

SUMMARY
G proteins play a central role in signal transduction and pharmacology. Signaling is initiated by cell-surface receptors, which promote GTP binding and the dissociation of Gα from the Gβγ subunits. Structural studies have revealed the molecular basis for subunit association with receptors, RGS proteins and downstream effectors. In contrast, the mechanism of subunit dissociation is poorly understood. We use cell signaling assays, MD simulations, biochemistry and structural analysis to identify a conserved network of amino acids that dictates subunit release. In the presence of the terminal phosphate of GTP, a glycine forms a polar network with an arginine and glutamate, putting torsional strain on the subunit binding interface. This “G-R-E motif” secures GTP and, through an allosteric link, discharges the Gβγ dimer. Replacement of network residues prevents subunit dissociation, regardless of agonist or GTP binding. These findings reveal the molecular basis for the final committed step of G protein activation.
Graphical Abstract
eTOC
G protein signaling involves binding of agonist to receptor and unbinding of GDP from the G protein. Using integrated molecular and computational approaches, Knight et al. investigate the second, committed step of G protein activation, involving an allosteric “Gly-Arg-Glu” network that links GTP binding to subunit dissociation and pathway activation.
INTRODUCTION
Many hormones, neurotransmitters, and clinically important drugs elicit their effects through G protein coupled receptors (GPCRs). Receptors of this class represent one of the largest gene families, and by far the largest class of drug targets. Upon receptor activation by agonist, the G protein α subunit releases GDP, binds GTP, and dissociates from the Gβγ dimer. The free Gα subunit then modulates the activity of downstream effector proteins such as adenylyl cyclase and phospholipase C, among others. The Gβγ dimer can regulate many of these same effectors as well as potassium channels, calcium channels, phosphatidylinositol-3 kinase and MAPKs. Signaling is terminated after Gα hydrolyzes GTP and the heterotrimer reassembles. In most cases this inactivation step is accelerated by RGS proteins. Thus, receptors serve as signal discriminators while RGS proteins serve as timing devices and G proteins serve as determinants of output specificity.
The effects of G protein activation -- such as vasodilation, pain reduction, euphoria as well as light and odor sensing -- are quite diverse depending on the cellular context. Additional diversity comes from differential expression of 800+ GPCRs, which includes members with little or no shared sequence similarity. In contrast, G proteins are highly conserved in structure and function, and in mammals consist of only four subfamilies (Gs, Gi/o, Gq/11 and G12/13) encoded by 16 individual genes.
The ability of such widely divergent receptors to activate a comparatively small number of G proteins indicates a shared mechanism of activation (Flock et al., 2015, Flock et al., 2017). Broadly speaking, this process can be broken down into three discrete protein transformations: (i) ligand-specific conformational changes in the receptor, (ii) nucleotide dependent conformational changes in Gα, and (iii) displacement of the Gβγ subunits. It had long been postulated that an activated receptor must induce an opening of the two domains within Gα, stabilizing the nucleotide-free state and facilitating GTP-GDP exchange (Reviewed in (Mahoney and Sunahara, 2016)). Direct evidence of such inter-domain opening was first provided through double electron-electron resonance experiments on rhodopsin-activated Gαi (Van Eps et al., 2011) and on Gαs (Dror et al., 2015), hydrogen-deuterium-exchange studies (Chung et al., 2011) and through X-ray crystallographic studies of the β2-adrenergic receptor bound to the Gs protein heterotrimer (Rasmussen et al., 2011, Westfield et al., 2011). In all of these structures however, Gβγ remains associated with Gα, even in the nucleotide-free and agonist-bound complex. Thus, the conformational changes associated with the release of nucleotide are necessary but not sufficient for the subsequent release of Gβγ.
The third and final step of G protein activation begins with GTP binding. This entails conformational changes in three segments of Gα known as Switch I, II and III (Lambright et al., 1994). In addition, there are interactions between the αN helix of Gαi and Gβ that need to be disrupted in order to cause dissociation of the heterotrimer. Crystal structures revealed that Switch I connects the two domains of the Gα subunit, comprised of a RAS-homologous domain and an all-helical domain. Switches II and III are located within the RAS-like domain and are disordered in the GDP-bound structure. Upon GTP binding Switch II forms a well-ordered helix and makes multiple contacts with residues in the α3 helix. Switches I and II interact directly with the γ-phosphate of GTP and form part of the Gβγ-interacting surface (Wall et al., 1995, Lambright et al., 1996). Therefore, to form the active state, residues critical to maintaining Gβγ binding must retract inward toward the newly bound GTP molecule, creating a “molecular tug-of-war.” Less is known about the subsequent cascade of events linking the γ-phosphate to Switch I and II and to the displacement of Gβγ.
Here, by combining cross-species sequence analysis, Molecular Dynamics (MD) simulations, thermostability measurements, direct Gα and Gβγ binding assays, NMR spectroscopy and cell-based functional assays we have uncovered the dynamic and transient effect of GTP binding leading to Gβγ dissociation. These models were also tested functionally, in biological contexts including the model organism S. cerevisiae. Our analysis reveals the existence of an evolutionarily conserved Gly-Arg-Glu triad (“G-R-E motif”) that underpins all G protein activation. In particular, we show that these residues form an allosteric link between GTP and Gβγ. Mutations in the central arginine impedes subunit dissociation, most likely due to a loss of a polar network between the terminal (γ) phosphate of GTP and the Gβγ-binding interface. Given that this motif is perfectly conserved among known Gα subunits, it is likely to be a universal feature of G protein activity. Thus, our findings reveal how a single phosphate group controls G protein subunit dissociation and signaling, processes that underlie much of human physiology and a substantial fraction of medical pharmacology.
RESULTS
Triad hypothesis
The active state of a receptor is that which binds simultaneously to agonist and nucleotide-free G protein. Agonist binding stabilizes the empty form of the G protein heterotrimer, thereby facilitating the exchange of GTP for GDP. Binding to either nucleotide favors release of G protein from receptor and of receptor from agonist (De Lean et al., 1980, DeVree et al., 2016). However, it is GTP alone that triggers the release of Gβγ from Gα (Figure 1). Thus, the active state of the G protein can be defined as the conformation stabilized by the terminal phosphate of GTP. Our goal here was to determine the molecular rearrangements responsible for this third and final committed step of G protein activation.
Figure 1. Schematic of the G protein triad hypothesis.
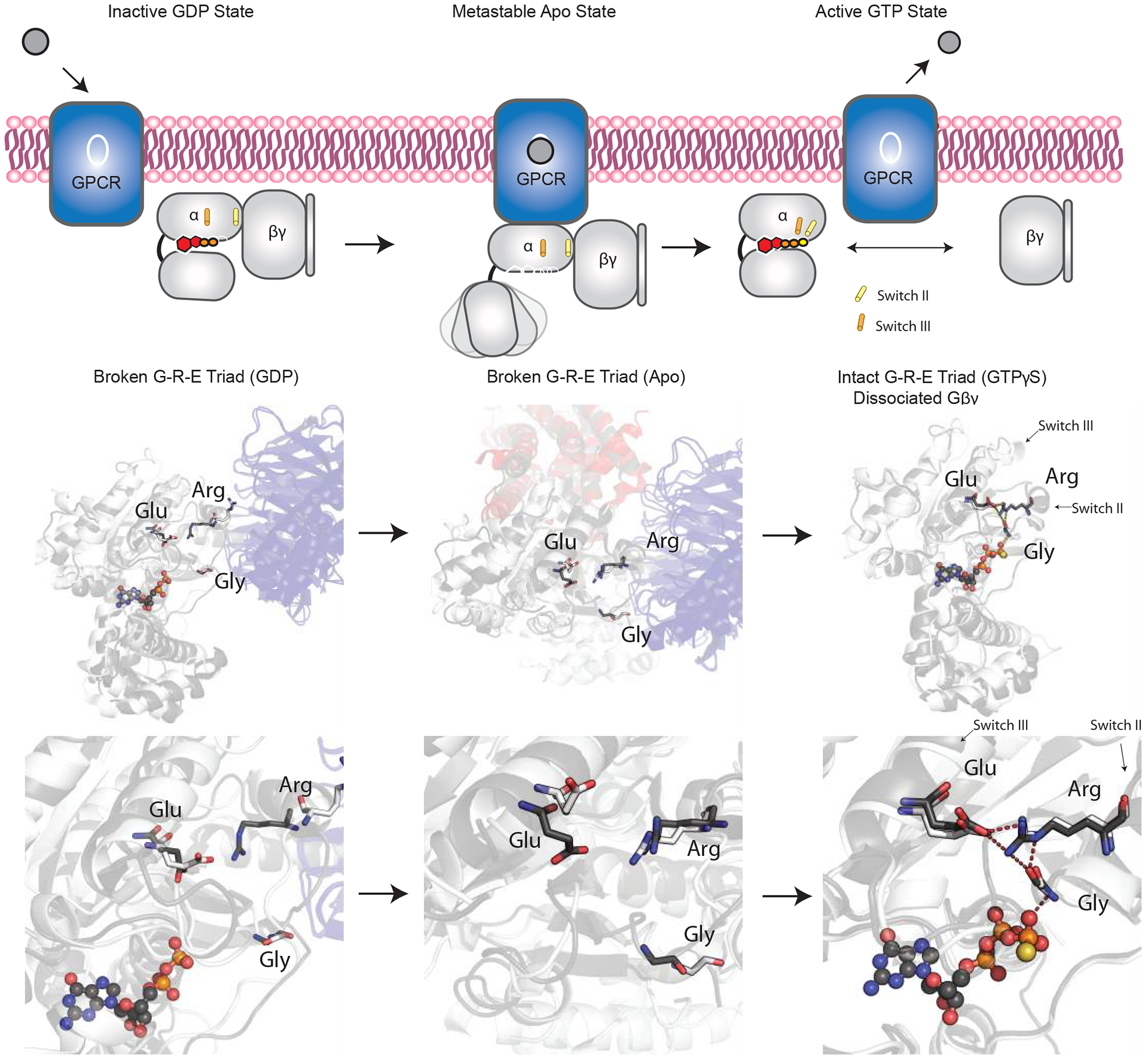
Top, schematic representation of the G protein (grey) bound to GDP (left), none (center), and GTP (right). Cylinders represent the movements of Switch II and Switch III.
Bottom, overlaid structural representation of Gαi1 and Gαs (grey) bound to GDP (PDB: 1GP2 6EG8) (Wall et al., 1995, Liu et al., 2019), none (apo state, PDB:6DDE 3SN6) (Koehl et al., 2018, Rasmussen et al., 2011), and GTP (PDB:1GIA and 1AZT) (Coleman et al., 1994, Sunahara et al., 1997). The overlaid residues for the conserved triad are highlighted in black. See also Figure S1
To guide our experimental analysis, we began with a detailed examination of the known Gα structures. In particular we focused on the amino acids that bridge the γ-phosphate of GTP to the Gβγ binding domain. This analysis revealed a triad of amino acids (in Gαi1 numbering: Gly203, Arg208, and Glu245) that are conserved in all Gα proteins (Figure S1, Supplemental Information). On GTP binding, the glycine repositions the arginine and allows it to form a salt bridge with the glutamate. This Arg-Glu pair has been described previously as a molecular “hasp” or latch that fastens Switch II to the α3 helix and locks Gα in the active conformation (Figure 1) (Iiri et al., 1997). Accordingly, the conserved glycine functions as a lock or pin that secures the latch and prevents access of Gβγ to Gα. The conserved triad exists in all heterotrimeric G proteins; in contrast, only the glycine (but not the arginine or glutamate) is present in monomeric G proteins such as RAS. Moreover, replacement of the conserved glycine in Gαs (G226A) or Gαi1 (G203A) disrupts formation of the Glu-Arg salt bridge (Berghuis et al., 1996), impairs binding to GTP-Mg2+, and as a consequence preserves binding to Gβγ (Miller et al., 1988, Lee et al., 1992, Berghuis et al., 1996). Mutations in the arginine (Gαs-R231H and yeast Gpa1-R327S) likewise enhance binding to Gβγ, but by a mechanism that remains poorly understood (Iiri et al., 1997, Apanovitch et al., 1998, Miller et al., 1988, Lee et al., 1992, Berghuis et al., 1996). Based on our modeling and previous observations, we hypothesized that this triad of amino acids acts in a coordinated fashion to sense the presence of the γ-phosphate of GTP and allosterically controls subunit dissociation. More specifically, we hypothesized that the conserved Arg links the conserved Gly, which dictates nucleotide binding, and the conserved Glu, which dictates Gβγ binding.
Triad residues regulate Gβγ signaling in vivo.
