

Abstract
RAS gene mutations are the most frequent oncogenic event in lung cancer. They activate multiple RAS-centric signaling networks among them the MAPK, PI3K and RB pathways. Within the MAPK pathway ERK1/2 proteins exert a bottleneck function for transmitting mitogenic signals and activating cytoplasmic and nuclear targets. In view of disappointing anti-tumor activity and toxicity of continuously applied MEK inhibitors in patients with KRAS mutant lung cancer, research has recently focused on ERK1/2 proteins as therapeutic targets and on ERK inhibitors for their ability to prevent bypass and feedback pathway activation. Here we show that intermittent application of the novel and selective ATP-competitive ERK1/2 inhibitor LY3214996 exerts single-agent activity in patient-derived xenograft (PDX) models of RAS mutant lung cancer. Combination treatments were well tolerated and resulted in synergistic (ERKi plus PI3K/mTORi LY3023414) and additive (ERKi plus CDK4/6i abemaciclib) tumor growth inhibition in PDX models. Future clinical trials are required to investigate if intermittent ERK inhibitor-based treatment schedules can overcome toxicities observed with continuous MEK inhibition and - equally important - to identify biomarkers for patient stratification.
Keywords: RAS, ERK inhibitor, LY3214996, LY3023414, abemaciclib, FOXO3a, Bim, lung cancer, Bliss synergy, PDX model
Introduction
Non-small cell lung cancers (NSCLC) with aberrations in the rat sarcoma oncogene family (H-, N-, KRAS) represent 30% of all lung tumors (1). RAS mutations activate multiple downstream signaling pathways, among them MAPK and PI3K signaling both of which converge on the Cyclin D1/CDK4/6-RB axis (2,3). Besides this direct mechanistic link to mutant RAS, RB signaling is activated by co-occurring genetic events such as loss of CDKN2A (encoding for p16/p14ARF) or amplification of CDK4 or CCND1 (encoding for Cyclin D1) (4-6). These effector pathways play pivotal roles for cell cycle progression, proliferation and apoptotic resistance of cancer cells (7). Yet, abemaciclib - a CDK4/6 inhibitor - had only limited single-agent activity in clinical trials (8,9) and MEK inhibitors administered on an uninterrupted schedule exhibited toxicity and poor anti-tumor activity in lung cancer patients (10,11). Ultimately, MEK and PI3K inhibitor combinations caused significant toxicity in humans (2,6,12,13).
Despite these setbacks, it remains an attractive strategy to inhibit RAS dependent effector pathways, since direct KRASG12C and immune checkpoint inhibitor (ICI) activity is limited to subgroups of patients with RAS mutant cancers (14-16). ERK1/2 proteins exert a bottleneck function in activating cytoplasmic and nuclear targets and therefore, ERK inhibitors are considered to be more potent in preventing bypass and feedback activation than MEK inhibitors (17-19). So far, however, only few ERK inhibitors have been tested in clinical trials for solid tumors despite demonstrating efficacy in preclinical models of solid tumors and in BRAF and/or MEK inhibitor-refractory melanoma patients (20-24).
In the present study, we used models of RAS mutant lung cancer derived from patients pretreated with multiple treatment modalities (including chemo+/−radiotherapy and ICIs) and from genetically engineered mouse models (GEMM) to investigate the efficacy of the novel ERK1/2 inhibitor LY3214996 alone and in combination with a PI3K/mTOR (LY3023414) (25) or CDK4/6 inhibitor (abemaciclib) (26). LY3214996 is effective in preclinical models across several cancer types (27,28) and currently being evaluated in Phase I trials as monotherapy or in combination treatments for patients with advanced cancer (NCT02857270) (29).
Materials and Methods
Detailed information is presented in Supplementary Materials and Methods.
Patient derived cell lines were generated from malignant pleural effusions or ascites collected under a Dana-Farber Cancer Institute IRB approved protocol. All patients provided written informed consent and the studies were conducted in accordance with the declaration of Helsinki. A targeted next-generation sequencing (NGS) cancer genomic assay (“Dana-Farber Cancer Institute (DFCI) OncoPanel”) was used to detect cell line specific somatic mutations, copy number variations and structural variants in tumor DNA. All cell lines were kept at 37 °C in complete media supplemented with 10% fetal bovine serum (FBS). DFCI168 (30), DFCI316, DFCI366, DFCI516 cells were grown in RPMI1640 media, DFCI24, DFCI298 and DFCI332 in ACL4 media. Murine cell lines were derived from previously described genetically engineered mouse models and grown in DMEM (GEMMs) (31,32). All cell lines tested negative for Mycoplasma throughout the study, were last tested on October 8th, 2020 and used until passage twenty-five for functional assays. LY3214996 (example #1) (33), abemaciclib (example #1) (34) and LY3023414 (example #1) (35) were discovered at the Lilly Research Laboratories and synthesized as described in the respective patents. Selumetinib/AZD6244 (36), SCH772984 (37), afatinib/BIBW2992 (38) and linsitinib/OSI-906 (39) were purchased from SelleckChem. Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. The list of antibodies used can be found in Supplementary Materials and Methods. Western blot band intensities were quantified with ImageJ. All in vitro experiments were performed under adherent cell culture condition and live cell imaging was performed with an IncuCyte ZOOM™ real-time imaging system. Bliss synergy was determined and visualized with Combenefit (40). The Firefly-luciferase expressing FHRE-Luc reporter plasmid was a gift from Michael Greenberg (Addgene plasmid #1789; http://n2t.net/addgene:1289 ; RRID:Addgene_1789). A renilla luciferase-expressing pRL-CMV control plasmid was used for normalization. Luciferase activities were quantified with the Dual-Glo® Luciferase Assay system. Nuclear-cytoplasmic fractionation was performed with the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents. For patient-derived xenograft (PDX) models, female NSG (NOD SCID Gamma, Bar Harbor, Maine) and NCr nude mice (Taconic Biosciences, Rensselaer, NY), were housed and treated in accordance with protocols approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. Tumor growth delay was calculated as previously described (41). MAPK pathway-dependent gene expression was quantified with a previously published 6-gene signature (DUSP4, DUSP6, ETV4, ETV5, SPRY2, PHLDA1) (42). A p-value less than 0.05 was considered statistically significant for all datasets. “*” indicates p<0.05, “**” p<0.01, “***” p<0.001 and “****” p<0.0001. Statistical analyses were performed with GraphPad Prism 8 and SAS (Version 9.3).
Results
Single agent activity of LY3214996 in RAS-mutant patient-derived lung cancer cell lines
We sought to establish cancer cell lines reflecting the clinical distribution of RAS mutations among lung cancer patients. In the TCGA Pan-Lung cancer dataset, about 30% of tumors exhibit aberrations in the RAS gene family including activating gene mutations (Suppl. Fig. 1). KRAS is overall more frequently affected (23%) than N- (2.6%) or HRAS (1.7%) (43). Six out of seven cancer cell lines from patients who had previously undergone treatment for metastatic RAS mutant lung cancer (Fig. 1A: bright-field microscopy images; Suppl. Fig. 2: patient histories) had KRAS mutations, one (DFCI168) had an NRAS mutation (30) and none had HRAS mutations. All patient-derived cell lines were genetically characterized by “OncoPanel” next-generation sequencing (NGS) at DFCI (44). We observed a high degree of concordance of genetic events between the available initial tumor biopsies and the established cell lines (Suppl. Table 1). Initially, we determined the absolute inhibitory concentrations (IC50 values) of single-agent LY3214996 after 72h. DFCI168NRASQ61K (1.1μM) and DFCI516KRASG12C (1.5μM) exhibited the highest, DFCI316KRASQ61H (3.6μM), DFCI24KRASG12C (4.9μM) and DFCI366KRASG12D (9.1μM) intermediate and DFCI298KRASG12C and DFCI332KRASG12D the lowest sensitivity (>10μM) to LY3214996 (Fig. 1B). Despite a paradoxical dose-dependent increase in pERK1/2Thr202/Tyr204 and pMEK1/2Ser217/221 following treatment with LY3214996, signaling downstream of pERK1/2 remained occluded as indicated by potent inhibition of phosphorylation of p90RSKThr359/Ser363 and S6 ribosomal proteinSer217/221 as well as reduction of c-MYC, DUSP4 and SPRY2 protein levels (Fig. 1C). We next established that compensatory ERK1/2 phosphorylation which was not induced in response to the structurally different ERK inhibitor SCH772984 (45) could be prevented by selumetinib (MEK inhibitor) pretreatment (Suppl. Fig. 3). Afatinib (pan-ErbB inhibitor) and linsitinib (IGFR inhibitor) had no effect on early ERK1/2 phosphorylation excluding a major involvement of these upstream RTKs in early ERK phosphorylation. Importantly, ERK target inhibition with LY3214996 (1μM) increased proportionally to treatment duration as evidenced by decreasing effector levels over 48 hours (Fig. 1D). Pathway inhibition was accompanied by stronger accumulation of pro-apoptotic BIM in sensitive (DFCI168NRASQ61K) and intermediate sensitive cell lines (DFCI316KRASQ61H) which also exhibited more prominent PARP cleavage. LY3214996 treatment activated compensatory PI3K signaling (pAKTSer473) over baseline in all cell lines to varying extents (most evident in resistant cell lines and DFCI316KRASQ61H cells).
