

Abstract
Regulatory T (Treg) cells have an essential role in maintaining immune homeostasis, in part by suppressing effector T (Teff) cell functions. Phosphoinositide-dependent kinase 1 (PDK1) is a pleiotropic kinase that acts as a key effector downstream of phosphatidylinositol-3-OH kinase (PI3K) in many cell types. In T cells, PDK1 has been shown to be critical for activation of NF-κB and AKT signaling upon T cell receptor ligation and is, therefore, essential for Teff cell activation, proliferation and cytokine production. Using Treg-specific conditional deletion, we now demonstrate that PDK1 is also essential for Treg suppressive activity in vivo. Ablation of Pdk1 specifically in Treg cells led to systemic, lethal, scurfy-like inflammation in mice. Genome-wide analysis confirmed that PDK1 is essential for the regulation of key Treg signature gene expression and, further, suggested that PDK1 acts primarily to control Treg gene expression through regulation of the canonical NF-κB pathway. Consistent with these results, the scurfy-like phenotype of mice lacking PDK1 in Treg cells was rescued by enforced activation of NF-κB downstream of PDK1. Therefore, PDK1-mediated activation of the NF-κB signaling pathway is essential for regulation of Treg signature gene expression and suppressor function.
Introduction
Regulatory T (Treg) cells maintain immune homeostasis and peripheral tolerance by suppressing immune responses by conventional effector T (Teff) cells and other immune cells. Treg and Teff cells develop from the same progenitors in the thymus and both require the engagement of the T cell receptor (TCR) complex for their development and function. However, the genetic programs activated by TCR conjugation in these two cells types and the function of these cells in the immune response are diametrically opposed. Given the importance of restoring and manipulating immune homeostasis in a variety of clinical settings, it is vital to understand how the components of the TCR signaling pathway function in regulatory and effector T cells.
Upon TCR and CD28 co-stimulation in conventional T cells, activation of phosphatidylinositol-3-OH kinase (PI3K) leads to accumulation of phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) and translocation of PDK1 to the plasma membrane. Activation of PDK1 leads to AKT phosphorylation (1–4), as well as activation of the protein kinase A-protein kinase G-protein kinase C (AGC) family of kinases (5). In T cells, the AGC kinase PKCθ is a crucial signaling intermediate downstream of TCR/CD28 stimulation and is required for full activation of the NF-κB pathway (6–8). PDK1 phosphorylates PKCθ, and brings together PKCθ and the CARMA1-Bcl10-Malt1 (CBM) complex (9, 10). Recruitment of phosphorylated PKCθ to the CBM leads to phosphorylation of CARMA1 (11, 12) and subsequent activation of NF-κB through IKK. Hence, in the absence of PDK1, NF-κB is not activated by TCR/CD28 co-stimulation (10). Despite these defects in conventional CD4 T cell (Tconv) activation, Cd4cre Pdk1f/f mice spontaneously developed colitis (13). This colitis resulted from activation of γδT cells due to defective regulatory T cell (Treg) function in the absence of PDK1. These results strongly suggested that PDK1 was required for either Treg development or suppressive activity, although it was not clear exactly how PDK1-dependent signaling contributed to Treg function. Given the pleiotropic activities of PDK1 in Teff cells, we suspected that PDK1 likely affected multiple aspects of Treg biology.
In the current study, we set out to examine the role of PDK1 in Tregs independent of thymic development, which could have been impacted in our previous studies that used Cd4cre (13). Conditional deletion of Pdk1 in lineage-committed Treg cells using Foxp3-driven Cre recombinase expression resulted in systemic, lethal inflammation due to complete loss of Treg function. There was significant overlap in genes whose expression was dysregulated upon deletion of Pdk1 in Tregs, and upon deletion of NF-κB Rela and Rel (14). Concordant gene expression changes, and Treg functional similarities, strongly suggested that the loss of PDK1 and canonical NF-κB influenced Treg function through the same mechanism. Remarkably, and in support of this hypothesis, the phenotype of mice lacking PDK1 in Tregs was rescued by constitutively activating NF-κB specifically in Treg cells. Taken together, these results demonstrate that PDK1 plays a critical role in the expression of the Treg transcriptome through its effect on NF-κB activation in Tregs, and hence in Treg function.
Materials and Methods
Mice
Pdk1-floxed mice (15) were crossed to Foxp3YFP-cre (Jackson laboratory). R26-StopFLikk2ca mice were purchased from Jackson laboratory and crossed to Foxp3YFP-cre and Foxp3YFP-cre Pdk1f/f mice.
Histology
Mouse tissues were fixed in 10% formalin and stored in 70% ethanol until staining. All fixed tissues were embedded in paraffin, sliced and stained with H&E by the Histology Core at the Columbia University Irving Medical Center. The disease severity score was determined by severity of immune cell infiltration: >4 (most severe), 2 to 3 (severe), 1 (mild), and 0. For ears, thickness of the epidermis was measured in AxioVision software.
Flow Cytometry and Sorting
Isolated cells were stained with αCD4, αCD8, αCD3, αTCRβ, αCD44, αGITR, αCTLA4, αKlrg1, αCD25, αCD62L, αFoxP3, αKi67, p-mTOR, p-S6, αIL- 10, αIL- 17 and αIFN-γ antibodies (eBioscience, Biolegend, BD biosciences or Tonbo Bioscience). For FoxP3, p-mTOR, p-S6, CTLA4 or Ki67 staining, cells were stained with the FoxP3 staining kit according to the manufacturer’s protocol (eBioscience). For cytokine staining, cells were stimulated with 50 ng/ml PMA and 1 μg/ml ionomycin in the presence of GolgiPlug (BD Bioscience) for 4 hours at 37°C and stained for surface markers followed by fixation with 4% paraformaldehyde or fixation buffer from the eBioscience kit. Then, the cells were permeabilized and stained for cytokines. Stained cells were acquired with LSRII or LSR Fortessa cytometers; data was analyzed using FlowJo (Treestar) (BD Bioscience). Magnetically-enriched (Miltenyi) CD4+ T cells were sorted into regulatory T cells (CD4+ YFP+) and conventional T cells (CD4+ YFP−) populations using a FACSAria (BD Bioscience).
In Vitro Suppression Assay
Tconv cells were sorted from WT mice and Treg cells were sorted from WT (Foxp3CRE-YFP) or conditional knockout mice. Tconv and Treg cells were co-cultured at different ratios in the presence of αCD3 and αCD28 antibodies. Cells were cultured for 3 days and 3H-thymidine was added to each well 8 hours before harvest. T cell proliferation was observed by measuring the incorporation of 3H-thymidine by the scintillation counter.