While RAS and Gα share the ability to bind and hydrolyze GTP, and are evolutionarily related, the acquisition of the arginine-glutamate latch may account for the ability of Gα to regulate Gβγ release. As an initial test of our model, we replaced the conserved arginine and glutamate with all 19 other amino acids and tested their functionality in a systematic fashion. Given the large number of mutants and, given the need to isolate the effects of each mutant from other functionally similar Gα subtypes, we conducted our preliminary analysis in yeast. The yeast system has some unique advantages that would help in our interpretation of the data. Most notably, yeast express a single canonical GPCR and G protein, greatly simplifying analysis of structure-function relationships. This is in marked contrast to humans where multiple G proteins receive signals from hundreds of distinct receptors. Moreover, G proteins in yeast and animals have conserved structures, and heterologous expression studies indicate that the receptors and G protein subunits are functionally interchangeable (Dowell and Brown, 2009). Therefore, any perturbation in the yeast Gα are usually predictive of perturbations in mammalian G proteins.
We first compared pheromone signaling in cells that express wild-type or mutant forms of Gpa1, expressed using the native promoter in a single copy plasmid. All of our functional assays used the extensively-studied G322A mutant as a positive control. This substitution has been characterized mechanistically for Gαs (G226A) and Gαi1 (G203A), and demonstrated to reduce the binding affinity for magnesium and GTP (Miller et al., 1988, Lee et al., 1992, Berghuis et al., 1996). These mutants retain the ability to bind to receptors, GDP, and Gβγ. While the Gs mutant fails to activate adenylyl cyclase in cells, as shown originally in the S49 cyc− cell line, purified Gαs-G226S can nevertheless stimulate adenylyl cyclase in vitro, and this effect is partially reversed by the addition of purified Gβγ (Lee et al., 1992). Based on these previous studies, and given the proximity of this residue to the guanine nucleotide, we predicted that Gpa1-G322A would exhibit the strongest suppression of G protein dissociation.
We measured two outcomes of pheromone signaling. The first was Gβγ-mediated cell growth arrest (halo assay). Pheromone spotted onto a filter disk produces a zone of growth arrest; the size and turbidity of which correlates with pheromone sensitivity. As shown in Figure 2, substitutions of Arg327 with Cys and Ser conferred the most effective inhibition (Figure 2A, bottom and Figure S2, Supplemental Information). Inhibition was absent when the arginine was replaced with glutamine. Other replacements yielded intermediate results. Most substitutions of the triad glutamate likewise yielded intermediate effects. None of the arginine or glutamate substitutions were as potent as the G322A mutant.
Figure 2. Triad residues regulate Gβγ signaling in vivo.
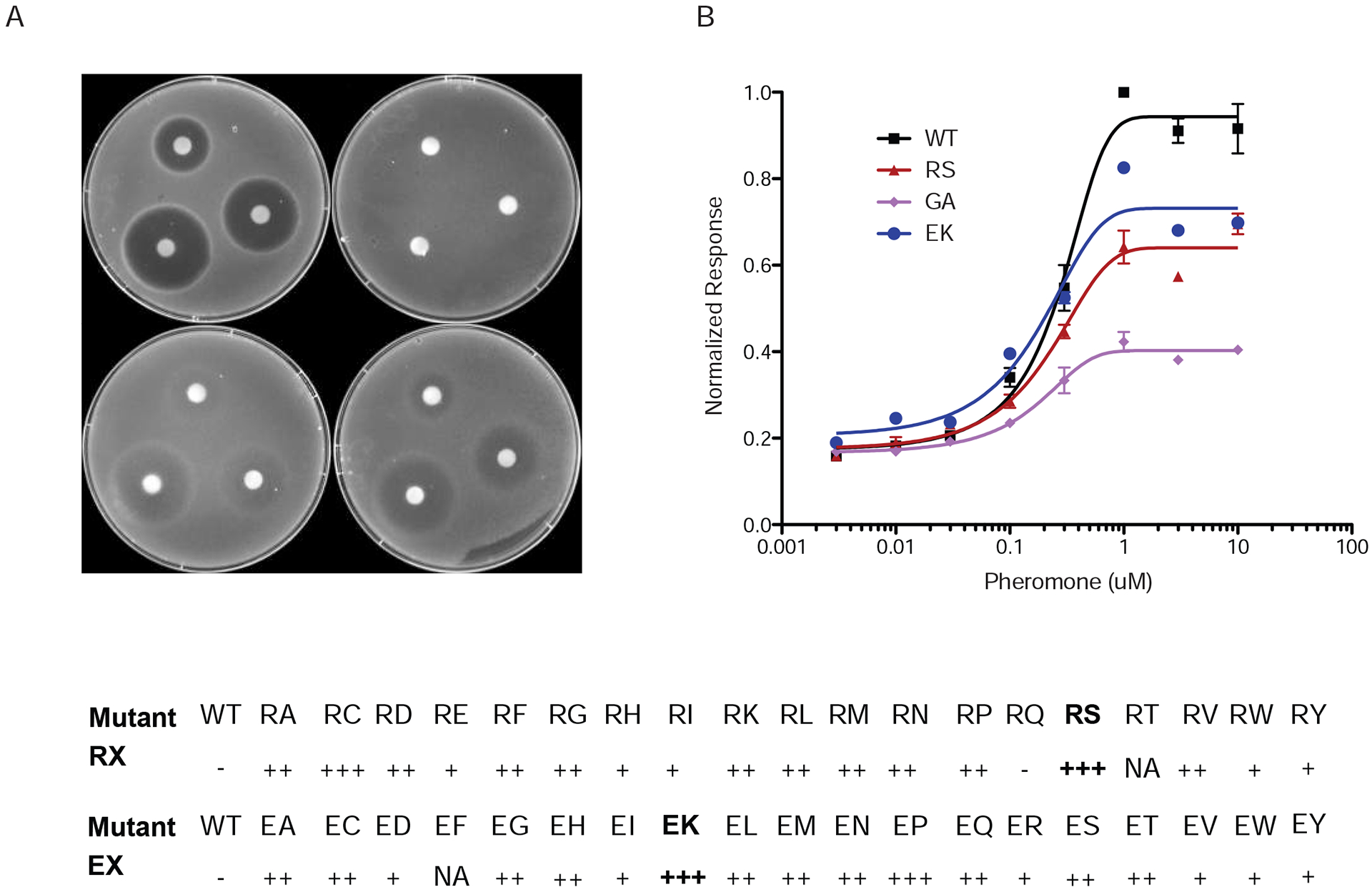
Wild-type yeast cells transformed with a single copy plasmid containing GPA1 or the indicated mutant G322A (GA), R327S (RS) or E364K (EK).
Left, plate assay with filter disks containing 75, 25, or 8 μg α-factor pheromone.
Right, transcription reporter assay after treatment with the indicated concentration of α factor pheromone. Data are averages of three independent experiments (+/−SE).
Bottom, quantitation of mutant phenotypes. Values indicate extent of growth inhibition, ranging from least (−) to most (+++) turbidity.
See also Figure S2
We then examined the effects of the triad mutations using a Gβγ-dependent gene transcription assay. In this method, induction of a pheromone-inducible promoter (from FUS1) leads to increased expression of a reporter protein. As shown in Figure S2B (Supplemental Information), expression of Gpa1-R327S caused a substantial reduction in reporter activity, but not to the extent of the G322A mutant. Thus, the two functional assays are largely in agreement and together show that pheromone signaling is diminished upon expression of some, but not all, triad mutants. Given that almost any substitution should disrupt the salt bridge that defines the hasp, we conclude that arginine and glutamate have functional properties that go beyond their ion-pair interaction.
Triad residues control subunit dissociation.
Our yeast-based measurements indicate that many, but not all, triad substitutions exhibit inhibitory effects on Gβγ release. To determine if these functions are universally conserved, we introduced our best-performing mutations into human Gα subunits and tested their activity in a mammalian expression system. To this end we used a quantitative protein proximity assay (Hollins et al., 2009, Lambert et al., 2010, Masuho et al., 2015), one that has been optimized to detect receptor-stimulated release of Gβγ from Gα. In this bioluminescence resonance energy transfer (BRET) assay, a Gα-Renilla luciferase (RLuc) fusion is co-transfected with Gβ3 and a Gγ9-GFP fusion (Olsen et al., 2020). For these experiments we used the neurotensin (NTSR1), β2-adrenergic (ADRB2), and μ-opioid (MOR1) receptors to activate Gα subtypes Gαi, Gαq and Gαs. When agonist stimulates the receptor, dissociation of the Gα-RLuc (energy donor) and Gβγ-GFP (energy acceptor) is measured as the ratio of the emission of GFP to the emission of RLuc. As shown in Figure 3A, activation of all three G proteins was substantially diminished by the triad substitutions, with the exception of the Arg-to-Gln replacement in Gαi3, (but not Gαs or Gαq, discussed below). Signaling by the μ-opioid receptor was likewise dampened by mutations in Gαi3, indicating that the inhibitory effects are shared by multiple receptors and agonists (Figure S3A, Supplemental Information). We conclude from these data that the triad residues are not only structurally conserved but are functionally conserved as well. These findings also demonstrate the utility of the yeast system for prioritizing mutants suitable for follow up analysis in a more complex but physiologically informative system.
Figure 3. Triad residues control subunit dissociation.
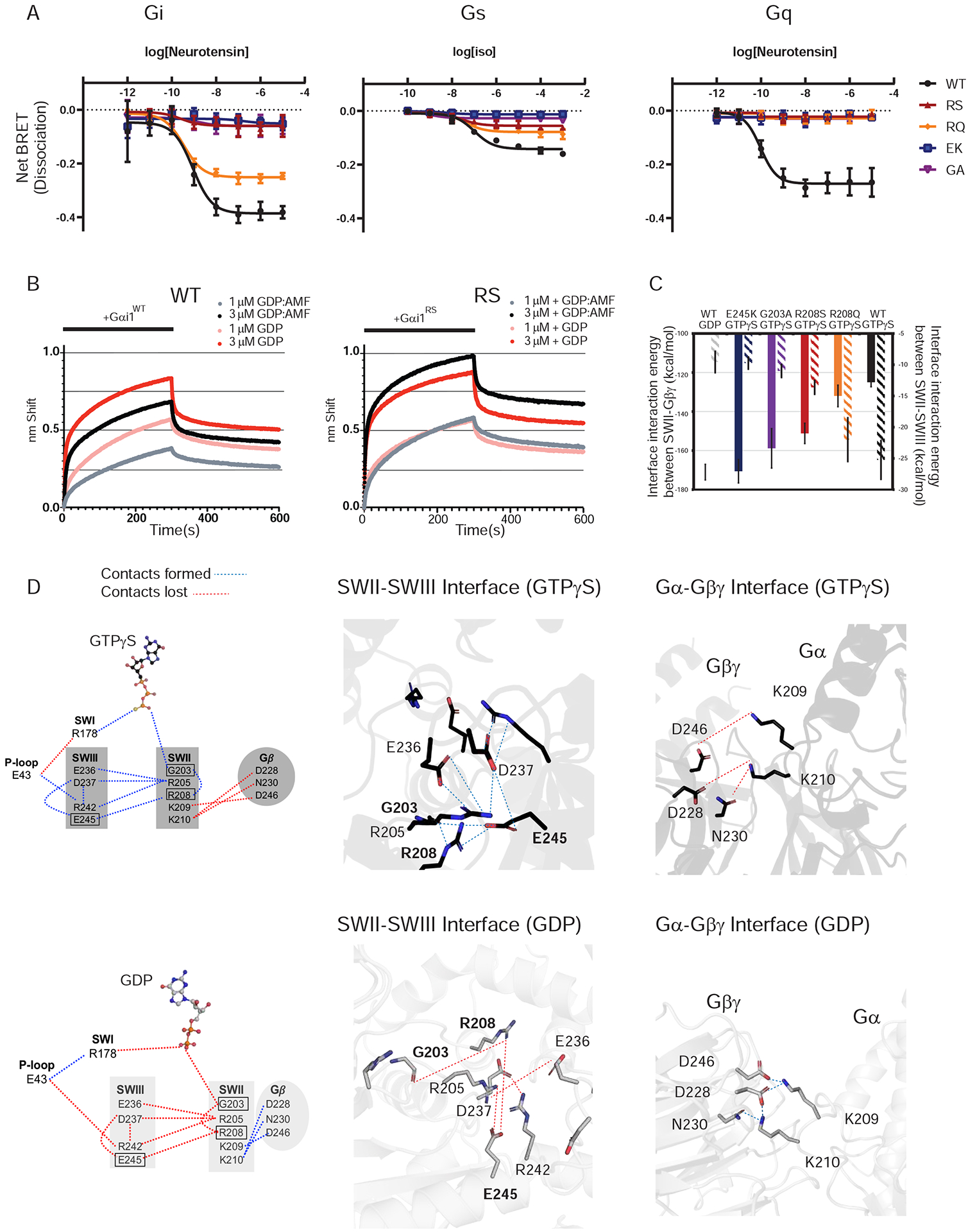
(A) HEK293 cells transfected with the indicated wildtype or mutant Gα-RLuc8 donor and Gγ-GFP acceptor proteins. Concentration-response measurements using the neurotensin or β2-adrenergic receptor are presented as fold-decrease in dynamic range (dissociation, Net BRET). Data are averages of two independent experiments in duplicate (+/−SE).
