

Abstract
While opioids constitute the major component of perioperative analgesic regimens for surgery in general, a variety of evidence points to an association between perioperative opioid exposure and longer-term oncological outcomes. The mechanistic details underlying these effects are not well understood. In this study, we focused on clear cell renal cell carcinoma (ccRCC) and utilized RNAseq and outcomes data from both TCGA as well as a local patient cohort to identify survival-associated gene coexpression networks. We then projected drug-induced transcriptional profiles from in vitro cancer cells to predict drug effects on these networks and recurrence-free, cancer-specific, and overall survival. The opioid receptor agonist leu-enkephalin was predicted to have anti-survival effects in ccRCC, primarily through Th2 immune and NRF2-dependent macrophage networks. Conversely, the antagonist naloxone was predicted to have pro-survival effects, primarily through angiogenesis, fatty acid metabolism, and hemopoesis pathways. Eight coexpression networks associated with survival endpoints in ccRCC were identified, and master regulators of the transition from the normal to disease state were inferred, a number of which are linked to opioid pathways. These results are the first to suggest a mechanism for opioid effects on cancer outcomes through modulation of survival-associated coexpression networks. While we focus on ccRCC, this methodology may be employed to predict opioid effects on other cancer types and to personalize analgesic regimens in cancer patients for optimal outcomes.
Keywords: Opioids, Cancer, Transcription, Surgery, Renal Carcinoma
INTRODUCTION
Retrospective clinical evidence suggests that perioperative opioid exposure may be associated with cancer recurrence and survival(1–4), and its specific effects are likely cancer-specific. For instance, while opioids are thought to have a negative effect on immune-mediated cancers(5) like lung adenocarcinoma(6) and renal cell carcinoma(7), they may play a protective role in esophageal cancer(8). Despite numerous studies indicating an association, causal evidence and biological rationale are both lacking in human populations.
Molecular evidence links opioids to pathways known to influence cancer. Opioids are immunomodulators(9), and a growing literature indicates the therapeutic importance of immune regulation and antigen presentation in mediating cancer progression(10). Both cancer cells and immune cells express opioid receptors(11), and activation of these receptors can alter cell-cycle pathways. Opioid receptor gene mutation and expression have been correlated with survival endpoints in a few cancer types(12–14). These experiments provide intriguing evidence for the functional impact of opioids on receptor-dependent cascades and suggest that these drugs may have broad effects across the cancer transcriptome.
This study tests the hypothesis that opioids modulate survival-associated coexpression networks in clear cell renal cell carcinoma (ccRCC). Kidney cancer is the seventh most commonly diagnosed solid tumor in the United States(15), with clear cell being its most common subtype. Genetic and molecular changes are associated with survival(16), and gene network expression changes distinguish renal cell carcinoma subtypes(17). Opioids play a critical role in perioperative analgesia in oncologic surgery, and recent in vitro evidence implicates opioids in proliferation, invasion and metastasis of ccRCC specifically(18). We study RNAseq and cancer-specific outcomes data in both the TCGA KIRC cohort and an independent ccRCC replication cohort at Memorial-Sloan Kettering Cancer Center (MSKCC) who underwent nephrectomy. We construct undirected and directed gene networks and correlate each with recurrence-free survival (RFS), cancer-specific survival (CSS), and overall survival (OS). We compare the expression and network connectivity of opioid- and survival-related pathways between ccRCC and controls. We also project drug-induced transcriptional profiles from in vitro cancer cells onto ccRCC gene networks to characterize pathways through which opioids may influence survival.
MATERIALS AND METHODS
Measuring gene expression and clinical outcomes in human cohorts
This study uses data from publicly available TCGA cohort, as well as an independent cohort who underwent nephrectomy at Memorial Sloan Kettering Cancer Center. The Firebrowse and LinkedOmics portals were used to access TCGA-KIRC data(19). Level 3 normalized RSEM gene expression was extracted for cases and controls, as well as relevant clinical measures and metadata. For the MSKCC validation cohort, gene expression was measured using RNA-sequencing (See Supplemental Methods). Cancer-specific and recurrence-free survival were calculated by extracting relevant clinical data from internal clinical records at MSKCC and linking them with previous data contributions to the larger TCGA cohort.
Analyzing gene and network level variation in RNAseq data
Differential gene expression was calculated in a subpopulation (N=72) for which ccRCC and neighboring healthy renal tissue were biopsied and sequenced. Median expression was calculated for each gene and genes in the bottom 20% were filtered out to remove bias from genes with low gene expression. Voom was used to estimate differential expression (See Supplemental Methods). The Kruskal Wallis test was used to associate pathologic stage and opioid pathway gene expression for samples with high and low opioid pathway expression. Samples with high and low opioid gene expression were defined for each gene as those samples in the top and bottom quartile of gene expression and expression-stage associations were determined for these sample subsets.
To estimate gene coexpression in the TCGA KIRC and BLCA cohorts, gene expression data was log2 transformed, and linear regression was used to correct gene expression for age, race, gender, and tumor purity. Genes in the bottom 20% percentile in variance and median expression were filtered to reduce noise, and samples with an interarray correlation greater than two standard deviations away from the mean were considered outliers and removed. Weighted gene coexpression analysis was used to determine correlation gene networks (See Supplemental Methods). The first principal component of each module was calculated (“module eigengene”) and univariate and multivariate Cox model were used to correlate eigengene expression with overall survival, cancer-specific survival, and recurrence-free survival. For multivariate testing, two models were used for the sake of comprehensiveness: one that included age, race, gender, and tumor purity, and a second model that included the aforementioned covariates, as well as stage.
Internal and external validation of gene networks
The robustness of each module was empirically calculated by repeatedly splitting the gene expression data in training and tests sets and calculating a module preservation score between each new network(20). A composite preservation statistic (Z) was calculated by integrating several measures of connectivity and network preservation and previously characterized threshold (Z>10) was used to assess for preservation. Empirical p-values were also calculated and Bonferroni p-value threshold < 0.05 was used to confirm these results. Previous evidence suggests that Z scores and p values have a strong inverse correlation, so this approach simply utilizes two statistics that reflect the same measure of robustness. As an external validation, gene expression was measured in an independent cohort and corrected for batch, sex, and age using linear regression. Gene coexpression was calculated independently in this cohort as previously described, without explicit reference or parameterization from the TCGA population. Module membership was directly compared between TCGA-KIRC and MSKCC-KIRC networks, and Fisher’s exact test was used to calculate enrichment. Modules were considered preserved if enrichment odds ratio > 1 and Bonferroni p-value < 0.05.
Calculating enrichment and connectivity measures in differentially expressed genes and networks
The R package, goSeq, was used to estimate biologic pathway enrichment for differentially expressed genes, with gene length bias correction and multiple testing correction. The anRichment R package was used to estimate gene ontology enrichment for each module, and p-values were corrected with the Benjamini-Hochberg method. Enrichr was used to calculate module overrepresentation for experimental datasets, including ChIP-seq and gene knockout data, for a variety of cell lines and animal models, and Q values were calculated to account for multiple hypothesis testing(21). Previously published immune cell type signatures were also used for enrichment testing(17). Module enrichment for differentially expressed genes, immune signatures, and previously published ccRCC networks was estimated with Fisher’s exact test and p-values were corrected with the Bonferroni method when appropriate. ICGC Data Portal was used to identify mutational burden and frequency in the Reactome pathway(22).