In Vivo Suppression Assay
Tconv cells were sorted from wildtype mice and Treg cells were sorted from wildtype or conditional knockout mice. 4 × 105 Tconv cells were transferred to Rag1−/− mice by i.v. injection alone or together with WT or knockout Treg cells (1 × 105). Weight was monitored weekly after adoptive transfer to observe progression of disease. Mice were euthanized when they reached 20% reduction of initial weight, and colons and other lymphoid organs were collected for H&E staining and flow cytometric analyses.
RNA-seq
Treg cells were sorted and RNA samples were prepared by using the RNeasy Mini Kit (Qiagen). Quantity and quality of RNA samples were measured by Bioanalyzer (Molecular Pathology core at Herbert Irving Comprehensive Cancer Center, Columbia University). Library construction and sequencing of Treg cells from Foxp3yfp-cre PDK1+/+ or Foxp3yfp-cre PDK1f/f were done by the Columbia Genome Center. Poly-A pull-down was used to enrich mRNAs from total RNA samples and libraries were prepared by using Illumina TruSeq RNA prep kit. Libraries were then sequenced using Illumina HiSeq2000. Library construction of Treg cells from Foxp3yfp-cre/+ PDK1+/+ or Foxp3yfp-cre/+ PDK1f/f was performed using NEBNext Ultra™ II RNA Library Prep Kit for Illumina in our lab and then sequenced by Hiseq2500. Upon sequencing, raw FASTQ files were aligned on the mm10 genome using STAR aligner with default parameters (16). Normalization and differential expression analysis were completed with the Bioconductor package DESeq2 after removing the batch effect using the ComBat function of the sva package v3.18.0. The data have been deposited in NCBI’s Gene Expression Omnibus and is available through GEO Series accession number GSE163663 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163663).
Results
Deletion of Pdk1 in Treg cells leads to lethal systemic inflammation
The development of colitis in aged Cd4cre Pdk1f/f mice suggested that PDK1 might have an important role in Treg cells (10). However, as Pdk1 was deleted early in thymopoiesis, it was not clear if the functional impairment was due to a defect in the development of Treg cells within the thymus (tTreg cells) or, potentially, extrathymic, peripheral generation of Treg (pTreg) cells. To investigate the role of PDK1 in mature Treg cells, we crossed Pdk1-floxed mice with Foxp3YFP-cre mice (17) to delete Pdk1 in Treg, but not Tconv cells (Figure 1A). Even though loss of Pdk1 did not affect the development of Treg cells in the thymus or proliferation of Treg cells in the non-inflammatory lymph nodes (supplementary Figure 1A, 1B and 1C), Foxp3YFP-cre/YFP-cre (referred to as Foxp3YFP-cre in the rest of the manuscript) Pdk1f/f mice died 2–4 weeks after birth. Foxp3YFP-crePdk1f/f mice were severely runted compared to their wildtype (WT) littermates (Figure 1B) and showed gross evidence of severe dermatitis (Figure 1C), splenomegaly and lymphadenopathy (Figure 1D). The length of the colon was shortened, consistent with severe colitis (Figure 1D). The large size of spleen and lymph nodes was accompanied by significantly increased cell numbers, indicative of lymphoproliferative diseases (Figure 1E). Lung and liver histology revealed significantly increased immune infiltration (Figure 1F). To further assess the systemic inflammatory phenotype of Foxp3YFP-crePdk1f/f mice, we analyzed the expression of activation markers on CD4+ and CD8+ T cells in the spleen and lymph nodes. Effector (CD44hiCD62Llo) and central (CD44hiCD62Lhi) memory CD4+ T cells were drastically increased compared to WT littermates (Figure 1G, H). Consistent with their inflammatory phenotype, production of IFNγ by CD4+ T cells was significantly increased in spleen and lymph nodes (Figure 1I, J). A similar trend was detected in CD4+ and CD8+ T cells from lung and colon lamina propria (LPL) (Figure 1I and data not shown). Moreover, we detected a trend towards increased IL-17A expression in the lymphoid tissues of Pdk1-deficient mice (Figure 1J). Taken together, these results demonstrate a requirement for PDK1 in maintaining immune tolerance.