(B) Purified biotinylated-Gβ and Gγ immobilized on streptavidin exposed for 300 sec (bar) to the indicated concentration of purified Gαi1 (WT, left) or Gαi1-R208S (RS, right), equilibrated in either GDP (red or pink) or GDP and aluminum fluoride (AMF, black or grey). Binding is reported as a shift in the interference pattern (nm). Data are representative of two independent experiments in duplicate.
(C) Plot of interaction energies between Gα and Gβ subunits versus interaction energies between residues in Switch II and Switch III regions in wildtype and the indicated Gαi1 mutants. The interaction energies are averaged over the snapshots of the MD simulation trajectories.
(D) A dynamic mechanism for the effect of the γ-phosphate group triggering the release of the Gβγ subunits. The dynamic pull-push effect from the γ-phosphate group of GTPγS, that forms the polar residue interaction network involving the P-loop, Switch I, Switch II and Switch III residues, derived from MD simulations for wildtype Gαi1 bound to GTPγS (top) or GDP (bottom). The nucleotides are shown in ball and stick representation. The blue and the red dotted lines indicate sustained and broken residue contacts, respectively. Right panels show residues involved in the polar network for the GTPγS- and GDP-bound Gαi1 switch regions (black and gray colors respectively).
See also Figure S3
The data provided above indicate that the conserved triad is needed for proper Gβγ release in yeast and in human cells. To delineate the mechanism by which the triad acts, we tested the ability of mutant Gα to bind Gβγ directly. We focused on the conserved arginine because it connects the Switch II glycine (binds γ-phosphate) and the Gβγ binding interface. In particular, the R208S mutant exhibited the strongest inhibitory effect of the 19 substitutions tested. For this analysis we chose bio-layer interferometry (BLI), which analyzes the interference pattern of white light reflected from two surfaces: a layer of immobilized protein on the biosensor tip, and an internal reference layer. We combined biotin-Gβ1γ2, immobilized onto Streptavidin-coated biosensor tips, with a solution containing either 1 μM, 3 μM, or 10 μM purified Gαi1 or Gαi1-R208S, and equilibrated the protein in either GDP or GDP plus aluminum fluoride. Aluminum fluoride mimics the pentavalent transition state for GTP hydrolysis (Sondek et al., 1994, Mixon et al., 1995). Thus, the structure of the AlF4− state is very similar to the active GTP-bound state and preserves the triad polar network, but activation by AlF4− does not depend on nucleotide exchange. After 5 min the tips were transferred to an identical solution but lacking Gαi1. By this measure, the wildtype (Figure 3B, left) and R208S mutant (Figure 3B, right) exhibited similar association and dissociation kinetics in the presence of GDP (red and pink traces) (Figure S3B–D, Supplemental Information). As expected, GDP-AlF4−-Gαi1 bound more poorly to Gβγ (black and grey traces), with a slower initial on rate and lower maximum binding at both concentrations of Gα subunit. In contrast, and in support of our model, Gβγ bound equally well to the “active” and “inactive” forms of the R208S mutant. The lack of an AlF4−-dependent alteration of Gαi1-R208S binding to Gβγ is consistent with the BRET data obtained in HEK293 cells (Figure 3A). Thus, the conserved arginine mediates the assembly of the triad and formation of an active state conformation leading to a lower affinity of Gα for Gβγ.
A dynamic mechanism for the γ-phosphate of GTP in coordinating the active-state conformation of Gα
To provide a dynamic view of events leading to dissociation of the Gβγ subunits, we performed all-atom MD simulations of Gαi1 in complex with Gβγ and bound to either GDP or GTPγS. We also simulated the dynamics of Gαi1-R208S, Gαi1-E245K and Gαi1-G203A, all bound to GTPγS. Finally, as a counter example to these loss-of-signaling mutants, we included in our analysis Gαi1-R208Q, which is unique in its ability to retain partial activity. Our analysis of the crystal structures of the GDP-bound trimeric Gαi1Gβγ (Wall et al., 1995) compared to GTPγS-bound Gαi1 (Coleman et al., 1994) showed that the residues in Switch II and Switch III form tighter interactions when bound to GTPγS and thereby enable Gβγ dissociation. To assess how mutants affect the strength of subunit interaction, we calculated the non-bond interaction energies between the Gα and the Gβ subunits, averaged over MD trajectories for each of the mutant and wild type proteins. We also calculated the interaction energies between the residues in the Switch II and the Switch III regions (see Methods section for more details). As anticipated, the Gα-Gβ subunit interaction energies varied inversely with the interaction energy between the Switch II and Switch III regions (Figure 3C). The non-signaling Gαi1 mutants showed Gα-Gβ interaction energies similar to that of GDP-bound wildtype Gαi1 even when bound to GTPγS.
Simulations showing the progression of Gα activation
In wild type Gs bound to GTP, we first observed salt bridge interactions for Arg231 and Lys233 with Gβ, after which Arg231 flips towards the triad Glu268 and away from Gβ (Figure S4A, Supplemental Information). In the R231Q mutant the non-native Gln, together with Lys233, forms hydrogen bond and salt bridge interactions with Asp residues in Gβ. Since there is no repulsion between Gln231 and Lys233, the network in R231Q remains intact at 200 ns but breaks down slower at around 500 ns (Figure S4A, Supplemental Information). Additionally, there is no triad interaction formed in the R231Q mutant. This explains why the Gln231 variant of Gαs shows weaker dissociation from Gβ despite being bound to GTPγS.
To understand the mechanism of this dynamic “pull-push” effect we analyzed the residue interaction network emerging from the γ-phosphate of GTP connecting to Switch I and cascading further to Switch III and the Switch II region, which forms the Gβ interface. As shown in the network diagrams in Figure 3D, we identified an extensive polar network linking the γ-phosphate group to Arg178 in Switch I. This pull breaks the interaction of Arg178 with Glu43 in the P-loop, which in turn strengthens the interaction of Glu43 with Arg242 in the Switch III region (Figure S4B–D, Supplemental Information). This interaction between Glu43 and Switch III leads to the triad formation, comprised of Glu245-Arg208 and Gly203 in the Switch II region. Additional polar interactions between Arg242, Asp237, Glu236 in Switch III with Arg205 in Switch II also strengthen the Switch II – Switch III interactions. This Switch III - Switch II pulling effect in turn weakens the polar network of interactions of Lys210 and Lys209 in Switch II with Asp246, Asn230 and Asp228 residues in the Gβ subunit. Breaking γ-phosphate interaction with Arg178 in the GDP-bound Gαi1 disrupts the cascade of polar interactions between Switch I, Switch III and Switch II, thereby breaking the triad and preserving the polar interaction network between Switch II and the Gβ interface, as shown in Figure 3D. The sustenance of these inter-residue polar contacts over the course of different MD simulation runs has also been investigated (Figure S4B–D, Supplemental Information). It can be seen that the contact between Gly203 and the γ-thiophosphate of the nucleotide is initiated in the last 100 ns of our simulations. We observed that the time at which the interactions between Switch II and Switch II regions are formed are either simultaneous or close to formation of the Gly203-γ-PO4 group of GTPγS interaction. A comparison between the left and the right panels in Figure S4A (Supplemental Information) demonstrates how the inter-residue contacts show transitions on going from the GDP to the GTPγS bound states that triggers Gβγ dissociation. This was captured successfully by our all-atom MD simulations. A similar interaction network mechanism was also evident for Gαs MD simulations (Figure S3E and G, Supplemental Information). These results reveal a common mechanism leading to Gβγ dissociation. Specifically, we have identified an allosteric link between the terminal phosphate of GTP and the Gα-Gβγ interface.
Triad mutants confer a GDP-like conformation on GTP-bound Gαi1
Our cell-based assays indicate that mutations in the triad arginine and glutamate confer sustained binding to Gβγ. The experimental BRET (Figure 3A) agrees reasonably well with the subunit interface interaction energy calculated from MD simulations. Our MD simulations suggest that these mutants lack important conformational changes associated with GTP binding, specifically changes in Switch II and Switch III, and that these structural differences are propagated by the γ-phosphate of GTP. To that end, we monitored the conformational stability of all three mutants when bound to guanine nucleotides.
We have previously shown that Gαi1 is thermodynamically more stable when bound to GTPγS than to GDP (Isom et al., 2013). This difference is consistent with structural studies showing that the GTP-bound state is considerably more rigid than the GDP-bound state, as dictated by regions near the γ-phosphate group and at the Gβγ interface (Sun et al., 2015). Since the triad mutants function as if they were bound to GDP, we anticipated that they would exhibit a reduced thermostability even if bound to GTP. To test this, we purified wildtype and several mutant forms of Gαi1 and determined the melting temperature of each using the quantitative cysteine reactivity (fQCR) assay. Briefly, on heating, protected cysteine residues of Gα become exposed and can be covalently labeled with a fluorogenic reagent (Isom et al., 2013). Using a two-state model of denaturation, we analyzed each Gαi1 mutant’s unfolding profile to quantify the midpoint of temperature unfolding (Tm). Representative thermostability profiles for the wildtype and triad mutant forms of Gαi1 are shown in Figure 4A. These experiments confirm that the wildtype protein undergoes a 9 °C increase in Tm when bound to GTPγS in place of GDP; in contrast, the mutants exhibited little or no change. We conclude that that the conserved triad confers critical conformational changes that favor the dissociated state of the G protein.
Figure 4. Triad residues coordinate the active-state conformation.
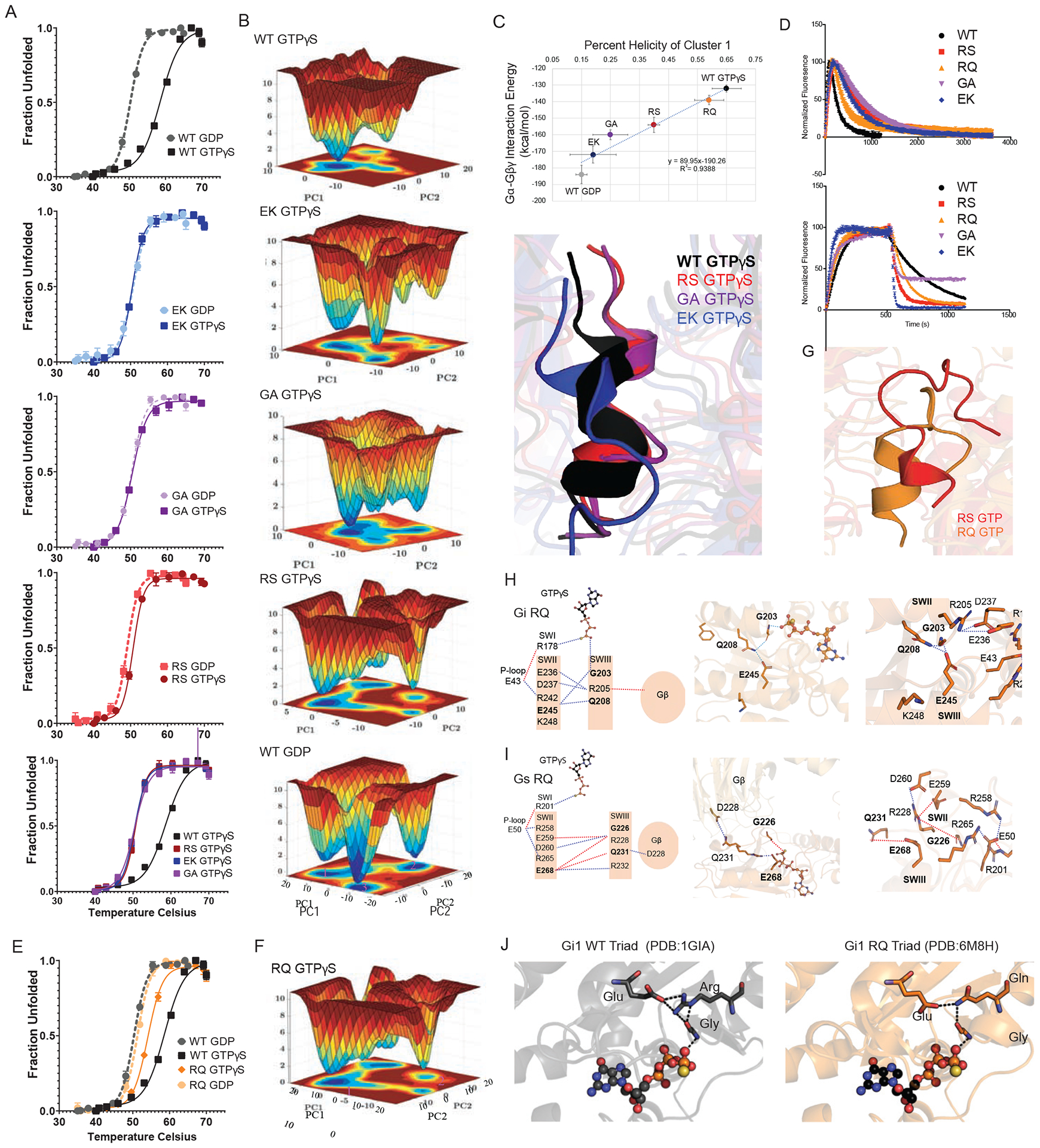
(A) Thermostability of purified wildtype and the indicated Gαi1 mutants equilibrated in either GTPγS (solid lines) or GDP (dashed lines). Tm values were quantified by fitting a two-state model of thermal unfolding to one of 3 independent experiments done in triplicate (+/− SEM).
