

Abstract
Huntington’s disease (HD) is a neurodegenerative disorder characterized by accumulation of mutant huntingtin protein and significant loss of neurons in striatum and cortex. Along with motor difficulties, the HD patients also manifest anxiety and loss of cognition. Unfortunately, the clinically approved drugs only offer symptomatic relief and are not free from side effects. This study underlines the importance of glyceryl tribenzoate (GTB), an FDA-approved food flavoring ingredient, in alleviating HD pathology in transgenic N171–82Q mouse model. Oral administration of GTB significantly reduced mutant huntingtin level in striatum, motor cortex as well as hippocampus and increased the integrity of viable neurons. Furthermore, we found the presence of sodium benzoate (NaB), a FDA-approved drug for urea cycle disorders and glycine encephalopathy, in the brain of GTB-fed HD mice. Accordingly, NaB administration also markedly decreased huntingtin level in striatum and cortex. Glial activation is found to coincide with neuronal death in affected regions of HD brains. Interestingly, both GTB and NaB treatment suppressed activation of glial cells and inflammation in the brain. Finally, neuroprotective effect of GTB and NaB resulted in improved motor performance of HD mice. Collectively, these results suggest that GTB and NaB may be repurposed for HD.
Keywords: Huntingtin, Inflammation, Gliosis, Glyceryl tribenzoate, Gait analysis
1. Introduction
Huntington’s disease (HD) is an extremely severe neurodegenerative disease caused by autosomal dominant mutation of the gene huntingtin (Htt). Clinically, the disease is manifested by progressive movement deficits, loss of cognition and psychiatric problems, which are basically caused by brain atrophy and loss of neurons in striatum and cerebral cortex (Bates et al., 2015; Beighton and Hayden, 1981). The prevalence of the disease is almost 5.5 per 100,000 individuals worldwide (Baig et al., 2016). The average age of onset of HD is around 30 years and patients generally die within 5–20 years from the time of disease manifestation. HD is also known as one of the polyglutamine disorders in which the protein huntingtin is found to contain several glutamine residues at the N-terminal of its structure (CAG repeats in the gene) (1993). Higher content of glutamine residues induces structural changes in the protein (mHtt), where the β-sheet secondary form predominates and that favors aggregation of mHtt in the neurons. In addition, mHtt is often cleaved by other proteases or partially transcribed by splicing errors to form N-terminal truncated form of the protein, which is even more potent in forming inclusion bodies in cells (Landles et al., 2010; Rubinsztein et al., 1999).
There are several cellular events which are found to be hampered by the presence of mHtt such as deficient ubiquitin-proteasomal system (Goswami et al., 2006), mitochondrial energy metabolism (Pandey et al., 2008), oxidative stress (La Fontaine et al., 2000), reduced axonal transport (Gunawardena and Goldstein, 2005), and also transcriptional dysregulation (Valor, 2015). Most importantly, inflammation is also unequivocally found in affected brain regions of HD and is assumed to be a major factor behind disease progression (Chakraborty et al., 2014; Valadao et al., 2020). Studies have shown that microglia of human patients express mHtt and abnormal microglial activation correlates well with disease severity (Tai et al., 2007). The protein mHtt influences microglia to secrete inflammatory cytokines like IL-6, IL-8 and TNF-α that can further promote astrocyte activation towards the A1 phenotype in the brain (Bjorkqvist et al., 2008; Lopez-Sanchez et al., 2020). In experimental studies performed in rodents, glial activation has been demonstrated even prior to the onset of neuronal death (Crotti and Glass, 2015; Sapp et al., 2001). Neuronal death induced by the toxic proteins can also further promote glial activation making the vicious cycle to continue in an irreversible manner (Kaushik and Basu, 2013). Accordingly, higher inflammatory cytokine profile is also observed in HD patients. Together these findings clearly suggest the involvement of glial activation and inflammation in HD pathogenesis and indicate that agents capable of inhibiting inflammation might offer neuroprotection in HD.
Glycerol tribenzoate (GTB) or tribenzoin belongs to the family of benzoic acid, which is used as a flavoring ingredient and a food preservative. Interestingly, this food additive was previously shown to inhibit inflammation and prevent demyelination in the spinal cord of mice with experimental allergic encephalomyelitis (EAE), an animal model for multiple sclerosis (Mondal et al., 2017). Therefore, in the present investigation, we evaluated the effect of GTB on inflammation and overall mHtt-induced pathology in a mouse model of HD. Here, we demonstrated that oral administration of GTB markedly lowered mHtt level, prevented brain atrophy and restored neuronal integrity in two major affected brain regions including striatum and motor cortex of transgenic N171–82Q mice. Moreover, GTB exerted the neuroprotective effect via formation of sodium benzoate (NaB) in HD animals. Reduction of mHtt level accompanied with significant inhibition of NF-κB activation, gliosis and inflammation in brain. These results indicate possible therapeutic importance of GTB and NaB in HD.
2. Materials and Methods
2.1. Reagents
GTB was purchased from Spectrum (New Brunswick, NJ) and NaB was procured from Sigma-Aldrich (St. Louis, MO). Crystal violet stain was bought from Sigma-Aldrich. Huntingtin and Iba1antibodieswere purchased from Abcam, (Cambridge, MA), GFAP antibody was procured from DAKO, whereas iNOS antibody was purchased from BD Bioscience (San Jose, CA). Details about the antibodies are mentioned in Supplementary Table 1.
2.2. Animals
Adult N171–82Q mice (B6C3-Tg(HD82Gln)81Gschi/J) were purchased from Jackson Laboratories. Experimental mice were housed under standard conditions with access to food and water ad libitum. Male N171–82Q mice were bred with female non-transgenic (nTg) B6C3 mice. Mice positive for the mHtt gene were selected by genotyping. Animal maintenance and experiments were performed in accordance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use committee of the Rush University Medical Center (Chicago, IL).
2.3. GTB and NaB treatment
GTB and NaB were solubilized in 0.1% methyl cellulose solution. NaB is completely soluble in aqueous solution, whereas GTB remains as colloidal particle. Transgenic (Tg) mice (3 months old) were treated with GTB or NaB at a dose of 50 mg/kg/day via gavage. Similarly, one group of Tg mice were also fed with vehicle, methyl cellulose (0.1%). Each mouse was fed with 100 µl of either GTB or NaB solution by gavage daily for the next 45 days. In general, any animal experiment is justified with 99% confidence interval that generates p = 0.99 and (1-p) = (1–0.99) =0.01; ε is the margin of error = 0.05. Based on these value, the resultant sample size is: Therefore, in most cases, six mice (n=6) were used in each group.
2.4. Open field test
Open field test was performed to monitor the locomotor abilities of the animals on a horizontal plane. Movement associated parameters were captured with a camera linked to Noldus system and EthoVision XT software (Netherlands). The instrument records the overall movement abilities of the animals such as total distance moved, velocity, moving time, resting time, center time, and frequencies of movement. Before recording the movement, all experimental mice were placed inside the open field arena for 10 min daily for 2 consecutive days for training and recording baseline values. Next day, animals were given rest and the following day each mouse was gently placed in the middle of the open field arena. After releasing the animal, data acquisition was started by the software for the next 5 min (Dutta et al., 2019).
2.5. Rotarod
Animals were placed on the rotating road against the direction of rotation. The machine was set to run at a gradual increasing speed of 4–40 rpm. The time for spending on the rotating rod was recorded and the experiment was ended once the animal slips from the rod to the base of the instrument (Dutta et al., 2019).
2.6. Grip test
Grip test was performed to measure the muscle strength of fore and hind limbs of the animals. The test was conducted using a square platform made of metal wires. Mouse was placed on the middle of the metal platform and then the whole platform was reversed allowing the animal to hang from that platform by clasping. Animals were initially trained for two days and then after a gap of one day the experiment was performed. Time taken by each mouse to fall from the metal platform was recorded (Castro and Kuang, 2017).
2.7. Gait analyses
Mice were acclimatized by making them walk on a slanting platform for consecutive two days. Each mouse was given five trials each day to walk on the platform to the ascending direction. After a gap of one day, the experiment was performed. The gangway was covered with a long white paper and the limbs of the animals were painted with non-toxic black colored ink to get the impression of the footprints of each animal. Following the experiment, based on the footprints different gait parameters such as stride length, stride width, foot length and toe spread were measured. If any animal stopped or started walking in reverse direction, experiment for that animal was repeated (Chakraborty et al., 2014).
2.8. Western blotting
Striatum and motor cortex region was isolated from mouse brain and homogenized in RIPA buffer containing 50 mM Tris-HCl, 1 mM EDTA sodium salt, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and protease inhibitor cocktail. Tissue homogenate was centrifuged at 17500xg for 10 min at 4°C, the resulting supernatant was collected, protein concentration was measured by BCA method and samples for Western blotting were prepared. Protein samples were run in 8% or 10% SDS-PAGE followed by transfer to the nitrocellulose membrane. The membrane was probed with primary antibodies overnight at 4°C. Next day, infrared fluorophore-tagged secondary antibodies (1:10,000; Jackson Immuno-Research) were added. Blots were scanned with an Odyssey infrared scanner (Li-COR, Lincoln, NE). Band intensities were quantified using ImageJ software (NIH, USA) (Dutta et al., 2018).
