

Abstract
PURPOSE:
The World Trade Center (WTC) attack of September 11, 2001 created an unprecedented environmental exposure to known and suspected carcinogens. High incidence of multiple myeloma (MM) and precursor conditions has been reported among first responders to the WTC disaster. To expand on our prior screening studies, and to characterize the genomic impact of the exposure to known and potential carcinogens in the WTC debris, we were motivated to perform whole genome sequencing (WGS) of WTC first responders and recovery workers who developed a plasma cell disorder after the attack.
PATIENTS AND METHODS:
We performed WGS of 9 CD138-positive bone marrow mononuclear samples from patients who were diagnosed with plasma cell disorders after the WTC disaster.
RESULTS:
No significant differences were observed in comparing the post-WTC driver and mutational signatures landscape with 110 previously published WGS from 56 patients with MM and the CoMMpass WGS cohort (n=752). Leveraging constant activity of the single base substitution mutational signatures 1 and 5 over time, we estimated that tumor-initiating chromosomal gains were windowed to both pre- and post-WTC exposure.
CONCLUSIONS:
Although limitations in sample size preclude any definitive conclusions, our findings suggest that the observed increased incidence of plasma cell neoplasms in this population is due to complex and heterogeneous effects of the WTC exposure that may have initiated or contributed to progression of malignancy.
Keywords: Multiple Myeloma, Mutational Signatures, World Trade Center, Timing
Introduction
Multiple myeloma, a clonal neoplasm of post-germinal center B cells, is one of the most prevalent hematologic malignancies among adults in the United States. It is always preceded by an asymptomatic precursor condition [i.e. monoclonal gammopathy of undetermined significance (MGUS) or smoldering myeloma (SMM)].(1,2) The evolution from a B cell’s first germinal center encounter to multiple myeloma is driven by the acquisition of different genomic drivers and is shaped by the activity of different mutational processes.(1,2) Using whole genome sequencing (WGS) data, seven main mutational processes (i.e. single base substitution signatures) have been described in newly diagnosed multiple myeloma - six of which are associated with a recognized etiology: SBS1 and SBS5 (aging), SBS2 and SBS13 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; APOBEC), SBS18 (damage by reactive oxygen species), and SBS9 (non-canonical activity of Activation-induced cytidine deaminase; nc-AID).(3–8)
Although multiple myeloma and myeloma precursor condition appear to occur sporadically without a known underlying etiology, an increased incidence of the disease has been reported in those exposed to a variety of carcinogens including Agent Orange (with dioxin contaminant), polychlorinated biphenyls, and polycyclic aromatic hydrocarbons (PAHs).(2,9,10)
On September 11th, 2001, the World Trade Center (WTC) attacks created an environmental exposure of unprecedented scale. Firefighters of the Fire Department of the City of New York were among the first responders met with aerosolized dust, gases, and debris containing many of the above known and other potential carcinogens.(11) Some of these substances (e.g. PAHs) have been reported to leave unique mutational signatures on the genome of human cells, allowing for quantification of the mutational burden for which they are responsible.(12) It has since been documented that first responders and recovery workers with exposure to the WTC attack have higher than expected rates of malignancy,(13–15) and, specifically, there are reports of increased incidence of multiple myeloma and precursor conditions following WTC exposure.(16) However, the mutational impact of this unique exposure has never been investigated or quantified in any cancer type so far.
Whole genome, exome and target sequencing have been extensively used to characterize the genomic landscape of multiple myeloma cases diagnosed in the general population. Discoveries have included a description of 81 potential driver genes, and the cataloguing of structural variations (SVs) including complex events such as chromothripsis, and copy number abnormalities with negative prognostic implications.(7,8,17–21) However, potential common themes in the genomic characteristics of plasma cell neoplasms developing following a shared carcinogenic event, such as the WTC attack, have never been investigated.
To expand on our prior screening study(16) we were motivated to perform WGS for 9 first responders and recovery workers who developed a plasma cell disorder after being exposed to the disaster. The key aims of this study were specifically: 1) to identify distinct genetic signatures (i.e. mutational signatures) of increased risk of multiple myeloma and precursor conditions among WTC survivors; 2) to establish whether the WTC disaster initiated the pre-neoplastic clone or accelerated the progression of a preexisting one. While we did not observe any unifying distinct genomic events, using mutational signatures and the molecular clock,(7) we were able to estimate that the exposure to the WTC attack may have had a role either in initiating the first aberrant cell, or by promoting the progression of a pre-existing clone.
Methods
Thirty-one WTC-exposed patients who developed plasma cell disorders after the WTC attack are currently in follow up in the Myeloma Service at the Memorial Sloan Kettering Cancer Center (Supplementary Table 1). Samples and data were obtained and managed in accordance with the Declaration of Helsinki. We performed whole genome sequencing of 9 cases that had sufficient CD138-positive bone marrow mononuclear cells: 4 MGUS, 2 SMM, 2 MMs, and 1 patient with plasma cell leukemia (PCL) (Table 1).(16) Eight patients (88%) were first responders and one was a recovery worker (IID_H135225). The study involved the use of human samples, which were collected after written informed consent was obtained (IRB 14–276). Plasma cell selection was performed by CD138-positive magnetic bead-selected bone marrow mononuclear cells. All sequencing investigations were performed at Memorial Sloan Kettering Cancer Center’s Integrated Genomics Operation (IGO).(22) Peripheral blood mononuclear cells were used as a normal match. For the only PCL sample, peripheral blood granulocytes were used as normal match to avoid tumor plasma cell contamination.