(B) Free energy surfaces computed using population densities show the conformational heterogeneity of Gαi1 and its mutant systems. The projections of the surfaces are shown on the principal components 1 and 2 (PC1 and PC2) axes; defined as the two vectors that recapitulate the dominant motions of the Gα protein during the simulations. The various conformational clusters are numbered (magenta) ordered by the population of each conformation cluster. Cluster 1 for example, is the most populated conformation cluster. The Z axis is the Gibb’s free energy obtained by inverting the population of these microstates and is shown as a colored heatmap.
(C) Helicity of residues in the Switch II regions of wild type and the indicated Gαi1 mutants, bound to GTPγS, calculated as an average over the snapshots in the most occupied conformational cluster I from the free energy surfaces.
(D) Association of 6 μM purified wildtype and the indicated Gαi1 mutants with 100 nM BODIPY-GTP, to monitor binding and hydrolysis as the increase and decrease in fluorescence, respectively, or with 100 nM BODIPY-GTPγS, followed after 600 s by addition of 50 μM unlabeled GTPγS, to monitor dissociation of the labeled nucleotide. Data are representative of 2 or more independent experiments.
(E) Thermostability of purified wildtype Gαi1 and R208Q mutant, as detailed above (A).
(F) Free energy surfaces computed using population densities calculated for principal components PC1 and PC2 for Gαi1-R208Q, as detailed above (B).
(G) Structural representation of the Switch II regions of Gαi1-R208Q bound to GTPγS, as detailed above (C).
(H) A dynamic mechanism for the effect of the γ-phosphate group triggering the release of the Gβγ subunits for Gαi1-R208Q as detailed above in Figure 3D.
(I) A dynamic mechanism for the effect of the γ-phosphate group triggering the release of the Gβγ subunits for Gαs-R231Q as detailed above in Figure 3D.
(J) Crystal model of Gαi1 and Gαi1-R208Q triad residues and GTPγS (ball and stick) from PDB: 1GIA and 6M8H.
See also Figure S4
Structural dynamics studies have shown that thermostable mutants exhibit conformational homogeneity, meaning fewer distinct conformations in the ensemble during dynamics simulations (Vaidehi et al., 2016). The conformational homogeneity manifests as fewer energy minima in the free energy surface of thermostable mutants (Ghosh et al., 2018). To understand the structural basis for the observed thermostability differences, we generated the free energy surface of GDP- and GTPγS-bound Gαi1 using MD simulation trajectories. Figure 4B shows the free energy surface of Gαi1 bound to GTPγS and GDP, along with the Gαi1 mutants bound to GTPγS. In particular, we observed that Gαi1-GTPγS had fewer energy minima and was more conformationally homogeneous compared to the mutant forms of Gαi1. Thus, there is a correlation between conformational heterogeneity and thermal instability.
A comparison of crystal structures of GDP- and GTPγS-bound Gαi1 shows that residues in Switch II fold into a helix leading to the dissociation of Gβγ. While the MD simulations were done on the heterotrimer, the thermostability measurements were done on Gα alone. Despite these differences, we infer that changes in Switch II determine the interaction strength between Gα and Gβ. We speculated that formation of an ordered helical region in Switch II in GTPγS-bound Gαi1 could lead to its enhanced thermostability. Therefore, we calculated the average helicity of the residues in the Switch II region averaged over all the MD snapshots in the most occupied conformation cluster, extracted from the free energy surfaces for the wildtype and mutant forms of Gαi1. As shown in Figure 4C, Switch II transitioned from about 15% to 63% helicity in going from GDP-bound to GTPγS-bound Gαi1. It is important to note that we started the MD simulations from the same disordered conformation in Switch II observed in the crystal structure of GDP-bound Gαi1. MD simulations recapitulate the loop-to-helical transitions that are triggered by the γ-phosphate group of the GTPγS engaging the dynamic mechanism shown in Figure 3D. To examine if the loop-to-helix transitions lead to increased thermostability, we calculated the average interaction energies between the Gα and Gβ subunits averaged over the MD snapshots in the most occupied conformation cluster, extracted from free energy surfaces. The calculated helicity of Switch II correlates inversely with the Gβγ interaction energies for wildtype Gαi1, bound to GDP and GTPγS, as well as the Gαi1 mutants bound to GTPγS (Figure 4C). In summary, these findings indicate that increased helicity could bring a more ordered structure to the Switch II region and confer thermostability and conformational homogeneity for Gαi1 bound to GTPγS.
An alternative interpretation is that the mutants bind to GDP but not GTP. To test this experimentally we determined the rate of nucleotide exchange and the rate of GTP hydrolysis, measured as a gain of fluorescence as BODIPY-GTP bound to protein (McEwen et al., 2001). As shown in Figure 4D, the labeled analog bound to mutants as well (or faster) than the wildtype protein. We then added an excess of unlabeled GTPγS and determined that the rate of dissociation was substantially faster for all of the mutants as compared to that of wildtype. We infer that the triad mutants confer a more dynamic binding pocket, which results in faster dissociation of nucleotide. The mutants were fully competent to bind and hydrolyze BODIPY-GTP, as indicated by the rapid increase and subsequent decrease (owing to the lower quantum yield of the hydrolysis product) in fluorescence respectively. More broadly, these data indicate that loss of the triad residues does not impede GTP binding or dissociation, but prevents the conformational changes required for Gα activation and Gβγ dissociation. Taken together, our MD simulations and biophysical assays confirm that mutations in the triad arginine and glutamate place the protein in a permanently “inactive” conformation, compatible with sustained binding to Gβγ.
The results presented above reveal the importance of the triad arginine. Generally speaking, substitutions at this site lead to diminished signaling by Gβγ. However, as detailed above (Figures 3 and 4), substitution of the arginine with glutamine partially preserves function, particularly in Gαi1. In accordance with these observations, our calculations of interaction energies (Figure 4C), thermal stability measurements (Figure 4E), computed free energy surfaces (Figure 4F), and helicity calculations (Figures 4C and G) revealed that the R208Q mutant is more similar to wildtype than any of the other arginine mutants. Weakening of interaction between Gαi1 and the Gβγ subunits of the G protein is caused by the enhanced interaction between the Switch II and Switch III regions within the Gαi1 subunit. These simulations are the theoretical basis for the proposed competition between Gβγ and Switch III for Switch II (Figure 4H). The calculated interaction energies for Gα and Gβγ subunits become less favorable as the interaction energies for Switch II and Switch III become more favorable, and vice versa (primary and secondary y-axis respectively, Figure 3C). The same paradigm holds for Gαs (Figure 4I). The analysis of the root mean square fluctuations (RMSF) indicates that out of the six Gαi systems simulated in this study the WT and its R208Q mutant bound to GTPγS exhibit the maximum degree of flexibility in the switch regions. In contrast, the other four mutants show less flexibility in the switch regions (Figure S4E, Supplemental Information). This observed flexibility might be counter-intuitive given the high thermal stability of the WT and R208Q mutant of Gαi. However, it is worth mentioning that the experimental thermostability measurements were conducted with the fully dissociated, monomeric constructs whereas the simulations we performed could capture signatures of the early events of Gαβ dissociation within the given timescale. Thus, our molecular dynamics simulations were able to predict substitution- and subtype-specific dissociation events with exquisite accuracy and mechanistic detail.
We then sought to determine the structural basis for the unique ability of Gαi1-R208Q to sustain signaling. To that end, we compared the structure of wild type Gαi1 with a new X-ray crystal structure of Gαi1-R208Q, both in the GTPγS-bound state (Figure 4J, compare left and right panels). This analysis revealed that Gln208, like the native arginine of Gαi1, is close enough to form a H-bond network with the conserved glycine and glutamate. In contrast, and as anticipated by our MD simulations, the same substitution would not restore the network in Gαs or Gαq. Whereas Arg208 of Gαi1 faces Switch III in both the active and inactive states, the corresponding residue in Gαs must rotate ~180° before it can interact with the conserved glycine and glutamate.
Our simulations appear to account for the distinct functional properties of the G-R-E motif, as determined by thermostability measurements, nucleotide and protein binding measurements, as well as the signaling outputs obtained in yeast and animal cells. Most strikingly, the atomistic MD simulations anticipated the unexpected differences between Gi and Gs, and the differences between R208Q and other Arg208 substitutions. The differences between Gαi1-R208Q and Gαs-R231Q in particular support our model that the arginine has functions beyond simply forming a salt bridge with the conserved glutamate. More broadly, these results highlight the predictive power of MD simulations, particularly when integrated with molecular and cellular experimental analysis.
The triad arginine controls the final committed step of G protein activation.
Finally, we sought to determine the structural basis for the functional properties of the Gαi1-R208S mutation. Based on the mechanism derived from MD simulations, we postulated that the mutant retains Gβγ binding by disrupting the conserved polar network that dictates the response to GTP. To test this directly, we collected 1H-15N 2D heteronuclear NMR spectra of wild-type and mutant Gαi1, both in the presence of GDP and GTPγS. This method allows for the detection of backbone and side-chain NH resonances. As an NH resonance can be detected for every residue with the exception of proline, the spectrum contains a “fingerprint” of the protein backbone and perturbations resulting from changes in intramolecular interactions. We consider this a definitive method for detecting the conformational changes that accompany nucleotide binding and G protein activation. In contrast to some other Gα isoforms, and most members of the RAS family of GTPases, Gαi1 undergoes nucleotide exchange within minutes, even in the absence of exchange factor or receptor (Figure 4D).
As shown previously (Goricanec et al., 2016), a substantial number of Switch II peaks (Figure 5 arrows) for wildtype Gαi1 were resolved in the presence of GTPγS (black) but not GDP (blue). These data are consistent with X-ray structural data, indicating that Switch II is more ordered in the activated state as compared with the inactive state. Likewise, Gαi1-R208S exhibited multiple peak shifts when GDP was replaced with GTPγS, indicating that nucleotide exchange had occurred (Figure S5 top, Supplemental Information). Consistent with our fast exchange data for R208S, the mutant GTPγS spectrum contains a shifted tryptophan that indicates GTPγS binding (Figure 5A circle). In contrast to wildtype, Gαi1-R208S appears to be missing multiple peaks, consistent with a more dynamic GTPγS state. In support of our observations from MD simulations (Figure S3, Supplemental Information), many resonances missing in the mutant spectrum were in the Switch regions for wildtype bound to GTPγS (Figure 5B). These results indicate that Gαi1-R208S is in an alternate, more dynamic conformational ensemble, even when bound to GTPγS. More broadly, these results indicate that the conserved arginine is necessary to detect the presence of the γ-phosphate and undergo the structural changes necessary to maintain the active state.
Figure 5. NMR analysis of wildtype Gαi1 and Gαi1-R208S.