2.9. Immunostaining
Immunohistochemistry was performed as described earlier (Raha et al., 2020; Dutta et al., 2019). Mice were perfused transcardially with 4% paraformaldehyde and the brains kept in 30% sucrose solution at 4°C. Coronal sections (30 μm thickness) were cut from the forebrain containing striatum and motor cortex. Sections were blocked with 3% normal horse serum and 2% BSA made in PBST containing 0.5% Triton X-100 (Sigma-Aldrich) for 1 h. Then the sections were kept in primary antibodies and incubated at 4°C temperature overnight under shaking conditions. Next day, the samples were washed with PBST for at least three times, 10 min each, and further incubated with Cy2- or Cy5-labeled secondary antibodies (all 1:500; Jackson Immuno-Research) for 1 h under similar shaking conditions. Following several washes with PBST, sections were incubated for 5 min with 4’,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma-Aldrich) for immunofluorescence. Whereas for immunohistochemistry, samples were kept in solution containing biotin-tagged secondary antibodies for 1 h followed by incubation in Vectastain A and B (Jackson Immuno-Research) mixture solution at RT. Sections were developed by 3,3’-diaminodenzidine (DAB; Sigma-Aldrich) solution containing peroxide. The sections were run in an ethanol and xylene (Fisher) gradient, mounted, and observed under confocal microscope (Zeiss). Mean fluorescence intensity (MFI) was measured using ImageJ and relative optical density of mHtt staining was conducted using Fiji (ImageJ2) (Dutta et al., 2019; Raha et al., 2020).
2.10. Nissl staining
Nissl staining was performed on coronal brain sections of 30 μm width. Staining was carried out in 0.1% cresyl violet solution and the sections were submerged in the solution for 30–60 s followed by dehydration in different percentages of ethanol (30% to 100%). Finally, sections were put in xylene for 1 min. Image of stained cells in striatum and cortex was captured under bright field microscope. Optical density of Nissl staining was conducted using Fiji (ImageJ2) and the mean O.D. values obtained from cells in each section were compared among groups. Striatal area of brain sections were measured following Nissl staining by using ImageJ. Striatum region from each section was marked manually using the freehand selection method and the area was obtained by Analyze-Measure option provided in the software. The mean area of each group was compared for the analysis.
2.11. HPLC
After 45 d of either vehicle or GTB (50 mg/kg body wt/d) feeding via gavage, mice were sacrificed followed by isolation of cortical tissues from brain. Brain lysates were analyzed for the detection of NaB in Waters 2695 separation module HPLC system with the help of “Em-power pro” software and Phenomenex Luna 5μm C18 100A column (250 × 4.6 mm; 280 nm UV wavelength). The mobile phase consisted of phase A (acetonitrile with 0.01M H3PO4) and phase B (H2O with 0.01M H3PO4) (1:4) at the flow rate of 0.5 ml/min. After GTB feeding, cortical tissue collected in chloroform: methanol (2:1) extraction solvent containing 0.05M perchloric acid. Aspirin was added as an internal standard. Tissues were homogenized and centrifuged at 20,000xg for 10 min. Organic and aqueous phases were separated carefully and 10 μl aqueous phase was analyzed for the detection of NaB by HPLC (Jana et al., 2013).
2.12. Statistical Analyses
Statistics were performed using GraphPad Prism v7.0. One-way ANOVA followed by Tukey’s multiple comparison test was performed for analyzing statistical significance among multiple samples, whereas unpaired two tailed t-test was performed to compare two samples. Values are expressed as mean ± S.E.M. The criterion for statistical significance was p< 0.05.
3. Results
3.1. GTB treatment lowered mHtt level in striatum, motor cortex and hippocampus.
The N171–82Q transgenic mouse, which expresses mHtt protein having 82 glutamine repeats at the N-terminal (Masuda et al., 2008), starts developing protein aggregates and behavioral symptoms when animals are approaching 4 months of age and generally die around 5–6 months of their lifespan. Therefore, in this study, we started GTB administration in the animals when the animals were 3 months old and start showing certain HD symptoms such as tremor of limbs. In our prior study involving EAE mice, we have shown that GTB at a dose of 50 mg/kg/d decreases clinical symptoms and inhibits inflammation and demyelination in the CNS(Mondal et al., 2017). Accordingly, the dose of GTB was kept at 50 mg/kg/d for 45 days and then mHtt pathology and other biochemical assays were conducted from brain tissues. The results demonstrated that Tg mice have pronounced mHtt inclusions in neurons of striatum and motor cortex as revealed by immunostaining (Fig. 1A-D). Optical density (O.D.) analysis showed almost 6–8 fold increase in mHtt level in vehicle-fed Tg mice brain as compared to nTg mice (Fig. 1B, D). We also found significant up-regulation of mHtt in the hippocampus of Tg brains (Supplementary Fig. 1A, B). Interestingly, neuronal mHtt aggregation remarkably decreased in Tg mice after GTB administration in all the brain regions analyzed (Fig. 1A-D and Supplementary Fig. 1A, B).
Fig. 1. GTB reduces mutant huntingtin (mHtt) level in brain and retains neuronal integrity.
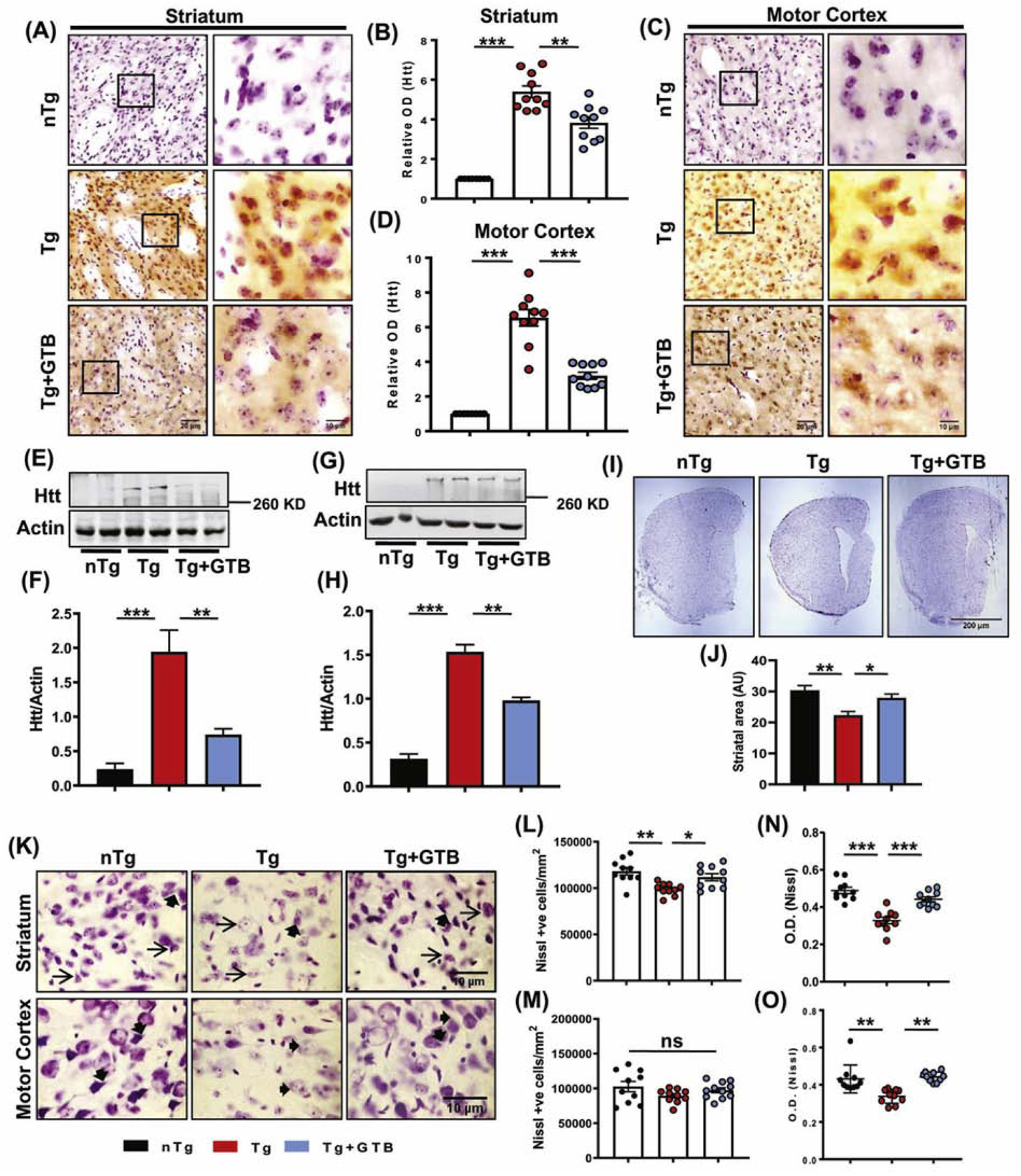
N171–82Q transgenic Huntington’s disease (HD) mice of 3 months old were administered with 50 mg/kg GTB daily for 45 days and mHtt level was monitored in striatum and motor cortex by immunohistochemistry (A, C). Optical density (O.D.) of mHtt was measured using Fiji and the relative fold change with respect to non-transgenic (nTg) control is expressed (B, D). Scale bars for lower (20X) and higher (60X) magnification images were kept as 20 µm and 10 µm respectively. Whole tissue expression of mHtt was assessed by immunoblotting, where expression of mHtt is shown relative to the loading control Actin (E-H). Mouse brain sections corresponding to mediolateral striatum region were processed for Nissl staining and the striatal area was measured using ImageJ and the average area is shown as arbitrary units (AU). Two brain sections from each brain were considered for the analysis (I, J). Nissl positive cells in striatum and cortex was counted from at least three fields (60X magnification) of each section and two sections from each brain were included for the counting analysis. The number of cells is expressed as number/mm2 of tissue (K-M). The mean value of Nissl staining obtained from neurons of each section of mice brains was calculated. Darker staining means lower is the mean value. The mean value of white is the highest (255), and therefore the formula used for calculating O.D. is log10 (255/mean of each cell). For staining purpose, at least two sections from each brain were used (N, O). One-way ANOVA followed by Tukey’s multiple comparison tests was performed for statistical analyses. *p<0.05, **p<0.01 and ***p<0.001 compared to respective groups. Data are represented as mean ± SEM (n=4 for immunoblotting and n=5 for staining).