Differential network connectivity was calculated by comparing the mean intramodular connectivity for each disease network with those same network genes in the control cohort(23). The ratio of average network connectivity between cases and controls was used as an estimate of differential connectivity. For example, a measure of 2 signifies that the average correlation strength for a group of genes in a network is two times greater in disease than in controls. We estimated two separate false discovery rates (FDR) by randomly shuffling samples and genes of disease and control networks. Shuffling samples creates networks with random edges and shuffling genes creates networks with random nodes. We quantified the final FDR by selecting the larger estimate and used a conservative FDR threshold to assess significance (FDR < 0.001).
Projecting drug-induced transcriptional profiles onto survival networks
Connectivity scores were calculated between network hubs and drug-induced transcriptional profiles for leu-enkephalin, naloxone, and the VEGF-inhibitor class. The drug profiles were catalogued by Connectivity Map and the CLUE Research platform was used to calculate connectivity scores(24). Connectivity Map has catalogued gene expression profiles for thousands of chemical and genetic perturbations across nine cell lines, and connectivity scores between all reference perturbations were calculated based on a weighted Kolmogorov Smirnov statistic, normalized for cell line and perturbation type(25). A non-parametric weighted connectivity score and an enrichment score, τ, was then calculated for each module-drug pair of interest, ranging from −100 to +100(26). τ measures the fraction of reference connectivity scores greater than the tested module-drug pair. A positive score shows that hub expression and drug-induced expression are in the same direction, while negative scores reflect expression in the opposite direction. A score of 90 indicates that only 10% of the reference set had a stronger score. Unlike a null distribution generated by random permutation, this empirical test avoids strong assumptions about the distribution of gene expression data under perturbed conditions. Instead, it tests module-drug connectivity directly against an expansive and diverse gene set under biologic and pharmacologic perturbation and provides a useful corresponding effect size. Empirical validation has demonstrated that τ > |90| also pass p-value and FDR thresholds < 0.05 based on permutation-based null distribution methods, but lower τ estimates may also pass those thresholds(26). Hubs positively correlated with survival were considered upregulated and hubs negatively correlated with survival were considered downregulated. Each module was studied independently and each gene in the corresponding hub set was considered as a binary, either upregulated or downregulated. Drug-hub pairs with τ > +90 indicate pro-survival relationship in hubs positively correlated with survival. Drug-hub pairs with τ > +90 indicate anti-survival relationship in hubs negatively correlated with survival.
Calculating master regulators of directed gene-gene networks
Directed transcriptional relationships were retrieved using the “aracne.network” R library, derived from the ARACNe algorithm. ARACNe first calculates pairwise gene expression mutual information to identify candidate relationship and then uses data processing inequality to trim edges representing indirect relationships between genes that are strongly co-regulated without being directly dependent(27). This two-step procedure recovers gene expression dependencies with high fidelity. Directed networks describing relationships between modules were constructed by calculating edges between genes in different modules. For each pair of modules, edge weight was calculated by summing the total number of edges between the module pair, scaled by the product of their respective module sizes. Permutation testing (N=1000) was performed to calculate a null distribution of edge weights between each pair of modules, and an edge was only kept if Benjamini-Hochberg p value < 0.05. Within each module, master regulators were inferred using MARINa, leveraging a phenotype transition signature derived from t-test analysis comparing gene expression between cases and controls(28).
RESULTS
Characterizing opioid pathway gene expression changes in clear cell renal cell carcinoma
We first characterized gene expression changes associated with ccRCC across the transcriptome. Using TCGA data, we compared tissue from ccRCC with adjacent normal renal tissue in 72 individuals (Figure 1A). Six thousand three hundred twenty one genes (6,321) were upregulated and six thousand three hundred sixty six (6,366) genes were downregulated (P < 0.05). Cell migration, biologic adhesion, and immune regulation gene ontology pathways are most robustly overrepresented in these differentially expressed genes (P < 0.05, Figure S1), consistent with previous literature. Notably, many genes involved in opioid metabolism, regulation, and signaling are also differentially expressed in ccRCC (Figure 1B–1F), including IL4R, OGFR, OPRL1, OGFRL1, ARRB1, ARRB2, POMC, FOS, CYP3A4, PTGS2, and BDNF (p < 0.05). Given the differential expression of several opioid-related genes, we specifically investigated enrichment testing for the Reactome opioid signaling pathway and found suggestive evidence for its overrepresentation in differentially expressed genes (Fisher’s exact test, nominal P = 0.03, OR = 1.7(1.04–2.81)). Further analysis also demonstrates that sixty-two of the top one hundred genes predicted to be functionally associated with opioids(29) are also differentially expressed. Opioid signaling is not nearly as overrepresented as large pathways more proximal to pathogenesis, like immune regulation and cell migration, but these analyses provide evidence for its association with ccRCC. Canonical opioid receptor genes OPRM1, OPRD1, OPRK1 are all poorly expressed in renal tissue and there is no evidence in our analysis that they are differentially expressed in ccRCC, though the expression of each shows greater variability in the disease state (Figure S2). At the same time, OPRL1 (nociception receptor), OGFR, and TLR4 receptor genes are upregulated in ccRCC (Figure 1C–1F). These receptors are known to bind opioids and are implicated in ccRCC progression, in which OGFR signaling is likely protective and TLR4 contributes to cancer pathogenesis(30,31).
Figure 1. Differential expression of opioid pathway genes in clear cell renal cell carcinoma.
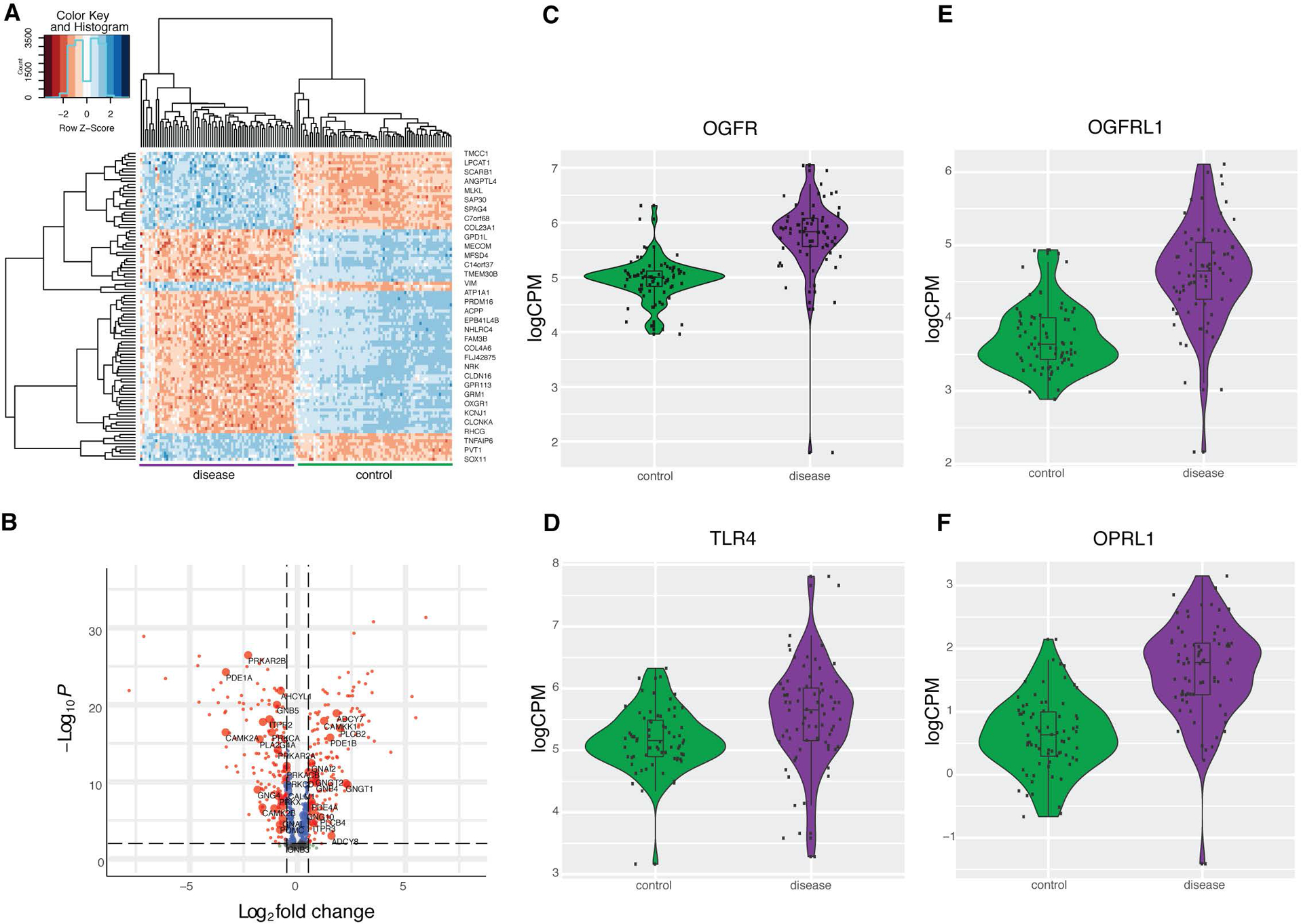
(A) Top fifty differentially expressed genes between cases and controls in clear cell renal cell carcinoma, clustered by gene expression. Shades of red represents lower expression, and blue expression represents greater expression. (B) P value and log2 fold change for a subset of five hundred differentially expressed genes. Horizontal dotted line represents P threshold of 0.001, and the vertical dotted line represents fold change threshold of 0.5. Genes with a log2 fold change > 0.5 are labeled in red and log2 fold change < 0.5 are labeled in blue. Genes in the opioid signaling pathway are represented by large labeled nodes. (C-F) Comparing gene expression distributions between cases and controls for OGFR, OGFRL1, TLR4, OPRL1, all of which P < 0.05.