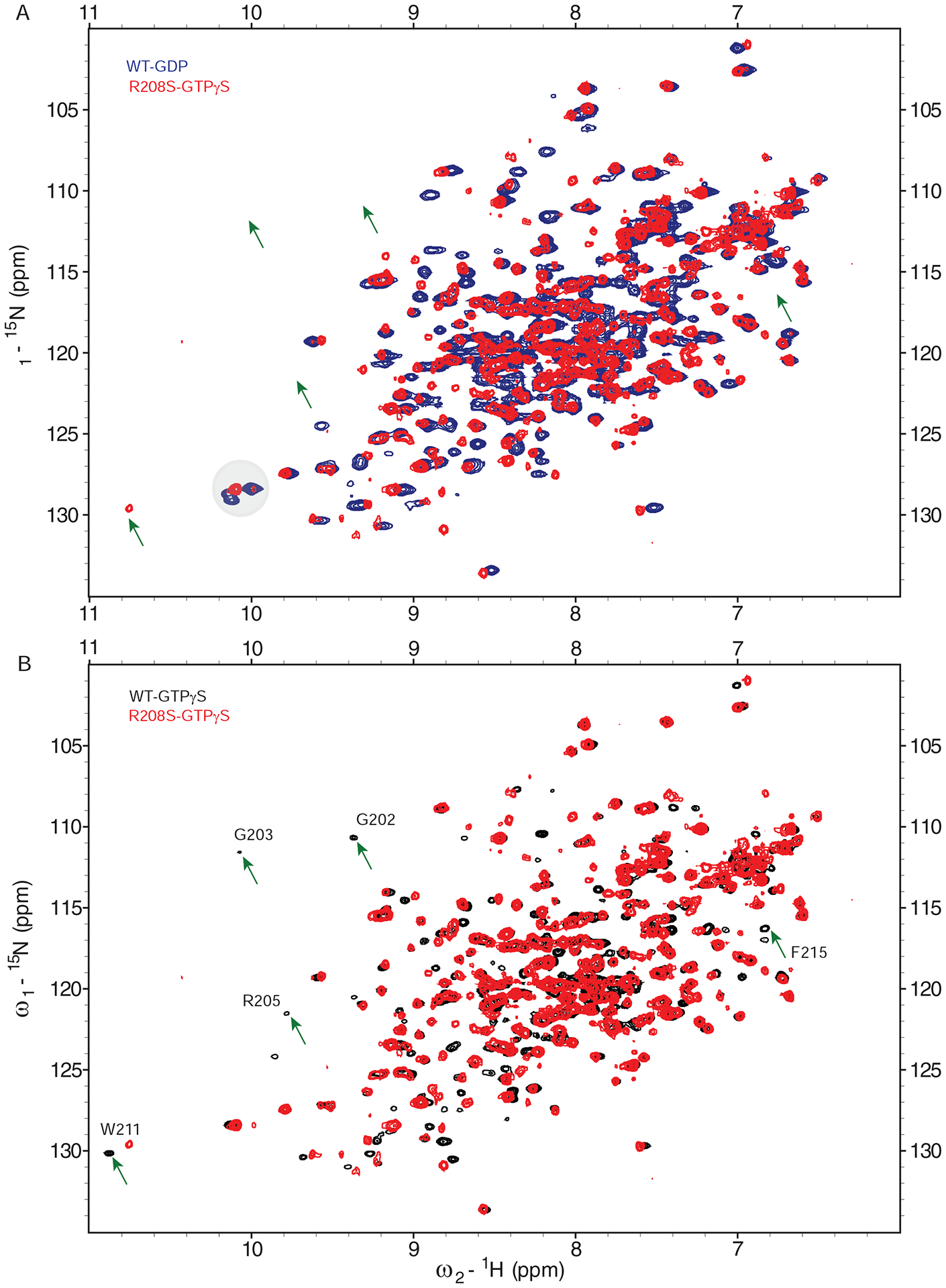
2D [15N, 1H]-HSQC NMR of Gαi1Δ31 wildtype and R208S mutant.
(A) Overlay of wildtype Gαi1Δ31 in complex with GDP (Blue) and R208S Gαi1 in complex with GTPγS (Red).
(B) Overlay of wildtype (Black) and R208S (Red) Gαi1, each in complex with GTPγS (Black). Switch II specific peaks are labeled (arrows).
See also Figure S5
DISCUSSION
Heterotrimeric G proteins are present in a wide variety of organisms and exist in multiple distinct subclasses. Recent findings, including structure determinations of receptor-bound G proteins, have revealed the molecular motions leading to the release of GDP. Here we considered the subsequent molecular events leading to the release of Gβγ. Our approach combined cross-species sequence analysis, cell-based functional assays, MD simulations, thermostability measurements, direct Gα and Gβγ binding measurements, and NMR spectroscopy.
Our study revealed the existence of a network of highly conserved amino acids that dictates the release of Gβγ by Gα. In the presence of the terminal phosphate of GTP, the conserved glycine forms a polar network with the arginine and glutamate, disrupting the Gα-Gβγ binding interface. By systematically replacing triad residues, and measuring the functional consequences of those changes, we demonstrated their importance in G protein activation in cells. Whereas a single phosphate can initiate subunit dissociation, we show that mutation of a single amino acid can override the process entirely. We then determined the molecular basis of activity through biochemical and biophysical experiments conducted in vitro. Finally, we established the atomic interactions responsible for dissociation of the G protein subunits in silico.
Our findings also provide the molecular basis for important functional behaviors documented through cellular genetic studies conducted over the last four decades. The S49 cyc− cell line, which bears the Gαs-G226A mutation and is deficient in cAMP production (Bourne et al., 1981, Salomon and Bourne, 1981), was instrumental in the discovery of G proteins (Gilman, 1987). Functional and structural characterization of Gαi-G203A, and the corresponding G226A mutation in Gαs, have shown that this substitution does not influence GTPase activity, at least at high magnesium concentrations, but prevents other residues (Thr181 and Ser47 in Gαs) from forming the magnesium binding site and thereby prevents the conformational changes in Switch II that lead to Gβγ dissociation (Lee et al., 1992, Miller et al., 1988, Mixon et al., 1995, Berghuis et al., 1996, Wall et al., 1995). In yeast, mutations of the G-R-E motif confer strong resistance to pheromone stimulation (Stratton et al., 1996, Apanovitch et al., 1998). Replacement of the arginine is even sufficient to block unrestrained signaling in the complete absence of GTP hydrolysis (Apanovitch et al., 1998).
The importance of the G-R-E motif has also been revealed through human genetic studies. The conserved arginine is one of two sites in Gαs mutated in patients with pseudohypoparathyroidism 1a, a syndrome characterized by Albright hereditary osteodystrophy and resistance to hormone stimulation of cAMP production (Farfel et al., 1996, Iiri et al., 1997, Iiri et al., 1999). More recent studies indicate that the triad arginine and glutamate are mutation “hotspots” in Gαo, frequently altered in individuals with neurodevelopmental disorders including involuntary movement and seizures (https://monarchinitiative.org/gene/HGNC:4389#overview). Thus, many of the phenotypes associated with loss of the G-R-E motif, in yeast and in humans, can be attributed to sustained subunit association.
Finally, our analysis of G protein activation complements recent advances in our understanding of receptor activation. The first crystal structure of a receptor-G protein complex provided detailed insights into the conformational changes leading to nucleotide release (Rasmussen et al., 2011). Most notably, that structure revealed a dramatic displacement of the Gα helical domain from the RAS-like domain, thereby exposing the nucleotide binding site. A major impediment to obtaining such structures is the difficulty of assembling an agonist-bound receptor with a nucleotide-free G protein. The first structures used fusions and single chain antibodies to stabilize this otherwise ephemeral state. Subsequent studies used variants of Gα containing a panel of up to 8 mutations, including substitutions of the triad glycine and glutamate (Liang et al., 2018b, Draper-Joyce et al., 2018, Liang et al., 2018a). Based on our findings, substitutions of the triad arginine are likely to be particularly useful in obtaining stabilized heterotrimer complexes suitable for structural analysis in the future.
Taken together, our analysis reveals events leading to the final committed step of G protein activation and provides a mechanistic basis for human disease. The structural rearrangements leading to subunit dissociation are more subtle than those affecting nucleotide exchange, but they are just as consequential. In both cases, conserved residues form a network of non-covalent contacts that link a bound ligand – either agonist or GTP - to a network of contacts needed to release Gβγ. Given that the G-R-E motif is conserved in all G proteins, the allosteric mechanism detailed here is likely to be universal for all subtypes and species. Just as a detailed understanding of the molecular basis for ligand binding has led to important advances in pharmacology, a better understanding of subunit dissociation could reveal strategies to bypass or compensate for the human genetic defects that cause disease.
Limitations
The G-R-E motif is conserved in all G proteins including animals, fungi, and plants. Our analysis was limited to human and yeast G proteins; the function of the G-R-E motif in plants remains untested. While our MD simulations explain the available experimental data, they are limited in the length of the simulation time. Hence the observations made here describe only the early atomic level events that ultimately lead to dissociation of Gβγ subunits from the Gα subunit. Whereas mutations in triad residues have been used to form stabilized receptor-G protein complexes, suitable for structural studies, substitutions of the critical triad arginine have not yet been used in this manner. While our analysis points to G proteins as potential drug targets, there are very few cell-permeable inhibitors of G proteins and so far these exist only for the Gq/11/14 subfamily (Kostenis et al., 2020).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Henrik G. Dohlman (hdohlman@med.unc.edu).
Materials Availability:
TRUPATH/BRET reagents are publicly available from Addgene (#1000000163). All other unique reagents generated in the study will be available without restrictions.
Data and Code Availability:
Data are available from H.G.D. and N.V. The study did not generate new unique code. MD simulations full trajectories will be made available at https://welcome.gpcrmd.org/. NMR data will be made available at: http://www.bmrb.wisc.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Insect cells.
High Five insect cells infected with recombinant baculovirus were used for production of recombinant rat Gβ1 (Ghosh et al., 2003) and bovine Gγ2 (Kozasa and Gilman, 1995). Cells were grown in SF900 II serum-free medium (Thermo-Fisher) in suspension culture at 27 °C with continuous shaking. Cells were infected with baculoviruses at a density 2×106 cells/mL.
Human cell lines.
HEK293T (ATCC, #CRL-11268) cells overexpressing human Gγ9-GFP2 and wild-type or mutant human Gα-RLuc8 donors were used for BRET assays. Cells were grown at 37 °C in Dulbecco’s Modified Eagle Medium (Corning, 10–017-CV) + 1% dialyzed fetal bovine serum (GIBCO A3382001).
Yeast strains.
Yeast Saccharomyces cerevisae strain BY4741 bar1Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 bar1::KanMX) (Research Genetics #95401.H2) was used for halo, transcription-reporter, and western blot analysis of Gpa1. Cells were grown at 30 °C in selective medium, comprised of yeast nitrogen base w/o amino acids and w/o ammonium sulfate (1.7 g/l), ammonium sulfate (5 g/l), complete synthetic media powder (MP Biomedicals) and + 2% dextrose.
METHOD DETAILS
Mutagenesis.
Primestar Max (Takara Bio) mutagenesis was performed using primers containing 5 bp 5’ to the codon and 20–30 bp 3’ to the codon (Table S1, Supplemental Information).
Halos.
Pheromone-induced growth inhibition was measured as described previously (Hoffman et al., 2002). Briefly, 75, 25, or 8 μg synthetic α factor was spotted onto paper disks. Agar (0.5%) was melted and then kept at 55 °C before mixing with 100 μL saturated culture of S. cerevisiae strain BY4741 bar1::KanMX (bar1Δ), transformed with pRS316 containing either no insert (vector), GPA1 or mutant GPA1 (Song et al., 1996), and grown at 30 °C in selective medium. After spreading the agar/yeast mixture, each disk was applied to one of three predefined locations on the surface of the agar. Cell growth was imaged after 24 and 48 hr.
Transcription.
Pheromone-induced gene induction was monitored as described previously (Shellhammer et al., 2019). Briefly, BY4741 bar1Δ cells cotransformed with plasmids pRS426-pFUS1-YeGFP3 (Shellhammer et al., 2019) and pRS316-GPA1 (wildtype or mutant) were grown to saturation then diluted to OD600 < 0.001. After reaching OD600 of 0.8, synthetic α factor was added at defined concentrations. After 1.5 hr, GFP fluorescence was measured using a Molecular Devices Spectramax i3x plate reader at an excitation wavelength of 483 nm and emission wavelength of 518 nm.
Immunoblotting.
Extracts of yeast BY4741 bar1Δ, transformed with the single copy plasmid pRS316-ADH1-GPA1, pRS316-ADH1-GPA1-Flag, or the indicated mutants (Table S1, Supplemental Information), were resolved by 10% acrylamide gel electrophoresis and immunoblotting, as described in (Cappell et al., 2010) except that blots were treated with 0.01% fish gelatin. Gpa1 was detected using mouse anti-Flag (Sigma-Aldrich, 1:3000) and horseradish peroxidase-conjugated goat-anti-mouse (Jackson ImmunoResearch Laboratories, 1:10,000) antibodies, and visualized with ECL-plus reagent (Life Technologies) on a ChemiDoc Touch Imaging System (Bio-Rad).
Bioluminescence Resonance Energy Transfer (BRET) assay.