Figure 1. Early lethal autoimmune syndrome upon Treg cell-specific deletion of Pdk1.
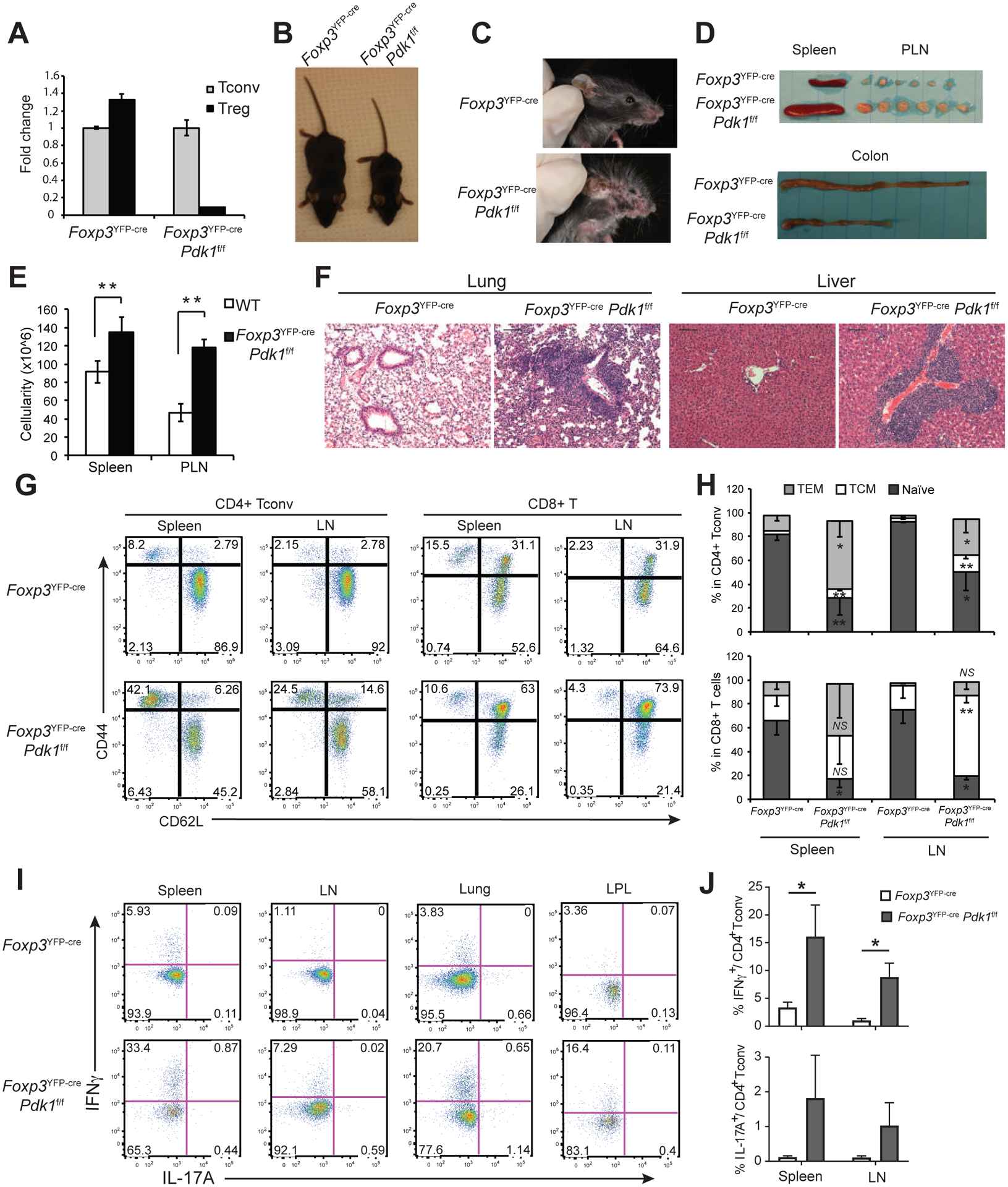
(A) Treg and Tconv cells were sorted from the spleen and LN of WT (FoxP3YFP-crePdk1+/+) and FoxP3YFP-crePdk1f/f, and Pdk1 expression was measured by RT-qPCR. (B, C) Representative photographs of 3-week-old mice. (D) Representative spleen, peripheral lymph nodes (PLN) and colon of 3-week-old mice. (E) Total cell count in spleen and PLN. (F) H&E staining of lung and liver sections from 3-week old mice. Bars=100μm; original magnification: 100X. (G-J) Lymphoid and non-lymphoid tissues of 3-week old mice were analyzed by flow cytometry. (G) Representative CD44 and CD62L expression in spleen T cells. (H) % of naïve (CD44lowCD62Lhi), central memory (TCM) (CD44hiCD62Lhi) and effector memory (TEM) (CD44hiCD62Llow) in spleen CD4+Foxp3− (Tconv) and CD8+ T cells. (I) Representative IL-17A and IFNγ expression in CD4+ Tconv in the indicated organs. Numbers indicate the % in quadrants. (J) Cumulative % of IFNγ+ and IL-17A+ in CD4+ T conv. Mean +/− S.D. is shown. Data is representative or cumulative of 3 to 6 mice per group from at least >3 experiments *p<0.05, **p<0.005, NS: non-significant.
PDK1 is required for Treg suppressive function
The early mortality and the systemic inflammatory phenotype of Foxp3YFP-crePdk1f/f mice resembled the phenotype of Scurfy mice, which lack functional FoxP3+ Treg cells. However, when we analyzed Foxp3YFP-crePdk1f/f mice we observed normal numbers of Treg cells in most tissues including thymus and peripheral LN (PLN), but a slightly decreased percentage in spleen (Figure 2A, B). Given this disconnect between Treg cell numbers and the systemic lymphoproliferative and inflammatory phenotype, we next examined the expression of Treg signature gene products, such as Cd44, Gitr, Ctla4, and Klrg1 (Supplementary Figures 2A and 2B). The expression of all of these proteins were downregulated in PDK1-deficient Foxp3yfp+ Treg cells from Foxp3YFP-cre/+Pdk1f/f mice, compared to WT Foxp3yfp+ Treg cells from Foxp3YFP-cre/+Pdk1+/+ control mice, while CD62L and CD25 were highly expressed. Moreover, Treg cells from Foxp3YFP-crePdk1f/f mice expressed reduced levels of the suppressive cytokine IL-10 (supplementary Figure 2C). Next, we tested the function of PDK1-deficient Treg cells by in vitro or in vivo suppression assay. Surprisingly, in in vitro suppression assays, Treg cells from Foxp3YFP-crePdk1f/f mice inhibited proliferation of conventional CD4+ T cells (Figure 2C). We then investigated the ability of PDK1-deficient Tregs to prevent weight loss in Rag1−/− mice following adoptive transfer of Tconv cells (Figure 2D). Colon histology confirmed that Pdk1−/− Treg cells were incapable of suppressing the development of colitis (Figure 2E). Taken together with the Scurfy-like phenotype of Foxp3YFP-crePdk1f/f mice, these results demonstrate that PDK1 is essential for the suppressive function of Treg cells in vivo.
Figure 2. Defective Treg cell homeostasis in FoxP3YFP-crePdk1f/f animals.