Figure 1: LY3214996 single agent activity in vitro.

(A) Bright-field microscopy images of the seven patient-derived RAS mutant lung cancer cell lines used in this study (scale bar = 150μM). (B) Absolute IC50 values for LY3214996 (in μM) in patient-derived cell lines after 72 h. (C) Western blot analysis of cell lysates from patient-derived cell lines treated for 24 h with increasing doses of LY3214996 (dose range 10-10,000nM). (D) Western blot analysis of lysates from patient-derived cell lines treated for up to 48 h with 1μM of LY3214996. (E) Overall ERK-dependent transcriptional output (6-gene signature) of cell lines treated for up to 48 h with 1μM of LY3214996. (F) Growth kinetics of patient-derived cell lines treated with increasing doses of LY3214996 over 72 h. (G) FOXO3a reporter activity in transiently transfected patient-derived cancer cell lines after 24 h of LY3214996 (1μM) treatment. (H) Western blot analysis of cytoplasmic and nuclear protein fractions of LY3214996-sensitive DFCI168 and -resistant DFCI332 cells after 24 h of treatment with 1μM of LY3214996.
Subsequently, we quantified the expression of MAPK pathway-dependent effector genes during LY3214996 (1μM) treatment, using a previously published MAPK pathway activation gene set (42) and observed a decrease in mean pathway-dependent transcription in all cell lines over 48 hours (Fig. 1E). In DFCI24 and DFCI332 cells, mean expression levels increased slightly after 48 hours over levels observed at 24 hours but did not reach levels of untreated cells. Despite similar pathway suppression, dose-dependent inhibition of cancer cell proliferation with LY3214996 differed in cell lines over 72 hours and DFCI322KRASG12D cells were left unaffected with drug concentrations of up to 10μM (Fig. 1F). To investigate potential mechanisms of differential drug responses, we performed FOXO3a luciferase reporter assays since FOXO3a nuclear translocation and transcriptional activity has been shown to influence MEK inhibitor sensitivity (46). The more sensitive cell lines (DFCI168, DFCI24) exhibited increased FOXO3a reporter activity after 24 hours of LY3214996 (1μM) treatment, whereas reporter activity remained unchanged in resistant cell lines (DFCI366, DFCI298, DFCI332) and was reduced in intermediate sensitive DFCI316 cells (Fig. 1G). Cytoplasmic-nuclear fractionation experiments corroborated increased nuclear accumulation of FOXO3a in sensitive DFCI168 cells while FOXO3a levels were left unaffected in resistant DFCI332 cells (Fig. 1H).
Single agent activity of LY3214996 in RAS mutant lung cancer PDX models
To test the in vivo single agent activity, we treated DFCI168NRASQ61K tumor-bearing NSG mice with LY3214996 (100mg/kg, PO QD) and observed a significant reduction in tumor growth compared to tumors of vehicle treated animals (Fig. 2A, n=8 animals/group, p<0.0001; Two-way Repeated Measures ANOVA with standard post hoc t-tests) which translated into a survival benefit despite treatment cessation after 21 days (median survival time: 49 days vs. 29.5, Fig. 2B, p=0.0003, Log-Rank test). Pharmacodynamic analyses (PD) demonstrated inhibition of ERK targets at 4 hours, including p90RSKThr359/Ser363 - a reliable PD biomarker for the extent of ERK inhibition (22) - of S6 ribosomal proteinSer235/236 and reduction of total c-MYC, DUSP4 and SPRY2 protein levels (Fig. 2C). However, MAPK pathway activity recovered by 16 and 24 hours post-treatment. These pharmacodynamic effects correlated with plasma concentrations of LY3214996. At 4 hours, the LY3214996 plasma concentration was 3840±654 nM resulting in 66% inhibition of pRSK. As the plasma concentrations declined at 16 (189±267 nM) and 24 hours (9 nM), pRSK inhibition declined to 33% and 34%, respectively (Fig. 2D). AKTSer473 phosphorylation slightly increased at 4 hours indicating PI3K pathway activation and gradually decreased again over the next 20 hours. Consistent with strong MAPK pathway inhibition at 4 hours, we detected strong transcriptional suppression of individual genes within the 6-gene signature (60-80%) (Suppl. Fig. 4A), and of the mean overall ERK transcriptional output (~60%), whereas gene expression increased slightly over baseline after 16 and 24 hours (Fig. 2E).
Figure 2: LY3214996 single agent activity in RAS mutant lung cancer PDX models.

(A) Growth kinetics of DFCI168NRASQ61K tumors treated with vehicle or LY3214996 (100mg/kg, QD) for 21 days (n=8 in vehicle group, n=7 in LY3214996 group; p<0.0001, Two-way Repeated Measures ANOVA with standard post hoc t-tests). (B) Kaplan-Meier survival curves of vehicle- and LY3214996-treated NSG mice with xenotransplanted DFCI168NRASQ61K tumors (p=0.0003, Log-rank test). (C) Western blot analysis of lysates of xenotransplanted DFCI168NRASQ61K tumors treated with vehicle or LY3214996 (100mg/kg) for 4, 16 and 24 h (n=3 tumors/time point). (D) Time course of LY3214996 plasma concentrations (in nM, left y-axis) and of relative p-p90RSK levels (in %, right y-axis) 4, 16 and 24 h after the last drug application (100mg/kg, QD; n=3 animals/group; p=0.0013; Unpaired t-Test). In the 24 h group, two plasma samples were not analyzable for technical reasons. (E) Mean relative expression of ERK-dependent target genes in DFCI168NRASQ61K tumors 4, 16 and 24 h after the last LY3214996 dose (100mg/kg, pooled data of n=3 tumors/group). PHLDA1 transcripts were undetectable. (F) Growth kinetics of DFCI316KRASQ61H tumors treated with vehicle or LY3214996 (100mg/kg, PO QD) for 21 days (n=8/group, p=0.9, Two-way Repeated Measures ANOVA with standard post hoc t-tests). (G) Mean relative expression of ERK-dependent target genes in DFCI316KRASQ61H tumors 4, 16 and 24 h after the last LY3214996 dose (100mg/kg) (pooled data of n=3 tumors/group). (H) Western blot analysis of lysates of xenotransplanted DFCI316KRASQ61H tumors treated with vehicle or with LY3214996 (100mg/kg) for 4, 16 and 24 h (n=3 tumors/group). (I) Growth kinetics of DFCI168NRASQ61K tumors treated for 21 days with once- (100mg/kg, PO QD) or twice-daily (50mg/kg, PO BID) LY3214996 (n=8 NSG mice/group, p=0.0102, Two-way Repeated Measures ANOVA with standard post hoc t-tests).