HEK293T cells were used to measure basal association and receptor-mediated dissociation of Gα and Gβγ, as described previously (Olsen et al., 2020). This particular BRET method has been extensively validated and provides a highly specific measure of Gα-Gβγ association (Olsen et al., 2020). In addition, because of its low basal signal and high dynamic range, the method is well suited for a side-by-side comparison of our mutants, all of which impede subunit dissociation and thereby lower the measured output to varying degrees. Briefly, cells were transfected with plasmids encoding the neurotensin (NTSR1), β2-adrenergic (ADRB2) or μ-opioid receptor (MOR1), Gα fused to Renilla luciferase 8 (Rluc8), Gβ3 and γ9- fused to GFP in a 1:1:1:1 ratio (cell density 750,000 cells in 3 mL). After transfection (approximately 16 hr) cells were plated into poly-D-lysine coated 96-well white clear bottom plates in medium containing Dulbecco’s Modified Eagle Medium +1 % dialyzed fetal bovine serum and then incubated overnight. The following day, cells were washed twice with assay buffer (20 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), Hank’s Balanced Salt Solution, pH 7.4). 10 μL of Rluc8 substrate (coelentrazine 400a, Nanolight) was added per well at a final concentration of 5 μM. Cells were then incubated 5 min in the dark. 30 μL (3X) of agonist in drug buffer (contains assay buffer, 0.1% bovine serum albumin) was added per well then incubated for 5 min in the dark. Plates were read for luminescence at 485 nm and fluorescence emission at 530 nm for 1 sec per well using a Mithras LB940 multimode microplate reader. The BRET ratio was determined by dividing fluorescence by luminescence (GFP/Rluc). The receptor-catalyzed dissociation of the heterotrimer (net BRET) was measured by comparing the energy transfer from donor to acceptor and reported as ratios: GFP/Rluc per well - Basal BRET (GFP/Rluc at lowest dose of agonist) = Net BRET/ dissociation. The net BRET was plotted as the GFP/Rluc ratio as a function of neurotensin, DAMGO or isoproterenol and the curve was fit in Graphpad Prism 8 (Graphpad Software Inc., San Diego, CA) (Che et al., 2020).
Expression and Purification of Gαi1.
RIPL (Agilent) or Rosetta (Novagen) cells were transformed with PET-SUMO-Gαi1 plasmid (Maly and Crowhurst, 2012) and grown to saturation in YZ medium containing 0.02% (w/v) glucose and 0.2% (w/v) ala-lactose; autoinduction occurred via inhibition of the Lac repressor when ala-lactose becomes the primary fuel source (Isom et al., 2013). The culture temperature was lowered from 37 to 18 °C and allowed to rotate overnight. The cultures were harvested by centrifugation for 1 hr at 4 °C and then lysed by sonication on ice. Lysate was then clarified by centrifugation for 1 hr at 4 °C. Clarified lysate was mixed with 1 mL of ProBond Ni-chelating resin (Thermo Fisher Scientific, Invitrogen #R80101) per 15 mL of lysate for 1 hr. The resin was washed twice with phosphate buffer pH 7 containing 10 μM imidazole, then SUMO-tagged Gαi1 was eluted using the same buffer containing 400 mM imidazole. To remove the SUMO, eluate was dialyzed at 4 °C in 4 L of phosphate buffer without imidazole after adding 1 mg ULP1 protease to the dialysis cassette. After dialysis, the cleaved product had no tag and was efficiently purified by reverse metal affinity chromatography. The protein was further purified by passing over a Sepharose Q anion exchange column (GE Healthcare) equilibrated in 100 mM potassium phosphate pH 7. The purified yield was typically 3–30 mg of Gα/L of cell culture.
Expression and Purification of Gβ1γ2.
High 5 cells (Invitrogen; 2 × 106 cells/mL) were infected with high titer Gβ1 and Gγ2 baculoviruses. Gβ1γ2 was purified according to (Kozasa and Gilman, 1995), with modifications. All steps were carried out at 4 °C. Cells were harvested 60 hr postinfection by centrifugation at 2600xg and then resuspended in 50 mL of lysis buffer (20 mM HEPES, pH 8, 150 mM NaCl, 5 mM 2-mercaptoethanol (2-ME), 1 mM EDTA, 1 mL of protease inhibitor cocktail (P-2714, Sigma-Aldrich) per liter of cell culture. Cells were lysed by sonication and centrifuged at 2600xg to collect the membranes. Resuspension of membranes were accomplished by dounce homogenization in 100 mL of lysis buffer. The membranes were solubilized by adding 1% Lubrol (C12E10; Sigma-Aldrich) with stirring, and the resultant solution was clarified by ultracentrifugation at 125000xg. The supernatant was loaded onto Ni-NTA agarose (Qiagen) equilibrated with lysis buffer + 1% Lubrol. The resin was washed and the Lubrol exchanged for sodium cholate using buffers Ni-A (20 mM HEPES, pH 8, 0.4 M NaCl, 5 mM 2-ME, 0.5% Lubrol, 0.15% cholate) and Ni-B (20 mM HEPES, pH 8, 0.1 M NaCl, 5 mM 2-ME, 0.25% Lubrol, 0.3% cholate). Gβ1γ2 was eluted in Ni-C (20 mM HEPES, pH 8, 0.01 M NaCl, 5 mM 2-ME, 1% cholate, 200 mM imidazole). The purified yield was typically 1 mg of Gβ1γ2/L of cell culture (Kozasa and Gilman, 1996).
Thermostability Assays (fQCR).
Tm values were determined using the fast Quantitative Cysteine Reactivity (fQCR) assay as described previously (Isom et al., 2013). Briefly, 10 μL of 40 μM protein was added into 12 strip PCR tubes with 170 μL phosphate buffer pH 7 + 10 μL of 1 mM guanine nucleotide (GDP or GTPγS) + 10 μL of 500 mM 4-fluoro-7-sulfamoylbenzofurazan (ABDF) for 5 min on ice. The protein was subjected to a temperature gradient in a standard thermocycler for 3 min. The reaction was quenched with ice cold 0.1 N (final concentration) HCl. The ABDF reacts with exposed cysteine residues and emits light detected on a PHERAstar (BMG Labtech) plate reader using wavelengths using excitation and emission bandpass filters of 400 and 500 nm (Isom et al., 2013).
Nucleotide loading and hydrolysis.
BODIPYFL-GTPγS or BODIPYFL-GTP (100 nM) was equilibrated in buffer (50 mM HEPES, 10 mM MgCl2, 25 mM NaCl, pH 7) for 60 sec. Purified Gα protein (100 nM) was added to a 1 mL cuvette. The fluorescence of the BODIPYFL group (502 nm excitation, 511 nm emission) was measured over 600 sec. 20 μM (final concentration) unlabeled GTPγS was then added to displace BODIPYFL-GTPγS. All measurements were made with a Perkin-Elmer Luminescence Spectrometer and the FLWinLab software package (McEwen et al., 2001, Jones et al., 2012).
Biolayer Interferometry.
Binding between Gα and Gβγ was determined by Biolayer Interferometry (BLI) using Octet Red96 (Fortebio) as described previously (Seneviratne et al., 2011). Briefly, purified biotinylated Gβγ (3 mg/mL) was incubated for 15 min with streptavidin biosensors in either PBST-NGM (25 mM KPO4, pH 7, 50 mM NaCl, 0.1% Tween, and 50 μM GDP + 5 mM MgCl2) or PBST-NGA (50 μM GDP, 5 mM MgCl2, 30 μM AlCl3, and 1 mM NaF).
Purified Gαi1 (untagged) was diluted to 10 μM, 3 μM or 1 μM in PBST-NGM or PBST-NGA and then mixed with Gβγ-loaded sensors for 5 min (association) and then protein-free buffer for 5 min (dissociation) at 25 °C. Nonspecific binding was measured using biosensors that were exposed to buffer alone. Baseline subtraction and Gβγ loading normalization were done in Excel. Kinetic Analysis was done using GraphPad Prism.
NMR sample preparation and spectroscopy.
15N-enriched wildtype and R208S Gαi1-Δ31 (PET-SUMO-Gαi1-Δ31 plasmid) were expressed and purified as described above and as detailed previously (Maly and Crowhurst, 2012). The purified proteins were exchanged into NMR buffer (20 mM sodium phosphate, pH 7.0, 50 mM NaCl, 2 mM MgCl2, 200 μM GDP, 5% D2O). To exchange GDP to GTPγS, the proteins were gently exchanged into a GDP-free solution with 1 mM MgCl2 and 10-fold excess of GTPγS were added to the protein. The sample was incubated on ice for 30 min and then MgCl2 concentration was increased to 5 mM for another 30 min. GTPγS-loaded samples were finally exchanged into NMR buffer with 200 μM GTPγS. Each NMR sample contained 100 μM Gαi1-Δ31. NMR spectra were acquired at 25°C on a Bruker Avance 850 NMR spectrometer. Two-dimensional 1H–15N HSQC experiments were recorded with 1024 and 128 complex points in the direct and indirect dimensions, respectively, 44 scans per increment and a recovery delay of 1.0 sec. Spectral widths used were 13586.957 Hz (1H) and 3015.682 (15N) Hz. Spectra were processed and analyzed using NMRPipe (NIDDK, NIH) and Sparky (University of California San Francisco). Backbone assignments for wildtype Gαi1-Δ31 were transferred from BMRB 30078 for GDP and 26746 for GTPγS (Goricanec et al., 2016). The backbone assignment for Gαi1-Δ31-R208S were transferred from the wildype Gαi1-Δ31. All ambiguously correlated or resolved peaks were omitted during the transfer. We were unable to quantify chemical shift perturbation as a function of residue number as we have not obtained backbone assignments for the mutant. In the GDP-bound state of Gαi, a number of resonances in the Switch regions are not visible in the NMR spectrum due to the dynamic nature of these key regions, so these peaks cannot be highlighted.
Starting structural models and the details of molecular dynamics simulations.
The initial coordinates of the heterotrimeric stimulatory and inhibitory human G proteins, Gαs and Gαi1, were taken from their crystal structures (PDB IDs: 6EG8 and 1GP2 respectively) in the inactive GDP-bound states (Liu et al., 2019, Wall et al., 1995). The initial coordinates of both GTPγS nucleotide as well as the counterion Mg2+ were obtained from the crystal structures of the two G proteins in the active state (PDB IDs: 1AZT and 1GIA for Gαs and Gαi1 respectively) (Sunahara et al., 1997, Coleman et al., 1994). For building the GTPγS/Mg2+-bound heterotrimeric G proteins of each kind the two crystal structures of opposite states were overlaid and GDP/Mg2+ in the inactive states were swapped by the GTPγS/Mg2+ in the heterotrimeric states. We removed the N-terminus from both the GDP and the GTPγS bound G proteins since this region was found to be missing in the monomeric active state crystals of both G proteins. The GTPγS bound heterotrimeric G proteins of each kind were simulated to capture the early events of dissociation in the Gα and Gβ subunits upon replacing GDP with GTPγS. The GDP-bound G proteins in their resting (inactive) states were also simulated as controls. We used CHARMM-GUI (Jo et al., 2008) for creating input structures for simulating G proteins (Gs and Gi) in aqueous medium. The nucleotides (GDP and GTPγS) and the counterions (Mg2+) were parameterized using the CGENFF (Vanommeslaeghe et al., 2012) force field as implemented in CHARMM. Point mutations either disrupting or fostering the proposed triad network in G-proteins were incorporated using the MAESTRO software in Schrodinger (https://www.schrodinger.com/maestro). Each of these heterotrimeric protein-nucleotide complexes was solvated in explicit TIP3P water molecules in a cubic box (approximate dimension of 11.30 nm × 11.30 nm × 11.30 nm) separately and sodium and chloride counterions were added for maintaining the physiological salt concentration of each system at 150 mM. We used the software GROMACS (Hess et al., 2008) (version 2019.4) in combination with the all-atom CHARMM36 (Brooks et al., 2009) force field for performing MD simulations at 310 K coupled to a temperature bath with a relaxation time of 0.1 ps (Berendsen et al., 1984). Pressure was calculated using molecular virial and held constantly by weak coupling to a pressure bath with a relaxation time of 0.5 ps. Each system was first subjected to a 5000 step steepest descent energy minimization for removing bad contacts (Petrova and Solov’ev, 1997). Then, the systems were heated for 100 ps in steps of ramping up the temperature to 310 °K under constant temperature-volume ensemble (NVT). Equilibrium bond length and geometry of water molecules were constrained using the SHAKE algorithm (Andersen, 1983). We used a time step of 2 fs. The short range electrostatic and van der Waals (VDW) interactions were estimated per time step using a charge group pair list with cut-off radius of 8 Å between the centers of geometry of the charged groups. Long range VDW interactions were calculated using a cut-off of 14 Å and long-range electrostatic interactions were treated using the particle mesh Ewald (PME) method (Darden et al., 1993). Temperature was kept constant by applying the Nose-Hoover thermostat (Evans and Holian, 1985). Parrinello-Rahman barostat (Parrinello and Rahman, 1981) with a pressure relaxation time of 2 ps was used for attaining the desired pressure for all simulations. The simulation trajectories were saved each 200 ps for analysis. The protein, nucleotide and counterion atoms were position restrained using a harmonic force constant of 1000 kJ mol-1 nm-2 during the NVT equilibration stage while the water molecules were allowed to move freely around the protein. The system was further equilibrated using the constant pressure NPT ensemble by reducing the force constant on protein, counterion and nucleotide atoms from 5 kJ mol−1 nm−2 to zero in a gradual manner for 3 ns each while having the pressure coupling on. We also performed an additional 10 ns of unrestrained simulation before beginning the actual production run. This accounts for a total 25 ns of NPT equilibration prior to the production run. We performed three productions runs each 400 ns long starting from three independent sets of initial velocities for each system. Three independent simulations (each 400 ns long) were performed for each system. Thus, we had 1.2 μs long MD trajectory for each of the wildtype Gαs and Gαi1 bound to GDP and GTPγS, four Gαi1 mutants (R208S, R208Q, E245K and G203A) and four Gαs mutants (R231S, R231Q, E268K and G226A) bound to GTPγS. Full trajectories and analysis methods are available at https://welcome.gpcrmd.org/.