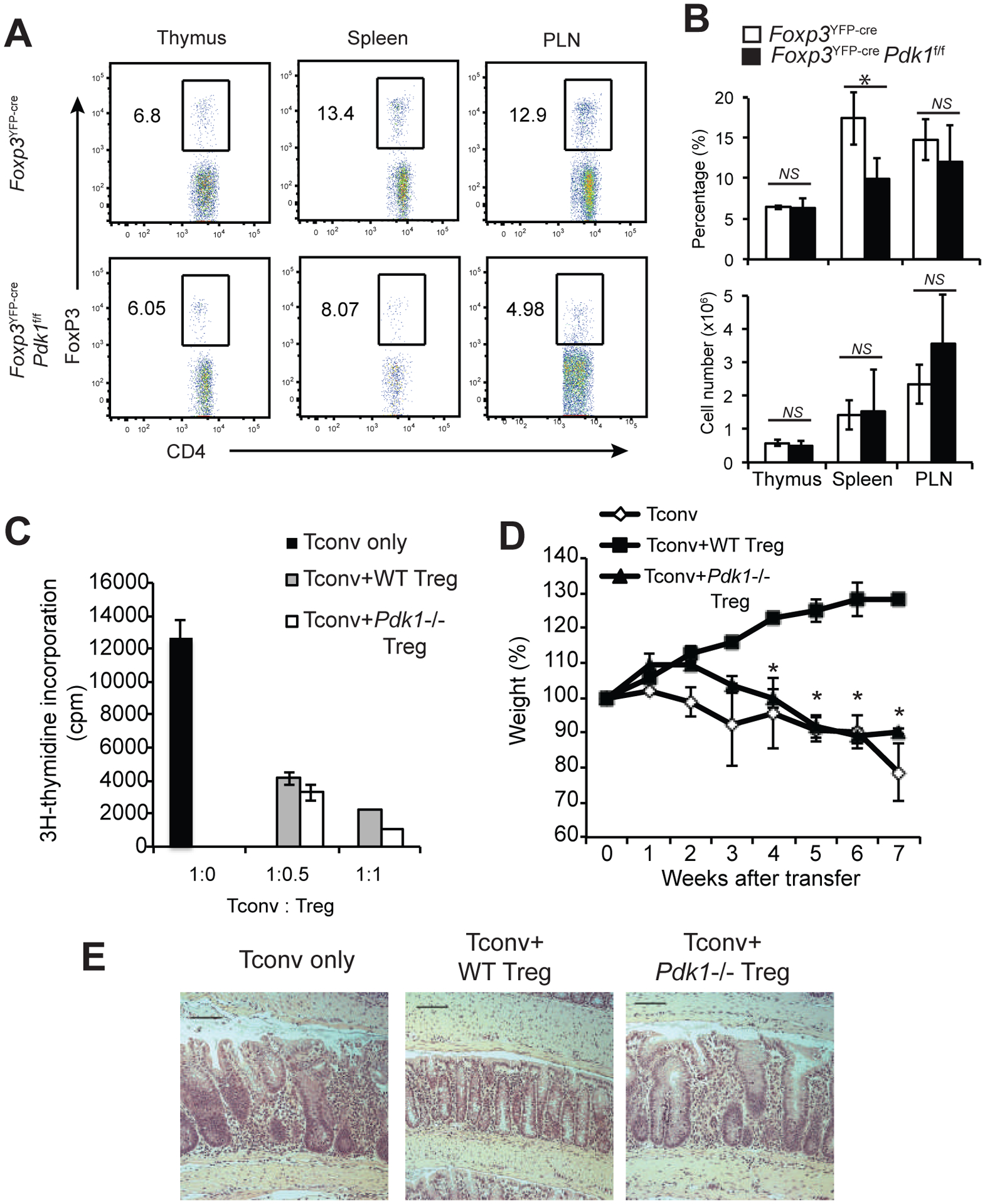
(A-B) Lymphoid and non-lymphoid tissues of 3-week old mice were analyzed by flow cytometry. (A) Representative Foxp3 expression in CD4+ T cells. Numbers indicate the % in the gate. (B) Cumulative % and number of Treg cells in the indicated tissues. (C) In vitro Treg suppression assay. (D-E) In vivo colitis suppression assay. (D) Weight curves, shown as % of original weight. (E) Representative colon histology 7 weeks after transfer. Bars=100μm; original magnification: 100X. Mean +/− S.D. is shown. Data is representative or cumulative of five mice per group from at least three experiments *p<0.05, NS: non-significant.
Dysregulation of the Treg transcriptome in the absence of PDK1
PDK1 regulates multiple signaling pathways in T cells including the canonical NF-κB pathway and Akt/mTOR pathways. To investigate the contribution of these, and other, PDK1-regulated pathways, to the phenotype observed in PDK1-deleted Treg cells, we analyzed their gene expression profiles. We performed RNA-seq using Tregs isolated from Foxp3YFP-cre (WT) and Foxp3YFP-crePdk1f/f mice. This analysis revealed widespread dysregulation of gene expression in the absence of PDK1 (Figure 3A). In Teff cells, PDK1 plays a key role in integrating CD28 co-receptor and TCR signaling. Consistent with this established role, pathway analysis (Gene Set Enrichment Analysis, GSEA) revealed significant effects of PDK1 deletion in Treg cells on the expression of genes associated with the CD28 pathway (Figure 3B). GSEA also revealed effects on the expression of genes involved in glucose transport (Figure 3C), consistent with the previously described role of PDK1 in T-cell metabolic reprogramming. Finally, and as predicted based on the loss of in vivo suppressive function of Treg cells lacking PDK1, we also observed significant dysregulation of Treg signature genes in the absence of PDK1 (Figure 3D). Taken together, these results are consistent with an essential role of PDK1 in TCR-associated signaling in Tregs. When we performed motif analysis using the DiRE database (18) to identify transcription factors (TF) that may have altered activity upon loss of PDK1, we found that NF-κB was the most significant TF associated with the differential gene expression in Treg cells lacking PDK1 (Figure 3E). Together with the phenotypic similarities resulting from loss of PDK1 and loss of canonical NF-κB in Treg cells (Oh et al., 2017), these results suggest that PDK1 may have an important role in regulating canonical NF-κB in Treg cells.
Figure 3. Dysregulation of the Treg transcriptome in the absence of PDK1.
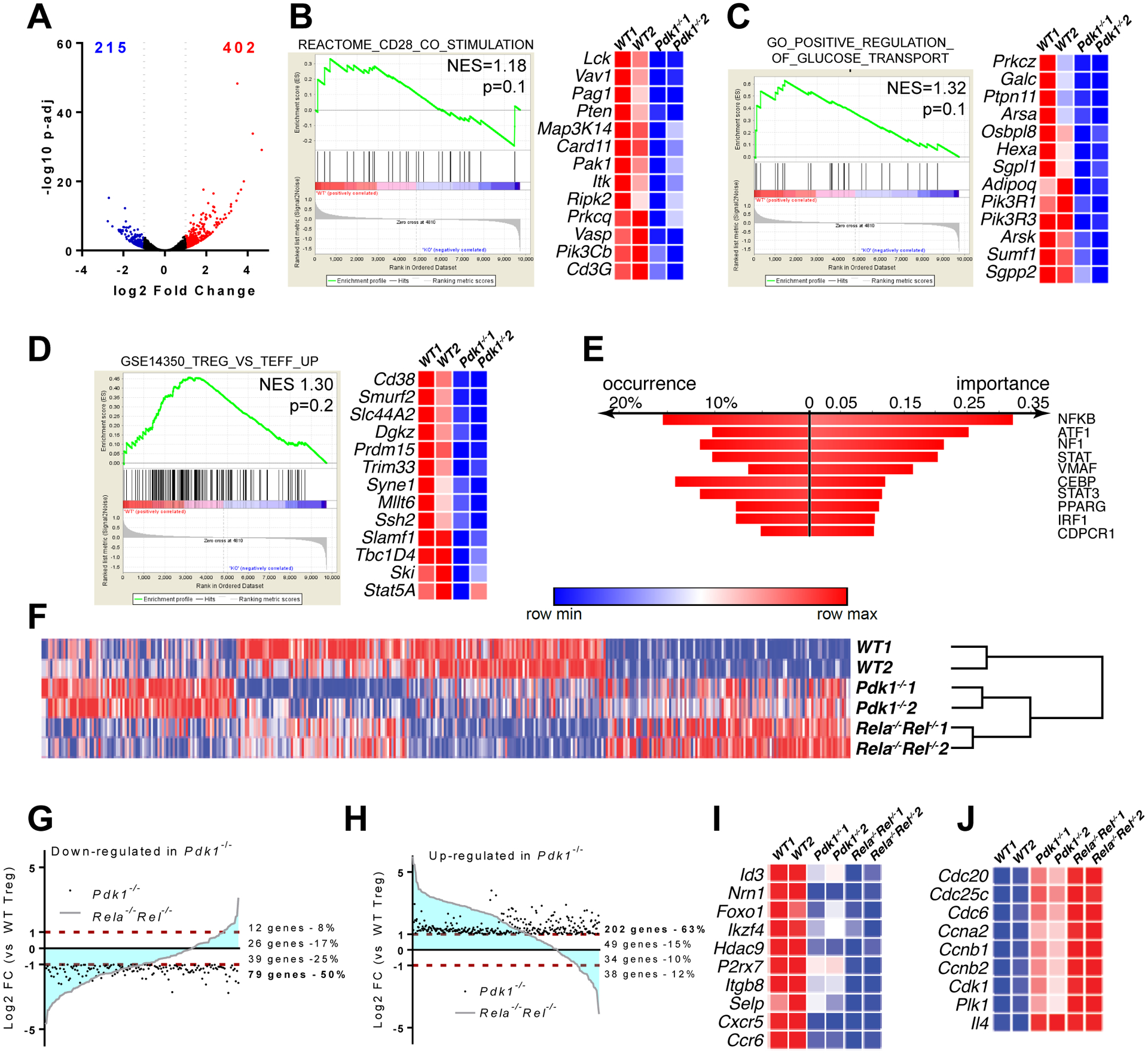
CD4+YFP+ Treg cells from spleen and PLN of (FoxP3YFP-cre and FoxP3YFP-crePdk1f/f were FACS-sorted and submitted to deep RNA-sequencing analysis. (A) Volcano plot showing differentially expressed genes (DEGs) (down-regulated log2 FC<−1 in blue, up-regulated log2 FC>1 in red). (B-D) Datasets were analyzed for signature enrichment using GSEA packages C2, C5 and C7. Representative Enplot graphs and expression heatmaps with selected genes are shown. (E) Transcription factor binding motif enrichment analysis using DiRE, in differentially expressed genes in Pdk1-deficient versus WT Treg cells. (F) Heatmap showing the expression of DEGs from (A), in WT, Pdk1−/− and Rela−/−Rel−/− Treg cells (data from Oh et al., 2017) upon batch correction and hierarchical clustering. (G, H) Plots showing the correlation of changes in gene expression between Pdk1−/− and Rela−/−Rel−/− Treg cells. (G) Genes significantly downregulated in Pdk1−/− cells. (H) Genes significantly upregulated in Pdk1−/− cells. (I, J). Expression heatmaps with selected Treg signature genes (I) and genes involved in cell cycle (J) are shown.