Furthermore, to convincingly demonstrate the effect of GTB on mHtt down-regulation, we also performed mHtt immunostaining with the EM48 antibody, which is well-known to detect Htt inclusion bodies. Unlike nTg brains, we found clear presence of mHtt puncta containing cells in Tg brains. Interestingly, the number of mHtt puncta containing cells was greatly reduced in striatum and cortex of GTB-treated mice (Supplementary Fig. 1C-E). The data obtained from histochemical analyses were also supported by immunoblotting, which exhibited significant decrease in the pathogenic aggregated mHtt level corresponding to molecular weight of 348 KD in striatum (Fig. 1E-F & Supplementary Fig. 2A-B) as well as in cortex (Fig. 1G-H and Supplementary Fig. 2C-D) of GTB-treated brains. Next, we addressed other important pathological features of HD brain including brain atrophy and loss of neuronal number or integrity by Nissl staining of coronal sections obtained from mediolateral striatum region of brain. As expected, Tg brains showed loss of striatal area compared to the age-matched nTg brains, whereas following GTB administration loss of striatal area was significantly attenuated (Fig. 1I-J). The findings on brain atrophy led us to investigate the number of viable cells in striatum and motor cortex of experimental mice. Interestingly, we found significant loss of neurons only in striatum, but not in the cortical region of Tg brains, suggesting that at the experimental time point the effect of mHtt aggregation in striatum is more severe than motor cortex (Figure 1K-M). However, when we analyzed the neuronal integrity in both these regions of the experimental animals, we found presence of normal neuronal pyramidal morphology of striatal neurons (shown with arrows) as well as extensive neuropils (shown in arrow heads) in both striatum and cortex in nTg animals (Fig. 1K). In contrast, the shape and projections were found to be deformed and the Nissl staining intensity was greatly reduced in majority of the striatal and cortical neurons in Tg mice. Interestingly, GTB treatment prevented loss of neuronal integrity in Tg mice as the data showed proper neuronal shape, presence of neuropils and increased staining intensity after GTB administration (Fig. 1K, N, O). Altogether, the findings clearly demonstrate the neuroprotective effect of GTB against mHtt-induced pathology.
3.2. Oral GTB improved motor behavior of HD animals.
The neuroprotective effect of GTB against HD-associated brain pathology necessitated evaluation of the behavioral parameters of the experimental mice. In the present investigation, for the biochemical analyses we have focused on striatum and motor cortex, the regions that are known to regulate movement and motor coordination. Therefore, motor behavioral performance of the animals was analyzed in the present study. As expected, vehicle-fed Tg mice showed extremely poor performance in the open field test, and the movement parameters such as velocity of movement (Fig. 2A, B), distance moved (Fig. 2C), moving frequency (Fig. 2D), cumulative duration of moving (Fig. 2E) in the arena were significantly less than the nTg mice. In addition, the lower latency time taken by Tg animals in rotarod (Fig. 2F) and grip test (Fig. 2G) indicates that coordination of feet movement as well as muscle strength are hugely impaired. In contrast, Tg mice administered with GTB exhibited significantly improved performance in all the behavioral experiments and this is evident by higher velocity, distance moved and moving abilities in the open field arena and also by higher latency time taken by these animals in rotarod and grip tests. To look into the motor coordination in greater details, we also conducted footprint analysis for measuring gait. While the stride length of Tg animals was significantly lower than the nTg animals (Fig. 2H, I), the toe spread increased when affected by HD pathology (Fig. 2H, J). It indicates that Tg animals, due to the compromised movement ability, take longer time to cross the gangway. Interestingly, after GTB treatment both stride length and toe spread of Tg animals turn more towards the value of nTg animals showing improvement in motor skills. We also measured stride width and foot length of the experimental animals, but these values were not found to be significantly altered in Tg mice (data not shown).
Fig. 2. GTB attenuates motor behavioral impairment of HD mice.
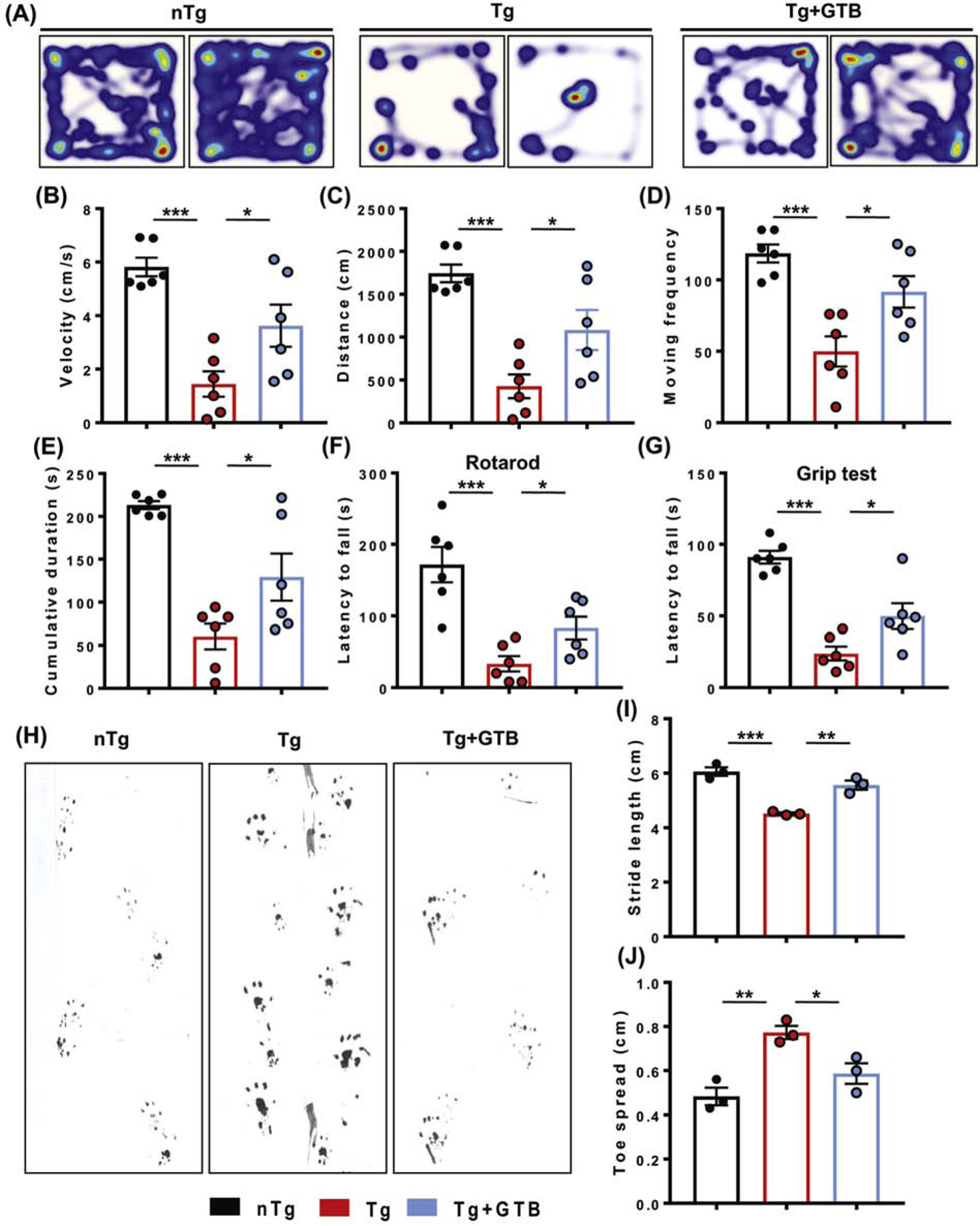
Tg mice were administered with vehicle or GTB at 50 mg/kg/d for 30 days and motor behavioral performance was analyzed by open field test (A) to obtain movement parameters including velocity (B), distance moved (C), moving frequency (D), cumulative duration of moving (E). Feet movement of animals was evaluated by rotarod test (F), whereas muscle strength was assessed by grip test (G). Motor coordination of experimental animals was evaluated by gait analysis and the footprints of animals on the gangway are shown (H). Stride length (I) and toe spread (J) of mice obtained from the gait analysis are calculated manually. One-way ANOVA followed by Tukey’s multiple comparison test was performed for statistical analyses. *p< 0.05, **p< 0.01 and ***p< 0.001 indicate statistical significance compared to respective groups. Data are represented as mean ± SEM (n=6 for open field, rotarod and grip test and n=3 for gait analysis).