Calculation of root mean square fluctuations (RMSF).
Per residue RMSF for each Gαi1 system was computed using the gmx rmsf utility with -res option as implemented in GROMACS. The per residue RMSFs were inserted as B-factor columns of the representative snapshots of the Gα subunits of each Gαi1 system and shown as cartoons.
Calculation of interaction energy.
Short range electrostatic and van der Waals interactions were estimated per time step using a charge group pair list with cut-off radius of 8 Å between the centers of geometry of the charged groups. The residue pairs of the G proteins engaged in van der Waals or electrostatic interactions for > 40% of the whole simulation were considered as sustained contacts and were identified using the GetContacts script available at the GitHub (https://getcontacts.github.io/). These residues of interest were indexed and tagged as energy groups. The total non-bonded interactions energies (van der Waals + Coulombic) between two protein subunits, Gα and Gβ or between Gα and nucleotide were extracted using the gmx energy module of GROMACS. The energies were calculated for each snapshot and averaged over the last 300 ns in each of the three simulation runs for each system. For calculating the interaction energies between Switch II and Switch III regions we used the following definition of Switch II and Switch III regions. Residues Gly203 to Glu216 make up the Switch II region for Gαi1 and residues Gly226 to Asn239 for Gαs. Switch III is comprised of residues Glu236 to Asn255 for Gαi1 and Glu259 to Asn278 for Gαs.
Representative structure calculation from MD simulation trajectories.
Representative snapshots from the most occupied conformation cluster for each system were chosen for structure representation in the figures. The conformation clustering of the snapshots in the MD trajectories was done using the RMSD-based clustering method using the GROMACS modules gmx rms and gmx cluster with a 1.5 Å cutoff on the concatenated trajectory from three simulations (a total of 1.2 μs simulation time) for each system. The most representative structure of the most populated cluster was calculated as the frame that has the smallest RMSD to the center of this cluster conformation. Snapshots were rendered using VMD (Humphrey et al., 1996) and PYMOL (https://www.schrodinger.com/pymol).
Principal component analysis and free energy surface generation.
For each system in this study, we merged the three independent MD runs into one concatenated trajectory. We then performed the principal component analysis using the gmx covar module of GROMACS for each system using covariance matrix of the Cα atoms of all of the residues. The gmx sham module of GROMACS was used to compute the probability of the microstates and convert it to free energy. The free energy surface thus generated was projected on the principal component space covered by principal component 1 and 2 (PC1 and PC2). We then clustered the MD snapshots by the values of PC1 and PC2 using the in-house extractcluster.py script in MATLAB and calculated the population of each of these conformational clusters (Figure 4). The population of the most populated cluster 1 was compared across the various systems under consideration for Gαi1. The MD snapshots under each conformational cluster were concatenated to obtain trajectories for each of these clusters and then the VMD script helicity.tcl was used to calculate the helicity of the Switch II regions in Gαi1. The eigenvectors corresponding to higher PCs decay faster in variance hinting at their lower contributions in the overall atomic motions as compared to the first two eigenvectors in each of the system under consideration (Figure S4F, Supplemental Information). The cumulative fraction of variance in the eigenvalues of the PCs (Figure S4G, Supplemental Information) accounts for about 95% of the variance captured by the first ten PCs for each Gαi1 system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitation and statistical analysis (standard error or standard error of the mean, SEM) was done using GraphPad Prism. Independent (biological) experiments are parallel measurements of biologically distinct samples obtained on different days. Technical replicates (duplicate, triplicates) are repeated measurements of the same sample and represent random noise associated with protocols or equipment. For Biolayer Interferometry, baseline subtraction and Gβγ loading normalization were done in Excel. Kinetic Analysis was done using GraphPad Prism (Graphpad Software Inc., San Diego, CA). The net BRET was fit in Graphpad Prism.
OLIGONUCLEOTIDES
Table S1 (Related to Key Resources Table). Primers Masterlist.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-Flag | Sigma-Aldrich | F3165 |
| horseradish peroxidase-conjugated goat-anti-mouse | Jackson ImmunoResearch Laboratories | 62–6520 |
| Bacterial and Virus Strains | ||
| RIPL | Agilent | 230280 |
| Rosetta DE3 | Novagen | 70954 |
| High 5 | Thermo Fisher | B85502 |
| Sf9 | Thermo Fisher | 11496015 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ULP1 | Maly and Crowhurst, 2012) | N/A |
| Critical Commercial Assays | ||
| ECL-plus reagent | Life Technologies | 1705061 |
| coelentrazine 400a | Nanolight | 340 |
| Streptavidin (SA) Dip and Read Biosensors | ForteBio | 18–5019 |
| Deposited Data | ||
| NMR data | This paper | http://www.bmrb.wisc.edu |
| MDS full trajectories | This paper | https://welcome.gpcrmd.org/ |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae BY4741 bar1Δ | Research Genetics | 95401.H2 |
| Oligonucleotides | ||
| See Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pFUS1‐YeGFP3 | (Shellhammer et al., 2019) | N/A |
| pRS316-GPA1 | (Song et al., 1996) | N/A |
| pRS316-ADH-GPA1 | (Song et al., 1996) | N/A |
| pRS316-ADH-GPA1-Flag | (Cappell et al., 2010) | N/A |
| NTSR1 human | (Olsen et al., 2020) | N/A |
| ADRB2 human | (Olsen et al., 2020) | N/A |
| MOR1 human | (Olsen et al., 2020) | N/A |
| GNAI3-Rluc8 (Gαi3 fused to Rluc8) human | (Olsen et al., 2020) | N/A |
| GNAS-Rluc8 (Gαs fused to Rluc8) human | (Olsen et al., 2020) | N/A |
| GNAQ-Rluc8 (Gαq fused to Rluc8) human | (Olsen et al., 2020) | N/A |
| GB3 (Gβ3) human | (Olsen et al., 2020) | N/A |
| GNG9-GFP2 (Gγ9- fused to GFP) human | (Olsen et al., 2020) | N/A |
| PET-SUMO-Gαι1-Δ31 | (Maly and Crowhurst, 2012) | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| FLWinLab | (McEwen et al., 2001, Jones et al., 2012) | N/A |
| CHARMM-GUI | (Jo et al., 2008) | N/A |
| CHARMM36 | (Brooks et al., 2009) | N/A |
| MAESTRO | N/A | https://www.schrodinger.com/maestro |
| SHAKE | (Andersen, 1983) | N/A |
| GROMACS | (Hess et al., 2008) | N/A |
| GetContacts | N/A | https://getcontacts.github.io/ |
| VMD | (Humphrey et al., 1996) | N/A |
| PYMOL | N/A | https://www.schrodinger.com/pymol |
List of mutagenesis primers containing 5 bp 5’ to the codon and 20–30 bp 3’ to the codon.
Supplementary Material
HIGHLIGHTS.
Receptors promote GTP-GDP exchange and dissociation of G protein α and βγ subunits
An allosteric Gly-Arg-Glu (GRE) network links the γ phosphate of GTP to release of Gβγ
Gly-Arg-Glu mutations prevent subunit dissociation regardless of agonist or GTP binding
Gly-Arg-Glu mutations are responsible for human endocrine and neurological disorders
ACKNOWLEDGEMENTS
We thank Bryan Roth for comments and access to laboratory resources, as well as Peter Cooke for expert technical assistance. Funded by NIH grants R35-GM134962 (S.L.C.), R35-GM127303 (A.V.S.), R01-GM117923 (N.V.), R35-GM118105 (H.G.D), F31NS093917 (R.H.J.O.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
R.H.J.O., is a co-inventor of the TRUPATH technology and could receive royalties. This relationship has been disclosed to and is under management by UNC-Chapel Hill. The remaining authors declare no competing interests.
REFERENCES
- ANDERSEN HC 1983. Rattle: A “Velocity” Version of the Shake Algorithm for Molecular Dynamics Calculations. Journal of Computational Physics, 52, 24–34. [Google Scholar]
- APANOVITCH DM, IIRI T, KARASAWA T, BOURNE HR & DOHLMAN HG 1998. Second site suppressor mutations of a GTPase-deficient G-protein a- subunit. Selective inhibition of bg-mediated signaling. J Biol Chem, 273, 28597–602. [DOI] [PubMed] [Google Scholar]
- BERENDSEN HJC, POSTMA JPM, GUNSTEREN WFV, DINOLA A & HAAK JR 1984. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81, 3684–3690. [Google Scholar]
- BERGHUIS AM, LEE E, RAW AS, GILMAN AG & SPRANG SR 1996. Structure of the GDP-Pi complex of Gly203-->Ala gialpha1: a mimic of the ternary product complex of galpha-catalyzed GTP hydrolysis. Structure, 4, 1277–90. [DOI] [PubMed] [Google Scholar]
- BOURNE HR, KASLOW D, KASLOW HR, SALOMON MR & LICKO V 1981. Hormone-sensitive adenylate cyclase. Mutant phenotype with normally regulated beta-adrenergic receptors uncoupled with catalytic adenylate cyclase. Mol Pharmacol, 20, 435–41. [PubMed] [Google Scholar]
- BROOKS BR, BROOKS CL III, MACKERELL AD JR., NILSSON L, PETRELLA RJ, ROUX B, WON Y, ARCHONTIS G, BARTELS C, BORESCH S, CAFLISCH A, CAVES L, CUI Q, DINNER AR, FEIG M, FISCHER S, GAO J, HODOSCEK M, IM W, KUCZERA K, LAZARIDIS T, MA J, OVCHINNIKOV V, PACI E, PASTOR RW, POST CB, PU JZ, SCHAEFER M, TIDOR B, VENABLE RM, WOODCOCK HL, WU X, YANG W, YORK DM & KARPLUS M 2009. CHARMM: The biomolecular simulation program. Journal of Computational Chemistry, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAPPELL SD, BAKER R, SKOWYRA D & DOHLMAN HG 2010. Systematic analysis of essential genes reveals important regulators of G protein signaling. Mol Cell, 38, 746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHE T, ENGLISH J, KRUMM BE, KIM K, PARDON E, OLSEN RHJ, WANG S, ZHANG S, DIBERTO JF, SCIAKY N, CARROLL FI, STEYAERT J, WACKER D & ROTH BL 2020. Nanobody-enabled monitoring of kappa opioid receptor states. Nat Commun, 11, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG KY, RASMUSSEN SG, LIU T, LI S, DEVREE BT, CHAE PS, CALINSKI D, KOBILKA BK, WOODS VL JR. & SUNAHARA RK 2011. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature, 477, 611–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLEMAN DE, BERGHUIS AM, LEE E, LINDER ME, GILMAN AG & SPRANG SR 1994. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science, 265, 1405–12. [DOI] [PubMed] [Google Scholar]
- DARDEN T, YORK D & PEDERSEN L 1993. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98, 10089–10092. [Google Scholar]
- DE LEAN A, STADEL JM & LEFKOWITZ RJ 1980. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem, 255, 7108–17. [PubMed] [Google Scholar]
- DEVREE BT, MAHONEY JP, VELEZ-RUIZ GA, RASMUSSEN SG, KUSZAK AJ, EDWALD E, FUNG JJ, MANGLIK A, MASUREEL M, DU Y, MATT RA, PARDON E, STEYAERT J, KOBILKA BK & SUNAHARA RK 2016. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature, 535, 182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOWELL SJ & BROWN AJ 2009. Yeast assays for G protein-coupled receptors. Methods Mol Biol, 552, 213–29. [DOI] [PubMed] [Google Scholar]