Table 1.
Sequencing, demographic and clinical profile of the 9.11 WTC cohort.
| Sample | WTC exposure | Age at Diagnosis | Year of diagnosis | Stage | Sex | Isotype | IGH translocation | Coverage | Purity | Ploidy |
|---|---|---|---|---|---|---|---|---|---|---|
| IID_H196059 | First responder | 67 | 2019 | MGUS | M | IgG kappa/ lambda% | HRD* | 49.19 | 0.35 | 2.45 |
| IID_H135336 | First responder | 50 | 2014 | SMM | M | IgG lambda | HRD | 76.54 | 0.40 | 2.18 |
| IID_H196060 | First responder | 48 | 2017 | PCL | M | IgG lambda | t(14;16) | 48.76 | 0.96 | 1.9 |
| IID_H130588 | First responder | 57 | 2018 | MM | M | IgG kappa | HRD | 50.39 | 0.86 | 2.29 |
| IID_H196061 | First responder | 52 | 2019 | MGUS | M | IgA kappa | t(11;14) | 51.73 | 0.45 | 2.48 |
| IID_H196062 | First responder | 58 | 2019 | SMM | M | IgG lambda | t(6;14) | 47.57 | 0.31 | 1.99 |
| IID_H196063 | First responder | 61 | 2019 | MGUS | M | IgG lambda | HRD | 47.85 | 0.5 | 2.81 |
| IID_H196064 | First responder | 65 | 2019 | MGUS | M | IgG lambda | - | 51.46 | 0.86 | 1.9 |
| IID_H135225 | Rescue worker | 47 | 2015 | MM | M | IgG kappa | t(2;16)(IGL-MAF) | 76.22 | 0.98 | 1.89 |
HRD: hyperdiploid
biclonal
Whole genome sequencing
After PicoGreen quantification and quality control by Agilent BioAnalyzer, 500ng of genomic DNA were sheared using a LE220-plus Focused-ultrasonicator (Covaris catalog # 500569) and sequencing libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with modifications. Briefly, libraries were subjected to a 0.5X size select using aMPure XP beads (Beckman Coulter catalog # A63882) after post-ligation cleanup. Libraries not amplified by PCR (07652_C) were pooled equivolume and were quantitated based on their initial sequencing performance. Libraries amplified with 5 cycles of PCR (07652_D, 07652_F, 07652_G) were pooled equimolar. Samples were run on a NovaSeq 6000 in a 150bp/150bp paired end run, using the NovaSeq 6000 SBS v1 Kit and an S4 flow cell (Illumina), as previously described22.
Whole genome analysis pipeline
The median coverage for tumor and normal samples was 50.9X (range 47–76) and 37X (range 35–41) respectively (Table 1). Short-insert paired-end reads were aligned to the reference genome (GRCh37) using the Burrows Wheeler Aligner (BWA) (v0.5.9) [17]. Somatic mutations were identified by CaVEman.(23) Copy number analysis and tumor purity (i.e. cancer cell fraction) were evaluated using Battenberg (https://github.com/Wedge-Oxford/battenberg). SVs were defined by merging calls from SvABA,(24) BRASS (https://github.com/cancerit/BRASS), and GRIDSS (https://github.com/PapenfussLab/gridss). Complex events (i.e. templated insertion, chromothripsis and chromoplexy) were defined and annotated as previously described.(20,21) The phylogenetic tree of each case was reconstructed using the Dirichlet process (https://github.com/Wedge-Oxford/dpclust).