An analysis of mutational burden in ccRCC in the TCGA cohort provides further orthogonal evidence of the relationship between ccRCC and opioid signaling. This study identified 149 mutations across 76% (67/88) of opioid signaling pathways genes in ccRCC (Supplementary Table S1), but the functional impact and clinical significance of most of these mutations are unknown. Further analysis showed that pathologic stage is associated with whether the sample is high or low expression of particular Reactome opioid signaling pathway genes (P < 0.05, Figure S3), suggesting tumor heterogeneity in opioid pathway expression may be associated with clinical features.
Calculating ccRCC gene networks relevant to survival
Gene expression is organized into networks that can respond to genetic, pharmacologic, and environmental perturbations. We identified gene networks in renal cell carcinoma (using the N=533 individuals in the TCGA KIRC data set) using weighted gene coexpression network analysis (WGCNA)(32). This analysis revealed 15 distinct gene networks, each labeled by an arbitrary color (Figure 2A). Module membership is reported in Supplementary Table S2. We used a resampling technique to confirm these networks were internally robust and reproducible (Figure 2B)(20). Lastly, we showed that these gene networks map onto known functional pathways, confirming their biologic coherence (Supplementary Table S3).
Figure 2. Characterization of the 15 gene coexpression networks in ccRCC and association with survival endpoints.
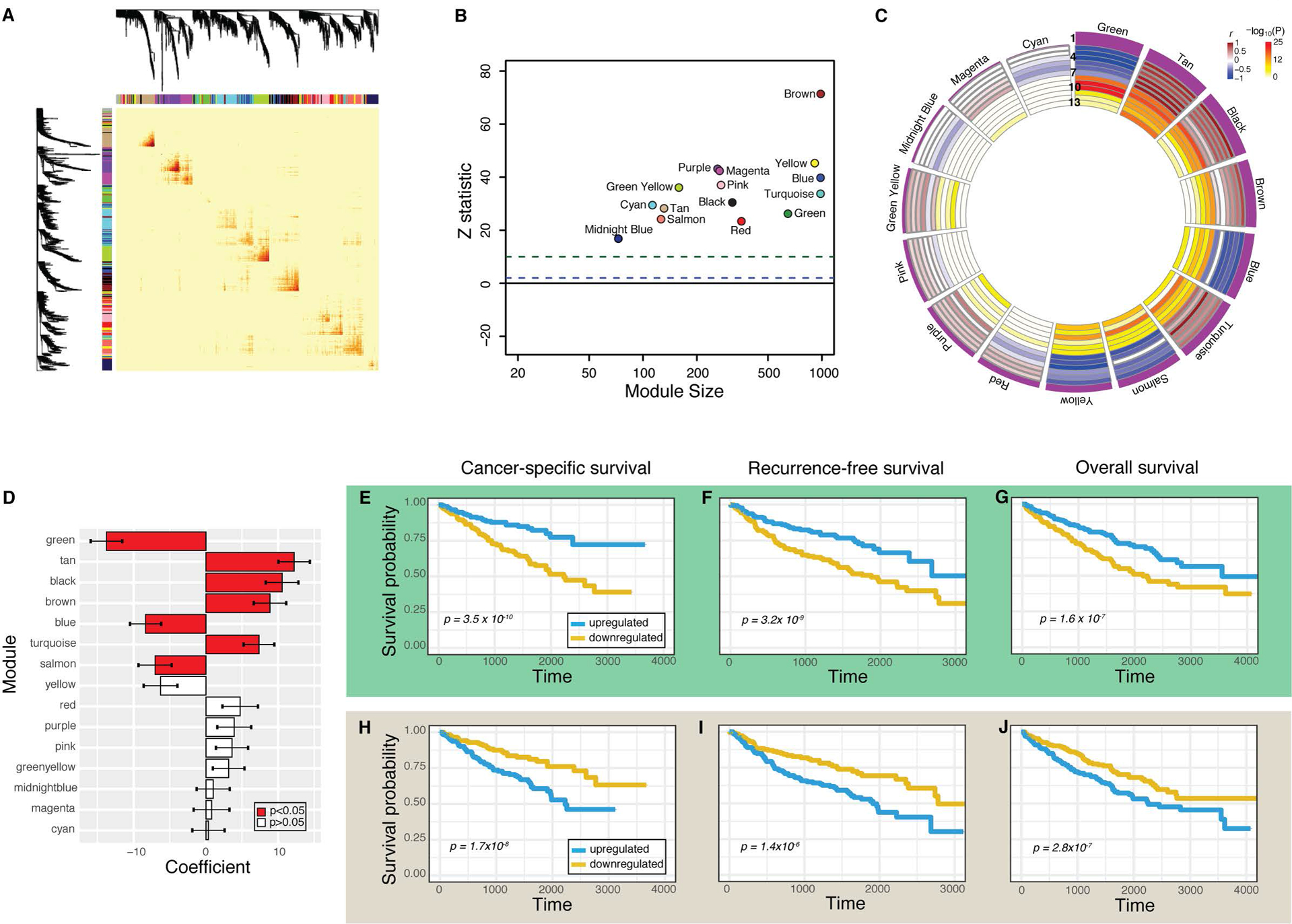
(A) Topological overlap matrix plot depicts gene coexpression. Darker yellow and red represents stronger correlations between genes. (B) Z statistic represents reproducibility of each module. Z > 10 (green dotted line) represents strong evidence of robustness and Z >1 (blue dotted line) represents weak evidence. (C) Circos plot depicts module eigengene correlations with survival and pathology measurements. Modules are ranked by the strength of their association with cancer-specific survival, reflected by the height of the purple histogram in Row 1. Rows 2–4 reflect the Cox model beta coefficient for overall survival, cancer-specific survival, and recurrence-free survival, respectively. Rows 5–7 reflect rho values for T, N, and M stage, respectively. Bluer values reflect negative values, while brown values reflect strong positive values. Rows 8–13 depict the −log10(P) values for the same survival (8–10) and pathology variables (11–13). (D) Cox model beta coefficient and 95% confidence interval for each module and its association with cancer-specific survival. Red bars depict P < 0.05. (E-J) Survival curves comparing individuals with upregulated (blue) and downregulated (gold) network expression for green module (E-G) and tan module (H-J). Cancer-specific survival (E,H), recurrence-free survival (F, I), and overall survival (G,J) are depicted, and their respective Cox P values are each less than 0.05.