- DRAPER-JOYCE CJ, KHOSHOUEI M, THAL DM, LIANG YL, NGUYEN ATN, FURNESS SGB, VENUGOPAL H, BALTOS JA, PLITZKO JM, DANEV R, BAUMEISTER W, MAY LT, WOOTTEN D, SEXTON PM, GLUKHOVA A & CHRISTOPOULOS A 2018. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature, 558, 559–563. [DOI] [PubMed] [Google Scholar]
- DROR RO, MILDORF TJ, HILGER D, MANGLIK A, BORHANI DW, ARLOW DH, PHILIPPSEN A, VILLANUEVA N, YANG Z, LERCH MT, HUBBELL WL, KOBILKA BK, SUNAHARA RK & SHAW DE 2015. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science, 348, 1361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS DJ & HOLIAN BL 1985. The Nose–Hoover thermostat. The Journal of Chemical Physics, 83, 4069–4074. [Google Scholar]
- FARFEL Z, IIRI T, SHAPIRA H, ROITMAN A, MOUALLEM M & BOURNE HR 1996. Pseudohypoparathyroidism, a novel mutation in the betagamma-contact region of Gsalpha impairs receptor stimulation. J Biol Chem, 271, 19653–5. [DOI] [PubMed] [Google Scholar]
- FLOCK T, HAUSER AS, LUND N, GLORIAM DE, BALAJI S & BABU MM 2017. Selectivity determinants of GPCR-G-protein binding. Nature, 545, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLOCK T, RAVARANI CNJ, SUN D, VENKATAKRISHNAN AJ, KAYIKCI M, TATE CG, VEPRINTSEV DB & BABU MM 2015. Universal allosteric mechanism for Galpha activation by GPCRs. Nature, 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHOSH M, PETERSON YK, LANIER SM & SMRCKA AV 2003. Receptor- and nucleotide exchange-independent mechanisms for promoting G protein subunit dissociation. J Biol Chem, 278, 34747–50. [DOI] [PubMed] [Google Scholar]
- GHOSH S, BIERIG T, LEE S, JANA S, LOHLE A, SCHNAPP G, TAUTERMANN CS & VAIDEHI N 2018. Engineering Salt Bridge Networks between Transmembrane Helices Confers Thermostability in G-Protein-Coupled Receptors. J Chem Theory Comput, 14, 6574–6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILMAN AG 1987. G proteins: transducers of receptor-generated signals. Annu Rev Biochem, 56, 615–49. [DOI] [PubMed] [Google Scholar]
- GORICANEC D, STEHLE R, EGLOFF P, GRIGORIU S, PLUCKTHUN A, WAGNER G & HAGN F 2016. Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc Natl Acad Sci U S A, 113, E3629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HESS B, KUTZNER C, VAN DER SPOEL D & LINDAHL E 2008. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput, 4, 435–47. [DOI] [PubMed] [Google Scholar]
- HOFFMAN G, GARRISON TR & DOHLMAN HG 2002. Analysis of RGS proteins in Saccharomyces cerevisiae. Methods Enzymol, 344, 617–631. [DOI] [PubMed] [Google Scholar]
- HOLLINS B, KURAVI S, DIGBY GJ & LAMBERT NA 2009. The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal, 21, 1015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUMPHREY W, DALKE A & SCHULTEN K 1996. VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- IIRI T, BELL SM, BARANSKI TJ, FUJITA T & BOURNE HR 1999. A Gsalpha mutant designed to inhibit receptor signaling through Gs. Proc Natl Acad Sci U S A, 96, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IIRI T, FARFEL Z & BOURNE HR 1997. Conditional activation defect of a human Gsalpha mutant. Proc Natl Acad Sci U S A, 94, 5656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISOM DG, SRIDHARAN V, BAKER R, CLEMENT ST, SMALLEY DM & DOHLMAN HG 2013. Protons as second messenger regulators of G protein signaling. Mol Cell, 51, 531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JO S, KIM T, IYER VG & IM W 2008. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem, 29, 1859–65. [DOI] [PubMed] [Google Scholar]
- JONES JC, JONES AM, TEMPLE BR & DOHLMAN HG 2012. Differences in intradomain and interdomain motion confer distinct activation properties to structurally similar Gα proteins. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOEHL A, HU H, MAEDA S, ZHANG Y, QU Q, PAGGI JM, LATORRACA NR, HILGER D, DAWSON R, MATILE H, SCHERTLER GFX, GRANIER S, WEIS WI, DROR RO, MANGLIK A, SKINIOTIS G & KOBILKA BK 2018. Structure of the micro-opioid receptor-Gi protein complex. Nature, 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSTENIS E, PFEIL EM & ANNALA S 2020. Heterotrimeric Gq proteins as therapeutic targets? J Biol Chem, 295, 5206–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOZASA T & GILMAN AG 1995. Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of alpha 12 and inhibition of adenylyl cyclase by alpha z. J Biol Chem, 270, 1734–41. [DOI] [PubMed] [Google Scholar]
- KOZASA T & GILMAN AG 1996. Protein kinase C phosphorylates G12 alpha and inhibits its interaction with G beta gamma. J Biol Chem, 271, 12562–7. [DOI] [PubMed] [Google Scholar]
- LAMBERT NA, JOHNSTON CA, CAPPELL SD, KURAVI S, KIMPLE AJ, WILLARD FS & SIDEROVSKI DP 2010. Regulators of G-protein signaling accelerate GPCR signaling kinetics and govern sensitivity solely by accelerating GTPase activity. Proc Natl Acad Sci U S A, 107, 7066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMBRIGHT DG, NOEL JP, HAMM HE & SIGLER PB 1994. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein [see comments]. Nature, 369, 621–8. [DOI] [PubMed] [Google Scholar]
- LAMBRIGHT DG, SONDEK J, BOHM A, SKIBA NP, HAMM HE & SIGLER PB 1996. The 2.0 A crystal structure of a heterotrimeric G protein [see comments]. Nature, 379, 311–9. [DOI] [PubMed] [Google Scholar]
- LEE E, TAUSSIG R & GILMAN AG 1992. The G226A mutant of Gs alpha highlights the requirement for dissociation of G protein subunits. J Biol Chem, 267, 1212–8. [PubMed] [Google Scholar]
- LIANG YL, KHOSHOUEI M, DEGANUTTI G, GLUKHOVA A, KOOLE C, PEAT TS, RADJAINIA M, PLITZKO JM, BAUMEISTER W, MILLER LJ, HAY DL, CHRISTOPOULOS A, REYNOLDS CA, WOOTTEN D & SEXTON PM 2018a. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature, 561, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG YL, KHOSHOUEI M, GLUKHOVA A, FURNESS SGB, ZHAO P, CLYDESDALE L, KOOLE C, TRUONG TT, THAL DM, LEI S, RADJAINIA M, DANEV R, BAUMEISTER W, WANG MW, MILLER LJ, CHRISTOPOULOS A, SEXTON PM & WOOTTEN D 2018b. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature, 555, 121–125. [DOI] [PubMed] [Google Scholar]
- LIU X, XU X, HILGER D, ASCHAUER P, TIEMANN JKS, DU Y, LIU H, HIRATA K, SUN X, GUIXA-GONZALEZ R, MATHIESEN JM, HILDEBRAND PW & KOBILKA BK 2019. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell, 177, 1243–1251 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAHONEY JP & SUNAHARA RK 2016. Mechanistic insights into GPCR-G protein interactions. Curr Opin Struct Biol, 41, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALY J & CROWHURST KA 2012. Expression, purification and preliminary NMR characterization of isotopically labeled wild-type human heterotrimeric G protein alphai1. Protein Expr Purif, 84, 255–64. [DOI] [PubMed] [Google Scholar]
- MASUHO I, OSTROVSKAYA O, KRAMER GM, JONES CD, XIE K & MARTEMYANOV KA 2015. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal, 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCEWEN DP, GEE KR, KANG HC & NEUBIG RR 2001. Fluorescent BODIPY-GTP analogs: real-time measurement of nucleotide binding to G proteins. Anal Biochem, 291, 109–17. [DOI] [PubMed] [Google Scholar]
- MILLER RT, MASTERS SB, SULLIVAN KA, BEIDERMAN B & BOURNE HR 1988. A mutation that prevents GTP-dependent activation of the alpha chain of Gs. Nature, 334, 712–5. [DOI] [PubMed] [Google Scholar]
- MIXON MB, LEE E, COLEMAN DE, BERGHUIS AM, GILMAN AG & SPRANG SR 1995. Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis [see comments]. Science, 270, 954–60. [DOI] [PubMed] [Google Scholar]
- OLSEN RHJ, DIBERTO JF, ENGLISH JG, GLAUDIN AM, KRUMM BE, SLOCUM ST, CHE T, GAVIN AC, MCCORVY JD, ROTH BL & STRACHAN RT 2020. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARRINELLO M & RAHMAN A 1981. Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52, 7182–7190. [Google Scholar]
- PETROVA SS & SOLOV’EV AD 1997. The Origin of the Method of Steepest Descent. Historia Mathematica, 24, 361–375. [Google Scholar]
- RASMUSSEN SG, DEVREE BT, ZOU Y, KRUSE AC, CHUNG KY, KOBILKA TS, THIAN FS, CHAE PS, PARDON E, CALINSKI D, MATHIESEN JM, SHAH ST, LYONS JA, CAFFREY M, GELLMAN SH, STEYAERT J, SKINIOTIS G, WEIS WI, SUNAHARA RK & KOBILKA BK 2011. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature, 477, 549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALOMON MR & BOURNE HR 1981. Novel S49 lymphoma variants with aberrant cyclic AMP metabolism. Mol Pharmacol, 19, 109–16. [PubMed] [Google Scholar]
- SENEVIRATNE AM, BURROUGHS M, GIRALT E & SMRCKA AV 2011. Direct-reversible binding of small molecules to G protein betagamma subunits. Biochim Biophys Acta, 1814, 1210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHELLHAMMER JP, POMEROY AE, LI Y, DUJMUSIC L, ELSTON TC, HAO N & DOHLMAN HG 2019. Quantitative analysis of the yeast pheromone pathway. Yeast, 36, 495–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONDEK J, LAMBRIGHT DG, NOEL JP, HAMM HE & SIGLER PB 1994. GTPase mechanism of G proteins from the 1.7-A crystal structure of transducin a-GDP-AIF-4. Nature, 372, 276–9. [DOI] [PubMed] [Google Scholar]
- SONG J, HIRSCHMAN J, GUNN K & DOHLMAN HG 1996. Regulation of membrane and subunit interactions by N-myristoylation of a G protein a subunit in yeast. J Biol Chem, 271, 20273–83. [DOI] [PubMed] [Google Scholar]
- STRATTON HF, ZHOU J, REED SI & STONE DE 1996. The mating-specific Ga protein of Saccharomyces cerevisiae downregulates the mating signal by a mechanism that is dependent on pheromone and independent of Gbg sequestration. Mol Cell Biol, 16, 6325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN D, FLOCK T, DEUPI X, MAEDA S, MATKOVIC M, MENDIETA S, MAYER D, DAWSON R, SCHERTLER GFX, MADAN BABU M & VEPRINTSEV DB 2015. Probing Galphai1 protein activation at single-amino acid resolution. Nat Struct Mol Biol, 22, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNAHARA RK, TESMER JJ, GILMAN AG & SPRANG SR 1997. Crystal structure of the adenylyl cyclase activator Gsalpha. Science, 278, 1943–7. [DOI] [PubMed] [Google Scholar]
- VAIDEHI N, GRISSHAMMER R & TATE CG 2016. How Can Mutations Thermostabilize G-Protein-Coupled Receptors? Trends Pharmacol Sci, 37, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN EPS N, PREININGER AM, ALEXANDER N, KAYA AI, MEIER S, MEILER J, HAMM HE & HUBBELL WL 2011. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci U S A, 108, 9420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANOMMESLAEGHE K, RAMAN EP & MACKERELL AD JR. 2012. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model, 52, 3155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALL MA, COLEMAN DE, LEE E, INIGUEZ-LLUHI JA, POSNER BA, GILMAN AG & SPRANG SR 1995. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell, 83, 1047–58. [DOI] [PubMed] [Google Scholar]
- WESTFIELD GH, RASMUSSEN SG, SU M, DUTTA S, DEVREE BT, CHUNG KY, CALINSKI D, VELEZ-RUIZ G, OLESKIE AN, PARDON E, CHAE PS, LIU T, LI S, WOODS VL JR., STEYAERT J, KOBILKA BK, SUNAHARA RK & SKINIOTIS G 2011. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci U S A, 108, 16086–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from H.G.D. and N.V. The study did not generate new unique code. MD simulations full trajectories will be made available at https://welcome.gpcrmd.org/. NMR data will be made available at: http://www.bmrb.wisc.edu.