3.3. Oral administration of GTB produced NaB in the cortex of HD mice.
GTB is a benzoic acid ester, which might be metabolized to benzoate in the body. Therefore, to examine whether NaB is present in the brain after oral GTB treatment, we conducted HPLC analysis from cortical tissue homogenates of both vehicle fed and GTB fed animals. Data showed the presence of a sharp NaB peak in the tissue fraction isolated from GTB fed animals, which was absent in vehicle treated mice (Supplementary Fig. 3B-C). In this experiment, we used aspirin as an internal control for all the analyzed samples. The retention times of aspirin and NaB obtained from experimental brain tissue samples were matched with the standards (Supplementary Fig. 3A). Overall, it clearly shows the presence of NaB, which is a FDA-approved drug for urea cycle disorders and glycine encephalopathy, in the brain of GTB treated HD mice.
3.4. Oral GTB prevents activation of microglia, astroglia and neuroinflammation in HD mice.
Our results clearly demonstrate the presence of NaB in the brain following GTB administration. As NaB was previously shown to inhibit glial activation and inflammation in microglia and astrocytes (Brahmachari et al., 2009), in the next segment of the study, we sought to investigate the effect of GTB on the suppression of gliosis and inflammation. Immunostaining of microglia specific marker, ionized calcium binding adaptor molecule 1 (Iba1), exhibited that the Tg mice had higher number of microglia in both striatum (Fig. 3A, E) and cortex (Fig. 3B, G) compared to the nTg mice. Moreover, the expression of nitrosative stress marker inducible nitric oxide synthase (iNOS) was up-regulated significantly in Iba1+ve microglia in striatum (Fig. 3C, F) and cortex (Fig. 3D, H) of Tg mice. The enhanced protein expression of Iba1 and iNOS was further confirmed by immunoblot analysis from these brain tissues (Fig. 3I-N). Interestingly, microglial number and expression of inflammatory markers greatly decreased after GTB administration in Tg mice suggesting that GTB profoundly exerts anti-inflammatory effect in HD brains. Along with microglial activation, astrogliosis was also reported to be prevalent in HD brains (Lopez-Sanchez et al., 2020) and in the present study, we also found up-regulation of astrogliosis in striatum and cortex of Tg mice as evidenced by immunostaining of astrocytes by its marker glial fibrillary acidic protein (GFAP) (Fig. 4A, B and Fig. 4F, G). Similarly, total GFAP expression was also increased in these brain regions (Fig. 4D, E and Fig. 4I, J). Furthermore, astroglial inflammation is substantiated by the elevated expression and colocalization of iNOS in the activated astrocytes of Tg brains (Fig. 4C, H). Interestingly, parallel to the inhibition of microglial activation, GTB treatment significantly reduced the number of GFAP positive cells in striatum and cortex and iNOS expression in astrocytes.
Fig. 3. GTB inhibits microglial activation and inflammation in striatum and cortex of HD animals.
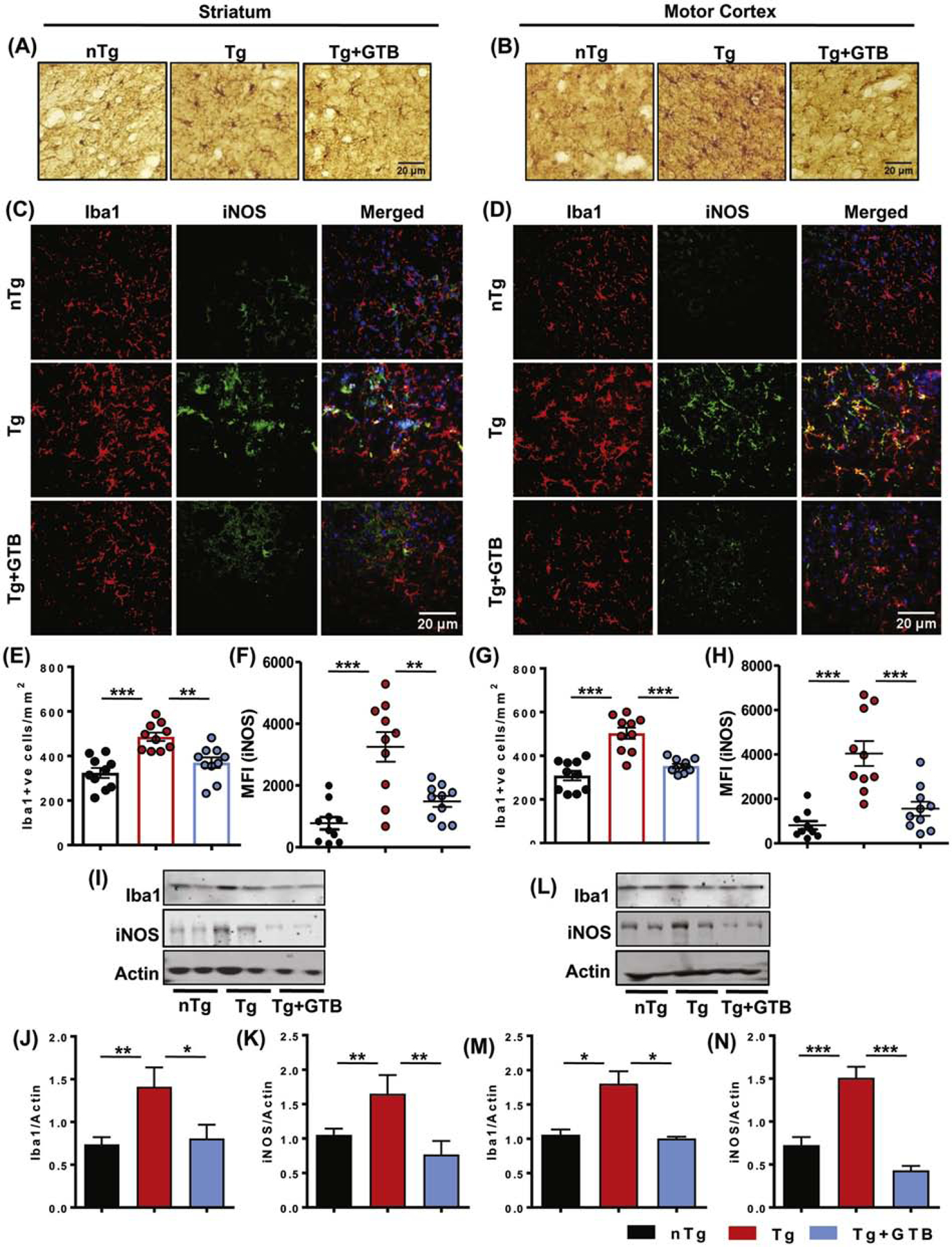
Microglial activation in nTg and different groups of Tg animals was evaluated by microglial marker Iba1 staining in brain sections (A, B). Counting of Iba1+ve microglia was performed in striatum (E) and cortex region (G) of brain and demonstrated as number of microglia/mm2 of region. Microglial inflammation was assessed by immunofluorescence colocalization analysis of nitrosative stress marker iNOS in Iba1+ve cells in both striatum (C) and cortex (D). Mean fluorescent intensity of iNOS in microglia was measured using Image J (F, H). Two sections from each brain were analyzed for counting and MFI analysis. Protein expression of Iba1 and iNOS from striatum and cortex was assessed by immunoblotting (I, L) and the ratio of band intensities of Iba1 (J, M) and iNOS (K, N) was calculated with respect to actin. One-way ANOVA followed by Tukey’s multiple comparison tests was performed for statistical analyses. *p< 0.05, **p< 0.01 and ***p< 0.001 indicate statistical significance compared to respective groups. Results are represented as mean ± SEM (n=5 for immunostaining and n=4 for immunoblotting).