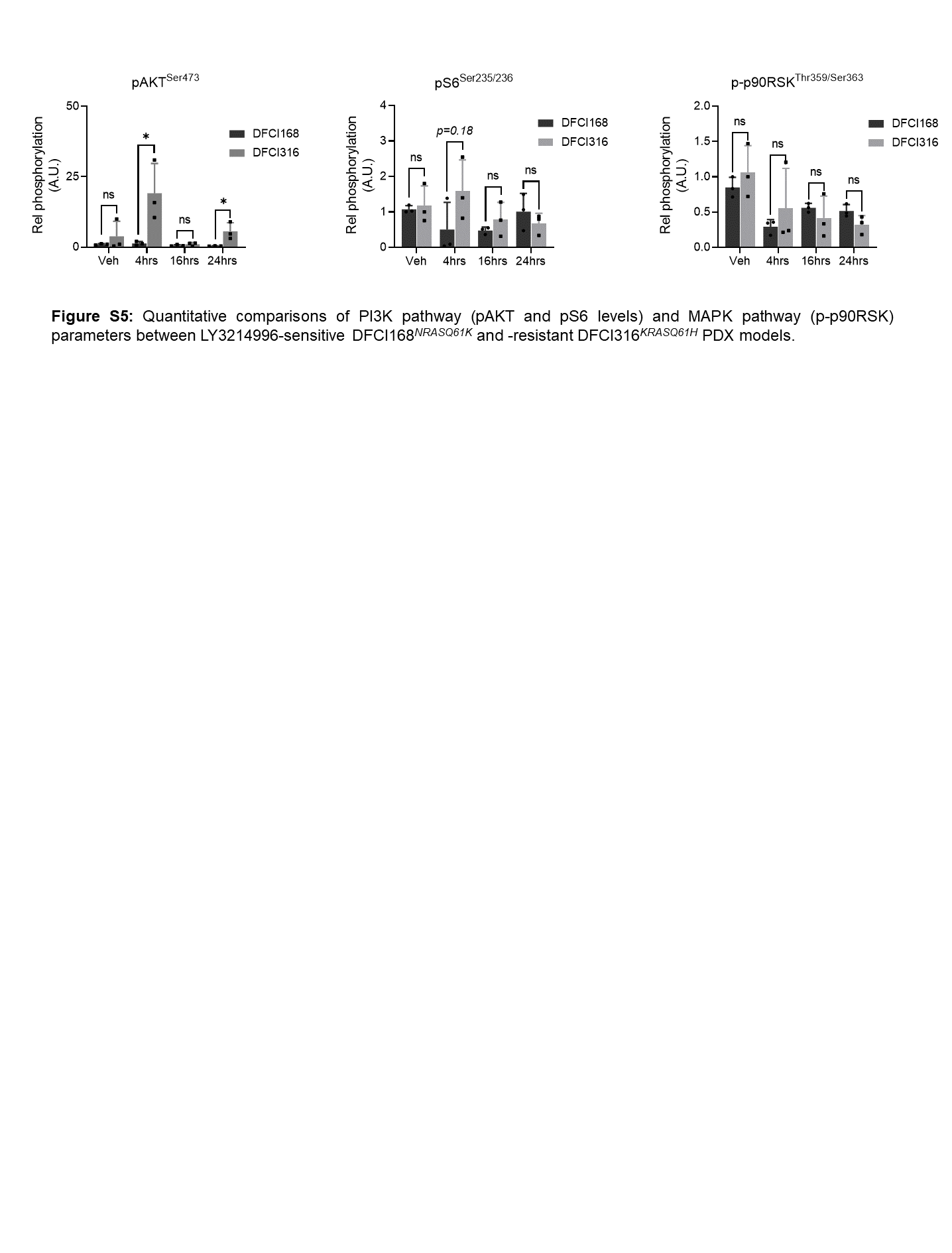
Despite good anti-tumor activity in DFCI168 tumors, LY3214996 did not affect growth of DFCI316KRASQ61H tumors when dosed once-daily (100mg/kg, Fig. 2F, Two-way Repeated Measures ANOVA with standard post hoc t-tests) despite similar suppression of ERK-dependent gene expression as in DFCI168NRASQ61K tumors (Fig. 2G, Suppl. Fig. 4B). Interestingly, in contrast to LY3214996-sensitive DFCI168NRASQ61K tumors, LY3214996-resistant DFCI316KRASQ61H tumors exhibited markedly increased ERKThr202/Tyr204 and AKTSer473 phosphorylation levels 4 hours after LY3214996 treatment compared to tumors of vehicle treated animals (Fig. 2H; DFCI316 vs. DFCI168, p<0.05, Student’s t-test, Suppl. Fig. 5). S6Ser235/236 phosphorylation levels in DFCI316 tumors slightly increased over baseline compared to DFCI168NRASQ61K tumors (Fig. 2H, Suppl. Fig. 5, p>0.05, Student’s t-test).
Due to the lack of anti-proliferative activity in DFCI316KRASQ61H tumors, the rapid MAPK pathway reactivation in two PDX models and the decrease of LY3214996 plasma levels with strong PK/PD relationship with once-daily drug dosing (Fig. 2C-H, Suppl. Fig. 4A,B), we subsequently tested if twice daily dosing at 50 mg/kg is superior to once daily dosing at 100 mg/kg. Indeed, this was the case in the DFCI168NRASQ61K PDX model (Fig. 2I, n=8 animals/group, p<0.0001, Two-way Repeated Measures ANOVA with standard post hoc t-tests). Twice daily application of LY3214996 was well tolerated by NSG mice over 21 days (Suppl. Fig. 6).
In vitro efficacy of combined ERK and PI3K/mTOR inhibition
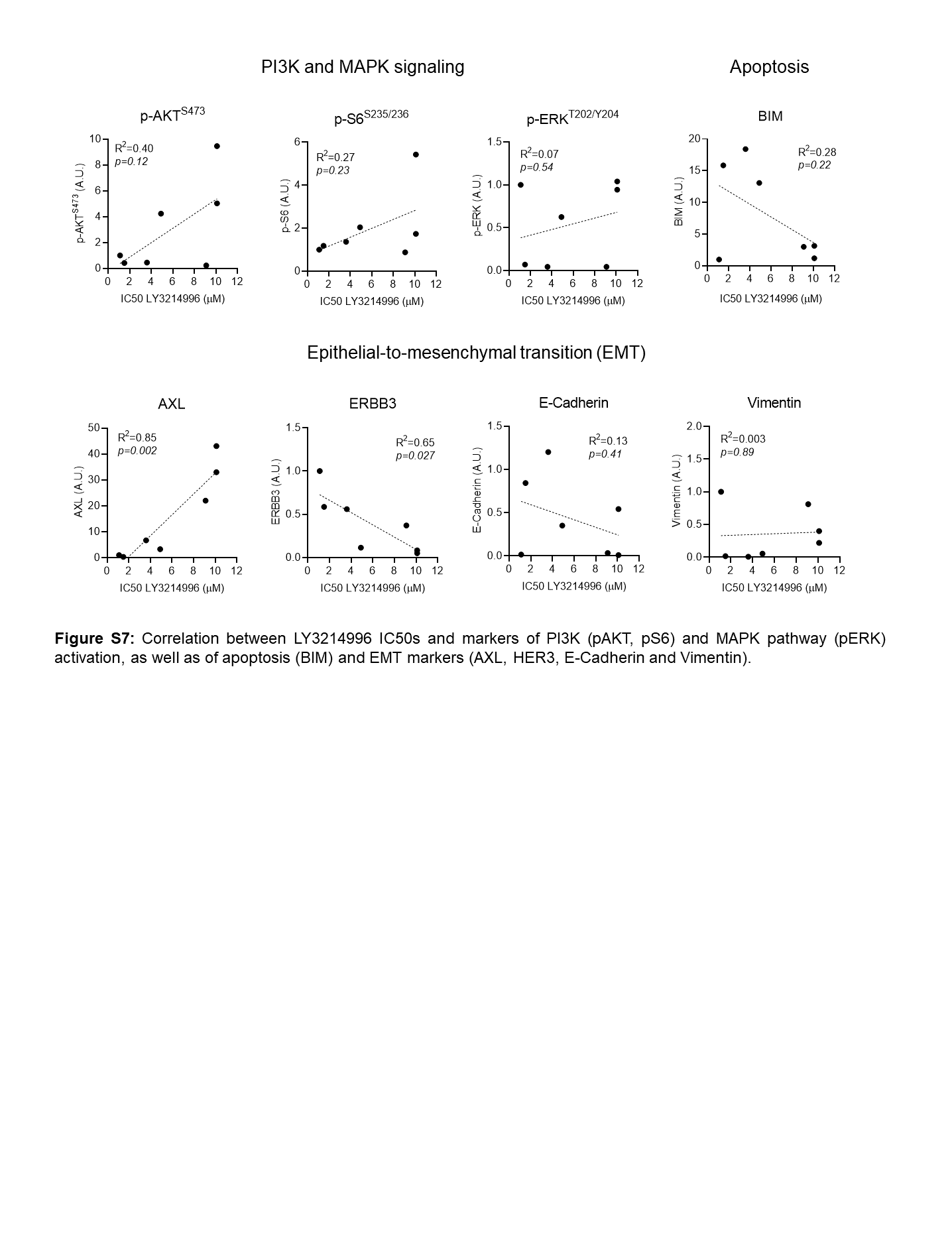
Since LY3214996-induced FOXO3a reporter activities differed substantially in the patient-derived cancer cell lines (Fig. 1G), suggestive of differences in PI3K pathway activity, we next investigated the baseline characteristics of these cell lines. We observed that resistant cell lines (DFCI366, DFCI298, DFCI332) exhibited a more mesenchymal phenotype (AXLhigh, ERBB3low, BIMlow) than sensitive cell lines (DFCI168, DFCI316 and DFCI24; Fig. 3A, Suppl. Fig 7). Since mesenchymal cells also exhibited increased PI3K pathway activation (pAKTS473, pS6S235/236), we next tested the effect of combined ERK1/2 and PI3K/mTOR inhibition with LY3214996 and LY3023414. First, we determined Bliss synergy after 72 hours in all patient-derived cell lines and observed mostly additive effects (color-coded in green) with some synergy (color-coded in blue) in DFCI366KRASG12A and DFCI516KRASG12C cells (Fig. 3B). Western blot analyses of DFCI24KRASG12C and DFCI316KRASQ61H protein lysates indicated, that PI3K inhibition with 1μM of LY3023414 slightly increased protein levels of ERK targets c-MYC, DUSP4 and SPRY2 compared to DMSO treated cells, whereas 1μM of LY3214996 and the combination of both drugs (both 1μM) reduced ERK target levels over 72 hours, respectively (Fig. 3C). In both models, combined ERK plus PI3K inhibition also reduced S6Ser235/236 phosphorylation more profoundly than either drug alone. Levels of pro-apoptotic BIM increased slightly in both cell lines and PARP cleavage was detectable after 48 and 72 hours in DFCI316 cells with combined drug treatment.
Figure 3: Anti-proliferative activity of combined ERK1/2 and PI3K/mTOR inhibition in vitro.