Mutational signatures were investigated by combining and comparing the WTC cohort with 110 WGS from 56 patients with multiple myeloma and myeloma precursor condition.(4,7,18,20–22) All WGSs were characterized using the same pipeline described above. To estimate the activity of mutational signatures we followed our recently published workflow based on three steps: de novo extraction, assignment and fitting.(5) For the first step we ran SigProfiler and hierarchical Dirichlet process.(3) All extracted signatures were then compared to the latest COSMIC reference (https://cancer.sanger.ac.uk/cosmic/signatures/SBS/) to define which known mutational processes were active in our cohort. Finally, we applied mmsig (https://github.com/evenrus/mmsig), a fitting algorithm designed for MM, to confirm the presence and estimate the contribution of each mutational signature in each sample.(7) Confidence intervals were generated by drawing 1000 mutational profiles from the multinomial distribution, each time repeating the signature fitting procedure, and finally taking the 2.5th and 97.5th percentile for each signature. To analyze the contribution of each mutational signature over time, we explore all Dirichlet process clusters with more than 50 mutations using mmsig as described above.(4,7)
To exclude the contribution of environmental agents detected in the WTC debris with recognized mutational signatures,(12) we ran mmsig in each post-WTC case, including and forcing the extraction of these mutational signatures.(7)
The landscape of recurrent genomic drivers and complex events was then compared to 752 multiple myeloma patients enrolled in the CoMMpass trial with available whole exome and low-coverage long-insert whole genome sequencing data (IA15; NCT01454297).(7,21) As recurrent and driver genomic events we selected the most relevant copy number changes, translocations, complex events and a catalogue of 81 driver genes involved by mutations derived from combining 2 large driver analyses.(6,19,20)
Molecular time
The relative timing of each multi-chromosomal gain event was estimated using the R package mol_time (https://github.com/nicos-angelopoulos/mol_time).(7,20) Correcting the ratio between duplicated mutations (VAF~66%, acquired before the chromosomal duplication) and non-duplicated mutations (VAF 33%, acquired on either the non-duplicated allele or on one of the two duplicated ones) this approach allows to estimate the relative timing of acquisition of all large (>1Mb) chromosomal gains (e.g. trisomy in hyperdiploid myeloma patients) with more than 50 clonal mutations as estimated by the Dirichlet process.(6,7,20,25) Tetrasomies with both alleles duplicated were removed given the impossibility of defining whether the two chromosomal gains occurred in close temporal succession, or in two discrete time-windows.(2)
Overall, the molecular time approach allowed the definition of chromosomal gains that were acquired in the same time window. Next, to convert the relative molecular time estimate into an absolute estimate, we combined chromosomal gains acquired in the same time window and calculated the molecular time based only on the mutational burden of single base SBS1 and SBS5.(3,7,26) Considering that these mutational processes are known to be constant in multiple myeloma (as in all cancers and normal tissues),(3,7,26) we could convert the SBS1 and SBS5 molecular clock into an absolute time estimate for the acquisition of these events in each patient’s life. Confidence of intervals were generated by bootstrapping the molecular time estimate. Only multi-gain events with more than 50 SBS1 and SBS5 mutations were included. The plasma cell leukemia case was excluded due to its mutational profile characterized by high mutational burden (>10,000) and hyper-APOBEC contribution.(7)
Data analysis and statistics
Data analysis was carried out in R version 3.6.1. Standard statistical tests are mentioned consecutively in the manuscript while more complex analyses are described above. All reported p-values are two-sided, with a significance threshold of < 0.05.
Results
A total of 56,682 single nucleotide variants (SNVs) were detected, with a median of 5,115 SNVs per sample (range 1,164–19,658). Among 433 nonsynonymous mutations detected [median 40 per sample (range 4–198)], only 12 involved known multiple myeloma drivers [median 1 per sample (range 0–4)] (Figure 1). Across 9 patients, a total of 277 SVs were called with a median 17 SVs per patient (range 0–93). Deletions were the most common SV type (31%), followed by inversions (29%), tandem duplications (21%) and translocations (19%). One hundred and seventy-four of 277 SVs (63%) were part of a complex event (i.e. chromothripsis, chromoplexy, templated insertion, or unclassifiable complex event).(20,21) Four patients had a chromothripsis and 2 had a templated insertion involving 2 or more chromosomes. Overall, 5 patients had at least one complex event. We did not observe any significant differences in multiple myeloma driver SNVs, SVs, complex events or CNAs between the WTC cohort and the CoMMpass series (Figure 1). Of interest, three out of four MGUS were characterized by low SV burden, absence of complex events and all followed an indolent clinical course. In contrast, one patient with MGUS, having intermediate-high risk for progression at diagnosis, a monoclonal protein spike of 2.1 g/dL, and a bone marrow plasma cell infiltration of 5%, was characterized by chromothripsis, high APOBEC signature contribution, and biallelic TP53 inactivation; within 2 years, this patient had progression into MM.
Figure 1.