Next, we hypothesized that a subset of these networks may be associated with survival. To test this hypothesis, we estimated the relationship between recurrence-free survival, cancer-specific survival, and overall survival, and pathologic characteristics to the first principal component (“eigengene”) of each module, which captures the predominant variation of gene expression of each respective network (Figure 2C, 2D). Each individual sample exhibited very high or low expression in relatively few modules, and eigengenes show modest correlation across networks (Figure S4A–C). We reasoned that increased or decreased expression in a gene network may be related to the length of survival. We analyzed these data using the Cox proportional hazard model, accounting for multiple hypothesis testing. Univariate and multivariate modeling showed that eight networks were associated with survival (P < 0.05), seven of which were also associated with cancer-specific survival (P < 0.05) and recurrence-free survival (P < 0.05) (Figure 2D–2J, Supplementary Table S4). Notably, many of these networks were also associated with tumor stage and grade as expected (Figure 2C) and Fisher’s exact testing of sample subgroups corroborated this finding, showing samples with high eigengene expression were associated with stage (Supplementary Table S5). These results reflect the well-known strong correlation between stage, grade, and survival in ccRCC. Lastly, we validated that each of these 8 networks was reproduced in an independent cohort of individuals (N=34) with ccRCC from Memorial-Sloan Kettering Cancer Center (ccRCC-MSKCC) (Figures S5 and S6A–H).
Tumor microenvironment and oncogenesis pathways associated with survival-associated gene networks
To examine the relationship between ccRCC pathogenesis and survival-related networks, we integrated our network analysis with publicly available experimental genomic, ChIP-seq, gene expression, and gene ontology data(21). Survival networks include modules overrepresented for gene signatures related to T helper type 2 cells (“tan”), angiogenesis (“yellow”), fatty acid metabolism (“green”), and mitochondrial ATP synthesis (“turquoise”), pathways significantly altered in ccRCC. CSS- and RFS-networks are also downstream targets for known ccRCC transcriptional regulators, including NRF2 (“green”, q=5.4×10–7, OR=4.6), JUN (“black”, q=1.7×10–4, OR=1.6), JAK2 (“brown”, q=1.6×10–4, OR=2.3), and MET (“blue”, q=4.9×10–4, OR=1.83). Survival-related networks also include approximately 40% (9 out of 22) of the intOGen RCC mutational driver genes(33). Notably, all survival networks are strongly overrepresented for differentially expressed genes, and four networks (black, brown, salmon, yellow) also gain stronger connections in ccRCC compared to controls (Figure 3A). Together, these results show that the survival-associated networks are intimately tied to oncogenesis, progression, and pathophysiology in ccRCC. We further probed possible localization of these networks within the tumor microenvironment by examining each network for enrichment of 24 empirically derived cell-type specific gene markers(17). These analyses revealed that the “green” network is strongly associated with macrophage-specific signatures, the “tan” network is overrepresented with genes specific to Th2 cells, and the yellow network is associated with B cells T follicular helper cells (P < 0.05, Figure S7A–D)
Figure 3. Prediction of drug effects on the survival-associated networks.
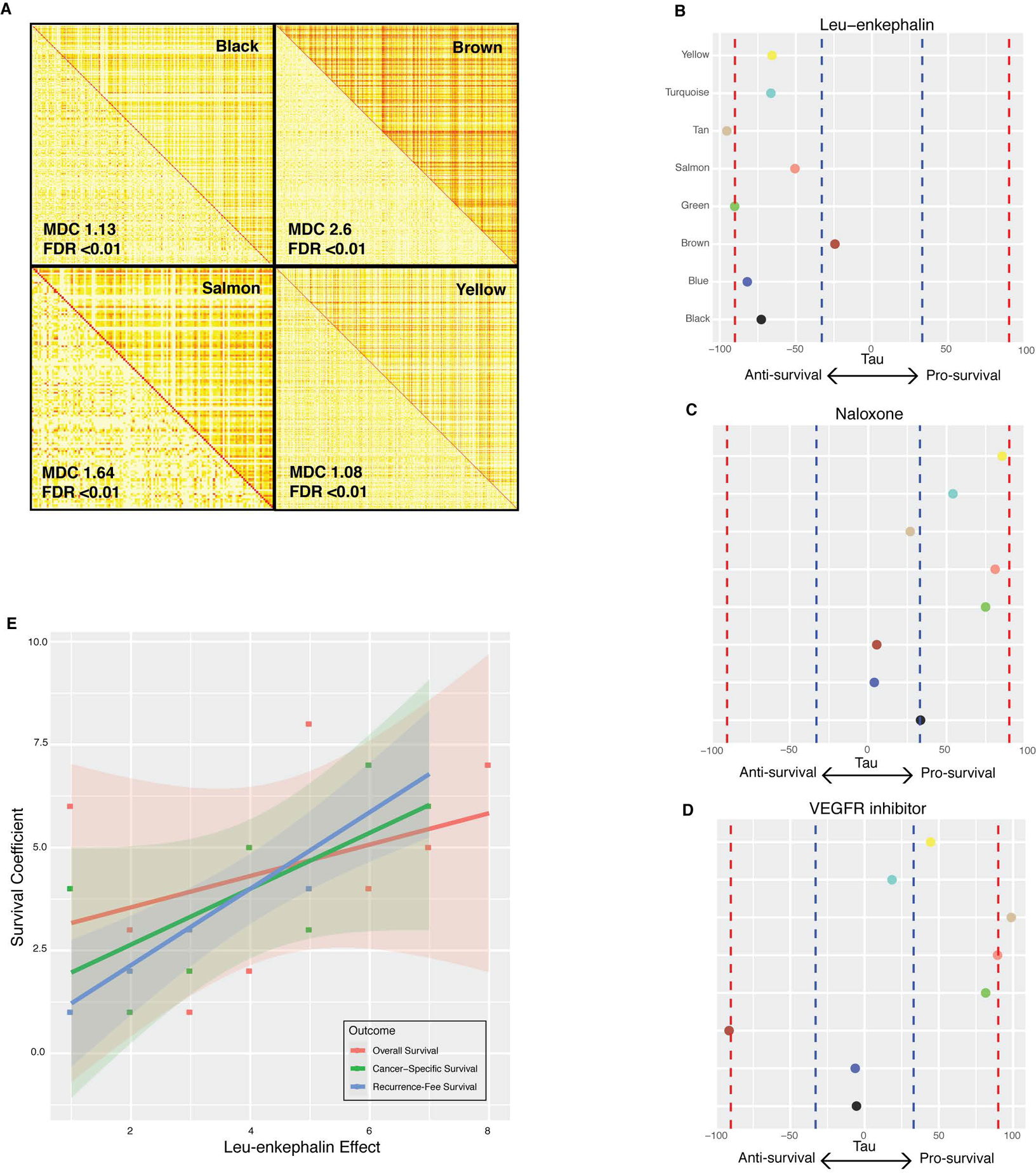
. (A) A comparison of topological overlap matrices in cases (top right triangle) versus controls (bottom left triangle) for four modules. Greater coexpression is colored in dark yellow and red, while less coexpression is colored in light yellow and white. Module differential connectivity (MDC) and FDR values are depicted for each module. Differential connectivity was considered significant by FDR < 0.01. (B-D) Tau scores representing modulation of each survival network by leu-enkephalin (B), naloxone (C), and VEGF-inhibitor (D). Red dotted lines represent strong evidence of drug modulation, |tau| > 90. Blue dotted lines represent suggestive evidence, |tau| >30. (E) The association between leu-enkephalin tau score and Cox model survival coefficients for overall survival, recurrence-free survival, and cancer-specific survival.