(A) Western blot analysis of signaling (MAPK and PI3K pathway) and EMT baseline characteristics of serum starved (0.1% FBS) RAS-mutant patient-derived cancer cell lines. (B) Bliss synergy between LY3214996 and LY3023414 (dose range for each drug 2-10,000nM) in patient-derived cancer cell lines after 72 h of treatment. Synergism: blue, antagonism: red (n=6 replicates/dose combination). (C) Western blot analysis of lysates of DFCI24KRASG12C (left panel) and DFCI316KRASQ61H (right panel) cells treated for up to 72 h with DMSO, LY3214996 (1μM), LY3023414 (100nM) or a combination of both drugs. (D) Relative (compared to DMSO control) pooled ERK-dependent gene expression in DFCI24KRASG12C (left graph) and DFCI316KRASQ61H (right graph) cells treated for up to 72 h with LY3214996 (1μM), LY3023414 (100nM) or a combination of both (Student’s t-Test). (E) Relative FOXO3a reporter activity in transiently transfected DFCI24 (left) and DFCI316 (right) cells after 24 h of treatment with LY3214996 (1μM), LY3023414 (100nM) or a combination of both drugs (Student’s t-test). (F) Western blot analysis of cytoplasmic and nuclear protein fractions of DFCI24 cells treated for 24 h with 1μM of LY3214996, LY3023414 (100nM) or a combination of both drugs. (G) BLISS synergy between LY3214996 and LY3023414 (dose range 40-10,000nM) in GEMM-derived cancer cell lines with KrasG12V (K), KrasG12V;Tp53fl/fl (KP) and KrasG12V;Tp53fl/fl;Lkb1fl/fl (KPL) genotype after 72 h (n=6 replicates/dose combination). Western blots show the loss of Lkb1 and TP53 in GEMM-derived cancer cell lines. “*” indicates treatment of cell lines for 24 h with 5μM of cisplatin to induce TP53.
Next, we quantified transcriptional changes during simultaneous ERK1/2 and PI3K/mTOR inhibition. While LY3023414 (1μM) significantly increased mean overall MAPK pathway-dependent gene expression over 72 hours in both models compared to DMSO-treated cells (Fig. 3D; p<0.001 for DFCI24 and p<0.01 for DFCI316; Unpaired t-Test), LY3214996 alone (1μM) or the combination of PI3K/mTOR and ERK1/2 inhibitor reduced the expression (p<0.0001; Unpaired t-Test). The reduction of overall gene expression in cells treated with single-agent LY3214996 and with the drug combination was comparable (p=0.83 for DFCI24 and p=0.068 for DFCI316; Unpaired t-Test). Stronger transcriptional pathway suppression with the drug combination was accompanied by increased FOXO3a reporter activity in DFCI24 and DFCI316 cells compared to LY3214996 treatment alone (Fig. 3E) and resulted in increased nuclear FOXO3a accumulation in DFCI24 cells (Fig. 3F).
To determine the effect of co-occurring Tp53 or Lkb1 mutations (47) on efficacy of combined ERK1/2 and PI3K/mTOR inhibition, we determined Bliss synergy in cell lines derived from genetically engineered mouse models (GEMM) (KrasG12V= ”K”-genotype: CHA14.1 and CHA14.2 (48); KrasG12V;Tp53fl/fl= ”KP”-genotype: CHA9.1 and CHA9.3; KrasG12V;Tp53fl/fl;Lkb1fl/fl= ”KPL”-genotype: CHA487 and CHA496) after 72 hours (Fig. 3G). We observed mostly additive effects (color-coded in green), increased synergy (color-coded in blue) was detected in the presence of Tp53 and Lkb1/Stk11 co-mutations. Overall, the surface area indicating synergy was increased in GEMM-derived cell lines across genotypes compared to human-derived cancer cell lines (Fig. 3B).
Anti-tumor effect of combined ERK and PI3K/mTOR inhibition in PDX models
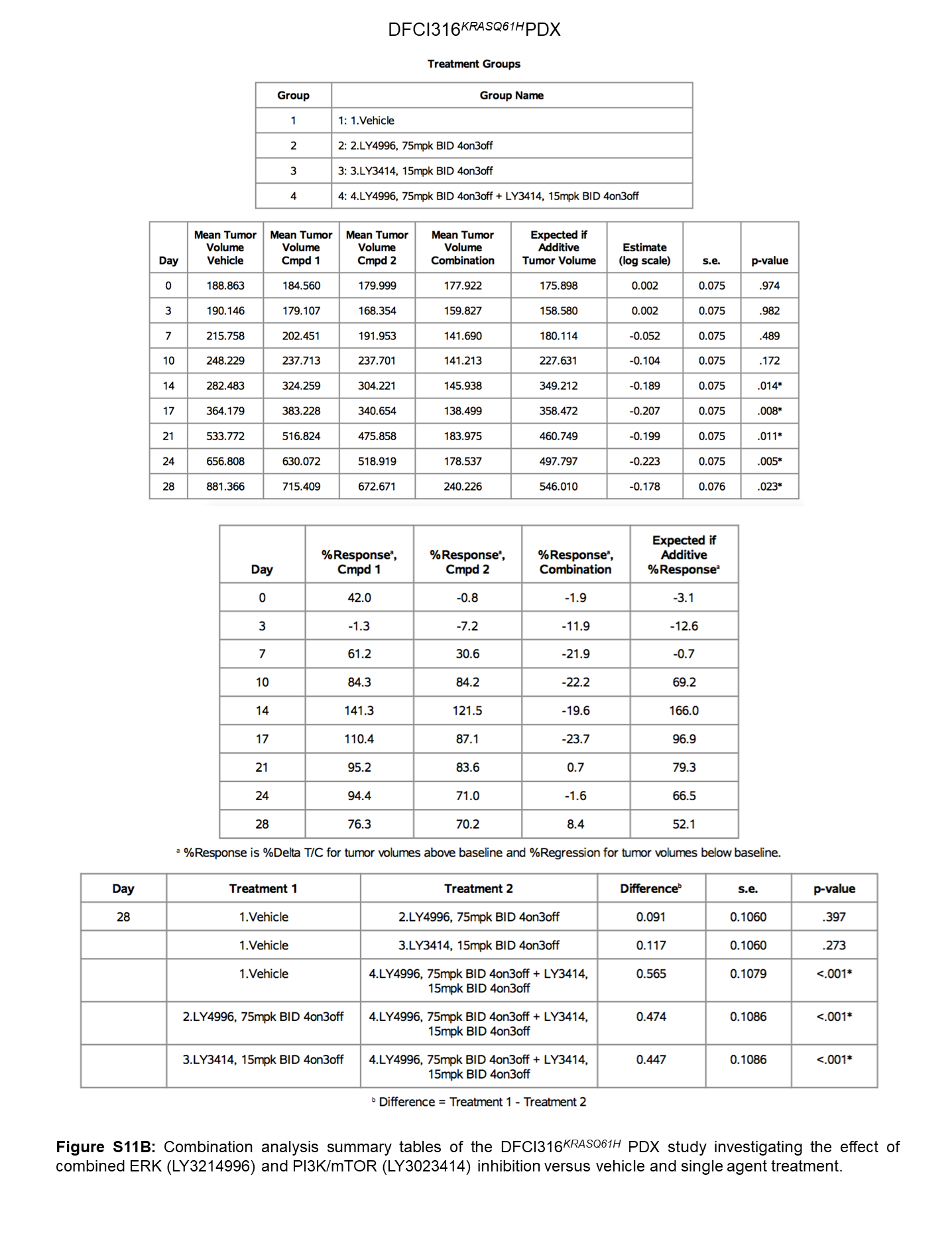
To test the in vivo efficacy of combined ERK1/2 and PI3K inhibition, we treated NSG mice with xenotransplanted DFCI24KRASG12C tumors for 4 weeks with vehicle, high-dose continuous LY3214996 (150mg/kg, PO QD), intermittent split-dose LY3214996 (75mg/kg, PO BID, 4d ON, 3d OFF), intermittent split-dose LY3023414 (15mg/kg, PO BID, 4d ON, 3d OFF) or a combination of both drugs. Anti-tumor effects of single agent treatments were comparably modest, whereas the inhibitor combination had a relatively robust anti-tumor effect (Fig. 4A and Suppl. Fig. 8A; p<0.0001; Two-way Repeated Measures ANOVA with standard post hoc t-tests). We also treated the DFCI316KRASQ61H PDX model with the same treatment schedules. We observed significant tumor growth inhibition in all treatment groups after 4-weeks, but tumor regressions were only achieved in the combination group (Fig. 4B and Suppl. Fig. 8B; p<0.0001; Two-way Repeated Measures ANOVA with standard post hoc t-tests). All treatment schedules were well tolerated (Suppl. Fig. 9) and no differences in plasma drug concentrations from day 1/2 to day 14/15 were observed thus excluding changes in drug metabolism over time (Suppl. Fig. 10). With most animals for both PDX models having progressive disease with single agent treatment, nearly half of the animals with DFCI24KRASG12C tumors and all animals with DFCI316KRASQ61H tumors achieved at least stable disease after 4-weeks of treatment in the combination group. Partial responses (PR) were observed in 3/8 animals (DFCI24) and 5/9 animals (DFCI316) in the combination group, respectively. Overall, treatment effects were synergistic in both PDX models (Bliss Independence method; Suppl. Fig. 11A,B) and translated into a stronger tumor growth delay in mice treated with the drug combination compared (Fig. 4C; DFCI24: 15.8 (combination) vs. 2.3 (LY3214996 BID) vs. 0.9 days (LY3023414); DFCI316: 26.8 (combination) vs. 12.2 (LY3214996 BID) vs. 17.5 days (LY3023414) compared to vehicle control).