Fig. 4. GTB inhibits astrogliosis and inflammation in striatum and cortex of HD animals.
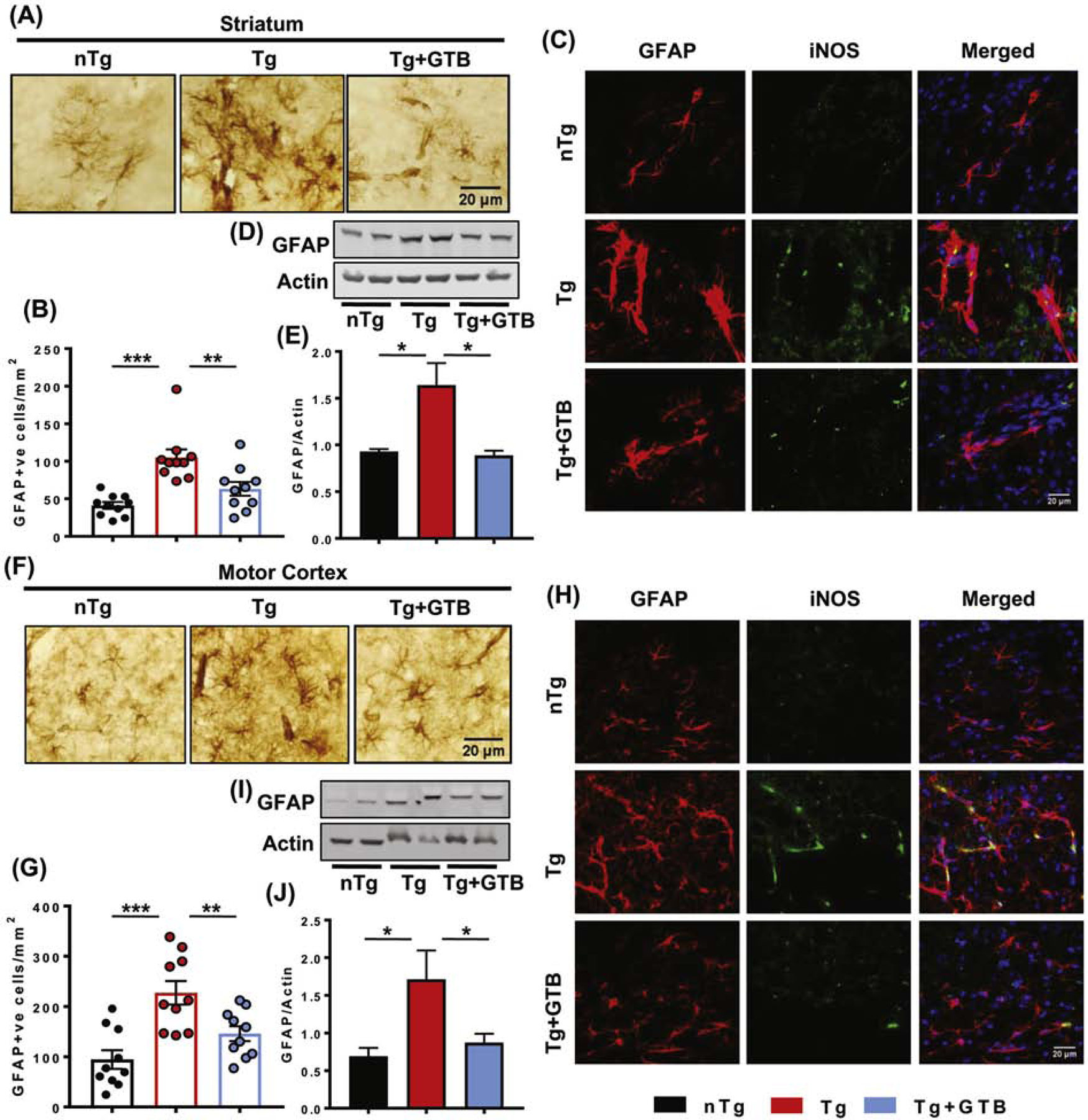
Astrogliosis in the brain of animals was evaluated by immunostaining of astroglial marker GFAP followed by counting of GFAP positive cells in striatum (A, B) and motor cortex (F, G) of nTg and all groups of Tg animals and represented as number of GFAP positive cells/mm2 of tissue. Two sections from each brain were analyzed for counting. Protein expression of GFAP in striatal and cortical tissues was monitored by immunoblotting (D, I) and GFAP expression was shown with relative to the loading control actin (E, J). Induction of astroglial inflammation was shown by immunofluorescence of iNOS in GFAP positive astrocytes in striatum (C) and cortex (H). One-way ANOVA followed by Tukey’s multiple comparison tests was performed for statistical analyses. *p< 0.05, **p< 0.01 and ***p< 0.001 indicate statistical significance compared to respective groups. Results are represented as mean ± SEM (n=5 for immunostaining and n=4 for immunoblotting).
3.5. Oral NaB treatment reduces mHtt level and improves motor skills of HD mice.
To further prove the involvement of NaB in GTB-mediated reduction of HD pathology, we treated Tg mice with NaB via gavage. The results demonstrated significant down-regulation of mHtt accumulation in striatal (Fig. 5A, B) and cortical neurons (Fig. 5C, D) upon NaB treatment. Furthermore, immunoblotting exhibited lower level of toxic mHtt in these tissue homogenates of NaB treated group compared to the vehicle treated Tg mice (Fig. 5E-H & Supplementary Fig. 2E-G). The findings firmly indicate that NaB, the metabolite of GTB, can alone ameliorate mHtt level in brain. We also evaluated the effect of NaB on improving motor behavior of the Tg mice. As expected, Tg mice demonstrated severe impairment in open field test as shown by reduction in movement parameters such as distance covered (Fig. 5I, J), velocity (Fig. 5K), cumulative duration of moving (Fig. 5L) and central frequency (Fig. 5M). In addition, the Tg mice also exhibited poor performance in rotarod (Fig. 5N) and grip test (Fig. 5O), affirming the compromised motor coordination and muscle strength experienced by these animals. However, NaB treatment reversed the poor motor performance of the Tg animals and improved locomotor activities in open field arena, rotarod and grip tests. Based on these data, it can be surmised that reduction of HD pathology positively correlates with the improved motor function in NaB-treated Tg animals.
Fig. 5. NaB treatment reduces mHtt level in brain and ameliorates behavioral impairments in HD mice.
