SEM. No statistical difference was observed as calculated by unpaired Student t test. (I) GSEA of the iCAF gene signature in FACS-sorted CAFs from 2-week LDE225-treated KPC tumors (n=2) compared to FACS-sorted CAFs from vehicle-treated controls (n=3). The iCAF gene signature was defined from the study by Öhlund et al. [27]. (J) RNA-seq expression of iCAF markers (Dpt, Clec3b, C3, Cxcl12) in FACS-sorted CAFs from vehicle- (n=3) and LDE225- (n=2) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student’s t-test.
We then investigated whether HH pathway inhibition resulted in an alteration of the iCAF and myCAF signatures, using previously established gene signatures [27], in the bulk RNA-seq dataset of FACS-sorted CAFs from 2-week vehicle- and LDE225- treated KPC tumors (Fig. 3C; S4E). In accordance with the flow cytometric data, this analysis revealed downregulation of the myCAF gene expression signature, as well as downregulation of specific myCAF markers upon LDE225 treatment (Fig. 4G–4H). Additionally, we observed an upregulation of the iCAF signature and iCAF markers in LDE225-treated KPC tumors compared to vehicle-treated controls (Fig. 4I–4J).
These data indicate that HH signaling is a key pathway for the maintenance of the myCAF phenotype, and its inhibition alters the ratio of myCAFs and iCAFs in PDAC.
HH pathway inhibition alters the immune infiltration in pancreatic tumors
As iCAFs are a source of inflammatory signals [27, 29, 30], we investigated the consequences of increased iCAF numbers, induced by HH pathway inhibition, on immune composition. Cytometry by time of flight (CyTOF) analysis of dissociated KPC tumors allowed for evaluation of multiple immune populations (Fig. 5A; Table S1). We observed no change in total B cells (CD45+CD19+), NK cells (CD45+CD161+), myeloid cells (CD45+CD11b+), granulocytic immature myeloid cells (Ly6G+Ly6C+), and macrophages (CD11b+F4/80+) upon 2-week HH pathway inhibition (Fig. 5B; S5A). However, Ly6C+Ly6G- monocytic immature myeloid cells, PD-L1+ macrophages and CD206+ macrophages significantly increased upon LDE225 treatment (Fig. 5B; S5A). Among different populations of dendritic cells, only total CD11b+CD11c+CD206+CD45+Lin-F4/80- were significantly upregulated (Fig. S5B). Total CD3+, CD4+ and CD8+ T cell infiltration was variable across samples, and while we observed a trend towards fewer T cells, there was no significant difference (Fig. S5C). However, the relative abundance of T cell subsets, when measured as a percentage of total CD3+ T cells, was altered, as HH pathway inhibition significantly decreased CD8+ T cells and increased CD4+ T cells and CD25+CD4+ T cells (potential regulatory T cells) (Fig. 5C). Accordingly, by immunohistochemistry, we observed fewer CD8+ T cells (Fig. 5D–5E) and more abundant FOXP3+ regulatory T cells (Fig. 5F–5G) in 2-week LDE225-treated KPC tumors compared to vehicle-treated controls.
Figure 5.

Two-week HH pathway inhibition alters the immune infiltration in pancreatic tumors. (A) Top: schematic of 2-week treatment of tumor-bearing KPC mice with 20 mg/kg LDE225 or vehicle prior to CyTOF (Cytometry by time of flight) analysis. U/S, ultrasound. Bottom: table of CyTOF panel including metal tag, antibody target, and cell type predominantly expressed on. See Table S1 for detailed antibody information. (B) Manual gating of CyTOF data for total myeloid cells (CD45+CD11b+), macrophages (CD11b+F4/80+), PDL1+ macrophages (F4/80+PD-L1+) and CD206+ macrophages (F4/80+CD206+) in 2-week vehicle- (n=7) and LDE225- (n=6) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student’s t-test. (C) Manual gating of CyTOF data for CD8+, CD4+ and CD4+CD25+ T cells as a percentage of total CD3+ T cells. Results show mean ± SEM. * P < 0.05, ** P < 0.01, unpaired Student’s t-test. (D) Representative IHC of CD8A in 2-week vehicle- and LDE225- treated KPC tumors. Inserts, magnifications. Scale bar, 50 μm. (E) Quantitation of CD8A stain in 2-week vehicle- (n=12) and LDE225- (n=11) treated KPC tumors. Results show mean ± SEM. * P < 0.05, unpaired Student’s t-test. (F) Representative IHC of FOXP3 in 2-week vehicle- and LDE225- treated KPC tumors. Inserts, magnifications. Scale bar, 50 μm. (G) Quantitation of FOXP3 stain in 2-week vehicle- (n=12) and LDE225- (n=11) treated KPC tumors. Results show mean ± SEM. ** P < 0.01, unpaired Student’s t-test. (H) Model explaining the role of HH signaling and the effects of HH inhibition in the PDAC microenvironment. Cancer-secreted HH ligands, such as SHH and IHH, activate HH signaling in surrounding fibroblasts (arrow), especially in myCAFs (left panel). HH inhibition leads to a reduction in myCAFs and an increase in iCAFs, and to decreased CD8+ T cells and more abundant regulatory T cells (right panel).