Figure 4: Anti-tumor activity of combined ERK1/2 and PI3K inhibition in PDX models.

Growth kinetics of DFCI24KRASG12C (A) and DFCI316KRASQ61H (B) tumors treated with vehicle, LY3214996 (75mg/kg BID, 4d ON 3d OFF), LY3214996 (150mg/kg, PO QD), LY3023414 (15mg/kg, PO BID, 4d ON 3d OFF) or a combination of both drugs for 28 days (n=8-9/group, Two-way Repeated Measures ANOVA with standard post hoc t-tests). Waterfall plots of individual tumor responses are depicted for day 24 (DFCI24) and day 28 (DFCI316), respectively (PD - progressive disease (solid columns); SD - stable disease (pattern columns); PR - partial response (open columns)). (C) Tumor growth delay (in days) in mice xenotransplanted with DFCI24 and DFCI316 tumors by treatment regimen compared to vehicle treated animals. (TGI – Tumor Growth Inhibition) (D) Western blot analysis of lysates of DFCI24KRASG12C tumors treated with vehicle, LY3214996 (75mg/kg, BID), LY3023414 (15mg/kg, BID) or a combination of both 4, 8 and 24 h after the last drug application. (E) Pooled mean ERK-dependent gene expression (compared to vehicle) in DFCI24KRASG12C tumors treated for 4, 8 and 24 h with LY3214996 (75mg/kg), LY3023414 (15mg/kg) or a combination of both drugs (n=3 tumors/group, Student’s t-Test).
Lysates of DFCI24KRASG12C tumors which had been treated with vehicle, LY3214996 (75mg/kg), LY3023414 (15mg/kg) or a combination of both drugs for 4, 8 and 24 hours showed reduced levels of DUSP4, SPRY2, c-MYC and phosphorylation of p90RSKThr359/Ser363 as early as 4 hours after the application of LY3214996 or of the drug combination. Contrariwise, phosphorylation and total levels of these proteins were increased in tumors treated with single agent LY3023414 (Fig. 4D). Whereas signaling parameters (phospho-p90RSK and phospho-S6) returned to baseline after 24 hours with single-agent treatment, the ERK/PI3K inhibitor combination induced continuous suppression of MAPK and PI3K downstream targets. ERK1/2Thr202/Tyr204 phosphorylation was increased in LY3214996-treated tumors at all time points and slightly increased in tumors of LY3023414-treated mice. More potent pathway inhibition with the drug combination coincided with stronger BIM accumulation and PARP cleavage at all time points. Analysis of MAPK pathway-dependent gene expression from the same tumors showed good overall pathway suppression with single agent LY3214996 4 and 8 hours after the last drug application, whereas PI3K/mTOR inhibition alone induced pathway hyperactivation at 4 hours (Fig. 4E). The drug combination not only prevented the early PI3K/mTOR inhibitor-induced transcriptional pathway hyperactivation but was also more efficacious in counteracting pathway reactivation with single agent ERK1/2 inhibition at 24 hours (Fig. 4E, dotted red line, p<0.05, Unpaired t-Test).
In vitro efficacy of combined ERK and CDK4/6 inhibition
Since inputs from the MAPK and PI3K signaling pathways promote G1/S cell cycle transition via the cyclin D1-CDK4/6-RB pathway (3), we also tested the combination of LY3214996 and abemaciclib (LY2835219), a specific cyclin-dependent kinase (CDK) 4/6 inhibitor. First, we determined Bliss synergy by treating all patient-derived cell lines for 72 hours with different concentrations of both drugs. We observed mostly additive effects (color-coded in green) across the tested dose range with some synergy (color-coded in blue) in DFCI316KRASQ61H, DFCI332KRASG12D and DFCI516KRASG12C cells (Fig. 5A). Treatment effects were independent of the drug sequence (Suppl. Fig. 12) and of the used ERK inhibitor as combination partner (SCH772984 in Suppl. Fig. 13). Lysates of DFCI24KRASG12C and DFCI332KRASG12D cells which had been treated for up to 72 hours with DMSO, LY3214996 (1μM), abemaciclib (1μM) or the combination of both drugs showed more profound suppression of RB, ERK1/2 and S6 phosphorylation as well as of c-MYC protein levels than with either drug alone (Fig. 5B). DUSP4 and SPRY2 protein levels were equally suppressed with LY3214996 and the drug combination. Single agent abemaciclib suppressed S6 phosphorylation which is due to inhibition of CDK4-activated mTOR activity (49). Neither treatment led to PARP cleavage and BIM accumulation in response to LY3214996 was counteracted by the addition of abemaciclib.
Figure 5: Anti-proliferative activity of combined ERK1/2 and CDK4/6 inhibition in vitro.

(A) Bliss synergy between LY3214996 and abemaciclib (dose range 40-10,000nM) in patient-derived RAS-mutant cancer cell lines after 72 h (n=6 replicates/dose combination). Synergism: blue, antagonism: red. (B) Western blot analysis of DFCI24KRASG12C (left panel) and DFCI332KRASG12D (right panel) cells treated with DMSO, LY3214996 (1μM), abemaciclib (1μM) or a combination of both drugs for up to 72 h. (C) BLISS synergy between LY3214996 and LY30323414 (dose range 40-10,000nM) in GEMM-derived cancer cell lines with KrasG12V (K), KrasG12V;Tp53fl/fl (KP) and KrasG12V;Tp53fl/fl;Lkb1fl/fl (KPL) genotype after 72 h (n=6 replicates/dose combination).
To determine the impact of co-occurring Tp53 and Lkb1 mutations (47) on the efficacy of combined ERK1/2 and CDK4/6 inhibition, we determined drug synergy in GEMM-derived KRASG12V mutant lung cancer cell lines with K, KP and KPL genotype after 72 hours and observed some drug synergy (color-coded in blue) across genotypes which was increased in the presence of co-mutations in Tp53 and STK11/Lkb1 (Fig. 5C). Overall, the synergy of combined ERK1/2 and CDK4/6 inhibition was more pronounced in GEMM-derived cell lines across genotypes compared to patient-derived cell lines (Fig. 5A).
Anti-tumor effect of combined ERK1/2 and CDK4/6 inhibition in PDX models
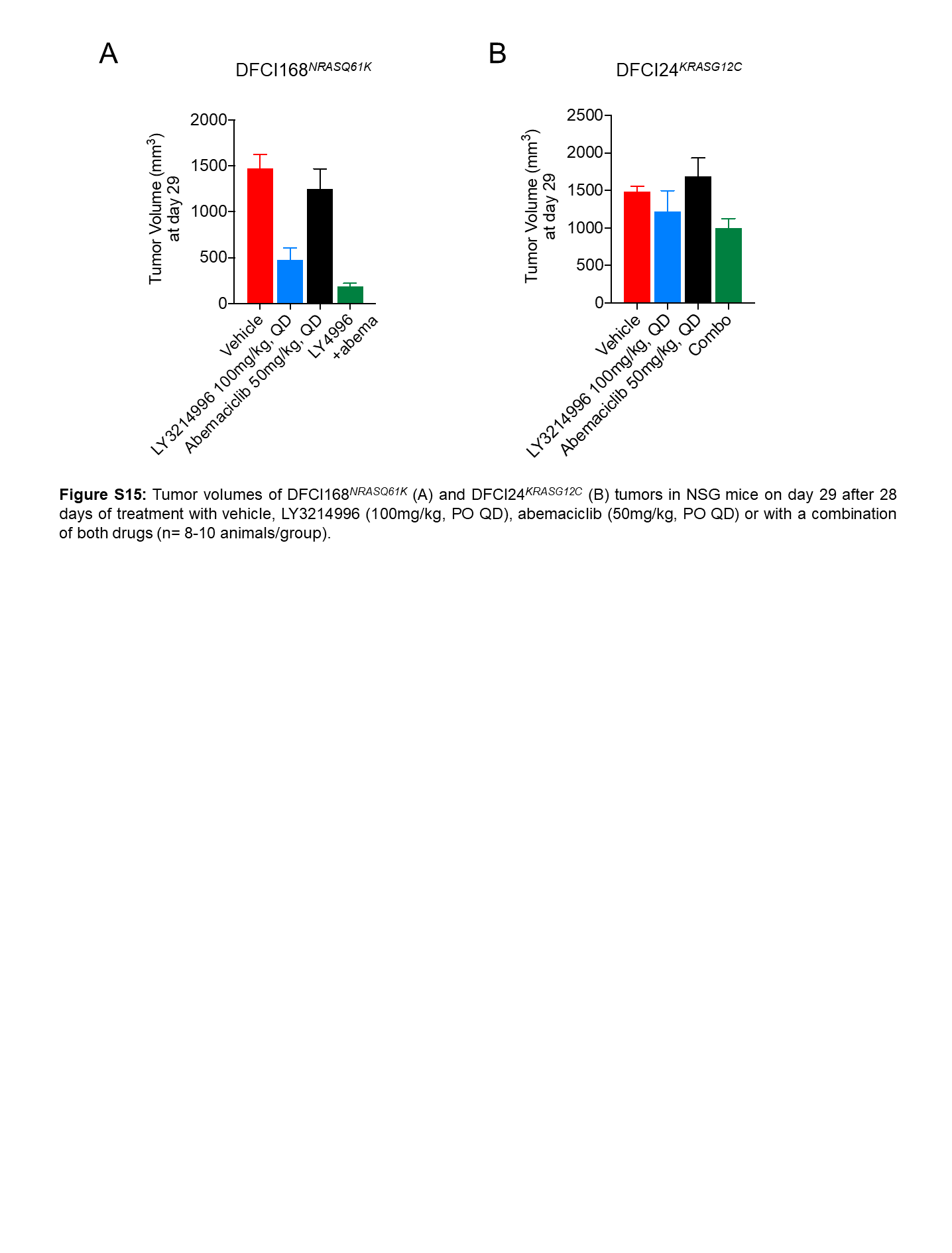
To test the in vivo efficacy of combined ERK1/2 and CDK4/6 inhibition, we treated the DFCI168NRASQ61K (Fig. 6A) and DFCI24KRASG12C PDX models (Fig. 6B) for 28 days with vehicle, LY3214996 (100mg/kg, PO QD), abemaciclib (50mg/kg, PO QD) or the combination of both drugs. We established, that the well-tolerated split-dose intermittent application of LY3214996 (50mg/kg, PO BID, 4d ON 3d OFF) in combination with abemaciclib (50mg/kg, PO QD) was not superior over once-daily drug application (Suppl. Fig. 14). Therefore, we chose a continuous once-daily dosing schedule for the efficacy studies. We observed tumor growth inhibition in both PDX models with LY3214996, whereas abemaciclib itself was ineffective (p<0.001, Two-way Repeated Measures ANOVA with standard post hoc t-tests). The strongest anti-tumor effect was observed in animals which received both drugs (DFCI168: Fig. 6A and Suppl. Fig. 15A; DFCI24: Fig. 6B and Suppl. Fig. 15B; p<0.0001, Two-way Repeated Measures ANOVA with standard post hoc t-tests). The effect of combining LY3214996 and abemaciclib was additive in the DFCI168NRASQ61K and less than additive in the DFCI24KRASG12C model (Bliss Independence method; Suppl. Fig. 