We therefore compared the Pdk1−/− Treg RNA-seq data to RNA-seq data from Treg cells isolated from Foxp3YFP-creRelaf/fRelf/f mice (Rela−/−Rel−/− Treg). Hierarchical clustering suggested that PDK1-deficient and cRel/p65-deficient Tregs shared common transcriptional changes, when compared to WT Treg cells (Figure 3F). Furthermore, we observed that the majority of genes that were significantly dysregulated in PDK1 deficient Treg cells (up- or downregulated) were similarly impaired in Treg cells lacking canonical NF-κB (Figure 3G,H). Pdk1−/− and Rela−/−Rel−/− Treg cells had decreased expression of genes implicated in Treg suppressive function (Figure 3I) including Id3 (19, 20); Nrn1 (encodes Neuritin) (21); and Foxo1 (22, 23). Furthermore, expression of Treg signature genes including Ikzf4, Hdac9 and P2rx7 were significantly reduced (Figure 3I). Genes encoding cell adhesion molecules such as Itgb8 and Selp were also downregulated in both Pdk1−/− and Rela−/−Rel−/−Tregs in addition to chemokine receptors (e.g. Cxcr5 and Ccr6), suggesting that defects in cell migration in the absence of PDK1 or canonical NF-κB may contribute to the defective suppressive function of PDK1-deficient Tregs in vivo (Figure 3I). Of note, at odds with Rela−/−Rel−/− Treg cells, expression of inflammatory cytokines was not increased in Pdk1−/− cells (not shown). Finally, both Pdk1−/− and Rela−/−Rel−/− Tregs displayed increased expression of genes involved in cell cycle progression and cell division (Figure 3J), which was likely a consequence of surrounding inflammatory mediators that are potent stimulators of Treg cell division (24). Considering that PDK1 is also involved in mammalian target of rapamycin (mTOR) signaling pathway, we analyzed phosphorylated mTOR and its downstream key player phosphorylated S6 (Supplementary Figure 1D and 1E). The results showed that there is no significant difference between WT and PDK1-deficient (KO) Treg cells. Overall, our genome-wide analysis of gene expression suggests that PDK1 signaling in Treg cells controls a pattern of gene sets involved in cell metabolism and Treg function that largely overlap with canonical NF-κB-regulated genes.
To eliminate any confounding effects of the spontaneous inflammation observed in Foxp3YFP-crePdk1f/f mice, we performed RNA-seq using CD4+Foxp3ypf+Treg cells isolated from Foxp3YFP-cre/+ (WT) and Foxp3YFP-cre/+Pdk1f/f (KO) mice. Due to X-chromosome inactivation, these mice do not develop an inflammatory phenotype, as only half of the Treg cells express Cre and are thus PDK1-deficient, while the remaining Tregs remain intact and functional. There were 774 differentially expressed genes (DEGs), among which 524 genes were downregulated, while 250 genes were upregulated in PDK1-deficient Treg cells (Supplementary Figures 3A and 3B and Supplementary Table1). Among the DEGs were 66 Treg signature genes, confirming that PDK1 expression is essential for normal Treg function (Supplementary Figure 3C). Expression of Ctla4, Klrg1, Cd44, IL-10 and Gitr were downregulated in the RNA-seq data set, which we confirmed by flow cytometry (Supplementary Figure 2). Importantly, GSEA identified an enrichment of known NF-κB binding sites in the DEG set (Supplementary Figure 3D, 3E, 3F and 3G). These data further corroborate the notion that PDK1 deficiency disrupts Treg cell function at least partly due to impaired NF-κB activation.