Leu-enkephalin modulates gene networks relevant to cancer-specific survival
Next, we examined whether opioid receptor agonism and antagonism affect the expression of these survival-relevant networks. First, we calculated intramodular connectivity and identified hub genes for each of the eight networks (Supplementary Table S6). Hub genes are the most highly connected nodes in each network, making them potent targets and important predictors of disease(34,35). We hypothesized that leu-enkephalin, a non-selective opioid receptor agonist, would downregulate pro-survival gene network hubs and upregulate anti-survival network hubs. To perform this analysis, we projected gene expression changes induced by all compounds catalogued by Connectivity Map onto each network(24). This analysis showed that leu-enkephalin has significant anti-survival effects on seven survival-related networks, upregulating modules negatively correlated with survival and downregulating modules positively correlated with survival (Figure 3B). Leu-enkephalin most strongly modulates the Th2 immune network (“tan”) and NRF2-dependent macrophage network (“green”) (τ >= |90|).
Next, we examined the molecular effects of naloxone, hypothesizing that opioid receptor antagonism would shift networks towards pro-survival expression (Figure 3C). This analysis showed that the effect of naloxone on survival-related networks opposes that of leu-enkephalin. It most strongly influences angiogenesis (“yellow”), NRF2-dependent macrophage network (“green”), and hemopoesis (“salmon”) networks and drives them towards pro-survival expression patterns, though naloxone’s effect doesn’t pass strict statistical threshold. As a positive control, we investigated the effect that VEGF receptor inhibitors, a pharmacologic class shown to have some survival benefits in renal cell carcinoma patients. As anticipated, it has strong pro-survival effects on Th2 immunity (“tan”, τ >= |90|) and hemopoesis (“salmon”, τ >= |90|), and a moderate effect on NRF2-dependent macrophage network (“green”, τ > |80|). Interestingly, it also has a strong anti-survival effect on the brown network (τ >= |90|) and minimal effect on three networks, pointing to a potential mechanism for its therapeutic limitations (Figure 3D).
Lastly, we tested the hypothesis that leu-enkephalin preferentially affects networks more strongly associated with survival (Figure 3E). The leu-enkephalin effect size is positively correlated with Cox model regression coefficients for survival-associated networks and significantly associated with recurrence-free survival (rho=0.93, P=0.007). This suggests that the clinical relationship between opioids and ccRCC survival may depend on the preferential effect of opioids on networks most strongly associated with survival.
In order to further validate these results, we applied this methodology to bladder cancer, where evidence also suggests that opioids promote tumor progression(36). Bladder cancer is a relevant comparison to ccRCC given that it is also a urological cancer where opioid excretion in urine could enable direct effects on tumorigenesis. Similar analysis in the TCGA BLCA cohort identified that one network (“pink”) was associated with overall survival. Leu-enkephalin showed anti-survival expression effects on the pink network, while naloxone and numerous chemotherapeutic drugs and classes showed pro-survival expression effects (Figure S8A–B).
Reconstructing directed transcriptional networks and master regulators of RFS, CSS, and OS
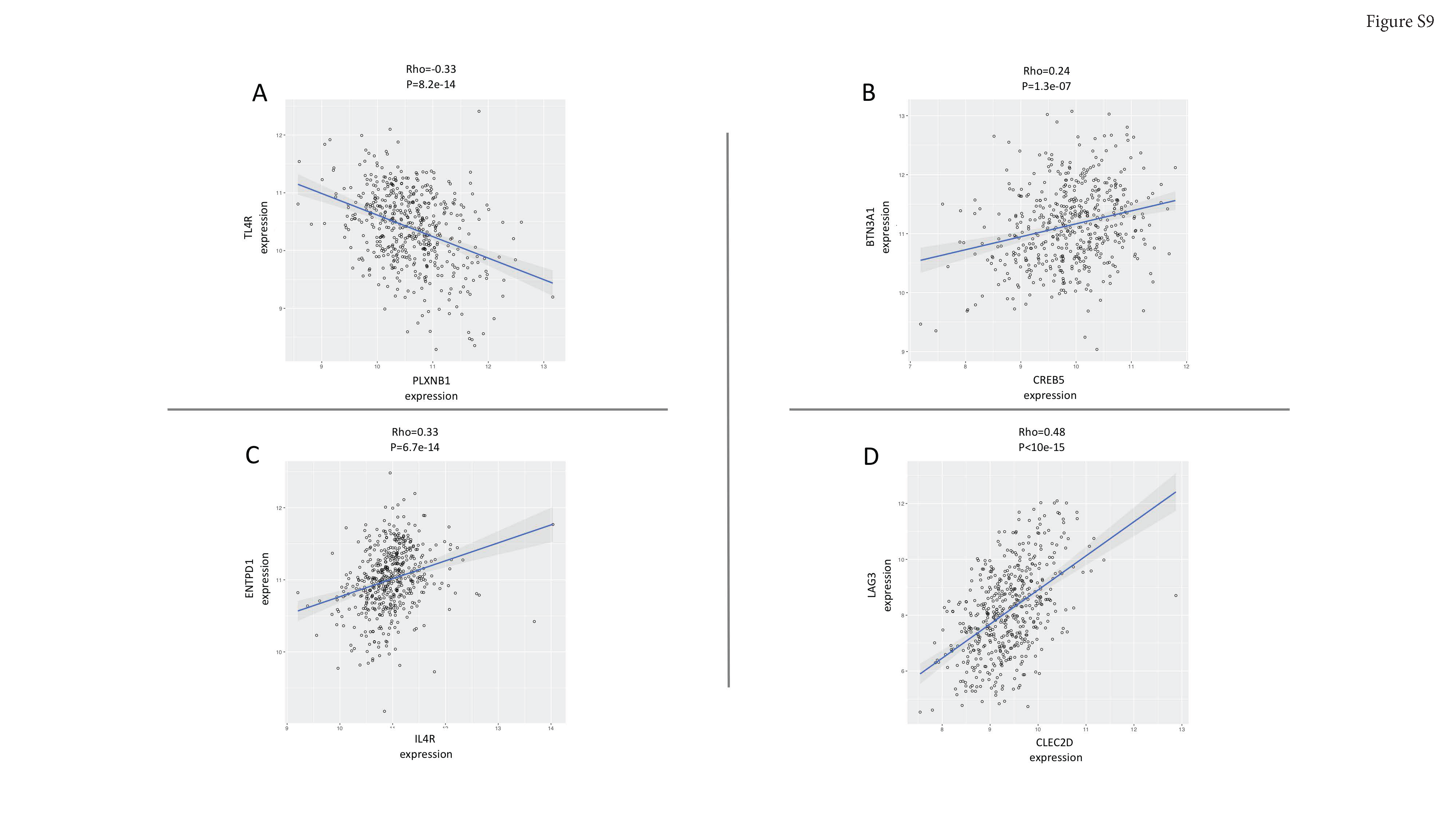
Finally, we reasoned that causal networks regulators may provide mechanistic and functional insight into the downstream transcriptional effects of opioids. An information theoretic approach was used to calculate direct transcriptional relationships between genes, and master regulators (MRs) of the transition from normal to disease state were inferred(27,28). This analysis revealed 211 MRs (FDR < 0.05) across the transcriptional network (Supplementary Table S7). The NRF2-dependent macrophage network (“green”) and Th2 (“tan”) networks, most robustly influenced by leu-enkephalin, had 27 MRs (Figure 4). Several of their regulators have been experimentally validated drivers of oncogenesis, tumor progression, and metastasis in ccRCC, suggesting possible transcriptional mechanisms through which opioids may affect ccRCC development and metastasis. Notably, CDH2, the MR with the strongest enrichment score, is known to be involved in the opioid pathway, and CDH2 variants influence methadone response(37,38). MRs associated with other survival-related networks also are tightly linked with opioid pathway (Figure 4), including genes involved in the opioid signaling cascade (GIT2(39), PLD2(40), RALGDS(41)) and opioid-related modulation of the immune system (CREB5(42), IL4R(43), CLEC2D(44), PLXNB1(45)). Given that opioid-related immunomodulation is generally considered central to opioid effects in cancer, we further examined correlation of gene expression of these four latter MRs with expression of immune signature genes(46) (Figure S9A–D and Supplementary Table S8). Taken together, the evidence suggests that ccRCC progression and opioid regulation converge onto these survival networks and provides a biologic rationale for how these seemingly divergent processes may transcriptionally influence each other in humans. The mechanistic details of this convergence remain unclear and require further study.