Thus, the observed increase in iCAFs upon HH inhibition correlated with changes in immune infiltration that are consistent with a more immunosuppressive pancreatic cancer microenvironment (Fig. 5H).
Discussion
A distinguishing feature of pancreatic cancer is an extensive stromal reaction. Among these stromal components, heterogeneous fibroblast subtypes have potentially distinct contributions to pancreatic cancer progression [24, 25, 27–30, 52, 53]. HH signaling is a key pathway activated in fibroblasts in pancreatic cancer; however, contradictory genetic and pharmacologic studies in mouse models, as well as disappointing clinical trials with HH pathway inhibitors indicate a complicated and potentially multifactorial contribution of the HH pathway to pancreatic cancer progression. Here, we confirm that HH pathway activation, as evidenced by expression of HH targets, such as GLI1, is restricted to the fibroblast compartment. Notably, we demonstrate that HH signaling is enriched in the myCAF subset compared to other CAF subtypes in both mouse models of PDAC and human pancreatic cancer. Moreover, we show that short-term HH pathway inhibition with LDE225 results in myCAF depletion and iCAF enrichment, altering fibroblast composition towards a more fibro-inflammatory stroma.
A particularly vexing question is why different studies reached distinct conclusions about the role of HH signaling in pancreatic cancer. One potential answer lies in the approaches used to target the HH pathway. Prior work demonstrated a tumor-restrictive role for HH signaling through genetic ablation of epithelial Shh [22, 23]. However, as noted previously [6], and confirmed here, pancreatic cancer cells express both Shh and Ihh. Further, genetic deletion of Shh in a GEMM of PDAC resulted in a corresponding, and potentially compensatory, increase in Ihh expression [22]. Here we demonstrate that Ihh KO results in decreased tumor size when implanting PDAC cells that do not produce significant levels of Shh and in the context of a genetically sensitized background (i.e. in Gli1lacz/+ mice that lack one copy of Gli1). Together, these data indicate that targeting of both Shh and Ihh is required to fully discern the contribution of HH signaling to pancreatic cancer growth.
An additional explanation for discord in the field may lie with the use of different pharmacologic HH pathway inhibitors. While previous studies employed Vismodegib (Genentech) and IPI-926 (Infinity Pharmaceuticals) [19, 22, 23], we utilized LDE225 (Sonidegib, Novartis) [48], which is still being clinically evaluated for PDAC (NCT02358161). Notably, although all three inhibitors target SMO, these compounds display distinct pharmacokinetics in mice [49, 50], which could result in different degrees of HH pathway inhibition in the pancreatic stroma. In support of this notion, Vismodegib treatment only partially reduced Gli1 expression in a mouse model of pancreatic cancer [23], whereas LDE225 treatment results in near complete abrogation of Gli1 expression in both orthotopic transplantation models of PDAC and KPC tumors. An important consideration in previous studies, as well as in our current work, is the potential impact of different levels of HH pathway activity on pancreatic tumor growth. HH ligands are best known as morphogens, where specific HH ligand concentrations induce distinct cellular outcomes [54]. The duration of HH pathway activity is also essential in determining cellular responses [54]. The outcome of HH signaling is regulated by the dynamic and overlapping contributions of various cell surface receptors [15]. Furthermore, GLI proteins, the transcriptional effectors of the HH pathway, also impact pancreatic tumor growth [45, 55]. Finally, in Gli1lacz/+ mice, HH signaling is lowered systemically [32] prior to tumor formation, which may explain the different outcomes observed upon further HH inhibition by treatment with LDE225, when compared to BL/6J transplantation models. Altogether, previous work from our group [15] and our current data support the notion that HH signaling plays dosage-dependent, and perhaps timing-dependent, roles in pancreatic cancer with the best outcome, in terms of inhibition of tumor growth, when the pathway is completely inactivated, rather than partially inhibited.