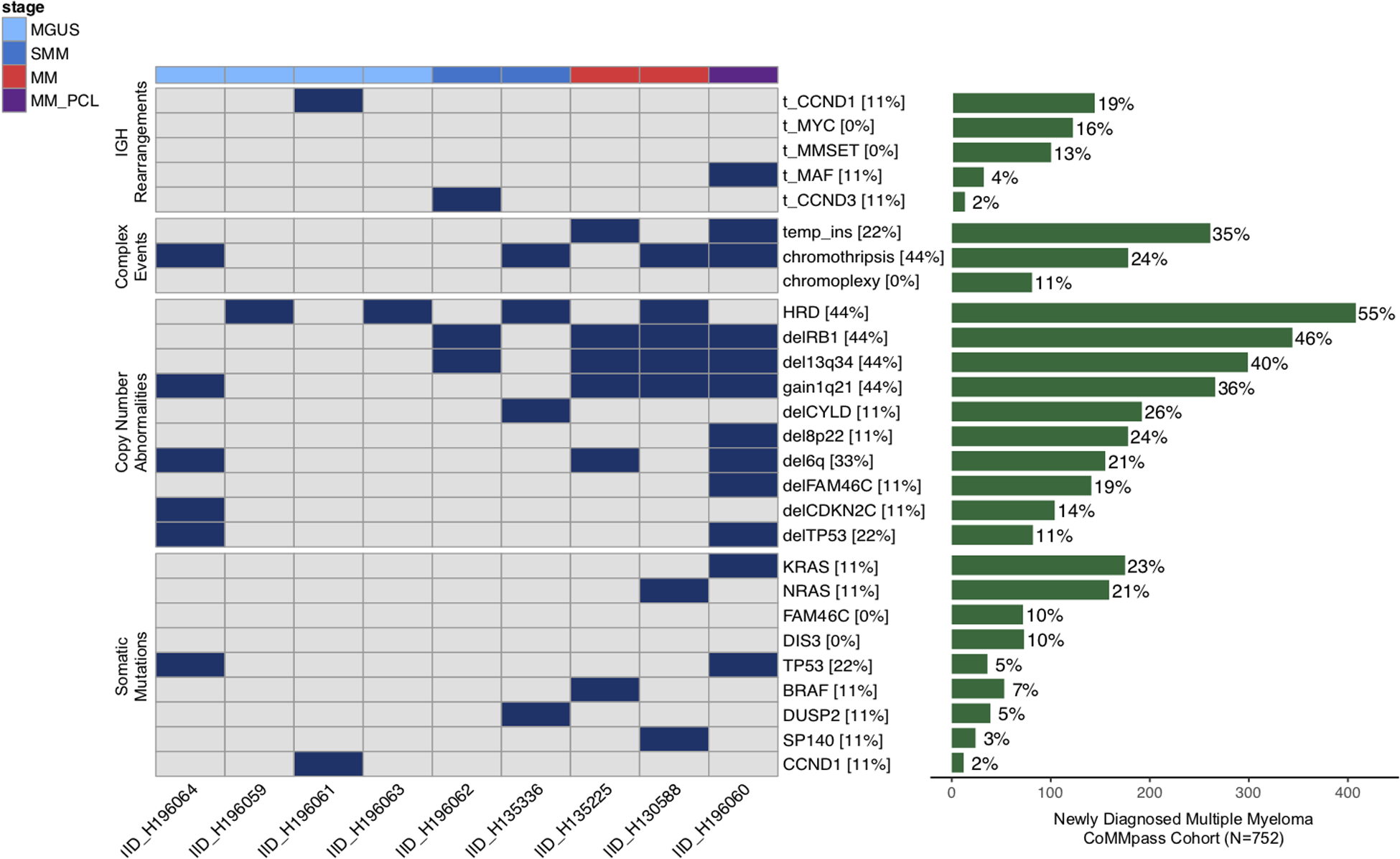
Multiple myeloma genomic driver landscape in the World-Trade Center (WTC) cohort. The prevalence of each genomic driver is compared to that observed in 752 multiple myeloma patients enrolled in the CoMMpass trial with whole exome and low-coverage long-insert whole genome sequencing data available. HRD = hyperdiploid; MGUS = Monoclonal gammopathy of undetermined significance; SMM = smoldering multiple myeloma; MM = multiple myeloma; PCL = plasma cell leukemia. Only fully clonal copy number were reported for the WTC cohort.
Mutational signature landscape in WTC-exposed patients
In comparison to 110 previously published WGS from 56 patients with multiple myeloma and precursor conditions,(7,18,20) we did not observe any new mutational signatures among WTC-exposed patients. Three WTC cases (one MGUS, one multiple myeloma and the one PCL) showed relative high APOBEC contribution with only the PCL having t(14;16)(MAF;IGH).(8,27) To rule out any undetected or low mutational contribution from exposure to WTC toxic substances, we used the mmsig mutational signature fitting approach, including and forcing the extraction of 5 mutational signatures associated with environmental agents detected in the WTC debris. (e.g. PAHs; Supplementary Table 2).(12) There were no significant contributions from any of these described mutational signatures. When re-fitting mutational signatures in the absence of those related to environmental exposure, the signature profile of the WTC cohort recapitulated previous observations for multiple myeloma and precursor conditions (Figure 2A).
Figure 2.