Figure 4. Reconstructing directed transcriptional networks and master regulators in ccRCC.
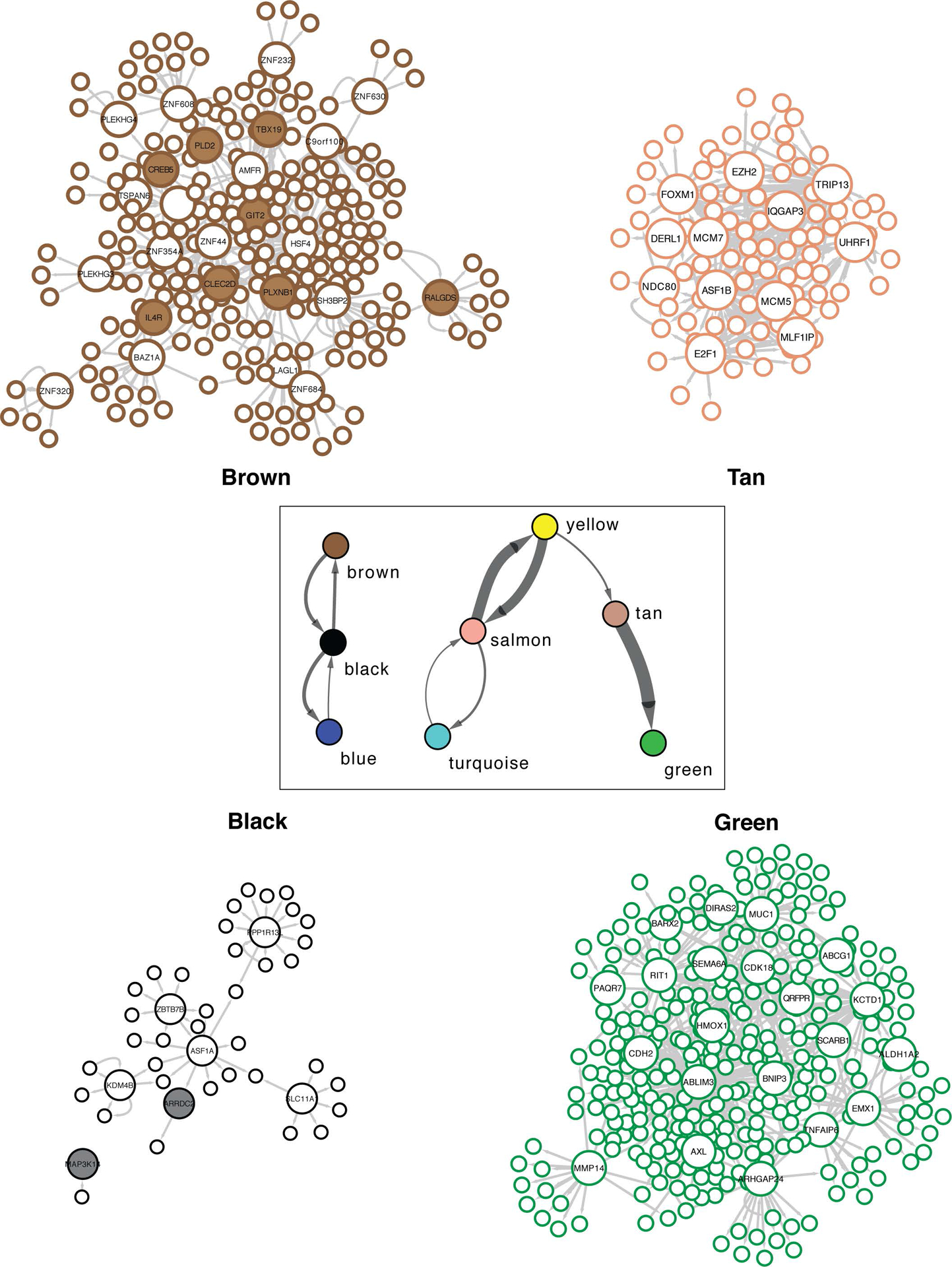
Directed networks representing relationships between modules (boxed), as well as gene-gene relationships within four separate modules. Each node is outlined based on its module color. Key drivers are represented by large nodes, and shaded key drivers are those with known associations to the opioid pathway.
DISCUSSION
This analysis explored the relationship between opioid-related transcriptional pathways and survival-relevant gene networks in ccRCC. This study identified opioid-related genes differentially expressed in ccRCC and characterized the effect of leu-enkephalin on ccRCC gene networks. While our analysis is focused on opioid pathways in ccRCC, we also highlight novel transcriptional features of ccRCC. Previous large-scale systems approaches have focused on networks that distinguish RCC subtypes(17) and networks correlated with pathology and overall survival(47–49). Our study focuses specifically on clear cell RCC coexpression networks, validates these networks in an independent cohort, and studies their relationship with recurrence-free survival, cancer-specific survival, and overall survival. We also integrate our analysis with experimental gene knockout and ChIP-seq data to characterize survival-relevant networks, and we calculate transcriptional master regulators in directed gene networks to help dissect drivers of disease and their relationship to opioid pathways.
We find that leu-enkephalin most strongly mediates fatty acid metabolism and Th2 immune networks, two processes central to the oncogenesis and progression of clear cell RCC. The clear cell subtype is characterized histologically by clear cytoplasm secondary to lipid accumulation, so fatty acid metabolism is a fundamental feature of clear cell RCC. Experimental evidence suggests that these mechanisms are not simply a byproduct of oncogenesis, but are critical to tumorigenesis and malignant transformation of renal cells(50). The tumor microenvironment is also critical to the development and progression of ccRCC, leading to the development of PD1 inhibitors. Both CD8+ and CD4+ T cells mediate tumor development by targeting antigenic components of renal cell carcinoma, and tumor-associated macrophages influence tumor development and angiogenesis(51). Our study suggests that leu-enkephalin affects ccRCC networks through both its effects on immune cells within the microenvironment and by direct action on primary tumor cells. This double-hit may be an important molecular feature distinguishing opioid agonists that can clinically influence cancer recurrence and survival.