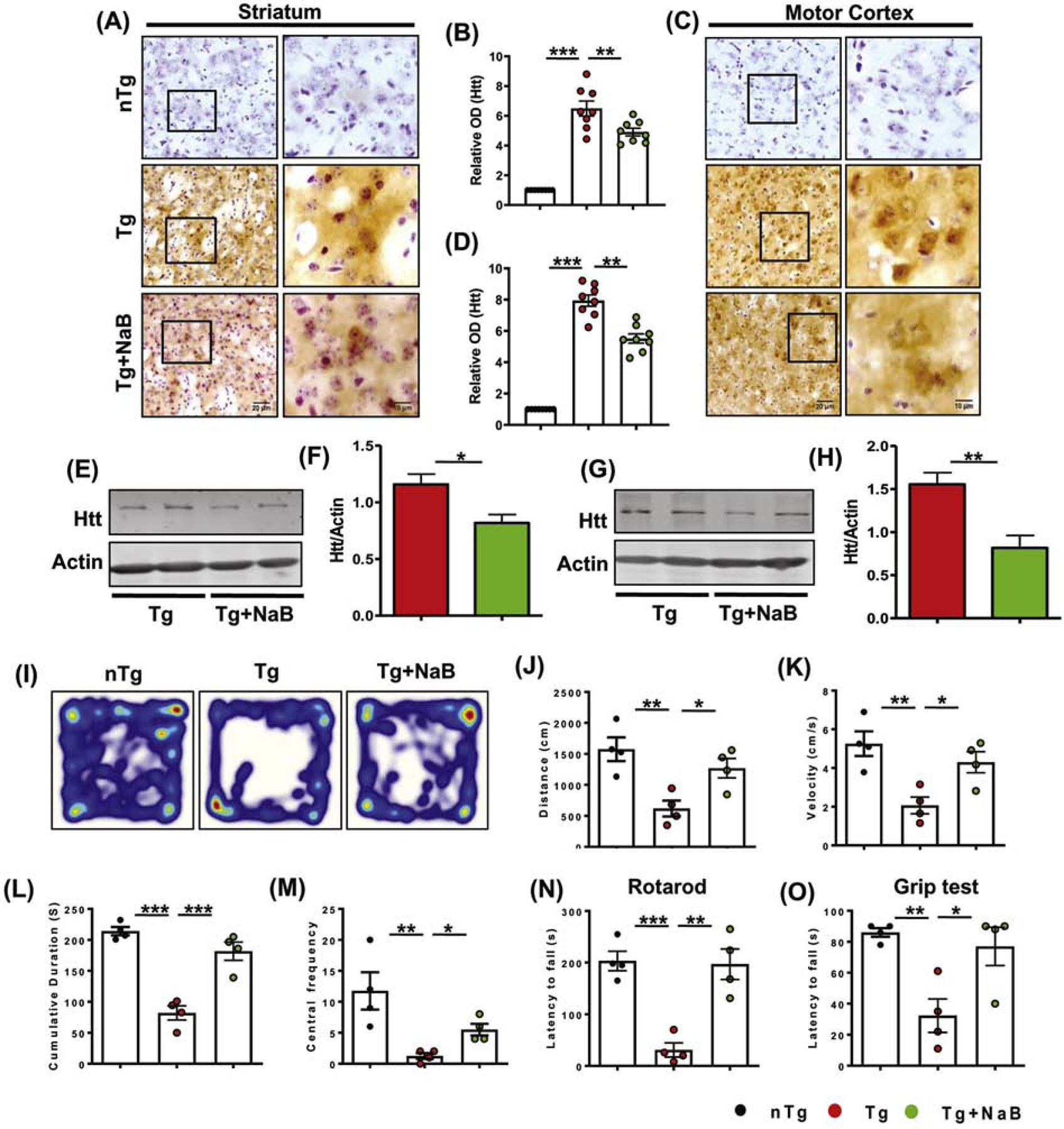
N171–82Q Tg mice of 3 months old were administered with 50 mg/kg sodium benzoate (NaB) daily for the next 45 days and mHtt level was monitored in striatum and motor cortex by immunohistochemistry (A, C). O.D. of mHtt was measured using Fiji and the relative fold change with respect to nTg control is expressed (B, D). Scale bars for lower (20X) and higher (60X) magnification images were kept as 20 µm and 10 µm respectively. Sections were counterstained with cresyl violet and two sections from each brain were used for immunostaining. Protein expression of mHtt was monitored from striatal and cortical tissues by immunoblotting (E, F) and the mHtt band intensity is presented with respect to that of actin (G, H). Locomotor abilities of animals were analyzed by open field test (I) and the behavioral parameters such as distance moved (J), velocity (K), cumulative duration of moving (L) and central frequency (M) are shown. Motor coordination and muscle strength of animals were performed by rotarod test (N) and grip test (O) respectively. Unpaired two-tailed t-test was performed for statistical analysis between two samples, whereas one-way ANOVA followed by Tukey’s multiple comparison tests was performed for analyzing multiple samples. *p< 0.05, **p< 0.01 and ***p< 0.001 indicate statistical significance compared to respective groups. Results are represented as mean ± SEM (n=4).
3.6. Oral NaB inhibits microglial activation and inflammation in HD mice.
After establishing the neuroprotective effect of NaB, it was necessary to examine the level of inflammation in the brain of NaB-treated Tg mice. To evaluate inflammation in brain, we carried out immunofluorescence analysis of microglial marker Iba1 and nitrosative marker iNOS. The results showed sustained increase in microglia number and microglial iNOS expression in striatum (Fig. 6A, B) and cortex (Fig. 6F, G) of Tg brains. Similar to earlier results, immunoblotting also confirmed the up-regulation of Iba1 and iNOS in Tg mice compared to the nTg mice (Fig. 6C-E and Fig. 6H-J). However, oral administration of NaB remarkably reduced the level of Iba1 and iNOS in striatum and cortex and also decreased iNOS expression in microglia. These findings clearly exhibited anti-inflammatory effect of NaB in the context of experimental HD.
Fig. 6. NaB inhibits microglial inflammation in HD mice.
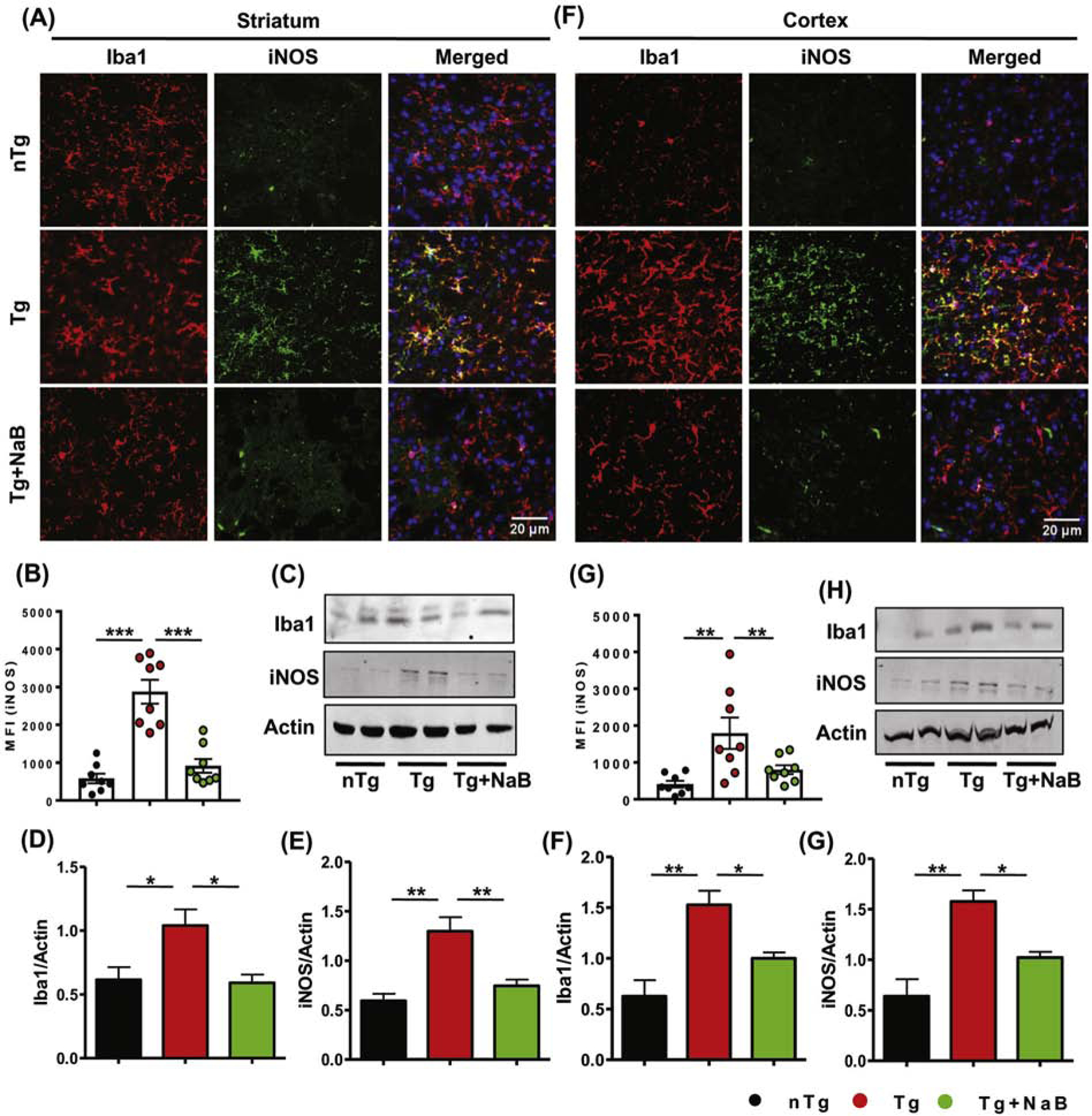
Microglial activation and inflammation in vehicle and NaB-treated HD brains was evaluated by immunofluorescence staining of Iba1 and iNOS in striatal (A) and cortical (F) sections. MFI of iNOS expression in microglia was measured by ImageJ (B, G), where two sections from each brain were used for immunostaining. Expression of Iba1 and iNOS in striatum (C) and cortex (H) was monitored by immunoblotting analysis. Band intensity of each protein is shown with respect to the loading control actin in both striatum (D, E) and cortex (I, J). One-way ANOVA followed by Tukey’s multiple comparison tests was performed for statistical analyses. *p< 0.05, **p< 0.01 and ***p< 0.001 indicate statistical significance compared to respective groups. Results are represented as mean ± SEM (n=4).