16A, B) but translated into a longer tumor growth delay compared to single agent treatment (Fig. 6C; DFCI168: 24.8 (combination) vs. 15.7 (LY3214996) vs. 2.9 days (abemaciclib); DFCI24: 7.2 (combination) vs. 3.2 (LY3214996) vs. 3.1 days (abemaciclib) compared to vehicle control). In tumor lysates we observed inhibition of RB phosphorylation with LY3214996 or abemaciclib at early timepoints (4 and 8 hours) but partial pathway reactivation after 24 hours (DFCI168: Fig. 6D; DFCI24: Suppl. Fig. 17). This rebound was effectively reduced by the addition of LY3214996 to abemaciclib. Single-agent abemaciclib increased phosphorylation of p90RSK (which is in contrast to p-p90RSK inhibition which we observed in vitro (Fig. 5B)) and AKT as well as protein levels of Cyclin D1 compared to vehicle which was pronounced at 24 hours in both models, suggesting compensatory MAPK and PI3K pathway hyperactivation. MAPK pathway hyperactivation could be counteracted by adding LY3214996 to abemaciclib. While BIM protein levels increased in LY3214996-treated DFCI168NRASQ61K tumors, treatment with abemaciclib or the drug combination had the opposite effect on BIM overall and neither treatment increased PARP cleavage. Analysis of MAPK pathway-dependent gene expression from the same tumors showed good overall pathway suppression with single agent LY3214996 4 and 8 hours after the last drug application, whereas abemaciclib induced early (4 and 8 hours) pathway hyperactivation which could be prevented by the drug combination (Fig. 6E).
Figure 6: Anti-tumor activity of combined ERK1/2 and CDK4/6 inhibition in PDX models.

Growth kinetics of DFCI168NRASQ61K (A) and DFCI24KRASG12C (B) tumors treated with vehicle, LY3214996 (100mg/kg, PO QD), abemaciclib (50mg/kg, PO QD) or a combination of both drugs for 28 days (n=8-10/group; Two-way Repeated Measures ANOVA with standard post hoc t-tests). Waterfall plots of individual tumor responses are depicted for day 29 (DFCI168) and day 15 (DFCI24), respectively (PD - progressive disease (solid columns); SD - stable disease (pattern columns); PR - partial response (open colums); CR - complete response (pink column)). (C) Tumor growth delay (in days) in mice xenotransplanted with DFCI168NRASQ61K and DFCI24 tumors by treatment regimen compared to vehicle treated animals. (TGI – Tumor Growth Inhibition) (D) Western blot analysis of lysates of DFCI168NRASQ61K tumors treated with vehicle, LY3214996 (100mg/kg, QD), abemaciclib (50mg/kg, QD) or a combination of both drugs 4, 8 and 24 h after the last drug application. (E) Pooled mean expression of MAPK pathway-dependent genes (compared to vehicle) in DFCI168NRASQ61K tumors treated for 4, 8 and 24 h with LY3214996 (100mg/kg), abemaciclib (50mg/kg) or a combination of both drugs (n=3 tumors/group, Student’s t-Test). (F) Schematic summary of the different treatment approaches investigated in this study: single agent LY3214996 treatment for ERK inhibitor sensitive cell lines, combined ERK plus PI3K inhibitor treatment for PI3K pathway-activated ERK inhibitor-resistant cell lines and combined ERK plus CDK4/6 inhibition. The figure was created with BioRender.com.
Discussion
Activating mutations in RAS proteins occur in about 30% of lung cancers (Suppl. Fig. 1) (1). Advances have been made in the clinical development of direct KRAS and immune checkpoint inhibitors (ICB) but these strategies are so far restricted to lung cancers with KRASG12C mutations and to immunologically “hot” tumors, respectively (14-16,50). Therefore, inhibiting RAS effector pathways which promote malignant behavior of cancer cells remains an attractive alternative. Unfortunately, single agent treatment approaches have had no (MEK inhibitors) or only limited (CDK4/6 inhibitor) anti-tumor activity in clinical trials (8,10,11). Based on high cancer cell plasticity and pathway redundancy, potential resistance mechanisms include the loss of ERK-mediated negative feedback inhibition and PI3K pathway activation via receptor tyrosine kinases (RTKs) for MEK (51,52) and compensatory MAPK pathway activation for CDK4/6 inhibitors (53). Additionally, co-occurring loss of function mutations of tumor suppressor genes (e.g. TP53 and STK11) increase the genetic heterogeneity of RAS mutant tumors and impede therapeutic interventions (47). The major obstacle, however, is the toxicity observed with continuous blockade of MAPK pathway activity alone or in combination with other pathways (e.g. MEK plus PI3K inhibition) hence strongly indicating an unmet need to develop alternative combination strategies (2,6,10,11,13).
Since ERK1/2 inhibitors are more potent in preventing feedback and bypass activation compared to MEK1/2 inhibitors, toxicities intrinsic to continuous MEK inhibition could potentially be overcome by intermittent application (i.e. to include drug-free periods) of ERK inhibitors without compromising on anti-tumor efficacy (17,18). Therefore, in the present study, we investigated the efficacy of the ATP-competitive ERK1/2 inhibitor LY3214996 as single agent and in combination with a PI3K/mTOR (LY3023414) inhibitor (25) or CDK4/6 inhibitor (abemaciclib) (26). LY3214996 has anti-neoplastic activity in commercially available cell lines and tumor models with MAPK pathway aberrations - many of them with KRAS mutations (27). Since for these models no or only very limited clinical annotation is available, we sought to study genetically well-characterized cell lines established from tumors of pre-treated patients which are more reflective of the clinical setting in which an ERK inhibitor or ERK inhibitor-based drug combination would be initially clinically evaluated.
We established seven RAS mutant cancer cell lines (n=6 KRAS, n=1 NRAS, Fig. 1A, Suppl. Table 1) from malignant effusions of in part heavily pre-treated patients (Suppl. Fig. 2), in which single agent LY3214996 treatment profoundly suppressed MAPK pathway activity (Fig. 1C-E). With the exception of DFCI332 cells, this translated into growth inhibition in all cell lines including those with STK11/LKB1 co-mutations (Fig. 1F), a genotype with relative intrinsic insensitivity to MEK inhibition (54), hence supporting pre-clinical data indicating, that ERK inhibitors may have the potential to overcome this limitation of MEK inhibitors (55). LY3214996 also exhibited good anti-tumor activity in various PDX models (Fig. 2A; Fig. 4A and B, Fig. 6A and B) in which – similar to other ERK inhibitors – we observed a strong PK/PD relationship with once-daily drug application due to rapid plasma elimination (Fig. 2D). This pharmacokinetic trait led to an early recovery of MAPK pathway activity (Fig. 2C-E, 2G-H; Suppl. Fig. 4A, B) and consequently, the anti-tumor potency of LY3214996 increased with a split-dose approach without affecting the plasma drug clearance over longer treatment periods (Fig. 2I; Fig. 4B versus Fig. 2F, Suppl. Fig. 6 and 10).