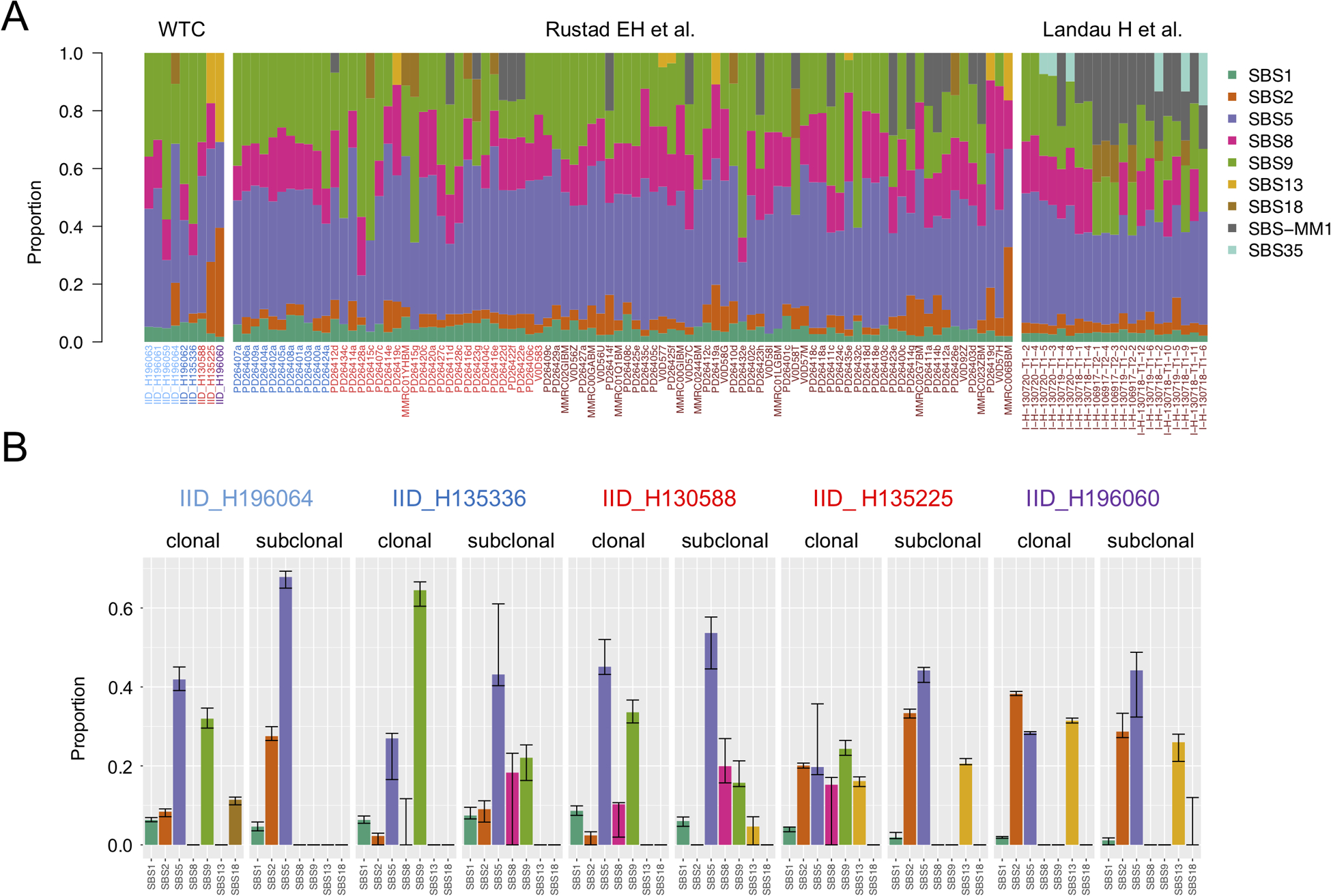
Mutational signature landscape in first responders and recovery workers exposed to the WTC disaster. A) Relative contribution of each mutational signature in the WTC cohort and in a validation set including 110 available WGS from 56 patients. B) Mutational signatures differences between clonal and subclonal CNVs in each case with more than 50 subclonal mutations. The confidence interval of each mutational signature estimate was generated by drawing 1000 mutational profiles from the multinomial distribution, each time repeating the signature fitting procedure (mmsig), and finally taking the 2.5th and 97.5th percentile for each signature.
We recently defined four main temporal patterns of mutational signatures activity in multiple myeloma:(7) 1) where AID activity is limited to the first phase of cancer development, and APOBEC is only active during later phases; 2) where APOBEC is active since the beginning without any significant AID contribution; 3) where AID activity is prolonged over time contributing to the subclonal diversification; and 4) where AID is active only during the first phase of cancer development, and APOBEC is always absent. Reconstructing the temporal activity of each mutational process, we sought to explore whether WTC-exposed patients had differing patterns in mutational signature timelines (Methods; Figure 2B).(7) Among the 5 cases where the subclonal mutational load allowed for a reliable mutational signature estimation,(4,7) four cases showed a reduction in AID from clonal to subclonal mutations. In three cases APOBEC increased from clonal to subclonal mutations. In the PCL case, AID was not detectable and APOBEC was the major mutational process in both clonal and subclonal variants, in line with the temporal pattern of hyper-APOBEC mutational signatures (#2 above).(7) Overall, these data revealed that the mutational signature activity over time in post-WTC plasma cell dyscrasia is heterogenous and reflects what has been previously observed in multiple myeloma without WTC-exposure.
Timing the initiation of plasma cell dyscrasias in WTC-exposed patients
We, and others, have recently shown that a cancer-initiating event can be acquired 30–40 years before its clinical diagnosis, often between the second and third decade of life. (7,26,28) These estimates are made possible due to distinct mutational processes that are stably active over time (i.e. clock-like).(29) Without mutational contributions directly linked to exposure, we leveraged the cancer molecular clock concept to ascertain whether exposure to the WTC attack might have either accelerated progression of a pre-existing clonal entity, served as an initiating carcinogenic event, or a combination of both.