Here we demonstrate that HH pathway inhibition alters the ratio of fibroblast subtypes in pancreatic cancer by altering myCAF abundance. Although all CAF subtypes appear to express the necessary receptors to respond to HH ligands, we observe preferential HH pathway activation in myCAFs. This observation could be due to the tumor-proximal location of myCAFs [27], which positions them closer to the source of HH ligands. This raises the question of whether genetic depletion of αSMA+ myofibroblasts [56] may result in inappropriate activation of HH signaling in iCAFs and apCAFs, which may promote disease progression.
Our observations indicate that detrimental effects associated with long-term HH pathway inhibition may not be due to the depletion of a tumor-restraining myofibroblastic CAF population, as previously suggested [22, 42], but instead may be depend on the enrichment of a potentially tumor-promoting inflammatory CAF subtype. Indeed, previous work from our laboratories have suggested a tumor-promoting role of iCAFs [27, 29, 36, 57]. Although the change in fibroblast composition observed upon a 2-week period of HH inhibition did not lead to more aggressive PDAC, in the long term it may cause the detrimental effects observed in previous studies, as iCAFs secrete ligands responsible for conditions such as immunosuppression and cachexia [27, 30, 58–60]. In support of this notion, we find that HH inhibition leads to a more immune-suppressive microenvironment, as evidenced by fewer CD8 T cells and more abundant regulatory T cells, which might result in worse long-term prognosis. Notably, we cannot exclude that HH pathway inhibition may also have direct effects on immune cells, as previously reported [61]. Alternatively, deleterious effects associated with long-term HH pathway inhibition may be associated with the loss of well differentiated PDAC and the increase in poorly differentiated PDAC that we observe following a 2-week LDE225 treatment, which is consistent with previous studies [22, 23]. Overall, the effects of HH pathway inhibition on PDAC progression may not just be due to alterations in specific CAF subtypes, but to how these alterations affect signaling within and between these cells and to how specific signals integrate with other components of the tumor microenvironment. A deeper understanding of the biology and functions of distinct CAF subtypes, including studies examining tumor/CAF/immune cell cross-talk, could lead to future combinatorial therapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Translational Relevance.
A better understanding of the roles of signaling pathways differentially activated in distinct fibroblast populations will shed light on the complexity of fibroblast heterogeneity in pancreatic cancer. Here, we show that HH pathway inhibition alters fibroblast composition towards a more inflammatory microenvironment, opening the possibility for future combination therapies.
Acknowledgements
The results shown here are in part based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This project was supported by NIH/NCI grants R01CA151588, R01CA198074, U01CA224145, and the American Cancer Society to M. Pasca di Magliano. This work was also supported by the University of Michigan Cancer Center Support Grant (P30CA046592), including an Administrative Supplement to H.C. Crawford and M. Pasca di Magliano.