The relationship between opioids and cancer outcomes is controversial. Some retrospective studies have failed to replicate earlier findings and reported effect sizes are variable and often specific to cancer subtypes(3,52–55). In some cases, this may point to truly null effects or confounders. It may also indicate that opioids have cancer subtype-specific effects that are masked in larger heterogeneous cohorts. Without prospective experiments, quasi-experimental studies, or a molecular understanding of opioids in humans with cancer, it is difficult to adjudicate between these possibilities. Our experiments address the molecular effects of opioids in humans with ccRCC by partly elucidating the regulation of opioid transcriptional pathways and the effect of opioid agonism in ccRCC. Our analysis provides a molecular rationale for this clinical relationship and supports the hypothesis that opioids negatively influence survival in clear cell renal cell carcinoma. Our results also suggest a possible therapeutic role for the peripherally acting opioid antagonist methylnaltrexone in RCC, analogous to the pro-survival effect observed in other cancer types(56). Future studies can model network perturbations of various opioid agonists on different cancer subtypes in order to predict and corroborate clinical associations between opioid exposure and cancer recurrence. These survival-associated networks may also function as potential pharmacogenomic biomarkers, helping to risk-stratify individual patients and predict individual response to the drug of interest. By better modeling underlying cancer biology and its response to pharmacologic perturbations, integrative systems-based models may use individual gene expression profiles to guide personalized anesthetic and analgesic plans and to optimize cancer-specific outcomes for individual cancer patients.
These analyses focused on leu-enkephalin as a model opioid, which has strengths and weaknesses. As an endogenous opioid peptide, it is a natural, non-specific ligand. It is not currently used in clinical practice, but recent work has focused on targeting endogenous opioid pathways in the development of novel analgesic agents(57), and leu-enkephalin specifically(58). Furthermore, endogenous opioids have been specifically implicated in cancer progression(59). The transcriptional profile induced by leu-enkephalin also has been detailed across numerous cancer cell lines, facilitating an analysis of opioid agonism in gene networks associated with recurrence-free and cancer-specific survival. At the same time, extrapolation to the exogenous opioids commonly used in the perioperative setting, like morphine, morphine-derivatives (hydromorphone), and the phenylpiperidines (fentanyl) is limited. Comparisons between opioid agents in general are limited since clinical effects are affected by differences in structure, pharmacokinetic profiles, and relative selectivity for the different opioid receptors. Our study is also limited by evidence suggesting transcriptional profiles can differ between in vitro and in vivo cancer cells(60). While our analysis of naloxone and VEGF inhibitors help support our general interpretation, the effect of other opioid receptor agonists should be tested directly in vivo.
An intriguing finding of our study is that the mu, delta, and kappa opioid receptor genes are poorly expressed in renal tissue and ccRCC, though greater variability was noted in the disease state. This implies that opioids may act through other mechanisms in ccRCC, like TLR4, OPRL1, and OGFR, which are known to bind opioids and are relevant to cancer progression in general and ccRCC in particular(30,31,61). In fact, both TLR4 and OGFR are found in our analysis to be members of survival-associated ccRCC networks – TLR4 in the MET-dependent (“blue”) network and OGFR in the JUN-dependent (“black”) network. Another explanation may be that our bulk tissue analysis masks important single-cell variability in mu, delta, and kappa opioid receptor gene expression, which plays a critical role in both cancer tissue and its microenvironment. Several studies have already demonstrated that intratumoral and microenvironment heterogeneity at the single cell level is associated with clinical outcomes and have possible therapeutic implications(62–64). Single cell RNAseq studies have also demonstrated that acute opioid administration has widespread suppression of antiviral pathways across the immune system and particularly in monocytes(65). These data point to ways in which single cell variability may play a critical role in understanding the relationship between opioid administration and clinical outcomes in ccRCC. Our results highlight the molecular relationship between ccRCC and opioid pathways, but future work is required to determine what are likely multiple mechanisms mediating this relationship.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF SIGNIFICANCE.
This study suggests a possible molecular mechanism for opioid effects on cancer outcomes generally, with implications for personalization of analgesic regimens.
ACKNOWLEDGMENTS
JSM acknowledges Professor John Chodera of the Sloan Kettering Institute for his support and insightful discussions. JSM also acknowledges Sahrena London for helpful conversations.
FINANCIAL SUPPORT: This research was supported by the National Cancer Institute Core Grant (P30 CA008748).
Footnotes
CONFLICT OF INTERESTS: The authors declare no potential conflicts of interest.
REFERENCES
- 1.Wigmore T, Farquhar-Smith P. Opioids and cancer. Curr Opin Support Pa. 2016;10:109–18. [DOI] [PubMed] [Google Scholar]
- 2.Kim R Effects of surgery and anesthetic choice on immunosuppression and cancer recurrence. J Transl Med. 2018;16:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wall T, Sherwin A, Ma D, Buggy DJ. Influence of perioperative anaesthetic and analgesic interventions on oncological outcomes: a narrative review. Brit J Anaesth. 2019;123:135–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekandarzad MW, Zundert AAJ van, Lirk PB, Doornebal CW, Hollmann MW. Perioperative Anesthesia Care and Tumor Progression. Anesthesia Analgesia. 2017;124:1697–708. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. Jama Oncol. 2019;5:1411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cata JP, Keerty V, Keerty D, Feng L, Norman PH, Gottumukkala V, et al. A retrospective analysis of the effect of intraoperative opioid dose on cancer recurrence after non-small cell lung cancer resection. Cancer Med-us. 2014;3:900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silagy AW, Hannum ML, Mano R, Attalla K, Scarpa JR, DiNatale RG, et al. Impact of intraoperative opioid and adjunct analgesic use on renal cell carcinoma recurrence: role for onco-anaesthesia. Brit J Anaesth 2020;125:e402–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du KN, Feng L, Newhouse A, Mehta J, Lasala J, Mena GE, et al. Effects of Intraoperative Opioid Use on Recurrence-Free and Overall Survival in Patients With Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Anesthesia Analgesia. 2018;127:210–6. [DOI] [PubMed] [Google Scholar]
- 9.Eisenstein TK. The Role of Opioid Receptors in Immune System Function. Front Immunol. 2019;10:2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Gene Dev. 2018;32:1267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boland JW, Pockley AG. Influence of opioids on immune function in patients with cancer pain: from bench to bedside. Brit J Pharmacol. 2017;175:2726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bortsov AV, Millikan RC, Belfer I, Boortz-Marx RL, Arora H, McLean SA. μ-Opioid receptor gene A118G polymorphism predicts survival in patients with breast cancer. Anesthesiology. 2012;116:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lennon FE, Mirzapoiazova T, Mambetsariev B, Salgia R, Moss J, Singleton PA. Overexpression of the μ-Opioid Receptor in Human Non-Small Cell Lung Cancer Promotes Akt and mTOR Activation, Tumor Growth, and Metastasis. Anesthesiology. 2012;116:857–67. [DOI] [PubMed] [Google Scholar]