Forced activation of NF-κB in Treg cells rescues the phenotype of Foxp3YFP-cre Pdk1f/fmice
We have found that canonical NF-κB activity is essential for the suppressive function of Treg cells (14). In contrast, several reports suggest AKT signaling may oppose Treg cell suppressive function and Treg signature genes (25, 26). In this context, the results of our RNA-seq analysis strongly suggest that the loss of suppressive function in PDK1-deficient Tregs is due to loss of canonical NF-κB activation. If NF-κB is the key downstream effector of PDK1 in Treg cells, then loss of PDK1 in Treg cells would be overcome by activating NF-κB in a PDK1-independent manner. Therefore, we crossed Foxp3YFP-cre Pdk1f/f mice to R26-StopFLikk2ca, a transgenic strain that conditionally expresses a constitutively active form of IκB kinase 2 (IKKβ). IKKβCA would function downstream of PDK1 to directly drive activation of the canonical NF-κB pathway. Strikingly, the lethal inflammatory phenotype seen in the Foxp3YFP-cre Pdk1f/f mice was rescued by activation of NF-κB in Treg cells. Histological evidence of inflammatory infiltrates in lung and liver (Figure 4A) and epidermal thickening of ears seen in Foxp3YFP-cre Pdk1f/f mice were both dramatically reduced upon conditional expression of constitutively active IKKβ-CA (Figure 4B, C). Quantification of disease severity in lung, liver and skin shows that the inflammatory infiltrate resulting from Treg cell PDK1 deficiency is significantly reversed by constitutive activation of NF-κB (Figure 4D). These results therefore demonstrate that loss of suppressive function in PDK1 deficient Treg cells in vivo can be rescued by downstream activation of the canonical NF-κB pathway. These studies, placed in the context of previous analyses of NF-κB and AKT signaling in Tregs, strongly support a model in which the primary function of PDK1 in Treg cells, in contrast to Tconv cells, is to mediate TCR induced activation of NF-κB and thereby help maintain the Treg signature transcriptome.
Figure 4. Constitutive activation of canonical NF-κB in Treg cells rescues the inflammatory phenotype in Pdk1-deficient mice.
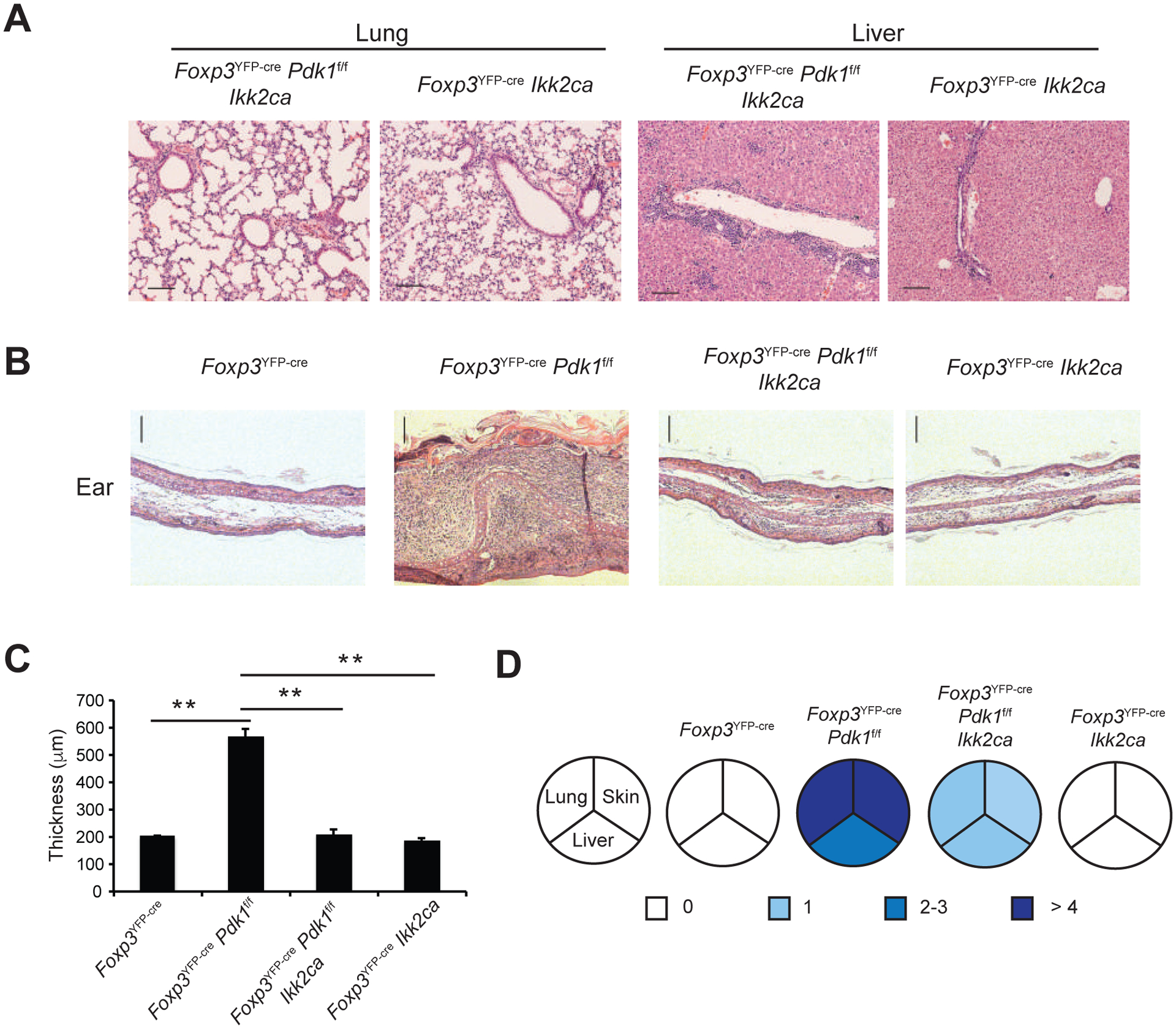
Foxp3YFP-cre Pdk1f/f mice were crossed to R26-StopFLikk2ca (Ikk2ca) mice. H&E staining of lung and liver sections from 3-week old mice. Bars=100μm; original magnification: 100X. (B,C) Representative H&E staining (B) and cumulative ear thickness of 3-week old mice of the indicated genotypes (C, Mean +/− S.D. is shown). (D) Inflammatory features in the lung, skin and liver of each genotype, as described in the Methods section. Data is representative or cumulative of five mice per group from five experiments. **p<0.005.