Across various cell lines and PDX models, however, the anti-neoplastic effect of LY3214996 differed substantially (Fig. 1B and F, Fig. 2A and F) despite comparable suppression of MAPK pathway activity (Fig. 1C-E; Fig. 2E and G). LY3214996 increased pro-apoptotic BIM in sensitive and intermediate sensitive cell lines in vitro (Fig. 1D), but failed to do so in resistant cell lines and - despite reducing tumor growth in various PDX models - tumor regressions were not observed (Fig. 2A; Fig. 4A and B; Fig. 6A and B). This possibly indicated compensatory mechanisms counteracting full execution of apoptosis. We subsequently established that cancer cell lines with reduced LY3214996 sensitivity exhibit a more mesenchymal phenotype with PI3K pathway activation and BIM suppression at baseline (DFCI298, DFCI332) (Fig. 3A, Suppl. Fig. 7) or activate PI3K signaling in response to LY3214996 (DFCI316, Fig. 1C and D, Fig. 2H, Suppl. Fig. 5). PI3K pathway activation is known to prevent nuclear translocation of FOXO3a upon MEK inhibition thus preventing BIM induction and apoptosis execution (46,56). Consistently, we observed a lack of FOXO3a nuclear translocation and transcriptional activation in resistant (DFCI298, DFCI332, DFCI366) versus sensitive (DFCI168) cell lines upon LY3214996 treatment (Fig. 1G, H).
We subsequently show that PI3K pathway-mediated LY3214996 resistance can be overcome by combined ERK1/2 plus PI3K/mTOR inhibition. The combination with LY3023414 increased downstream pathway suppression, BIM induction and anti-proliferative activity in human and GEMM-derived cancer cell lines (Fig. 3B-D, G) by promoting nuclear FOXO3a accumulation and transcriptional activition (Fig. 3D,E). This translated into synergistic growth inhibition and stronger tumor growth delay in PDX models with actual tumor regressions indicating an in vivo apoptotic response (Fig. 4A-D; Suppl. Fig. 8, Suppl. Fig. 11A and B). Tumor growth delay is a critically important measure of antitumor efficacy since it most closely mimics clinical endpoints that required observation of mice through the time of disease progression (41). Intermittent dosing of both drugs (i.e. 4 days treatment followed by a 3 day treatment-free period) was well tolerated by NSG mice (Suppl. Fig. 9) and did not compromise treatment efficacy which is important for the transfer into clinical applications even though toxicity profiles of ERK plus PI3K inhibitor combinations in humans remain unknown today. In light of significant toxicity observed with MEK or BRAF plus PI3K inhibitor combinations on uninterrupted schedules (6,57), future clinical trials are therefore required to investigate if the LY3214996 plus LY3023414 drug combination is better tolerated. Compared to MEK inhibitors, ERK inhibitors lack a subset of toxicities (e.g. retinopathy) (58) and LY3023414 differentiates from other PI3K pathway inhibitors in showing a short half-life of approximately two hours in clinical PK studies (59) thus potentially making the LY3214996 plus LY3023414 combination more applicable to humans to overcome overlapping toxicities such as nausea, vomiting, diarrhea, fatigue or rash. The pronounced drug synergy that we observed in GEMM-derived cell lines with subtype-defining Tp53 and Stk11/Lkb1 co-mutations (Fig. 3G) may hold promise for this drug combination also in these otherwise hard to treat lung cancer subtypes (47,54).
Whereas tumor regressions were observed with the ERK/PI3K inhibitor combination, combining LY3214996 with the CDK4/6 inhibitor abemaciclib (26) – a concept supported by mechanistic studies which showed synthetic lethality between KRAS and CDK4 and identified MAPK pathway activation as a CDK4/6 inhibitor resistance mechanism (5,60) – lacked the potential to induce an apoptotic response in PDX models despite more sustained pathway inhibition and anti-neoplastic activity in vitro and in vivo (Fig. 5B and C; Fig. 6A-E, Suppl. Fig. S12A and B, Suppl. Fig. 13). This is not unexpected since a key feature of CDK4/6 inhibition is the induction of a cell cycle inhibitory response that mimics the intrinsic senescence phenotype (5). Failure to induce cancer cell death, however, could compromise treatment efficacy, especially if rapid tumor shrinkage is required in symptomatic patients with high tumor burden.
Therefore, one major aspect that needs to be addressed in future pre-clinical and clinical studies is a survey to identify predictive biomarkers to aid patient stratification especially for the ERK plus CDK4/6 inhibitor combination. Here, we identify epithelial differentiation as a correlative marker for LY3214996 sensitivity. We hypothesize that apart from epithelial-to-mesenchymal transition, mutations affecting PI3K signalling (e.g. loss of PTEN or PIK3CA mutations) may exert similar effects. Interestingly, resistant DFCI332 cells which harbor a Noonan syndrome-associated SHP2/PTPN11 (c.923A>G, p.N308S) missense mutation exhibited the highest level of baseline PI3K pathway activation of all cell lines (Fig. 3A). SHP2 controls PI3K (and MAPK) pathway activity (61) and the PTPN11 c.923A>G mutation is considered pathogenic (FATHMM prediction score 1.0) in COSMIC. Furthermore of interest, DFCI168 cells with the highest LY3214996 single agent sensitivity harbor a NRASQ61K mutation. Zhou et al. reported recently, that KRASQ61H preferentially signals through the MAPK pathway conferring MEK inhibitor sensitivity (62). Even though our study is limited to draw general conclusions, certain RAS mutations may also predict increased LY3214996 sensitivity. Of note, single agent abemaciclib was ineffective in both PDX models (Fig. 6A and B), even though both cell lines harbor non-functional p16 proteins which have been associated with CDK4/6 inhibitor sensitivity (63). DFCI168 cells present with a p16INK4A R80* mutation predicted to be pathogenic (FATHMM prediction score 0.88 in COSMIC) and reported to produce a truncated, non-functional p16 protein (64); DFCI24 cells exhibit a two-copy deletion of CDKN2A conferring loss of p16 (Suppl. Table 1). The predictive value of p16 loss, however, remains unclear (65) and in DFCI24 cells, mesenchymal differentiation with associated PI3K pathway activation (Fig. 3A) may have prevented abemaciclib from being effective (66). Overall, our experimental findings in combination with the available clinical data (8,9) suggest that isolated CDK4/6 inhibition may be insufficient to achieve satisfactory anti-tumor activity in RAS mutant lung cancer and that combined CDK4/6 plus ERK1/2 inhibition has the potential to overcome this limitation to a certain extent especially in tumors with TP53 and STK11/LKB1 co-mutations (Fig. 3E; Fig. 5C) (47,54).
In summary, we demonstrate the efficacy of LY3214996-based drug combinations in PDX models of RAS mutant lung cancer (Fig. 6F summarizes the therapeutic approaches). Intermittent ERK inhibition on a “4-days-On-3-days-OFF” schedule is well tolerated in mice without compromising on anti-tumor activity. Currently ongoing (NCT02857270, NCT03454035) and future clinical trials should aim to determine whether intermittent ERK inhibitor-based drug combinations can overcome toxicities associated with continuous MEK inhibition and to validate treatment predictors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
This study was funded by Eli Lilly and Company. We thank Johannes Köster for supporting this project scientifically.
Research support: This study was supported in part by Eli Lilly and Company, by the National Cancer Institute grant R35CA220497 (PAJ) and by a Mildred-Scheel postdoctoral research fellowship of the German Cancer Aid Foundation (70111755 to JK).
Footnotes
Conflicts of Interest:
JK: received research support from Eli Lilly and Company and from the German Cancer Aid Foundation. He previously served as a consultant and on advisory boards for Boehringer Ingelheim.
YZ: None
JL: None
PCG: Sponsored research agreement with Elstar Therapeutics.
HLT: None
ARK: None
MKW: None
KMS: None
ML: None
CA: None
MM: None
AO: None
JC: None
MB: None
AAB: None
ESC: None
CPP: received honoraria from Bio-Rad, is a co-founder of XSphera Biosciences, and is on the scientific advisory board of Dropworks and XSphera Biosciences; sponsored research agreements with Daiichi Sankyo, Bicycle Therapeutics, Transcenta, Bicara Therapeutics, AstraZeneca, Intellia Therapeutics, and Constellation pharmaceuticals.
SVB: is an employee and stockholder of Eli Lilly and Company.
JRM: is an employee and stockholder of Eli Lilly and Company.
RVT: was a Senior Medical Advisor at Eli Lilly and Company at the inception of this work. He has an ownership interest (stocks and patents) in Eli Lilly and Company and is currently employed as Senior Medical Director at Astellas US Pharma.
PAJ: reports consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, Loxo Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals and Biocartis; receiving post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines and Astellas Pharmaceuticals; and stock ownership in Loxo Oncology and Gatekeeper Pharmaceuticals.