This work was also supported by the National Institutes of Health [R01 DC014428, R01 CA198074, R01 118751 to B. Allen]. Research reported in this publication was also supported by the University of Michigan Cancer Center Support Grant [P30 CA046592] by the use of the following Cancer Center Shared Resource: Cell and Tissue Imaging. F. Bednar was funded by the Association of Academic Surgery Joel Roslyn Award. T. Frankel was funded by K08CA201581. Research reported in this publication was supported by the National Cancer Institute Award Number P30CA046592 by the use of the following Rogel Cancer Center Shared Resource: Transgenic Animal Models (A. Dlugosz). This project was supported by NIH/NCI grants R01 CA118875 (L. Syu), P01 DK062041, 5P30 CA046592 to A. Dlugosz. S. Kemp was supported by T32 GM113900. N. Steele was supported by T32 CA009676 and is a recipient of the American Cancer Society Postdoctoral Award PF-19–096-01 and the Michigan Institute for Clinical and Healthy Research (MICHR) Postdoctoral Translational Scholar Program fellowship award. E. Carpenter is supported by the American College of Gastroenterology Clinical Research Award and by T32 DK094775. A. Rao and S. The were supported by institutional startup funds from the University of Michigan, a gift from Agilent Technologies, NCI grant R37 CA214955 and a Research Scholar Grant from the American Cancer Society (RSG-16–005-01). C. Halbrook was supported by K99 CA241357 and P30 DK034933 NIH awards. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The Tuveson laboratory is supported by the NIH Cancer Center Support Grant P30 CA045508 and the Lustgarten Foundation, where D.A. Tuveson is a distinguished scholar and Director of the Lustgarten Foundation–designated Laboratory of Pancreatic Cancer Research. D.A. Tuveson is also supported by the Thompson Foundation, the Cold Spring Harbor Laboratory and Northwell Health Affiliation, the Northwell Health Tissue Donation Program the Cold Spring Harbor Laboratory Association and the National Institutes of Health (NIH P30 CA45508, U01 CA210240, R01 CA229699, U01 CA224013, R01 CA188134, and R01 CA190092). This work was also supported by a gift from the Simons Foundation (552716 to D.A. Tuveson). G. Biffi was a fellow of the Human Frontiers Science Program (LT000195/2015-L) and EMBO (ALTF 1203–2014) and is supported by a Cancer Research UK (CRUK) core funding (A27463) and by the Pancreatic Cancer and CMB Programmes of the Cancer Research UK Cambridge Centre. E. Elyada was a fellow of the Human Frontiers Science Program (LT000403/2014-L). J. Preall is supported by the Cold Spring Harbor Laboratory and Northwell Health Affiliation. Y. Park is supported by R50 CA211506. The authors would like to thank Tricia Tamsen and Judy Opp from the University of Michigan Advanced Genomics Core, and the Tissue Procurement Center at the University of Michigan. We would also like to thank Matthew Cochran and Terry Wightman at the University at the Rochester Medical Center Flow Cytometry Shared Resource Laboratory. We would like to thank Dr. Christ Wright for sharing the iKC (Ptf1a-CreERT; KrasLSL-G12D/+) mouse model. This work was performed with assistance from the University of Michigan Shared Resources: Biostatistics, Analytics & Bioinformatics; Flow Cytometry; Transgenic Animal Models; Tissue and Molecular Pathology; Structure & Drug Screening; Cell & Tissue Imaging; Experimental Irradiation; Preclinical Imaging & Computational Analysis; Health Communications; Immune Monitoring; Pharmacokinetics. This work was also performed with assistance from the Cold Spring Harbor Laboratory (CSHL) shared resources, which are supported by the NIH Cancer Center Support Grant (CCSG) P30 CA045508: Bioinformatics, Flow Cytometry, Animal, and Cell and Tissue Imaging Shared Resources.
Footnotes
Conflict of interest statement: D.A.T. discloses membership in the scientific advisory board for Leap Therapeutics, Surface Oncology, Cyngnal Therapeutics, Mestag Therapeutics, stock in Leap Therapeutics and Surface Oncology, Co-Founder of Mestag Therapeutics, Honorarium from Merck and sponsored research with ONO, Fibrogen, and Mestag Therapeutics. The other authors declare no conflict of interests.
References
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2020. CA Cancer J Clin, 2020. 70(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R, The biology and function of fibroblasts in cancer. Nat Rev Cancer, 2016. 16(9): p. 582–98. [DOI] [PubMed] [Google Scholar]
- 3.Helms E, Onate MK, and Sherman MH, Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov, 2020. 10(5): p. 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahai E, et al. , A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer, 2020. 20(3): p. 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thayer SP, et al. , Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature, 2003. 425(6960): p. 851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman DM, et al. , Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature, 2003. 425(6960): p. 846–51. [DOI] [PubMed] [Google Scholar]
- 7.Bailey JM, Mohr AM, and Hollingsworth MA, Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene, 2009. 28(40): p. 3513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fendrich V, et al. , Ectopic overexpression of Sonic Hedgehog (Shh) induces stromal expansion and metaplasia in the adult murine pancreas. Neoplasia, 2011. 13(10): p. 923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao J, et al. , A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res, 2006. 66(20): p. 10171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morton JP, et al. , Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci U S A, 2007. 104(12): p. 5103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nolan-Stevaux O, et al. , GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev, 2009. 23(1): p. 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey JM, et al. , Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res, 2008. 14(19): p. 5995–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian H, et al. , Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A, 2009. 106(11): p. 4254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yauch RL, et al. , A paracrine requirement for hedgehog signalling in cancer. Nature, 2008. 455(7211): p. 406–10. [DOI] [PubMed] [Google Scholar]
- 15.Mathew E, et al. , Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep, 2014. 9(2): p. 484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingham PW and McMahon AP, Hedgehog signaling in animal development: paradigms and principles. Genes Dev, 2001. 15(23): p. 3059–87. [DOI] [PubMed] [Google Scholar]