Discussion
PDK1 is a pleiotropic kinase, responsible for the regulation of multiple signaling pathways downstream of the numerous events that influence PI3K activity (27). In lymphocytes, there has been significant interest in dissecting the contributions of PDK1 to the multiple pathways activated upon antigen receptor signaling. Using a variety of genetic models and tools, PDK1 has been definitively shown to have a key role in both B and T cell development (28–32). The contributions of PDK1, and the downstream signaling pathways regulated by PDK1, to the functional response of B and T cells following antigen-receptor ligation, however has remained more controversial. We previously demonstrated that PDK1 is required for the activation of NF-κB by TCR/CD28 ligation in CD4+ T cells and that PDK1 is, therefore, important for T cell activation and cytokine production (10). Other studies demonstrated that PDK1 was also required for AKT activation downstream of the T cell receptor, and that this signaling was essential for regulation of mTOR (1), Foxo1 (33) and NOTCH (34) signaling pathways in T cells. In CD8+ T cells, it has been shown that in CTLs, PDK1 maintains IL-2 driven proliferation and controls glucose metabolism and glycolysis through an mTOCR1-HIF1 pathway-regulated transcriptional program (35, 36). Hence, loss of PDK1 results in substantive disruption of conventional T cell functions.
Despite the established role of PDK1 in T cell responses, we previously observed that Cd4cre Pdk1f/f mice, which have defective conventional T cells (10), develop colitis (13). TCRγδ+ T cells, which are not affected by CD4-mediated deletion of PDK1, were found to be driving colitis in this model (13). In this model, PDK1 expression was lost in all TCRαβ+ T cells, including Tconv and Treg cells. As PDK1 is essential for TCR signaling and thereby Tconv function, Tconv cells could not have contributed to the development of spontaneous colitis in Cd4cre Pdk1f/f mice. These results therefore suggested that PDK1 might have an important role in the suppressive function of Treg cells, which possess the ability to suppress activation of TCRγδ+ T cells in intestines (13). However, the exact role of PDK1 in Treg cells had remained unclear. For example, PKCθ, a well described downstream target of PDK1, is involved in Treg development (37), whereas inhibition of PKCθ surprisingly augments Treg function (38). A recent study showed that Treg cells lacking PKCθ are normally suppressive in vitro (37, 39), but the activity of PKCθ-deficient Tregs in vivo (e.g. in the colitis transfer model) was not tested. Of note, we recently established that canonical the NF-κB subunits, p65 and c-Rel, play critical roles in the maintenance of Treg identity and function (Oh et al, 2017). However, despite the NF-κB-deficient Tregs being completely defective in in vivo suppression of transfer colitis, they were fully active in in vitro assays of suppression. Therefore, it is possible that absence of PKCθ, similar to the absence of PDK1, leads to functionally inactive Tregs.
To definitively determine the role of PDK1 in mature Treg cells, we deleted Pdk1 specifically in Tregs using Foxp3YFP-cre. The resulting severe, systemic inflammatory phenotype in Foxp3YFP-crePdk1f/f mice resembles the Scurfy phenotype, which is observed in the absence of FoxP3+ Treg cells, suggesting that PDK1 is critical for the suppressive function of Treg cells in vivo (Figure 1, 2). These results are consistent with the phenotype of mice lacking the canonical NF-κB subunits p65 and c-Rel (14), or those lacking IKKβ (40), in Treg cells. Given that the roles of AKT and mTOR in Treg function are far less definitive, and are unlikely to account for the observed phenotype (41, 42), it is more likely that it a lack of PDK1-induced NF-κB activation is responsible for the observed effects on Treg function. Consistent with this hypothesis, genome-wide gene expression analysis of Pdk1−/− Tregs demonstrated enrichment of genes with NF-κB motifs in their promoters amongst the differentially expressed genes, and showed a significant overlap between Pdk1−/− and Rela−/−Rel−/− Treg cells (Figure 3). In contrast, gene signatures related to cholesterol/lipid metabolism, which are the accepted targets for AKT/mTOR pathways, were not significantly perturbed in Pdk1−/− Tregs. Taken together, both the phenotypic, and genomic data suggest that NF-κB is the primary downstream target of PDK1 in Treg cell function. To clearly demonstrate that loss of canonical NF-κB accounts for the Scurfy phenotype of Cd4cre Pdk1f/f mice, we utilized conditional expression of a constitutively active form of IKKβ to activate NF-κB downstream of PDK1 (Figure 4). Strikingly, these mice no longer exhibited the Scurfy-like phenotype, demonstrating that PDK1 mediated activation of NF-κB is essential for Treg function. These results definitively establish that PDK1 is essential for Treg suppressive function in vivo and that this function is likely manifested through signaling to NF-κB. These results also suggest the need for further efforts to understand the signaling pathways leading to NF-κB activation in Treg cells. The severe, Scurfy-like phenotype exhibited by mice lacking canonical NF-κB (14, 40) or PDK1 in Treg cells is far more dramatic than that observed upon loss of CD28 (43) or TCR (44) in Treg cells. Taken together with the still unclear role of PKCθ in Treg cells, these results suggest that PDK1-mediated NF-κB activation may occur downstream of additional Treg signaling pathways. Complete elucidation of the signals that regulate PDK1 and NF-κB in Treg cells will therefore be essential for the continued development of immunomodulatory therapies in autoimmune diseases and cancer.
Supplementary Material
Key points:
PDK1 is required for Treg suppressive function
PDK1-deficient Treg cells have impaired NF- κB signaling
Forced activation of NF-κB rescues the phenotype of Foxp3YFP-cre Pdk1f/f mice
Acknowledgments
This work was supported by National Institute of Allergy and Infectious Diseases, National Institutes of Health Grant R01AI068977 (to S.G.).
Footnotes
Supplementary Table: Differentially expressed genes in WT vs KO Treg cells
CD4+YFP+ Treg cells from spleens and PLNs of FoxP3YFP-cre/+ Pdk1+/+(WT) and FoxP3YFP-cre/+Pdk1f/f (KO) were sorted and RNA was isolated and RNA-seq libraries were constructed and sequenced by Hiseq2500. Differentially expressed genes (DEGs) (down-regulated log2 FC<−1 in blue, up-regulated log2 FC>1 in red, p adj<0.05) were included in the table.