- 14.Zylla D, Gourley BL, Vang D, Jackson S, Boatman S, Lindgren B, et al. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer. 2013;119:4103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. Ca Cancer J Clin. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 16.Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Robertson AG, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricketts CJ, Cubas AAD, Fan H, Smith CC, Lang M, Reznik E, et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Reports. 2018;23:313–326.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Ren Z, Ma S, Yan W, He M, Wang D, et al. Morphine enhances renal cell carcinoma aggressiveness through promotes survivin level. Renal Failure. 2016;39:258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2017;46:D956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? Plos Comput Biol. 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. Bmc Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, et al. International Cancer Genome Consortium Data Portal—a one-stop shop for cancer genomics data. Database. 2011;2011:bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science. 2006;313:1929–35. [DOI] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc National Acad Sci. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171:1437–1452.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Margolin AA, Nemenman I, Basso K, Klein U, Wiggins C, Stolovitzky G, et al. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. Bmc Bioinformatics. 2004;7:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lachmann A, Schilder BM, Wojciechowicz ML, Torre D, Kuleshov MV, Keenan AB, et al. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019;47:W571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Zheng Y, Yang Y, Wang J, Li B, Wei F, et al. Nociceptin Receptor Is Overexpressed in Non-small Cell Lung Cancer and Predicts Poor Prognosis. Frontiers Oncol. 2019;9:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maher DP, Walia D, Heller NM. Morphine decreases the function of primary human natural killer cells by both TLR4 and opioid receptor signaling. Brain Behav Immun. 2019;83:298–302. [DOI] [PubMed] [Google Scholar]
- 32.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol. 2005;4:Article17. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langfelder P, Mischel PS, Horvath S. When is hub gene selection better than standard meta-analysis? Ravasi T, editor. Plos One. 2013;8:e61505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barabási A-L, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–13. [DOI] [PubMed] [Google Scholar]
- 36.Chipollini J, Alford B, Boulware DC, Forget P, Gilbert SM, Lockhart JL, et al. Epidural anesthesia and cancer outcomes in bladder cancer patients: is it the technique or the medication? A matched-cohort analysis from a tertiary referral center. Bmc Anesthesiol. 2018;18:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y-L, Kuo H-W, Fang C-P, Tsung J-H, Chen ACH. Adhesion Molecules as Potential Novel Biomarkers for Opioid Dependence. Curr Pharm Design. 2020;26. [DOI] [PubMed] [Google Scholar]
- 38.Kuo H-W, Shih C-L, Tsung J-H, Liu S-W, Chu S-K, Yang H-C, et al. Pharmacogenomics study on cadherin 2 network with regard to HIV infection and methadone treatment outcome. Plos One. 2017;12:e0174647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoefen RJ, Berk BC. The multifunctional GIT family of proteins. J Cell Sci. 2006;119:1469–75. [DOI] [PubMed] [Google Scholar]
- 40.Koch T, Brandenburg L-O, Liang Y, Schulz S, Beyer A, Schröder H, et al. Phospholipase D2 modulates agonist-induced mu-opioid receptor desensitization and resensitization. J Neurochem. 2004;88:680–8. [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharya M, Babwah AV, Godin C, Anborgh PH, Dale LB, Poulter MO, et al. Ral and Phospholipase D2-Dependent Pathway for Constitutive Metabotropic Glutamate Receptor Endocytosis. J Neurosci. 2004;24:8752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long X, Li Y, Qiu S, Liu J, He L, Peng Y. MiR-582–5p/miR-590–5p targeted CREB1/CREB5-NF-κB signaling and caused opioid-induced immunosuppression in human monocytes. Transl Psychiat. 2016;6:e757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Busch-Dienstfertig M, González-Rodríguez S. IL-4, JAK-STAT signaling, and pain. Jak-stat. 2013;2:e27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Liang Y, Levran O, Randesi M, Yuferov V, Zhao C, et al. Alterations of expression of inflammation/immune-related genes in the dorsal and ventral striatum of adult C57BL/6J mice following chronic oxycodone self-administration: a RNA sequencing study. Psychopharmacology. 2017;234:2259–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuferov V, Zhang Y, Liang Y, Zhao C, Randesi M, Kreek MJ. Oxycodone Self-Administration Induces Alterations in Expression of Integrin, Semaphorin and Ephrin Genes in the Mouse Striatum. Frontiers Psychiatry. 2018;9:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T-HO, et al. The Immune Landscape of Cancer. Immunity. 2018;48:812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao H, Chen P, Zeng G, Xu D, Wang X, Zhang X. Three novel hub genes and their clinical significance in clear cell renal cell carcinoma. J Cancer. 2019;10:6779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Chen L, Wang G, Cheng S, Qian K, Liu X, et al. Fifteen hub genes associated with progression and prognosis of clear cell renal cell carcinoma identified by coexpression analysis. J Cell Physiol. 2018;234:10225–37. [DOI] [PubMed] [Google Scholar]
- 49.Chen L, Yuan L, Qian K, Qian G, Zhu Y, Wu C-L, et al. Identification of Biomarkers Associated With Pathological Stage and Prognosis of Clear Cell Renal Cell Carcinoma by Co-expression Network Analysis. Front Physiol. 2018;9:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8:1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cakmakkaya OS, Kolodzie K, Apfel CC, Pace NL. Anaesthetic techniques for risk of malignant tumour recurrence. Cochrane Db Syst Rev. 2014;11:CD008877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoo S, Lee H-B, Han W, Noh D-Y, Park S-K, Kim WH, et al. Total Intravenous Anesthesia versus Inhalation Anesthesia for Breast Cancer Surgery. Anesthesiology. 2019;130:31–40. [DOI] [PubMed] [Google Scholar]
- 54.Wuethrich PY, Thalmann GN, Studer UE, Burkhard FC. Epidural analgesia during open radical prostatectomy does not improve long-term cancer-related outcome: a retrospective study in patients with advanced prostate cancer. Plos One. 2013;8:e72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scavonetto F, Yeoh TY, Umbreit EC, Weingarten TN, Gettman MT, Frank I, et al. Association between neuraxial analgesia, cancer progression, and mortality after radical prostatectomy: a large, retrospective matched cohort study. Brit J Anaesth. 2013;113 Suppl 1:i95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janku F, Johnson LK, Karp DD, Atkins JT, Singleton PA, Moss J. Treatment with methylnaltrexone is associated with increased survival in patients with advanced cancer. Ann Oncol. 2016;27:2032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sitbon P, Elstraete AV, Hamdi L, Juarez-Perez V, Mazoit J-X, Benhamou D, et al. STR-324, a Stable Analog of Opiorphin, Causes Analgesia in Postoperative Pain by Activating Endogenous Opioid Receptor–dependent Pathways. Anesthesiology. 2016;125:1017–29. [DOI] [PubMed] [Google Scholar]
- 58.Feng J, Lepetre-Mouelhi S, Gautier A, Mura S, Cailleau C, Coudore F, et al. A new painkiller nanomedicine to bypass the blood-brain barrier and the use of morphine. Sci Adv. 2019;5:eaau5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lennon FE, Moss J, Singleton PA. The μ-opioid receptor in cancer progression: is there a direct effect? Anesthesiology. 2012;116:940–5. [DOI] [PubMed] [Google Scholar]
- 60.Liu K, Newbury PA, Glicksberg BS, Zeng WZD, Paithankar S, Andrechek ER, et al. Evaluating cell lines as models for metastatic breast cancer through integrative analysis of genomic data. Nat Commun. 2019;10:2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bisignani GJ, McLaughlin PJ, Ordille SD, Beltz MS, Jarowenko MV, Zagon IS. Human Renal Cell Cancer Proliferation in Tissue Culture is Tonically Inhibited By Opioid Growth Factor. J Urology. 1999;162:2186–91. [DOI] [PubMed] [Google Scholar]
- 62.Kim K-T, Lee HW, Lee H-O, Song HJ, Jeong DE, Shin S, et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016;17:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsieh JJ, Miheecheva N, Ramachandran A, Lyu Y, Galkin I, Svekolkin V, et al. Integrated single-cell spatial multi-omics of intratumor heterogeneity in renal cell carcinoma. J Clin Oncol. 2020;38:e17106–e17106. [Google Scholar]
- 64.Young MD, Mitchell TJ, Braga FAV, Tran MGB, Stewart BJ, Ferdinand JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. 2018;361:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karagiannis TT, Cleary JP, Gok B, Henderson AJ, Martin NG, Yajima M, et al. Single cell transcriptomics reveals opioid usage evokes widespread suppression of antiviral gene program. Nat Commun. 2020;11:2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.