For each patient with at least one large chromosomal gain in the WTC cohort (8/9), we applied our recently published molecular time workflow(20) to estimate the relative order and time window of its acquisition (Methods; Figure 3A). Then, following the molecular-clock concept,(7,26,29) we collapsed together chromosomal gains acquired in the same time window and used the pre- and post-gain SBS1 and SBS5 mutation burden to convert relative time estimates into absolute ones (i.e. the age at which these events were acquired in each patient’s life). One case was excluded due to low SBS5 mutational burden (IID_H196064) and another due to high mutational burden and hyper-APOBEC activity (H196060; PCL) each of which affect the accuracy of timing predictions (Methods; Figure 3B).(7) According to our time estimates, one multiple myeloma case (IID_H130588), one smoldering myeloma case (IID_H135336), and one MGUS (IID_H196059) show evidence of a pre-existing clone before the WTC attack. The remaining two MGUS showed evidence of multi-chromosomal gain acquisition after the attack (H196061 and H196063). Finally, in the last multiple myeloma case (IID_H13225), the 1q gain was acquired around the time of the WTC attack. Overall, these data are consistent with the recently proposed model that the first genomic driver precedes myeloma diagnosis by decades.(7)
Figure 3.
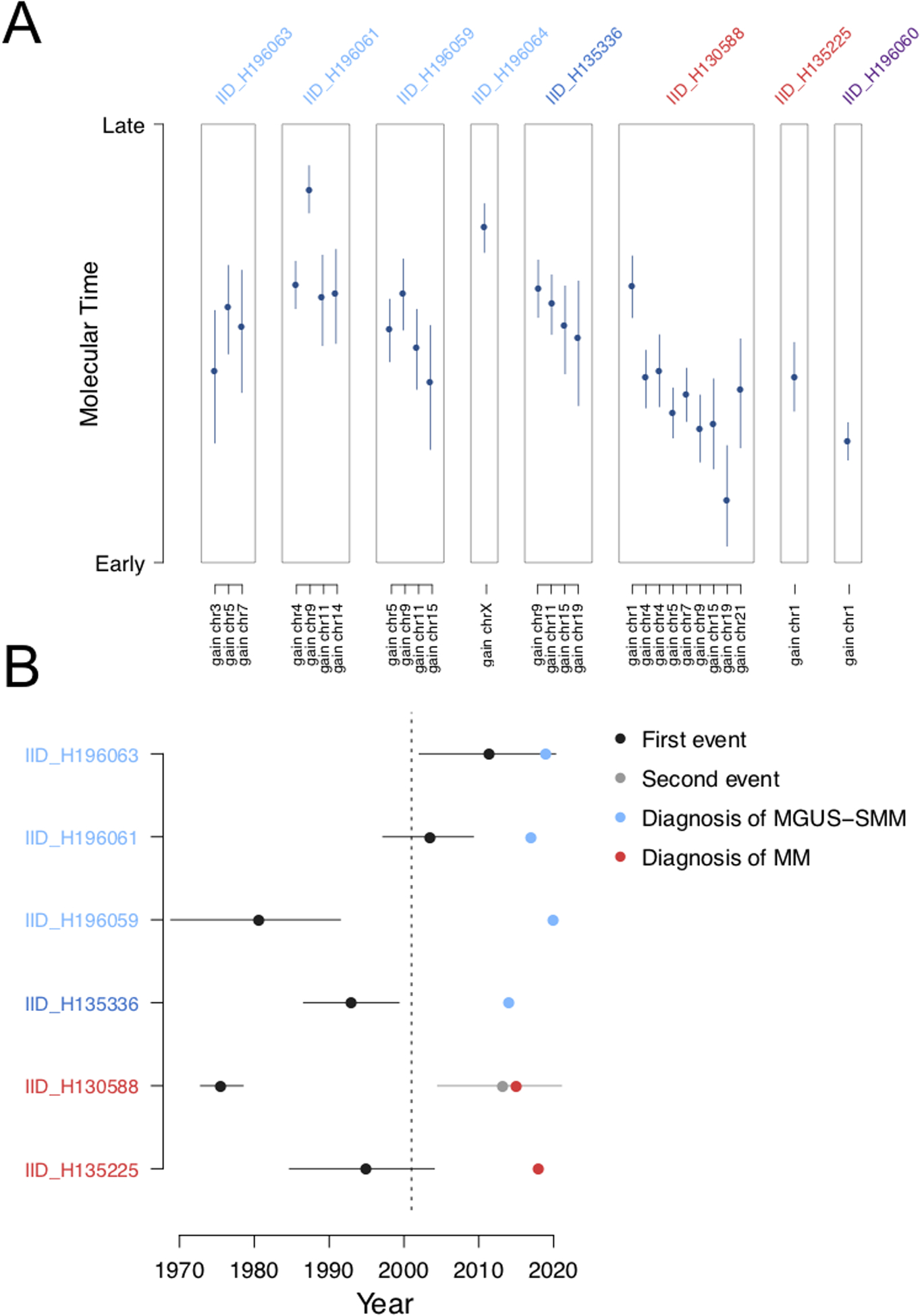
Timing multi-chromosomal gain events in first responders and recovery workers exposed to the WTC disaster with multiple myeloma and precursor conditions. A) Molecular time estimated for each clonal gain and copy-neutral loss of heterozygosity in the WTC cohort. Blue dots and lines represent the molecular time estimates and the 95% confidence intervals, respectively. Only large chromosomal gains (>1 Mb) with more than 50 clonal single nucleotide variants were considered. B) Absolute timing of each multi-gain event in relation to the WTC attack and the patient’s age at diagnosis for 6 evaluable patients. Dark blue and grey dots represent the first and second multi-gain events, respectively, with 95% confidence intervals. Blue dots represent MGUS diagnosis and red dots represent multiple myeloma diagnosis. The dotted line represents September 11, 2001.