3.7. Oral GTB inhibits NF-κB activation in microglia in HD mice.
Since activation of NF-κB plays an important role in inflammation, for any drug, it is almost mandatory to suppress the activation of NF-κB to exhibit anti-inflammatory effect (Ghosh and Hayden, 2008; Kim et al., 2014). Therefore, we examined the effect of oral GTB on the activation of NF-κB in vivo in the brain of HD mice. Since the Ser536 phosphorylation of p65, the RelA subunit of NF-κB heterodimer, leads to nuclear translocation and induction of several inflammatory genes (Christian et al., 2016; Pahan et al., 1997; Saha and Pahan, 2006), we monitored the level of p-Ser536 p65 in the brain. The results demonstrated that the level of p-Ser536 p65 is significantly higher in microglia of both striatum (Fig. 7A, B) and cortex (Fig. 7C, D) of Tg mice than nTg animals. Interestingly, the level of p-Ser536 p65 in microglia was diminished markedly following GTB administration. These results made it clear that prevention of NF-κB activation by GTB underlies its anti-inflammatory effect in microglia.
Fig. 7. Oral GTB prevents microglial NF-κB activation in HD mice.
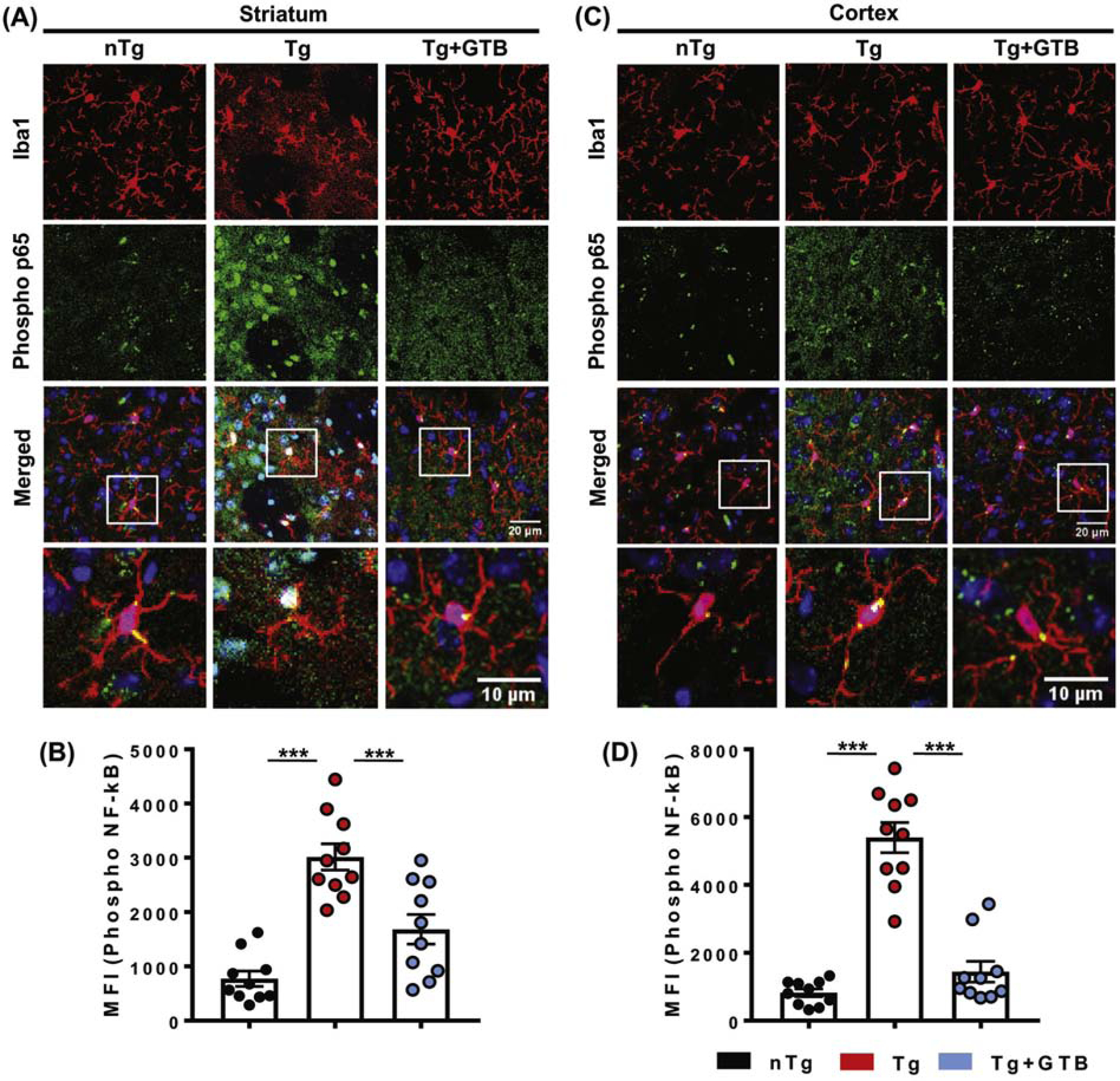
NF-κB activation in microglia of different groups of experimental mice was monitored by evaluating the expression of phospho-Ser 536 form of p65. Immunofluorescence analysis was performed to visualize the expression of phospho-Ser 536 p65 in Iba1 positive microglia in both striatum (A) and cortex (C). Images are shown in 20X magnification and further zoomed to shown individual microglia. Two sections from each brain were taken for the immunostaining and the MFI value of phospho-p65 obtained from each section is presented in the diagram. One-way ANOVA followed by Tukey’s multiple comparison tests was performed for statistical analyses. ***p< 0.001 indicates statistical significance compared to respective groups. Results are represented as mean ± SEM (n=5).
4. Discussion
To date, there is no available drugs to prevent the onset or slow the progression of HD pathology (Troncoso-Escudero et al., 2020). Current therapies provide only symptomatic relief to temporarily reduce chorea, anxiety and psychosis. Moreover, in long-term, the medications are associated with severe side effects. Therefore, the significance of the present study is established by the first evidence that oral administration of GTB, which is a non-toxic food preservative, remarkably alleviates HD associated pathologies in mice. The main highlights of our study are, first, GTB reduces pathological mHtt level and retains neuronal integrity in striatum and cortex, second, GTB mediates its neuroprotective activity via its metabolite NaB, an FDA-approved drug for urea cycle disorders and glycine encephalopathy or nonketotic hyperglycinemia, and therefore NaB administration mitigates mHtt level in brain, third, GTB is capable of significantly suppressing microglial NF-κB activation resulting in prevention of glial activation and inflammation, fourth, neuroprotective effect of both GTB and NaB attenuates behavioral impairment in HD mice and delay the onset of the disease.
The protein mHtt is known to impart cytotoxicity of specific subsets of neurons where brain regions like dorsal and medial striatum and cortex are found to be most vulnerable (Gil and Rego, 2008; Roos, 2010). This observation has been well manifested in human patients and also in multiple experimental animal models expressing different lengths of PolyQ in the mHtt protein (Aggarwal et al., 2012; Cirillo et al., 2019). Similar to other neurodegenerative diseases, glial activation (Crotti and Glass, 2015) and synthesis of inflammatory molecules coupled with the generation of oxidative or nitrosative stress is conspicuously found in affected regions of HD brains (Khoshnan et al., 2004; Pandey et al., 2009; Tai et al., 2007). Although from the mechanistic viewpoint, it is still unclear how microglial activation occurs in HD brain, experimental studies indicate that microglial activation can either be a consequence of mHtt-induced neuronal pathology or be a prerequisite for potentiating mHtt toxicity in brain. It suggests that targeting glial inflammation may be beneficial in reducing HD pathology.
Considering the point of exaggerated inflammation in HD pathogenesis, the present study focused on evaluation of therapeutic efficacy of GTB. We started our experiment with this molecule based on the earlier notion that GTB inhibits inflammation and stops demyelination in CNS of EAE mice (Mondal et al., 2017). In the HD model as well, the anti-inflammatory effect of GTB is further established as it prevents glial proliferation and expression of stress marker iNOS in microglia and astrocytes. Concomitant down-regulation of mHtt level in neurons and retention of neuronal integrity are also indicative of the beneficial effect of GTB against HD pathology. Since GTB contains three benzoic acid residues bound to the glycerol backbone by carboxyl groups, in the body, GTB can be readily hydrolyzed to form benzoic acid to be further converted into NaB. We found the abundance of NaB in the brain of GTB-fed animals, supporting our earlier report that NaB can cross the blood-brain-barrier (Jana et al., 2013). To prove whether neuroprotective effect of GTB is mediated by NaB, we further administered NaB in HD animals and demonstrated reduced pathology and significantly better behavioral performance of HD animals. Therefore, it clearly suggests that NaB is the principal component which helps reduce inflammation and protects neurons against mHtt toxicity.