References
- 1.Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 2012;72:210–9 [DOI] [PubMed] [Google Scholar]
- 3.Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, et al. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein's C-Terminal Helix. Mol Cell 2019;74:758–70 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol 1997;17:3850–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010;18:63–73 [DOI] [PubMed] [Google Scholar]
- 6.Shapiro GI, LoRusso P, Kwak E, Pandya S, Rudin CM, Kurkjian C, et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Invest New Drugs 2020;38:419–32 [DOI] [PubMed] [Google Scholar]
- 7.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell 2017;170:17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldman JW, Mazieres J, Barlesi F, Koczywas M, Dragnev KH, Göksel T, et al. A randomized phase 3 study of abemaciclib versus erlotinib in previously treated patients with stage IV NSCLC with KRAS mutation: JUNIPER. Journal of Clinical Oncology 2018;36:9025- [Google Scholar]
- 9.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 2016;6:740–53 [DOI] [PubMed] [Google Scholar]
- 10.Blumenschein GR Jr., Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann Oncol 2015;26:894–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017;317:1844–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramanathan RK, Von Hoff DD, Eskens F, Blumenschein G Jr., Richards D, Genvresse I, et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer. Target Oncol 2020;15:163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23 [DOI] [PubMed] [Google Scholar]
- 15.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10:54–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015;373:1627–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nissan MH, Rosen N, Solit DB. ERK pathway inhibitors: how low should we go? Cancer Discov 2013;3:719–21 [DOI] [PubMed] [Google Scholar]
- 18.Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 2017;23:929–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther 2012;11:1143–54 [DOI] [PubMed] [Google Scholar]
- 20.Blake JF, Burkard M, Chan J, Chen H, Chou KJ, Diaz D, et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-y l)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J Med Chem 2016;59:5650–60 [DOI] [PubMed] [Google Scholar]
- 21.Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ, et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther 2017;16:2351–63 [DOI] [PubMed] [Google Scholar]
- 22.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 2013;3:742–50 [DOI] [PubMed] [Google Scholar]
- 23.Moschos SJ, Sullivan RJ, Hwu WJ, Ramanathan RK, Adjei AA, Fong PC, et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov 2018;8:184–95 [DOI] [PubMed] [Google Scholar]
- 25.Smith MC, Mader MM, Cook JA, Iversen P, Ajamie R, Perkins E, et al. Characterization of LY3023414, a Novel PI3K/mTOR Dual Inhibitor Eliciting Transient Target Modulation to Impede Tumor Growth. Mol Cancer Ther 2016;15:2344–56 [DOI] [PubMed] [Google Scholar]
- 26.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014;32:825–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhagwat SV, McMillen WT, Cai S, Zhao B, Whitesell M, Shen W, et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther 2020;19:325–36 [DOI] [PubMed] [Google Scholar]
- 28.Weisberg E, Meng C, Case A, Sattler M, Tiv HL, Gokhale PC, et al. Evaluation of ERK as a therapeutic target in acute myelogenous leukemia. Leukemia 2020;34:625–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pant S, Bendell JC, Sullivan RJ, Shapiro G, Millward M, Mi G, et al. A phase I dose escalation (DE) study of ERK inhibitor, LY3214996, in advanced (adv) cancer (CA) patients (pts). Journal of Clinical Oncology 2019;37:3001- [Google Scholar]
- 30.Ogino A, Choi J, Lin M, Wilkens MK, Calles A, Xu M, et al. Genomic and pathological heterogeneity in clinically diagnosed small cell lung cancer in never/light smokers identifies therapeutically targetable alterations. Mol Oncol 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambrogio C, Carmona FJ, Vidal A, Falcone M, Nieto P, Romero OA, et al. Modeling lung cancer evolution and preclinical response by orthotopic mouse allografts. Cancer Res 2014;74:5978–88 [DOI] [PubMed] [Google Scholar]
- 32.Ambrogio C, Gomez-Lopez G, Falcone M, Vidal A, Nadal E, Crosetto N, et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med 2016;22:270–7 [DOI] [PubMed] [Google Scholar]
- 33.Cortez GS, Joseph S, McLean JA, McMillen WT, Rodriguez MJ, Zhao G. Erk inhibitors. United States Patent US 9,469,652 B2. 2016. October 18.
- 34.Coates DA, Gelbert LM, Knobeloch JM, De Dios Magana A, De Prado Gonzalez A, Del Prado Catalina MF, et al. Protein Kinase Inhibitors. United States Patent US 7,855,211 B2 2010. December 21.
- 35.Barda DA, Mader MM. PI3 Kinase/mTor Dual Inhibitor. United States Patent US 8,658,668 B2 2014. February 25.
- 36.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 2007;13:1576–83 [DOI] [PubMed] [Google Scholar]
- 37.Chaikuad A, Tacconi EM, Zimmer J, Liang Y, Gray NS, Tarsounas M, et al. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol 2014;10:853–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eskens FA, Mom CH, Planting AS, Gietema JA, Amelsberg A, Huisman H, et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer 2008;98:80–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, Cooke A, Feng L, Mak G, et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther 2007;6:2158–67 [DOI] [PubMed] [Google Scholar]
- 40.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016;32:2866–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teicher BA. Tumor models for efficacy determination. Mol Cancer Ther 2006;5:2435–43 [DOI] [PubMed] [Google Scholar]
- 42.Brant R, Sharpe A, Liptrot T, Dry JR, Harrington EA, Barrett JC, et al. Clinically Viable Gene Expression Assays with Potential for Predicting Benefit from MEK Inhibitors. Clin Cancer Res 2017;23:1471–80 [DOI] [PubMed] [Google Scholar]
- 43.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016;48:607–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 2016;1:e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kidger AM, Munck JM, Saini HK, Balmanno K, Minihane E, Courtin A, et al. Dual-Mechanism ERK1/2 Inhibitors Exploit a Distinct Binding Mode to Block Phosphorylation and Nuclear Accumulation of ERK1/2. Mol Cancer Ther 2020;19:525–39 [DOI] [PubMed] [Google Scholar]
- 46.Yang JY, Chang CJ, Xia W, Wang Y, Wong KK, Engelman JA, et al. Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res 2010;70:4709–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 2003;4:111–20 [DOI] [PubMed] [Google Scholar]
- 49.Litchfield LM, Boehnke K, Brahmachary M, Mur C, Bi C, Stephens JR, et al. Combined inhibition of PIM and CDK4/6 suppresses both mTOR signaling and Rb phosphorylation and potentiates PI3K inhibition in cancer cells. Oncotarget 2020;11:1478–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020;383:1207–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Transl Med 2018;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 2014;25:697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Leeuw R, McNair C, Schiewer MJ, Neupane NP, Brand LJ, Augello MA, et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin Cancer Res 2018;24:4201–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012;483:613–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caiola E, Iezzi A, Tomanelli M, Bonaldi E, Scagliotti A, Colombo M, et al. LKB1 Deficiency Renders NSCLC Cells Sensitive to ERK Inhibitors. J Thorac Oncol 2020;15:360–70 [DOI] [PubMed] [Google Scholar]
- 56.Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS One 2010;5:e13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yam C, Xu X, Davies MA, Gimotty PA, Morrissette JJD, Tetzlaff MT, et al. A Multicenter Phase I Study Evaluating Dual PI3K and BRAF Inhibition with PX-866 and Vemurafenib in Patients with Advanced BRAF V600-Mutant Solid Tumors. Clin Cancer Res 2018;24:22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de la Cruz-Merino L, Di Guardo L, Grob JJ, Venosa A, Larkin J, McArthur GA, et al. Clinical features of serous retinopathy observed with cobimetinib in patients with BRAF-mutated melanoma treated in the randomized coBRIM study. J Transl Med 2017;15:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bendell JC, Varghese AM, Hyman DM, Bauer TM, Pant S, Callies S, et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res 2018;24:3253–62 [DOI] [PubMed] [Google Scholar]
- 60.Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang H, Wang P, et al. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin Cancer Res 2016;22:122–33 [DOI] [PubMed] [Google Scholar]
- 61.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, et al. Receptor-specific regulation of phosphatidylinositol 3'-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 2002;22:4062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou ZW, Ambrogio C, Bera AK, Li Q, Li XX, Li L, et al. KRAS(Q61H) Preferentially Signals through MAPK in a RAF Dimer-Dependent Manner in Non-Small Cell Lung Cancer. Cancer Res 2020;80:3719–31 [DOI] [PubMed] [Google Scholar]
- 63.Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res 2014;27:590–600 [DOI] [PubMed] [Google Scholar]
- 64.Horn S, Leonardelli S, Sucker A, Schadendorf D, Griewank KG, Paschen A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J Natl Cancer Inst 2018;110:677–81 [DOI] [PubMed] [Google Scholar]
- 65.Gopalan PK, Villegas AG, Cao C, Pinder-Schenck M, Chiappori A, Hou W, et al. CDK4/6 inhibition stabilizes disease in patients with p16-null non-small cell lung cancer and is synergistic with mTOR inhibition. Oncotarget 2018;9:37352–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pandey K, An HJ, Kim SK, Lee SA, Kim S, Lim SM, et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int J Cancer 2019;145:1179–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.