Discussion
Exposure to environmental carcinogens can promote cancer development through various mechanisms. As a canonical example, tobacco smoke stimulates hundreds of mutations in directly exposed cells; leaving evidence of its effect through signature SBS4.(3,30,31) While this evidence is seen directly in virtually all tobacco smoke-related lung cancers, there are other tobacco-associated cancers without any evidence of SBS4 contribution, suggesting that tobacco-mediated carcinogenesis is also promoted through other mechanisms.(30) The exposure to the WTC debris is known to promote several cancers, including multiple myeloma and precursor conditions. The lack of a homogeneous and distinct mutational signatures among WTC-exposed patients who subsequently developed either multiple myeloma or myeloma precursor condition suggest that the carcinogens present in the WTC debris may promote myelomagenesis through alternate evolutionary trajectories and combination of drivers, without leaving direct mutagenic evidence. While the small sample size might have limited the power of genomic driver discovery, likely it did not affect the analysis of mutational signatures. In fact, 9 patients, all exposed to the same carcinogenic event, had enough power to potentially detect a new WTC-related mutational process.
While we did not observed any distinct genomic link between the WTC exposure and multiple myeloma or precursor conditions, leveraging the molecular time concept, we were able to show that the WTC exposure might, in some cases, have had a role in promoting a pre-existing clonal entity (i.e., progression from pre-existing myeloma precursor condition to frank malignancy) and, in others, it may have contributed to creating the conditions required for initiating the clonal entity. It is also possible that (at least some) myeloma precursors were acquired independent of the WTC attack, as in patient IID_H196061, where the first datable event was acquired approximately 12 years after the attack. Due to the limited sample size, if all patients had had the same temporal pattern (i.e. first multi-gain events uniformly acquired either before or after the WTC-attack), we would not have been able to claim a universal WTC-related temporal model due to power limitation. However, the observation of different/dichotomous patterns supports the concept that WTC-exposure affected individuals in different ways.
Overall, in our study, we provided the first WGS characterization of first responders and recovery workers exposed to the WTC attack, who developed multiple myeloma and myeloma precursor conditions. The observed genomic and temporal heterogeneity herein suggests that the observed increased incidence of plasma cell neoplasm in WTC-exposed patients is due to complex and heterogenous effects that may have initiated or promoted subsequent disease development.
Supplementary Material
Translational relevance:
The World Trade Center (WTC) attack of September 11, 2001 created an unprecedented environmental exposure to known and potential carcinogens and first responders have demonstrated a higher risk of developing multiple myeloma. We were motivated to identify distinct genetic signatures and temporal patterns responsible for this increased risk and so we performed the first whole genome sequencing characterization of plasma cell neoplasms in first responders and recovery workers exposed to the WTC attack. While we did not observe any unifying genomic events or mutational signatures, we were able to estimate that the exposure to the debris may have had a role either in initiating the first aberrant cell, or in promoting the progression of a pre-existing clone. The existence of pre-malignant clonal entities at time of WTC exposure may therefore be relevant for future WTC-related studies.
Acknowledgements
This work is supported by the Society of Memorial Sloan Kettering and by the Memorial Sloan Kettering Cancer Center NCI Core Grant (P30 CA 008748).
FM is supported by the American Society of Hematology, the International Myeloma Foundation and The Society of Memorial Sloan Kettering Cancer Center.
Benjamin Diamond is supported by Conquer Cancer, the ASCO Foundation.
KHM is supported by the Haematology Society of Australia and New Zealand New Investigator Scholarship and the Royal College of Pathologists of Australasia Mike and Carole Ralston Travelling Fellowship Award.
We thank Integrated Genomics Operation (IGO) for the sequencing support.