It must be mentioned that NaB is a component of Ucephan, a FDA-approved drug used for the treatment for urea cycle disorders involving deficiencies of carbamoyl phosphate synthetase, ornithine transcarbamylase, or argininosuccinic acid synthetase (Bridges et al., 1970; Toth, 1984). Moreover, NaB is the only FDA-approved drug for glycine encephalopathy (Nihon-Yanagi et al., 2013). Interestingly, past several studies have also demonstrated the anti-inflammatory and neuroprotective function of NaB in major neurodegenerative diseases like Alzheimer’s disease (Modi et al., 2015), Parkinson’s disease (Khasnavis and Pahan, 2014; Raha et al., 2020) as well as in auto-immune diseases like MS (Brahmachari and Pahan, 2007). At the molecular level, NaB is known to suppress the activation of NF-κB via inhibition of p21ras (Brahmachari et al., 2009). As NF-κB is one of the essential factors required for the transcriptional activation of numerous inflammatory genes (Ghosh and Hayden, 2008), reduction of NF-κB activation firmly suggests the mechanism behind reduced expression of inflammatory molecules in GTB- and NaB-treated HD mice. Furthermore, decrease in iNOS expression in microglia indicates inhibition in NO synthesis, which is also known to stimulate microgliosis (Roy et al., 2006) and astrogliosis (Brahmachari et al., 2006; Kim et al., 2013). The anti-inflammatory effect of NaB is also reproduced in astroglia and this finding supports our previous report where we have shown that NaB inhibits IL-1β-induced production of NO and the expression of iNOS protein in primary human astroglia (Brahmachari et al., 2009). Collectively, the results of the present study firmly demonstrate the beneficial role of food additive GTB and a FDA-approved drug NaB in ameliorating pathological mHtt level and protecting neuronal integrity. Therefore, GTB and NaB may be repurposed for HD as primary and/or adjunct therapy to halt or delay the disease progression.
Supplementary Material
Acknowledgements
This study was supported by Curyx Bio and grants (AG050431 and NS108025) from NIH to KP. Moreover, KP is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs.
Abbreviations:
- GTB
Glyceryl tribenzoate
- NaB
Sodium benzoate
- iNOS
induced nitric oxide synthase
- Iba1
Ionized calcium binding adaptor molecule 1
- GFAP
Glial fibrillary acidic protein
- HD
Huntington’s disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–83. [DOI] [PubMed] [Google Scholar]
- Aggarwal M, et al. , 2012. Spatiotemporal mapping of brain atrophy in mouse models of Huntington’s disease using longitudinal in vivo magnetic resonance imaging. Neuroimage 60, 2086–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig SS, et al. , 2016. The global prevalence of Huntington’s disease: a systematic review and discussion. Neurodegener Dis Manag 6, 331–43. [DOI] [PubMed] [Google Scholar]
- Bates GP, et al. , 2015. Huntington disease. Nat Rev Dis Primers 1, 15005. [DOI] [PubMed] [Google Scholar]
- Beighton P, Hayden MR, 1981. Huntington’s chorea. S Afr Med J 59, 250. [PubMed] [Google Scholar]
- Bjorkqvist M, et al. , 2008. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 205, 1869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S, et al. , 2006. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci 26, 4930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S, et al. , 2009. Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J Immunol 183, 5917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S, Pahan K, 2007. Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol 179, 275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges JW, et al. , 1970. The fate of benzoic acid in various species. Biochem J 118, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro B, Kuang S, 2017. Evaluation of Muscle Performance in Mice by Treadmill Exhaustion Test and Whole-limb Grip Strength Assay. Bio Protoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty J, et al. , 2014. Quercetin improves behavioral deficiencies, restores astrocytes and microglia, and reduces serotonin metabolism in 3-nitropropionic acid-induced rat model of Huntington’s Disease. CNS Neurosci Ther 20, 10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian F, et al. , 2016. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo G, et al. , 2019. Selective Vulnerability of Basal Ganglia: Insights into the Mechanisms of Bilateral Striatal Necrosis. J Neuropathol Exp Neurol 78, 123–129. [DOI] [PubMed] [Google Scholar]
- Crotti A, Glass CK, 2015. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol 36, 364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, et al. , 2018. Low Levels of Prohibitin in Substantia Nigra Makes Dopaminergic Neurons Vulnerable in Parkinson’s Disease. Mol Neurobiol 55, 804–821. [DOI] [PubMed] [Google Scholar]
- Dutta D, et al. , 2019. RANTES-induced invasion of Th17 cells into substantia nigra potentiates dopaminergic cell loss in MPTP mouse model of Parkinson’s disease. Neurobiol Dis 132, 104575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS, 2008. New regulators of NF-kappaB in inflammation. Nat Rev Immunol 8, 837–48. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC, 2008. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci 27, 2803–20. [DOI] [PubMed] [Google Scholar]
- Goswami A, et al. , 2006. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem Biophys Res Commun. 342, 184–90. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS, 2005. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol 62, 46–51. [DOI] [PubMed] [Google Scholar]
- Jana A, et al. , 2013. Up-regulation of neurotrophic factors by cinnamon and its metabolite sodium benzoate: therapeutic implications for neurodegenerative disorders. J Neuroimmune Pharmacol 8, 739–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik DK, Basu A, 2013. A friend in need may not be a friend indeed: role of microglia in neurodegenerative diseases. CNS Neurol Disord Drug Targets 12, 726–40. [DOI] [PubMed] [Google Scholar]
- Khasnavis S, Pahan K, 2014. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9, 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnan A, et al. , 2004. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci 24, 7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. , 2013. Effects of polymorphisms of innate immunity genes and environmental factors on the risk of noncardia gastric cancer. Cancer Res Treat 45, 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, et al. , 2014. Rotundarpene inhibits toll-like receptor 2 activation-induced production of inflammatory mediators in keratinocytes by suppressing the Akt and NF-kappaB pathways. Int Immunopharmacol 18, 325–32. [DOI] [PubMed] [Google Scholar]
- La Fontaine MA, et al. , 2000. 3-nitropropionic acid induced in vivo protein oxidation in striatal and cortical synaptosomes: insights into Huntington’s disease. Brain Res 858, 356–62. [DOI] [PubMed] [Google Scholar]
- Landles C, et al. , 2010. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem 285, 8808–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez C, et al. , 2020. Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda N, et al. , 2008. Tiagabine is neuroprotective in the N171–82Q and R6/2 mouse models of Huntington’s disease. Neurobiol Dis 30, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi KK, et al. , 2015. Cinnamon and Its Metabolite Sodium Benzoate Attenuate the Activation of p21rac and Protect Memory and Learning in an Animal Model of Alzheimer’s Disease. PLoS One 10, e0130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, et al. , 2017. Glyceryl Tribenzoate: A Flavoring Ingredient, Inhibits the Adoptive Transfer of Experimental Allergic Encephalomyelitis via TGF-beta: Implications for Multiple Sclerosis Therapy. J Clin Cell Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihon-Yanagi Y, et al. , 2013. beta-2 microglobulin is unsuitable as an internal reference gene for the analysis of gene expression in human colorectal cancer. Biomed Rep 1, 193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, et al. , 1997. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest 100, 2671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey M, et al. , 2009. Striatal dopamine level contributes to hydroxyl radical generation and subsequent neurodegeneration in the striatum in 3-nitropropionic acid-induced Huntington’s disease in rats. Neurochem Int 55, 431–7. [DOI] [PubMed] [Google Scholar]
- Pandey M, et al. , 2008. Mitochondrial NAD+-linked State 3 respiration and complex-I activity are compromised in the cerebral cortex of 3-nitropropionic acid-induced rat model of Huntington’s disease. J Neurochem 104, 420–34. [DOI] [PubMed] [Google Scholar]
- Raha S, et al. , 2020. Reduction of Lewy Body Pathology by Oral Cinnamon. J Neuroimmune Pharmacol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos RA, 2010. Huntington’s disease: a clinical review. Orphanet J Rare Dis 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, et al. , 2006. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem 281, 14971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, et al. , 1999. Intracellular inclusions, pathological markers in diseases caused by expanded polyglutamine tracts? J Med Genet 36, 265–70. [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K, 2006. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal 8, 929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, et al. , 2001. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60, 161–72. [DOI] [PubMed] [Google Scholar]
- Tai YF, et al. , 2007. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 130, 1759–66. [DOI] [PubMed] [Google Scholar]
- Toth B, 1984. Lack of tumorigenicity of sodium benzoate in mice. Fundam Appl Toxicol 4, 494–6. [DOI] [PubMed] [Google Scholar]
- Troncoso-Escudero P, et al. , 2020. On the Right Track to Treat Movement Disorders: Promising Therapeutic Approaches for Parkinson’s and Huntington’s Disease. Front Aging Neurosci 12, 571185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadao PAC, et al. , 2020. Inflammation in Huntington’s disease: A few new twists on an old tale. J Neuroimmunol 348, 577380. [DOI] [PubMed] [Google Scholar]
- Valor LM, 2015. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: a genome-wide perspective. Mol Neurobiol 51, 406–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.