References
- 1.Calleja V, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, Downward J, Parker PJ, and Larijani B. 2007. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol 5: e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costello PS, Gallagher M, and Cantrell DA. 2002. Sustained and dynamic inositol lipid metabolism inside and outside the immunological synapse. Nat Immunol 3: 1082–1089. [DOI] [PubMed] [Google Scholar]
- 3.Garcon F, Patton DT, Emery JL, Hirsch E, Rottapel R, Sasaki T, and Okkenhaug K. 2008. CD28 provides T-cell costimulation and enhances PI3K activity at the immune synapse independently of its capacity to interact with the p85/p110 heterodimer. Blood 111: 1464–1471. [DOI] [PubMed] [Google Scholar]
- 4.Bayascas JR, Wullschleger S, Sakamoto K, Garcia-Martinez JM, Clacher C, Komander D, van Aalten DM, Boini KM, Lang F, Lipina C, Logie L, Sutherland C, Chudek JA, van Diepen JA, Voshol PJ, Lucocq JM, and Alessi DR. 2008. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol Cell Biol 28: 3258–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mora A, Komander D, van Aalten DM, and Alessi DR. 2004. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15: 161–170. [DOI] [PubMed] [Google Scholar]
- 6.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, and Littman DR. 2000. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404: 402–407. [DOI] [PubMed] [Google Scholar]
- 7.Lin X, O’Mahony A, Mu Y, Geleziunas R, and Greene WC. 2000. Protein kinase C-theta participates in NF-kappaB activation induced by CD3-CD28 costimulation through selective activation of IkappaB kinase beta. Mol Cell Biol 20: 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coudronniere N, Villalba M, Englund N, and Altman A. 2000. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci U S A 97: 3394–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KY, D’Acquisto F, Hayden MS, Shim JH, and Ghosh S. 2005. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science 308: 114–118. [DOI] [PubMed] [Google Scholar]
- 10.Park SG, Schulze-Luehrman J, Hayden MS, Hashimoto N, Ogawa W, Kasuga M, and Ghosh S. 2009. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat Immunol 10: 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Wang D, and Lin X. 2005. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity 23: 575–585. [DOI] [PubMed] [Google Scholar]
- 12.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, and Rawlings DJ. 2005. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23: 561–574. [DOI] [PubMed] [Google Scholar]
- 13.Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS, and Ghosh S. 2010. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity 33: 791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, Hayden MS, Klein U, Rabadan R, and Ghosh S. 2017. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 47: 450–465 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto N, Kido Y, Uchida T, Asahara S, Shigeyama Y, Matsuda T, Takeda A, Tsuchihashi D, Nishizawa A, Ogawa W, Fujimoto Y, Okamura H, Arden KC, Herrera PL, Noda T, and Kasuga M. 2006. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat Genet 38: 589–593. [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., Muller W, and Rudensky AY. 2008. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28: 546–558. [DOI] [PubMed] [Google Scholar]
- 18.Gotea V, and Ovcharenko I. 2008. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic acids research 36: W133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, Varki N, Chang AN, Broide DH, and Murre C. 2014. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol 15: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rauch KS, Hils M, Lupar E, Minguet S, Sigvardsson M, Rottenberg ME, Izcue A, Schachtrup C, and Schachtrup K. 2016. Id3 Maintains Foxp3 Expression in Regulatory T Cells by Controlling a Transcriptional Network of E47, Spi-B, and SOCS3. Cell Rep 17: 2827–2836. [DOI] [PubMed] [Google Scholar]
- 21.Barbi JJ, Vignali PDA, Yu H, Pan F, and Pardoll D. 2016. The Neurotrophic Factor Neuritin Maintains and Promotes the Function of Regulatory T cells in Autoimmunity and Cancer. The Journal of Immunology 196: 58.12–58.12. [Google Scholar]
- 22.Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, Meijer D, Zhao K, Rudensky AY, Atwal G, Zhang MQ, and Li MO. 2012. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491: 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, Katayama CD, and Hedrick SM. 2010. Foxo transcription factors control regulatory T cell development and function. Immunity 33: 890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, Martin GH, Elhage R, Derian N, Carpentier W, Marodon G, Klatzmann D, Piaggio E, and Salomon BL. 2010. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. The Journal of clinical investigation 120: 4558–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitz A, de Marcken M, Gautron AS, Mitrovic M, Hafler DA, and Dominguez-Villar M. 2016. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep 17: 1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, Shokat KM, Fisher AG, and Merkenschlager M. 2008. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proceedings of the National Academy of Sciences of the United States of America 105: 7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearce LR, Komander D, and Alessi DR. 2010. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11: 9–22. [DOI] [PubMed] [Google Scholar]
- 28.Kelly AP, Hinton HJ, Clarke RG, and Cantrell DA. 2006. Phosphoinositide-dependent kinase l (PDK1) haplo-insufficiency inhibits production of alpha/beta (alpha/beta) but not gamma delta (gamma/delta) T lymphocytes. FEBS Lett 580: 2135–2140. [DOI] [PubMed] [Google Scholar]
- 29.Hinton HJ, Alessi DR, and Cantrell DA. 2004. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat Immunol 5: 539–545. [DOI] [PubMed] [Google Scholar]
- 30.Baracho GV, Cato MH, Zhu Z, Jaren OR, Hobeika E, Reth M, and Rickert RC. 2014. PDK1 regulates B cell differentiation and homeostasis. Proc Natl Acad Sci U S A 111: 9573–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park SG, Long M, Kang JA, Kim WS, Lee CR, Im SH, Strickland I, Schulze-Luehrmann J, Hayden MS, and Ghosh S. 2013. The kinase PDK1 is essential for B-cell receptor mediated survival signaling. PLoS One 8: e55378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venigalla RK, McGuire VA, Clarke R, Patterson-Kane JC, Najafov A, Toth R, McCarthy PC, Simeons F, Stojanovski L, and Arthur JS. 2013. PDK1 regulates VDJ recombination, cell-cycle exit and survival during B-cell development. EMBO J 32: 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, and Cantrell DA. 2009. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med 206: 2441–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, and Cantrell DA. 2007. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J 26: 3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, Feijoo C, Okkenhaug K, and Cantrell DA. 2011. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, and Cantrell DA. 2012. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med 209: 2441–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, and Sun Z. 2008. Differential requirement of PKC-theta in the development and function of natural regulatory T cells. Mol Immunol 46: 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, Blazar BR, Abramson SB, Lafaille JJ, and Dustin ML. 2010. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science 328: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegmund K, Thuille N, Wachowicz K, Hermann-Kleiter N, and Baier G. 2017. Protein kinase C theta is dispensable for suppression mediated by CD25+CD4+ regulatory T cells. PLoS One 12: e0175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heuser C, Gotot J, Piotrowski EC, Philipp MS, Courreges CJF, Otte MS, Guo L, Schmid-Burgk JL, Hornung V, Heine A, Knolle PA, Garbi N, Serfling E, Evaristo C, Thaiss F, and Kurts C. 2017. Prolonged IKKbeta Inhibition Improves Ongoing CTL Antitumor Responses by Incapacitating Regulatory T Cells. Cell Rep 21: 578–586. [DOI] [PubMed] [Google Scholar]
- 41.Haxhinasto S, Mathis D, and Benoist C. 2008. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 205: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng H, Yang K, Cloer C, Neale G, Vogel P, and Chi H. 2013. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499: 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, and Turka LA. 2013. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest 123: 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine AG, Arvey A, Jin W, and Rudensky AY. 2014. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.