Footnotes
Data availability
- EGAD00001003309: 67 WGS data from 30 multiple myeloma patients
- EGAS00001004404: 21 WGSs from 4 patients
- phs000748.v1.p1: Whole exome sequencing (WXS) and low coverage/long insert WGS sequencing data from 752 newly diagnosed multiple myeloma patients included in this study (CoMMpass trial; IA 15)
- phs000348.v2.p1 WGS data from 22 multiple myeloma patients
- EGAS00001004467: WTC WGS data
Conflicts of interest
- Grant support: NIH, FDA, MMRF, IMF, LLS, Perelman Family Foundation, Rising Tide Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, Karyopharm
- Honoraria/ad boards: Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, Pfizer
- Independent Data Monitoring Committee (IDMC): Takeda, Merck, Janssen, Theradex
References
- 1.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 2017;14(2):100–13 doi 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 2.Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA Oncol 2019. doi 10.1001/jamaoncol.2019.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun 2018;9(1):3363 doi 10.1038/s41467-018-05058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maura F, Degasperi A, Nadeu F, Leongamornlert D, Davies H, Moore L, et al. A practical guide for mutational signature analysis in hematological malignancies. Nat Commun 2019;10(1):2969 doi 10.1038/s41467-019-11037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maura F, Rustad EH, Yellapantula V, Luksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia 2019. doi 10.1038/s41375-019-0689-0. [DOI] [PubMed] [Google Scholar]
- 7.Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nat Commun 2020;11(1):1917 doi 10.1038/s41467-020-15740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun 2015;6:6997 doi 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajkumar SV, Landgren O, Mateos MV. Smoldering multiple myeloma. Blood 2015;125(20):3069–75 doi 10.1182/blood-2014-09-568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landgren O, Shim YK, Michalek J, Costello R, Burton D, Ketchum N, et al. Agent Orange Exposure and Monoclonal Gammopathy of Undetermined Significance: An Operation Ranch Hand Veteran Cohort Study. JAMA Oncol 2015;1(8):1061–8 doi 10.1001/jamaoncol.2015.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landrigan PJ, Lioy PJ, Thurston G, Berkowitz G, Chen LC, Chillrud SN, et al. Health and environmental consequences of the world trade center disaster. Environ Health Perspect 2004;112(6):731–9 doi 10.1289/ehp.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019;177(4):821–36 e16 doi 10.1016/j.cell.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Brackbill RM, Liao TS, Qiao B, Cone JE, Farfel MR, et al. Ten-year cancer incidence in rescue/recovery workers and civilians exposed to the September 11, 2001 terrorist attacks on the World Trade Center. Am J Ind Med 2016;59(9):709–21 doi 10.1002/ajim.22638. [DOI] [PubMed] [Google Scholar]
- 14.Solan S, Wallenstein S, Shapiro M, Teitelbaum SL, Stevenson L, Kochman A, et al. Cancer incidence in world trade center rescue and recovery workers, 2001–2008. Environ Health Perspect 2013;121(6):699–704 doi 10.1289/ehp.1205894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeig-Owens R, Webber MP, Hall CB, Schwartz T, Jaber N, Weakley J, et al. Early assessment of cancer outcomes in New York City firefighters after the 9/11 attacks: an observational cohort study. Lancet 2011;378(9794):898–905 doi 10.1016/S0140-6736(11)60989-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landgren O, Zeig-Owens R, Giricz O, Goldfarb D, Murata K, Thoren K, et al. Multiple Myeloma and Its Precursor Disease Among Firefighters Exposed to the World Trade Center Disaster. JAMA Oncol 2018;4(6):821–7 doi 10.1001/jamaoncol.2018.0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolli F, Maura M, Minvielle M, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun 2018;9(1)(3363) doi 10.1038/s41467-018-05058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014;25(1):91–101 doi 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018;132(6):587–97 doi 10.1182/blood-2018-03-840132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun 2019;10(1):3835 doi 10.1038/s41467-019-11680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rustad E, Yellapantula V, Glodzik D, Maclachlan K, Diamond B, Boyle E, et al. Revealing the impact of recurrent and rare structural variants in multiple myeloma. Blood Cancer Discovery 2020; 1(3)258–273 doi 10.1158/2643-3230.BCD-20-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landau H, Yellapantula V, Diamond B, Rustad E, Maclachlan K, Gundem G, et al. Accelerated single cell seeding in relapsed multiple myeloma. Nat Commun 2020; 11: 3617 doi 10.1038/s41467-020-17459-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, Teague JW, et al. cgpCaVEManWrapper: Simple Execution of CaVEMan in Order to Detect Somatic Single Nucleotide Variants in NGS Data. Curr Protoc Bioinformatics 2016;56:15 0 1–0 8 doi 10.1002/cpbi.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 2018;28(4):581–91 doi 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maura F, Rustad EH, Boyle EM, Morgan GJ. Reconstructing the evolutionary history of multiple myeloma. Best Pract Res Clin Haematol 2020;33(1):101145 doi 10.1016/j.beha.2020.101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature 2020;578(7793):122–8 doi 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maura F, Petljak M, Lionetti M, Cifola I, Liang W, Pinatel E, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2017. doi 10.1038/leu.2017.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018;173(3):611–23 e17 doi 10.1016/j.cell.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet 2015;47(12):1402–7 doi 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354(6312):618–22 doi 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Degasperi A, Amarante TD, Czarnecki J, Shooter S, Zou X, Glodzik D, et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancer 2020;1(2):249–63 doi 10.1038/s43018-020-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.