- 17.Feldmann G, et al. , An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther, 2008. 7(9): p. 2725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldmann G, et al. , Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut, 2008. 57(10): p. 1420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olive KP, et al. , Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science, 2009. 324(5933): p. 1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim EJ, et al. , Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res, 2014. 20(23): p. 5937–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko AH, et al. , A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas, 2016. 45(3): p. 370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhim AD, et al. , Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell, 2014. 25(6): p. 735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JJ, et al. , Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A, 2014. 111(30): p. E3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dominguez CX, et al. , Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov, 2020. 10(2): p. 232–253. [DOI] [PubMed] [Google Scholar]
- 25.Hosein AN, et al. , Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight, 2019. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia PE, et al. , Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell Mol Gastroenterol Hepatol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohlund D, et al. , Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med, 2017. 214(3): p. 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernard V, et al. , Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res, 2019. 25(7): p. 2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biffi G, et al. , IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov, 2019. 9(2): p. 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elyada E, et al. , Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov, 2019. 9(8): p. 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long KB, et al. , IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov, 2016. 6(4): p. 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai CB, et al. , Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development, 2002. 129(20): p. 4753–61. [DOI] [PubMed] [Google Scholar]
- 33.Hingorani SR, et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell, 2005. 7(5): p. 469–83. [DOI] [PubMed] [Google Scholar]
- 34.Peng J, et al. , Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res, 2019. 29(9): p. 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steele NG, et al. , Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nature Cancer, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, et al. , Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov, 2020. 10(3): p. 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oni TE, et al. , SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med, 2020. 217(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boj SF, et al. , Organoid models of human and mouse ductal pancreatic cancer. Cell, 2015. 160(1–2): p. 324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanjana NE, Shalem O, and Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods, 2014. 11(8): p. 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiriac H, et al. , Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov, 2018. 8(9): p. 1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stone DM, et al. , The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature, 1996. 384(6605): p. 129–34. [DOI] [PubMed] [Google Scholar]
- 42.Allen BL, et al. , Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell, 2011. 20(6): p. 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen BL, Tenzen T, and McMahon AP, The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev, 2007. 21(10): p. 1244–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopp JL, et al. , Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell, 2012. 22(6): p. 737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathew E, et al. , The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J Biol Chem, 2014. 289(40): p. 27727–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lees CW, et al. , Analysis of germline GLI1 variation implicates hedgehog signalling in the regulation of intestinal inflammatory pathways. PLoS Med, 2008. 5(12): p. e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin K, et al. , Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature, 2011. 472(7341): p. 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan S, et al. , Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med Chem Lett, 2010. 1(3): p. 130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie H, et al. , Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells, 2019. 8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauressergues E, et al. , Pharmacological evaluation of a series of smoothened antagonists in signaling pathways and after topical application in a depilated mouse model. Pharmacol Res Perspect, 2016. 4(2): p. e00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mills LD, et al. , Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J Biol Chem, 2013. 288(17): p. 11786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waghray M, et al. , GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov, 2016. 6(8): p. 886–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathew E, et al. , Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia, 2016. 18(3): p. 142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dessaud E, McMahon AP, and Briscoe J, Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development, 2008. 135(15): p. 2489–503. [DOI] [PubMed] [Google Scholar]
- 55.Mills LD, et al. , Inactivation of the transcription factor GLI1 accelerates pancreatic cancer progression. J Biol Chem, 2014. 289(23): p. 16516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozdemir BC, et al. , Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell, 2014. 25(6): p. 719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Somerville TD, et al. , Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flint TR, et al. , Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab, 2016. 24(5): p. 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feig C, et al. , Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A, 2013. 110(50): p. 20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, et al. , Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity, 2018. 49(1): p. 178–193 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de la Roche M, et al. , Hedgehog signaling controls T cell killing at the immunological synapse. Science, 2013. 342(6163): p. 1247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.